CN102477008A - Method for synthesizing ezetimibe - Google Patents
Method for synthesizing ezetimibe Download PDFInfo
- Publication number
- CN102477008A CN102477008A CN2010105530701A CN201010553070A CN102477008A CN 102477008 A CN102477008 A CN 102477008A CN 2010105530701 A CN2010105530701 A CN 2010105530701A CN 201010553070 A CN201010553070 A CN 201010553070A CN 102477008 A CN102477008 A CN 102477008A
- Authority
- CN
- China
- Prior art keywords
- reaction
- phenyl
- fluorophenyl
- fluorine
- dissolved
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 0 C*(CC1)CCC1(*)O Chemical compound C*(CC1)CCC1(*)O 0.000 description 7
- VWQAPIJXLQYLHA-UHFFFAOYSA-N CC(C)C(C)(C)F Chemical compound CC(C)C(C)(C)F VWQAPIJXLQYLHA-UHFFFAOYSA-N 0.000 description 1
Abstract
The invention belongs to the technical field of medicines and relates to a method for synthesizing ezetimibe. The method comprises the steps of taking parahydroxybenzaldehyde and R-fluoromandelic acid as starting raw materials and synthesizing the ezetimibe through the conventional methods, such as protection, condensation, cyclization, reduction, bromination, protection, butt joint, deprotection and the like, under the action of a catalyst. The method disclosed by the invention has the advantages of high yield, greatly reduced cost because expensive chiral ligands or chiral catalysts are prevented from being used, and less side reactions and is applied to industrialized production.
Description
Technical field:
The invention belongs to medical technical field, relate to the compound method of ezetimibe, be specifically related to a kind of method of utilizing the synthetic ezetimibe of midbody.
Background technology:
Ezetimibe (Ezetimibe) is people such as Harry Davis, Margaret Van Heek and the Kevin Alton research and development in Schering Plough (Schering-Plough) company research centre; In October, 2002; Ezetimibe (Zetia) obtains the approval of FDA; And at first go on the market trade(brand)name ezetrol in Germany November in the same year.Ezetimibe is first selectivity cholesterol absorption inhibitor, and it can disturb in SUV and the enterohepatic circulation of food source by liver synthetic absorption of cholesterol simultaneously, and the absorption of other nutritive ingredient is not exerted an influence.Its pharmacology only acts on small intestine, reduces intestinal cholesterol through the inhibition absorption of cholesterol and turns to liver, reduces its storage; Can strengthen the removing of SUV in the blood, thereby reduce blood plasma cholesterol level.Itself and Statins are united the frequency of utilization that use can reduce the statins high dosage, and drug effect is single 8 times with the effect of statins deposits yields decreasing cholesterol.Ezetimibe is individually dosed or all good with HMG-CoA reductase inhibitor administation of combination tolerance, and adverse reaction rate is similar with placebo.
The chemistry of ezetimibe is by name: (3R, 4S)-1-(4-fluorophenyl)-3-[ (3S)-3-(4-fluorophenyl)-3-hydroxypropyl ]-4-(4-phenylor)-2-azetidinone) has following structure:

In the prior art, the synthetic route of ezetimibe has a variety of, but all exists many deficiencies:
Patent WO2000/34240 (U.S. Schering Plough company) discloses the method for the synthetic ezetimibe of a kind of improved chirality; At first make (S)-3-oxy-compound in this method through the chiral reduction agent; Then through single step reaction when carbonyl α position connects side chain (E)-N-(4-fluorophenyl)-4-hydroxybenzene methylene amine with two hydroxyls in the product structure by the silica-based protection of front three; Then Cheng Huan, remove blocking group, obtain ezetimibe.This compound method is when the carbonyl of chiral reduction, and owing to the group with chiral structure is far away apart from carbonyl, the content of the activity chiral carbon that obtains after the reduction is low, and the amount ratio of chiral reduction agent is bigger; And the yield that step that connects side chain reacts is very low, so this synthetic route cost is high, is not suitable for suitability for industrialized production.
Patent WO2007072088 discloses the synthetic route of another kind of preparation ezetimibe, and this compound method reactions step is few, and concrete synthetic route is following:
Among the patent WO2007072088 in the route of synthetic ezetimibe the hydroxyl on the chiral carbon of 3 side chains be get by the carbonyl chiral reduction, before chiral reduction not, this carbonyl protects spent glycol so that it is destroyed when carrying out other chemical reactions; The midbody compound IV of gained, compound VI are thinner solid matters, in last handling process, and difficult crystallization; And, in crystallisation process, there is impurity to separate out extremely easily, in the filtration procedure because its granularity is very thin; Few portion of product can flow out along with filtrating, causes the second-rate of product, and subsequent reactions difficulty carries out fully; Whole yield is lower, has increased synthetic cost.
Other is US:5739321; US:5886171 has reported route 1: with (4
S)-hydroxyl tetrahydrofuran-2-ketone and N-(4-fluorophenyl)-4-benzyloxy benzene methylene amine is that starting raw material prepares ezetimibe, and its reaction scheme is following:
This compound method is: N-(4-fluorophenyl)-4-benzyloxy benzene methylene amine (1) and (4
S)-hydroxyl tetrahydrofuran-2-ketone (2) at low temperatures, (LDA) carries out cyclization with lithium diisopropylamine, reaction is carried out recrystallization after finishing can obtain compound (3).Compound (3) obtains product (4) after the oxidation of NaIO, obtain compound (6) with 4-fluoro acetophenone and chloro trimethyl silane (TMS-Cl) reaction, uses TiCl
4Carry out condensation and generate compound (7), obtain compound (8) behind the TsOH catalytic dehydration.Behind the Pd/C shortening, two key reduction are sloughed benzyl simultaneously and are carried out asymmetric reduction with chiral catalyst CBS and reductive agent boronation hydrogen at low temperatures at last, obtain title product again.This reaction process can produce cis-isomeride, and content can reach about 5%, can in subsequent disposal, remove with recrystallization.Making title product by compound (8) reduction also can be through chlorinated triphenyl phosphorus base rhodium [(PPh
3)
3RhCl] the two keys of catalytic hydrogenation, again through BH
3/ CBS asymmetric reduction, catalytic hydrogenation debenzylation obtain compound (9).This method is except that cyclization reaction yield 60%, and other respectively goes on foot yield all more than 70%.
In the route 1, (4
S)-hydroxyl tetrahydrofuran-2-ketone cost is higher, and character is unstable, and certain difficulty is arranged in the production.
Other reports route 2: with (5S)-acetoxyl group-5-(4-fluorophenyl) valeric acid is that reaction intermediate prepares ezetimibe, and its synthesis technique is following:

At first with (5
S)-acetoxyl group-5-(4-fluorophenyl) valeric acid (27) and (4
S)-4-phenyl-oxazolidones (13) carry out condensation, obtain compound (28).Product is at TiCl
4With obtain compound (30) with N-(4-fluorophenyl) 4-acetyloxy phenyl methylene amine (29) through condensation in the dichloromethane solution of DIPEA, this step reaction yield can reach 51%.Compound (30) carries out cyclization in the toluene solution that adds BSA and catalyzer TBAF, generate compound (31), and reaction yield can reach 91%.Compound (31) obtains title product after falling ethanoyl via the Lithium Hydroxide MonoHydrate hydrolysis.
The raw material of route 2 is all relatively more expensive, and the technology more complicated, and reaction conditions is relatively harsher, and in the fractionation to isomer, uses column chromatography, and industriallization is difficulty very.
Reported route 3: with Pyroglutaric acid and N-(4-fluorophenyl)-4-benzyloxy benzene methylene amine is that starting raw material prepares ezetimibe; It is through following prepared in reaction: Pyroglutaric acid and methyl alcohol effect generate monomethyl glutarate, through sulfur oxychloride reaction back generation 4-(chloroformyl) methyl-butyrate (21).Compound (21) in the dichloromethane solution of triethylamine and DMAP with (4S)-amidate action takes place, obtains (4 in 4-phenyl-oxazolidones (13)
S)-3-(4-methyl-formiate base-1-oxo butyl)-4-phenyl-2-oxazolidone (22); Under the effect of alkaline condition and titanium tetrachloride, titanium tetraisopropylate and DIPEA with N-(4-fluorophenyl)-4-benzyloxy benzene methylene amine condensation; Obtain compound (24) by BSA/TBAF (tetrabutyl ammonium fluoride) effect closed loop, after the LiOH hydrolysis, obtain compound (25).Compound (25) is prepared into acyl chlorides (26) by oxalyl chloride, through to fluorophenyl magnesium bromide (27) at ZnCl
2And Pd (PPh
3)
4Effect generates compound (17) down, again through CBS/BH
3The asymmetric reduction carbonyl, Pd/C and H
2Remove benzyl and obtain title product.Its reaction formula is following:

The low in raw material cost of route 3 is easy to get, and the midbody productive rate is relatively all higher, is fit to suitability for industrialized production, but it uses column chromatography in the fractionation of optical isomer, it is had a greatly reduced quality in practicality.
Reported route 4: with 5-(4-fluorophenyl)-4-pentenoic acid is that reaction intermediate prepares ezetimibe, and it prepares through following method: 5-(4-fluorophenyl)-4-pentenoic acid is after oxalyl chloride is prepared into acyl chlorides, with chiral reagent (4
S)-4-phenyl-2-oxazolidone carries out amidate action under DMAP catalysis, product obtains compound (A) through recrystallization.With TiC1
4, DPEA and imines add in the dichloromethane solution, drips compound (A) at low temperatures, keeps low temperature to reaction to finish, and with acetate quencher reaction, bullion carried out recrystallization and obtains compound (B) after it was handled.Compound (B) cyclization in the toluene solution of TBAF obtains compound (C), and it is splashed into Pd (OAc)
2, HClO
4In the acetonitrile solution of benzoquinones, reaction is used organic solvent extraction after finishing dilute with water again, removes catalyzer, behind the separating filtrate evaporate to dryness, uses the column chromatography separating optical isomers to obtain compound (D), obtaining target compound through preceding method operation again.Its reaction formula is following:

Route 4 is identical with 2, and raw material is somewhat expensive, complex process, and industriallization is difficulty relatively.
US:627882B; WO:2005-IB2393 has reported route 5: with 5-(4-fluorophenyl)-5-oxopentanoic acid is that reaction intermediate prepares ezetimibe; This route is starting raw material with the fluorobenzene; Under ice-water bath, use aluminum chloride catalysis with Pyroglutaric acid; Friedel-Crafts reaction takes place, and generates 5-(4-fluorophenyl)-5-oxopentanoic acid (33).5-(4-fluorophenyl)-5-oxopentanoic acid (33) adds the dichloromethane solution that is dissolved with triethylamine, and the pivaloyl chloride after that is added dropwise to equivalent becomes mixed acid anhydride (34).Mixed acid anhydride (34) under the catalysis of DMAP with (4
SAmidate action takes place in)-4-phenyl-2-oxazolidone (13), thick product behind the Virahol recrystallization purifying, obtain 4 (
S)-3-[5-(4-fluorophenyl)-1,5-dioxo amyl group]-4-phenyl-2-oxazolidone (35).Compound (35) is a catalyzer generation asymmetric reduction reaction with [(R)-MeCBS)] in the low temperature tetrahydrofuran solution, and the reaction back that finishes is used ydrogen peroxide 50 processing is made (4 with sulphuric acid soln
S)-3-[(5
S)-(4-fluorophenyl)-5-hydroxyl-1-oxo amyl group]-4-phenyl-2-oxazolidone (36).Compound (36) adds and contains DIPEA, TMS-Cl and TiCl
4Dichloromethane solution in; At low temperatures, slowly add 4-(4-fluorophenyl imido grpup) phenol (37), condensation reaction finishes the back and handles with glacial acetic acid, 7% tartaric acid solution and 20% sodium sulfite solution; Add the abundant back flow reaction of BSA again; Then product is concentrated recrystallization and obtain compound (38), compound (38) carries out cyclization under BSA and TBAF effect, and removes (4
S)-4-phenyl-2-oxazolidone.With the cyclization product in Virahol and H
2SO
4The protection of desiliconization alkane promptly obtains title product (10) in the mixing solutions.Its reaction formula is following:
?
Route 5 is a patent in 2005, owing to relate to patent protection, its research is restricted.
The problem that all exists in the compound method of above-mentioned ezetimibe is that synthetic route is long; Perhaps yield, purity are low, and the synthetic route that has has been wasted the midbody more than 50%; The amount of the chiral reduction agent of perhaps using is many, solvent toxicity is big, and in sum, the cost of the compound method of ezetimibe brings very big inconvenience for its suitability for industrialized production than higher in the prior art.
Summary of the invention:
Technical problem to be solved by this invention provides a kind of compound method of new ezetimibe, thereby it is high to overcome the compound method cost that exists in the prior art, is not suitable for shortcomings such as industriallization.
The present invention realizes through following technical scheme:
The synthetic route of ezetimibe of the present invention is:
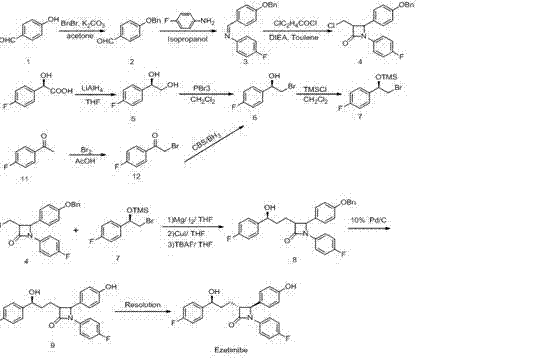
It prepares through following method:
(1) PARA HYDROXY BENZALDEHYDE and benzyl under the alkali effect, generate P-benzyloxybenzaldehyde 2 through the benzyl protection reaction in solvent;
(2) in anhydrous protic solvent, add para-fluoroaniline and P-benzyloxybenzaldehyde 2, condensation reaction generates N-(4-fluorophenyl)-4-benzyloxy benzene methylene amine 3;
(3) N-(4-fluorophenyl)-4-benzyloxy benzene methylene amine (3) with organic base catalytic, obtains Trans-1-(4-fluorophenyl)-3-(2-chloromethyl)-4-(4-benzyloxy phenyl)-2-azetidinone 4 with the cyclization of 3-chlorpromazine chloride in toluene;
(4) R-obtains (2R)-4-fluorophenethyl glycol 5 to the fluorine racemic melic acid through reduction;
(5) (2R)-4-fluorophenethyl glycol (5) obtains Alpha-hydroxy (4-fluorine) phenyl-bromide ethane 6 through bromo;
(6) protection obtains (R) α-trimethyl silicon based oxygen base (4-fluorine) phenyl-bromide ethane 7 to Alpha-hydroxy (4-fluorine) phenyl-bromide ethane 6 through TMSCl;
(7) (R) α-trimethyl silicon based oxygen base (4-fluorine) phenyl-bromide ethane 7 and Trans-1-(4-fluorophenyl)-3-(2-chloromethyl)-4-(4-benzyloxy phenyl)-2-azetidinone 4 generate Trans-1-(4-fluorophenyl)-3-[3R-hydroxyl-3-(4-fluorophenyl) propyl group]-4-(4-benzyloxy phenyl)-2-azetidinone 8 through grignard reaction, metal linked reaction, deprotection base in THF;
(8) Trans-1-(4-fluorophenyl)-3-[3R-hydroxyl-3-(4-fluorophenyl) propyl group]-4-(4-benzyloxy phenyl)-2-azetidinone 8 deprotection base under the Pd/C condition obtains formula 9;
(9) formula 9 obtains the title product ezetimibe through recrystallization purifying.
Solvent described in the step (1) be among acetone, the DMF any one; Alkali is selected from Na
2CO
3, K
2CO
3, NaOH, KOH, the benzyl of protection usefulness is selected from bromobenzyl and benzyl chloride, and temperature of reaction is a reflux temperature.
Anhydrous protic solvent described in the step (2) is selected from anhydrous methanol, absolute ethyl alcohol, anhydrous isopropyl alcohol; P-benzyloxybenzaldehyde is that the 2-4 of para-fluoroaniline doubly measures, reflux time 2-5h.
The benzyloxy phenyl)-2-azetidinone (4) prepares through following method:
N-(4-fluorophenyl)-4-benzyloxy benzene methylene amine (3) is dissolved in the toluene, adds alkali, add the 3-chlorpromazine chloride, finish 80 ℃-120 ℃ reactions of back temperature control 8-12 h; Be cooled to room temperature, add the hydrochloric acid cancellation, add water stratification; Water layer is used dichloromethane extraction, saturated sodium bicarbonate washing, saturated common salt water washing; Anhydrous sodium sulfate drying concentrates, and described alkali is selected from TEA, DIEA, 3-n-butylamine.
Described (2R)-4-fluorophenethyl glycol (5) prepares through following method
:
Get R-4-fluorine racemic melic acid and be dissolved in THF or the diethyl ether solution, ice bath drips borine tetrahydrofuran solution or LAH down, finishes; Stirring at room 2h, ice bath drip down the shrend reaction of going out, decompression concentrated solution; Add the extraction of ETHYLE ACETATE or methylene dichloride separatory, drying concentrates.
(R) Alpha-hydroxy (4-fluorine) phenyl-bromide ethane (6) prepares through following method:
(2R)-4-fluorophenethyl glycol (5) is dissolved in the methylene dichloride, adds NBS, PBr
3, any one brominated reagent in the lithiumbromide, drip the vitriol oil or glacial acetic acid, stirring at room, stopped reaction after 2-4 hour adds entry, separatory, a small amount of dichloromethane extraction merges organic phase, the saturated sodium bicarbonate washing, the saturated common salt washing, drying concentrates.
The following method of passing through of monobromethane (7) prepares:
Get (R) Alpha-hydroxy (4-fluorine) phenyl-bromide ethane (6) and be dissolved in the methylene dichloride, ice bath adds DIEA down, TMSCl or TBSCl, and stirring at room 2-4h, stopped reaction adds entry, and separatory, water layer merge organic phase with a small amount of dichloromethane extraction, washing, drying concentrates.
The benzyloxy phenyl)-2-azetidinone (8) prepares through following method:
With magnesium chips, catalytic amount iodine grain, be suspended in THF or the ether, (R) α-trimethyl silicon based oxygen base (4-fluorine) phenyl-bromide ethane (7) is dissolved in and drops in the reaction system in the dry tetrahydrofuran; The observation solution colour is decorporated; Behind the stirring at room 1-2h, make Grignard reagent, under the ice bath compound (4) is dissolved in the dry tetrahydrofuran; Add coupling reagent, described coupling reagent is selected from Pd (PPh
3)
4, CuI, PdCl
2(dppf), ZnCl
2Ice bath stirs down, will make Grignard reagent to join in the reaction solution room temperature reaction 8-12h, stopped reaction at present; Ice bath adds aqueous ammonium chloride solution cancellation reaction down, separatory, and a small amount of dichloromethane extraction of water, saturated common salt water washing organic phase, drying concentrates.Bullion is dissolved in the exsiccant tetrahydrofuran solution, adds tetrabutyl ammonium fluoride stirring at room 3-6h, concentration of reaction solution adds entry, the extraction of methylene dichloride separatory, and washing, drying concentrates.
Compound (8) is dissolved in any one solvent of methyl alcohol, ethanol, Virahol, adds ammonium formiate, formic acid, catalytic amount palladium carbon, reaction 3-6h, reaction finishes.Suction filtration, filtrating concentrates, silica gel column chromatography; Obtain the compound trans ezetimibe, after be dissolved in ETHYLE ACETATE or the methylene dichloride, drip that any one two optical isomers split in dioxane, THF, MTBE, the anhydrous diethyl ether; Insolubles is filtered; After getting concentrated the doing of filtrating, carry out recrystallization, obtain ezetimibe with ethyl acetate/petroleum ether.
Described (R) α-trimethyl silicon based oxygen base (4-fluorine) phenyl-bromide ethane (7) to be starting raw material to fluoro acetophenone (11), obtains α-bromo to fluoro acetophenone (12) under bromine, Glacial acetic acid min. 99.5 condition.Formula (12) is dissolved in exsiccant ether or the tetrahydrofuran solution, and room temperature condition drips down slowly the CBS/ hexane solution, drips borine-tetrahydrofuran solution under the condition of ice bath, stirred 2 hours, and the dropping shrend reaction of going out, decompression concentrated solution gets formula (6).Formula (6) obtains formula (7) through the TMSCl protection.
Invention provides a kind of new compound method of ezetimibe.Present method is starting raw material with PARA HYDROXY BENZALDEHYDE, R-to the fluorine racemic melic acid, under the effect of catalyzer, through the synthetic ezetimibe of ordinary methods such as protection, condensation, cyclization, reduction, bromo, protection, butt joint, deprotection.This method yield is high, avoids the use of expensive chiral ligand or chiral catalyst, reduces cost on a large scale, and side reaction is few, is fit to industrialized production.
Embodiment:
With PARA HYDROXY BENZALDEHYDE 5 g, Na
2CO
36.2 g is dissolved in the 150 mL acetone, adds cylite 6.7 mL back flow reaction 3.5 h, is cooled to room temperature, reaction solution is poured into separated out solid in the frozen water; Stirred 10 minutes, suction filtration, filter cake is with a small amount of washing, drying; Get white solid 4-benzyloxy phenyl aldehyde (2), 8.5 g, yield 98%.
Embodiment two
8.5 g are dissolved in the 120 mL anhydrous isopropyl alcohols with 4-benzyloxy phenyl aldehyde, are heated to 85 degrees centigrade, stir to drip 4-fluoroaniline 11.5 mL down; Back flow reaction 4h is cooled to room temperature and separates out solid, suction filtration; Filter cake washs with small amount of ethanol, and oven dry gets white solid N-(4-fluorophenyl)-4-benzyloxy benzene methylene amine (3); Meter 11g, yield 90%.
Embodiment three
Compound (3) 11 g are dissolved in the 100 mL toluene, and nitrogen protection adds DIEA 18 mL down and is warming up to backflow.Slowly drip 3-chlorpromazine chloride 6.3 mL, dropwise 100 ℃ of reactions of back temperature control, 12 h, be cooled to room temperature, add the 2 mL cancellation of 1 M hydrochloric acid; Add 20 mL water stratifications, water layer merges organic phase with dichloromethane extraction (2 * 10 mL); The saturated sodium bicarbonate washing, saturated common salt water washing, anhydrous sodium sulfate drying; Filter, concentrate to such an extent that Off-white solid Trans-1-(4-fluorophenyl)-3-(2-chloromethyl)-4-(4-benzyloxy phenyl)-2-azetidinone (4) is counted 12.5 g, yield 85%.
Embodiment four
Get R-4-fluorine racemic melic acid 4.7 g and be dissolved in the 20 mL exsiccant tetrahydrofuran solutions, ice bath adds down 2.4gLAH, and stirring at room 3h, ice bath drip the 10 mL shrends reaction of going out down.Concentrating under reduced pressure reclaims tetrahydrofuran solution, adds ETHYLE ACETATE separatory extraction (2 * 10 mL), the saturated common salt washing, and drying concentrates, and obtains colourless liquid (2R)-4-fluorophenethyl glycol (5) meter 4.2 g, yield 97.6%.
Embodiment five
Compound (5) 4.2 g are dissolved in the 20 mL methylene dichloride, drip PBr
3Solution 7.8mL, stirring at room, stopped reaction after 3 hours adds 10 mL water; Separatory, a small amount of dichloromethane extraction merges organic phase, the saturated sodium bicarbonate washing; The saturated common salt washing, drying concentrates; Silica gel column chromatography obtains colorless oil (R)-α hydroxyl (4-fluorine) phenyl-bromide ethane (6) meter 4.8 g, yield 80%.
Embodiment six
Get compound (6) 4.8 g and be dissolved in the 20 mL dry methylene chloride, ice bath adds DIEA 3.3mL, TMSCl4.2 mL, stirring at room 3h, stopped reaction down.Add 10 mL water, separatory, water layer merge organic phase with a small amount of dichloromethane extraction, washing, and drying concentrates, and silica gel column chromatography obtains white solid (R)-α-trimethyl silicon based oxygen base (4-fluorine) phenyl-bromide ethane (7) meter 5.1 g, yield 80 %.
Embodiment seven
With magnesium chips 620 mg catalytic amount iodine grains, be suspended in the exsiccant 10 mL THFs, compound (7) 5.1 g are dissolved in the 10mL dry tetrahydrofuran drop to reaction system, observe solution colour and decorporate, behind the continuation stirring at room 2h, make Grignard reagent.Under the ice bath compound (4) 7.1 g are dissolved in the dry tetrahydrofuran, add anhydrous cuprous iodide 570mg, ice bath will be made Grignard reagent to join in the reaction solution room temperature reaction 10h, stopped reaction after stirring 20 min down at present.Ice bath adds aqueous ammonium chloride solution cancellation reaction down.Stirring at room 10 minutes, separatory, a small amount of dichloromethane extraction of water, saturated common salt water washing organic phase; Drying concentrates, and bullion 2.3 g are dissolved in the exsiccant tetrahydrofuran solution, adds tetrabutyl ammonium fluoride 1.5 g; Stirring at room 4h, concentration of reaction solution adds entry, the extraction of methylene dichloride separatory; The saturated common salt water washing, anhydrous sodium sulfate drying concentrates; Silica gel column chromatography obtains Off-white solid Trans-1-(4-fluorophenyl)-3-[3R-hydroxyl-3-(4-fluorophenyl) propyl group]-4-(4-benzyloxy phenyl)-2-azetidinone (8) meter 2.0 g, yield 30%.
Embodiment eight
(8) 2.0 g are dissolved in the absolute ethyl alcohol with compound, add ammonium formiate 820 mg, formic acid 2.5 mL, and catalytic amount palladium carbon, 60 ℃ of reactions of temperature control 3h, reaction finishes.Suction filtration, filtrating concentrates, silica gel column chromatography; Obtain compound trans ezetimibe 1.3 g, after be dissolved in the ETHYLE ACETATE, drip anhydrous diethyl ether and split two optical isomers; Insolubles is filtered, getting after filtrating concentrating do, carry out recrystallization with ethyl acetate/petroleum ether; Obtain ezetimibe (1) meter 600 mg, yield 40%.
Embodiment nine
To be dissolved in the 20ml Glacial acetic acid min. 99.5 fluoro acetophenone (11) 5ml 0.037mol, room temperature condition slowly drips bromine 6.25g 0.039mol down, the clarification of question response liquid, and reaction promptly finishes.Steam and remove Glacial acetic acid min. 99.5, obtain the yellowish white solid, obtain white solid α-bromo to fluoro acetophenone (12) 6.69g yield 79.6% through toluene/normal hexane recrystallization.
Embodiment ten
Getting α-bromo is dissolved in the 20 mL exsiccant tetrahydrofuran solutions fluoro acetophenone 2.16g; Ice bath drips 1mol/L CBS hexane solution 1ml down, and borine-tetrahydrofuran solution 22 mL of 2.5 mol/L are after dropwising; Stirring at room 3h, ice bath drip down the 10 mL shrends reaction of going out.The concentrating under reduced pressure dry thf solution adds ETHYLE ACETATE separatory extraction (2 * 10 mL), the saturated common salt washing, and drying concentrates, colorless oil (R)-α hydroxyl (4-fluorine) phenyl-bromide ethane (6) meter 2.17 g, yield 99.6%.
Claims (10)
1. the compound method of ezetimibe is characterized in that, prepares through following method:
(1) PARA HYDROXY BENZALDEHYDE and benzyl under the alkali effect, generate P-benzyloxybenzaldehyde 2 through the benzyl protection reaction in solvent;
(2) in anhydrous protic solvent, add para-fluoroaniline and P-benzyloxybenzaldehyde 2, condensation reaction generates N-(4-fluorophenyl)-4-benzyloxy benzene methylene amine 3;
(3) N-(4-fluorophenyl)-4-benzyloxy benzene methylene amine (3) with organic base catalytic, obtains Trans-1-(4-fluorophenyl)-3-(2-chloromethyl)-4-(4-benzyloxy phenyl)-2-azetidinone 4 with the cyclization of 3-chlorpromazine chloride in toluene;
(4) R-obtains (2R)-4-fluorophenethyl glycol 5 to the fluorine racemic melic acid through reduction;
(5) (2R)-4-fluorophenethyl glycol (5) obtains Alpha-hydroxy (4-fluorine) phenyl-bromide ethane 6 through bromo;
(6) protection obtains (R) α-trimethyl silicon based oxygen base (4-fluorine) phenyl-bromide ethane 7 to Alpha-hydroxy (4-fluorine) phenyl-bromide ethane 6 through TMSCl;
(7) (R) α-trimethyl silicon based oxygen base (4-fluorine) phenyl-bromide ethane 7 and Trans-1-(4-fluorophenyl)-3-(2-chloromethyl)-4-(4-benzyloxy phenyl)-2-azetidinone 4 generate Trans-1-(4-fluorophenyl)-3-[3R-hydroxyl-3-(4-fluorophenyl) propyl group]-4-(4-benzyloxy phenyl)-2-azetidinone 8 through grignard reaction, metal linked reaction, deprotection base in THF;
(8) Trans-1-(4-fluorophenyl)-3-[3R-hydroxyl-3-(4-fluorophenyl) propyl group]-4-(4-benzyloxy phenyl)-2-azetidinone 8 deprotection base under the Pd/C condition obtains formula 9;
(9) formula 9 obtains the title product ezetimibe through recrystallization purifying;
Its reaction formula is following:

2. compound method according to claim 1 is characterized in that, the solvent described in the step (1) be among acetone, the DMF any one; Alkali is selected from Na
2CO
3, K
2CO
3, NaOH, KOH, the benzyl of protection usefulness is selected from bromobenzyl and benzyl chloride, and temperature of reaction is a reflux temperature.
3. compound method according to claim 1 is characterized in that, the anhydrous protic solvent described in the step (2) is selected from anhydrous methanol, absolute ethyl alcohol, anhydrous isopropyl alcohol; P-benzyloxybenzaldehyde is that the 2-4 of para-fluoroaniline doubly measures, reflux time 2-5h.
4. compound method according to claim 1 is characterized in that, described Trans-1-(4-fluorophenyl)-3-(2-chloromethyl)-4-(4-benzyloxy phenyl)-2-azetidinone (4) prepares through following method:
N-(4-fluorophenyl)-4-benzyloxy benzene methylene amine (3) is dissolved in the toluene, adds alkali, add the 3-chlorpromazine chloride, finish 80 ℃-120 ℃ reactions of back temperature control 8-12 h; Be cooled to room temperature, add the hydrochloric acid cancellation, add water stratification; Water layer is used dichloromethane extraction, saturated sodium bicarbonate washing, saturated common salt water washing; Anhydrous sodium sulfate drying concentrates, and described alkali is selected from TEA, DIEA, 3-n-butylamine.
5. compound method according to claim 1 is characterized in that, described (2R)-4-fluorophenethyl glycol (5) prepares through following method
:
Get R-4-fluorine racemic melic acid and be dissolved in THF or the diethyl ether solution, ice bath drips borine tetrahydrofuran solution or LAH down, finishes; Stirring at room 2h, ice bath drip down the shrend reaction of going out, decompression concentrated solution; Add the extraction of ETHYLE ACETATE or methylene dichloride separatory, drying concentrates.
6. compound method according to claim 1 is characterized in that, described (R) Alpha-hydroxy (4-fluorine) phenyl-bromide ethane (6) prepares through following method:
(2R)-4-fluorophenethyl glycol (5) is dissolved in the methylene dichloride, adds NBS, PBr
3, any one brominated reagent in the lithiumbromide, drip the vitriol oil or glacial acetic acid, stirring at room, stopped reaction after 2-4 hour adds entry, separatory, a small amount of dichloromethane extraction merges organic phase, the saturated sodium bicarbonate washing, the saturated common salt washing, drying concentrates.
7. compound method according to claim 1 is characterized in that, the following method of passing through of described (R) α-trimethyl silicon based oxygen base (4-fluorine) phenyl-bromide ethane (7) prepares:
Get (R) Alpha-hydroxy (4-fluorine) phenyl-bromide ethane (6) and be dissolved in the methylene dichloride, ice bath adds DIEA down, TMSCl or TBSCl, and stirring at room 2-4h, stopped reaction adds entry, and separatory, water layer merge organic phase with a small amount of dichloromethane extraction, washing, drying concentrates.
8. compound method according to claim 1 is characterized in that, described Trans-1-(4-fluorophenyl)-3-[3R-hydroxyl-3-(4-fluorophenyl) propyl group]-4-(4-benzyloxy phenyl)-2-azetidinone (8) prepares through following method:
With magnesium chips, catalytic amount iodine grain, be suspended in THF or the ether, (R) α-trimethyl silicon based oxygen base (4-fluorine) phenyl-bromide ethane (7) is dissolved in and drops in the reaction system in the dry tetrahydrofuran; The observation solution colour is decorporated; Behind the stirring at room 1-2h, make Grignard reagent, under the ice bath compound (4) is dissolved in the dry tetrahydrofuran; Add coupling reagent, described coupling reagent is selected from Pd (PPh
3)
4, CuI, PdCl
2(dppf), ZnCl
2Ice bath stirs down, will make Grignard reagent to join in the reaction solution room temperature reaction 8-12h, stopped reaction at present; Ice bath adds aqueous ammonium chloride solution cancellation reaction, separatory, a small amount of dichloromethane extraction of water, saturated common salt water washing organic phase down; Drying concentrates, and bullion is dissolved in the exsiccant tetrahydrofuran solution, adds tetrabutyl ammonium fluoride stirring at room 3-6h; Concentration of reaction solution adds entry, the extraction of methylene dichloride separatory; Washing, drying concentrates.
9. compound method according to claim 1 is characterized in that, compound (8) is dissolved in any one solvent of methyl alcohol, ethanol, Virahol, adds ammonium formiate; Formic acid, catalytic amount palladium carbon, 30 ℃-75 ℃ reactions of temperature control 3-6h, reaction finishes; Suction filtration, filtrating concentrates, and silica gel column chromatography obtains the compound trans ezetimibe; After be dissolved in ETHYLE ACETATE or the methylene dichloride, drip that any one two optical isomers split in dioxane, THF, MTBE, the anhydrous diethyl ether, insolubles is filtered; After getting concentrated the doing of filtrating, carry out recrystallization, obtain ezetimibe with ethyl acetate/petroleum ether.
10. compound method according to claim 1 is characterized in that, described (R) α-trimethyl silicon based oxygen base (4-fluorine) phenyl-bromide ethane (7); To be starting raw material to fluoro acetophenone (11), under bromine, Glacial acetic acid min. 99.5 condition, obtain α-bromo to fluoro acetophenone (12), formula (12) is dissolved in exsiccant ether or the tetrahydrofuran solution; Room temperature condition slowly drips the CBS/ hexane solution down; Drip borine-tetrahydrofuran solution under the condition of ice bath, stirred 2 hours, drip the shrend reaction of going out; Decompression concentrated solution gets formula (6), and formula (6) obtains formula (7) through the TMSCl protection.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201010553070.1A CN102477008B (en) | 2010-11-22 | 2010-11-22 | Method for synthesizing ezetimibe |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201010553070.1A CN102477008B (en) | 2010-11-22 | 2010-11-22 | Method for synthesizing ezetimibe |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN102477008A true CN102477008A (en) | 2012-05-30 |
| CN102477008B CN102477008B (en) | 2014-05-21 |
Family
ID=46089799
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201010553070.1A Expired - Fee Related CN102477008B (en) | 2010-11-22 | 2010-11-22 | Method for synthesizing ezetimibe |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN102477008B (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105272852A (en) * | 2014-07-16 | 2016-01-27 | 浙江九洲药物科技有限公司 | Ezetimibe intermediate and preparation method |
| CN106397292A (en) * | 2016-09-20 | 2017-02-15 | 苏州普罗达生物科技有限公司 | Ezetimibe intermediate, synthesis method of intermediate and synthesis method of ezetimibe |
| CN112574055A (en) * | 2019-09-30 | 2021-03-30 | 天津天药药业股份有限公司 | Preparation method and application of formoterol and medicinal salt thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997045406A1 (en) * | 1996-05-31 | 1997-12-04 | Schering Corporation | 3-hydroxy gamma-lactone based enantioselective synthesis of azetidinones |
| WO2008089984A2 (en) * | 2007-01-24 | 2008-07-31 | Krka | Process for the preparation of ezetimibe and derivatives thereof |
| CN101346349A (en) * | 2005-12-20 | 2009-01-14 | 吉瑞工厂 | Process for the production of ezetimibe and intermediates used in this proces |
-
2010
- 2010-11-22 CN CN201010553070.1A patent/CN102477008B/en not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997045406A1 (en) * | 1996-05-31 | 1997-12-04 | Schering Corporation | 3-hydroxy gamma-lactone based enantioselective synthesis of azetidinones |
| US5739321A (en) * | 1996-05-31 | 1998-04-14 | Schering Corporation | 3-hydroxy γ-lactone based enantionselective synthesis of azetidinones |
| CN101346349A (en) * | 2005-12-20 | 2009-01-14 | 吉瑞工厂 | Process for the production of ezetimibe and intermediates used in this proces |
| WO2008089984A2 (en) * | 2007-01-24 | 2008-07-31 | Krka | Process for the preparation of ezetimibe and derivatives thereof |
Non-Patent Citations (1)
| Title |
|---|
| 黄伟 等: "Ezetimibe的合成", 《中国医药工业杂志》, vol. 37, no. 6, 31 December 2006 (2006-12-31), pages 364 - 366 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105272852A (en) * | 2014-07-16 | 2016-01-27 | 浙江九洲药物科技有限公司 | Ezetimibe intermediate and preparation method |
| CN105272852B (en) * | 2014-07-16 | 2019-04-23 | 浙江九洲药物科技有限公司 | A kind of Ezetimible intermediate and preparation method thereof |
| CN106397292A (en) * | 2016-09-20 | 2017-02-15 | 苏州普罗达生物科技有限公司 | Ezetimibe intermediate, synthesis method of intermediate and synthesis method of ezetimibe |
| CN112574055A (en) * | 2019-09-30 | 2021-03-30 | 天津天药药业股份有限公司 | Preparation method and application of formoterol and medicinal salt thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102477008B (en) | 2014-05-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Luo et al. | Chemoenzymatic Synthesis and Application of Bicyclo [2.2. 2] octadiene Ligands: Increased Efficiency in Rhodium-Catalyzed Asymmetric Conjugate Additions by Electronic Tuning We acknowledge Dr. John Whittall for initial inspiration, Dr. Neil Berry for preliminary modeling and the EPSRC for a Dorothy Hodgkin Postgraduate Award to YL | |
| Marshall et al. | Stereoselective total synthesis of the pseudopterolide kallolide A | |
| CN102702067B (en) | Novel intermediate for synthesizing silodosin as well as preparation method and purpose of novel intermediate | |
| CN101935309B (en) | Method for preparing ezetimibe and intermediate thereof | |
| CN101941927A (en) | Method for analyzing (1R, 2R)-2-amino-1-(4-(methylsulfonyl)-phenyl)-1,3-propylene glycol as intermediate of florfenicol | |
| Wang et al. | Synthesis of chiral ferrocenyl aziridino alcohols and use in the catalytic asymmetric addition of diethylzinc to aldehydes | |
| CN111423394B (en) | Synthesis method of 1,3, 4-oxadiazole heterocyclic compound | |
| CN102477008B (en) | Method for synthesizing ezetimibe | |
| CN102112430B (en) | Process for synthesis of ezetimibe and intermediates useful therefor | |
| CN101367746B (en) | Novel method for synthesis of (S)-propisochlor | |
| Abrahams et al. | Addition to activated imines of enolates from chiral N-acyloxazolidinones | |
| CN105130999A (en) | Synthesis method of Sitagliptin impurities | |
| CN101735134A (en) | Chiral 3-hydroxy pyrrolidone compound and preparation method and application thereof | |
| JPS63258838A (en) | Optically active alpha-aminoaldehyde | |
| US8912345B2 (en) | Method for preparing optically pure (−)-clausenamide compound | |
| CN103923030B (en) | Synthesis method of key intermediate of anacetrapib | |
| CN105646285B (en) | One kind dimension Lactel sieve intermediate and its preparation method and application | |
| Koide et al. | Asymmetric synthesis of axially chiral benzamides and anilides utilizing planar chiral arene chromium complexes | |
| CA2642388C (en) | Process for preparing optically active alcohols | |
| CN102320984A (en) | Preparation method of (R)-3-(3-methoxy phenyl)-N,N,2-trimethylpent-3-ene-1-amine | |
| CN104418797A (en) | Preparation methods of methylphenidate and dexmethylphenidate, intermediates and preparation methods of intermediates | |
| CN101952242B (en) | Convergent synthesis of renin inhibitors and intermediates useful therein | |
| CN102134208B (en) | [R,R]-methylphenidate intermediate and analogue thereof and their synthesis and application | |
| CN107382858B (en) | Series of 1,2,3, 4-tetrahydroisoquinoline-4-ketone compounds, and synthetic method and application thereof | |
| CN1978442A (en) | (R,R,R,S)2,2'-[imino-di(methylen)] di-(6-fluoro-3,4-dihydro-2H-1-benzopyran-2-methanol) nebivolo chloride |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20140521 Termination date: 20201122 |
|
| CF01 | Termination of patent right due to non-payment of annual fee |