CN1751027A - 杀精子和/或杀真菌组合物和使用它们的方法 - Google Patents
杀精子和/或杀真菌组合物和使用它们的方法 Download PDFInfo
- Publication number
- CN1751027A CN1751027A CNA2004800047483A CN200480004748A CN1751027A CN 1751027 A CN1751027 A CN 1751027A CN A2004800047483 A CNA2004800047483 A CN A2004800047483A CN 200480004748 A CN200480004748 A CN 200480004748A CN 1751027 A CN1751027 A CN 1751027A
- Authority
- CN
- China
- Prior art keywords
- hydrogen
- compound
- formula
- propyl
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 238000000034 method Methods 0.000 title claims abstract description 59
- 230000001150 spermicidal effect Effects 0.000 title abstract description 5
- 239000012871 anti-fungal composition Substances 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 177
- 239000000203 mixture Substances 0.000 claims abstract description 79
- 241000233866 Fungi Species 0.000 claims abstract description 5
- 229910052739 hydrogen Inorganic materials 0.000 claims description 80
- 239000001257 hydrogen Substances 0.000 claims description 80
- OKKJLVBELUTLKV-UHFFFAOYSA-N methanol Natural products OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 78
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 55
- -1 methylol ester Chemical class 0.000 claims description 53
- 229910052736 halogen Inorganic materials 0.000 claims description 39
- 150000002367 halogens Chemical class 0.000 claims description 37
- 150000002431 hydrogen Chemical class 0.000 claims description 34
- 239000000934 spermatocidal agent Substances 0.000 claims description 33
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 30
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 27
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 27
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 25
- 239000003433 contraceptive agent Substances 0.000 claims description 21
- 230000002254 contraceptive effect Effects 0.000 claims description 17
- 241000124008 Mammalia Species 0.000 claims description 14
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims description 14
- 150000001733 carboxylic acid esters Chemical class 0.000 claims description 14
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 claims description 13
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 claims description 13
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 claims description 13
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 13
- 230000008569 process Effects 0.000 claims description 12
- 230000004888 barrier function Effects 0.000 claims description 11
- 230000004962 physiological condition Effects 0.000 claims description 11
- CFHIDWOYWUOIHU-UHFFFAOYSA-N oxomethyl Chemical compound O=[CH] CFHIDWOYWUOIHU-UHFFFAOYSA-N 0.000 claims description 8
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 7
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 7
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 7
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 7
- 230000001857 anti-mycotic effect Effects 0.000 claims description 5
- 239000002543 antimycotic Substances 0.000 claims description 4
- 239000002674 ointment Substances 0.000 claims description 4
- 230000000843 anti-fungal effect Effects 0.000 claims description 2
- 229940121375 antifungal agent Drugs 0.000 claims description 2
- 239000006260 foam Substances 0.000 claims description 2
- 239000000499 gel Substances 0.000 claims description 2
- 235000015110 jellies Nutrition 0.000 claims description 2
- 239000008274 jelly Substances 0.000 claims description 2
- 238000002560 therapeutic procedure Methods 0.000 claims description 2
- 230000000699 topical effect Effects 0.000 claims description 2
- 125000004185 ester group Chemical group 0.000 claims 8
- 230000002147 killing effect Effects 0.000 abstract description 38
- 230000000855 fungicidal effect Effects 0.000 abstract description 8
- 239000000417 fungicide Substances 0.000 abstract description 3
- CINJODFGIBPBSD-UHFFFAOYSA-N 2,3,4,4a,4b,5-hexahydro-1h-indeno[2,1-b]pyridine Chemical class C1=CCC2C(CCCN3)C3=CC2=C1 CINJODFGIBPBSD-UHFFFAOYSA-N 0.000 abstract 1
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 69
- 239000000243 solution Substances 0.000 description 64
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 36
- AOJFQRQNPXYVLM-UHFFFAOYSA-N pyridin-1-ium;chloride Chemical compound [Cl-].C1=CC=[NH+]C=C1 AOJFQRQNPXYVLM-UHFFFAOYSA-N 0.000 description 34
- 230000000694 effects Effects 0.000 description 29
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 26
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 25
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 24
- 150000003839 salts Chemical class 0.000 description 24
- 239000007787 solid Substances 0.000 description 20
- 230000004899 motility Effects 0.000 description 18
- 210000001550 testis Anatomy 0.000 description 17
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 16
- 239000011550 stock solution Substances 0.000 description 16
- 229940125904 compound 1 Drugs 0.000 description 15
- 238000010790 dilution Methods 0.000 description 15
- 239000012895 dilution Substances 0.000 description 15
- 210000004116 schwann cell Anatomy 0.000 description 15
- 238000005160 1H NMR spectroscopy Methods 0.000 description 14
- 241001465754 Metazoa Species 0.000 description 14
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 14
- 239000000460 chlorine Substances 0.000 description 14
- 238000002360 preparation method Methods 0.000 description 14
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 13
- 210000004027 cell Anatomy 0.000 description 13
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 13
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 13
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 12
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 12
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 12
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 12
- 238000005406 washing Methods 0.000 description 12
- 239000012043 crude product Substances 0.000 description 11
- 238000000605 extraction Methods 0.000 description 11
- 238000001704 evaporation Methods 0.000 description 10
- 239000002583 male contraceptive agent Substances 0.000 description 10
- 239000006187 pill Substances 0.000 description 10
- 239000000047 product Substances 0.000 description 10
- 230000002829 reductive effect Effects 0.000 description 10
- 239000002904 solvent Substances 0.000 description 10
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 239000007864 aqueous solution Substances 0.000 description 9
- 238000013016 damping Methods 0.000 description 9
- 239000012530 fluid Substances 0.000 description 9
- 239000003921 oil Substances 0.000 description 9
- 238000003756 stirring Methods 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- 241000699666 Mus <mouse, genus> Species 0.000 description 8
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 8
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 8
- 230000008859 change Effects 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 8
- 230000035558 fertility Effects 0.000 description 8
- 150000002576 ketones Chemical class 0.000 description 8
- 238000012360 testing method Methods 0.000 description 8
- 210000001177 vas deferen Anatomy 0.000 description 8
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 7
- 241001225321 Aspergillus fumigatus Species 0.000 description 7
- 239000012981 Hank's balanced salt solution Substances 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 229910052799 carbon Inorganic materials 0.000 description 7
- 239000012141 concentrate Substances 0.000 description 7
- 239000003814 drug Substances 0.000 description 7
- 230000012010 growth Effects 0.000 description 7
- 229910052751 metal Inorganic materials 0.000 description 7
- 239000002184 metal Substances 0.000 description 7
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 7
- 238000001953 recrystallisation Methods 0.000 description 7
- 238000010992 reflux Methods 0.000 description 7
- 230000001568 sexual effect Effects 0.000 description 7
- 125000001424 substituent group Chemical group 0.000 description 7
- 210000001519 tissue Anatomy 0.000 description 7
- AYJJTPLDSZAGGA-UHFFFAOYSA-N 2-ethyl-7-methyl-5-(4-methylphenyl)-1,3,4,4a,5,9b-hexahydroindeno[1,2-c]pyridine Chemical compound C1N(CC)CCC2C1C1=CC=C(C)C=C1C2C1=CC=C(C)C=C1 AYJJTPLDSZAGGA-UHFFFAOYSA-N 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- 206010041349 Somnolence Diseases 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 229940091771 aspergillus fumigatus Drugs 0.000 description 6
- 210000004369 blood Anatomy 0.000 description 6
- 239000008280 blood Substances 0.000 description 6
- 238000001035 drying Methods 0.000 description 6
- 238000005516 engineering process Methods 0.000 description 6
- 210000002570 interstitial cell Anatomy 0.000 description 6
- 238000001819 mass spectrum Methods 0.000 description 6
- 239000011541 reaction mixture Substances 0.000 description 6
- 238000011160 research Methods 0.000 description 6
- 230000003068 static effect Effects 0.000 description 6
- 230000002381 testicular Effects 0.000 description 6
- 229960003604 testosterone Drugs 0.000 description 6
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 239000002872 contrast media Substances 0.000 description 5
- 230000008020 evaporation Effects 0.000 description 5
- 238000003818 flash chromatography Methods 0.000 description 5
- 239000000543 intermediate Substances 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 150000004702 methyl esters Chemical class 0.000 description 5
- 239000012074 organic phase Substances 0.000 description 5
- 239000012266 salt solution Substances 0.000 description 5
- 230000035946 sexual desire Effects 0.000 description 5
- 239000011734 sodium Substances 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 241000283973 Oryctolagus cuniculus Species 0.000 description 4
- 230000002378 acidificating effect Effects 0.000 description 4
- 238000009395 breeding Methods 0.000 description 4
- 230000001488 breeding effect Effects 0.000 description 4
- 238000004364 calculation method Methods 0.000 description 4
- 238000001816 cooling Methods 0.000 description 4
- 210000000981 epithelium Anatomy 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 239000012458 free base Substances 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 4
- 229910052740 iodine Inorganic materials 0.000 description 4
- 239000002243 precursor Substances 0.000 description 4
- 230000000750 progressive effect Effects 0.000 description 4
- 210000000582 semen Anatomy 0.000 description 4
- 229960001866 silicon dioxide Drugs 0.000 description 4
- 230000001954 sterilising effect Effects 0.000 description 4
- 238000004659 sterilization and disinfection Methods 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 229920001817 Agar Polymers 0.000 description 3
- 241000222122 Candida albicans Species 0.000 description 3
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 3
- 241000282994 Cervidae Species 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 239000007993 MOPS buffer Substances 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 description 3
- 208000011622 Testicular disease Diseases 0.000 description 3
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 3
- 229960000583 acetic acid Drugs 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 239000008272 agar Substances 0.000 description 3
- 239000003513 alkali Substances 0.000 description 3
- 125000000217 alkyl group Chemical group 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 230000003509 anti-fertility effect Effects 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 210000002937 blood-testis barrier Anatomy 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 229940095731 candida albicans Drugs 0.000 description 3
- 239000006285 cell suspension Substances 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 229940126214 compound 3 Drugs 0.000 description 3
- 230000004069 differentiation Effects 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 238000004817 gas chromatography Methods 0.000 description 3
- XLXSAKCOAKORKW-AQJXLSMYSA-N gonadorelin Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 XLXSAKCOAKORKW-AQJXLSMYSA-N 0.000 description 3
- 230000036541 health Effects 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 150000003840 hydrochlorides Chemical class 0.000 description 3
- 238000011081 inoculation Methods 0.000 description 3
- 239000011630 iodine Substances 0.000 description 3
- UBJFKNSINUCEAL-UHFFFAOYSA-N lithium;2-methylpropane Chemical compound [Li+].C[C-](C)C UBJFKNSINUCEAL-UHFFFAOYSA-N 0.000 description 3
- 239000002207 metabolite Substances 0.000 description 3
- 229940031815 mycocide Drugs 0.000 description 3
- 210000002381 plasma Anatomy 0.000 description 3
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 3
- 230000002199 spermatogenetic effect Effects 0.000 description 3
- 210000004336 spermatogonium Anatomy 0.000 description 3
- 230000002194 synthesizing effect Effects 0.000 description 3
- 229910052722 tritium Inorganic materials 0.000 description 3
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical class C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 2
- NVKAWKQGWWIWPM-ABEVXSGRSA-N 17-β-hydroxy-5-α-Androstan-3-one Chemical compound C1C(=O)CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@H]21 NVKAWKQGWWIWPM-ABEVXSGRSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- PFWJFKBTIBAASX-UHFFFAOYSA-N 9h-indeno[2,1-b]pyridine Chemical class C1=CN=C2CC3=CC=CC=C3C2=C1 PFWJFKBTIBAASX-UHFFFAOYSA-N 0.000 description 2
- 241000894006 Bacteria Species 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 2
- XBLVHTDFJBKJLG-UHFFFAOYSA-N Ethyl nicotinate Chemical compound CCOC(=O)C1=CC=CN=C1 XBLVHTDFJBKJLG-UHFFFAOYSA-N 0.000 description 2
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- 101150003085 Pdcl gene Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 241000282898 Sus scrofa Species 0.000 description 2
- 150000001263 acyl chlorides Chemical class 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 229960003473 androstanolone Drugs 0.000 description 2
- 239000003429 antifungal agent Substances 0.000 description 2
- 238000011888 autopsy Methods 0.000 description 2
- 230000031709 bromination Effects 0.000 description 2
- 238000005893 bromination reaction Methods 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 244000309464 bull Species 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- 230000001332 colony forming effect Effects 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 230000007850 degeneration Effects 0.000 description 2
- YDVNLQGCLLPHAH-UHFFFAOYSA-N dichloromethane;hydrate Chemical compound O.ClCCl YDVNLQGCLLPHAH-UHFFFAOYSA-N 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 229910001873 dinitrogen Inorganic materials 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 210000000918 epididymis Anatomy 0.000 description 2
- 201000010063 epididymitis Diseases 0.000 description 2
- ZKQFHRVKCYFVCN-UHFFFAOYSA-N ethoxyethane;hexane Chemical compound CCOCC.CCCCCC ZKQFHRVKCYFVCN-UHFFFAOYSA-N 0.000 description 2
- 230000003203 everyday effect Effects 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 238000005194 fractionation Methods 0.000 description 2
- 230000001408 fungistatic effect Effects 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 238000003304 gavage Methods 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 239000012362 glacial acetic acid Substances 0.000 description 2
- 239000003163 gonadal steroid hormone Substances 0.000 description 2
- VHHHONWQHHHLTI-UHFFFAOYSA-N hexachloroethane Chemical compound ClC(Cl)(Cl)C(Cl)(Cl)Cl VHHHONWQHHHLTI-UHFFFAOYSA-N 0.000 description 2
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 230000000394 mitotic effect Effects 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 230000003076 paracrine Effects 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- NHKJPPKXDNZFBJ-UHFFFAOYSA-N phenyllithium Chemical compound [Li]C1=CC=CC=C1 NHKJPPKXDNZFBJ-UHFFFAOYSA-N 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 230000009257 reactivity Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 230000001624 sedative effect Effects 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 235000012239 silicon dioxide Nutrition 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 238000007619 statistical method Methods 0.000 description 2
- 150000003431 steroids Chemical class 0.000 description 2
- 239000010902 straw Substances 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 210000000538 tail Anatomy 0.000 description 2
- 125000003944 tolyl group Chemical group 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 210000002700 urine Anatomy 0.000 description 2
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- UCCUXODGPMAHRL-UHFFFAOYSA-N 1-bromo-4-iodobenzene Chemical compound BrC1=CC=C(I)C=C1 UCCUXODGPMAHRL-UHFFFAOYSA-N 0.000 description 1
- JIIHSVKNPYDPLN-UHFFFAOYSA-N 1-ethyl-3,6-dihydro-2h-pyridine-4-carboxylic acid;hydrochloride Chemical compound Cl.CCN1CCC(C(O)=O)=CC1 JIIHSVKNPYDPLN-UHFFFAOYSA-N 0.000 description 1
- VOXZDWNPVJITMN-ZBRFXRBCSA-N 17β-estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 VOXZDWNPVJITMN-ZBRFXRBCSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 241000283708 Capra aegagrus Species 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical class [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- JEJXVQHBGCUMJN-UHFFFAOYSA-N Cl.C1=CC=CC2=C3CN=CC=C3C=C21 Chemical compound Cl.C1=CC=CC2=C3CN=CC=C3C=C21 JEJXVQHBGCUMJN-UHFFFAOYSA-N 0.000 description 1
- 102000003780 Clusterin Human genes 0.000 description 1
- 108090000197 Clusterin Proteins 0.000 description 1
- 229940126657 Compound 17 Drugs 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- 241001600609 Equus ferus Species 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 239000000579 Gonadotropin-Releasing Hormone Substances 0.000 description 1
- 239000007818 Grignard reagent Substances 0.000 description 1
- 206010062767 Hypophysitis Diseases 0.000 description 1
- 208000001953 Hypotension Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010061217 Infestation Diseases 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 102000009151 Luteinizing Hormone Human genes 0.000 description 1
- 108010073521 Luteinizing Hormone Proteins 0.000 description 1
- 206010027336 Menstruation delayed Diseases 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 108010046685 Rho Factor Proteins 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 201000001880 Sexual dysfunction Diseases 0.000 description 1
- 229910004298 SiO 2 Inorganic materials 0.000 description 1
- BNRNXUUZRGQAQC-UHFFFAOYSA-N Sildenafil Natural products CCCC1=NN(C)C(C(N2)=O)=C1N=C2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(C)CC1 BNRNXUUZRGQAQC-UHFFFAOYSA-N 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- 101000857870 Squalus acanthias Gonadoliberin Proteins 0.000 description 1
- 208000025865 Ulcer Diseases 0.000 description 1
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 1
- KYCJGUYSNMURAO-UHFFFAOYSA-N [Li]C1=CC=C(Br)C=C1 Chemical compound [Li]C1=CC=C(Br)C=C1 KYCJGUYSNMURAO-UHFFFAOYSA-N 0.000 description 1
- NWETWFLOVUFJNR-UHFFFAOYSA-N [Li]C1=CC=CC=C1Br Chemical compound [Li]C1=CC=CC=C1Br NWETWFLOVUFJNR-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- CSCPPACGZOOCGX-UHFFFAOYSA-N acetone Substances CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 102000001307 androgen receptors Human genes 0.000 description 1
- 108010080146 androgen receptors Proteins 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 239000003005 anticarcinogenic agent Substances 0.000 description 1
- 239000003420 antiserotonin agent Substances 0.000 description 1
- 235000019789 appetite Nutrition 0.000 description 1
- 230000036528 appetite Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 150000004792 aryl magnesium halides Chemical class 0.000 description 1
- 230000003305 autocrine Effects 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 210000002469 basement membrane Anatomy 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- WTEOIRVLGSZEPR-UHFFFAOYSA-N boron trifluoride Chemical class FB(F)F WTEOIRVLGSZEPR-UHFFFAOYSA-N 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 125000004799 bromophenyl group Chemical group 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- DPJYJNYYDJOJNO-NRPADANISA-N camphorsultam Chemical compound C1S(=O)(=O)N[C@H]2C[C@H]3C(C)(C)[C@@]12CC3 DPJYJNYYDJOJNO-NRPADANISA-N 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 230000007012 clinical effect Effects 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 230000002508 compound effect Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 229940124558 contraceptive agent Drugs 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 239000002254 cytotoxic agent Substances 0.000 description 1
- 231100000599 cytotoxic agent Toxicity 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 238000006356 dehydrogenation reaction Methods 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 208000002925 dental caries Diseases 0.000 description 1
- 239000007933 dermal patch Substances 0.000 description 1
- 239000002274 desiccant Substances 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- YQGOJNYOYNNSMM-UHFFFAOYSA-N eosin Chemical compound [Na+].OC(=O)C1=CC=CC=C1C1=C2C=C(Br)C(=O)C(Br)=C2OC2=C(Br)C(O)=C(Br)C=C21 YQGOJNYOYNNSMM-UHFFFAOYSA-N 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 229960005309 estradiol Drugs 0.000 description 1
- 229930182833 estradiol Natural products 0.000 description 1
- 235000019439 ethyl acetate Nutrition 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 229940064982 ethylnicotinate Drugs 0.000 description 1
- 230000004720 fertilization Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 210000004392 genitalia Anatomy 0.000 description 1
- 229940035638 gonadotropin-releasing hormone Drugs 0.000 description 1
- 150000004795 grignard reagents Chemical class 0.000 description 1
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 230000026030 halogenation Effects 0.000 description 1
- 238000005658 halogenation reaction Methods 0.000 description 1
- 230000012447 hatching Effects 0.000 description 1
- 239000008241 heterogeneous mixture Substances 0.000 description 1
- 230000036732 histological change Effects 0.000 description 1
- 238000010562 histological examination Methods 0.000 description 1
- 239000012456 homogeneous solution Substances 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- QWPPOHNGKGFGJK-UHFFFAOYSA-N hypochlorous acid Chemical compound ClO QWPPOHNGKGFGJK-UHFFFAOYSA-N 0.000 description 1
- 208000021822 hypotensive Diseases 0.000 description 1
- 230000001077 hypotensive effect Effects 0.000 description 1
- 230000002267 hypothalamic effect Effects 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 239000002054 inoculum Substances 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- ICIWUVCWSCSTAQ-UHFFFAOYSA-M iodate Chemical compound [O-]I(=O)=O ICIWUVCWSCSTAQ-UHFFFAOYSA-M 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical class CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 1
- ACKFDYCQCBEDNU-UHFFFAOYSA-J lead(2+);tetraacetate Chemical class [Pb+2].CC([O-])=O.CC([O-])=O.CC([O-])=O.CC([O-])=O ACKFDYCQCBEDNU-UHFFFAOYSA-J 0.000 description 1
- 150000002642 lithium compounds Chemical class 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 230000013011 mating Effects 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- 238000010907 mechanical stirring Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004660 morphological change Effects 0.000 description 1
- 239000010813 municipal solid waste Substances 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 230000002371 mycocidal effect Effects 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- 230000035764 nutrition Effects 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 230000010765 pachytene Effects 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- QQVIHTHCMHWDBS-UHFFFAOYSA-N perisophthalic acid Natural products OC(=O)C1=CC=CC(C(O)=O)=C1 QQVIHTHCMHWDBS-UHFFFAOYSA-N 0.000 description 1
- 150000003016 phosphoric acids Chemical class 0.000 description 1
- 230000001817 pituitary effect Effects 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 239000002574 poison Substances 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Substances [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- 239000001965 potato dextrose agar Substances 0.000 description 1
- 230000003334 potential effect Effects 0.000 description 1
- 239000010970 precious metal Substances 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 230000035935 pregnancy Effects 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 230000005180 public health Effects 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 230000006340 racemization Effects 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 238000000163 radioactive labelling Methods 0.000 description 1
- 238000002601 radiography Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000009933 reproductive health Effects 0.000 description 1
- 230000000452 restraining effect Effects 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 230000001932 seasonal effect Effects 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 238000010956 selective crystallization Methods 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 231100000872 sexual dysfunction Toxicity 0.000 description 1
- 229910000077 silane Inorganic materials 0.000 description 1
- DEIYFTQMQPDXOT-UHFFFAOYSA-N sildenafil citrate Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.CCCC1=NN(C)C(C(N2)=O)=C1N=C2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(C)CC1 DEIYFTQMQPDXOT-UHFFFAOYSA-N 0.000 description 1
- 238000002603 single-photon emission computed tomography Methods 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical class [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000002798 spectrophotometry method Methods 0.000 description 1
- 230000019100 sperm motility Effects 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 125000002345 steroid group Chemical group 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 150000003509 tertiary alcohols Chemical class 0.000 description 1
- PIZNQHDTOZMVBH-UHFFFAOYSA-N thionylimide Chemical compound N=S=O PIZNQHDTOZMVBH-UHFFFAOYSA-N 0.000 description 1
- 229940125725 tranquilizer Drugs 0.000 description 1
- 239000003204 tranquilizing agent Substances 0.000 description 1
- 230000002936 tranquilizing effect Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- 231100000397 ulcer Toxicity 0.000 description 1
- 210000001215 vagina Anatomy 0.000 description 1
- 229940094720 viagra Drugs 0.000 description 1
- 238000004073 vulcanization Methods 0.000 description 1
- 238000013316 zoning Methods 0.000 description 1
Images




Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/473—Quinolines; Isoquinolines ortho- or peri-condensed with carbocyclic ring systems, e.g. acridines, phenanthridines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/16—Masculine contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/18—Feminine contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/02—Local antiseptics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Abstract
提供了充当杀精子剂和/或杀真菌剂的六氢茚并吡啶化合物、含有它们的杀精子和/或杀真菌组合物和使用这些化合物与组合物杀死能动的精子和/或真菌的方法。
Description
发明背景
发明领域
本发明涉及充当杀精子剂和/或杀真菌剂的六氢茚并吡啶化合物、含有它们的杀精子或杀真菌组合物和使用这些化合物与组合物杀死能动的精子或真菌的方法。
背景讨论
在美国和多数西方国家,不断增长的对避孕药的高需求是生活方式优化的结果,而在很多发展中国家,人口控制是非常紧迫的公共健康问题。鉴于避孕是全球健康的需要,尽管在世界的不同地区出于不同的原因,男性避孕药的总体市场可能大大超过单独美国的数字。
在西方国家,避孕药市场在过去50年间的变化相当微小,1951年即已开发出“药丸”,至今仍然是避孕所普遍的不二选择。避孕研究的进展仅仅提供稍微更多的选择,都是针对妇女的,她们在历史上承担避孕所带来的责任、代价和健康风险的冲击(特别是心血管疾病和某些类型癌症的风险,与长期使用激素类避孕药有关)。在16世纪发明的安全套是唯一可用于男性避孕的有效方式(除了“退出”和输精管切除术)。唯一发生于安全套的真正创新性改进是19世纪的橡胶硬化法(From New Scientist,20April 1994,Vol.142,No.1923)。
随着药业公司开发用于性功能障碍的化合物(例如用于勃起功能障碍的万艾可),预计对避孕药的需求会有增长。安全套在美国的销量在1999年上升了5.8%,产生了两亿六千万美元的税收,这被一些人称为新的美国性革命(Drug Store News,Nov 29,1999 v21 il9p29)。尽管大多数育龄妇女已经采取避孕措施(世界上所有处于育龄期的已婚妇女有58%采用某种避孕方法(The Population Division ofthe United Nations Department of Economic and Social Affairs2000),不过仍然有一半的妊娠不是有意的(NICHD,Contraceptionand Reproductive Health Branch:Report to the NACHHD CouncilSeptember 1999)。健康消费群一直呼吁更多的替代选择,特别是使男性承担更大比例的避孕责任。全球性关注已经引起一些组织、例如World Health Organization和Family Health International发起行动,目的在于鼓励开发男性避孕药。至少有两家公司Schering和Organon正在投入巨资使一种激素类男性避孕药在未来十年内上市。
很多年来一直寻求安全有效的口服活性男性避孕药。不过,能够安全阻断精子发生而不影响性欲、由此发挥男性避孕药功能的药物的开发已被证实是一项困难的任务。
理想的男用避孕药将有效阻止精子的产生,阻滞它们受精的能力,而不影响性欲或附属性器官与它们的功能,和/或杀死能动的精子。另外,它应当在有效与毒性剂量之间具有较大的距离,其方法应当是可逆的。这样一种理想的男性避孕药目前是没有的。
一些通用的细胞毒性剂、例如抗癌药和烷基化剂影响精子发生,但是作为避孕药显然不是可接受的。干扰细胞能量过程的化合物、例如硫代糖类也干扰精子发生,不是充分选择性的。雄激素、例如睾酮及其类似物在以充分高剂量给药时,很可能通过一种牵涉下丘脑-垂体轴的机理干扰精子发生。这些甾族化合物已经成功地用于临床研究。不过,这些甾类的合成代谢性质可能引起不可取的副作用。
促性腺素释放激素(GNRH)类似物的研究比较活跃,它们是有效阻滞精子发生的化合物。不过,GNRH类似物干扰内源性睾酮产生,因而降低性欲,除非补充给以雄激素。
男性避孕的一种方法基于男性生殖过程生物化学的鉴别和利用。睾丸由三个功能腔组成。第一个负责精子的产生,由输精管组成,其中含有发育中的精细胞。第二个是塞尔托立氏细胞,也位于输精管内部,它有助于精子发生过程的组织与功能协调,很可能具有旁分泌和自分泌角色。由于在塞尔托立氏细胞与发育中的精细胞之间存在复杂的组织关系,以及在相邻塞尔托立氏细胞之间存在紧密连接,构成血液睾丸屏障,将输精管分成若干区域,血液所携带的化学品或营养物不能直接接近。在间质组织中围绕输精管的是莱迪希氏细胞,它们具有若干内分泌和旁分泌功能,睾酮的产生是最好的诠释。
胚细胞不断分裂和分化,随着成熟从基膜移动至管腔。精原细胞位于基底腔,选择性募集的精原细胞有丝分裂成为精原细胞或者分化为初级精母细胞。初级精母细胞移行通过塞尔托立氏细胞之间的接合处,有丝分裂成次级精母细胞。次级精母细胞分裂成精细胞。精细胞然后分化为成熟的精子。精细胞的分化经常被称为精子发生。不过,出于本申请的目的,“精子发生”被定义为涵盖精子的整个生成与成熟(分化)过程,“抗精子发生化合物”是破坏这种过程的任意部分的化合物。
塞尔托立氏细胞的功能总结如下:(a)支持和供养输精管上皮,(b)释放晚期精细胞至管腔,(c)形成形态学与生理学上的血液睾丸屏障,(d)吞噬退化的生殖细胞,和(e)调节输精管上皮的周期。
莱迪希氏细胞也支持精子发生。来自垂体的黄体化激素(LH)刺激莱迪希氏细胞产生睾酮。睾酮及其代谢产物二氢睾酮是支持正常精子发生所必需的。睾酮受体存在于各种生殖细胞类型上。睾酮被递送通过血液睾丸屏障,有可能通过转运进入塞尔托立氏细胞,在那里被代谢为雌二醇、二氢睾酮或者保持不变。
生殖细胞类型即使不是全部也有一些与莱迪希氏细胞和/或塞尔托立氏细胞相互作用。这些相互作用是化学信使的形式,它们是由塞尔托立氏细胞、莱迪希氏细胞和生殖细胞所产生的。例如,粗线期精母细胞调控塞尔托立氏细胞蛋白质因子的分泌,该因子进而刺激莱迪希氏细胞的甾类生成。精细胞的结合仅发生于塞尔托立氏细胞,它们因暴露于FSH而被赋予有效性或功能性。大鼠塞尔托立氏细胞以周期方式分泌数种蛋白质,最大产生发生在输精管上皮的特定阶段;也就是在它与特定的生殖细胞缔合时。当输精管上皮处于阶段VII或VIII构型时,塞尔托立氏细胞最大地产生clusterin,这独立于FSH刺激,提示了生殖细胞对塞尔托立氏细胞分泌功能的局部调节。
由Sandoz Ltd.开发的六氢茚并吡啶化合物no.20-438(图1中的化合物1)在对动物口服给药时提供可逆的精子发生抑制作用。参见Arch.Toxicol.Suppl.,1984,7:171-173;Arch.Toxicol.Suppl.,1978,1:323-326和Mutation Research,1979,66:113-127。
多种茚并吡啶化合物外消旋混合物的合成是已知的,例如参见美国专利Nos.2,470,108;2,470,109;2,546,652;3,627,773;3,678,057;3,462,443;3,408,353;3,497,517;3,574,686;3,678,058和3,991,066。这些茚并吡啶化合物具有多种用途,包括用作表现抗炎与止痛性质的血清素拮抗剂、成血细胞聚集抑制剂、镇静剂和精神抑制性化合物以及溃疡保护性、降血压与减食欲性化合物。
美国专利Nos.5,319,084和5,952,336公开了具有抗精子发生活性的六氢茚并吡啶化合物,其中5-位被具有对位取代基的苯基环取代。
尽管本领域已经进行了广泛的研究,不过仍然需要具有有限副作用的活性可逆性男性抗生育药。仍然存在的问题是需要在可能导致副作用的剂量水平下给以已知化合物。本领域中的其他问题是缺乏适合的造影剂,它们在睾丸之上或之中具有特定结合位点。仍然需要这样的化合物,它们可以在睾丸功能的研究中和睾丸机能障碍的诊断中用作造影剂。
除了男性口服避孕药以外,还需要更有效的杀精子组合物,用作传统的局部/外用避孕措施。
发明概述
因此,本发明的一个目的是提供口服活性男性避孕药,它不会影响性欲,具有较高的效力和活性,具有微小的副作用或毒性。
本发明的另一目的是提供抑制精子发生的口服活性男性避孕药和利用这种药物抑制精子发生的方法。
本发明的另一目的是提供充当杀精子剂的组合物,杀死能动的精子,从而是有效的外用避孕药。
本发明的另一目的是提供充当抗真菌组合物的组合物。
借助下列发现已经达到本发明的这些和其他目的,即发现了本发明的六氢茚并吡啶化合物,并且发现这些化合物是非常有力的,阻断精子发生,对能动的精子充当杀精子剂,并且表现有效的抗真菌性质。
本发明化合物解决了上述问题。本发明化合物在比已知化合物1更低的剂量下表现很高的效力,减少了副作用的发生,例如利用这种化合物所观察到的镇静作用。进而,本发明化合物与睾丸中的大分子位点相互作用。含有一种标记、例如放射性标记的本发明化合物克服了不当造影剂的问题,提供了可用于睾丸功能研究和睾丸机能障碍诊断的造影剂。本发明化合物也被发现充当杀精子剂,以有效和高效的方式杀死能动的精子,这提示了它们在多种杀精子组合物中的用途。还发现本发明化合物充当抗真菌剂。
附图的简要说明
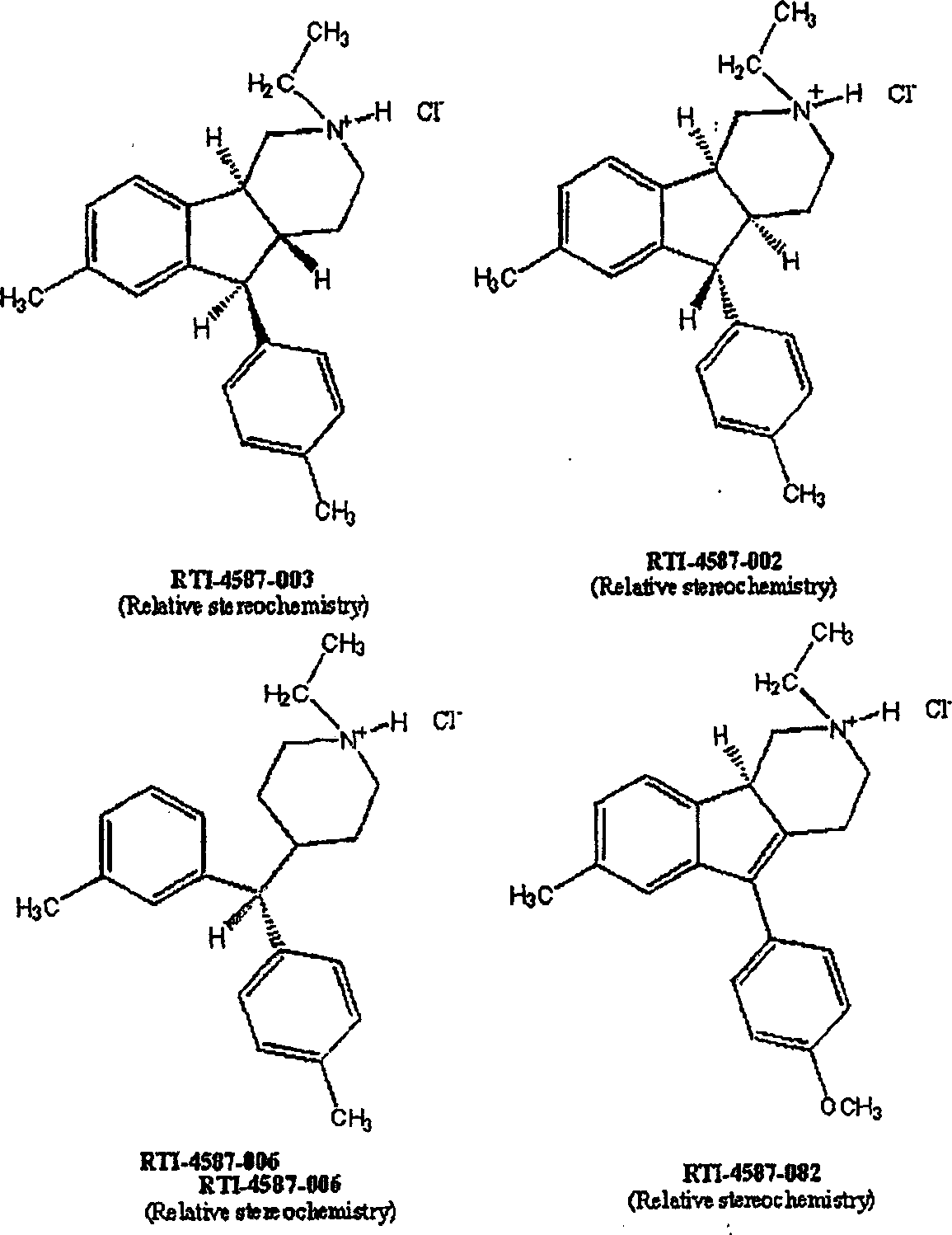
图1显示三种六氢茚并吡啶化合物的结构,标明这些化合物的编号系统。
图2显示制备本发明化合物前体的过程。
图3显示从前体化合物到本发明化合物的对映选择性合成。
图4显示碘化如图2和3所示制备的前体化合物以及转化该碘化化合物为其他本发明化合物的合成流程。
发明的详细说明
现已发现,具有下示结构I(a)、优选具有下示结构I(b)的六氢茚并吡啶化合物:

其中4a、5和9b位氢原子具有所显示的相对立体化学(4a和5位氢彼此为反式,4a和9b氢彼此为顺式;式I(b));或者9b位相对立体化学可以是相反的,以便4a和5位氢彼此为反式,4a与9b氢彼此为反式;或者所有三个氢都可以彼此为顺式;或者
其中4a与5之间的虚线表明该化合物也可以是4a,5-脱氢化合物,从而在4a与5碳之间具有双键;
其中R1是氢或者直链或支链C1-6烷基,优选C1-3烷基,或者C3-8环烷基;
R2是氢、直链或支链C1-6烷基,优选C1-3烷基;
R3和R5独立地是氢、卤素、SO3H、直链或支链C1-6烷基、CH2OH、CH2OMe、直链或支链C1-6烷氧基、羧基(COOH)或者可以在哺乳动物生理条件下转化为羧基的基团、羧酸酯(COOR,其中R是C1-10烷基、C6-10芳基、C7-10芳烷基)、羟甲基酯(CH2OC(O)--R,其中R是如上所定义的)、CONH2、CONHR、CONR2、CH2OCONHR、CN、CH=NHNHCONH2和卤素;
R4是氢、卤素、R3Si或COR,
不仅是抗精子发生的,其活性多达以前美国专利5,319,084所报道的最好的已知化合物的口服抗精子发生活性的约40倍,而且充当杀精子剂,以杀死能动的精子,还充当抗真菌剂。
在本发明的上下文中,术语“抗精子发生”涉及破坏睾丸中精子产生的能力,而术语“杀精子剂”或“杀精子的”涉及在产生之后、更优选在射精之后杀死能动的精子的能力。
本发明化合物具有结构(I)所示的相对立体化学。本发明包括两种个别的对映异构形式(本质上为旋光纯的)以及这些形式的任意混合物,例如外消旋混合物。
在本发明内也包括具有上示结构(I)的化合物的药学上可接受的盐。药学上可接受的盐包括但不限于碱性官能团(例如但不限于胺基)与无机酸的盐,例如盐酸盐、氢碘酸盐、硫酸盐、磷酸盐、二磷酸盐、氢溴酸盐和硝酸盐,或者碱性官能团与有机酸的盐,例如乙酸盐、苹果酸盐、马来酸盐、富马酸盐、酒石酸盐、琥珀酸盐、柠檬酸盐、乳酸盐、甲磺酸盐、对-甲苯磺酸盐、扑酸盐、水杨酸盐和硬脂酸盐;或者酸性官能团与金属离子的盐,例如(但不限于)Na、K、Ca的盐,或者酸性官能团与铵离子的盐,或者酸性官能团与有机离子的盐,例如(但不限于)胺和四取代的铵离子的盐。
取代基R1优选地是直链烷基(正烷基)、异烷基或环烷基,例如甲基、乙基、正丙基、异丙基、正丁基、异丁基、正戊基、异戊基、正己基、异己基、环丙基、环戊基、环己基。最优选地,R1是乙基。
取代基R2也优选地是直链烷基或异烷基,如上R1所述。
取代基R3优选地位于环的对位或4位,优选地选自羟甲基(CH2OH)、甲酰基(CHO)、羧基(COOH)、羧酸酯(COOR,其中R是C1-10烷基、C6-10芳基、C7-10芳烷基)、羟甲基酯(CH2OC(O)--R,其中R是如上所定义的)、CONH2、CONHR、CONR2、CH2OCONHR、CN、CH=NHNHCONH2和卤素。
取代基R4优选地是卤素,包括I、Br、Cl和F。这些化合物的潜在活性是惊人的。卤素可以是放射性同位素,例如123I、125I或131I。其他放射性同位素、例如11C、氚(3H)或18F或者溴和氯的放射性同位素可以取代上述化合物中的普通(非放射性)同位素。
化合物1是外消旋混合物。化合物1的结构如图1化合物1所示。六氢茚并吡啶具有三个不对称中心,它们可以利用已知命名法加以定义。作为替代选择,相对立体化学可以借助与三环系统4a、5和9b位碳系键合的氢原子的顺-反式关系加以定义,得到立体化学赋值。按照Cahn-Ingold-Prelog命名法,化合物1的立体化学和名称为(4aRS,5SR,9bRS)-2-乙基-2,3,4,4a,5,9b-六氢-7-甲基-5-(4-甲基苯基)-1H-茚并[1,2-c]吡啶。
化合物1在5-苯基上具有疏水性甲基取代基,对应于上示结构(I)中的取代基R3。化合物1的抗精子发生活性本质上仅仅存在于(+)异构体中(当在Cook et al.,1995所述条件下测量时旋光性为右旋),它在小鼠中是有效的抗精子发生药。这一系列其他化合物的抗精子发生活性也本质上仅仅存在于一种旋光异构体中。不过,所测量的这些化合物的旋光性可以是(+)或(-),这依赖于取代方式和测量条件,正如立体化学技术人员所已知的。另一方面,本发明化合物的抗真菌性质不是立体特异性的,(+)和(-)异构体都有活性,尽管它们的相对活性可能不同。
本发明化合物的杀精子作用已经见于抗精子发生异构体中,据信也存在于其他异构体中。
强极性羧基或者能够在哺乳动物生理条件下代谢为羧基的基团可以存在于本发明化合物的5-苯基环的任意位置,优选对位,同时保留杀精子和/或抗真菌活性。例如,其中对位被羟甲基(CH2OH)、甲酰基(CHO)、羧基(COOH)和甲氧羰基(C(O)OCH3)取代的化合物保留潜在的活性。这些化合物表现活性,尽管在5-苯基环对位存在极性取代基。
“在哺乳动物生理条件下代谢”表示当结构(I)化合物对需要抗精子发生治疗的活哺乳动物给药时转化为羧基的官能团R3。给药可以借助任意常规手段或途径,包括但不限于口服、腹膜内、静脉内、皮下、肌内、吸入、颊膜和皮肤渗透。这些给药途径以及局部给药同样可用于杀精子和/或抗真菌治疗。通过监测结构(I)化合物在血液或尿液中的代谢产物,容易测定基团R3向羧基的转化。代谢产物可以利用常规分析方法加以监测,例如质谱(MS)、气相色谱(GC)等。
优选地,至少50%、更优选至少80%、进而更优选90%、95%或100%的官能团R3在对哺乳动物给药后被代谢为羧基,尽管这不是为获得杀精子和/或杀真菌性质所必需的。转化百分比可以这样测定,利用上述常规分析方法之一定量分析血液或尿液样品,以测定含有官能团R3的未转化化合物相对于其中R3已被转化为羧基的化合物的相对含量。
在对大鼠单次口服30mg/kg之后观察到化合物1的抗精子发生活性,在24小时内急剧减少睾丸的重量。观察到输精管的退化改变。精细胞变为固缩,偶尔形成多核缔合。塞尔托立氏细胞似乎在细胞学上是正常的。似乎化合物1靶向精细胞或者与这些精细胞有关的塞尔托立氏细胞,因为首先在这些精细胞中观察到组织学改变。
化合物1在30mg/kg口服剂量下导致小鼠一定的嗜睡和镇静,在皮下给予的相同剂量下导致极度嗜睡。嗜睡和镇静显然在避孕药中是不可取的副作用。与利用化合物1所观察到的嗜睡和镇静相反,本发明化合物产生轻微的嗜睡。
本发明化合物可以区分抗生育活性与利用化合物1所观察到的镇静活性。本发明化合物因此是有效的抗生育药,其中明显减少了不可取的镇静和嗜睡副作用。
按照下述Cook et al(1995)所述工艺,在单次口服后三天测试本发明化合物对小鼠精子发生的作用。在这种试验中有活性的化合物也是抗生育化合物。
如下筛选化合物的抗精子发生活性,在第1天给雄性小鼠服用管饲剂量的对照载体、阳性对照(化合物1)或本发明化合物。服用后72小时,杀死动物,切除睾丸,剪去脂肪,称重。对一只睾丸进行组织学检查,利用精子发生指数评价精子发生潜力(J.M.Whitsett,P.F.Noden,J.Cherry and A.D.Lawton,J.Reprod.Fertil.,72,277(1984)),这是睾丸产生精子能力的一种半定量估计。该指数基于输精管中精子发生细胞的组织学外观。得分为1至6,5至6为正常状态。第二种评估基于睾丸的重量。
表1和2显示有关的生物学结果,以睾丸重量(TW)和精子发生指数(SI)相对于仅含给药载体而无茚并吡啶的对照而言的改变表示。在表1和2中,R3和R4表示结构Ib,其中R3处于对位;R1是乙基;R2是甲基;R5是氢。
利用8-碘-7-甲基-4’-羧基或4’-甲酯基取代方式,口服剂量为2μmol/kg(1mg/kg)的外消旋物导致精子发生指数降低57-67%,至少与剂量为79μmol/kg(30mg/kg)的没有8-碘取代基的对应类似物是同样有效的。在8-溴或8-氯类似物的情况下,所测试的最低剂量(6或2μmol/kg;3或1mg/kg)也至少与剂量为79μmol/kg(30mg/kg)的非卤化类似物是同样有效的(见表1)。比较8-碘-7-甲基-4’-甲酯基类似物的活性(左旋)对映体与8-H-7-甲基-4’-甲酯基类似物的活性对映体(表2),显示前者化合物在0.6和2μmol/kg(0.3和1mg/kg)下具有等于或大于后者化合物在25和75μmol/kg(10和30mg/kg)下的作用。因而,8位卤化实现摩尔效力增加大约40倍。
表1
| 外消旋茚并吡啶化合物对成年雄性瑞士小鼠的抗精子发生作用a | |||||
| 化合物 | R3 | R4 | 剂量(mg/kg) | TWb改变(%) | SIc改变(%) |
| 122181818171717191919202020 | MeCO2HCO2HCO2MeCO2MeCO2MeCO2HCO2HCO2HCO2HCO2HCO2HCO2HCO2HCO2H | HHHIIIIIIBrBrBrClClCl | 30103013101310310301310 | 19%*2%-7%-16%-27%*-36%*-18%-9%-32%*-8%-28%*-39%*-16%-23%-22% | -55%*-24%*-52%*-57%*-69%*-74%*-67%*-66%*-76%*-69%*-71%*-72%*-55%*-66%*-72%* |
a数值从[100(试验对照)/(对照)]计算而来,为平均值(n=5)。仅显示化合物无活性的最高剂量。按照10ml/kg通过管饲法对小鼠给以单剂茚并吡啶或载体。载体为90%水、7%吐温20和3%乙醇。在第3天进行尸检,开始于给药后约72小时。
b睾丸重量(载体对照的改变%为217.8+/-46.0(S.E.)mg)
c精子发生指数(载体对照的改变%为5.8+/-0.2(S.E.))
*显著不同于载体对照;Dunnett氏单侧T检验p<0.05
在转化为改变%之前,对原始数据进行统计学分析。
表2
| 8-碘化对手性茚并吡啶化合物在成年雄性瑞士小鼠a中抗精子发生作用的影响(化合物1为外消旋物;化合物3和18为左旋(1)异构体) | |||||
| 化合物 | R3 | R4 | 剂量(mg/kg) | TWb改变(%) | SIc改变(%) |
| 13(1)3(1)3(1)3(1)18(1)18(1)18(1)18(1) | MeCO2MeCO2MeCO2MeCO2MeCO2MeCO2MeCO2MeCO2Me | HHHHHIIII | 30d1310e300.31310e | -24%*8%-12%-13%-13%*-11%*-21%*-27%*-31%* | -61%*3%-2%-33%*-64%*-34%*-66%*-71%*-72%* |
a数值从[100(试验对照)/(对照)]计算而来,为平均值(n=5)。按照10ml/kg通过管饲法对小鼠给以单剂茚并吡啶或载体。在第3天进行尸检,开始于给药后约72小时。载体为含1%吐温20的水。
b睾丸重量(载体对照的改变%为227.5+/-8.6mg)
c精子发生指数(载体对照的改变%为5.7+/-0.2)
dn=6
en=4
*显著不同于载体对照;Dunnett氏单侧T检验p<0.05
在转化为改变%之前,对原始数据进行统计学分析。
组合物单次应用于精液,本发明化合物的杀精子活性显著,浓度仅为3μm的组合物显著减少能动性,在浓度仅为100μm的本发明化合物组合物应用之后能动性下降至零。本发明杀精子组合物可以具有任意足以降低精子能动性至不足以导致受精的水平的浓度,优选1-500μm,更优选3-300μm,最优选10-200μm的浓度。杀精子活性是借助下述方法测定的。
测定药物对精子能动性的直接作用的方法
利用下列方案测定药物对精子能动性的直接作用。大致而言,从附睾尾得到精子,此为大鼠的情况,或者利用人工阴道收集射出的精子,此为兔子的情况。手工或者利用Hamilton Thorn IVOS精子分析仪测定最初的精子能动性。然后将精子保持在34℃恒温下,稀释至10×106/ml的恒定浓度,加入到3ml缓冲液或培养基中。此时再次测定能动性,记录任何改变。然后向精子制备物加入不同浓度的供试药物。然后将精子样品在相同温度下保持1小时,测定能动性。记录结果,为样品中能动精子的百分比。
药物对精子能动性的直接作用的测定方案
一般方法:
在全部实验期间将精子保持在34℃下。精子浓度为大约10×106/ml(注:样品可能不得不用缓冲液或培养基稀释过,以达到这种浓度)。
就大鼠研究而言(使用来自附睾尾的精子):
制备1mM茚并吡啶在HBSS缓冲液+BSA中的储备溶液(5mg/10=0.5μ/μl;0.5μg/ml=1μm),如下加入:
1μm=1μl储备溶液+949μl HBSS缓冲液+BSA+50μl稀释的精子
3μm=3μl储备溶液+947μl HBSS缓冲液+BSA+50μl稀释的精子
10μm=10μl储备溶液+940μl HBSS缓冲液+BSA+50μl稀释的精子
30μm=30μl储备溶液+920μl HBSS缓冲液+BSA+50μl稀释的精子
100μm=100μl储备溶液+850μl HBSS缓冲液+BSA+50μl稀释的精子
300μm=300μl储备溶液+650μl HBSS缓冲液+BSA+50μl稀释的精子
1000μM=1000μl储备溶液+50μl稀释的精子
1小时后测定精子能动性。
就兔子研究而言(使用射出的精子):
制备1mM茚并吡啶在含BSA的M-199培养基中的储备溶液(2.5mg/5=0.5μg/μl;0.5μg/ml=1μM),如下加入:
1μm=1μl储备溶液+949μl含BSA的M-199培养基+50μl稀释的精子
3μm=3μl储备溶液+947μl含BSA的M-199培养基+50μl稀释的精子
10μm=10μl储备溶液+940μl含BSA的M-199培养基+50μl稀释的精子
30μm=30μl储备溶液+920μl含BSA的M-199培养基+50μl稀释的精子
100μM=100μl储备溶液+850μl含BSA的M-199培养基+50μl稀释的精子
300μm=300μl储备溶液+650μl含BSA的M-199培养基+50μl稀释的精子
1000μM=1000μl储备溶液+50μl稀释的精子
1小时后测定精子能动性。
制备切片以测定形态学改变,加入曙红以测定细胞死亡。
杀精子试验的结果如下表3(兔子精液)和表4(大鼠附睾尾精子)所示。
表3:兔子精子能动性
| 精液ID | 精液体积(ml) | 精液浓度(×106) | 最初能动性(%) | 60分钟时的能动性(%) |
| 净样品 | 0.7+plug | 132 | 78.8 | 76.8 |
| 对照(净稀释比1∶10) | 78.5 | 76.6 | ||
| +1μM 073L | 73.0 | |||
| +3μM 073L | 68.4 | |||
| +10μM 073L | 67.6 | |||
| +30μM 073L | 61.9 | |||
| +100μM 073L | 15.2* | |||
| +300μM 073L | 0 | |||
| +1000μM 073L | 0** |
*能动性没有前进-颤搐
**精子似乎“崩解”-精子头含有很多空泡,膜不总是完整的
表4:大鼠附睾尾精子能动性
| 60分钟时的能动性(%) | |
| 对照0μM | 有前进的能动性 |
| +1μM 073L | 有前进的能动性-等于对照 |
| +3μM 073L | 有前进的能动性减少 |
| +10μM 073L | 没有前进的能动性-颤搐 |
| +30μM 073L | 与10μM相同 |
| +100μM 073L | 死亡 |
在任意足以减少真菌水平至不足以导致真菌感染的水平的浓度下可以获得本发明化合物的杀真菌活性,浓度优选为1-500μM,更优选20-300μM,最优选20-200μM。
按照NCCLS指南进行抗真菌敏感性试验,并进行微小调整。简而言之,在35℃下在酵母-麦芽提取物(YM)琼脂上生长过夜后,从纯培养物制备白色念珠菌细胞悬液。从YM琼脂平板上除去若干小的菌落,转移至5ml 0.85%盐水。涡旋15秒使细胞悬浮,利用分光光度计测定所得悬液的细胞密度。加入0.85%盐水调节细胞密度,直至在530nm波长下测量时透光度匹配0.5麦克法兰标准。将这种悬液的等分试样在RPMI-MOPS中稀释至1∶1000,得到工作悬液。使用贮存在40℃0.85%盐水中的烟曲霉孢子得到相似的孢子悬液。在0.85%盐水中稀释这些孢子至匹配麦克法兰标准透光度。然后在RPMI-MOPS中进一步稀释这种悬液至1∶50,得到工作接种悬液。
在含有2%DMSO的RPMI-MOPS中制备供试化合物的系列稀释液。将每种稀释液的等分试样一式两份加入到无菌96孔平底微量滴定板的小孔中。然后向每孔加入白色念珠菌细胞悬液或烟曲霉孢子悬液的等分试样,最终体积为200μl。然后在35℃下孵育平板。孵育19小时后,利用裸眼观察白色念珠菌接种平板,以测定最小抑制浓度(MIC),在该浓度下没有可观察到的生长。48小时后对烟曲霉平板进行相似的测定。就所有表现没有供试生物生长的稀释液而言,为了测定最小杀真菌浓度(MFC),从代表性小孔划取100μl培养基至YM琼脂平板上供白色念珠菌生长,或者至马铃薯葡萄糖琼脂平板上供烟曲霉生长。统计菌落形成单位(CFU)的数量,这些数值用于计算接种物中细胞或孢子存活百分比,相对于工作接种悬液中的CFU数而言。最小杀真菌浓度(MFC)是存活百分比小于2%的最低供试化合物浓度。
杀真菌剂试验的结果如下表4-6所示。
表4:对白色念珠菌的杀真菌活性
| 白色念珠菌(19小时) | ||||
| 化合物 | 最低 | MFC的CFU | ||
| - | [活性] | 300 | 200 | 100 |
| 4587-006 | 300 | Static | ||
| 4587-029 | 200 | Cidal | Static | |
| 4587-055 | 300 | Static | ||
| 4587-054 | N/A | |||
| 4587-054d | N/A | |||
| 4587-073d | 100 | Cidal | Cidal | Cidal |
| 4587-0731 | 100 | Cidal | Cidal | Cidal |
| 4587-040 | 100 | Cidal | Cidal | Cidal |
| 4587-074 | N/A | |||
| 4587-0561 | 200 | Cidal | Cidal | |
| 4587-060 | N/A | |||
| 4587-037 | 300 | Cidal | ||
| 4587-065 | N/A | |||
| 4587-057 | N/A | |||
| 4587-064 | 100 | Cidal | Cidal | Static |
表5:对烟曲霉的杀真菌活性
| 烟曲霉(48小时) | ||||
| 化合物 | 最低 | MFC的CFU | ||
| - | [活性] | 300 | 200 | 100 |
| 4587-006 | N/A | |||
| 4587-029 | 100 | Cidal | Cidal | Cidal |
| 4587-055 | N/A | |||
| 4587-054 | N/A | |||
| 4587-054d | N/A | |||
| 4587-073d | 100 | Cidal | Cidal | Cidal |
| 4587-0731 | 100 | Cidal | Cidal | Cidal |
| 4587-040 | 100 | Cidal | Cidal | Cidal |
| 4587-074 | N/A | |||
| 4587-0561 | 100 | Cidal | Cidal | Cidal |
| 4587-060 | 200 | 未测 | Cidal | |
| 4587-037 | 200 | Static* | Cidal | |
| 4587-065 | N/A | |||
| 4587-057 | N/A | |||
| 4587-064 | 100 | Cidal | Cidal | Cidal |
表4-5注:
MFC=最小杀真菌浓度
CFU=菌落形成单位
Cidal=杀死>98%
Static=看上去良好,但是杀死<98%
*菌落太接近以致不能进行良好的统计。有可能接近杀真菌的。
注意到在073d与1孔中似乎存在一定程度的沉淀。也许PPT“俘获”孢子。没有其他化合物显示这种PPT。
表6:最大杀真菌活性的比较
| 白色念珠菌 | 烟曲霉 | |||
| 最低 | 最低 | 最低 | 最低 | |
| 化合物 | MIC | MFC | MIC | MFC |
| 4587-002d | 50 | fungistatic | 50 | 50 |
| 4587-0021 | 75 | 100 | 50 | 75 |
| 4587-003 | 50 | 75 | 25 | 20 |
| 4587-056d* | >100 | N/A | 100 | 100 |
| 4587-061 | 50 | 75 | 25 | 25 |
| 4587-062* | 20 | 20 | 10 | 10 |
| 4587-064 | 100 | 100 | 50 | 75 |
| 4587-082 | >100 | N/A | 100 | 100 |
| AMP B | 0.5 | 2 | ||
| 酮康唑 | 32 | 8 | ||
MIC=似乎没有生长的最低浓度(μg/ml)
MFC=杀死小孔中>=98%细胞的最低浓度
Fungistatic=具有MIC的化合物,但是杀死<98%细胞
Bold=仅对烟曲霉有活性的化合物
062的浓度:假定在50μl中有0.1mg,因此最终测定浓度将是所报道的1/5。尽管如此,这些浓度是粗略的估计。
用在上表试验中的各种化合物如下所示:
茚并吡啶类似物


利用美国专利No.3,678,057所公开的方法改进,借助美国专利No.5,319,084所公开的方法可以制备本发明化合物的前体。这些专利全文引用在此作为参考。利用适当的格利雅试剂或苯基锂试剂向分子中引入R3取代基。借助盐的生成、继之以选择性结晶或色谱,将由这种过程所生成的对映体混合物拆分为纯的对映体。例如,与S(+)和R(-)-2,2’-(1,1’-联萘)磷酸生成盐可以实现化合物1的拆分,与R-和S-扁桃酸生成盐可以实现化合物3的拆分,如C.E.Cook et al,J.Med.Chem.,38:753(1995)所述。借助CHIRACEL-OD柱上的高效液相色谱(HPLC)测定光学纯度。
本发明化合物的制备可以开始于羧酸2或其酯之一(例如3)。诸如2和3等化合物是如美国专利No.5,319,084所述制备的。作为替代选择,它们可以借助图2所示过程加以制备,其中将N-取代的-3-芳基六氢吡啶-4-羧酸酯(4)水解为羧酸5,然后用亚硫酰氯处理,得到酰氯6。将这种化合物用AlCl3处理,将化合物环化为三环酮7。使酮7与对-卤素取代的苯基卤化镁或对-卤素取代的苯基锂(4-溴苯基锂)反应,生成叔醇8,用三烷基硅烷、例如三-C1-6-烷基硅烷、例如三乙基硅烷和BF3处理后,还原为化合物9,然后与强碱(例如KOH)在醇溶剂中回流,优选高沸点醇,例如正丁醇,得到具有所需立体化学的溴苯基化合物10。转化溴苯基为锂苯基,例如用C1-6烷基锂化合物,再用已知试剂进行羧基化(CO2),得到羧酸2,后者可以借助本领域熟知的常规手段酯化,例如与C1-6烷醇反应,得到酯3。
调整上述合成,可以提供化合物2和3的活性对映体的对映选择性合成,然后可以用于合成本发明的活性对映体,如图3所示。因而,将N-取代的-1,2,5,6-四氢吡啶-4-羧酸(例如12)转化为它的酰氯,后者化合物用于酰化1R(+)-(2,10)-樟脑磺内酰胺或1S(-)-(2,10)-樟脑磺内酰胺。当将所得烯酰基磺内酰胺(13)用芳基卤化镁处理时,它经历高非对映选择性的1,4-加成,以高对映体过量在3-位引入芳基。环化得到纯的对映体14。水解酰胺官能,然后可以回收手性助剂。然后将羧酸转化为三环酮7,如上所述。用溴苯基锂处理,随后进行图2所示步骤,可以将这种化合物转化为本质上对映体纯的2和3。作为替代选择,借助美国专利No.5,319,084中外消旋物合成所述工艺可以将手性酮7转化为对映体富集的2和3。富集程度依赖于对映异构四氢茚并吡啶类似物向中间体5还原的催化剂和温度。参见美国专利No.5,319,084的图3。因而,在23℃、PdCl2/NaBH4/3atm H2下,存在73%对映体过量(ee),但是在55℃完全外消旋化;而在60℃、Pt/C/H2下的ee相当于23℃下的ee(分别为67%和70%)。
在氧化条件下与碘反应,或者与碘的氧化形式反应,可以碘化羧酸2或其酯、例如甲基酯3,得到8-碘类似物17或18(图4)。例如,在氧化汞的存在下,3与约1mol碘反应,得到高收率的8-碘化合物18。借助本领域熟知的标准化学技术,酯和酸是可相互转化的。可以使用外消旋物或对映体。还可以使用碘的放射性同位素,例如125I、123I或131I,得到放射性标记的17或18类似物。这类化合物可用于测定这些化合物作用的定位和位点,可以用作男性生殖障碍的诊断造影剂。
碘代化合物、特别是8-碘酸17可以这样转化为溴代和氯代化合物,生成酸的金属盐,例如钠盐,然后生成8-金属中间体,其中该金属是一种金属,例如锂,或者被已知试剂取代的金属,例如t-BuLi。8-金属中间体与卤素源反应,例如六氯乙烷或1,2-二溴乙烯,得到对应的8-取代的类似物,例如化合物19或20,如图4所示。相应的氟代化合物可以这样制备,使8-金属中间体与氯代三甲基硅烷反应,生成对应的8-三甲代甲硅烷基化合物,然后在BF3-Et2O的存在下使这种化合物与四乙酸铅反应。参见De Mio et al,1993,Tetrahedron,49:8129-8138。
例如将8-金属中间体用含有亲电性卤原子作为其放射性同位素的试剂处理,可以得到各种主题化合物的放射性类似物,或者正如前面所指出的,在上述化合物的合成中取代碘的放射性同位素,可以制备化合物17或18的放射性类似物。例如在贵金属的催化下,例如钯或铂,将8-碘化合物用氚气还原,可以得到氚标记的本发明化合物。例如在图2所示化合物2合成的步骤“g”中使用14C标记的二氧化碳,可以制备碳-14类似物。还可以采用放射化学合成领域中常用的其他同位素标记化合物的方法。
本发明化合物可用作男性抗生育药,用于控制哺乳动物、包括人类的生育。除了它们在计划生育中的潜在用途以外,本发明化合物还可用于控制驯养、野生或凶猛动物的生育,其中致命措施不是实用的或可取的。例如,鹿群的控制在美国一些地区是个问题。本发明化合物对季节性繁殖动物、例如鹿借助含有这些化合物的食饵在适当时间口服给药将基本上减少繁殖能力。其他目标动物包括啮齿动物,例如小鼠、大鼠、草原犬鼠等,以及野生的山羊、猪、马等。对捕捉动物园动物给以本发明化合物提供了控制过多物种繁殖的手段。
本文所用的“控制生育”表示减少所处置的哺乳动物的繁殖能力或生育力。不育的长度是剂量的函数,利用充足的剂量可以延长不育的时间,以便在本质上利用本发明化合物进行绝育;因而,本发明化合物可以代替输精管切除术,作为雄性绝育的手段。在实施这类绝育时,本发明化合物是按单次剂量或多次(两次或更多)剂量给药的,其中这些剂量足以减少哺乳动物精子产生能力(精子发生指数)至不育的水平。也就是说,本发明化合物给药的剂量和时间长度足以减少精子数至不足以繁殖的水平。
就上述用途而言,本发明化合物的剂量自然将因所采用的具体化合物、给药方式和所需不育的长度而异。不过,在动物中取得令人满意的结果的口服剂量为约0.02至约10mg/kg、优选约0.1-3mg/kg体重每天。就更大的动物而言,可以给予约10-100mg的每日剂量,此为单次口服单元剂量或者分次剂量单元,其中含有约0.1-10mg本发明化合物。当给以单一的活性对映体时,一般可以给予较小的剂量,与当给以外消旋化合物时相比。如果需要或必要的话,本发明化合物可以与固体或液体载体或稀释剂一起或者在缓释剂型中给药。这些药物剂型的配制是本领域所熟知的,任意常规的制备固体、液体和缓释制剂的方法都可以用于本发明化合物。本发明化合物还可以借助常规植入物或皮肤贴剂给药,这些是本领域所熟知的。
本发明化合物可以用于男性避孕,既可逆地阻滞精子发生,又非手术地绝育。在后者用途中,适当地大的剂量给药可实现输精管切除术的效果,无需使用手术,同时消除输精管切除术的潜在副作用。
本发明化合物还可用于控制驯养、野生、凶猛或动物园动物的繁殖。例如,所述化合物可以用于控制动物园动物的繁殖。通过选择性喂饵,无需使用致命手段,例如射杀或投毒,可以控制靠近人类居所的野生与凶猛动物群,例如鹿,或者强烈影响自然生态的动物群,例如野马和野猪。在这种过程中,动物行为不受影响,只有生育力。
当R4是放射性标记时,本发明化合物可用于研究睾丸功能和诊断睾丸机能障碍。以上述剂量给药的化合物与睾丸组织结合。
在它们的抗精子发生性质中,化合物的高度化学-、立体-与对映选择性以及它们缺乏全身作用、例如对性欲的作用,表明它们相互作用于睾丸中的特定大分子。用所述化合物的放射性衍生物处置睾丸或睾丸部分,继之以借助放射化学领域熟知的技术检测放射性,能够定位和鉴别睾丸的区域和参与抗精子发生作用的大分子。这可以用于检测和鉴别睾丸的重要成分,它的破坏能够引起抗生育作用。比较其他化合物(例如当前化合物类似物或者来自组合型文库的那些)抑制放射性标记化合物结合的能力,可以进而得到更多选择性和更有力的抗精子发生性化合物。此外,对动物或人类受治疗者给以小剂量(太小以致不能对生育力产生临床作用)的放射性标记化合物,然后测量睾丸中或睾丸特定区域中的放射量,能够显示现有不育问题是否与这种大分子的缺乏相关。可以借助诸如PET和SPECT等技术在活动物或人体中测量放射性,这些都是生物组织造影领域所熟知的。
所述化合物还可用作分析用内标。因而例如,可以向来自服用化合物17的动物或人体的血液、血浆或组织样品加入已知量的化合物诸如20。然后可以将血液、血浆或组织样品用有机溶剂萃取,对萃取液进行分析型高效液相色谱或气相色谱处理,这需要或者无需转化为衍生物,例如甲基酯。测量与17和20有关的色谱峰面积,与受相同条件处理的已知量17和20的面积比相比,能够测定血液、血浆或组织样品中17的浓度。由于17与20之间的密切结构相似性,这两种化合物的物理化学性质就萃取而言将是相似的,从而一种几乎是另一种的理想标准。
就用作杀精子剂而言,本发明化合物可以被制成多种给药剂型。常规杀精子组合物剂型是容易利用已知方法制备的。这类杀精子组合物可以采取凝胶、泡沫、胶冻、霜剂、软膏、油膏等剂型。使用常规载体制备组合物。本发明杀精子组合物可以单独给药,或者与一种或多种避孕屏障方法联合给药,例如隔膜、海绵或安全套。组合物可以在使用前不久直接涂在隔膜、海绵或安全套上,或者可以与海绵或安全套预包装在一起(甚至隔膜,尽管大多数隔膜是多次使用的,在两次使用之间进行清洁)。
就用作杀真菌剂而言,本发明组合物可以被制成任意适合于对需要的区域给药的剂型。给药剂型包括但不限于上面关于杀精子组合物所列举的那些,以及液体混合物。杀真菌与杀精子性质也有可能联合用于一种普通的给药剂型。
在下列示范性实施方式的说明过程中,本发明的其他特征将变得显而易见,它们仅供阐述发明,不打算限制发明。
实施例
实施例1
(4aRS,5SR,9bRS)2-乙基-7-甲基-2,3,4,4a,5,9b-六氢-5-(对-羧基苯基-1H-茚并[1,2-c]吡啶盐酸盐的合成
将碘乙烷(540g,3.41mol)的甲醇(500ml)溶液加入到异烟酸乙酯(500g,3.31mol)中。将混合物轻微回流过夜。在冷却(冰浴)下向上述溶液分批加入硼氢化钠(140g)。NaBH4的加入完成后,将混合物在室温下搅拌过夜。蒸发大多数甲醇,向溶液加入水和乙醚,分离醚层。将醚层干燥(Na2SO4),蒸发,得到油。蒸馏这种红色的油,得到微黄色油(470g,78%):bp 160℃,0.5mm。
将上述化合物(146g,0.8mol)的无水乙醚(200ml)溶液滴加到1M对-甲苯基溴化镁的乙醚溶液(600ml,1.6mol,-10℃)中。搅拌3小时后,将反应混合物倒入10%NH4Cl水溶液(200ml)中。水层用乙醚萃取。将醚层干燥(Na2SO4),蒸发,得到微黄褐色油。将该油溶于18%HCl水溶液(500ml),用乙醚萃取。将HCl水溶液回流2小时。蒸发溶剂,得到对应的氨基酸(181g,收率80%),将一部分(32g)与多磷酸(500g)混合,在140℃下剧烈搅拌3小时。将反应混合物冷却,小心地加入50%KOH水溶液。经过碱化的溶液用乙醚萃取。将醚层干燥(Na2SO4),蒸发,得到2-乙基-7-甲基-2,3,4,4aα,5,9bα-六氢-1H-茚并[1,2-c]吡啶-5-酮,为油(22.6g,87%)。通过小柱SiO2,用含MeOH的CHCl3梯度(0-5%)洗脱,得到分析样品:
1H NMR(90MHZ,CDCl3)δ7.5(1H,s,H-6),7.3(2H,m,H-8,H-9),3.5(1H,m),3.0(1H,m),2.6(2H,m),2.3(3H,s,7-Me),2.2(3H,m),1.9-1.7(3H,m),1.1(3H,t,Me);HRMS(M+):Calcd.for C15H19NO:m/z 229.1467.Found:m/z229.1466.
在-78℃下,向机械搅拌着的对-溴苯甲酸(1.6g,8.0mmol)的四氢呋喃(THF)(15ml)溶液历经45分钟滴加正丁基锂(16.2mmol.6ml2.5M己烷溶液)。将混合物搅拌另外1.5小时后,历经30分钟滴加三环酮(1.1g,5.1mmol)的THF(5ml)溶液,继续在-78℃下搅拌2.5小时。将混合物倒入冰冷的1M HCl(75ml)中,用乙醚萃取(2×30ml)。将酸性水层在室温下搅拌15小时,在减压下浓缩,得到固体。该固体经由二氧化硅快速柱色谱纯化,用含10-20%MeOH的CHCl3梯度洗脱,得到2-乙基-7-甲基-2,3,4,9b-四氢-5-(对-羧基苯基)-1H-茚并[1,2-c]吡啶盐酸盐,为黄色固体(1.1g,58%):
1H NMR(250MHZ,CDCl3),δ1.54(3H,t,J=7.2Hz),2.35(3H,bs),2.25-2.42(1H,m),2.50-2.72(1H,m),2.94-3.0(1H,m),3.15-3.30(2H,m),3.50-3.80(2H,m),4.17-4.30(1H,m),4.40-4.52(1H,m),7.0-7.12(2H,m),7.32(1H,d,J=7.5Hz),7.45(2H,d,J=8.4Hz),8.20(2H,d,J=8.4Hz).HRMS(M+)Calcd.MW for C22H23NO2:m/z 333.1729.Found:m/z 333.1725.
向上述化合物(379mg,1.03mmol)的乙醇/水(40ml 1∶1混合物)溶液加入NaCl(81mg)、PdCl2(98mg)、NaBH4(100mg)和浓HCl(10滴)。将混合物在50℃氢气氛(45psi)下、在Parr装置上摇动15小时后,通过C盐过滤,在减压下浓缩。将所得固体悬浮在绝对乙醇中,通过C盐过滤,在减压下浓缩滤液,得到(4aRS,5RS,9bRS)2-乙基-7-甲基-2,3,4,4a,5,9b-六氢-5-(对-羧基苯基)-1H-茚并[1,2-c]吡啶盐酸盐:
1H NMR(250MHZ,CDCl3):δl.4(3H,t,7.2Hz),1.50-1.60(1H,m),1.85-2.00(1H,m),2.20(3H,s),2.20-2.40(1H,m),2.70-2.90(3H,m)2.90-3.15(2H,m),3.50-3.65(1H,m),3.90-4.10(1H,m),4.50(1H,d,J=7.3Hz),6.95,(1H,bs),7.10(1H,d,J=7.5Hz),7.20(1H,d,J=7.5Hz),7.30,(2H,d,J=8.0Hz),8.00(2H,d,J=8.0Hz).HRMS(M+)Calcd.MW for C22H25NO2:m/z 335.18853.Found:m/z 335.1887.
向氢氧化钾(15g)的正丁醇(60ml)溶液一次性加入上述化合物(2.99g,8.0mmol)。回流20小时后,将暗褐色混合物冷却至0℃,用18%HCl酸化至pH=1。在真空中除去溶剂,得到黄色固体。将该固体溶于CHCl3,通过C盐过滤,在真空中浓缩滤液,得到粗的(4aRS,5SR,9bRS)2-乙基-7-甲基-2,3,4,4a,5,9b-六氢-5-(对-羧基苯基)-1H-茚并[2,2-c]吡啶盐酸盐,为灰白色固体。该固体经由快速柱色谱纯化,用含10%MeOH的CHCl3洗脱,得到1.23g(41%)标题化合物,为白色固体。m.p.=280℃(dec.)
1H NMR(250MHZ,CDCl3-CD3OD).δ1.45(3H,t,J=7.3Hz),1.8(1H,bd,J=14.7Hz),2.2(3H,s),2.4-2.7(2H,m),3.0-3.4(4H,m),3.4-3.7(2H,m),3.7-4.0(1H,m),4.2(1H,d,11Hz),6.6(1H,bs),7.0-7.2(4H,m),8.0(1H,d,J=7.7Hz).HRMS(M+)Calcd.MW for C22H25NO2:m/z 335.18853.Found:m/z 335.18830.
C22H26ClNO2H2O的分析计算值:C,69.37;H,7.14;N,7.14;N,3.68。实测值:C,69.72;H,7.15;n,3.55。
实施例2
(4aRS,5SR,9bRS)2-乙基-7-甲基-2,3,4,4a,5,9b-六氢-5-(对-甲酯基苯基)-1H-茚并[1,2-c]吡啶盐酸盐
在-10℃下,向实施例1羧酸(3.6g,9.69mmol)的甲醇(50ml)溶液历经10分钟加入亚硫酰氨(1.1ml,14.5mmol)。将所得溶液在5℃冰箱中放置68小时,在此期间产物开始结晶出微细白色针晶。得到三批,合并,得到2.65g标题化合物:mp=204℃(升华)。
1H NMR(250MHz,CDCl3):δ1.1(3H,t,J=7.2Hz),1.6(1H,bd,J=14.2Hz),1.80-2.00(2H,m),2.1-2.2(1H,m),2.2(3H,s),2.4(2H,q,J=7.2Hz),2.5-2.6(1H,m),2.7-2.8(1H,m),2.9(1H,dd,J=5.94,11.64Hz),3.3-3.4(1H,m),3.9(3H,s),4.2(1H,d,J=10.0Hz),6.7(1H,bs),7.0(1H,d,J=7.5Hz),7.2(1H,d,J=7.5Hz),7.3(2H,d,J=8.0Hz),8.0(2H,d,8.0Hz).
C23H28ClNO2H2O的分析计算值:C,70.75;H,7.36;N,3.59;实侧值:C,70.67;H,7.36;N,3.59。
实施例3
(4aRS,5SR,9bRS)-2-乙基-2,3,4,4a,5,9b-六氢-8-碘-7-甲基-5-(4-甲酯基苯基)-1H-茚并[1,2-c]吡啶盐酸盐(18)及其(1)-对映体((1)-18)的合成
向搅拌着的(4aRS,5SR,9bRS)-2-乙基-2,3,4,4a,5,9b-六氢-7-甲基-5-(4-甲酯基苯基)-1H-茚并[1,2-c]吡啶(341mg,0.88mmol)的冰乙酸(2ml)溶液加入62%HClO4(1ml),继之以HgO(205mg,0.95mmol)。将混合物略加声波处理,目的是得到均匀的溶液。历经15分钟滴加碘(235mg,0.925mmol)的冰乙酸(17ml)溶液,将所得混合物在室温下搅拌过夜。将橙-红色混合物倒入水(100ml)中,冷却至5℃,用30%NaOH碱化至pH 12,用乙醚萃取(3×75ml)。合并澄清的无色乙醚萃取液,连续用水(20ml)和盐水(30ml)洗涤,干燥(MgSO4),过滤,在真空中浓缩,得到18的粗游离碱(448mg)。将该产物用3%氯化氢的甲醇溶液转化为HCl盐,从EtOAc/MeOH中重结晶。收率=400mg(89%)。
m.p.=>190℃(dec.)
1H NMR(250MHz,CDCl3,asfree base);δ1.15(3H,t,J=7.2Hz),1.65(1H,bd),1.8-2.1(3H,m),2.32(3H,s),2.48(3H,q,J=7.2Hz,+m),2.80(1H,bd),2.97(1H,dd,J=11.8,5.8Hz),3.41(1H,m),3.91(3H,s),4.19(1H,d,J=9.8Hz),6.78(1H,s),7.22(2H,d,J=8.3Hz),7.73(1H,s),8.00(2H,d,J=8.3Hz).HRMS:Calcd.for C23H26NO2I,(corresponding to the free base):m/z 475.1008.Found:m/z475.1004.Anal.Calcd.for C23H27ClINO2H2O:C,53.04;H,5.42;N,2.69.Found:C,52.70;H,5.60;N,2.57.
从(1)-3开始按照相似方式合成活性对映体(1)-18。[α]D=-5.6(c=1.18,CHCl3).
实施例4
(4aRS,5SR,9bRS)-2-乙基-2,3,4,4a,5,9b-六氢-8-碘-7-甲基-5-(4-羧基苯基)-1H-茚并[1,2-c]吡啶盐酸盐(17)的合成
向(4aRS,5SR,9bRS)-2-乙基-2,3,4,4a,5,9b-六氢-7-甲基-5-(4-羧基苯基)-1H-茚并[1,2-c]吡啶盐酸盐(250mg,0.673mmol)的2ml乙酸溶液加入6ml乙酸与高氯酸的1∶1混合物。加入HgO(1.35mmol),将反应混合物在室温下搅拌,直至HgO溶解。利用加液漏斗向反应混合物滴加I2(427mg,1.68mmol)在4ml乙酸与6ml CH2Cl2中的溶液。将反应混合物在室温下搅拌过夜,然后通过C盐过滤。将红色固体用水和CH2Cl2洗涤。合并两相滤液,用分液漏斗分离。将有机相用饱和亚硫酸氢钠溶液洗涤,经硫酸钠(无水)干燥,过滤,浓缩,得到234mg黄褐色固体,按照通常的方式转化为盐酸盐。
1H NMR(250MHz,CDCl3/CD3OH)δ1.28(3H,t,J=7.2Hz),2.0-2.1(1H,m),2.3(3H,s),2.56(2H,m),3.04(3H,m),3.24(1H,m),3.46(2H,m),4.18(1H,d,J=11Hz),6.73(1H,s),7.13(2H,d,J=8.2Hz),7.71(1H,s),7.89(2H,d,J=8.2Hz).HRMS Calcd for C22H24NO2I(corresponding to the free base):m/z 461.0852.Found:m/z 461.0857.
实施例5
(4aRS,5SR,9bRS)-2-乙基-8-溴-7-甲基-2,3,4,4a,5,9b-六氢-5-(4-羧基苯基)-1H-茚并[1,2-c]吡啶盐酸盐(19)的合成
将(4aRS,5SR,9bRS)-2-乙基-8-碘-7-甲基-2,3,4,4a,5,9b-六氢-5-(4-羧基苯基)-1H-茚并[1,2-c]吡啶盐酸盐(200mg,0.402mmol)溶于20ml THF和0.4ml六甲基磷酰胺。向该溶液加入50mg氢化钠(含60%的矿物油)。将混合物回流1小时,然后冷却至-78℃。缓慢加入叔丁基锂溶液(0.73ml,1.1M戊烷溶液,0.804mmol)。加入后,将混合物在-78℃下搅拌20分钟。加入1,2-二溴乙烯(1ml)。将混合物在-78℃下搅拌另外30分钟,然后温热至室温。向溶液加入5%盐酸,直至溶液变为酸性。混合物用二氯甲烷萃取。将二氯甲烷溶液用盐水洗涤,经MgSO4干燥。粗产物用快速柱色谱纯化(二氧化硅;二氯甲烷和甲醇10∶1),得到标题化合物:30mg,17%收率。
m.p.>169.6-170.3℃
1H NMR(250MHZ,D2O--CDCl3),δ1.25(3H,t,J=7.0Hz),1.72(1H,d,J=15Hz),1.90-2.15(1H,m),2.19(3H,s),2.36(1H,t,J=12.5Hz),2.5-2.65(1H,m),2.7-3.0(3H,m),3.2-3.4(4H,m),3.4-3.6(1H,m),4.13(1H,d,J=10.5Hz),6.71(1H,s),7.11(2H,d,J=8.0Hz),7.43(1H,s),7.89(2H,d,J=8.0Hz).MS:413(M).Anal.(C22H25O2BrClN·1.8H2O):Calculated C 54.68,H 5.22,N 2.90;Found C 54.77,H 5.52,N2.57.HRMS Calcd for C22H24NO2Br(corresponding to the free base):m/z 413.0990.Found:m/z 413.0994.
实施例6
(4aRS,5SR,9bRS)-2-乙基-8-氯-7-甲基-2,3,4,4a,5,9b-六氢-5-(4-羧基苯基)-1H-茚并[1,2-c]吡啶盐酸盐(20)的合成
将(4aRS,5SR,9bRS)-2-乙基-8-碘-7-甲基-2,3,4,4a,5,9b-六氢-5-(4-羧基苯基)-1H-茚并[1,2-c]吡啶盐酸盐(250mg,0.5mmol)溶于25ml THF和0.5ml HMPA。向该溶液加入60mg氢化钠(含60%的矿物油)。将混合物回流1小时,然后冷却至-78℃。缓慢加入叔丁基锂溶液(0.91ml,1.1M戊烷溶液,1.04mmol)。加入后,将混合物在-78℃下搅拌20分钟。加入六氯乙烷(2.46g,10.4mmol)的2ml THF溶液。将混合物在-78℃下搅拌另外30分钟,然后温热至室温。向溶液加入5%盐酸,直至溶液变为酸性。混合物用二氯甲烷萃取。将二氯甲烷溶液用盐水洗涤,经MgSO4干燥。粗产物用快速柱色谱纯化(二氯甲烷和甲醇10∶1),得到标题化合物:60mg,30%收率。
1HNMR(250MHZ,D2O-CDCl3)δ1.35(3H,t,J=7.25Hz),1.75-1.95(1H,m),2.30(3H,s),2.45-2.75(2H,m),2.80-3.15(2H,m),3.20-3.50(4H,m),3.50-3.70(1H,m),4.25(1H,d,J=10Hz),6.80(1H,s),7.25(2H,d,J=7.5Hz),7.32(1H,s),8.0(2H,d,J=7.5Hz).MS:370(M).Anal.(C22H25O2Cl2N):Calculated C65.50,H 6.20,N 3.45;Found C 65.65,H 6.73,N 3.59.HRMS Calcd for C22H24NO2Cl(corresponding to the free base):m/z 369.1495.Found:m/z 369.1494.
实施例7
(4aRS,9bRS)-2-乙基-1,2,3,4,4a,9b-六氢-1H-茚并[1,2-c]吡啶-5-酮(7)的合成
将从165g 1-乙基-1,2,5,6-四氢吡啶羧酸甲酯制备的粗1-乙基-3-(4-甲基苯基)-4-吡啶羧酸甲酯(如美国专利No.5,319,084关于类似的乙基酯所述制备)溶于1L 18%HCl水溶液,用乙醚(300ml)萃取除去作为合成副产物的双甲苯基化合物。然后将水溶液回流48小时,然后在减压下浓缩,加入乙腈(共沸),得到粗的1-乙基-1,2,5,6-四氢吡啶羧酸盐酸盐(283g),在100℃高真空下充分干燥。由于该产物是非常吸湿的,贮存在氮下。在5℃下,向净7(45g,159mmol)小心地加入亚硫酰氯(150ml)。加入后,除去冰浴,将所得均匀溶液在室温下搅拌4小时。在真空中除去过量SOCl2,得到暗色浓稠的糊状物。向该产物加入1,2-二氯乙烷(250ml),在真空中除去30ml溶剂,目的是除去任何残留的SOCl2。向浑浊的混合物历经45分钟分批加入AlCl3(53g,397mmol)。借助水浴控制温度在大约25℃。加入后,将暗红-褐色溶液在35-40℃下搅拌1小时,然后倒入含有大约400g碎冰和50ml浓HCl的烧杯中。将水层用30%NaOH(约350ml)碱化至pH约12,同时在冰水浴中冷却。在冷却于冰水浴的情况下萃取所得混合物。将所得混合物用乙醚萃取(3×400ml),合并醚层,连续用水和盐水洗涤,干燥(MgSO4),过滤,在减压下浓缩,得到橙-红色油。将该油用Kugelrohr仪器蒸馏(125-135℃,0.5mmHg),得到21.6g(59%)酮7,为浅黄色固体,NMR性质等同于公认的产物。
实施例8
(4aRS,5SR,9bRS)-2-乙基-7-甲基-5-(4-甲酯基苯基)-2,3,4,4a,5,9b-六氢茚并[1,2-c]吡啶和(4aRS,5SR,9bRS)-2-乙基-7-甲基-5-(4-羧基苯基)-2,3,4,4a,5,9b-六氢茚并[1,2-c]吡啶的对映体的合成
对映体被描述为(d)或(1),基于钠D线在给定溶剂中的旋光度而言。具有相同旋光符号的化合物不一定具有相同的绝对构型。
1-乙基-4-羧基-1,2,5,6-四氢吡啶盐酸盐
将1-乙基-1,2,5,6-四氢吡啶羧酸甲酯(11)在250ml 1.5M HCl中回流4小时。利用外加加热和氮气流将混合物浓缩至干,得到高度结晶性固体。使固体从MeOH中重结晶,得到19.6g 12的HCl盐:m.p.=265(dec.),C8H14ClNO2的分析计算值:50.14;H,7.36;N,7.31.Found:C,50.23;H,7.36;N,7.28.
(1)-烯酰基磺内酰胺(从1S(-)-(2,10)樟脑磺内酰胺衍生的(1)-13)
向12的盐酸盐(1.3g,6.79mmol)加入亚硫酰氯(15ml),将所得混合物加热至回流达2小时。在真空中除去过量SOCl2,将残余物用10ml无水甲苯研制,在真空中浓缩。研制过程重复两次,得到黄色粉状固体。在单独的容器中,在5℃下向1S-(-)-2,10-樟脑磺内酰胺(3.16g,14.7mmol)的THF(30ml)溶液滴加正丁基锂(15mmol,6.0ml 2.5M己烷溶液)。加入后,使澄清的无色溶液恢复至室温,搅拌另外45分钟。然后在5℃下将磺内酰胺阴离子溶液借助套管加入到含有氨基酰氯盐酸盐的烧瓶中。加入后,使橙色混合物恢复至室温,搅拌18小时。向反应物加入饱和NH4Cl(约1ml)进行猝灭,在真空中浓缩至褐色焦油状残余物。使残余物在乙醚与水之间分配,醚层再用水洗涤一次。然后将醚层用稀HCl水溶液(约5%)洗涤,分离。从绝对EtOH中重结晶,得到游离磺内酰胺(醚层)(1.2g)。将酸性水层用浓NH4OH碱化至pH 12,用乙醚萃取,蒸发醚层,使残余物从正己烷中重结晶,得到产物[(1)-13]。这得到1.9g(1)-13,为白色粗针晶:m.p.=120℃,[α]D 21=-74.8°(c=1.0,CHCl3),1H NMR等同于它的对映体(见下)。C8H28N2O3S的分析计算值:C,61.33;H,8.01;N,7.95.Found:C,61.35;H,8.06;N,7.89.
从1R(+)-(2,10)-樟脑磺内酰胺衍生的13的(d)-烯酰基磺内酰胺对映体
按照与对映体所述相似的工艺(见上),从氨基酸12的盐酸盐(6.5g,34.1mmol)和1R-(+)-2,10-樟脑磺内酰胺(15.4g,71.4mmol)制备,收率86%。m.p.=118.5℃-119.6℃(从己烷中重结晶,为浓密的稻草色叶状晶体)
[α]D 21=+74.1°(c=1.0,CHCl3);1H NMR(250MHZ,CDCl3):δ1.00(3H,s),1.12(3H,t,J=7.1Hz),1.22(3H,s),1.3-1.5(2H,m),1.8-2.1(5H,m),2.2-2.4(1H,m),2.55(2H,q,J=7.1Hz),2.6-2.7(3H,m),3.1-3.3(2H,m)3.38(1H,d,J=13.6Hz),3.50(1H,d,J=13.6Hz),4.0-4.1(1h,m),6.5-6.6(1H,m);Anal.Calcd.for C18H28N2O3S:C,61.33;H,8.01;N,7.95.Found:C,61.48;H,8.02;N,7.98.
该产物的晶型因从己烷中沉淀出来的速率和纯化步骤期间的浓度而异。
从(1)-13衍生的1,4-加合物(1)-14
在-78℃下,向烯酰基磺内酰胺(1)-13(5.6g,16.0mmol)的甲苯(200ml)溶液历经10分钟加入对-甲苯基溴化镁(33.6mmol,33.6ml1.0M乙醚溶液)。在-78℃下搅拌另外30分钟后,将反应混合物置于冰箱(-10℃)中过夜,然后温热至+5℃达另外2小时。向混合物加入饱和NH4Cl(200ml)进行猝灭。用乙醚(400ml)萃取水层后,醚层用3%HCl萃取(3×200ml)。合并酸性层,用浓NH4OH调至碱性(pH=12),用乙醚萃取(3×200ml),将醚层用盐水洗涤,干燥(MgSO4),过滤,在减压下浓缩,得到橙色固体(7.12g)。使该固体从乙醚-己烷(约40ml的大约1∶2混合物)中重结晶。收率=3.64g。第二批得到另外1.24g。总计=4.68g(66%)。m.p.=150.5-151.7℃(乙醚-己烷;浓密的稻草色棱晶)
[α]D 21=26.2°(c=1.14,CHCl3);1H NMR(500MHZ,CDCl3);δ0.44(3H,s),0.82(3H,s),1.13(3H,t,J=7.16Hz),1.20-1.30(2H,m),1.40-1.55(1H,m),1.62-1.65(1H,m),1.70-1.85(3H,m),1.95-2.05(1H,m),2.05-2.10(1H,m),2.27(3H,s),2.55(2H,q,J=7.16Hz),2.55-2.62(1H,m),2.68-2.72(1H,m),2.82(1H,dd,J=10.64,3.47Hz),3.12(1H,t,J=10.8Hz),3.24-3.28(1H,m),3.30(1H,d,J=14.0Hz),3.32(1H,d,J=14.0Hz),3.55-3.60(1H,m),3.67-3.71(1H,m),7.02(2H,d,J=7.96Hz),7.15(2H,d,J=7.96Hz);Anal.Calcd.forC25H36N2O3S:C,67.53;H,8.16;N,6.30.Found:C,67.58;H,8.15;N,6.30.
从(1)-14衍生的对映体纯的酮(d)-7
向1,4-加合物(1)-14(6.86g,15.45mmol)的THF(40ml)溶液加入新鲜制备的LiOH.H2O(6.43g,153mmol)的水(40ml)溶液。将所得不均匀混合物在轻微回流下剧烈搅拌26小时。将混合物冷却至约+5℃,用浓HCl酸化至pH=0,引导中等强度氮气流经过混合物表面,同时浸在温水浴(50℃)中,以除去大多数挥发性组分。将剩余固体在高真空下充分干燥。使用亚硫酰氯和然后的AlCl3的1,2-二氯乙烷溶液,按照与外消旋产物相似的方式(见上)将所得粗产物环化为酮(d)-7。这得到1.12g游离碱酮(d)-7,为油,放置过夜后固化。从下一步回收后纯化一部分该产物,目的是获得物理数据。[a]D 20=+95.9°(游离碱,c=1.2,CHCl3);[α]D 20=+71.9°(HCl盐,c=1.1,CHCl3).
从酮(d)-7衍生的对映体纯的烯烃(d)-15
按照与外消旋工艺相似的方式(参见美国专利No.5,319,084),从酮(d)-7(1.12g,4.89mmol)得到该产物。收率为850mg(47%)。[α]D 19=+21.2°(c=1.24,CHCl3).
(1)-2-乙基-7-甲基-2,3,4,4a,5,9b-六氢-5-(4-溴苯基)-5-羟基-1H-茚并[1,2-c]吡啶的合成
在-78℃下,向剧烈搅拌着的4-溴碘苯(13.8g,48.9mmol)的160mlTHF溶液非常缓慢地加入正丁基锂溶液(19.6ml,2.5M戊烷溶液,49mmol)。加入后,将溶液在-78℃下搅拌10分钟。溶液变为黄色和浑浊。加入(d)-2-乙基-7-甲基-1,2,3,4,4a,5,9b-六氢茚并[1,2-c]吡啶-5-酮(8g,34.9mM)的40ml THF溶液。然后将混合物在-78℃下搅拌2小时。除去冷却浴,混合物用水猝灭。分离有机相,水相用二氯甲烷萃取。合并有机相,用盐水洗涤,经MgSO4干燥。蒸发溶剂,得到粗产物,从二氯甲烷中重结晶,得到标题化合物(10.8g,80%)。m.p.,169.6-170.3℃.1H NMR(250MHZ,CDCl3),δ1.00(3H,t,J=7.3Hz),1.70-2.00(2H,m),2.15-2.30(1H,m),2.29(3H,s),2.38(2H,q,J=7.3Hz),2.5-2.7(2H,m),2.70-2.85(1H,m),2.85-3.00(1H,m),3.30-3.50(1H,m),6.84(1H,s),7.17(2H,q,J=7.5Hz),7.31(2H,d,J=11Hz),7.43(2H,d,J=11Hz).MS:386(M),230(100%).[α]D=-11.5°(c=1.03,CHCl3).Anal.(C22H24OBrN):Calculated C,65.28,H 6.26,N 3.62;Found C 65.11,H 6.21,N 3.64.
(4aSR,5RS,9bSR)-2-乙基-7-甲基-2,3,4,4a,5,9b-六氢-5-(4-溴苯基)-1H-茚并[1,2-c]吡啶(1)的(1)-对映体的合成
将(1)-10-2-乙基-7-甲基-2,3,4,4a,5,9b-六氢-5-(4-溴苯基)-5-羟基-1H-茚并[1,2-c]吡啶(4,5g,13mmol)与100ml三乙基硅烷的300ml无水二氯甲烷溶液冷却至-78℃。向溶液通入三氟硼烷气体达10分钟。无色溶液变为橙色。使混合物温热至室温,加入10g碳酸钾,继之以水。分离有机相,水相用二氯甲烷萃取。合并有机相,用盐水洗涤,经MgSO4干燥。蒸发溶剂,得到粗产物。
将粗产物溶于40ml正丁醇。加入氢氧化钾(9g)。将混合物加热至回流,同时搅拌。回流20小时后,将混合物冷却至室温,倒入冰中。混合物用二氯甲烷萃取。将二氯甲烷溶液用盐水洗涤,经MgSO4干燥。蒸发溶剂,使粗产物在二乙醚与18%盐酸溶液之间分配。分离各层,水溶液再用二乙醚洗涤一次。将水溶液冷却至0℃,用50%氢氧化钠溶液碱化至pH>14。混合物用二氯甲烷萃取三次。将有机溶液用盐水洗涤,经MgSO4干燥。蒸发溶剂,得到粗产物,用快速柱色谱纯化(硅胶,CH2Cl2和MeOH,100∶3),得到标题化合物(1)-10,3.2g,67%收率(经过两步)。按照通常的方式制备盐酸盐。m.p.240℃(分解)。
1H NMR(250MHZ,CDCl3),δ1.12(3H,t,J=7.25),1.6-1.8(1H,m),1.80-2.05(2H,m),2.15-2.40(2H,m),2.26(3H,s),2.70-2.85(1H,m),2.90-3.10(1H,m),3.30-3.45(1H,m),4.12(1H,d,J=10.25Hz),6.72(1H,s),7.00-7.30(4H,m),7.44(2H,d,J=9.0Hz).MS:370(M).[α]D=-7.8°(c=0.83,MeOH).Anal.(C21H24BrN·HCl):Calculated C 62.00,H 6.19,N 3.44;Found C 61.96,H 6.23,N 3.35.
(4aRS,5SR,9bRS)-2-乙基-2,3,4,4a,5,9b-六氢-7-甲基-5-(4-羧基苯基)-1H-茚并[1,2-c]吡啶盐酸盐的(1)-对映体[(1)-2]的合成
将100mg(0.27mmol)(1)-10化合物的5ml THF溶液冷却至-78℃。向该溶液加入0.54ml正丁基锂溶液(2.5M戊烷溶液,1.35mmol)。将溶液在-78℃下搅拌30分钟。通过针头向溶液通入二氧化碳气体达10分钟。将混合物在-78℃下搅拌另外十多分钟,温热至室温。蒸发THF,残余物用18%盐酸酸化。混合物用二氯甲烷萃取。将二氯甲烷溶液用盐水洗涤,经MgSO4干燥。过滤干燥剂,浓缩溶液,得到粗产物。粗产物经过柱色谱处理(硅胶,CH2Cl2和MeOH 10∶1至1∶1),得到72mg(71%收率)(-)-2。[α]D=-15.5°(c=1.24,MeOH).
(4aRS,5SR,9bRS)-2-乙基-2,3,4,4a,5,9b-六氢-7-甲基-5-(4-甲酯基苯基)-1H-茚并[1,2-c]吡啶盐酸盐的(d)-对映体[(d)-3]的合成
将(1)-2(20mg)的1ml甲醇溶液冷却至-10℃(冰-丙酮)。加入过量亚硫酰氯。加入后,使混合物温热至室温,搅拌过夜。用氮吹去过量亚硫酰氯和溶剂,在真空下干燥残余物。用HPLC分析粗产物(Sumichiral,QA-4900,4mm×25cm;溶剂:53.8%1,2-二氯乙烷,44%己烷,2.2%乙醇和0.1%TFA;流速:0.8mL/min,λ=254nm),显示有>97%ee的(d)-3。
(4aRS,5SR,9bRS)-2-乙基-2,3,4,4a,5,9b-六氢-7-甲基-5-(4-羧基苯基)-1H-茚并[1,2-c]吡啶盐酸盐的(d)-对映体[(d)-2]和(4aRS,5SR,9bRS)-2-乙基-2,3,4,4a,5,9b-六氢-7-甲基-5-(4-甲酯基苯基)-1H-茚并[1,2-c]吡啶盐酸盐的(1)-对映体[(1)-3]的合成
从上述烯酰基磺内酰胺(d)-13开始,随后进行上面关于它们对映体的合成所用步骤,可以合成这两种化合物。它们的性质以前已有描述。参见Cook et al.,J.Med.Chem.,38:753-763(1995)。显然,鉴于上述教导,本发明的大量修改和变化都是可能的。因此不言而喻的是,在所附权利要求的范围内,本发明在实施时可以不同于本文的具体描述。
Claims (44)
1、杀死能动的精子的方法,包含:
使含有精子的组合物与包含式I(a)化合物的杀精子组合物接触:

其中R1是氢或者直链或支链C1-6烷基或者C3-8环烷基;
R2是氢、直链或支链C1-6烷基;
R3和R5各自独立地是氢、SO3H、直链或支链C1-6烷基、CH2OH、CH2OMe、直链或支链C1-6烷氧基、羧基(COOH)、可以在哺乳动物生理条件下转化为羧基的非酯基团、羧酸酯(COOR,其中R是C1-10烷基、C6-10芳基、C7-10芳烷基)、羟甲基酯(CH2OC(O)--R,其中R是如上所定义的)、CONH2、CONHR、CONR2、CH2OCONHR、CN、CH=NHNHCONH2和卤素;
R4是氢、卤素、R3Si或COR。
2、权利要求1的方法,其中该化合物是式I(b)化合物:
3、权利要求1的方法,其中R1选自氢、甲基、乙基、正丙基、异丙基、正丁基、异丁基、正戊基、异戊基、正己基、异己基、环丙基、环戊基和环己基;
R2选自氢、甲基、乙基、正丙基、异丙基、正丁基、异丁基、正戊基、异戊基、正己基和异己基;
R3位于4位,选自氢、SO3H、直链或支链C1-6烷基、CH2OH、CH2OMe、直链或支链C1-6烷氧基、羧基(COOH)、可以在哺乳动物生理条件下转化为羧基的非酯基团、羟甲基(CH2OH)、甲酰基(CHO)、羧基(COOH)、羧酸酯(COOR,其中R是C1-10烷基、C6-10芳基、或C7-10芳烷基)、羟甲基酯(CH2OC(O)--R,其中R是如上所定义的)、CONH2、CONHR、CONR2、CH2OCONHR、CN、CH=NHNHCONH2和卤素;
R5是氢;
R4选自氢和卤素。
4、权利要求3的方法,其中R1是甲基、乙基、正丙基或异丙基;
R2是甲基、乙基、正丙基或异丙基;
R3是4-COOH或4-COOR,其中R是如上所定义的;
R5是氢;
R4是卤素。
5、杀精子组合物,包含有效杀精子剂量的式I(a)化合物:
其中R1是氢或者直链或支链C1-6烷基或者C3-8环烷基;
R2是氢、直链或支链C1-6烷基;
R3和R5各自独立地是氢、SO3H、直链或支链C1-6烷基、CH2OH、CH2OMe、直链或支链C1-6烷氧基、羧基(COOH)、可以在哺乳动物生理条件下转化为羧基的非酯基团、羧酸酯(COOR,其中R是C1-10烷基、C6-10芳基、C7-10芳烷基)、羟甲基酯(CH2OC(O)--R,其中R是如上所定义的)、CONH2、CONHR、CONR2、CH2OCONHR、CN、CH=NHNHCONH2和卤素;
R4是氢、卤素、R3Si或COR,
和适合的载体。
6、权利要求5的杀精子组合物,其中该组合物是选自凝胶、胶冻、霜剂、泡沫、软膏和油膏的形式。
7、权利要求5的杀精子组合物,其中该化合物是式I(b)化合物:

8、权利要求5的组合物,其中R1选自氢、甲基、乙基、正丙基、异丙基、正丁基、异丁基、正戊基、异戊基、正己基、异己基、环丙基、环戊基和环己基;
R2选自氢、甲基、乙基、正丙基、异丙基、正丁基、异丁基、正戊基、异戊基、正己基和异己基;
R3位于4位,选自氢、SO3H、直链或支链C1-6烷基、CH2OH、CH2OMe、直链或支链C1-6烷氧基、羧基(COOH)、可以在哺乳动物生理条件下转化为羧基的非酯基团、羟甲基(CH2OH)、甲酰基(CHO)、羧基(COOH)、羧酸酯(COOR,其中R是C1-10烷基、C6-10芳基、或C7-10芳烷基)、羟甲基酯(CH2OC(O)--R,其中R是如上所定义的)、CONH2、CONHR、CONR2、CH2OCONHR、CN、CH=NHNHCONH2和卤素;
R5是氢;
R4选自氢和卤素。
9、权利要求8的组合物,其中R1是甲基、乙基、正丙基或异丙基;
R2是甲基、乙基、正丙基或异丙基;
R3是4-COOH或4-COOR,其中R是如上所定义的;
R5是氢;
R4是卤素。
10、杀精子的避孕装置,包含:
有效杀精子量的式I(a)化合物:

其中R1是氢或者直链或支链C1-6烷基或者C3-8环烷基;
R2是氢、直链或支链C1-6烷基;
R3和R5各自独立地是氢、SO3H、直链或支链C1-6烷基、CH2OH、CH2OMe、直链或支链C1-6烷氧基、羧基(COOH)、可以在哺乳动物生理条件下转化为羧基的非酯基团、羧酸酯(COOR,其中R是C1-10烷基、C6-10芳基、C7-10芳烷基)、羟甲基酯(CH2OC(O)--R,其中R是如上所定义的)、CONH2、CONHR、CONR2、CH2OCONHR、CN、CH=NHNHCONH2和卤素;
R4是氢、卤素、R3Si或COR,
载体;和
避孕屏障装置。
11、权利要求10的杀精子避孕装置,其中该化合物是式I(b)化合物:
12、权利要求10的杀精子避孕装置,其中R1选自氢、甲基、乙基、正丙基、异丙基、正丁基、异丁基、正戊基、异戊基、正己基、异己基、环丙基、环戊基和环己基;
R2选自氢、甲基、乙基、正丙基、异丙基、正丁基、异丁基、正戊基、异戊基、正己基和异己基;
R3位于4位,选自氢、羟甲基(CH2OH)、甲酰基(CHO)、羧基(COOH)、羧酸酯(COOR,其中R是C1-10烷基、C6-10芳基、或C7-10芳烷基)、羟甲基酯(CH2OC(O)--R,其中R是如上所定义的)、CONH2、CONHR、CONR2、CH2OCONHR、CN、CH=NHNHCONH2和卤素;
R5是氢;
R4选自氢和卤素。
13、权利要求12的杀精子避孕装置,其中R1是甲基、乙基、正丙基或异丙基;
R2是甲基、乙基、正丙基或异丙基;
R3是4-COOH或4-COOR,其中R是如上所定义的;
R5是氢;
R4是卤素。
14、权利要求10的杀精子避孕装置,其中所述避孕屏障装置选自隔膜、避孕海绵和安全套。
15、杀死真菌的方法,包含:
对需要抗真菌治疗的受治疗者给以组合物,所述组合物包含抗真菌有效量的式I(a)化合物:

其中R1是氢或者直链或支链C1-6烷基或者C3-8环烷基;
R2是氢、直链或支链C1-6烷基;
R3和R5各自独立地是氢、SO3H、直链或支链C1-6烷基、CH2OH、CH2OMe、直链或支链C1-6烷氧基、羧基(COOH)、可以在哺乳动物生理条件下转化为羧基的非酯基团、羧酸酯(COOR,其中R是C1-10烷基、C6-10芳基、C7-10芳烷基)、羟甲基酯(CH2OC(O)--R,其中R是如上所定义的)、CONH2、CONHR、CONR2、CH2OCONHR、CN、CH=NHNHCONH2和卤素;
R4是氢、卤素、R3Si或COR,
和可接受的载体。
16、权利要求15的方法,其中该化合物是式I(b)化合物。

17、权利要求15的方法,其中R1选自氢、甲基、乙基、正丙基、异丙基、正丁基、异丁基、正戊基、异戊基、正己基、异己基、环丙基、环戊基和环己基;
R2选自氢、甲基、乙基、正丙基、异丙基、正丁基、异丁基、正戊基、异戊基、正己基和异己基;
R3位于4位,选自氢、羟甲基(CH2OH)、甲酰基(CHO)、羧基(COOH)、羧酸酯(COOR,其中R是C1-10烷基、C6-10芳基、或C7-10芳烷基)、羟甲基酯(CH2OC(O)--R,其中R是如上所定义的)、CONH2、CONHR、CONR2、CH2OCONHR、CN、CH=NHNHCONH2和卤素;
R5是氢;
R4选自氢和卤素。
18、权利要求17的方法,其中R1是甲基、乙基、正丙基或异丙基;
R2是甲基、乙基、正丙基或异丙基;
R3是4-COOH或4-COOR,其中R是如上所定义的;
R5是氢;
R4是卤素。
19、权利要求15的方法,其中所述给药是局部给药。
20、权利要求15的方法,其中所述给药步骤是对受治疗者内部给药。
21、避孕方法,包含:
对受治疗者口服给以组合物,所述组合物在药理学上可接受的载体中包含有效杀精子量的第一个式I(a)化合物:

其中R1是氢或者直链或支链C1-6烷基或者C3-8环烷基;
R2是氢、直链或支链C1-6烷基;
R3和R5各自独立地是氢、SO3H、直链或支链C1-6烷基、CH2OH、CH2OMe、直链或支链C1-6烷氧基、羧基(COOH)、可以在哺乳动物生理条件下转化为羧基的非酯基团、羧酸酯(COOR,其中R是C1-10烷基、C6-10芳基、C7-10芳烷基)、羟甲基酯(CH2OC(O)--R,其中R是如上所定义的)、CONH2、CONHR、CONR2、CH2OCONHR、CN、CH=NHNHCONH2和卤素;
R4是氢、卤素、R3Si或COR,
所述受治疗者同时使用杀精子的避孕装置,所述装置包含:
有效杀精子量的第二个式I(a)化合物:

其中R1是氢或者直链或支链C1-6烷基或者C3-8环烷基;
R2是氢、直链或支链C1-6烷基;
R3和R5各自独立地是氢、SO3H、直链或支链C1-6烷基、CH2OH、CH2OMe、直链或支链C1-6烷氧基、羧基(COOH)、可以在哺乳动物生理条件下转化为羧基的非酯基团、羧酸酯(COOR,其中R是C1-10烷基、C6-10芳基、C7-10芳烷基)、羟甲基酯(CH2OC(O)--R,其中R是如上所定义的)、CONH2、CONHR、CONR2、CH2OCONHR、CN、CH=NHNHCONH2和卤素;
R4是氢、卤素、R3Si或COR,
载体;和
避孕屏障装置;
其中所述第一个式I(a)化合物与所述第二个式I(a)化合物可以是相同或不同的。
22、权利要求21的方法,其中所述第一个化合物是第一个式I(b)化合物:

23、权利要求21的方法,其中在所述第一个式I(a)化合物中,R1选自氢、甲基、乙基、正丙基、异丙基、正丁基、异丁基、正戊基、异戊基、正己基、异己基、环丙基、环戊基和环己基;
R2选自氢、甲基、乙基、正丙基、异丙基、正丁基、异丁基、正戊基、异戊基、正己基和异己基;
R3位于4位,选自氢、羟甲基(CH2OH)、甲酰基(CHO)、羧基(COOH)、羧酸酯(COOR,其中R是C1-10烷基、C6-10芳基、或C7-10芳烷基)、羟甲基酯(CH2OC(O)--R,其中R是如上所定义的)、CONH2、CONHR、CONR2、CH2OCONHR、CN、CH=NHNHCONH2和卤素;
R5是氢;
R4选自氢和卤素。
24、权利要求23的方法,其中在所述第一个式I(a)化合物中,R1是甲基、乙基、正丙基或异丙基;
R2是甲基、乙基、正丙基或异丙基;
R3是4-COOH或4-COOR,其中R是如上所定义的;
R5是氢;
R4是卤素。
25、权利要求21的方法,其中所述避孕屏障装置选自隔膜、避孕海绵和安全套。
26、权利要求21的方法,其中所述第二个化合物是第二个式I(b)化合物:
27、权利要求21的方法,其中在所述第二个式I(a)化合物中,R1选自氢、甲基、乙基、正丙基、异丙基、正丁基、异丁基、正戊基、异戊基、正己基、异己基、环丙基、环戊基和环己基;
R2选自氢、甲基、乙基、正丙基、异丙基、正丁基、异丁基、正戊基、异戊基、正己基和异己基;
R3位于4位,选自氢、羟甲基(CH2OH)、甲酰基(CHO)、羧基(COOH)、羧酸酯(COOR,其中R是C1-10烷基、C6-10芳基、或C7-10芳烷基)、羟甲基酯(CH2OC(O)--R,其中R是如上所定义的)、CONH2、CONHR、CONR2、CH2OCONHR、CN、CH=NHNHCONH2和卤素;
R5是氢;
R4选自氢和卤素。
28、权利要求27的方法,其中在所述第二个式I(a)化合物中,R1是甲基、乙基、正丙基或异丙基;
R2是甲基、乙基、正丙基或异丙基;
R3是4-COOH或4-COOR,其中R是如上所定义的;
R5是氢;
R4是卤素。
29、权利要求21的方法,其中所述第一个式I(a)化合物与所述第二个式I(a)化合物是相同的。
30、权利要求21的方法,其中所述第一个式I(a)化合物与所述第二个式I(a)化合物是不同的。
31、权利要求22的方法,其中所述第一个式I(a)化合物与所述第二个式I(a)化合物是相同的。
32、权利要求22的方法,其中所述第一个式I(a)化合物与所述第二个式I(a)化合物是不同的。
33、权利要求23的方法,其中所述第一个式I(a)化合物与所述第二个式I(a)化合物是相同的。
34、权利要求23的方法,其中所述第一个式I(a)化合物与所述第二个式I(a)化合物是不同的。
35、权利要求24的方法,其中所述第一个式I(a)化合物与所述第二个式I(a)化合物是相同的。
36、权利要求24的方法,其中所述第一个式I(a)化合物与所述第二个式I(a)化合物是不同的。
37、权利要求25的方法,其中所述第一个式I(a)化合物与所述第二个式I(a)化合物是相同的。
38、权利要求25的方法,其中所述第一个式I(a)化合物与所述第二个式I(a)化合物是不同的。
39、权利要求26的方法,其中所述第一个式I(a)化合物与所述第二个式I(a)化合物是相同的。
40、权利要求26的方法,其中所述第一个式I(a)化合物与所述第二个式I(a)化合物是不同的。
41、权利要求27的方法,其中所述第一个式I(a)化合物与所述第二个式I(a)化合物是相同的。
42、权利要求27的方法,其中所述第一个式I(a)化合物与所述第二个式I(a)化合物是不同的。
43、权利要求28的方法,其中所述第一个式I(a)化合物与所述第二个式I(a)化合物是相同的。
44、权利要求28的方法,其中所述第一个式I(a)化合物与所述第二个式I(a)化合物是不同的。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/350,232 | 2003-01-24 | ||
| US10/350,232 US7544696B2 (en) | 2003-01-24 | 2003-01-24 | Spermicidal and/or antifungal composition and methods of using the same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1751027A true CN1751027A (zh) | 2006-03-22 |
Family
ID=32735516
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA2004800047483A Pending CN1751027A (zh) | 2003-01-24 | 2004-01-23 | 杀精子和/或杀真菌组合物和使用它们的方法 |
Country Status (20)
| Country | Link |
|---|---|
| US (2) | US7544696B2 (zh) |
| EP (1) | EP1585732B1 (zh) |
| JP (2) | JP4597136B2 (zh) |
| KR (1) | KR20050103282A (zh) |
| CN (1) | CN1751027A (zh) |
| AT (1) | ATE499914T1 (zh) |
| AU (1) | AU2004207436B2 (zh) |
| BR (1) | BRPI0406572A (zh) |
| CA (1) | CA2514047A1 (zh) |
| CO (1) | CO5670353A2 (zh) |
| DE (1) | DE602004031588D1 (zh) |
| EC (1) | ECSP055992A (zh) |
| ES (1) | ES2359814T3 (zh) |
| IL (1) | IL169784A0 (zh) |
| MX (1) | MXPA05007892A (zh) |
| NO (1) | NO20053913L (zh) |
| RU (1) | RU2346687C2 (zh) |
| UA (1) | UA82864C2 (zh) |
| WO (1) | WO2004066934A2 (zh) |
| ZA (1) | ZA200505870B (zh) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7795436B2 (en) | 2005-08-24 | 2010-09-14 | Bristol-Myers Squibb Company | Substituted tricyclic heterocycles as serotonin receptor agonists and antagonists |
| AU2007240384A1 (en) * | 2006-04-19 | 2007-11-01 | Research Triangle Institute | Antispermatogenic, spermicidal and/or antifungal composition and methods of using the same |
| WO2009026375A2 (en) * | 2007-08-21 | 2009-02-26 | Lehigh University | Spermicidal and microbicidal compositions |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2470109A (en) | 1949-05-17 | Heterocyclic amines | ||
| US2470108A (en) | 1949-05-17 | Heterocyclic amines | ||
| US2546652A (en) | 1949-07-30 | 1951-03-27 | Hoffmann La Roche | Pyridindenes and process for their manufacture |
| CH432518A (de) | 1964-01-17 | 1967-03-31 | Sandoz Ag | Verfahren zur Herstellung von neuen heterocyclischen Verbindungen |
| CH498120A (de) | 1968-03-28 | 1970-10-31 | Sandoz Ag | Verfahren zur Herstellung neuer Indenopyridinderivate |
| US3462443A (en) | 1967-04-19 | 1969-08-19 | Mcneilab Inc | Indeno(1,2-c)pyridine derivatives |
| DE77720C (de) | 1968-05-21 | FARBWERKE VORM. MEISTER LUCIUS & BRÜNING, Höchst a. M | Verfahren zur Darstellung eines grün bis schwarz färbenden Beizenfarbstoffs der Anthracenreihe | |
| CY819A (en) | 1969-05-07 | 1976-12-01 | Sandoz Ltd | Indenopyridine derivatives |
| US3627773A (en) | 1969-06-09 | 1971-12-14 | Sandoz Ltd | 1,3,4,4A,5,9B-HEXAHYDRO-5-PHENYL-2H-INDENO{8 1,2-c{9 -PYRIDINES |
| US3678057A (en) | 1970-03-26 | 1972-07-18 | Sandoz Ltd | 1,3,4,4A,5,9B-HEXAHYDRO-5-PHENYL-2H-INDENO{8 1,2-c{9 -PYRIDINES |
| US3991066A (en) | 1971-01-26 | 1976-11-09 | Sandoz Ltd. | 1,3,4,9B-Tetrahydro-5-methyl-2H-indeno[1,2-c]pyridines |
| US5314917A (en) * | 1991-03-22 | 1994-05-24 | E. B. Michaels Research Associates, Inc. | Method for inactivating enveloped viruses and sperm |
| US5319084A (en) * | 1993-08-16 | 1994-06-07 | Research Triangle Institute | Hexahydroindenopyridine compounds having antispermatogenic activity |
| US5952336A (en) * | 1997-01-31 | 1999-09-14 | Research Triangle Institute | Hexahydroindenopyridine compounds having antispermatogenic activity |
-
2003
- 2003-01-24 US US10/350,232 patent/US7544696B2/en not_active Expired - Fee Related
-
2004
- 2004-01-23 UA UAA200508141A patent/UA82864C2/uk unknown
- 2004-01-23 CA CA002514047A patent/CA2514047A1/en not_active Abandoned
- 2004-01-23 MX MXPA05007892A patent/MXPA05007892A/es not_active Application Discontinuation
- 2004-01-23 CN CNA2004800047483A patent/CN1751027A/zh active Pending
- 2004-01-23 JP JP2006536504A patent/JP4597136B2/ja not_active Expired - Fee Related
- 2004-01-23 BR BR0406572-7A patent/BRPI0406572A/pt not_active IP Right Cessation
- 2004-01-23 DE DE602004031588T patent/DE602004031588D1/de not_active Expired - Lifetime
- 2004-01-23 KR KR1020057013691A patent/KR20050103282A/ko not_active Application Discontinuation
- 2004-01-23 WO PCT/US2004/000206 patent/WO2004066934A2/en active Application Filing
- 2004-01-23 EP EP04704789A patent/EP1585732B1/en not_active Expired - Lifetime
- 2004-01-23 AT AT04704789T patent/ATE499914T1/de not_active IP Right Cessation
- 2004-01-23 ES ES04704789T patent/ES2359814T3/es not_active Expired - Lifetime
- 2004-01-23 RU RU2005126720/14A patent/RU2346687C2/ru not_active IP Right Cessation
- 2004-01-23 AU AU2004207436A patent/AU2004207436B2/en not_active Ceased
- 2004-01-23 ZA ZA200505870A patent/ZA200505870B/en unknown
-
2005
- 2005-07-20 IL IL169784A patent/IL169784A0/en unknown
- 2005-08-22 CO CO05083226A patent/CO5670353A2/es not_active Application Discontinuation
- 2005-08-22 NO NO20053913A patent/NO20053913L/no not_active Application Discontinuation
- 2005-08-31 EC EC2005005992A patent/ECSP055992A/es unknown
-
2008
- 2008-11-25 US US12/277,708 patent/US8193213B2/en not_active Expired - Fee Related
-
2010
- 2010-06-22 JP JP2010141878A patent/JP2010202671A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| RU2346687C2 (ru) | 2009-02-20 |
| US20090149490A1 (en) | 2009-06-11 |
| ECSP055992A (es) | 2006-01-16 |
| NO20053913L (no) | 2005-08-22 |
| IL169784A0 (en) | 2007-07-04 |
| EP1585732B1 (en) | 2011-03-02 |
| JP4597136B2 (ja) | 2010-12-15 |
| ATE499914T1 (de) | 2011-03-15 |
| WO2004066934A3 (en) | 2005-05-19 |
| MXPA05007892A (es) | 2005-11-17 |
| US20040147539A1 (en) | 2004-07-29 |
| WO2004066934A2 (en) | 2004-08-12 |
| EP1585732A4 (en) | 2009-01-28 |
| JP2007506806A (ja) | 2007-03-22 |
| UA82864C2 (en) | 2008-05-26 |
| RU2005126720A (ru) | 2006-02-10 |
| DE602004031588D1 (de) | 2011-04-14 |
| ZA200505870B (en) | 2006-10-25 |
| US8193213B2 (en) | 2012-06-05 |
| CA2514047A1 (en) | 2004-08-12 |
| EP1585732A2 (en) | 2005-10-19 |
| JP2010202671A (ja) | 2010-09-16 |
| ES2359814T3 (es) | 2011-05-27 |
| KR20050103282A (ko) | 2005-10-28 |
| AU2004207436B2 (en) | 2009-08-06 |
| BRPI0406572A (pt) | 2005-12-20 |
| AU2004207436A1 (en) | 2004-08-12 |
| US7544696B2 (en) | 2009-06-09 |
| CO5670353A2 (es) | 2006-08-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20240091184A1 (en) | Modulators of sestrin-gator2 interaction and uses thereof | |
| TWI306859B (en) | Muscarinic agonists | |
| JP2021528404A (ja) | Oga阻害剤化合物 | |
| CN1077574C (zh) | 托烷衍生物,其制备和应用 | |
| CN104662021B (zh) | 新的双环吡啶酮类 | |
| JP3538431B2 (ja) | 腫瘍細胞増殖予防薬を調製するためのアミンの使用 | |
| CN110234638A (zh) | 杂芳基苯氧基苯甲酰胺kappa阿片类配体 | |
| JPH06293758A (ja) | 抗鬱病および抗パーキンソン氏病化合物 | |
| CN1751027A (zh) | 杀精子和/或杀真菌组合物和使用它们的方法 | |
| CN1110497C (zh) | 双-或三氮杂-螺旋[4,5]癸烷衍生物 | |
| CN1216604C (zh) | 具有抗精子生成活性的六氢茚并吡啶化合物 | |
| JPH06293759A (ja) | アルキル置換複素環式化合物 | |
| WO2008049996A2 (fr) | Nouveaux derives indoliques, leur procede de preparation et les compositions pharmaceutiques qui les contiennent | |
| EP2167460A2 (fr) | Nouveaux derives naphtaleniques, leur procede de preparation et les compositions pharmaceutiques qui les contiennent | |
| JP2007513136A (ja) | 治療用化合物としてのムスカリン作用剤 | |
| EP1873139A1 (fr) | Nouveaux dérivés naphtaleniques, leur procédé de préparation et les compositions pharmaceutiques qui les contiennent | |
| WO2005012282A1 (fr) | Nouveaux derives du benzothiofene 2-thiosubstitues, leur procede de preparation et les compositions pharmaceutiques qui les contiennent | |
| KR20040111473A (ko) | 강력한 알파 2-아드레노셉터 길항제인 폴리사이클릭 화합물 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C12 | Rejection of a patent application after its publication | ||
| RJ01 | Rejection of invention patent application after publication |
Open date: 20060322 |