The application requires the priority of the U.S. Patent application 11/143,535 submitted on June 2nd, 2005, and described application has required the priority of the U.S. Patent application 60/576,939 submitted on June 4th, 2004, its two incorporated into this paper in full as a reference.
Summary of the invention
In one embodiment, the present invention relates to the method that may further comprise the steps:
A., composition is provided, and it comprises:
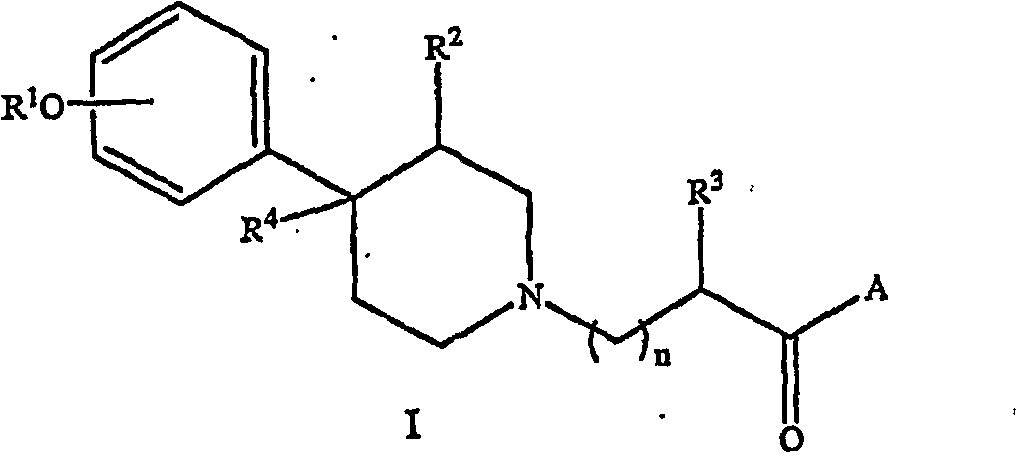
(i) the pharmaceutically acceptable slaine of at least a formula I compound:
Wherein:
R
1Be hydrogen or alkyl;
R
2Be hydrogen, alkyl or alkenyl;
R
3Be the alkyl of hydrogen, alkyl, thiazolinyl, aryl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
4Be hydrogen, alkyl or alkenyl;
A is OR
5Or NR
6R
7
R
5Be the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
6Be hydrogen or alkyl;
R
7The B that alkyl, aralkyl, aralkyl or the alkylidene that replaces for the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, aryl, cycloalkyl substituted, cycloalkenyl group, cycloalkenyl group replaces, or R
6And R
7Form heterocycle with the nitrogen-atoms that they connected;
B is
C (=O) W or NR
8R
9
R
8Be hydrogen or alkyl;
R
9The alkyl, aryl or the aralkyl that replace for the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl substituted, cycloalkyl, cycloalkenyl group, cycloalkenyl group, or R
8And R
9Form heterocycle with the nitrogen-atoms that they connected;
W is OR
10, NR
11R
12Or OE;
R
10Be the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
11Be hydrogen or alkyl;
R
12C (=O) the Y, or R that alkyl, aralkyl or the alkylidene that replaces for the alkyl of hydrogen, alkyl, thiazolinyl, aryl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, cycloalkenyl group replaces
11And R
12Form heterocycle with the nitrogen-atoms that they connected;
E is
(C=O) D that alkylidene replaces or-R
13OC (=O) R
14
R
13Alkylidene for the alkyl replacement;
R
14Be alkyl;
D is OR
15Or NR
16R
17
R
15Be the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
16Be the alkyl of hydrogen, alkyl, thiazolinyl, aryl, aralkyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted or the alkyl of cycloalkenyl group replacement;
R
17Be hydrogen or alkyl, or R
16And R
17Form heterocycle with the nitrogen-atoms that they connected;
Y is OR
18Or NR
19R
20
R
18Be the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
19Be hydrogen or alkyl;
R
20Be the alkyl of hydrogen, alkyl, thiazolinyl, aryl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces, or R
19And R
20Form heterocycle with the nitrogen-atoms that they connected;
R
21Be hydrogen or alkyl;
With n be 0 to 4;
The (ii) incremental agent of at least a crystallization;
(iii) at least a weak base; With
(iv) water;
Wherein said composition has the initial pH at least about 10.5; With
B. the pH that regulates described composition arrives the final pH that arrive in about 11 scopes about 9;
Wherein, to patient's administration the time, described composition has the dissolution rate and the bioavilability of the improvement that is used for oral or parenterai administration.
In another embodiment, the method that partly relates to of the present invention comprises described composition dries with the described water of removing at least a portion in addition to form the partially or completely step of dry products.
In another embodiment, the method that the present invention relates to comprises in addition by making it organize the step of structure again with the solution that forms described dry products described dry products and the merging of pharmaceutically useful solvent.
In other embodiments, the present invention relates to the product produced by each said method.
In another embodiment, the method that the present invention relates to comprises in addition with the described solution of the described dry products step to patient's administration.
In another embodiment, the present invention relates to composition, it comprises:
The pharmaceutically acceptable slaine of a. at least a formula I compound:
Wherein:
R
1Be hydrogen or alkyl;
R
2Be hydrogen, alkyl or alkenyl;
R
3Be the alkyl of hydrogen, alkyl, thiazolinyl, aryl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
4Be hydrogen, alkyl or alkenyl;
A is OR
5Or NR
6R
7
R
5Be the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
6Be hydrogen or alkyl;
R
7The B that alkyl, aralkyl, aralkyl or the alkylidene that replaces for the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, aryl, cycloalkyl substituted, cycloalkenyl group, cycloalkenyl group replaces, or R
6And R
7Form heterocycle with the nitrogen-atoms that they connected;
B is
C (=O) W or NR
8R
9
R
8Be hydrogen or alkyl;
R
9The alkyl, aryl or the aralkyl that replace for the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl substituted, cycloalkyl, cycloalkenyl group, cycloalkenyl group, or R
8And R
9Form heterocycle with the nitrogen-atoms that they connected;
W is OR
10, NR
11R
12Or OE;
R
10Be the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
11Be hydrogen or alkyl;
R
12C (=O) the Y, or R that alkyl, aralkyl or the alkylidene that replaces for the alkyl of hydrogen, alkyl, thiazolinyl, aryl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, cycloalkenyl group replaces
11And R
12Form heterocycle with the nitrogen-atoms that they connected;
E is
(C=O) D that alkylidene replaces or-R
13OC (=O) R
14
R
13Alkylidene for the alkyl replacement;
R
14Be alkyl;
D is OR
15Or NR
16R
17
R
15Be the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
16Be the alkyl of hydrogen, alkyl, thiazolinyl, aryl, aralkyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted or the alkyl of cycloalkenyl group replacement;
R
17Be hydrogen or alkyl, or R
16And R
17Form heterocycle with the nitrogen-atoms that they connected;
Y is OR
18Or NR
19R
20
R
18Be the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
19Be hydrogen or alkyl;
R
20Be the alkyl of hydrogen, alkyl, thiazolinyl, aryl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces, or R
19And R
20Form heterocycle with the nitrogen-atoms that they connected;
R
21Be hydrogen or alkyl;
With n be 0 to 4;
B. the incremental agent of at least a crystallization;
Wherein said composition has and is lower than about 1.0g/cm
3Density;
Wherein, to patient's administration the time, described composition has the dissolution rate and the bioavilability of the improvement that is used for oral or parenterai administration.
In other embodiments, the present invention relates to composition, it comprises:
The pharmaceutically acceptable slaine of a. at least a formula I compound:
Wherein: R
1Be hydrogen or alkyl;
R
2Be hydrogen, alkyl or alkenyl;
R
3Be the alkyl of hydrogen, alkyl, thiazolinyl, aryl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
4Be hydrogen, alkyl or alkenyl;
A is OR
5Or NR
6R
7
R
5Be the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
6Be hydrogen or alkyl;
R
7The B that alkyl, aralkyl, aralkyl or the alkylidene that replaces for the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, aryl, cycloalkyl substituted, cycloalkenyl group, cycloalkenyl group replaces, or R
6And R
7Form heterocycle with the nitrogen-atoms that they connected;
B is
C (=O) W or NR
8R
9
R
8Be hydrogen or alkyl;
R
9The alkyl, aryl or the aralkyl that replace for the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl substituted, cycloalkyl, cycloalkenyl group, cycloalkenyl group, or R
8And R
9Form heterocycle with the nitrogen-atoms that they connected;
W is OR
10, NR
11R
12Or OE;
R
10Be the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
11Be hydrogen or alkyl;
R
12C (=O) the Y, or R that alkyl, aralkyl or the alkylidene that replaces for the alkyl of hydrogen, alkyl, thiazolinyl, aryl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, cycloalkenyl group replaces
11And R
12Form heterocycle with the nitrogen-atoms that they connected;
E is
(C=O) D that alkylidene replaces or-R
13OC (=O) R
14
R
13Alkylidene for the alkyl replacement;
R
14Be alkyl;
D is OR
15Or NR
16R
17
R
15Be the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
16Be the alkyl of hydrogen, alkyl, thiazolinyl, aryl, aralkyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted or the alkyl of cycloalkenyl group replacement;
R
17Be hydrogen or alkyl, or R
16And R
17Form heterocycle with the nitrogen-atoms that they connected;
Y is OR
18Or NR
19R
20
R
18Be the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
19Be hydrogen or alkyl;
R
20Be the alkyl of hydrogen, alkyl, thiazolinyl, aryl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces, or R
19And R
20Form heterocycle with the nitrogen-atoms that they connected;
R
21Be hydrogen or alkyl;
With n be 0 to 4;
B. the incremental agent of at least a crystallization;
C. based on composition total weight, be lower than the solubilizing surfactant of about 1 weight %;
D. based on composition total weight, be lower than the nonaqueous solvents of about 10 weight %; With
E. based on composition total weight, be lower than the cyclodextrin of about 500 weight %;
Wherein, to patient's administration the time, described composition has the dissolution rate and the bioavilability of the improvement that is used for oral or parenterai administration.
In another embodiment, the present invention relates to comprise the injectable dosage formulations of above-mentioned composition.
In another embodiment, the present invention relates to cartridge bag, it comprises:
A. the container that comprises injectable dosage formulations; With
B. be used to prepare the specification of Injectable solution.
In another embodiment, the present invention relates to prevent or treat the method for side effect relevant among the patient with opioid, it step that comprises is:
To the above-mentioned composition of the described patient's effective dosage that needs is arranged.
This method can be used for prevention and treatment intestinal obstruction, scratches where it itches, constipation, the retention of urine, courage spasm, opium bowel dysfunction, angina, n or V or its combination, particularly postoperative ileus, postpartum intestinal obstruction, opium bowel dysfunction, postoperative nausea or postoperative vomiting or its combination.
In other embodiments, the present invention relates to prevent or treat the method for the pain among the patient, it step that comprises is:
To the above-mentioned composition of the described patient's effective dosage that needs is arranged.
In preferred embodiments, composition comprises at least a opioid in addition.
Can make these and other aspect of the present invention become apparent by following detailed description.
Detailed description of the invention
As above-mentioned and to run through the disclosure employed, except as otherwise noted, it should be understood that following term has following implication.
As used in this article, " have the dissolution rate of the improvement that is used for oral or parenterai administration and the composition of bioavilability " and be meant that composition comprises at least a opioid antagonists that is suitable for oral or parenterai administration, owing to the method that forms said composition make said composition with respect to from identical active component and incremental agent preparation but the composition with different final physical character (as density) has higher levels of dissolution rate and bioavilability, preferably make not desired components as being used for prior art to improve the solubilizing surfactant of active component solvability, nonaqueous solvents, cyclodextrin etc. minimize or remove.
As used in this article, " parenterai administration " is meant drug administration outside intestines, and do not pass through digestive tract.The main path that the mammal main body is carried out parenterai administration is in intravenous, the muscle, in subcutaneous, the intracutaneous, intraocular, synovial membrane, in intracardiac, the backbone, in the joint, in the sheath, intra-arterial, through epithelium (comprise in transdermal, the peritonaeum, eye with, hypogloeeis and oral administration), local (comprise eye usefulness, skin, eye, by the snuffing of insufflation go into, aerosol and rectum system) administration.Preferred parenterai administration approach is for passing through in intravenous, the muscle and the hypodermic injection administration.
As used in this article, " bioavilability " be meant medicine or other material after administration by available ratio of target tissue and degree.In situation of the present invention, bioavilability is meant that opioid antagonists is by central nervous system or the available degree of its periphery.
As used in this article, " alkyl " is meant to have about 1 hydrocarbon to optional substituted saturated straight chain, side chain or the ring-type of about 20 carbon atoms (with all combinations and the recombinant and the concrete carbon number of scope in scope), preferably is referred to herein as the alkyl that arrive about 8 carbon atoms containing of " low alkyl group " about 1." side chain " is meant wherein has low alkyl group such as methyl, ethyl or propyl group to be connected in the alkyl of linear alkyl chain.In some preferred embodiment, alkyl is C
1-C
5Alkyl promptly, has 1 branched-chain or straight-chain alkyl to about 5 carbon.In other preferred embodiment, alkyl is C
1-C
3Alkyl promptly, has 1 branched-chain or straight-chain alkyl to about 3 carbon.Exemplary alkyl comprises methyl, ethyl, n-pro-pyl, isopropyl, butyl, isobutyl group, sec-butyl, the tert-butyl group, amyl group, hexyl, heptyl, octyl group, nonyl and decyl." low alkyl group " is meant to have 1 alkyl to about 6 carbon atoms.Preferred alkyl comprises 1 low alkyl group to about 3 carbon.Alkyl includes but not limited to methyl, ethyl, n-pro-pyl, isopropyl, normal-butyl, isobutyl group, the tert-butyl group, n-pentyl, cyclopenta, isopentyl, neopentyl, n-hexyl, isohesyl, cyclohexyl, ring octyl group, adamantyl, 3-methyl amyl, 2,2-dimethylbutyl and 2, the 3-dimethylbutyl.
As used in this article, " alkylidene " is meant and has general formula-(CH
2) divalent alkyl of n-, wherein n is 1 to 10, and all combinations and the recombinant of the scope in this scope.Alkylidene can be straight chain, side chain or ring-type.Non-limitative example comprises methylene, methylene (CH
2-), ethylidene (CH
2CH
2-), propylidene ((CH
2)
3-), trimethylene, pentamethylene and hexa-methylene.May randomly insert one or more oxygen, sulphur or optional substituted nitrogen-atoms along alkylidene, wherein the substituting group of nitrogen is foregoing alkyl.Alkylidene can be chosen wantonly and be substituted.Term herein " low-grade alkylidene " is meant to have about 1 those alkylidenes to about 6 carbon atoms.Preferred alkylidene has about 1 to about 4 carbon.
As used in this article, " thiazolinyl " is meant the monovalent alkyl that comprises at least one carbon-to-carbon double bond and have 2 to about 10 (and combination and the recombinant of the scope in this scope) carbon atoms in chain.Thiazolinyl can be chosen wantonly and be substituted.In some preferred embodiment, thiazolinyl is C
2-C
10Thiazolinyl promptly, has 2 side chain or straight-chain alkenyls to about 10 carbon.In other preferred embodiment, thiazolinyl is C
2-C
6Thiazolinyl promptly, has 2 side chain or straight-chain alkenyls to about 6 carbon.In other preferred embodiment, thiazolinyl is C
3-C
10Thiazolinyl promptly, has 3 side chain or straight-chain alkenyls to about 10 carbon.In other preferred embodiment, thiazolinyl is C
2-C
5Thiazolinyl promptly, has 2 side chain or straight-chain alkenyls to about 5 carbon.Exemplary thiazolinyl comprises for example vinyl, acrylic, cyclobutenyl, pentenyl, hexenyl, heptenyl, octenyl, nonene base and decene base.
As used in this article, " aryl " is meant to have about 5 to about 50 carbon atoms optional substituted monocycle of (with all combinations and the recombinant and the concrete carbon number of scope in scope), two rings, three rings or the fragrant loop systems of other many cyclophanes, preferably contains about 6 aryl that arrive about 10 carbon atoms.Non-limitative example comprises for example phenyl, naphthyl, anthryl and phenanthryl.
As used in this article, " aralkyl " is meant and has aryl substituent and have about 6 to about 50 carbon atoms alkyl of (with all combinations and the recombinant and the concrete carbon number of scope in scope), preferably contain about 6 aralkyl to about 10 carbon atoms.Aralkyl can randomly be substituted in aryl or moieties.Non-limitative example comprises for example phenyl methyl (benzyl), benzhydryl, trityl, phenylethyl, diphenyl-ethyl and 3-(4-aminomethyl phenyl) propyl group.
As used in this article, " heteroaryl " is meant and comprises that at least one and preferred 1 optional substituted monocycle, two rings, three that arrive the annular atoms of about 4 sulphur, oxygen or nitrogen heteroatoms encircle or the aromatic rings system of other many rings.Heteroaryl can have for example about 3 to about 50 carbon atoms (and all combinations and recombinant and the concrete carbon number of the scope in this scope), preferably contains about 4 heteroaryls to about 10 carbon.The non-limitative example of heteroaryl comprises for example pyrrole radicals, furyl, pyridine radicals, 1,2,4-thiadiazolyl group, pyrimidine radicals, thienyl, isothiazolyl, imidazole radicals, tetrazole radical, pyrazinyl, pyrimidine radicals, quinolyl, isoquinolyl, thienyl, benzothienyl, isobenzofuran-base, pyrazolyl, indyl, purine radicals, carbazyl, benzimidazolyl He isoxazolyl.
As used in this article, " cycloalkyl " is meant to have about 3 optional substituted, the alkyl that have one or more rings in its structure to about 20 carbon atoms (with all combinations and the recombinant and the concrete carbon number of scope in scope), preferably contain about 3 to about 10 carbon atoms, more preferably contain about 3 to about 8 carbon atoms, more preferably contain about 3 to about 6 carbon atoms.Multiring structure can be bridged ring or condensed ring structure.Cycloalkyl can be randomly by for example alkyl (preferred C
1-C
3Alkyl), alkoxyl (preferred C
1-C
3Alkoxyl) or halo replace.Non-limitative example comprises for example cyclopropyl, cyclobutyl, cyclopenta, cyclohexyl, suberyl, ring octyl group and adamantyl.
As used in this article, " alkyl of cycloalkyl substituted " is meant that carbon is by cycloalkyl (preferred C endways
3-C
8Cycloalkyl) straight chained alkyl of Qu Daiing (preferred low alkyl group).Non-limitative example comprises for example cyclohexyl methyl, cyclohexyl ethyl, cyclopenta ethyl, cyclopenta propyl group, cyclopropyl methyl or the like.
As used in this article, " cycloalkenyl group " is meant and has the about 4 undersaturated cycloalkyl of olefinic to about 10 carbon (and all combinations and the recombinant of the scope in this scope).In preferred embodiments, cycloalkenyl group is C
5-C
8Cycloalkenyl group promptly, has 5 cycloalkenyl groups to about 8 carbon.
As used in this article, " alkyl-cycloalkyl " is meant the optional substituted loop systems that comprises the cycloalkyl with one or more alkyl substituents.Non-limitative example comprises for example alkyl-cycloalkyl, comprises 2-methylcyclohexyl, 3,3-dimethylcyclopentyl, trans-2,3-diformazan basic ring octyl group and 4-methyl decahydronaphthalenes base.
As used in this article, " heteroarylalkyl " is meant to have the about 2 optional substituted alkyl that replaced by heteroaryl to about 50 carbon atoms (and all combinations and recombinant and the concrete carbon number of the scope in this scope), preferably contains about 6 to about 25 carbon atoms.Non-limitative example comprises 2-(1H-pyrroles-3-yl) ethyl, 3-pyridylmethyl, 5-(2H-tetrazole radical) methyl and 3-(pyrimidine-2-base)-2-methyl cyclopentane base.
As used in this article, " Heterocyclylalkyl " is meant and comprises that at least one and preferred 1 optional substituted monocycle, two rings, three that arrive the annular atoms of about 4 sulphur, oxygen or nitrogen heteroatoms encircle or the aliphatic loop systems of other many rings.Cycloalkyl can have for example about 3 to about 20 carbon atoms (and all combinations and recombinant and the concrete carbon number of the scope in this scope), and preferred about 4 to about 10 carbon.Heterocyclylalkyl can be undersaturated, and also can condense in aromatic rings.Non-limitative example for example comprises also [c] pyranose, 1 of tetrahydrofuran base, tetrahydro-thienyl, piperidyl, pyrrolidinyl, isoxazole alkyl, isothiazole alkyl, pyrazolidinyl, oxazole alkyl, thiazolidinyl, piperazinyl, morpholinyl, piperidyl, decahydroquinolyl, octahydro benzopyranyl, octahydro pentamethylene, 2,3,4-tetrahydric quinoline group, octahydro [2] pyridine radicals, octahydro cyclooctane be [c] furyl and imidazolidinyl also.
As used in this article, term " spirane base " is meant optional substituted alkylidene double-basis, and its two ends are incorporated into the same carbon atom of female group to form the volution group.As defined herein, spirane base group female with it has 3 to 20 annular atomses.Preferably, it has 3 to 10 annular atomses.The non-limitative example of group female with it spirane base together comprises 1-(1-methyl-cyclopropyl)-propane-2-ketone, 2-(1-phenoxy group-cyclopropyl)-ethamine and 1-methyl-spiral shell [4.7] dodecane.
As used in this article, term " alkoxyl " is meant optional substituted alkyl-O-group, and wherein alkyl defines as described above.Non-limitative example for example comprise methoxyl group, ethyoxyl, positive propoxy, isopropoxy, n-butoxy and heptan the oxygen base.
As used in this article, term " aryloxy group " is meant optional substituted aryl-O-group, and wherein aryl defines as described above.Non-limitative example comprises for example phenoxy group and naphthoxy.
As used in this article, term " aralkoxy " is meant optional substituted aralkyl-O-group, and wherein aralkyl defines as described above.Non-limitative example comprises for example benzyloxy, 1-phenyl ethoxy, 2-phenyl ethoxy and 3-naphthyl oxygen in heptan base.
As used in this article, term " aryloxy group aryl " is meant to have the substituent aryl of aryloxy group, and wherein aryloxy group and aryl define as described above.The aryloxy group aryl can be chosen wantonly and be substituted.Non-limitative example comprises for example Phenoxyphenyl and naphthoxy phenyl.
As used in this article, term " heteroaryl aryl " is meant to have the substituent aryl of heteroaryl, and wherein heteroaryl and aryl define as described above.The heteroaryl aryl can be chosen wantonly and be substituted.Non-limitative example comprises for example 3-pyridine radicals phenyl, 2-quinolyl naphthyl and 2-pyrrole radicals phenyl.
As used in this article, term " alkoxy aryl " is meant the aryl that has alkoxy substituent, and wherein alkoxyl and aryl define as described above.Alkoxy aryl can be chosen wantonly and be substituted.Non-limitative example comprises for example p-methoxyphenyl, tert-butoxy phenyl and methylenedioxyphenyl.
As used in this article, " carboxyl " is meant-C (=O) OH group.
As used in this article, " alkanoyl " be meant-C (=O)-and alkyl group, wherein alkyl defines as described above.Exemplary alkanoyl comprises acetyl group, positive propiono, positive bytyry, 2-methylpropionyl, positive valeryl, 2-methylbutyryl base, 3-methylbutyryl base, 2,2-dimethyl propylene acyl group, heptanoyl group, capryl and palmityl.
As used in this article, " heterocycle " is meant the carboatomic ring atom group of monocycle or multi-loop system, it comprises about 4 all combinations and recombinants to about 10 atoms and the scope in scope, and wherein one or more atoms are the atom that is different from carbon, as nitrogen, oxygen or sulphur.Heterocyclic group can be aromatic or non-aromatic.Non-limitative example comprises for example pyrroles and piperidines group.
As used in this article, " halo " is meant fluoro, chloro or bromo.
Typically, substituted chemical part comprises the substituting group of one or more replacement hydrogen.Exemplary substituting group for example comprise halo (as, F, Cl, Br, I), alkyl, cycloalkyl, alkyl-cycloalkyl, thiazolinyl, alkynyl, aralkyl, aryl, heteroaryl, heteroarylalkyl, spirane base, Heterocyclylalkyl, hydroxyl (OH), nitro (NO
2), cyano group (CN), amino (NH
2) ,-amino that N-replaces (NHR ") ,-N, the dibasic amino of N-(N (R ") R "), carboxyl (COOH) ,-C (=O) R " ,-OR " ,-C (=O) OR " ,-NHC (=O) R ", amino carbonyl (C (=O) NH
2) ,-amino carbonyl that N-replaces (C (=O) NHR ") ,-N, the dibasic amino carbonyl of N-(C (=O) N (R ") R "), mercaptan, sulfenyl (SR "), sulfonic acid (SO
3H), phosphonic acids (PO
3H), S (=O)
2R ", S (=O)
2NH
2, S (=O)
2NHR ", S (=O)
2NR " R ", NHS (=O)
2R ", NR " S (=O)
2R ", CF
3, CF
2CF
3, NHC (=O) NHR ", NHC (=O) NR " R ", NR " C (=O) NHR ", NR " C (=O) NR " R ", NR " C (=O) R " etc.For above-mentioned substituting group, each part R " can be any in H, alkyl, cycloalkyl, thiazolinyl, aryl, aralkyl, heteroaryl or the Heterocyclylalkyl for example independently.
As used in this article, " side effect " is meant the result that the result is used in described medicine or measure that is different from as the side effect that is produced by medicine, particularly to not being the tissue pursued of administration or the harmful effect of tract.In the situation of for example opioid, term " side effect " can be meant such as intestinal obstruction for example, scratches where it itches, the situation of constipation, the retention of urine, courage spasm, opium bowel dysfunction, angina, n or V or its combination.
As used in this article, " intestinal obstruction " is duodenum 12 or the pylorus obstruction to the intestines between rectum, particularly colon.Referring to for example, Dorland ' s Illustrated Medical Dictionary, p.816,27th ed. (W.B.Saunders Company, Philadelphia 1988).Intestinal obstruction should be different from constipation, and constipation is meant seldom or very difficult defecation.Referring to for example, Dorland ' s IllustratedMedical Dictionary, p.375,27th ed. (W.B.Saunders Company, Philadelphia 1988).Intestinal obstruction can cause intestinal contents to advance diagnosing malfunction by the destruction of the normal coordinated movement of various economic factors of digestive tract.Referring to for example, Resnick, J.Am.J.ofGastroenterology, 1992,751 and Resnick, J.Am.J.of Gastroenterology, 1997,92,934.In some cases, particularly to perform the operation after (comprising abdominal operation), bowel dysfunction may be quite serious, continues to surpass a week and influences GI more than a part.This situation often is called postoperative ileus, and the most continually (referring to Livingston, E.H.and Passaro, E.D.Jr., Digestive Diseases and Sciences, 1990,35,121) takes place after laparotomy.Similarly, postpartum, the intestinal obstruction common problem that is the women after childbirth in a period of time and thought that the similar fluctuation of the natural opioid level that stress be caused by childbirth causes.
As used in this article, " effective dose " is meant the amount of the described compound herein of the symptom that can suppress, prevent or treat specified disease, illness or side effect on acology effectively.Described disease, illness and side effect (for example include but not limited to those pathological conditions relevant with the administration opioid, with treatment of pain and/or prevent relevant), wherein the treatment or the prevention comprise for example by making compound exposing cell of the present invention, tissue or acceptor suppress its activity.Therefore, for example, term " effective dose " is being used for being meant when relevant with the opioid of for example treating pain treating and/or preventing the situation of pain.Term " effective dose " treats and/or prevents typically the side effect relevant with opioid being used for being meant when relevant with the peripheral mu opioid antagonists, comprises such as intestinal obstruction for example, scratches where it itches, the side effect of constipation, the retention of urine, courage spasm, opium bowel dysfunction, angina, n or V or its combination.
As used in this article, " with ... combination ", " therapeutic alliance " and " combination product " are meant simultaneously administration antiemetic and peripheral mu opioid antagonists (comprising for example compound of formula I) to the patient in certain embodiments, or to patient's while administration antiemetic, peripheral mu opioid antagonists and opioid.When combination medicine-feeding, the administration simultaneously of each component or at different time points with random order administration continuously.Therefore, each component discriminably but the time on fully closely administration, so that required curative effect to be provided.
As used in this article, " dosage unit " is meant the discrete unit of physics that is suitable as the unit dose that is used for the particular patient of being treated.Each unit can comprise the reactive compound that being intended for use in of combining with required pharmaceutical carrier produces the scheduled volume of required curative effect.The specification of unit dosage forms of the present invention can by the specific characteristic of (a) reactive compound and the specific therapeutical that will realize and (b) the intrinsic limitation of this in the art reactive compound stipulate.
As used in this article, term " pharmaceutically useful " be meant those compounds, material, composition and/or formulation in rational medical judgment scope, be suitable for contacting human and animal's tissue and do not have too much toxicity, excitant, allergy or with rational interests/risk than the other problem complication that matches.
As used in this article, " pharmaceutically acceptable slaine " is meant the derivative of compound disclosed herein, and wherein parent compound carries out modification by being prepared into its alkali salt.The example of officinal salt includes but not limited to the alkaline residue for example inorganic acid or the organic acid salt of amine, alkali etc.Officinal salt comprises the nontoxic salts of the routine that parent compound and for example nontoxic inorganic base or organic base form.The acceptable salt of these physiology prepares by method as known in the art, for example, and by will free amine alkali dissolving in aqueous alcohol with excessive acid or with alkali metal base such as hydroxide or with in the amine and the carboxylic acid that dissociates.
Running through the compound of describing herein can alternative use or preparation.Law of isomorphism type, all chirality and racemization shape, N-oxide, hydrate and solvate have also been considered within the scope of the invention.
Some acidity of the present invention or alkali compounds can be used as amphion and exist.The form of ownership of compound comprises that free acid, free alkali and amphion all are considered within the scope of the invention.As known in the art is that the compound that comprises amino and carboxyl simultaneously often exists with its zwitterionic form.Therefore, any compound described herein that runs through that for example comprises amino and carboxyl simultaneously also comprises their corresponding amphions.
As used in this article, " patient " is meant and comprises mammal by animal, and be preferred human.
As used in this article, " prodrug " is meant the maximized compound of amount of the active substance that is designed to reach required response location especially, itself for required activity normally non-activity or have lowest activity, but become the metabolite of biologic activity by biological transformation.
As used in this article, " stereoisomer " be meant have identical chemical composition but on the spatial arrangements of atom or group different compound.
As used in this article, " N-oxide " is meant that the oxidized formation of basic nitrogen atom of heteroaryl ring wherein or tertiary amine has the quaternary nitrogen of a form positive charge and the oxygen atom that has the form negative electrical charge that is connected.
When any variable occurs surpassing one time in any composition or in any formula, have nothing to do in its definition in each situation and other situation.The combination of substituting group and/or variable only is only permission when this combination results stable compound.
Formula I explanation can be used for 3-that the piperidine derivative in method of the present invention, composition and the cartridge bag can be used as at piperidine ring and the trans and cis three-dimensional chemical isomer of 4-position exists.In the compound of most preferred formula I, R
2Substituting group and R
4Substituting group is " trans " orientation on piperidines.
R except formula I
2Substituting group and R
4Outside substituent " cis " and " trans " orientation, have R among the formula I
2Substituting group and R
4The absolute stereo chemistry of substituent carbon atom also uses " R " of common employing and " S " to define (people such as Orchin, The Vocabulary of OrganicChemistry, John Wiley and Sons, Inc., page 126, and it is merged in this paper as a reference).R on the piperidine ring that preferred compound of the present invention is its Chinese style I
2Substituting group and R
4Substituent configuration all is those of " R ".
In addition, depend on R
4Structure, can in molecule, introduce asymmetric carbon atom.Therefore, these compounds categories can be used as at independent " R " or " S " of these chiral centres stereoisomer and exist, or exist as the racemic mixture of isomer, and it is considered within the scope of the invention all.Preferably, use the pure basically stereoisomer of compound of the present invention, that is, the isomer that is configured as " R " or " S " of chiral centre wherein, that is, wherein the configuration at three chiral centre I is preferably 3R, 4R, S or 3R, 4R, those compounds of R.
As used in this article, " periphery " or " periphery works " is meant that medicine works outside central nervous system.
As used in this article, " maincenter works " is meant that medicine works in central nervous system.
Method of the present invention, composition and cartridge bag relate to periphery opioid antagonists compound.Term " periphery " is meant that this compound mainly acts on department of physiology beyond the central nervous system key element of unifying.In a preferred form, the periphery opioid antagonists compound that is used for the inventive method shows the high level activity with respect to peripheral tissues such as stomach intestinal tissue, and shows the CNS activity of reduction and preferably do not have the CNS activity basically.As used in this article, phrase " does not have the CNS activity " and is meant to show about 20% of the pharmacological activity that is lower than the inventive method compound used therefor basically in CNS, preferably be lower than about 15%, more preferably less than about 10%, more preferably less than about 5%, most preferably in CNS, show about 1% of the pharmacological activity that is lower than the inventive method compound used therefor.
In addition, give therein in drug compound certain embodiments of the present invention with the periphery side effect of antagonism opioid, preferably this compound is gone up substantially not by blood-brain barrier, thereby does not reduce the favorable activity of opioid.As used in this article, phrase " basically not by " is meant that the about 20 weight % of being lower than of the inventive method compound used therefor pass through blood-brain barrier, preferably be lower than about 15 weight %, more preferably less than about 10 weight %, more preferably less than about 5 weight %, and most preferably the compound of 0 weight % passes through blood-brain barrier.By measuring the CNS permeability of the intravenous administration evaluation of the level in blood plasma and brain afterwards selected compounds.
US-B-6,451,806 and US-B-6,469,030 disclose such method and composition, and it comprises opioid and opioid antagonists, comprises the peripheral mu opioid antagonists, and it is open is incorporated into this paper as a reference in full.This method and composition especially can be used for preventing and treats pain and the side effect that is used for the treatment of and/or prevention is relevant with opioid, comprise intestinal obstruction, scratch where it itches, constipation, the retention of urine, courage spasm, opium bowel dysfunction, angina, feel sick or its combination, particularly postoperative ileus, postpartum intestinal obstruction, opium bowel dysfunction, postoperative nausea or postoperative vomiting.Method of the present invention, composition and cartridge bag relate to the peripheral mu opioid antagonists, and relate to the antiemetic that antiemetic that peripheral mu opioid antagonists and maincenter work and maincenter work and the combination of opioid, be used for the treatment of and prevent for example pain and/or the side effect relevant with opioid, comprise intestinal obstruction, scratch where it itches, constipation, the retention of urine, courage spasm, opium bowel dysfunction, angina, vomiting or feel sick its combination, particularly postoperative or postpartum intestinal obstruction, opium bowel dysfunction, postoperative nausea or postoperative vomiting.
Therefore, in one embodiment, the invention provides the method that may further comprise the steps:
A., composition is provided, and it comprises:
(i) the pharmaceutically acceptable slaine of at least a formula I compound:
Wherein:
R
1Be hydrogen or alkyl;
R
2Be hydrogen, alkyl or alkenyl;
R
3Be the alkyl of hydrogen, alkyl, thiazolinyl, aryl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
4Be hydrogen, alkyl or alkenyl;
A is OR
5Or NR
6R
7
R
5Be the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
6Be hydrogen or alkyl;
R
7The B that alkyl, aralkyl, aralkyl or the alkylidene that replaces for the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, aryl, cycloalkyl substituted, cycloalkenyl group, cycloalkenyl group replaces, or R
6And R
7Form heterocycle with the nitrogen-atoms that they connected;
B is
C (=O) W or NR
8R
9
R is a hydrogen or alkyl;
R
9The alkyl, aryl or the aralkyl that replace for the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl substituted, cycloalkyl, cycloalkenyl group, cycloalkenyl group, or R
8And R
9Form heterocycle with the nitrogen-atoms that they connected;
W is OR
10, NR
11R
12Or OE;
R
10Be the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
11Be hydrogen or alkyl;
R
12C (=O) the Y, or R that alkyl, aralkyl or the alkylidene that replaces for the alkyl of hydrogen, alkyl, thiazolinyl, aryl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, cycloalkenyl group replaces
11And R
12Form heterocycle with the nitrogen-atoms that they connected;
E is
(C=O) D that alkylidene replaces or-R
13OC (=O) R
14
R
13Alkylidene for the alkyl replacement;
R
14Be alkyl;
D is OR
15Or NR
16R
17
R
15Be the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
16Be the alkyl of hydrogen, alkyl, thiazolinyl, aryl, aralkyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted or the alkyl of cycloalkenyl group replacement;
R
17Be hydrogen or alkyl, or R
16And R
17Form heterocycle with the nitrogen-atoms that they connected;
Y is OR
18Or NR
19R
20
R
18Be the alkyl of hydrogen, alkyl, thiazolinyl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces;
R
19Be hydrogen or alkyl;
R
20Be the alkyl of hydrogen, alkyl, thiazolinyl, aryl, cycloalkyl, cycloalkenyl group, cycloalkyl substituted, alkyl or the aralkyl that cycloalkenyl group replaces, or R
19Form heterocycle with 20 with the nitrogen-atoms that they were connected;
R
21Be hydrogen or alkyl;
With n be 0 to 4;
The (ii) incremental agent of at least a crystallization;
(iii) at least a weak base; With
(iv) water;
Wherein said composition has the initial pH at least about 10.5; With
B. the pH that regulates described composition arrives the final pH that arrive in about 11 scopes about 9;
Wherein, to patient's administration the time, described composition has the dissolution rate and the bioavilability of the improvement that is used for oral or parenterai administration.
Method of the present invention especially can be used for formation and has the dissolution rate of improvement and the composition of bioavilability with respect to prior art combinations (but comprising the composition that contains identical or different component be not processed into final preparation (for example pH regulator, original place form pharmaceutically acceptable slaine, freeze drying and/or the kneading (anneal) of active component) in the same way or do not have same physical properties (for example density or porosity)), particularly by injectable formulation, as at those of intravenous administration.
In the preferred embodiment of this method, when organizing structure again, solution forms in less than about five minutes under environmental condition, more preferably, under environmental condition in less than about one minute, and more preferably, under environmental condition, in less than about 30 seconds, form, preferably by simply shaking, stir or mixing and organize structure again.As used in this article, " under environmental condition " is meant under normal atmospheric pressure and about 10 ℃ of room temperatures that arrive in about 50 ℃ of scopes, and not directly heating or cooling.The formation of solution can be by for example measuring, measure by Ku Erte suspended matter determination instrument mensuration or by the light scattering instrument by the existence of visualization (the preferred use by diaphragm projects microscope light in the solution) clear solution when mixing.
In the preferred embodiment of this method, initial pH is adjusted at least about 11.Preferably, initial pH is no more than about pH 12, because high like this pH can cause the composition instability.Initial pH can regulate with any suitable pharmaceutically acceptable pH regulator agent, comprise strong or weak acid or alkali, preferred pharmaceutically useful metal carbonate, pharmaceutically useful alkali metal bicarbonate salt, pharmaceutically useful metal hydroxides or, hydrochloric acid, more preferably wherein pharmaceutically useful metal is a sodium, and more preferably sodium carbonate or sodium bicarbonate, and more preferably sodium carbonate.Preferred sodium carbonate and sodium bicarbonate are because they produce carbonic acid gas, thereby help required lower composition density.
In the preferred embodiment of this method, regulating final pH is about 9.5 to about 10.5.Final pH can be regulated with any suitable pharmaceutically acceptable pH regulator agent, comprises strong or weak acid or alkali, preferred sodium hydroxide or hydrochloric acid.
In some preferred embodiment of this method, the original place prepares the pharmaceutically acceptable slaine of at least a formula I compound.
In some preferred embodiment of this method, the pharmaceutically acceptable slaine of the compound of formula I is formed by at least a weak base, and wherein said weak base is to join in the compound of described formula I at least about equimolar amounts.Preferably, do not use big excessive weak base, because this excessive high pH (greater than about 12) that does not expect that causes, it can help the composition instability.
In some preferred embodiment of this method,, add the compound of described formula I and prepare composition to described mixture then by at first the pharmaceutically acceptable slaine of described incremental agent and described weak base being mixed in water.
In some other preferred embodiment, by side by side the pharmaceutically acceptable slaine of the compound of described formula I, described incremental agent and described weak base being mixed with composition in water basically.As mixing under the background employedly, " basically side by side " be meant each other in about five minutes, preferably in about one minute, more preferably in about 30 seconds each component is mixed.
In a preferred embodiment of the invention, pharmaceutically acceptable metal is an alkali metal, as sodium, potassium or lithium; Or alkaline earth metal, as calcium or magnesium, or its combination.Preferred sodium, calcium and magnesium.More preferably sodium.For parenterai administration, preferably avoid using potassium.
This method can comprise described composition dries with the described water of removing at least a portion in addition to form the step of partially or completely dry product.In preferred embodiments, in the drying steps process, composition is mediated.As used in this article, " kneading " is meant the process that the material heating is slowly cooled off then, comprises the heating and cooling circulation of repetition.Suitable drying means comprises freeze drying (freeze-drying), atomized drying, vacuum drying and combination thereof.Preferred drying means is freeze drying.
This method can comprise in addition by making it organize the step of structure again with the solution that forms described dry products described dry products and the merging of pharmaceutically useful solvent.
In the preferred embodiment of this method, weak base is bicarbonate or carbonate, more preferably carbonate.Preferred these weak base are because they produce carbonic acid gas, thereby help required lower composition density.
In the preferred embodiment of this method, pharmaceutically useful solvent is moisture, preferred water, isotonic sodium chlorrde solution, ringer's solution (Ringer ' s solution), dextrose solution or Lactated Ringer'S Solution (lactated Ringer ' s solution).
In preferred embodiments, the inventive method comprises in addition with the described solution of the described dry products step to patient's administration.Composition can be before art, in the surgical procedure and/or administration under the situation that is not having operation.
In preferred embodiments, composition is by drug administration by injection, particularly hypodermic injection, intramuscular injection or intravenous injection.
In composition of the present invention, can use any pharmaceutically useful incremental agent of crystallization.As used in this article, " incremental agent " is meant inert diluent or the additive that plays as the carrier function of medicine (being the compound of formula I in situation of the present invention).Suitable incremental agent can be at Handbook of Pharmaceutical Excipients, 3rd Ed.Washington, and D.C:American Pharmaceutical Association finds in 1998, and it openly is merged in this paper as a reference.In some other preferred embodiment, incremental agent is a polyalcohol, as carbohydrate or sugar alcohol.Suitable carbohydrate comprises sucrose, trehalose, lactose, maltose and composition thereof.Suitable sugar alcohol comprises mannitol, xylitol, erythrite, lactitol, isomalt, polyalditol, maltitol and composition thereof.Preferred especially mannitol.
Though be not wishing to be bound by theory, believe that the incremental agent generation of crystallization has the required pie structure of good mechanical properties.These character are for guaranteeing that it is important organizing structure speed rapidly again.In addition, the incremental agent of crystallization in freezing dry process rapidly nucleation produce and have the more pie of high surface area, thereby produce higher diffusion flux and rate of sublimation faster.The drying of amorphous incremental agent needs high-energy and can not produce required pie structure.Because the primary crystallization size depends on nucleation rate and growth rate, the brilliant generation of the brief summary that forms in comprising the system of amorphous solid has the more hole of low surface area of every volume.This lower surface area produces lower diffusion flux and lower rate of sublimation.
In certain embodiments, the present invention relates to composition, it comprises:
The pharmaceutically acceptable slaine of a. at least a formula I compound;
B. the incremental agent of at least a crystallization;
Wherein said composition has and is lower than about 1.0g/cm
3Density;
Wherein, to patient's administration the time, described composition has the dissolution rate and the bioavilability of the improvement that is used for oral or parenterai administration.
Preferably, composition has the 0.5g/cm of being lower than
3Density, more preferably be lower than about 0.2g/cm
3, more preferably be lower than about 0.15g/cm
3, more preferably about 0.05g/cm
3To about 0.12g/cm
3Density in the scope, more preferably from about 0.06g/cm
3To about 0.08g/cm
3Density in the scope.
In certain embodiments, the present invention relates to composition, it comprises:
The pharmaceutically acceptable slaine of a. at least a formula I compound;
B. the incremental agent of at least a crystallization;
C. based on composition total weight, be lower than the solubilizing surfactant of about 1 weight %;
D. based on composition total weight, be lower than the nonaqueous solvents of about 10 weight %; With
E. based on composition total weight, be lower than the cyclodextrin of about 0.5 weight %;
Wherein, to patient's administration the time, described composition has the dissolution rate and the bioavilability of the improvement that is used for oral or parenterai administration.
These compositions provide for oral or dissolution rate and the bioavilability of parenterai administration for improving, because they allow the compound of formula I more easily to dissolve in other situation that is compared to the compound of formula I when dry products is organized structure again (it is low-down water-soluble owing to zwitterionic character has).
Preferably, the pharmaceutically acceptable slaine of the compound of formula I exists with the level at least about 0.1mg/mL, more preferably exists with the level at least about 1mg/mL, more preferably exists with the level at least about 2mg/mL.
Preferably, composition of the present invention comprises at least a pharmaceutically useful solvent in addition.In the preferred embodiment of said composition, pharmaceutically useful solvent is moisture, preferred water, isotonic sodium chlorrde solution, ringer's solution, glucose solution or Lactated Ringer'S Solution.
Preferably, composition has the pot-life at least about 18 months.As used in this article, " pot-life " be meant after the production of preparation and packing up to its chemistry or biologic activity and be no less than the time that predeterminated level (common about 90%) that mark renders a service and its physical property do not have appreciable or harmful variation.
In some preferred embodiment, composition of the present invention can comprise the prodrug and/or the pharmacological activity metabolite of opioid, opioid, and condition is dissolution rate or the bioavilability that its inclusion does not hinder formula I compound.Suitable opioid comprises alfentanil, buprenorphine, butorphanol, codeine, dezocine, Dihydrocodeine, fentanyl, hydrocodone, Hydromorphone, levorphanol, meperidine(pethidine), methadone, morphine, Nalbuphine, Oxycodone, Oxymorphone, pentazocine, propiram, dextropropoxyphene, sufentanil, tramadol and composition thereof.Preferred opioid comprises morphine, codeine, Oxycodone, hydrocodone, Dihydrocodeine, dextropropoxyphene, fentanyl and tramadol.
Composition of the present invention can comprise in addition that one or more are generally used for other active component of pain relieving and/or cough-flu-pectoral combination product, and condition is dissolution rate or the bioavilability that its inclusion does not hinder formula I compound.The component of this routine comprises for example aspirin, cox 2 inhibitor, paracetamol, phenylpropanolamine, neo-synephrine, chlorphenamine, caffeine and/or gualfenesin.Typical or the conventional composition that can comprise is at for example Physicians ' Desk Reference, describes in 2004, and it is open is incorporated into this paper as a reference in full.
In addition, composition of the present invention can comprise in addition that one or more are designed to can strengthen the pain relieving effectiveness of opioid and/or be designed to reduce the compound that forms the anodyne tolerance, and condition is dissolution rate or the bioavilability that its inclusion does not hinder formula I compound.This compound for example comprises dextromethorphan or other nmda antagonist, and (Pain 1996 for Mao, people such as M.J., 67,361), L-364,718 and other CCK antagonist (Dourish, CT. wait people, Eur.J.Pharmacol., 1988,147,469), no inhibitor (Bhargava, people such as H.N., Neuropeptides, 1996,30,219), pkc inhibitor (Bilsky, EJ. wait people, J.Pharmacol.Exp.Ther.1996,277,484) and dynorphin antagonist or antiserum (Nichols, M.L. wait people, Pain, 1997,69,317).The open quilt of aforementioned each document is incorporated this paper into as a reference in full.
Except that above-mentioned illustrational those, the pain relieving that can be used for being used in the method and composition of the present invention and increase opioid is renderd a service and/or the opioid component and the optional compound that are used to reduce other opioid of forming the anodyne tolerance, optional routine is conspicuous for the those skilled in the art that grasped instruction disclosed by the invention.
Preferred 4-aryl-piperidine derivative for example comprises at US-A-5,250,542, US-A-5,159,081, US-A-5,270,328 and US-A-5,434,171, US-B-6,451, disclosed compound in 806 and US-B-6,469,030, it is open is incorporated into this paper as a reference in full.
In preferred embodiments, the compound of formula I is trans 3, the 4-isomer.
In some embodiment of the compound that adopts formula I, preferred
R
1Be hydrogen;
R
2Be alkyl;
N is 1 or 2;
R
3Be benzyl, phenyl, cyclohexyl or cyclohexyl methyl; With
R
4Be alkyl.
In some embodiment of the compound that adopts formula I, preferred
A is OR
5With
R
5Be hydrogen or alkyl.
In some embodiment of the compound that adopts formula I, preferred
A is NR
6R
7
R
6Be hydrogen;
R
7B for the alkylidene replacement; With
B is C (O) W.
In some embodiment of the compound that adopts formula I, preferred
R
7Be (CH
2) q-B;
Q is about 1 to about 3;
W is OR
10With
R
10The alkyl of alkyl, cycloalkyl or the cycloalkyl substituted that replaces for hydrogen, alkyl, phenyl.
In some embodiment of the compound that comprises formula I, preferred
W is NR
11R
12
R
11Be hydrogen or alkyl; With
R
12The C that replaces for hydrogen, alkyl or alkylidene (=O) Y.
In some embodiment of the compound that adopts formula I, preferred
R
12Be (CH
2) mC (O) Y;
M is 1 to 3;
Y is OR
18Or NR
19R
20With
R
18, R
19And R
20Be hydrogen or alkyl independently.
In some embodiment of the compound that adopts formula I, preferred
W is OE;
E is CH
2C (=O) D;
D is OR
15Or NR
16R
17
R
15Be hydrogen or alkyl;
R
16Be methyl or benzyl; With
R
17Be hydrogen.
In some embodiment of the compound that adopts formula I, preferred
W is OE;
E is R
13OC (=O) R
14
R
13For-CH (CH
3)-or-CH (CH
2CH
3)-; With
R
14Be alkyl.
In some embodiment of the compound that adopts formula I, preferred
A is OR
5With
R
5Be hydrogen.
In some embodiment of the compound that adopts formula I, 3 of preferred piperidine ring and 4 s' the configuration R type of respectively doing for oneself.
The compound of preferred formula I comprises:
Q-CH
2CH(CH
2(C
6H
5))C(O)OH、
Q-CH
2CH
2CH(C
6H
5)C(O)NHCH
2C(O)OCH
2CH
3、
Q-CH
2CH
2CH(C
6H
5)C(O)NHCH
2C(O)OH、
Q-CH
2CH
2CH(C
6H
5)C(O)NHCH
2C(O)NHCH
3、
Q-CH
2CH
2CH(C
6H
5)C(O)NHCH
2C(O)NHCH
2CH
3、
G-NH(CH
2)
2C(O)NH
2、
G-NH(CH
2)
2C(O)NHCH
3、
G-NHCH
2C(O)NH
2、
G-NHCH
2C(O)NHCH
3、
G-NHCH
2C(O)NHCH
2CH
3、
G-NH(CH
2)
3C(O)OCH
2CH
3、
G-NH(CH
2)
3C(O)NHCH
3、
G-NH(CH
2)
2C(O)OH、
G-NH(CH
2)
3C(O)OH、
Q-CH
2CH(CH
2(C
6H
11))C(O)NHCH
2C(O)OH、
Q-CH
2CH(CH
2(C
6H
11))C(O)NH(CH
2)
2C(O)OH、
Q-CH
2CH(CH
2(C
6H
11))C(O)NH(CH
2)
2C(O)NH
2、
Z-NHCH
2C(O)OCH
2CH
3、
Z-NHCH
2C(O)OH、
Z-NHCH
2C(O)NH
2、
Z-NHCH
2C(O)N(CH
3)
2、
Z-NHCH
2C(O)NHCH(CH
3)
2、
Z-NHCH
2C(O)OCH
2CH(CH
3)
2、
Z-NH(CH
2)
2C(O)OCH
2(C
6H
5)、
Z-NH(CH
2)
2C(O)OH、
Z-NH(CH
2)
2C(O)NHCH
2CH
3、
Z-NH(CH
2)
3C(O)NHCH
3、
Z-NHCH
2C(O)NHCH
2C(O)OH、
Z-NHCH
2C(O)OCH
2C(O)OCH
3、
Z-NHCH
2C(O)O(CH
2)
4CH
3、
Z-NHCH
2C(O)OCH
2C(O)NHCH
3、
Z-NHCH
2C (O) O-(4-methoxyl group cyclohexyl),
Z-NHCH
2C (O) OCH
2C (O) NHCH
2(C
6H
5) and
Z-NHCH
2C(O)OCH(CH
3)OC(O)CH
3;
Wherein: Q represents
G represents
Z represents
The compound of preferred formula I comprises:
(3R,4R,S)-Z-NHCH
2C(O)OCH
2CH(CH
3)
2、
(+)-Z-NHCH
2C(O)OH、
(-)-Z-NHCH
2C(O)OH、
(3R,4R,R)-Z-NHCH
2C(O)-OCH
2CH(CH
3)
2、
(3S,4S,S)-Z-NHCH
2C(O)OCH
2CH(CH
3)
2、
(3S,4S,R)-Z-NHCH
2C(O)OCH
2CH(CH
3)
2、
(3R, 4R)-Z-NHCH
2C (O) NHCH
2(C
6H
5) and
(3R,4R)-G-NH(CH
2)
3C(O)OH。
Wherein Q, Z and G define as described above.
The compound of preferred formula I comprises (+)-Z-NHCH
2C (O) OH and (-)-Z-NHCH
2C (O) OH, wherein Z defines as described above.At described compound is (+)-Z-NHCH
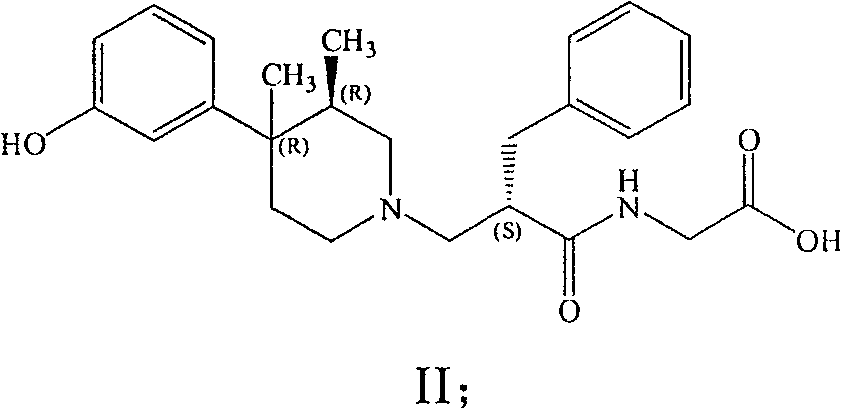
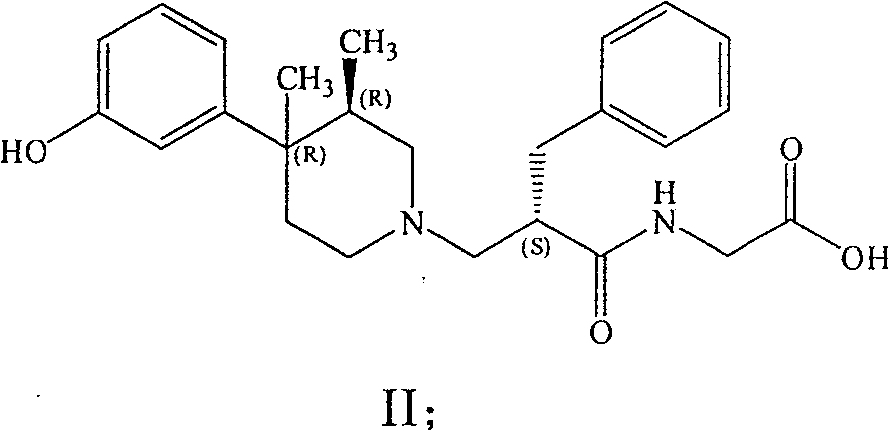
2Preferred especially during C (O) OH.Particularly preferred compound is [[2 (S)-[[4 (R)-(3-hydroxy phenyl)-3 (R), 4-dimethyl-piperidyl] methyl]-1-oxo-3-phenyl propyl] amino] acetate dihydrate (USAN title, Aiweimopan).
The compound of preferred formula I comprises Q-CH
2CH (CH
2(C
6H
5)) C (O) OH, wherein Q defines as described above.At described compound be (3R, 4R, S)-Q-CH
2CH (CH
2(C
6H
5)) preferred especially during C (O) OH.This compound is the active metabolite of Aiweimopan, but when oral administration, it has the tendency of the reverse analgesia do not expected more much bigger than Aiweimopan.When parenterai administration, particularly during intravenous administration, but the much lower dosage of administration is to reduce this tendency.
Work, have efficient and have the compound of the formula I of Orally active particularly preferably in the enteron aisle part.Particularly preferred embodiment of the present invention is (+)-Z-NHCH
2C (O) OH, that is, and with the compound of following formula (II):
The compound of formula (II) has low aqueous solubility, unless under low or high pH condition.Zwitterionic character may be that this compound is intrinsic, and the system that can produce the difference of desirable character after for example oral absorbs and in gastral lasting local action.
In particularly preferred embodiments, the compound of formula I is pure basically stereoisomer.
In another embodiment, the present invention relates to comprise the injectable dosage formulations of above-mentioned composition.In preferred embodiments, with respect to the pain that is caused by venipuncture, injectable dosage formulations helps to have minimum pain or does not have other pain, prepares under the pH higher than physiology pH although comprise composition of active components.
In another embodiment, the present invention relates to prevent or treat the method for side effect relevant among the patient with opioid, it step that comprises is:
To the above-mentioned composition of the described patient's effective dosage that needs is arranged.
This method can be used for prevention and treatment intestinal obstruction, scratches where it itches, constipation, the retention of urine, courage spasm, opium bowel dysfunction, angina, n or V or its combination, particularly postoperative ileus, postpartum intestinal obstruction, opium bowel dysfunction, postoperative nausea or postoperative vomiting.
In other embodiments, the present invention relates to prevent or treat the method for the pain among the patient, it step that comprises is:
To the above-mentioned composition of the described patient's effective dosage that needs is arranged.
In preferred embodiments, composition comprises at least a opioid in addition.
In another embodiment, the present invention relates to cartridge bag, it comprises:
A. the container that comprises injectable dosage formulations; With
B. be used to prepare the specification of Injectable solution.
Preferably, cartridge bag comprises syringe in addition.Preferably, injectable dosage formulations comprises at least a opioid in addition.Composition can randomly comprise conventional medicinal cartridge bag component.
The present invention relates to comprise method, composition and the cartridge bag of opioid compound.As mentioned above, this opioid compound can be used for for example treating and/or preventing pain.Yet, also as mentioned above, undesirable side effect for example comprises intestinal obstruction, scratches where it itches, constipation, the retention of urine, courage spasm, opium bowel dysfunction, angina, vomiting or feel sick or its combination, particularly postoperative and postpartum intestinal obstruction, opium bowel dysfunction, feel sick and/or vomiting and possible frequent other side effect that takes place in the patient who accepts the opioid compound.Rely on method of the present invention, composition and cartridge bag, can advantageously realize effective and desirable inhibition is carried out in undesirable side effect that may be relevant with the opioid compound.Therefore, wherein method, composition and the cartridge bag of opioid with the combination of combination of suitable peripheral mu opioid antagonists compound or co-administered can be provided than independent compound and the more favourable effectiveness of medicine.
In this respect, as mentioned above, often patient's administration opioid is used for the treatment of for example pain condition.Yet, as mentioned above, undesirable side effect such as intestinal obstruction for example, scratch where it itches, constipation, the retention of urine, courage spasm, opium bowel dysfunction, angina, n or V or its combination may cause by the administration opioid.But these undesirable side effects can be used as with to the amount relative restrictions factor of patient's administration opioid.That is to say, can the amount of the opioid of patient's administration can be restricted owing to the generation of undesirable above-mentioned side effect.The degree that can reduce pain relief again unfriendly that is restricted to the amount of the opioid of patient's administration.The method and composition of combination of the present invention can advantageously increase the amount to the opioid of patient's administration, thereby increases the degree of pain relief, thus make may be relevant with opioid undesirable side effect reduce, minimize and/or avoid.The peripheral mu opioid antagonists that is preferred for method and composition of the present invention does not have the central nervous system activity basically, and therefore, the pain relieving that does not influence opioid is as required renderd a service.
Though do not wish to be bound by any theory or theory of operation, estimate opioid side effect such as intestinal obstruction, scratch where it itches, constipation, the retention of urine, courage spasm, opium bowel dysfunction, angina, vomiting or feel sick or its combination can be caused by the interaction of not expecting of opioid and peripheral mu acceptor.The μ opioid antagonists that the method according to this invention administration is worked in periphery can be blocked the interaction of opioid compound and μ acceptor, thereby prevention and/or suppress side effect, particularly postoperative or postpartum intestinal obstruction, opium bowel dysfunction, feel sick and/or vomiting.
Except that above-mentioned illustrational those, other μ opioid antagonists compound that can be used for method and composition of the present invention is conspicuous for the those skilled in the art that learned instruction of the present invention.
The compound that is used for method of the present invention can be used as prodrug forms and exists.As used in this article, " prodrug " is intended to comprise any covalently bound carrier, and with this prodrug during to the administration of mammal main body, it discharges the active parent drug of formula I used in the inventive method for example in vivo.Because the character of many needs of known prodrug raising medicine (as, dissolution rate, bioavilability, production etc.), if expectation, the inventive method compound used therefor can be sent by prodrug forms.Therefore, the present invention has considered to send the method for prodrug.Be used for the functional group that the prodrug of compound of the present invention (suc as formula I) can exist by modified compound, make this be modified in the conventional treatment or be cracked into parent compound in vivo.
Therefore, prodrug for example comprises that wherein hydroxyl, amino or carboxyl are incorporated into the described compound of any group herein, in the described linking group cracking during to the administration of mammal main body with prodrug, forms free hydroxyl, free amino or carboxylic acid respectively.Its example includes but not limited to acetic acid esters, formic acid esters and the benzoic ether of alkohol and amine functional group; With ester alkyl, carbocyclic ring, aryl and alkylaryl, as the ester of methyl, ethyl, propyl group, isopropyl, butyl, isobutyl group, sec-butyl, the tert-butyl group, cyclopropyl, phenyl, benzyl and phenethyl, or the like.
Compound used in the inventive method can be by well known to a person skilled in the art the several different methods preparation.Compound can pass through method for example as described below or the method variant is synthetic as understood by a person skilled in the art.Estimate that all methods disclosed by the invention put into practice with any scale, comprise milligram, gram, number gram, kilogram, thousands of gram or commercial scale.
Go through as above-mentioned, the inventive method compound used therefor can comprise the carbon atom of one or more asymmetric replacements, and separable be the form of optically active or racemization.Therefore, be intended to comprise all chiralitys, diastereomer, racemization form and all geometrical isomerism forms, unless specifically show concrete spatial chemistry or isomeric form.Known in the artly how to prepare and separate this optical activity form.For example, can be by the mixture separation of standard technique with stereoisomer, described standard technique includes but not limited to the fractionation, positive of racemization form and anti-phase and chirality chromatography, preferential salt formation, recrystallization etc., or it is synthetic, or synthetic by the chirality of the careful target chiral centre of planning by the chirality that begins from active initiation material.
Can easily understand, the functional group of existence can comprise protecting group in building-up process.Protecting group is known chemical functional group itself, can be optionally removes with its affix with from degree of functionality such as hydroxyl and carboxyl.These groups are present in the compound, make that this degree of functionality is an inertia to the chemical reaction condition of compound contact.The present invention can use any in the kinds of protect base.Preferred protecting group comprises benzyloxycarbonyl and tertbutyloxycarbonyl.Can be used for other preferred protecting group of the present invention can be as Greene, T.W. and Wuts, P.G.M., Protective Groups inOrganic Synthesis 2d.Ed., Wiley ﹠amp; Sons is described in 1991.
4-aryl-piperidine derivative of formula I of the present invention can adopt at for example US-A-5, and 250,542, US-A-5,434,171, US-A-5,159,081, US-A-5,270,328, US-B-6,451,806, US-B-6,469,030 and WerneR, people's such as J.A. Journal ofOrganic Chemistry, 61, the method of instruction is synthetic among the 587-597 (1996), and its open quilt is separately incorporated this paper into as a reference in full.For example; be used as in The compounds of this invention synthetic that the 3-of initiation material replaces-4-methyl-4-(3-hydroxyl-or alkanoyl oxygen base phenyl) piperidine derivative can be by at US-A-4; 115; 400 and US-A-4; 891; the conventional method of instruction preparation in 379, its open quilt is separately incorporated this paper into as a reference in full.Be used for the synthetic initiation material (3R of described compound herein, 4R)-4-(3-hydroxy phenyl)-3, the 4-lupetidine can pass through at US-A-4,581, the method preparation of describing in 456 and US-A-5,136,040, it is openly incorporated into this paper as a reference in full, but is adjusted into preferred β-spatial chemistry as described.
The first step of this method can relate to by making the reaction of 3-alkoxyl bromobenzene and alkyl lithium reagents form 3-alkoxyl phenyl lithium reagent.This reaction can be carried out under inert conditions and in the presence of suitable non-reactive solvent such as absolute ether or preferred anhydrous tetrahydro furan.The preferred alkyl lithium reagents that is used for this method is n-BuLi, particularly s-butyl lithium.Usually, add the slight excessive alkyl lithium reagents of arriving of about equimolar amounts to reactant mixture.Reaction can be carried out under about-20 ℃ to about-100 ℃ temperature, preferably carries out under about-50 ℃ to about-55 ℃ temperature.
After forming 3-alkoxyl phenyl lithium reagent, to the 1-alkyl-4-piperidones of the about equimolar amounts of mixture adding, holding temperature is between-20 ℃ to-100 ℃ simultaneously.Reaction was typically finished after about 1 to 24 hour.At this moment, can make the reactant mixture room temperature of rising again gradually.The separation of product can be by adding saturated nacl aqueous solution with any residual lithium reagent of cancellation to reactant mixture.Separate organic layer, and if expect and can be further purified, suitable 1-alkyl-4-(3-alkoxyl phenyl) piperidines 01 derivatives obtained.
The dehydration of the 4-Phenylpiperidine alcohol of above-mentioned preparation can be finished with strong acid according to known method.Take place with different amounts though dewater, preferably dewater and carry out, particularly the p-methyl benzenesulfonic acid in toluene or benzene with phosphoric acid with any of several strong acid example hydrochloric acids, hydrobromic acid etc.This reaction typically can under refluxad be carried out, and is more typically between about 50 ℃ to 150 ℃.The product of Xing Chenging can be by with the acidic aqueous solution alkalization of the salt form of product and with suitable separating with the immiscible solvent extraction of water thus.The residue that will obtain after will evaporating as required then is further purified.
1-alkyl-4-methyl-4-(3-alkoxyl phenyl) tetrahydrochysene piperidine derivative can be by metal enamine alkylation preparation.Preferred this reaction uses the n-BuLi in oxolane (THF) to carry out under inert atmosphere such as nitrogen or argon gas.Usually, slight excessive n-BuLi is joined in the 1-alkyl-4-in THF (3-the alkoxyl phenyl)-tetrahydrochysene piperidines that is cooled to-50 ℃ to about 0 ℃ approximately (more preferably being cooled to-20 ℃ to-10 ℃ approximately) of stirring.With this mixture stir about 10 to 30 minutes, in solution, add the halide of about 1.0 to 1.5 equivalents subsequently, keep the temperature of reactant mixture to be lower than 0 ℃ simultaneously.After about 5 to 60 minutes, can add entry and collect organic facies to reactant mixture.Product is according to the standard method purifying, but preferred crude product by it distill under vacuum or with it at hexane: ethyl acetate (65: 35, v: v) with in the silica gel pulled an oar about two hours and carry out purifying.According to a kind of method in back, then can be by filtering and under reduced pressure filtrate being evaporated and separated product subsequently.
Next step of this method relates to the mannich reaction (Mannich reaction) that unconjugated endocyclic enamine is carried out amino methylization.Preferred this reaction is blended in the appropriate solvent by the suitable secondary amine with the water-containing acetal of about 1.2 to 2.0 equivalents and about 1.3 to 2.0 equivalents to be carried out.Though water may be preferred solvent, also other non-nucleophilicity solvent such as acetone and acetonitrile can be used for this reaction.Available provide the anionic acid of non-nucleophilicity with the pH regulator of this solution to about 3.0 to 4.0.The example of this acid comprises sulfuric acid, sulfonic acid such as methanesulfonic acid and p-methyl benzenesulfonic acid, phosphoric acid and tetrafluoro boric acid, preferably sulfuric acid.1-alkyl-4-methyl-4-(3-alkoxyl phenyl) tetrahydropyridine (typically being dissolved in the aqueous sulfuric acid) that in this solution, adds 1 equivalent, and available non-nucleophilicity acid or the suitable secondary amine pH of regulator solution once more.PH is maintained at about 1.0 to 5.0 in the preferred process, and more preferably pH is maintained at about 3.0 to 3.5.When about 50 ℃ to about 80 ℃, when more preferably from about carrying out under 70 ℃ the temperature, reaction was finished after about 1 to 4 hour basically, more typically finished at about 2 hours.Reaction can be cooled to about 30 ℃ and join in the sodium hydroxide solution then.This solution can be used then and the immiscible organic solvent of water for example hexane or ethyl acetate extraction, and organic facies fully can be washed with evaporated under reduced pressure after removing any residual formaldehyde at water.
1-alkyl-4-methyl-4-(3-alkoxyl phenyl)-3-tetrahydropyridine methylamine catalytic hydrogenation that next step of this method can relate to preparation is corresponding anti-form-1-alkyl-3,4-dimethyl-4-(3-alkoxyl phenyl) piperidines.In fact this reaction is carried out with two steps.The first step is wherein with the hydrogenolysis of external form C-N key reductive cleavage with generation 3-methyl tetrahydropyridine.In second step, 2 of tetrahydro pyridine ring, the two keys of 3-are reduced, and obtain required piperidine ring.
The reduction of the two keys of enamine is introduced critical relative spatial chemistry at 3 of piperidine ring with 4 carbon atoms.Reduction is not usually with stereoselectivity generation completely.The catalyzer that this method adopts can be selected from various palladiums, preferred platinum catalysts.
The catalytic hydrogenation step of preferred this method is carried out in acid reaction medium.Be used for this method appropriate solvent and comprise alcohol, as methyl alcohol or ethanol; And ethyl acetate, oxolane, toluene, hexane etc.
Suitable spatial chemistry output can be depending on the amount of the catalyzer of employing.The amount that produces the required catalyzer of required spatial chemistry result can be depending on the purity of initiation material, whether has the difference of different catalyst posions and difference.
Hydrogen Vapor Pressure in the reaction vessel is not vital, but can be under about 5 to about 200psi.The concentration of the initiation material of preferred by volume is the about 20mL liquid of every gram initiation material, though also can adopt higher or lower starting material concentration.Under the condition that specifies in this article, because the impossible over-reduction of molecule, the duration of catalytic hydrogenation is not crucial.Though reaction can continue up to about 24 hours or more of a specified duration, may not need continue reducing condition after two mol of hydrogen of absorption approach.Then can be by reactant mixture being filtered by diatomite and reduction vaporization filtrate to drying and separated product.May be dispensable to being further purified of product of separating thus, the mixture of preferred diastereomer can be directly used in subsequent reaction.
Can remove alkyl substituent from the 1-position of piperidine ring by the dealkylation method of standard.Preferably, can adopt chloroformate derivative, particularly vinyl or phenyl derivatives, and remove with acid.Then, the alkoxide compound dealkylation of preparation can be obtained corresponding phenol.This reaction can be undertaken by compound is reacted in 48% hydrobromic acid aqueous solution usually.About 50 ℃ under about 150 ℃ temperature, more preferably when the reflux temperature of reactant mixture carries out, this reaction can be finished after about 24 hours basically at about 30 minutes.Then can be with mixture by with solution cooling, carry out post processing with neutralize about 8 pH value of alkali subsequently.This aqueous solution can be used and the immiscible organic solvent extraction of water.The residue of evaporation organic facies can be directly used in later step.
As the compound of the initiation material of The compounds of this invention also can by with 1-alkyl-4-methyl-4-(3-alkoxyl phenyl)-3-tetrahydropyridine methylamine in 3-position bromination, the bromo compound lithiumation that will so prepare, and the intermediate of lithiumation and halide such as methyl bromide are reacted, obtain corresponding 1-alkyl-3,4-dimethyl-4-(3-alkoxyl phenyl) tetrahydropyridine methylamine.Can and be converted into aforesaid initiation material with this compound reduction then.
As mentioned above, compound of the present invention can be used as independent stereoisomer existence.Preferably, will be as US-A-4,581,456 or as US-A-5,250,542 embodiment 1 described reaction condition is adjusted into to be stereoselective basically and the racemic mixture of two enantiomers basically is provided.Then these enantiomers are split.The method that can be used for preparing the initiation material of fractionation used in synthetic these compounds comprise usefulness (+)-or (-)-dimethylamino benzophenone acyl group tartaric acid handle alkyl-3, the racemic mixture of 4-dimethyl-4-(3-alkoxyl phenyl) piperidines.Then can be with this compound with the chloro-carbonic acid vinyl esters at 1-position dealkylation and finally be converted into required 4-(3-hydroxy phenyl) piperidines isomer.
Perhaps, 3,4-alkyl-replacement-stereoselectivity of 4-(3-hydroxy phenyl) piperidines is synthetic can pass through WerneR, J.A. wait people's Journal of Organic Chemistry, 61,587-597 (1996) and US-A-5136,040 described method is used alkoxyl phenyl lithium (20 ℃ to-100 ℃) or corresponding Grignard reagent (40 ℃ to 60 ℃) and 1, and 3-dialkyl group-4-piperidones carries out.
With the alcohol that obtains ethyl chloroformate acidylate, obtain racemic carbonic ester, with it with (+)-two-toluyl groups-D-tartaric acid (DTTA) is split effectively.The heat of the free alkali of chiral purity carbonic ester is eliminated (170-200 ℃) and is obtained required alkene.
For example, in the presence of n-BuLi with alkene with the sulfuric acid dimethyl esters methylate obtain trans-3,4-dimethyl enamine.With the enamine sodium borohydride reduction,, obtain the compound of enamine purity>99.5% subsequently by (+)-DTTA purifying.Free alkali is removed protecting group subsequently with chloro-carbonic acid phenylester demethylation, obtain (3R, 4R)-3-(3,4-dimethyl-4-piperidine base) phenol, it is the key intermediate of preparation I compound.By at Journal of Organic Chemistry, 61,587-597 (1996) and US-A-5, the method described in 136,040 is produced Aiweimopan.
As understood by a person skilled in the art, also can use (+) or (-) dibenzoyl tartaric acid to separate independent enantiomer of the present invention as required from the racemic mixture of corresponding The compounds of this invention.Preferably, obtain (+)-trans enantiomer.
Though (+) trans-3,4 stereoisomers are preferred, all possible stereoisomer of described compound is all in the scope that the present invention considers herein.The racemic mixture of stereoisomer and pure basically stereoisomer are within the scope of the invention.As used in this article, term " pure basically " is meant with respect to other possible stereoisomer, exist at least about 90 moles of %, more preferably at least about 95 moles of %, most preferably at least about the required stereoisomer of 98 moles of %.
Intermediate can pass through 3, and the 4-alkyl replaces-4-(3-hydroxy phenyl) piperidines and formula LCH
2(CH
2)
N-1CHR
3The compound prepared in reaction of C (O) E, wherein L is leaving group such as chlorine, bromine or iodine, E is a carboxylic acid, ester or acid amides, R
3With n as mentioned in the definition.Preferably, L can be chlorine, and is reflected under the existence of alkali and carries out, thereby with the azanylization of piperidines.For example, can with 4-chloro-2-cyclohexyl ethyl butyrate with (3R, 4R)-4-(3-hydroxy phenyl)-3, the contact of 4-lupetidine,, 4R)-4-(3-hydroxy phenyl)-3,4-dimethyl-1-piperidines so that 4-[(3R to be provided] ethyl butyrate.Though but optimization acid's ester can use the acid amides of free acid itself or carboxylic acid.
In the synthetic method that substitutes, the piperidines that replaces can be contacted with the methylene Arrcostab, the azanylization of piperidines.For example, can use 2-methylene-required piperidines of 3-phenylpropionic acid contact, so that 2-benzyl-3-piperidines ethyl propionate to be provided.
Another synthetic route can relate to the reaction of the piperidines and the haloalkyl nitrile of replacement.The itrile group of the piperidines alkyl nitrile that obtains can be hydrolyzed to corresponding carboxylic acid.
For each synthetic route, ester or carboxylic acid and amine or the alcohol reaction that obtains can be obtained adorned chemical constitution to provide.In preparation during acid amides, piperidines-carboxylic acid or piperidines-carboxylate can be reacted with amine in the presence of coupling agent such as dicyclohexylcarbodiimide, boric acid, borine-trimethylamine etc.Ester can by with piperidines-carboxylic acid at coupling agent such as p-methyl benzenesulfonic acid, boron trifluoride etherate or N, the existence of N '-carbonyl dimidazoles down and suitable alcohol contact and prepare.Perhaps, can use preparation piperidines-acid chlorides such as reagent such as thionyl chloride, phosphorus trichloride, phosphorus pentachloride.This alkanoyl chlorine can react with suitable amine or alcohol, to pass through corresponding amide or ester.
Put into practice the pharmaceutically acceptable incremental agent combination of selecting with the compound of formula I and according to selected method of administration and standard pharmaceutical, as for example at Remington ' s Pharmaceutical Sciences (MackPublishing Co., Easton, PA, 1980) describe in, it is openly incorporated into this paper as a reference in full.
The compound of formula I can be suitable for the various ways of selected method of administration (as, oral or non-enteron aisle) to the administration of mammal main body.In this respect, parenterai administration comprises by following administration: intravenous, muscle are interior, subcutaneous, in the intraocular, synovial membrane, through epithelium (comprise transdermal, through eye, hypogloeeis with through the cheek administration); Local (comprise eye usefulness, skin, eye, rectum and by the snuffing of insufflation go into, aerosol and rectum system) administration.
The preferred amount of reactive compound in the useful composition of this treatment is to obtain suitable dosage.Can prepare preferred compositions of the present invention or preparation, make unit dosage forms comprise about 0.1 to the reactive compound of about 1000mg, 1 reactive compound that arrives 100mg more preferably from about.
According to predetermined application, these preparations can comprise preservative to prevent microbial growth.
The pharmaceutical dosage form that is suitable for injecting application comprises aseptic aqueous solution for example and is used for preparing the aseptic powdery of aseptic injectable solution and dispersion temporarily.In all situations, preferred dosage form is aseptic, and must be the liquid of the easy degree of injection.Preferred its is stable under the condition of producing and storing and preferably prevents microorganism such as the contamination of bacterium and fungi is saved.The effect of prophylaxis of microbial can be by multiple antibacterial agent and antifungal realization, for example parabens, anesin, phenol, sorbic acid, thimerosal etc.
Aseptic Injectable solution can combine with the above-mentioned various other components of enumerating in appropriate solvent as required by the reactive compound with requirement, subsequent filtration sterilization preparation.Usually, dispersion can be combined in the sterile media by the active component with sterilization and prepare, and described medium comprises above-mentioned other composition enumerated that is selected from of alkaline dispersion medium and needs.In the situation of sterile injectable solution preparation with aseptic powdery or pie, preferred manufacturing procedure can comprise atomized drying, vacuum drying and freeze-drying (freeze drying) technology, and it produces the powder that active component adds any other required composition that derives from aforementioned aseptic filtration solution.
Composition palatable clothes of the present invention administration.For example, dry compositions of the present invention can be packaged in the gelatine capsule of duricrust or soft shell, its boil down to tablet (for example fast dissolving orally tablet, orally disintegrating tablet comprise being used for through those of cheek administration) perhaps can directly can be combined it with food.
Be used for oral tablet, lozenge, pill, capsule etc. and also can comprise following one or more, condition is the dissolution rate and the bioavilability of their improvement of not hindering composition: binding agent, as bassora gum, gum Arabic, corn starch or gelatin; Excipient is as Dicalcium Phosphate; Disintegrant is as corn starch, potato starch, alginic acid etc.; Lubricant is as dolomol; Sweetener is as sucrose, lactose or asccharin; Or flavor enhancement, as peppermint, wintergreen or cherry flavor enhancement.When unit dosage forms was capsule, it can comprise the liquid-carrier except that the material of the above-mentioned type.Can have multiple other material as dressing or be used to change the profile of dosage unit.For example, tablet can be coated with shellac dressing, sweet tablet or its both.Syrup or elixir can comprise reactive compound, as the sucrose of sweetener, as methyl p-hydroxybenzoate and propylparaben, dyestuff and flavor enhancement such as the cherry or the orange flavor enhancement of preservative.Certainly, any material that is preferred for preparing any unit dosage forms be pharmacy pure and be nontoxic basically under usage amount.In addition, reactive compound can be combined in extended release preparation and the composition.
Method of administration and the pharmacy practice decision active component of standard and the relative scale of carrier of solvability that as mentioned above, can be by compound for example and chemical property, selection.
Being suitable for most of compound of the present invention, prevent or the dosage for the treatment of changes with the concrete patient's of the particular compound of form of medication, selection and experience treatment the different of physiological characteristic.Usually, can use low dose at first, if necessary, increase with little increment, up to producing a desired effect in this case.
Comprise with peripheral mu opioid antagonists compound (as, the compound of formula I) combination product of the present invention of the opioid of combination (as, Pharmaceutical composition) can be any non-parenteral dosage forms, for example described herein those non-parenteral dosage forms, and also administration in several ways, as described herein.In preferred embodiments, combination product of the present invention (that is, in a kind of liquid, combine, or the like) formulated together in single formulation.When combination product is not in single formulation when formulated together, opioid compound and peripheral mu opioid antagonists compound can be simultaneously or (that is, together) administration synchronously or with any order administration.When not being while or administration synchronously, promptly when successive administration, the administration of preferred peripheral mu opioid antagonists and opioid is carried out in being lower than about one hour interval, more preferably less than about 30 minutes interval, more preferably less than about 15 minutes interval, more preferably less than about 5 minutes interval.
Though preferred peripheral mu opioid antagonists and opioid administration in an identical manner are (promptly, for example all be parenterai administration), if but expectation, they can be separately administration is (promptly in a different manner, the opioid component Orally-administrable of combination product for example, but peripheral mu opioid antagonists component intravenous administration).The dosage of combination product of the present invention can change according to multiple factor, as pharmacodynamic profile and the administering mode and the approach of concrete medicine; Recipient's age, health status and body weight; The nature and extent of symptom; And the treatment kind of depositing; The frequency and the required curative effect of treatment.
Though the suitable dosage of combination product of the present invention can be determined after having learned of the present invention disclosing by those skilled in the art, but as general guide, when opioid compound and the combination of peripheral mu opioid antagonists, for example, typical daily dose can be every kg of patient body weight about 0.01 and arrives about 100 milligrams peripheral mu opioid antagonists (with all combinations and the recombinant of scope in this scope) to about 100 milligrams opioid (with all combinations and the recombinant of scope in this scope) and about 0.001.Preferably, daily dose can be every kg of patient body weight about 0.1 and arrives about 10 milligrams peripheral mu opioid antagonists to about 10 milligrams opioid and about 0.01.More preferably, daily dose can be the about 1.0 milligrams opioid of every kg of patient body weight and about 0.1 milligram peripheral mu opioid antagonists.Usually exist to about 200 milligrams amount with about 5 for the representative dosage forms opioid compound of this class combination product (as, morphine), the peripheral mu opioid antagonists is about 0.1 to about 12 milligrams.
Be used for for example treating opioid administration or pain therapy peripheral mu opioid antagonists compound side effect, comprise the opioid for the treatment of effective dose and treatment effective dose at one or more sterile chambers medicinal cartridge bag also within the scope of the invention.The sterilization of container can be used and well known to a person skilled in the art that conventional sterilizing methods carries out.The sterile chamber of material can comprise independent container as required, or one or more multichamber vessel, as UNTVIAL
TMTwo-compartment container (derive from Abbott Labs, Chicago, Illinois) illustrational.Optional opioid compound and peripheral mu opioid antagonists compound are separable, or make up in aforesaid single formulation.If expectation, this cartridge bag can further comprise the conventional medicinal cartridge bag component that one or more are different, and such as the other bottle that for example is used for component is mixed etc., this it will be apparent to those skilled in the art that.Show to the amount of component, be used for the guideline of administration and/or be used for specification (as insert or as label) the mixing guideline of component, also can be included in the cartridge bag.
The compound (compound that comprises formula I) that is used for method of the present invention, composition and cartridge bag is showing that the Opioid Receptors that preferentially is incorporated into [mu characterizes in testing.Shown that in chorista (GPI and mouse vas deferens) and the research in other vitro system (as, GTP γ S) these compounds can be used as antagonist and work, do not had detectable agonist activity.The compound of the present invention that studies show that in animal can reverse the constipation in the morphine dependence mouse with very low dose oral administration or parenterai administration the time, and does not block the analgesic effect of morphine, unless give with 100 times or higher dosage.Generally speaking, these data show that compound as herein described can have the periphery selectivity of very high degree.
Embodiment
Now by the present invention being described with reference to following concrete non-limiting example.Described embodiment is not used in and limits the scope of the invention.
Embodiment 1: Aiweimopan synthetic
Prepare Aiweimopan according to following synthetic method.
Synthesizing of 1-bromo-3-(1-methyl ethoxy) benzene (compound 1)
| Reagent |
MW |
Amount (kg) |
Kilomol |
Mol ratio |
| The 3-bromophenol |
173.01 |
80.0 |
0.4624 |
1.00 |
| 2-bromo propane |
123.0 |
85.6 |
0.6959 |
1.51 |
| Potash is through grinding |
138.2 |
96.0 |
0.6946 |
1.50 |
| Ethanol 1X
* |
46.07 |
144 |
-- |
-- |
| Water |
18.02 |
739 |
-- |
-- |
| Hydrochloric acid, 31% |
36.46 |
6.6 |
-- |
-- |
| Sodium hydroxide, 50%, w/w |
40.0 |
44.4 |
-- |
-- |
| Heptane |
100.2 |
185 |
-- |
-- |
*Ethanol 1X is with 0.5% toluene sex change.
To potash (96.0kg) and the ethanol 1X (134kg) of reactor adding through grinding.Reactant mixture is adjusted to 20 to 25 ℃.
Under agitation add 3-bromophenol (80.0kg) to reactor, keeping temperature simultaneously is 20 to 35 ℃.With transfer device ethanol 1X (5kg) rinsing rapidly.With adjustment to 20 to 25 ℃.Add 2-bromo propane (85.6kg) to reactor.With transfer device ethanol 1X (5kg) rinsing rapidly.(20L) joins in the reactor with water.
Solution in the reactor is heated to 60 to 65 ℃ and remained in this scope at least 16 hours.Mixture is cooled to 45 to 50 ℃, the 3-bromophenol of test mixture.When waiting for as a result, mixture is risen again 60 to 65 ℃.Mixture is cooled to 45 to 50 ℃ once more.
Add entry (303L) to reactor.Reactant mixture is concentrated to the amount of 400L by air-distillation.The mixture that concentrates is cooled to 20 to 25 ℃.
Add heptane (185kg) to reactor, under 20 to 25 ℃ temperature, stirred at least 20 minutes then.
Separate two phase liquid and with organic layer water (45L) and hydrochloric acid (31%, solution washing 6.6kg).Organic layer water (56L) washing, subsequently water (49L) and sodium hydroxide (50%, solution washing 6.6kg).Organic layer water (56L) washing is last.
Organic solution is by azeotropic distillation drying, up to no longer collecting water.Then reactant mixture is concentrated to 150 by air-distillation and arrives the amount of 170L, and be cooled to 20 to 25 ℃.The solution packing is used for next step.Product (compound 1) sampling to packing, detect: HPLC purity is no less than 98%a/a and the HPLC check is no less than 55%w/w.
Cis-(±)-1,3-dimethyl-4-[3-(1-methyl ethoxy) phenyl]-4-piperidines alcohol (compound 2) synthetic
| Reagent |
MW |
Amount (kg) |
Kilomol |
Mol ratio |
| Compound 1 |
215.1 |
27.9 |
0.07514
* |
1.21 |
| Magnesium chips |
24.3 |
2.1 |
0.08642 |
1.39 |
| 1,3-dimethyl-4-piperidine ketone |
127.2 |
7.9 |
0.06211 |
1.00 |
| Oxolane |
72.01 |
162 |
-- |
-- |
| Ammonium chloride |
53.5 |
6.6 |
-- |
-- |
| Water |
18.02 |
56 |
-- |
-- |
| Hyflo supercel |
-- |
4 |
-- |
-- |
| Heptane |
100.2 |
86.5 |
-- |
-- |
*Check according to reagent is calculated
Before using by the gross, to the oxolane sample analysis water content of using.
Add oxolane (18kg) and do not having to be heated to backflow under the condition of stirring to reactor.Solvent is kept refluxing 1 hour and being cooled to 30 ℃ or lower.Carrying out KF analyzes to guarantee that the water content in the reactor satisfies specification requirement.THF is discharged to the waste liquid cylinder and with the reactor drying.
Add magnesium (2.1kg) to reactor, add oxolane (80kg) subsequently.Under agitation, reactant mixture is concentrated to 40 to the amount of 44L by air-distillation.Concentrate is cooled to 40 to 45 ℃.
Add oxolane (18kg) and stirred at least 20 minutes to the stainless steel tank of portable stirring.Carrying out KF analyzes to guarantee that water content satisfies specification requirement in the reactor.THF is discharged to the waste liquid cylinder.
In groove, add 1-bromo-3-(1-methyl ethoxy) benzene (27.9kg) and oxolane (31kg).Solution was at room temperature stirred 20 minutes at least.
The 2.5kg of mixture in the groove partly transferred in 40 to 45 ℃ the reactor.Under agitation with mixture 40 to 60 ℃ of maintenances at least 30 minutes.
Second 2.5kg of mixture in the groove partly transferred in 40 to 45 ℃ the reactor.Under agitation, with mixture 40 to 60 ℃ of maintenances at least 30 minutes.
The 5kg of the mixture in the groove partly transferred in 40 to 45 ℃ the reactor.Under agitation, with mixture 40 to 60 ℃ of maintenances at least 30 minutes.
Add 1 to groove, 3-dimethyl-4-piperidine ketone (7.9kg) and with transfer device with oxolane (5kg) rinsing rapidly.
The 15kg of the mixture in groove part transferred at least 1 hour in 40 to 45 ℃ the reactor.Under agitation mixture was kept 15 to 30 minutes at 40 to 60 ℃.Reactant mixture is cooled to 40 to 45 ℃.
Second 15kg part of the mixture in the groove transferred at least 1 hour in 40 to 45 ℃ the reactor.Under agitation, mixture was kept 15 to 30 minutes at 40 to 60 ℃.Reactant mixture is cooled to 40 to 45 ℃.
The 3rd 15kg part of the mixture in the groove transferred at least 1 hour in 40 to 45 ℃ the reactor.Under agitation, mixture was kept 15 to 30 minutes at 40 to 60 ℃.Reactant mixture is cooled to 40 to 45 ℃.
The remainder of the mixture in the groove transferred at least 1 hour in 40 to 45 ℃ the reactor.With transfer device THF (5kg) rinsing rapidly.Under agitation, mixture was kept 15 to 30 minutes at 40 to 60 ℃.Reactant mixture is cooled to 40 to 45 ℃.
After reaction is finished, mixture is cooled to 20 to 25 ℃.
Add entry (40L) and ammonium chloride (6.6kg) to second reactor.Under the stirring of appropriateness, with mixture 20 to 25 ℃ of maintenances at least 20 minutes.
In case the solid dissolving adds Hyflo supercel (4kg) to second reactor.Moisture mixture is cooled to 0 to 5 ℃.
Under agitation the organic mixture in first reactor is transferred in second reactor.With transfer device THF (5kg) rinsing rapidly.Mixture is heated to 20 to 25 ℃ and kept at least 15 minutes.
Mixture is filled in first reactor, with the rapid rinsing of heptane (2 * 6kg), and 20 to 25 ℃ of maintenances at least 20 minutes.
Separate two phase liquid and organic layer water (16L) is washed.Organic solution is concentrated to 30 by air-distillation arrives the amount of 34L, and be cooled to 45 to 50 ℃.
Add heptane (54kg) and solution is concentrated to 69 to 73L amount by air-distillation to reactor.Solution is cooled to 30 to 35 ℃.Residual oxolane and water content in the check reactant mixture.To react with product crystal seeding and in minimum 1 hour, mixture will be cooled to 0 to 5 ℃ and also keep at least 3 hours.
By the isolated by filtration solid product, (2 * 10kg) washings are also dry with cold heptane.Collect product sample drying and packing.To product (compound 2) sampling of packing, detect: HPLC purity is no less than 97%a/a, unpacks before being used for next step.
Cis-(±)-1,3-dimethyl-4-[3-(1-methyl ethoxy) phenyl]-purifying of 4-piperidines alcohol (compound 2)
| Reagent |
MW |
Amount (kg) |
Kilomol |
Mol ratio |
| Compound 2 |
263.4 |
96.1 |
0.3648 |
1.00 |
| Heptane |
100.2 |
590 |
-- |
-- |
Add compound 2 (96.1kg) and heptane (328L) to reactor.Mixture is heated to 55 to 60 ℃ also to be kept 1 hour at least.Test mixture is to guarantee all solid dissolvings.
In at least 1 hour solution being cooled to 30 to 35 ℃ also kept 1 hour at least.Test mixture takes place to guarantee precipitation.In at least two hours mixture being cooled to 0 to 5 ℃ also kept 4 hours at least.
By isolated by filtration solid pure compound 2, with cold heptane wash (2 * 131kg) and dry.Collect that the product sample is used for drying and with its packing.Product sampling to packing detects HPLC purity, is no less than 97%a/a, unpacks before being used for next step.
Ethyl carbonate (3S, 4R)-1,3-dimethyl-4-[3-(1-methyl ethoxy) phenyl]-4-piperidyl ester compounds and (+)-D-2,3-two [(4-methyl benzoyl) oxygen base] succinic acid (1: 1) (compound 3) synthetic
| Reagent |
MW |
Amount (kg) |
Kilomol |
Mol ratio |
| Compound 2 |
263.4 |
10.8 |
0.04100 |
1.00 |
| The chloracetic acid ethyl ester |
108.52 |
5.6 |
0.05160 |
1.26 |
| Triethylamine, anhydrous |
101.19 |
0.4 |
0.003953 |
0.10 |
| (+)DTTA |
386.36 |
15.8 |
0.04089 |
1.00 |
| Sodium hydroxide, 50%w/w |
40.0 |
47.6 |
-- |
-- |
| Ethyl acetate |
88.11 |
52 |
-- |
-- |
| Ethanol 1X |
46.07 |
285 |
-- |
-- |
Add compound 2 (10.8kg) and ethyl acetate (48L) to reactor.Mixture 20 to 25 ℃ of maintenances 30 minutes, is dissolved up to all solids at least.Solution is cooled to 0 to 5 ℃.
To reactor adding triethylamine (0.4kg) and with transfer device ethyl acetate (1kg) rinsing rapidly.
Add ethyl chloroformate (5.6kg) to reactor, keep 0 to 15 ℃ temperature simultaneously.With transfer device ethyl acetate (3kg) rinsing rapidly.With mixture 20 to 25 ℃ of maintenances at least 3 hours.
Add 50% sodium hydroxide (7.6kg) to reactor, keep 0 to 38 ℃ temperature simultaneously.With transfer device water (17L) rinsing rapidly.With solution at 20 to 25 ℃ of maintenances at least 20 minutes and the pH that checks solution to guarantee that pH is greater than 10.
Separate two phase liquid and with organic layer water (22L) washed twice.Organic solution is concentrated to 20 to 24L amount then by azeotropic distillation drying by air-distillation.Solution is cooled to 40 to 50 ℃.
Add ethanol 1X (60kg) to reactor.Organic solution is concentrated to 30 by air-distillation arrives the amount of 34L, and be cooled to 55 ℃ to 60 ℃.
Adding (+)-two in having glass-lined reactor-to toluyl groups-D-tartaric acid (15.8kg) and ethanol 1X (51kg).Under the stirring of appropriateness, regulate temperature to 60 to 65 ℃.
Reactant mixture is transferred in the acid solution, kept 60 to 70 ℃ temperature simultaneously.With transfer device ethanol 1X (17kg) rinsing rapidly.Solution was kept 1 to 1.5 hour at 60 to 65 ℃.Suspension is cooled to 50 to 55 ℃ and kept 2 to 2.5 hours.In at least 3 hours suspension being cooled to 20 to 25 ℃ also kept 10 hours at least.
By the isolated by filtration solid, with ethanol 1X washing (17kg), dry and packing.To the crude product sampling of packing and the chiral purity of detection compound 3.
Add crude product and ethanol 1X (according to calculating) to reactor.Mixture is adjusted to 60 to 65 ℃ and kept 2 to 2.5 hours.In at least 2 hours, suspension is cooled to 20 to 25 ℃.Suspension is cooled to 0 to 5 ℃ also to be kept 3 hours at least.
By isolated by filtration solid chemical compound 3, with cold ethanol 1X (17kg) washing, dry and packing.Product sampling to packing detects, and HPLC purity is no less than 99.0%a/a; Chirality HPLC is no less than 99.5%, and unpacks before being used for next step.
(3R, 4R)-3-(3,4-dimethyl-4-piperidine base) phenol (compound 4) synthetic
| Reagent |
MW |
Amount (kg) |
Kilomol |
Mol ratio |
| Compound 3 |
647.8 |
18.3 |
0.02825 |
1.00 |
| Toluene |
92.14 |
50 |
-- |
-- |
| Water |
18.02 |
434 |
-- |
-- |
| Sodium hydroxide, 50%w/w |
40.0 |
110.7 |
-- |
-- |
| The carbonochloridic acid phenyl ester |
156.57 |
5.3 |
0.03385 |
1.20 |
| Hydrochloric acid, 31% |
36.46 |
2.8 |
-- |
-- |
| Glacial acetic acid |
60.05 |
17.6 |
0.2931 |
10.38 |
| Hydrobromic acid |
80.92 |
19 |
0.1127 |
4.00 |
| T-butyl methyl ether |
88.15 |
56 |
-- |
-- |
| Methyl alcohol |
32.04 |
8.7 |
-- |
-- |
Add compound 3 (18.3kg), toluene (48kg) and water (32L) to reactor.Mixture is adjusted to 20 to 25 ℃.
To reactor add sodium hydroxide (50%, 9.2kg), keep 20 to 30 ℃ temperature simultaneously.With transfer device water (4L) rinsing rapidly.Under agitation mixture is cooled to 20 to 25 ℃ and kept 1 hour.The pH that checks water layer is to guarantee that pH is greater than 12.
Separate two phase liquid and with organic layer water (14L) and sodium hydroxide (50%, solution washing 0.7kg).Organic layer water (15L) washed twice is also passed through azeotropic distillation drying.Solution is cooled to 80 to 85 ℃.
In at least 1.5 hours, phenyl chloroformate (5.3kg) is joined in the reactor, keep 80 to 85 ℃ temperature simultaneously.With transfer device toluene (2kg) rinsing rapidly.Solution is heated to refluxes and kept at least 3 hours, be cooled to 50 to 55 ℃ then.When waiting for as a result, mixture is kept refluxing.
Mixture is cooled to 20 to 25 ℃ and water (14L) joined in the reactor.In at least 1 hour to reactor add sodium hydroxide (50%, 2.3kg), keep 20 to 30 ℃ temperature simultaneously.With transfer device water (4L) rinsing rapidly.With solution 20 to 25 ℃ of maintenances at least 1 hour.
Separate two phase liquid and with organic layer water (15L) and hydrochloric acid (31%, solution washing 1.9kg).Organic solution is concentrated to 23 by air-distillation arrives the amount of 26L, and be cooled to 65 to 70 ℃.
Add entry (7L) and acetate (13.6kg) to reactor.With transfer device water (2L) rinsing rapidly.Organic solution is concentrated to 26 by air-distillation arrives the amount of 29L, and be cooled to 50 ℃ to 60 ℃.
Add hydrobromic acid (19kg) to reactor, add acetate (4kg) subsequently.Solution is heated to refluxes and kept at least 18 hours.Solution is cooled to 55 to 60 ℃.Solution is cooled to 10 to 15 ℃.
In at least 1 hour to reactor add sodium hydroxide (50%, 6kg), keep 10 to 30 ℃ temperature simultaneously.With transfer device water (5L) rinsing rapidly.With adjustment to 20 to 25 ℃ and check that pH is to guarantee that pH is less than 1.7.
Add t-butyl methyl ether (16kg) to reactor, keep 20 to 25 ℃ temperature simultaneously.To reactor add entry (27L) and with solution 20 to 25 ℃ of maintenances at least 30 minutes.
Separate two phase liquid and the aqueous solution is transferred in the reactor.Organic solution is transferred in the 200L glass recipient.Aqueous solution t-butyl methyl ether (16kg) washed twice.
Organic layer is transferred to the reactor from the glass recipient.Add entry (5L) to reactor, add subsequently hydrochloric acid (31%, 0.9kg), keep 20 to 25 ℃ temperature simultaneously.With transfer device water (2L) rinsing rapidly.With two phase liquid 20 to 25 ℃ of maintenances at least 20 minutes.
Separate two phase liquid and with the aqueous solution with t-butyl methyl ether (4kg) washed twice.
Acid solution is transferred to the 200L reactor from new PE bucket.With transfer device water (2L) rinsing rapidly.
In at least 30 minutes, add methyl alcohol (8.7kg), keep 20 to 25 ℃ temperature simultaneously to reactor.
In the stainless steel tank of portable stirring, add entry (41L) and sodium hydroxide (50%, 12.5kg).With transfer device water (4L) rinsing rapidly.Solution is transferred in the reactor, realized 10.0 to 10.5 pH, keep 20 to 35 ℃ temperature simultaneously.
Suspension is cooled to 0 to 5 ℃ also to be kept 4 hours at least.
By isolated by filtration, (2 * 9L), drying is also packed with cold water washing with compound 4.To the product sampling of packing, detect: HPLC purity is no less than 98.5%a/a; Chiral purity be no less than 99.0% and HPLC check be no less than 95%w/w, and before being used for next step, unpack.
(α S, 3R, 4R)-4-(3-hydroxy phenyl)-3,4-dimethyl-α-(phenyl methyl)-1-piperidines methyl propionate hydrochloride (compound 6) synthetic
Compound 4 compounds 5 compounds 6
| Reagent |
MW |
Amount (kg) |
Kilomol |
Mol ratio |
| Compound 4 |
205.3 |
19.2 |
0.09352 |
1.00 |
| Methyl acrylate |
86.09 |
8.5 |
0.09875 |
1.05 |
| Oxolane |
72.11 |
692 |
-- |
-- |
| N-BuLi |
64.06 |
87.4 |
0.2056 |
2.20 |
| Diisopropylamine |
101.19 |
21.8 |
0.2154 |
2.30 |
| Bromobenzyl |
171.04 |
32.0 |
0.1871 |
2.00 |
| Heptane |
100.21 |
209 |
-- |
-- |
| Methyl alcohol |
32.04 |
659 |
-- |
-- |
| Hydrochloric acid, 31% |
36.46 |
36.2 |
0.3078 |
3.29 |
| Sodium hydroxide, 50%, w/w |
40.0 |
4.9 |
0.06125 |
0.65 |
| Hydrogen chloride gas |
36.46 |
14.4 |
0.3950 |
4.23 |
| Hyflo supercel |
-- |
1.9 |
-- |
-- |
| Water |
18.02 |
566 |
-- |
-- |
Add compound 4 (19.2kg) and oxolane (222kg) to reactor.Under 50% stirring, mixture is heated to 40 to 45 ℃.
In at least 30 minutes, add methyl acrylate (8.5kg), keep 40 to 45 ℃ temperature simultaneously to reactor.With transfer device THF (17kg) rinsing rapidly.Reactant mixture was kept 18 to 19 hours at 40 to 45 ℃.Reactant mixture is cooled to 20 to 25 ℃.
Add Hyflo supercel (1.9kg) and heptane (13kg) to the stainless steel tank of portable stirring.Mixture was stirred five minutes at least.Mixture transferred in the reactor and with heptane (5kg) rinsing rapidly.With mixture 20 to 25 ℃ of maintenances at least 20 minutes.
Mixture is filled in the reactor so that clarification, with the rapid rinsing of heptane (26kg) and be cooled to-5 to 0 ℃.Solution is concentrated to 29 to the amount of 48L by vacuum distillation, obtains the solution of compound 5.
30 ℃ or more low temperature downhill reaction device add heptane (26kg).Solution is cooled to-5 to 0 ℃ and be concentrated to 29 to the amount of 48L by vacuum distillation.
Add oxolane (333kg) to reactor, add diisopropylamine (21.8kg) subsequently.With transfer device oxolane (12kg) rinsing rapidly.Solution is cooled to-15 to-10 ℃.
In at least 1 hour, add the hexane (87.4kg) that contains n-BuLi, keep-15 to-5 ℃ temperature simultaneously to reactor.With transfer device THF (2 * 5kg) rapid rinsings.Solution was kept 1 to 3 hour at-10 to-5 ℃, be cooled to-25 to-20 ℃ then.
Acrylate solution in the reactor is transferred in this reactor, kept-25 to-15 ℃ temperature simultaneously.With transfer device THF (8kg) rinsing rapidly.Suspension was kept 30 to 60 minutes at-25 to-20 ℃.
In at least 2 hours, add bromobenzyl (32.0kg), though keep-25 to-20 ℃ temperature to reactor.With transfer device THF (8kg) rinsing rapidly.With mixture-25 to-20 ℃ of maintenances at least 16 hours.
To portable storage tank add entry (61L) and hydrochloric acid (31%, 18.1kg), stirred at least two minutes then, to form solution.To second portable storage tank add entry (61L) and hydrochloric acid (31%, 18.1kg), stirred at least two minutes then, to form solution.In at least two hours, two kinds of acid solutions are transferred in the reactor, kept-25 to-15 ℃ temperature simultaneously.In at least three hours, solution is warmed up to 20 to 25 ℃.
To portable storage tank add entry (29L) and sodium hydroxide (50%, 4.9kg).Stir at least two minutes to form solution with the rapid rinsing of transfer device water (15L) and with mixture.
(29kg) transfers in the reactor with alkaline solution, keeps 20 to 25 ℃ temperature simultaneously, up to the pH that obtains 9.0 to 9.5.Separate two phase liquid and the aqueous solution is transferred in the 600L reactor.
The aqueous solution washs with heptane (26kg).The organic solution that obtains transferred in the 1500L reactor and with transfer device with heptane (17kg) rinsing rapidly.Solution is cooled to-30 to-20 ℃.
Add methyl alcohol (113kg) and be cooled to-30 to-20 ℃ to reactor.Add hydrogen chloride gas (14.4kg) to reactor, keep-30 to-10 ℃ temperature simultaneously.
Acid solution is joined in the above-mentioned reactor, keep-30 to-5 ℃ temperature simultaneously.With transfer device methyl alcohol (19kg) rinsing rapidly.Solution temperature is adjusted to-10 to-5 ℃.Solution is concentrated to 168 to the amount of 216L by vacuum distillation.
Solution is transferred in the 600L reactor also with methyl alcohol (48kg) rinsing rapidly.Solution is cooled to-10 to-5 ℃ and be concentrated to 48 to the amount of 68L by vacuum distillation.
In the 1500L reactor, add methyl alcohol (77kg) and be rinsed in the reactor.Then solution is cooled to-10 to-5 ℃ and be concentrated to 48 to the amount of 68L by vacuum distillation.
30 ℃ or more low temperature downhill reaction device add methyl alcohol (106kg), be heated to 40 to 45 ℃ then.Solution was kept 1 to 2 hour at 40 to 45 ℃.In at least 3 hours, solution is cooled to 20 to 25 ℃ and kept at least 1 hour in this scope.In at least 1 hour, solution is cooled to 2 to 7 ℃ and kept at least 1 hour in this scope.
By isolated by filtration crude product compound 6, with cold methanol washing (2 * 15kg), and detect purity.Keep filtrate and be used for further processing.
Add wet cake and methyl alcohol (60kg) to reactor.Mixture is heated to refluxes and under refluxing, kept 1 to 2 hour.In at least 4 hours, solution is cooled to 2 to 7 ℃ and kept at least 1 hour in this scope.
By the isolated by filtration crude product, with cold methanol washing (2 * 15kg), and detect purity.Keep filtrate and be used for further processing.
Add wet cake and methyl alcohol (60kg) to reactor.Mixture is heated to refluxes and under refluxing, kept 1 hour.In at least 4 hours, solution is cooled to 2 to 7 ℃ and kept at least 1 hour in this scope.
By the isolated by filtration crude product, with cold methanol washing (2 * 15kg), and detect purity and chirality HPLC.
Add wet cake and methyl alcohol (60kg) to reactor.Mixture is heated to refluxes and under refluxing, kept 1 hour.In at least 4 hours, solution is cooled to 2 to 7 ℃ and kept at least 1 hour in this scope.
By isolated by filtration product compound 6, (2 * 15kg), sampling is used to analyze HPLC purity, chirality HPLC and isomer and with its packing with cold methanol washing.Product sampling to packing detects: HPLC>99.0%a/a; Chirality HPLC, 3.0%: and before being used for next step, unpack.
(α S, 3R, 4R)-4-(3-hydroxy phenyl)-3,4-dimethyl-α-(phenyl methyl)-1-piperidines propionic acid monohydrate (compound 7) synthetic
Compound 6 compounds 7
| Reagent |
MW |
Amount (kg) |
Kilomol |
Mol ratio |
| Compound 6 |
417.97 |
9.9 |
0.02369 |
1.00 |
| Methyl alcohol |
32.04 |
107 |
-- |
-- |
| Hydrochloric acid, 31% |
36.46 |
9.4 |
0.07992 |
3.37 |
| Sodium hydroxide, 50%, w/w |
40.0 |
7.9 |
0.09875 |
4.16 |
| Water |
18.02 |
~244 |
-- |
-- |
Add compound 6 (9.9kg) and water (74L) to reactor.Mixture is adjusted to 20 to 25 ℃.
In at least 10 minutes to reactor add sodium hydroxide (50%, 7.9kg).With transfer device water (10L) rinsing rapidly.The pH that checks mixture is to guarantee that pH is greater than 12.
Solution remained under 20 to 25 ℃ the temperature and stirred at least 4 hours.Then reactant mixture is filled in the reactor, so that clarification.With product water (8L) rinsing rapidly.
Add methyl alcohol (84kg) and be adjusted to 20 to 25 ℃ to reactor.
To reactor add in batches hydrochloric acid (31%, 6.9kg) up to the pH that reaches 9.0 to 10.0.
To new PE bucket add entry (7.6L) and hydrochloric acid (31%, 2.5kg).With transfer device water (4.0L) rapidly rinsing and with solution stirring at least two minutes so that mix.
Add methyl alcohol (0.4kg), water (0.5L) and compound 7 (100g) to beaker.Mixture joined in the reactor and the rapid rinsing of solution of water (0.3L) and methyl alcohol (0.2kg), be the reactant mixture seeding.
With the pH of acid solution (10.4kg) the conditioned reaction mixture for preparing, up to the pH that obtains 5.8 to 6.2.Mixture 20 to 25 ℃ of maintenances 1 hour, and is tested to guarantee that crystallization takes place at least.Suspension is cooled to 0 to 5 ℃ and be concentrated to 107 to the amount of 124L by vacuum distillation.Suspension is adjusted to 20 to 25 ℃ and check that pH is to guarantee that pH is between 5.8 and 6.2.
Suspension is cooled to 2 to 7 ℃ and stirred at least 4 hours.
By isolated by filtration, (2 * 30L), dry, sample analysis water content is also packed with cold water washing with product.Product sampling to packing detects: HPLC purity 98.%%a/a; Chirality HPLC:98% and HPLC check 98.0%w/w, and before being used for next step, unpack.
Synthesizing of [[2 (S)-[[4 (R)-(3-hydroxy phenyl)-3 (R), 4-dimethyl-1-piperidyl] methyl]-1-oxo-3-phenyl propyl] amino] acetate dihydrate (Aiweimopan)
Compound 7 compounds 8 Aiweimopans
| Reagent |
MW |
Amount (kg) |
Kilomol |
Mol ratio |
| Compound 7 |
385.5 |
7.9 |
2.02049 |
1.00 |
| Glycine ethyl ester hydrochloride |
139.58 |
3.1 |
0.02254 |
1.10 |
| The I-hydroxybenzotriazole hydrate |
135.13 |
3.5 |
0.02562 |
1.25 |
| Triethylamine |
101.2 |
2.3 |
0.02254 |
1.10 |
| 1, the 3-dicyclohexylcarbodiimide |
206.33 |
4.7 |
0.02254 |
1.10 |
| Oxolane |
72.11 |
156 |
-- |
-- |
| Ethyl acetate |
88.11 |
858 |
-- |
-- |
| Sode ash (sodium carbonate) |
105.99 |
4.8 |
-- |
-- |
| Sodium bicarbonate |
84.00 |
3.1 |
-- |
-- |
| Salt solution |
-- |
112 |
-- |
-- |
| Ethanol 1X |
46.07 |
743 |
-- |
-- |
| Purify waste water |
18.02 |
1196 |
-- |
-- |
| Sodium hydroxide, 50%, w/w |
40.0 |
16.8 |
-- |
-- |
| Hydrochloric acid, 31% |
36.46 |
30.0 |
-- |
-- |
Add oxolane (15kg) and 1,3-dicyclohexylcarbodiimide (4.7kg) to the stainless steel tank of portable stirring (PAST).With transfer device THF (16kg) rinsing rapidly.
Add compound 7 (7.9kg), glycine ethyl ester hydrochloride (3.1kg), I-hydroxybenzotriazole hydrate (3.5kg), oxolane (99kg) and purify waste water (3.3kg) to reactor.Under 60% stirring, mixture is adjusted to 20 to 25 ℃.
Add triethylamine (2.3kg) to reactor.With transfer device oxolane (3kg) rinsing rapidly.Solution was kept 20 to 60 minutes at 20 to 25 ℃.
With 1,3-dicyclohexylcarbodiimide solution is transferred in the reactor, keeps 20 to 25 ℃ temperature simultaneously.With transfer device oxolane (23kg) rinsing rapidly.
Reactant mixture was kept 36 to 38 hours at 20 to 25 ℃ under 100% stirring.
Reactant mixture is cooled to 0 to 5 ℃.Mixture was kept in this scope 1 to 2 hour, be filled into then in another reactor.With reactant mixture ethyl acetate (20kg) rinsing rapidly.Mixture is cooled to 0 to 5 ℃ and be concentrated to 140 to the amount of 149L by vacuum distillation.
Add ethyl acetate (731kg) and be cooled to 0 to 5 ℃ to reactor.Solution is concentrated to 140 to the amount of 149L by vacuum distillation.The residual oxolane of check reactant mixture.
Add to the stainless steel tank of portable stirring purify waste water (94kg), ground caustic (4.8kg) and sodium bicarbonate (3.1kg).Mixture was stirred two minutes at least, dissolve up to solid.
To reactor add alkaline solution and with adjustment to 20 to 25 ℃.To stir 60% and keep 20 to 60 minutes.Check solution 60% guaranteeing that 60% be 9.5 to 10, and regulate as required.Separate two phase liquid and use salt solution (112kg) to wash organic solution.
Add ethyl acetate (87kg) and be cooled to 0 to 5 ℃ to reactor.Solution is concentrated to 140 by vacuum distillation arrives the amount of 149L, and be cooled to-25 to-20 ℃.Kept 10 to 11 hours in this temperature.
With suspension filtered in reactor, with rapidly rinsing and be warmed up to 0 to 5 ℃ of ethyl acetate (20kg).Filtrate is concentrated to 39 to the amount of 51L by vacuum distillation.
Add ethanol 1X (199kg) and be cooled to 0 to 5 ℃ to reactor.Solution is concentrated to 136 to the amount of 161L by vacuum distillation.The residual ethyl acetate that adds ethanol 1X (93kg) and test mixture to reactor.
To portable storage tank add purify waste water (83L) and sodium hydroxide (50%, 5.6kg).With transfer equipment with (19kg) the rapidly rinsing of purifying waste water.Mixture is stirred at least two minutes to form solution.Alkaline solution is transferred in the reactor and kept 1.5 to 3.5 hours at 20 to 25 ℃.With suspension filtered in reactor and be adjusted to 20 to 25 ℃.With the 600L reactor with (13kg) the rapidly rinsing of purifying waste water.
To portable storage tank add purify waste water (15L) and hydrochloric acid (31%, 11.2kg).With transfer equipment with (5kg) the rapidly rinsing of purifying waste water.Mixture is stirred at least two minutes to form solution.Acid solution is joined in the reactor up to the pH that realizes 5.8 to 6.2 in batches.
By the isolated by filtration crude product, (2 * 26kg), (2 * 26kg), drying is also packed with ethanol 1X washing with the washing of purifying waste water.
Crude product and purify waste water (according to calculating) are joined in the reactor.
To new PE bucket add purify waste water (1.9L) and sodium hydroxide (50%, 5.3kg).With transfer equipment with (1.0kg) the rapidly rinsing of purifying waste water.Mixture is stirred at least two minutes to form solution.
Use alkaline solution (7.5kg) reactant mixture to be adjusted to minimum 13 pH.Mixture was kept 20 to 60 minutes at 20 to 25 ℃.
Reactant mixture is filled in the reactor, so that clarification.With reactor with (10kg) the rapidly rinsing and add ethanol 1X (according to calculating) of purifying waste water.
To portable storage tank add purify waste water (14L) and hydrochloric acid (31%, 9.6kg).With transfer equipment with (4kg) the rapidly rinsing of purifying waste water.Mixture is stirred at least two minutes to form solution.Acid solution is joined in the reactor up to the pH that obtains 4.0 to 4.5 in batches.
To new PE bucket add purify waste water (1.9L) and sodium hydroxide (50%, 0.3kg).With transfer equipment with (1.0kg) the rapidly rinsing of purifying waste water.Mixture is stirred at least two minutes to form solution.Alkaline solution is joined in the reactor up to the pH that obtains 5.8 to 6.2 in batches.
Check whether mixture exists solid and with suspension 20 to 25 ℃ of maintenances at least 12 hours.
By the isolated by filtration product, at first with the washing of purifying waste water (according to calculating), then with ethanol 1X washing (according to calculating), once more with the washing of purifying waste water (according to calculating).With filtration cakes torrefaction and packing.
Crude product and purify waste water (according to calculating) are joined in the reactor.
To new PE bucket add purify waste water (1.9L) and sodium hydroxide (50%, 5.3kg).With transfer equipment with (1.0kg) the rapidly rinsing of purifying waste water.Mixture is stirred at least two minutes to form solution.Alkaline solution is joined in the reactor up to the pH that obtains minimum 13 in batches.
Mixture was stirred 20 to 60 minutes at 20 to 25 ℃.Reactant mixture is filled in another reactor, so that clarification.With reactor with (10kg) the rapidly rinsing of purifying waste water.Add ethanol 1X (according to calculating) to reactor.
To portable storage tank add purify waste water (13.5L) and hydrochloric acid (31%, 9.2kg).With transfer equipment with (3.9kg) the rapidly rinsing of purifying waste water.Mixture is stirred at least two minutes to form solution.Acid solution is joined in the reactor up to the pH that obtains 4.0 to 4.5 in batches.
To new PE bucket add purify waste water (1.9L) and sodium hydroxide (50%, 0.3kg).With transfer equipment with (1.0kg) the rapidly rinsing of purifying waste water.Mixture is stirred at least two minutes to form solution.Alkaline solution is joined in the reactor up to the pH that obtains 5.8 to 6.2 in batches.
Check whether mixture exists solid and with suspension 20 to 25 ℃ of maintenances at least 12 hours.
By the isolated by filtration product, at first with the washing of purifying waste water (according to calculating), then with ethanol 1X washing (according to calculating), once more with the washing of purifying waste water (according to calculating).Filter cake sampling is used for analyzing chloride, dry and with its packing.
Add over-drying product and purify waste water (2.0kg) to drier, purge and at room temperature leave standstill, up to realizing concrete hydration level with nitrogen.
Then hydrated product is packed and joined in the product blender of 50L L.Product was mixed 20 to 30 minutes and the drying of taking a sample.Product is mixed other 20 to 30 minutes and resample.
With the Aiweimopan packing, take a sample then, detect: HPLC purity is no less than 99.2%w/w; Chirality HPLC is no less than 99.0%; HPLC measures, and 98.0 to 102.0%w/w; Be no more than the 1.2%w/w total amount with residual solvent, and the packing that opens it.
Embodiment 2: the evaluation of injectable formulation (preparation ability and stability test)
Comprise the preparation of the injection preparation of Aiweimopan
Aiweimopan is [[(25)-2-[[(3R, 4R)-4-(3-hydroxy phenyl)-3,4-lupetidine-1-yl] methyl]-1-oxo-3-phenyl propyl] amino] the acetate dihydrate.It is used with its natural hydrated form.All weight measurement of Aiweimopan and concentration are all according to anhydrous basic representation.
The product formulation that cooperates four 100ml batch of materials:
| Preparation |
Describe |
| 1 (relatively using) |
Aiweimopan (1mg/ml), in 2% glycine (incremental agent), pH 10.5, regulate with 10N and 1N sodium hydroxide |
| 2 (relatively using) |
Aiweimopan (1mg/ml), in 5% sodium carbonate (buffer), pH 10.5, regulate with 1N hydrochloric acid |
| 3 |
Aiweimopan (1mg/ml), in having 3% mannitol (incremental agent) of 50mM sodium carbonate (buffer), pH 10.5, regulate with 1N hydrochloric acid |
| 4 (relatively using) |
Aiweimopan (1mg/ml), in having 3% mannitol (incremental agent) of 50mM boric acid (buffer), pH 10.5, regulate with 1N sodium hydroxide |
Buffer and incremental agent joined in 80% the final solution volume and regulate pH to 10.5 before adding active component (Aiweimopan).Then solution is adjusted to pH 10.5 again and be mixed into clear solution.Make solution reach final volume then and pH is adjusted to pH 10.5 with 1N sodium hydroxide.With final solution aseptic filtration.The equal portions aliquot of 12.5mL is assigned in the bottle, covers stopper then and place on the freeze-dryer shelf and be used for drying.With preparation 1 to 4 freeze drying, simultaneously the following kneading of preparation is handled then:
| Step |
Temperature, pressure |
Time |
| 1 |
Be set to 5 ℃ ± 3 ℃ |
|
| 2 |
At 5 ℃ ± 3 ℃ dippings |
At least 60 minutes |
| 3 |
The inclined-plane shelf temperature reaches-40 ℃ ± 3 ℃ |
In about 90 minutes |
| 4 |
-40 ℃ ± 3 ℃ maintenances |
About 120 minutes |
| 5 |
The inclined-plane shelf temperature reaches-20 ℃ ± 3 ℃ |
In about 60 minutes |
| 6 |
-20 ℃ ± 3 ℃ maintenances |
In about 120 minutes |
| 7 |
The inclined-plane shelf temperature reaches-40 ℃ ± 3 ℃ |
In about 60 minutes |
| 8 |
-40 ℃ ± 3 ℃ maintenances |
In about 120 minutes |
| 9 |
Freezer condenser is to-50 ℃ or lower and the chamber found time |
|
| 10 |
Purge the control vacuum at 100 ± 10 microns with nitrogen |
|
| 11 |
The inclined-plane shelf temperature reaches-15 ℃ ± 3 ℃ |
In about 50 minutes |
| 12 |
-15 ℃ ± 3 ℃ maintenances |
In about 3000 minutes |
| 13 |
The inclined-plane shelf temperature reaches-30 ℃ ± 3 ℃ |
In about 90 minutes |
| 14 |
When circulation finishes, slowly return to 11 ± 0.5 pounds/square inch with the nitrogen N .F. chamber of making, filter retentivity filter by kill microorganisms |
|
| 15 |
Accordion partition makes stopper in place |
|
| 16 |
Make the chamber return to environmental pressure |
|
The result that freeze drying and kneading are handled is as follows:
| Preparation |
The result |
| 1 (relatively using) |
The product of serious fusing |
| 2 (relatively using) |
Uniform cake, but slight haze is arranged when organizing structure again in water |
| 3 |
Have about packing volume and evenly white uniform cake; Again organize the colourless solution that structure produces clarification with 12.2ml water for injection or sodium chloride for injection, when visual inspection, do not find particle (organizing structure pH afterwards again is 10.6-10.7) |
| 4 (relatively using) |
The cake that subsides and dwindle |
Preparation 3 produces and seems physically stable and lyophilisation product organize structure easily again with 1mg/mL in water for injection or sodium chloride.
Embodiment 3: the bioavilability test
Preparation comprises the following preparation of Aiweimopan, is used for the bioavilability of comparison different dosage form (oral capsule, oral administration solution and intravenous injection liquid).
Oral capsule
Oral capsule (6mg) comprises Aiweimopan at polyethylene glycol (PEG; Molecular weight 3350) the homodisperse suspension in.According to analytical proof, the actual content of measuring capsule is 5.868mg (6mg 97.8%).
Oral administration solution
Oral administration solution comprises Aiweimopan, propane diols (USP), orange bland and purify waste water (USP).The every 50mL of the oral administration solution of unit dose comprises 12mg Aiweimopan (0.24mg/mL).According to analytical proof, the actual content of measuring oral administration solution is 11.868mg (12mg 98.9%).
Iv formulation
Injection preparation is configured to the freeze drying powder that comprises 12.5mg Aiweimopan (solvent-free, anhydrous alkali) and mannitol (USP) and natrium carbonicum calcinatum (NG).Sodium hydroxide (NP) and hydrochloric acid (NF) are used for regulating pH before freeze-drying forms the freeze drying powder.Add the 12.2ml sodium chloride for injection in the freeze drying powder after, the solution of organizing structure again comprises 11mg/mL.This product is stored in the bottle of 12mg.According to analytical proof, the actual content of measuring iv formulation is 11.928mg (12mg 99.4%).
Estimate bioavilability in the process in three cycles, oral capsule preparation is to the crossing research of the relative bioavailability of the Aiweimopan of oral administration solution and the oral capsule preparation crossing research to the absolute bioavailability of the Aiweimopan of iv formulation.
36 main bodys are divided into the masculinity and femininity main body that equal amount is arranged in program separately, and each sex number approximately equal under study for action.Main body is at three Aiweimopans of accepting single dose independent opportunity (in three cycles first day of each cycle), begins to experience the removing (washout) of least fourteen days from date of the dosed administration in each cycle.Every main body in each cycle in three cycles (a kind of preparation of each cycle) is accepted independent 12mg oral capsule dosage (2 * 6mg capsule), independent 12mg oral administration solution dosage and independent 12mg intravenous dosages.Each cycle comprises independent Aiweimopan dosage and 5 days research evaluation.The order that main body is accepted each single dose preparation is determined by processing sequence, is at random for main body.One in six processing sequences may be by daily schedule decision at random.
Main body each give with testing drug before fasting (NPO) at least 10 hours, and after giving, keeping NPO 1 hour with testing drug.Some preset time before at every turn giving with testing drug and after giving with testing drug in 96 hours the process is collected the blood sample that is used for the Aiweimopan pharmacokinetic analysis, according to whether deciding to using peroral dosage form (that is, capsule or oral administration solution) or iv formulation.In research process, obtain about altogether 460mL blood from every main body.For each cycle, main body is limited in the clinical mechanism, finishes up to all 3 days processes.Require main body to get back to clinical mechanism then, carry out the blood sample collection of 72 hours and 96 hours respectively at the 4th day and 5 days.
Sample collection
In the following time blood sample (7mL) is extracted into and comprises in the pipe that contains liquaemin: 0 hour (in 15 minutes before dosed administration) and after dosed administration 0.5,1,1.5,2,3,4,6,9,12,18,24,36,48,72 and 96 hour with respect to the dosed administration oral formulations.For intravenous solution, except deriving from those above-mentioned samples, 0.2 hour (when infusion finishes) after infusion begins, four other samples of extraction in 0.25,0.33 and 0.75 hour.After collecting each sample, immediately each pipe is reversed lightly and place ice.In 30 minutes of sample collection,, be used for going out cell from plasma separation with under 5 ℃ of the Guan Zaiyue under about 1200xg centrifugal 15 minutes.Do not use sepa-ration aid.Blood plasma shifts with clean pipette and places two polypropylene storage tubes with the volume that equates.Be the following information of storage tube mark: the relative time and the analyte of rules numbering, main body numbering, cycle numbering, sampling.Mark is wrapped up to guarantee that mark adheres to oolemma.Before transportation, storage tube is placed 70 ℃ or the freezer unit of low temperature more.Be sent to analysis institution and before analyzing, keep freezing sample is freezing.
Bioanalytical method
Use the Aiweimopan concentration of LC/MS/MS (liquid chromatography/mass spectrometry/mass spectrum) method mensuration plasma sample that confirm and sensitivity.Measure and use solid phase extractions technology, be used for extracting Aiweimopan, its metabolite and interior mark from blood plasma.Be dissolved in solvent with the solvent evaporation and with residue, its part is expelled in the liquid chromatographic system.By using SCIEX API
Mass spectrograph detects by MS/MS (mass spectrum/mass spectrum).In each run, comprise calibration curve and quality control sample.The measurement range of two kinds of analytes be 0.25ng/mL to 250ng/mL, the quantitative limit of two kinds of analytes is 0.25ng/mL.Analyze the quality control standard that comprises three concentration in service at each and (low be 0.75ng/mL; That middle is 25ng/mL; With high be 175ng/mL).Owing to derive from the high concentration of intravenous solution, increase another standard (800ng/mL).With the withinrun precision of coefficient of deviation (%CV) expression, for lower quality control standard, Aiweimopan is 12.9%; For the within-assay with average with theoretical deviation % (%DMT) expression, Aiweimopan is 1.2%.
Pharmacokinetics is measured
Aiweimopan concentration in the blood plasma is input in the spreadsheet.After spreadsheet data is fit to compatibility as input file, information is used
Professional (Version 3.3) handles, to obtain the pharmacokinetics variable and to produce concentration time curve and table.Use WinNonlin Validation Kit (Version 3.3) checking WinNonlin program.Use has the pharmacokinetics variable that the outer non-chamber type Model 200 that imports of blood vessel obtains Aiweimopan in the oral medication process.The non-chamber type Model 202 that use has constant infusion speed analyzes the intravenous administration plasma concentration of Aiweimopan afterwards.For all main bodys, the infusion time is 12 minutes.
For the pharmacokinetic analysis use standard sample time of Aiweimopan, it is significant that any deviation of the sample time and the scheduled time all is considered to not be.Handle as 0 with the concentration value that is lower than quantitative limit (BLQ) report, be used to calculate mean concentration and concentration time curve.
For the pharmacokinetic analysis of Aiweimopan, in input file, comprise BLQ result.Directly use the plasma concentration of report, but the concentration measured that obtains observing after the BLQ result is got rid of from analyzing.
Obtain the following pharmacokinetic parameter of Aiweimopan from the WinNonlin output file:
The maximum of the observed plasma drug level of Cmax
The plasma drug level of Cp point preset time
Tmax reaches the time of maximum plasma drug level, directly obtains from concentration-time data
T
1/
2λ z disposes the half life period of the end of a period part in stage
AUC (0-t
At last) from 0 time to the last plasma drug level-time that can measure the concentration time
Area under a curve
Area under the plasma drug level-time graph of AUC (0-∞) from 0 time to the infinitely great time
The health plasma clearance (mL/min and mL/min/kg) that CL is total only is used for I.V.
Preparation.
Vss apparent steady state distribution volume (L and L/kg) only is used for the I.V. preparation
The apparent oral clearance of CL/F (mL/min and mL/min/kg) is used for oral formulations
The apparent oral distribution volume of Vz/F (L and L/kg) is used for oral formulations
Apparent oral distribution volume (L and L/kg) in Vz index in the latter stage phase process is only used
In the I.V. preparation
CL
βBlood clearance (mL/min) only is used for the I.V. preparation
Tlag can measure the time of concentration from 0 hour to first
Clinical relevant t
1/
2β is based on the drug plasma of the manual time point of selecting of stage of first disposal of expression
The half life period in the disposal stage of the clinical relevant portion of concentration time curve (referring to
Hereinafter)
AUC (0-∞) ' describe plasma drug level-time graph clinical relevant portion from 0 time to
Area under plasma drug level-time graph of infinitely great time
AUC is than AUC (0-∞) '/AUC (0-∞)
In order to calculate clinical relevant t
1/
2β is used to judge that the manual range of choice of λ z is intended with second non-chamber pattern that WinNonlin carries out data.By determining it is not second and the time point range of choice of the part in back disposal stage.Whether relevant in order to measure this second simulation, calculate AUC (0-∞) '/AUC (0-∞) from the 1st day data of Aiweimopan.If the mean value of ratio 〉=85% thinks that then AUC (0-∞) ' is the clinical relevant portion of AUC (0-∞).Clinical relevant t
1/
2β also uses second simulation of manual range of choice based on this.
In order to measure relatively and absolute bioavailability, calculate specific AUC ratio.For relative bioavailability, with oral capsule as test (Test), with oral administration solution (Reference) as a reference.For absolute bioavailability, oral Preparation is as test (Test), and I.V. preparation (Reference) as a reference.Use AUC (0-t
At last) and AUC (0-∞) calculate AUC than (with 95% confidence interval).Main consideration AUC (0-∞).Calculating stems from the lowest mean square mean value (LS mean value) of variance analysis (ANOVA).Relatively be converted into original scale to obtain the bioavilability ratio with two then with the difference of the LS mean value of handling.Calculating following ratio is used for measuring relatively and absolute bioavailability:
Relative bioavailability: oral capsule (test) is to oral administration solution (reference)
Absolute bioavailability: oral capsule (test) is to I.V. preparation (reference)
Oral administration solution (test) is to I.V. preparation (reference)
AUC (0-∞) test
AUC (0-∞) reference
AUC (0-t
At last ) test
AUC (0-t
At last) reference
Use the total blood clearance (CL after following equation calculates intravenous administration
β):
CLB=CL*(Cp/Cβ)
Wherein: CL is the geometric mean CL (ml/min) after the I.V. administration, and Cp/C β is 0.68 blood: inverse (that is Cp=1, of blood plasma distribution ratio; C
β=0.68).
The result is as shown in following table:
Pharmacokinetic parameter summary at the iv formulation that comprises Aiweimopan 12mg (1mg/mL) Aiweimopan afterwards of the oral administration solution that comprises Aiweimopan 12mg (0.24mg/mL) of giving oral capsule Aiweimopan 12mg (2 * 6mg capsule) with single dose, single dose and single dose
aNumerical value is to body weight normalization.
The absolute bioavailability of the Aiweimopan of oral capsule and oral administration solution is respectively 6.0% (95% confidence interval: 4.7-7.7%) with 14.3% (95% confidence interval: 11.1-18.3%).The capsule of Aiweimopan is 41.9% (95% confidence interval: 32.6-53.7%) with respect to the bioavilability of its oral administration solution.System's exposure that the system that provides than oral capsule and oral administration solution exposes high respectively 6 times and 14 times is provided for the iv formulation of Aiweimopan.
When using the scope of expression physical property such as molecular weight or chemical property such as chemical formula in this article, be intended to be included in all combinations and the recombinant of the specific embodiments in this scope.
Each patent of quoting herein or describing, patent application and openly therefore incorporated into this paper in full as a reference.
It should be appreciated by those skilled in the art that and to carry out many changes and improvements to the preferred embodiments of the invention, and can carry out this changes and improvements and do not break away from spiritual essence of the present invention.Therefore, claim covers all these and drops on real spiritual essence of the present invention and the interior variant of equal value of scope.