-
GEBIET DER
ERFINDUNG
-
Die
vorliegende Erfindung bezieht sich auf die Verwendung eines Wirkstoffes
zur Herstellung eines Medikamentes zur Behandlung von weiblicher
Sexualstörung
(FSD), insbesondere weiblicher sexueller Erregungsstörung (FSAD).
Der Wirkstoff ist ein Inhibitor der neutralen Endopeptidase (I:NEP).
-
Zur
Vereinfachung ist eine Liste von Abkürzungen, welche im folgenden
Text verwendet werden, vor dem Abschnitt mit den Ansprüchen angegeben.
-
WEIBLICHES
SEXUALANSPRECHEN
-
Die
weibliche Sexualansprechphase der Erregung lässt sich nicht ohne weiteres
von der Phase des Begehrens unterscheiden bis die physiologischen
Veränderungen
in der Vagina und Klitoris sowie in anderen Sexualorganen sattzufinden
beginnen. Die sexuelle Erregung und Lust sind von einer Kombination
von vaskulären
und neuromuskulären
Ereignissen begleitet, welche zum Anschwellen der Klitoris, Schamlippen
und Vaginalwand, zu verstärkter
vaginaler Lubrikation und zur Dilatation des Vaginallumens führen (Levin,
1980; Ottesen, 1983; Levin, 1991; Levin, 1992; Sjoberg, 1992; Wagner,
1992; Schiavi et al., 1995; Masters et al., 1996; Berman et al.,
1999).
-
Die
vaginale Schwellung ermöglicht
eine Transsudation, und dieser Prozess ist für die verstärkte vaginale Lubrikation verantwortlich.
Die Transsudation erlaubt einen Fluss von Plasma durch das Epithel
und auf die Vaginaloberfläche,
wobei die antreibende Kraft ein erhöhter Blutfluss im vaginalen
Kapillarbett während
des Erregungszustandes ist. Ausserdem führt die Schwellung zu einer
Erhöhung
der Vaginallänge
und des Lumendurchmessers, insbesondere in den distalen 2/3 des
Vaginalkanals. Die Dilatation des Lumens der Vagina ergibt sich
aus einer Kombination einer Relaxation der glatten Muskulatur ihrer
Wandung und einer Relaxation der Skelettmuskulatur der Untelreibsmuskeln.
Von gewissen sexuellen Schmerzstörungen
wie Vaginismus wird angenommen, dass sie mindestens teilweise durch
eine ungenügende
Relaxation verursacht werden, welche eine Dilatation der Vagina
verhindert; es bleibt abzuklären,
ob dies primär
ein Problem der glatten oder der Skelettmuskulatur ist. (Levin,
1980; Ottesen, 1983; Levin, 1991; Levin, 1992; Sjoberg, 1992; Wagner,
1992; Schiavi et al., 1995; Master et al., 1996; Berman et al.,
1999).
-
Die
Gefässe
und Mikrogefässe
der Vagina sind von Nerven innerviert, welche Neuropeptide und andere
Neurotransmitter-Kandidaten
enthalten. Diese umfassen das Calcitonin-Genassoziierte Peptid (CGRP), das
Neuropeptid Y (NPY), die Stickstoffmonoxid-Synthase (NOS), die Substanz
P und das vasoaktive intestinale Peptid (VIP) (Hoyle et al., 1996).
Die in der Klitoris vorhandenen Peptide werden unten besprochen.
Die Stickstoffmonoxid-Synthase, welche oft mit dem VIP kolokalisiert
ist, zeigt immunologisch eine stärkere
Expression in den tiefen Arterien und Venen gegenüber den
Blutgefässen
der Propria (Hoyle et al., 1996).
-
WEIBLICHE
SEXUALSTÖRUNG
-
Es
ist bekannt, dass gewisse Individuen an weiblicher Sexualstörung (FSD)
leiden können.
-
FSD
wird am besten definiert als die Schwierigkeit oder Unfähigkeit
einer Frau, Befriedigung bei der sexuellen Entfaltung zu finden.
FSD ist ein kollektiver Begriff für mehrere unterschiedliche
weibliche Sexualstörungen
(Leiblum, 1998, Berman et al., 1999). Die Frau kann einen Mangel
an Begehren, Schwierigkeiten mit der Erregung oder dem Orgasmus,
Schmerzen beim Geschlechtsverkehr oder eine Kombination dieser Probleme
haben. Mehrere Arten von Erkrankungen, Medikationen, Verletzungen
oder psychologische Problemen können
FSD verursachen.
-
Studien,
welche Sexualstörungen
bei Ehepaaren untersuchten, ergaben, dass sich bis zu 76% der Frauen über Sexualstörungen beklagen
und dass 30–50%
der Frauen in den USA an FSD leiden.
-
Subtypen
von FSD umfassen eine Störung
mit hypoaktivem sexuellem Begehren, weibliche sexuelle Erregungsstörung, Orgasmusstörung und
Störung
des sexuellen Begehrens.
-
Die
sich in der Entwicklung befindenden Behandlungen haben zum Ziel,
spezifische Subtypen von FSD, vornehmlich Störungen des Begehrens und Erregungsstörungen zu
behandeln.
-
Die
Kategorien von FSD lassen sich am besten durch Abgrenzung gegenüber den
Phasen des normalen weiblichen Sexualansprechens: Begehren, Erregung
und Orgasmus (Leiblum 1998) definieren. Begehren oder Libido ist
der Antrieb für
sexuelle Entfaltung – und
die Manifestationen umfassen oft sexuelle Gedanken entweder beim
Zusammensein mit einem interessanten Partner oder unter dem Einfluss
anderer erotischer Stimuli. Im Gegensatz dazu ist die sexuelle Erregung
die vaskuläre
Antwort auf eine sexuelle Stimulation, wovon eine wichtige Komponente
die vaginale Lubrikation und die Ausdehnung der Vagina ist. Demnach ist
die sexuelle Erregung im Gegensatz zum sexuellen Begehren eine Antwort,
die mit dem genitalen (z.B. vaginalen und klitoralen) Blutfluss
zusammenhängt
und nicht notwendigerweise eine Sensibilität ist. Der Orgasmus ist die
Freisetzung von sexueller Anspannung, welche während der Erregung zu einem
Höhepunkt
gekommen ist. Demnach kommt FSD typischerweise vor, wenn eine Frau
eine inadäquate
oder unbefriedigende Antwort in irgend einer dieser Phasen, gewöhnlich Begehren,
Erregung oder Orgasmus hat. Die FSD-Kategorien umfassen eine Störung mit
hypoaktivem sexuellem Begehren, eine sexuelle Erregungsstörung, Orgasmusstörungen und
sexuelle Schmerzstörungen.
-
Eine
Störung
mit hypoaktivem sexuellem Begehren liegt vor, wenn eine Frau kein
oder wenig Begehren hat, sexuell aktiv zu sein, und keine oder wenige
sexuelle Gedanken oder Fantasien hat. Diese Art von FSD kann durch
niedrige Testosteronspiegel entweder infolge natürlicher Menopause oder infolge
chirurgischer Menopause verursacht werden. Andere Ursachen umfassen
Krankheit, Medikationen, Müdigkeit,
Depression und Angst.
-
Die
weibliche sexuelle Erregungsstörung
(FSAD) ist durch eine inadequate genitale Antwort auf sexuelle Stimulation
gekennzeichnet. Die Genitalien (z.B. die Vagina und/oder die Klitoris)
unterliegen nicht der Anschwellung, welche die normale sexuelle
Erregung kennzeichnet. Die Vaginalwände sind wenig lubriziert,
so dass der Geschlechtsverkehr schmerzhaft ist. Die Orgasmen können verhindert
sein. Die Erregungsstörung kann
durch reduziertes Östrogen
in der Menopause oder nach der Geburt eines Kindes und während des
Stillens sowie durch Erkrankungen mit vaskulären Komponenten wie Diabetes
und Atherosklerose verursacht sein. Andere Ursachen ergeben sich
aus der Behandlung mit Diuretika, Antihistaminika, Antidepressiva
z.B. SSRIs oder Antihypertensiva. FSAD wird unten detaillierter
besprochen.
-
Sexuelle
Schmerzstörungen
(welche Dyspareunie und Vaginismus umfassen) sind durch Schmerzen gekennzeichnet,
die sich bei der Penetration ergeben, und können durch lubrikationsvermindernde
Medikationen, eine Endometriose, eine Unterleibsentzündung, entzündliche
Darmerkrankungen oder Probleme im Harntrakt verursacht sein.
-
Die
Prävalenz
von FSD ist schwierig zu beurteilen, weil der Begriff mehrere Arten
von Problemen abdeckt, wovon einige schwierig zu messen sind, und
weil das Interesse zur Behandlung von FSD erst seit kurzem besteht.
Bei vielen Frauen hängen
die sexuellen Probleme entweder direkt mit dem weiblichen Alterungsprozess
oder mit chronischen Erkrankungen wie Diabetes und Hypertonie zusammen.
-
Bei
der angegebenen Inzidenz und Prävalenz
von FSD besteht eine grosse Variabilität, was teilweise durch die
Verwendung von unterschiedlichen Evaluationskriterien erklärt wird,
wobei aber die meisten Untersucher berichten, dass ein signifikanter
Anteil von andersweitig gesunden Frauen Symptome von einer oder mehreren
der FSD-Subgruppen haben. Beispielsweise zeigen Studien, welche
Sexualstörung
bei Paaren vergleichen, dass 63% der Frauen Erregungs- oder Orgasmusstörung hatten,
während
im Vergleich dazu 40% der Männer
Erektions- oder Ejakulationsstörung
haben (Frank et al., 1978).
-
Allerdings
variiert die Prävalenz
der weiblichen sexuellen Erregungsstörung beträchtlich von Erhebung zu Erhebung.
In einer kürzlich
veröffentlichten
National Health und Social Life Erhebung berichteten 19% der Frauen über Lubrikationsschwierigkeiten,
während
14% der Frauen in einer ambulanten gynäkologischen Klinik ähnliche
Schwierigkeiten mit der Lubrikation angaben (Rosen et al., 1993).
-
In
mehreren Studien wurde von sexuellen Erregungsstörungen bei diabetischen Frauen
(bis zu 47%) berichtet, wobei diese eine verringerte vaginale Lubrikation
umfassten (Wincze et al., 1993). Es bestand kein Zusammenhang zwischen
Neuropathie und Sexualstörung.
-
In
zahlreichen Studien wurde auch gezeigt, dass zwischen 11-48% der Frauen insgesamt
mit dem Altern ein verringertes sexuelles Begehren haben. Ähnlicherweis
geben zwischen 11-50%
der Frauen Probleme mit der Erregung und der Lubrikation an und
leiden demzufolge während
dem Geschlechtsverkehr an Schmerzen. Vaginismus ist weitaus weniger
häufig
und betrifft ungefähr
1% der Frauen.
-
In
Studien mit sexuell erfahrenen Frauen wurde gezeigt, dass 5–10% eine
primäre
Anorgasmie haben. Weitere 10% haben selten Orgasmen und noch weitere
10% erfahren diese inkonsistent (Spector et al., 1990).
-
Weil
FSD aus mehreren Subtypen besteht, welche Symptome in unterschiedlichen
Phasen des sexuellen Ansprechzyklus zeigen, gibt es keine einzelne
Therapie. Die gegenwärtige
Behandlung der FSD konzentriert sich im Prinzip auf psychologische
oder partnerschaftliche Aspekte. Die Behandlung der FSD entwickelt sich
allmählich,
indem mehr klinische und grundlagenwissenschaftliche Studien der
Erforschung dieses medizinischen Problems gewidmet werden. Nicht
alle weiblichen sexuellen Beschwerden haben eine psychologische
Pathophysiologie, insbesondere für
diejenigen Individuen, die eine Komponente einer vaskulogenen Störung (z.B.
FSAD) haben könnten,
welche zur gesamthaften weiblichsexuellen Beschwerde beitragen.
Es gibt zur Zeit keine Medikamente, die für die Behandlung der FSD zugelassen
sind. Die empirische medikamentöse Therapie
umfasst die Verabreichung von Östrogen
(topisch oder als Hormonersatztherapie), Androgenen oder stimmungsaufhellenden
Medikamenten wie Buspiron oder Trazodon. Diese Behandlungsoptionen
sind wegen der niedrigen Wirksamkeit oder den unannehmbaren Nebenwirkungen
oft unbefriedigend.
-
Da
das Interesse, FSD pharmakologisch zu behandeln, relativ neu ist,
besteht die Therapie aus Folgenden: - psychologische Beratung, rezeptfreie
Sexualgleitmittel und experimentelle Kandidaten, einschliesslich
Medikamente, die für
andere Krankheitszustände
zugelassen sind. Diese Medikationen umfassen hormonelle Wirkstoffe,
entweder Testosteron oder Kombinationen von Östrogen und Testosteron, und
seit kurzem vaskuläre
Medikamente, die sich bei der männlichen
erektilen Dysfunktion als wirksam erwiesen haben. Keiner dieser
Wirkstoffe hat sich bei der Behandlung von FSD als sehr wirksam
erwiesen.
-
WEIBLICHE SEXUELLE ERREGUNGSSTÖRUNG (FSAD)
-
Die
sexuelle Erregungsantwort besteht aus einer Vasokongestion im Unterleibsbereich,
einer vaginalen Lubrikation und einer Ausdehnung und Anschwellung
der äusserlichen
Genitalien. Die Störung
verursacht eine ausgeprägte
Belastung and/oder zwischenmenschliche Schwierigkeiten. Studien
zur Untersuchung von Sexualstörung
bei Paaren ergaben, dass es eine grosse Anzahl von Frauen gibt,
die an sexueller Erregungsstörung – anderweitig
als weibliche sexuelle Erregungsstörung (FSAD) bezeichnet – leiden.
-
Das
Diagnostic und Statistical Manual (DSM) IV der American Psychiatric
Association definiert weibliche sexuelle Erregungsstörung (FSAD)
als:
"eine
persistierende oder rezidivierende Unfähigkeit, bis zur Vollendung
der sexuellen Aktivität
eine adäquate Lubrikations-Anschwellungsantwort
auf die sexuelle Erregung zu erreichen oder zu erhalten. Die Störung muss
eine ausgeprägte
Belastung oder zwischenmenschliche Schwierigkeit verursachen."
-
FSAD
ist eine weit verbreitete sexuelle Störung, die prä-, peri-
und postmenopausale (+/–HRT)
Frauen betrifft. Sie ist mit begleitenden Störungen wie Depression, kardiovaskulären Erkrankungen,
Diabetes und UG-Störungen
vergesellschaftet.
-
Die
primären
Folgen der FSAD sind ein Mangel an Anschwellung/Schwellung, ein
Mangel an Lubrikation und ein Mangel an lustvollen Empfindungen
im Genitalbereich. Die sekundären
Folgen der FSAD sind vermindertes sexuelles Begehren, Schmerzen
während
des Geschlechtsverkehrs und Schwierigkeit zum Erreichen eines Orgasmus.
-
Kürzlich wurde
die Hypothese aufgestellt, dass zumindest für einen Teil der Patienten
mit Symptomen der FSAD eine vaskuläre Basis besteht (Goldstein
et al., 1998), wobei Tierdaten diese Ansicht unterstützen (Park
et al., 1997).
-
Wirkstoff-Kandidaten
zum Behandeln der FSAD, die sich in der Erprobung bezüglich der
Wirksamkeit befinden, sind primär
Therapien der erektile Dysfunktion, welche die Zirkulation in den
männlichen
Genitalien fördern.
Sie bestehen aus zwei Arten von Formulierungen, orale oder sublinguale
Medikationen (Apomorphin, Phentolamin, Sildenafil) und Prostaglandin
(PGE1 – Alprostadil),
die bei Männern
injziiert oder transurethral verabreicht werden und bei Frauen topisch
auf die Genitalien appliziert werden.
-
Die
vorliegende Erfindung bezweckt, einen effektiven Wirkstoff zur Behandlung
von FSD und insbesondere von FSAD bereitzustellen.
-
ZUSAMMENFASSUNG
VON ASPEKTEN DER VORLIEGENDEN ERFINDUNG
-
Ein
entscheidener Befund der vorliegenden Erfindung ist die Fähigkeit,
eine an FSD (bevorzugt FSAD) leidende Frau unter Verwendung eines
I:NEP zu behandeln.
-
Erfindungsgamäss wird
die Verwendung eines Inhibitors der neutralen Endopeptidase (I:NEP)
zur Herstellung eines Medikamentes für die Behandlung von weiblicher
Sexualströung
(FSD) bereitgestellt.
-
In Übereinstimmung
mit der vorliegenden Erfindung wird der erfindungsgemässe I:NEP
als der "erfindungsgemässe Wirkstoff" bezeichnet.
-
Der
erfindungsgemässe
Wirkstoff (d.h. I:NEP) kann auch in Kombination mit einem oder mehreren
zusätzlichen
pharmazeutisch aktiven Wirkstoffen verwendet werden. Der zusätzliche
pharmazeutisch aktive Wirkstoff, falls dieser vorhanden ist oder
zusammen mit dem erfindungsgemässen
Wirkstoff verwendet wird, kann als ein "zusätzlicher
Wirkstoff" bezeichnet
werden. Einer oder mehrere dieser zusätzlichen Wirkstoffe können eines
oder mehrere sein von: I:PDE, ein anderer I:NEP, ein I:NPY. Kombinationen
von Wirkstoffen werden unten näher
beschrieben.
-
Falls
der erfindungsgemässe
zusätzliche
Wirkstoff ein I:PDE ist, dann ist besagter PDE in gewissen Ausgestaltungen
ein cAMP hydrolysierender (und wahlweise cGMP-hydrolysierender)
PDE. Der Begriff "cAMP-hydrolysierend" umfasst auch das
Metabolisieren und/oder Zersetzen von cAMP. Der Begriff "cAMP-hydrolysierend
(und wahlweise cGMP)" bedeutet,
dass der zusätzliche
Wirkstoff fähig
sein kann, cGMP zusätzlich
zu cAMP zu hydrolysieren. Hier umfasst der Begriff "cGMP Hydrolysieren" auch das Metabolisieren und/oder
Zersetzen von cGMP. Allerdings gilt für gewisse Ausgestaltungen der
vorliegenden Erfindung, dass der zusätzliche Wirkstoff nicht notwendigerweise
fähig sein
muss, cGMP zu hydrolysieren.
-
Allgemeine
Bezugnahmen auf Wirkstoffe können
hier auf zusätzliche
Wirkstoffe wie auch auf erfindungsgemässe Wirkstoffe anwendbar sein.
-
Gemäss der vorliegenden
Erfindung wirkt der erfindungsgemässe Wirkstoff (d.h. I:NEP)
auf ein Wirkziel (nachfolgend: "Target"), vorzugsweise spezifisch
auf das besagte Target. Dieses Target wird gelegentlich als das "erfindungsgemässe Target" bezeichnet. Allerdings
kann der erfindungsgemässe
Wirkstoff auch an einem oder mehreren anderen Targets wirken. Diese
anderen Targets können
als ein "zusätzliches
Target" bezeichnet
werden. Ähnlicherweise
gilt, dass wenn ein zusätzlicher
Wirkstoff verwendet wird, dieser zusätzliche Wirkstoff an demselben
erfindungsgemässen
Target und/oder an einem zusätzlichen
Target (welches nicht dasselbe zusätzliche Target zu sein braucht,
an dem der erfindungsgemässe
Wirkstoff wirkt) wirken kann. Targets sind hier beschrieben. Es
versteht sich, dass hier allgemeine Bezugnahmen auf Targets ebenso
auf die zusätzlichen
Targets wie auf das erfindungsgemässe Target anwendbar sein können.
-
Ein
weiterer entscheidender Befund der vorliegenden Erfindung ist die
Fähigkeit,
unter Verwendung des erfindungsgemässen Wirkstoffes (d.h. I:NEP)
den weiblichen genitalen (z.B. vaginalen oder klitoralen) Blutfluss
zu verstärken.
-
In
unseren Experimenten wurde gefunden, dass FSAD mit einem verringerten
genitalen Blutfluss – insbesondere
einem verringerten Blutfluss in der Vagina und/oder der Klitoris
einhergeht. Demnach kann eine Behandlung von Frauen mit FSAD durch
Erhöhung
des genitalen Blutfluss mittels vasoaktiver Wirkstoffe erreicht
werden. In unseren Studien wurde gezeigt, dass cAMP die vaginale
und klitorale Vasorelaxation vermittelt und dass der genitale (z.B.
vaginale und klitorale) Blutfluss durch Erhöhung der cAMP-Spiegel verstärkt/potenziert
werden kann. Dies ist ein weiterer entscheidender Befund.
-
In
diesem Zusammenhang hat vorgängig
niemand vorgeschlagen, dass FSAD auf eine solche Weise – d.h. durch
direkte oder indirekte Erhöhung
der cAMP-Spiegel – behandelt
werden kann. Darüber
hinaus gab es keine Lehre im Fachgebiet, welche vorgeschlagen hätte, dass
FSAD mit einer nachteiligen Modulierung von cAMP-Aktivität und/oder
-Spiegeln zusammenhängt
oder dass cAMP für
die Vermittlung der vaginalen und klitoralen Vasorelaxation verantwortlich
ist. Demnach ist die vorliegende Erfindung sogar noch weiter überraschend.
-
Ausserdem
wurde gefunden, dass es unter Verwendung der erfindungsgemässen Wirkstoffe
möglich ist,
die genitale Anschwellung zu verstärken und die FSAD zu behandeln – z.B. die
Lubrikation in der Vagina und die Sensitivität in der Vagina und Klitoris
zu erhöhen.
-
Demnach
bezieht sich die vorliegende Erfindung in einem breiten Aspekt auf
die Verwendung eines cAMP-Potenziators zur Behandlung von FSD, insbesondere
FSAD.
-
Die
vorliegende Erfindung ist vorteilhaft, weil sie einen Wirkstoff
zum Wiederherstellen einer normalen sexuellen Erregungsantwort – nämlich eines
verstärkten
genitalen Blutflusses, der zur vaginalen, klitoralen und labialen Anschwellung
führt,
bereitstellt. Dies wird zu einer verstärkten vaginalen Lubrikation über Plasmatranssudation,
zu einer verstärkten
vaginalen Dehnbarkeit und einer verstärkten genitalen (z.B. vaginalen
und klitoralen) Sensitivität
führen.
Demnach bezieht sich die vorliegende Erfindung auf einen Wirkstoff
zur Wiederherstellung oder Potenzierung der normalen sexuellen Erregungsantwort.
-
DETAILLIERTE
ASPEKTE DER VORLIEGENDEN ERFINDUNG
-
In
einem ihrer Aspekte bezieht sich die vorliegende Erfindung auf die
Verwendung eines I:NEP zur Herstellung eines Medikamentes zur Behandlung
von FSD.
-
In
einem anderen Aspekt bezieht sich die vorliegende Erfindung auf
die Verwendung eines Wirkstoffs zur Herstellung eines Medikamentes
(wie einer pharmazeutischen Zusammensetzung) für die Behandlung von FSD, insbesondere
FSAD; wobei der Wirkstoff zur Potentierung von cAMP in den sexuellen
Genitalien einer an FSD, insbesondere FSAD; leidenden Frau befähig ist;
und wobei besagter Wirkstoff der hier definierte erfindungsgemässe Wirkstoff
(d.h. I:NEP) ist.
-
Der
Einfachheit halber werden diese und weitere Aspekte der vorliegenden
Erfindung nun unter geeigneten Abschnittstiteln besprochen. Allerdings
sind die Lehren unter jedem Abschnitt nicht notwendigerweise auf
den jeweiligen einzelnen Abschnitt beschränkt.
-
BEVORZUGTE ASPEKTE
-
Der
erfindungsgemässe
Wirkstoff (d.h. I:NEP) ist bevorzugt für die Behandlung von FSAD vorgesehen.
-
Der
erfindungsgemässe
Wirkstoff (d.h. I:NEP) ist bevorzugt ein Vermittler der weiblichen
genitalen (z.B. vaginalen oder klitoralen) Vasorelaxation.
-
In
einer der Ausgestaltungen ist der erfindungsgemässe Wirkstoff (d.h. I:NEP)
bevorzugt für
die orale Verabreichung vorgesehen.
-
In
einer weiteren Ausgestaltung kann der erfindungsgemässe Wirkstoff
(d.h. I:NEP) für
die topische Verabreichung vorgesehen sein.
-
Der
erfindungsgemässe
Wirkstoff ist ein I:NEP (gelegentlich als NEPi geschrieben), wobei
besagter NEP das EC 3.4.24.11 ist.
-
Für gewisse
Anwendungen ist der Wirkstoff bevorzugt ein selektiver I:NEP, wobei
besagter NEP das EC 3.4.24.11 ist.
-
Der
erfindungsgemässe
Wirkstoff (d.h. I:NEP) ist ein Inhibitor – d.h. er ist fähig, eine
Inhibitionsfunktion zu entfalten.
-
Der
erfindungsgemässe
Wirkstoff (d.h. I:NEP) hat bevorzugt einen indirekten Potenzierungseffekt
auf cAMP. Anders ausgedrückt,
hat der Wirkstoff für
gewisse Anwendungen bevorzugt keinen direkten Potenzierungseffekt
auf cAMP. Es versteht sich, dass der Wirkstoff einen indirekten
Potenzierungseffekt auf cAMP haben kann durch Wirkung auf natürlich vorkommende
und natürlich
gefundene direkt wirkende Wirkstoffe – wie auf natürlich vorkommendes
und gefundenes VIP.
-
Für gewisse
Anwendungen kann der erfindungsgemässe Wirkstoff (d.h. I:NEP)
in Kombination mit einem anderen pharmazeutisch aktiven Wirkstoff
verabreicht werden. Dabei muss die Mitverabreichung nicht zur selben
Zeit erfolgen, und schon gar nicht über denselben Weg. Beispielsweise
kann eine Mitverabreichungs-Zusammensetzung eine Zusammensetzung
sein, die ein Wirkstoff gemäss
der vorliegenden Erfindung (d.h. I:NEP) und einen zusätzlichen
Wirkstoff umfasst, wobei der zusätzliche
Wirkstoff einen direkten Potenzierungseffekt auf cAMP haben könnte. Kombinationsbeispiele
werden unten beschrieben.
-
Für gewisse
Anwendungen hat der zusätzliche
Wirkstoff bevorzugt einen indirekten Potenzierungseffekt auf cAMP.
Beispiele für
solche zusätzliche
Wirkstoffe umfassen I:NEP und/oder I:NPY. Anders ausgedrückt, hat
der zusätzliche
Wirkstoff für
gewisse Anwendungen bevorzugt keinen direkten Potenzierungseffekt auf
cAMP. Es versteht sich, dass der zusätzliche Wirkstoff einen indirekten
Potenzierungseffekt auf cAMP haben kann durch Wirkung auf natürlich vorkommende
und natürlich
gefundene direkt wirkende Wirkstoffe – wie auf natürlich vorkommendes
und gefundenes VIP.
-
Für gewisse
Anwendungen hat der zusätzliche
Wirkstoff bevorzugt einen direkten Potenzierungseffekt auf cAMP.
Beispiele für
solche zusätzlichen
Wirkstoffe umfassen I:PDE.
-
Für gewisse
Anwendungen ist der zusätzliche
Wirkstoff ein Inhibitor – d.h.
er ist fähig,
eine Inhibitionsfunktion zu entfalten.
-
Für gewisse
Anwendungen ist der zusätzliche
Wirkstoff ein Antagonist.
-
Für gewisse
Anwendungen ist der zusätzliche
Wirkstoff bevorzugt ein I:PDE (gelegentlich als PDEi bezeichnet).
-
Für gewisse
Anwendungen ist der zusätzliche
Wirkstoff bevorzugt eine selektiver I:PDE.
-
Für gewisse
Anwendungen ist der zusätzliche
Wirkstoff bevorzugt ein I:PDE1 oder l:PDE2 (gelegentlich als I:PDEII
oder PDEIIi oder PDE2i bezeichnet) oder I:PDE3 oder I:PDE4 oder
I:PDE7 oder I:PDE8, wobei der Wirkstoff besonders bevorzugt ein
I:PDE2 ist.
-
Für gewisse
Anwendungen ist der zusätzliche
Wirkstoff bevorzugt ein selektiver I:PDEII (gelegentlich als PDE2
bezeichnet).
-
Für gewisse
Anwendungen ist der zusätzliche
Wirkstoff bevorzugt ein I:NEP (gelegentlich als NEPi bezeichnet).
-
Für gewisse
Anwendungen ist der zusätzliche
Wirkstoff bevorzugt ein selektiver I:NEP.
-
Für gewisse
Anwendungen ist der zusätzliche
Wirkstoff bevorzugt ein I:NEP, wobei besagter NEP das EC 3.4.24.11
ist.
-
Für gewisse
Anwendungen ist der zusätzliche
Wirkstoff bevorzugt ein selektiver I:NEP, wobei besagter NEP das
EC 3.4.24.11 ist.
-
Für gewisse
Anwendungen ist der zusätzliche
Wirkstoff bevorzugt ein I:NPY (gelegentlich als NPYi bezeichnet).
-
Für gewisse
Anwendungen ist der zusätzliche
Wirkstoff bevorzugt ein I:NPY Y1 oder I:NPY Y2 oder I:NPY Y5, besonders
bevorzugt ist der Wirkstoff ein I:NPY Y1.
-
Für gewisse
Anwendungen ist der zusätzliche
Wirkstoff bevorzugt ein selektiver I:NPY.
-
Für gewisse
Anwendungen ist der zusätzliche
Wirkstoff bevorzugt ein I:NPY Y1.
-
Für gewisse
Anwendungen ist der zusätzliche
Wirkstoff bevorzugt ein selektiver I:NPY Y1.
-
Bei
gewissen Anwendungen verursacht der Wirkstoff keinen anhaltenden
Abfall des Blutdruckes – oder
er wird in einer solchen Weise verabreicht (z.B. über eine
Periode von ungefähr
5 Minuten oder mehr), dass er keinen anhaltenden Abfall des Blutdruckes
verursacht. In dieser Ausgestaltung, kann der Wirkstoff, wenn er
topisch zu verabreichen ist, die Fähigkeit haben, einen ausgedehnten
Abfall des Blutdruckes zu verursachen (so wie bei intravenöser Verabreichung),
vorausgesetzt, dass bei der topischen Applikation nur minime Mengen
des Wirkstoffs in den Blutstrom gelangen. Bei einem oralen Wirkstoff
ist es bevorzugt, dass der Wirkstoff keinen anhaltenden Abfall des
Blutdruckes verursacht.
-
In
einem bevorzugten Aspekt verursacht der erfindungsgemässe Wirkstoff
(d.h. I:NEP) keine grosse Veränderung
der Herzfrequenz – oder
er wird in einer solchen Weise verabreicht, dass es keine grosse
Veränderung der Herzfrequenz verursacht.
-
BEHANDLUNG
-
Es
versteht sich, dass hier alle Bezugnahmen auf eine Behandlung eine
oder mehrere kurative, palliative und prophylaktische Behandlungen
mit einschliessen. Der Begriff Behandlung umfasst bevorzugt mindestens
eine kurative Behandlung und/oder eine palliative Behandlung.
-
WEIBLICHE
GENITALIEN
-
Der
Begriff "weibliche
Genitalien" wird
in Übereinstimmung
mit der in Gray's
Anatomy, C.D. Clemente, 13th American Edition gegebenen Definition
verwendet – nämlich:
"Die Genitalorgane
bestehen aus einer inneren und äusseren
Gruppe. Die inenren Organe befinden sich innerhalb des Unterleibs
und bestehen aus den Eierstöcken,
den Eileitern, der Gebärmutter
und der Vagina. Die äusseren
Organe liegen ausserhalb des urogenitalen Diaphragmas und unterhalb
des Beckenbogens. Sie umfassen den Venusberg, die äusseren
und inneren Schamlippen, die Klitoris, den Scheidenvorhof, den Scheidenvorhofkörper und
die grossen Scheidenvorhofdrüsen".
-
ENDOGENES
cAMP
-
In
einer besonders bevorzugten Ausgestaltung potenziert der erfindungsgemässe Wirkstoff
das endogene cAMP – also
die endogenen cAMP-Spiegel werden erhöht.
-
Hier
bezieht sich der Begriff "endogenes
cAMP" auf cAMP,
welches aus der sexuellen Stimulation (sexuellen Erregung) stammt.
Demnach umfasst der Begriff nicht die cAMP-Spiegel, welche unabhängig von
sexuellen Antrieb erhöht
sind.
-
Demnach
wird die Behandlung von FSAD gemäss
der erfindungsgemässen
Verwendung durch direkte oder indirekte Potenzierung der endogenen
cAMP-Signalisierung erreicht, welche ihrerseits den vaginalen Blutfluss/die
vaginale Lubrikation und/oder den klitoralen Blutfluss erhöht; wodurch
die natürliche
sexuelle Erregungsantwort verstärkt
wird. Demnach führt
das Behandlungsverfahren zur Wiederherstellung oder Potenzierung
der normalen Erregungsantwort.
-
Das
Behandlungsverfahren entsprechend der erfindungsgemässen Verwendung
kann durch Verwendung eines Inhibitors von NEP (EC 3.4.24.11) erreicht
werden.
-
Ein
tierisches Testmodell ist hier angegeben. Dieses tierische Testmodell
kann zur Bestimmung von Erhöhungen
des genitalen Blutflusses als Folge der cAMP-Potenzierung verwendet
werden. In diesem Tiermodell wird ein Pelvisnerv stimuliert - was
ein Wirkung herbeiführt,
welche die Physiologie einer sexuellen Erregung/Antwort nachahmt.
In diesen Experimenten verursachen die erfindungsgemässen Wirkstoffe
eine Erhöhung
des Blutflusses, welche über
der Zunahme bei den Kontrollen liegt, nachdem der Nerv stimuliert
worden ist. Bei fehlender Stimulation haben die Wirkstoffe keine
(oder eine vernachlässigbare)
Wirkung bezüglich des
Verursachens eines erhöhten
Blutflusses. Typischerweise wird in diesen Experimenten zunächst der
Nerv stimuliert, um eine anfängliche
Erhöhung
des Blutflusses zu erhalten. Dann wird dem Tier ein Kandidaten- (oder
ein aktueller) Wirkstoff systemisch oder lokal verabreicht, beispielsweise über den
intravenösen,
topischen oder oralen Weg. Eine Erhöhung des Blutflusses im Vergleich
zur Erhöhung
bei den Kontrollen weist dann auf einen erfindungsgemässen Wirkstoff
hin.
-
SEXUELLE STIMULATION
-
Die
vorliegende Erfindung umfasst auch die Verabreichung des erfindungsgemässen Wirkstoffs
(d.h. I:NEP) vor und/oder während
der sexuellen Stimulation. Hier kann der Begriff "sexuelle Stimulation" mit dem Begriff "sexuelle Erregung" gleichbedeutend
sein. Dieser Aspekt der vorliegenden Erfindung ist vorteilhaft,
weil er systemische Selektivität
ergibt. Die natürliche
Kaskade findet nur in den Genitalien und nicht an anderen Orten – z.B. im
Herz usw. statt. Demnach wäre
es möglich,
eine selektive Wirkung an den Genitalien zu erreichen.
-
Demnach
ist es in gewissen Aspekten der vorliegenden Erfindung äusserst
wünschenswert,
dass es einen sexuellen Stimulationsschritt gibt. Es wurde gefunden,
dass dieser Schritt systemische Selektivität ergeben kann. Hier kann "sexelle Stimulation" eine oder mehrere
aus einer visuellen Stimulation, einer körperlichen Stimulation, einer
auditorischen Stimulation oder einer gedanklichen Stimulation sein.
-
Demnach
werden die erfindungsgemässen
Wirkstoffe bevorzugt vor oder während
der sexuellen Stimulation verabreicht, insbesondere wenn diese Wirkstoffe
zur oralen Verabreichung vorgesehen sind.
-
Demnach
beschreibt die vorliegende Erfindung in diesem bevorzugten Aspekt
die Verwendung eines erfindungsgemässen Wirkstoffes zur Herstellung
eines Medikamentes für
die Behandlung von FSAD; wobei der Wirkstoff zur Potenzierung von
cAMP in den sexuellen Genitalien einer an FSAD leidenden Frau befähigt ist;
und wobei besagte Frau vor oder während Verabreichung des besagten
Medikamentes sexuell stimuliert wird; und wobei besagter Wirkstoff
ein I:NEP ist.
-
Die
vorliegende Erfindung bezieht sich vorzugsweise auf die Verwendung
eines erfindungsgemässen Wirkstoffes
zur Herstellung eines Medikamentes für die Behandlung von FSAD;
wobei der Wirkstoff zur Potenzierung von cAMP in den sexuellen Genitalien
einer an FSAD leidenden Frau befähigt
ist; und wobei besagte Frau vor oder während Verabreichung des besagten
Medikamentes sexuell stimuliert wird; und wobei das besagte Medikament
der besagten Frau oral verabreicht wird; und wobei besagter Wirkstoff
ein I:NEP ist.
-
POTENZIERUNG VON cAMP
-
Im
Zusammenhang mit cAMP umfasst hier der Begriff "Potenzierung" irgend eines oder mehrere von: Erhöhung der
Wirksamkeit von cAMP, Erhöhung
der cAMP-Spiegel, Erhöhung
der Aktivität
von cAMP, Verminderung des Ausmasses des cAMP-Abbaus, Verminderung des Ausmasses der
cAMP-Inhibition.
-
Die
potenzierende Wirkung kann eine direkte Wirkung sein. Ein Beispiel
für eine
direkte Wirkung wäre die
Aufregulierung des cAMP-Spiegels durch einen Wirkstoff, welcher
dessen Expression erhöht.
-
Alternativ
könnte
die potenzierende Wirkung eine indirekte Wirkung sein. Ein Beispiel
für eine
solche Wirkung wäre
die Wirkung auf eine Substanz, welche sonst cAMP inhibieren und/oder
die Spiegel und/oder Aktivität
von cAMP verringern würde.
Ein anderes Beispiel für
eine solche Wirkung wäre
die Erhöhung
der Wirkung einer Substanz, welche die Wirksamkeit von cAMP erhöht, den
Spiegel von cAMP erhöht,
die Aktivität von
cAMP erhöht,
das Ausmass des cAMP-Abbaus verringert oder das Ausmass der cAMP-Inhibition
verringert.
-
Ein
Beispiel für
einen zusätzlichen
Wirkstoff, das als ein PcAMP wirken könnte, wäre ein I:PDE
wie beispielsweise I:PDEII.
-
cAMP-Mimetikum
-
In
gewissen Aspekten der vorliegenden Erfindung kann der zusätzliche
Wirkstoff als ein cAMP-Mimetikum wirken.
-
Der
hier verwendete Begriff "cAMP-Mimetikum" bezieht sich auf
einen Wirkstoff, der in einer ähnlichen Weise
(z.B. mit ähnlichem
biologischen Profil und Wirkung) auf cAMP in den weiblichen sexuellen
Genitalien wirken kann und dabei irgend eines oder mehrere der Folgenden
bewirkt: Erhöhung
der Wirksamkeit von cAMP-artigen Einheiten, Erhöhung des Spiegels von cAMP-artigen
Einheiten, Erhöhung
der Aktivität
von cAMP-artigen Einheiten, Verringerung des Ausmasses des Abbaus
von cAMP-artigen Einheiten, Verringerung des Ausmasses der Inhibition
von cAMP-artigen Einheiten.
-
Ein
Beispiel für
ein cAMP-Mimetikum wäre
Forskolin. Es wurde hier gefunden, dass Forskolin den vaginalen
und klitoralen Blutfluss erhöht,
und es kann auch als ein vaginales Relaxans wirken.
-
In
einem bevorzugten Aspekt wird das cAMP-Mimetikum oral verabreicht.
-
AKTIVATOR
VON cAMP
-
Der
hier verwendete Begriff "Aktivator
von cAMP" bedeutet
eine Substanz, welche cAMP in den weiblichen sexuellen Genitalien
kontrolliert oder freisetzt. Die Kontrolle kann direkt (z.B. auf
cAMP selber) oder indirekt (z.B. über die Aktivierung von cAMP)
erfolgen. Der Einfachheit halber werden diese Substanzen als AcAMp bezeichnet.
-
TARGET
-
Der
hier im Zusammenhang mit der vorliegenden Erfindung verwendete Begriff "Target" bedeutet jede Substanz,
die cAMP, ein AcAMP, ein IcAMP,
oder ein AMcAMP ist. Anders ausgedrückt, kann
das erfindungsgemässe Target
als PcAMP-Target bezeichnet werden.
-
Das
erfindungsgemässe
Target und/oder das zusätzliche
Target kann eine Aminosäurensequenz und/oder
eine für
dasselbe kodierende Nukleotid-Sequenz und/oder eine für die Expression
desselben verantwortliche Expressions-Einheit und/oder ein Modulator
desselben sein.
-
Das
Target kann sogar eine Kombination von solchen Targets sein.
-
WIRKSTOFF
-
Der
erfindungsgemässe
Wirkstoff ist ein I:NEP. Der erfindungsgemässe Wirkstoff kann jeder geeignete Wirkstoff
sein, der als PcAMP wirken kann.
-
Der
Wirkstoff (d.h. der erfindungsgemässe Wirkstoff und/oder der
zusätzliche
Wirkstoff) kann eine Aminosäurensequenz
oder ein chemisches Derivat davon sein. Die Substanz kann auch eine
organische Verbindung oder eine andere Chemikalie sein. Der Wirkstoff
kann sogar eine Nukleotid-Sequenz sein – wobei es sich um eine Sense-Sequenz
oder eine Antisense-Sequenz handeln kann. Der Wirkstoff kann sogar
ein Antikörper
sein.
-
Demnach
umfasst der Begriff "Wirkstoff" ohne darauf beschränkt zu sein
eine Verbindung, welche von irgend einer geeigneten Quelle, ob natürlich oder
nicht, erhältlich
ist oder daraus gebildet werden kann.
-
Der
Wirkstoff kann aus einer Bibliothek von Verbindungen, welche Peptide
sowie andere Verbindungen, beispielsweise kleine organische Moleküle wie Leitverbindungen
umfassen kann, erstellt oder erhalten werden.
-
Beispielsweise
kann der Wirkstoff eine natürliche
Substanz, ein biologisches Makromolekül oder ein aus biologischem
Material wie Bakterien, Pilzen oder Tierzellen oder -gewebe (insbesondere
von Säugern) hergestellter
Extrakt, ein organisches oder ein anorganisches Molekül, ein synthetischer
Wirkstoff, ein semi-synthetischer Wirkstoff, ein strukturelles oder
funktionelles Mimetikum, ein Peptid, ein Peptidomimetikum, ein derivatisierter
Wirkstoff, ein aus einem ganzen Protein abgespaltetes Peptid oder
ein synthetisch hergestelltes Peptid (beispielsweise unter Verwendung
eines Peptid-Synthesizers oder von rekombinanten Verfahren oder
Kombinationen davon), ein rekombinanter Wirkstoff, ein Antikörper, ein
natürlicher
oder ein nichtnatürlicher
Wirkstoff, ein Fusionsrotein oder ein Äquivalent davon sowie ein Mutant,
Derivat oder Kombination davon sein.
-
Der
hier verwendete Begriff "Wirkstoff" kann eine einzelne
Einheit sein oder er kann eine Kombination von Wirkstoffen sein.
-
Falls
der I:NEP eine organische Verbindung ist, dann kann diese organische
Verbindung bei einigen Anwendungen typischerweise eine Amid-Gruppe
(d.h.-N(H)-C(O)- oder sogar -C(O)-N(H)-) und eine oder mehrere Kohlenwasserstoffgruppen
umfassen. Hier bedeutet der Begriff "Kohlenwasserstoffgruppen" eine mindestens
C und H umfassende Gruppe, die wahlweise einen oder mehrere andere geeignete.
Substituenten umfassen kann. Beispiele von solchen Substituenten
können
Halogen-, Alkoxy-, Nitro-, eine Alkylgruppe, eine cyclische Gruppe
usw. umfassen. Zusätzlich
zur Möglichkeit,
dass die Substituenten eine cyclische Gruppe sein können, kann
eine Kombination von Substituenten eine cyclische Gruppe bilden.
Falls die Kohlenwasserstoffgruppe mehr als ein C umfasst, dann müssen diese
Kohlenstoffe nicht notwendigerweise miteinander verknüpft sein.
Beispielsweise können
mindestens zwei der Kohlenstoffe über ein(e) geeignete(s) Element
oder Gruppe verknüpft
sein. Demnach kann die Kohlenwasserstoffgruppe Heteroatome enthalten.
Geeignete Heteroatome sind einer Fachperson bekannt und umfassen
beispielsweise Schwefel, Stickstoff und Sauerstoff. Für gewisse
Anwendungen umfasst der Wirkstoff bevorzugt mindestens eine cyclische
Gruppe. Für
gewisse Anwendungen umfasst der Wirkstoff bevorzugt mindestens eine
mit einer anderen Kohlenwasserstoffgruppe über eine Amid-Bindung verknüpfte cyclische
Gruppe. Beispiele von solchen Verbindungen sind hier im Abschnitt mit
den Beispielen aufgeführt.
-
Falls
der zusätzliche
Wirkstoff eine organische Verbindung ist, dann kann diese organische
Verbindung bei gewissen Anwendungen – beispielsweise falls der
Wirkstoff ein I:PDE ist – typischerweise
zwei oder mehrere verknüpfte
Kohlenwasserstoffgruppen umfassen. Bei gewissen Anwendungen umfasst
der Wirkstoff bevorzugt zwei cyclische Gruppen – wobei eine dieser cyclischen
Gruppen eine fusionierte cyclische Ringstruktur sein kann. Bei gewissen
Anwendungen ist bevorzugt mindestens eine der cyclischen Gruppen eine
heterocyclische Gruppe. Bei gewissen Anwendungen umfasst die heterocyclische
Gruppe bevorzugt mindestens ein N im Ring.
-
Beispiele
von solchen Verbindungen sind hier im Abschnitt mit den Beispielen
aufgeführt.
-
Falls
der zusätzliche
Wirkstoff eine organische Verbindung ist, dann kann diese organische
Verbindung bei gewissen Anwendungen – beispielsweise falls der
Wirkstoff ein I:NPY ist – typischerweise
zwei oder mehrere verknüpfte
Kohlenwasserstoffgruppen umfassen. Bei gewissen Anwendungen umfasst
der Wirkstoff bevorzugt zwei cyclische Gruppen – wobei eine dieser cyclischen
Gruppen wahlweise eine fusionierte cyclische Ringstruktur sein kann.
Bei gewissen Anwendungen ist mindestens eine der cyclische Gruppen
eine heterocyclische Gruppe. Bei gewissen Anwendungen umfasst die
heterocyclische Gruppe bevorzugt mindestens ein N im Ring. Beispiele
von solchen Verbindungen sind hier im Abschnitt mit den Beispielen
aufgeführt.
-
Der
Wirkstoff kann Halogengruppen enthalten. Hier bedeutet "Halogen" Fluor, Chlor, Brom
oder Iod.
-
Der
Wirkstoff kann eine oder mehrere Alkyl-, Alkoxy-, Alkenyl-, Alkylen-
und Alkenylengruppen enthalten – welche
unverzweigt- oder verzweigtkettig sein können.
-
Der
Wirkstoff kann in der Form eines pharmazeutisch annehmbaren Salzes – wie eines
Säureadditionssalzes
oder eines Basensalzes – oder
eines Solvates davon, einschliesslich eines Hydrates davon vorliegen.
Für eine Übersicht über geeignete
Salze siehe Berge et al, J. Pharm. Sci., 1977, 66, 1–19.
-
Geeignete
Säureadditionssalze
werden aus Säuren
gebildet, welche nicht-toxische Salze bilden, und Beispiele sind
Hydrochlorid-, Hydrobromid-, Hydroiodid-, Sulfat-, Bisulfat-, Nitrat-, Phosphat-,
Hydrogenphosphat-, Acetat-, Maleat-, Fumarat-, Lactat-, Tartrat-,
Citrat-, Gluconat-, Succinat-, Saccharat-, Benzoat-, Methansulfonat-,
Ethansulfonat-, Benzolsulfonat-, p-Toluolsulfonat- und Pamoatsalze.
-
Geeignete
Basensalze werden aus Basen gebildet, welche nicht-toxische Salze
bilden, und Beispiele sind Natrium-, Kalium-, Aluminium-, Kalzium-,
Magnesium-, Zink- und Diethanolaminsalze.
-
Ein
pharmazeutisch annehmbares Salz eines erfindungsgemässen Wirkstoffes
kann ohne weiteres durch entsprechendes Zusammenmischen von Lösungen des
Wirkstoffes und der gewünschten
Säure oder Base
hergestellt werden. Das Salz kann aus der Lösung ausgefällt und durch Filtration gesammelt
werden oder durch Abdampfen des Lösungsmittels wiedergewonnen
werden.
-
Der
Wirkstoff kann in polymorpher Form vorliegen.
-
Der
Wirkstoff kann ein oder mehrer asymmetrische Kohlenstoffatome enthalten
und demnach in zwei oder mehreren stereoisomeren Formen vorliegen.
Wenn ein Wirkstoff eine Alkenyl- oder Alkenylengruppe enthält, kann
auch cis-(E)- und
trans-(Z)-Isomerie vorkommen. Die vorliegende Erfindung umfasst
die einzelnen Stereoisomeren des Wirkstoffes und gegebenenfalls
die einzelnen tautomeren Formen davon, zusammen mit Gemischen davon.
-
Die
Auftrennung von Diastereoisomeren oder cis- und trans-Isomeren kann durch
herkömmliche
Verfahren, z.B. durch fraktionierte Kristallisation, Chromatographie
oder H.P.L.C. eines Stereoisomeren-Gemisches des Wirkstoffes oder
eines geeigneten Salzes oder Derivatives davon erreicht werden.
Ein einzelnes Enantiomer des Wirkstoffs kann auch aus einem entsprechenden
optisch reinen Zwischenprodukt oder durch Auftrennen, beispielsweise
durch H.P.L.C. des entsprechenden Racemates unter Verwendung eines
geeigneten chiralen Trägers
oder durch fraktionierte Kristallisation des diastereoisomeren Salzes,
das durch Umsetzen des entsprechenden Racemates mit einer geeigneten
optisch aktiven Säure
oder Base gebildet wird, hergestellt werden.
-
Die
vorliegende Erfindung umfasst auch alle geeigneten Isotopenvariationen
des Wirkstoffes oder eines pharmazeutisch annehmbaren Salzes davon.
Eine Isotopenvariation eines erfindungsgemässen Wirkstoffes oder eines
pharmazeutisch annehmbaren Salzes davon ist definiert als eine solche,
in welcher mindestens ein Atom ersetzt ist durch ein Atom, welches
dieselbe Atomzahl hat, aber eine Atommasse aufweist, die sich von
der üblicherweise
in der Natur gefundenen Atommasse unterscheidet. Beispiele von Isotopen,
die in den Wirkstoff und die pharmazeutisch annehmbaren Salze davon
eingebaut werden können,
umfassen Isotope von Wasserstoff, Kohlenstoff, Stickstoff, Sauerstoff,
Phosphor, Schwefel, Fluor und Chlor wie 2H, 3H, 13C, 14C, 15N, 17O, 18O, 31P, 32P, 35S, 18F beziehungsweise 36Cl. Gewisse Isotopenvariationen des Wirkstoffes
und der pharmazeutisch annehmbaren Salze davon, beispielsweise diejenigen,
in welchen ein radioaktives Isotop wie 3H oder 14C eingebaut ist, sind für Wirkstoff- und/oder Substrat- Gewebeverteilungsstudien
nützlich.
Tritiierte, d.h. 3H-, und Kohlenstoff-14-,
d.h. 14C-Isotope sind wegen deren Einfachheit
zur Herstellung und Nachweisbarkeit besonders bevorzugt. Ausserdem
kann die Substitution mit Isotopen wie Deuterium, d.h. 2H
gewisse therapeutische Vorteile ergeben, die sich aus einer grösseren metabolischen
Stabilität,
beispielsweise aus einer erhöhten
in vivo-Halbwertszeit oder einem verringerten Dosierungsbedarf ergeben,
und kann demnach in gewissen Fällen
bevorzugt sein. Isotopenvariationen des Wirkstoffes und pharmazeutisch
annehmbare Salze davon können
im Allgemeinen durch konventionelle Verfahren unter Verwendung von
geeigneten Isotopenvariationen von geeigneten Reagenzien hergestellt
werden.
-
Es
wird Fachpersonen bekannt sein, dass der Wirkstoff aus einer Prodrug
abgeleitet werden kann. Beispiele von Prodrugs umfassen Einheiten,
die (eine) gewisse geschützte
Gruppe(n) aufweisen und welche keine pharmakologische Aktivität als solche
besitzen müssen,
aber die in gewissen Fällen
verabreicht werden können
(beispielsweise oral oder parenteral) und danach im Körper unter
Bildung des pharmakologisch aktiven Wirkstoffes metabolisiert werden.
-
Es
ist weiterhin bekannt, dass gewisse Anteile, bekannt als "Pro-Anteile", an geeignete Funktionalitäten der
Wirkstoffe angebracht werden können,
beispielsweise wie in "Design
of Prodrugs" by
H. Bundgaard, Elsevier, 1985 (dessen Inhalt hiermit durch Bezugnahme übernommen
wird) beschrieben. Solche Prodrugs gehören ebenfalls zum Umfang der
vorliegenden Erfindung.
-
Der
PcAMP kann eines oder mehrere der Folgenden
bewirken: direkte oder indirekte Erhöhung der Wirksamkeit von cAMP,
direkte oder indirekte Erhöhung
des Spiegels von cAMP, direkte oder indirekte Erhöhung der
Aktivität
von cAMP, direkte oder indirekte Verringerung des Ausmasses des
cAMP-Abbaus, direkte
oder indirekte Erhöhung
des Spiegels von cAMP, direkte oder indirekte Verringerung des Ausmasses
der cAMP-Inhibition.
-
Der
erfindungsgemässe
Wirkstoff (I:NEP) führt
vorzugsweise zu einer direkten oder indirekten Erhöhung des
cAMP-Spiegels in den sexuellen Genitalien einer an FSAD leidenden
Frau.
-
Der
erfindungsgemässe
Wirkstoff (I:NEP) führt
besonders bevorzugt zu einer direkten oder indirekten selektiven
Erhöhung
des cAMP-Spiegels in den sexuellen Genitalien einer an FSAD leidenden
Frau.
-
Der
erfindungsgemässe
Wirkstoff (d.h. I:NEP) führt
besonders bevorzugt zu einer direkten oder indirekten selektiven
Erhöhung
des cAMP-Spiegels, wobei besagtes cAMP durch sexuelle Erregung induziertes cAMP
ist.
-
In
einem besonders bevorzugten Aspekt erhöht der erfindungsgemässe Wirkstoff
(d.h. I:NEP) die relative Menge des durch sexuelle Erregung induzierte
cAMP.
-
Bei
gewissen Anwendungen führt
der erfindungsgemässe
Wirkstoff (d.h. I:NEP) zu einer selektiven Behandlung von FSAD.
-
In
einem Aspekt kann der Wirkstoff ein geeignetes Target inhibieren
oder antagonisieren und dabei die cAMP-Spiegel in den weiblichen
sexuellen Genitalien potenzieren. Im Text wird der Begriff Inhibitor
als Bezeichnung für
einen Inhibitor und/oder Antagonisten verwendet.
-
In
einem anderen Aspekt kann der Wirkstoff ein geeignetes Target aktivieren
oder agonisieren und dabei die cAMP-Spiegel in den weiblichen sexuellen
Genitalien potenzieren. Im Text werden die Begriffe Aktivator und
auf regulierender Inhibitor als Bezeichnung für einen Aktivator und/oder
Aufregulierer und/oder Agonisten verwendet.
-
Demnach
kann der Wirkstoff ein geeignetes Target agonisieren, antagonisieren,
auf regulieren, oder inhibieren.
-
Der
erfindungsgemässe
Wirkstoff (d.h. I:NEP) kann eine einzelne Einheit sein, die befähigt ist,
zwei oder mehrere dieser Eigenschaften zu entfalten. Alternativ
oder zusätzlich
kann der erfindungsgemässe
Wirkstoff (d.h. I:NEP) eine Kombination vom Wirkstoffen sein, die
befähigt
sind, zwei oder mehrere dieser Eigenschaften zu entfalten.
-
Vorzugsweise
kann der Wirkstoff ein geeignetes Target selektiv agonisieren, selektiv
antagonisieren, selektiv aufregulieren oder selektiv inhibieren.
-
Der
Wirkstoff kann auch befähig
sein, eine oder mehrere anderen vorteilhafte funktionelle Eigenschaften
zu zeigen. Beispielsweise kann der erfindungsgemässe Wirkstoff (d.h. I:NEP)
sowohl cAMP als auch cGMP potenzieren.
-
Für gewisse
Anwendungen (wie eine topischen Anwendung) kann der Wirkstoff auch
eine ACE-(Angiotensin-konvertierendes-Enzym)-Inhibitionswirkung zeigen. Ein
ACE-Assay ist hier im experimentellen Abschnitt vorgestellt. Für gewisse
Anwendungen (wie bei besonderen Patientenarten), sind solche Wirkstoffe (d.h.
diejenigen, die auch ACE-Inhibitionswirkung zeigen) nicht für die orale
Verabreichung geeignet.
-
Für gewisse
Anwendungen kann der Wirkstoff auch eine ECE-(Endothel-konvertierendes-Enzym)-Inhibitionswirkung
zeigen. ECE-Assays sind im Fachgebiet wohlbekannt.
-
PHARMAZEUTISCHE
KOMBINATIONEN
-
Der
erfindungsgemässe
Wirkstoff (d.h. I:NEP) kann in Kombination mit einem oder mehreren
anderen pharmazeutisch aktiven Wirkstoffen, beispielsweise mit einem
PcGMP (wie einem Phosphodiesterase-Typ-5-Inhibitor,
z.B. Sildenafil, oder einem Stickstoffmonoxid-Donor, oder einem
Stickstoffmonoxid-Vorläufer
z.B. L-Arginin oder Inhibitoren der Arginase) und/oder einem zentral
wirkenden Pharmazeutikum (z.B. mit einem Dopaminrezeptoragonisten
wie Apomorphin oder einem selektiven Dopamin-D2-Rezeptoragonisten wie PNU-95666 oder
einem Melanocortin-Rezeptoragonist
wie Melanotan II) verwendet werden. Lehren über die Verwendung von Apomorphinen
als Pharmazeutikum finden sich in US-A-5945117. In jenem Dokument
wird Apomorphin sublingual verabreicht. Zusätzlich oder alternativ kann
der Wirkstoff zusammen mit einem oder mehreren aus: einem PDE5-Inhibitor
(z.B. Sildenafil, Vardenafil (Bayer BA 38-9456) und IC351 (Cialis,
Icos Lilly)), einem oder mehreren Stickstoffmonoxid-Donoren (z.B. NMI-921),
einem oder mehreren Dopamin-Rezeptoragonisten (z.B. Apomorphin,
Uprima, Ixsene), einem oder mehreren heterocyclischen Aminen wie
dies allgemein und spezifisch in WO 00/40226, insbesondere in den
Beispielen Nummer 7, 8 und 9 beschrieben ist, einem oder mehreren
Melanocortin-Rezeptoragonisten
(z.B. Melanotan II oder PT14), einem oder mehreren Kaliumkanalöffner (z.B.
ein KATP-Kanalöffner (z.B. Minoxidil, Nicorandil)
und/oder einem Kalzium-aktivierten Kaliumkanalöffner (z.B. BMS-204352), einem
oder mehreren α1-Adrenozeptor-Antagonisten
(z.B. Phentolamin, Vasofem, Vasomax), einem oder mehreren VIP-Rezeptoragonisten
oder einem VIP-Analogon (z.B. Ro-125-1553) oder einem VIP-Fragment, einem oder
mehreren α-Adrenozeptor-Antagonisten
in Kombination mit VIP (z.B. Invicorp, Aviptadil), einem oder mehreren α2-Adrenozeptor-Antagonisten
(z.B. Yohimbine), einem oder mehreren Östrogenen, Östrogen und Medroxyprogesteron
oder Medroxyprogesteronacetat (MPA) oder Östrogen- und Methyltestosteron-Hormonersatztherapie-Wirkstoffen (z.B.
HRT, insbesondere Premarin, Cenestin, Oestrofeminal, Equin, Estrace,
Estrofem, Elleste Solo, Estring, Eastraderm, Eastraderm TTS, Eastraderm
Matrix, Dermestril, Premphase, Prempro, Prempak, Premique, Estratest,
Estratest HS, Tibolon), einem oder mehreren Testosteronersatz-Wirkstoffen
(einschl. DHEA (Dehydroandrostendion), Testosteron (Tostrelle) oder
einem Testosteronimplantat (Organon)), einem oder mehreren Testosteron/Öestradiol-Wirkstoffen,
einem oder mehreren Östrogenagonisten,
z.B. Lasofoxifen, einem oder mehreren Serotonin-Rezeptoragonisten
oder -antagonisten (z.B. 5HT1A-, 5HT2C-, 5HT2A- und 5HT3-Rezeptoragonisten
und -antagonisten; wie in WO2000/28993 beschrieben), einem oder
mehreren Prostanoid-Rezeptoragonisten (z.B. Muse, Alprostadil, Misoprostol),
einem oder mehreren Purinerg-Rezeptoragonisten (insbesondere P2Y2
und P2Y4) einem oder mehreren Antidepressiva (z.B. Bupropion (Wellbutrin),
Mirrtazapin, Nefazodon) verwendet werden.
-
Die
Struktur von IC351 ist:
-
-
Falls
eine Kombination von aktiven Wirkstoffen verabreicht wird, dann
können
diese gleichzeitig, getrennt oder nacheinander verabreicht werden.
-
VIP-KOMBINATION
-
Gemäss der vorliegenden
Erfindung ist der Wirkstoff bevorzugt nicht VIP (oder vorzugsweise
nicht ein Analogon davon oder nicht ein Fragment davon). Allerdings
kann der erfindungsgemässe
Wirkstoff (d.h. I:NEP) in gewissen Ausführungsformen zusammen mit einem
VIP oder einem Analogon davon oder einem Fragment davon verabreicht
werden.
-
In
einem besonders bevorzugten Aspekt wird kein VIP oder Analogon davon
oder Fragment davon verabreicht. Der Grund hierfür liegt darin, dass gemäss einem
Bericht VIP-Infusionen
zu signifikanten kardiovaskulären
Nebenwirkungen wie einem Anstieg der Herzfrequenz und einer Abnahme
des diastolischen arteriellen Blutdruckes führen (Ottesen 1983, 1987, 1995).
-
Ausserdem,
und trotz der Tatsache, dass Ottesen und Mitarbeiter gezeigt haben,
dass VIP bei gesunden Freiwilligen eine Erhöhung des vaginalen Blutflusses
und der Lubrikation induziert, sind die Mechanismen, durch die VIP
seine Wirkung entfaltet, unklar. In der Literatur gibt es eine Anzahl
von Beispielen wonach VIP über
verschiedene Second-Messenger-Systeme, z.B. cGMP/Guanylatcyclase
(Ashur-Fabian, 1999);
Kohlenstoffmonoxid-(CO)/Häm-Oxygenase
(Fan et al., 1998) und cAMP/Adenylatcyclase (Foda, 1995; Schoeffter, 1985;
Gu, 1992) signalisiert. Ein Beispiel hierfür findet sich in einem neueren
Bericht, der aufzeigt, wie die vasorelaxierenden Wirkungen von VIP
in der Gebärmutterarterie
durch die Freisetzung von Stickstoffmonoxid erklärt werden kann (Jovanovic,
1998). Weiterhin bestehen auch Hinweise, dass VIP bei der männlichen
urogenitalen Funktion Stickstoffmonoxid (NO)/cGMP moduliert (Kim,
1994).
-
Ausserdem
wurde in der Literatur berichtet, dass VIP in Kulturen von vaginalen
glatten Muskelzellen keine Wirkung auf die cAMP-Spiegel hat (siehe
Traish, A., Moreland, R.B., Huang, Y., et al. (1999). Development
of human und rabbit vaginal smooth muscle cell cultures: Effects
of vasoactive agents on intracellular level of cyclic nucleotides.
Mol. Cell Biol. Res. Comm., 2, 131–137).
-
Ausserdem
berichteten Ottesen und Mitarbeiter (siehe Palle, Bredkjaer, Ottesen
und Fahrenkrug 1990 Clinical und Experimental Pharmacology und Physiology
vol 17 61–68)
in Follow-up-Studien, dass die Wirkung von VIP auf den vaginalen
Blutfluss unabhängig
vom Verabreichungsweg Teil einer systemischen vasodilatorischen
Wirkung und nicht einer lokalen Antwort ist. Ausserdem berichteten
sie über
eine Anzahl von mit VIP zusammenhängenden vaskulären Nebenwirkungen – beispielsweise
Gesichtrötung,
Hypertonie und Tachykardie.
-
Ki -
WERTE
-
Für gewisse
Anwendungen hat der erfindungsgemässe Wirkstoff (und wahlweise
der mögliche
zusätzliche
Wirkstoff) vorzugsweise einen Ki-Wert von
weniger als ungefähr
100 nM, vorzugsweise weniger als ungefähr 75 nM, vorzugsweise weniger
als ungefähr
50 nM, vorzugsweise weniger als ungefähr 25 nM, vorzugsweise weniger
als ungefähr
20 nM, vorzugsweise weniger als ungefähr 15 nM, vorzugsweise weniger
als ungefähr
10 nM, vorzugsweise weniger als ungefähr 5 nM.
-
Kb -
WERTE
-
Für gewisse
Anwendungen hat der erfindungsgemässe Wirkstoff (d.h. I:NEP)
(und wahlweise der mögliche
zusätzliche
Wirkstoff) einen Kb-Wert von weniger als
ungefähr
100 nM, vorzugsweise weniger als ungefähr 75 nM, vorzugsweise weniger
als ungefähr
50 nM, vorzugsweise weniger als ungefähr 25 nM, vorzugsweise weniger
als ungefähr
20 nM, vorzugsweise weniger als ungefähr 15 nM, vorzugsweise weniger
als ungefähr
10 nM, vorzugsweise weniger als ungefähr 5 nM.
-
Ka -
WERTE
-
Für gewisse
Anwendungen hat der erfindungsgemässe Wirkstoff (d.h. I:NEP)
(und wahlweise der mögliche
zusätzliche
Wirkstoff) einen Ka-Wert von weniger als
ungefähr
100 nM, vorzugsweise weniger als ungefähr 75 nM, vorzugsweise weniger
als ungefähr
50 nM, vorzugsweise weniger als ungefähr 25 nM, vorzugsweise weniger
als ungefähr
20 nM, vorzugsweise weniger als ungefähr 15 nM, vorzugsweise weniger
als ungefähr
10 nM, vorzugsweise weniger als ungefähr 5 nM.
-
PHARMAKOKINETIK
-
Bei
einigen Ausgestaltungen der vorliegenden Erfindung haben die erfindungsgemässen Wirkstoffe (d.h.
I:NEP) (und wahlweise der mögliche
zusätzliche
Wirkstoff) vorzugsweise einen log D von –2 bis +4, besonders bevorzugt
von –1
bis +2. Der log D kann mittels im Fachgebiet bekannten Standardverfahren,
wie in J. Pharm. Pharmacol. 1990, 42:144 beschrieben, bestimmt werden.
-
Zusätzlich oder
alternativ haben die erfindungsgemässen Wirkstoffe (d.h. I:NEP)
in gewissen Ausgestaltungen (und wahlweise der mögliche zusätzliche Wirkstoff) vorzugsweise
einen caco-2-Fluss grösser
als 2 × 10-6 cms-1, besonders
bevorzugt grösser
als 5 × 10-6 cms-1. Der Wert
des caco-2-Flusses
kann mittels im Fachgebiet bekannten Standardverfahren, wie in J.
Pharm. Sci 79, 7, S. 595–600 (1990),
und Pharm. Res. vol 14, no. 6 (1997) beschrieben, bestimmt werden.
-
SELEKTIVITÄT
-
Für gewisse
Anwendungen hat der erfindungsgemässe Wirkstoff (d.h. I:NEP)
(und wahlweise der mögliche
zusätzliche
Wirkstoff) vorzugsweise eine mindestens ungefähr 100-fache Selektivität zum gewünschten
Target, vorzugsweise eine mindestens ungefähr 150-fache Selektivität zum gewünschten
Target, vorzugsweise eine mindestens ungefähr 200-fache Selektivität zum gewünschten
Target, vorzugsweise eine mindestens ungefähr 250-fache Selektivität zum gewünschten
Target, vorzugsweise eine mindestens ungefähr 300-fache Selektivität zum gewünschten
Target, vorzugsweise eine mindestens ungefähr 350-fache Selektivität zum gewünschten
Target.
-
Für gewisse
Anwendungen hat der erfindungsgemässe Wirkstoff (d.h. I:NEP)
(und wahlweise der mögliche
zusätzliche
Wirkstoff) vorzugsweise eine mindestens ungefähr 400-fache Selektivität zum gewünschten
Target, vorzugsweise eine mindestens ungefähr 500-fache Selektivität zum gewünschten
Target, vorzugsweise eine mindestens ungefähr 600-fache Selektivität zum gewünschten
Target, vorzugsweise eine mindestens ungefähr 700-fache Selektivität zum gewünschten
Target, vorzugsweise eine mindestens ungefähr 800-fache Selektivität zum gewünschten
Target, vorzugsweise eine mindestens ungefähr 900-fache Selektivität zum gewünschten
Target, vorzugsweise eine mindestens ungefähr 1000-fache Selektivität zum gewünschten
Target.
-
CHEMISCHE
SYNTHESEVERFAHREN
-
Typischerweise
wird der Wirkstoff durch chemische Syntheseverfahren hergestellt
werden.
-
Der
Wirkstoff oder das Target oder Varianten, Homologa, Derivate, Fragmente
oder Mimetika davon können
unter Verwendung von chemischen Verfahren zur Synthese des Wirkstoffes
als ganzes oder als Teil hergestellt werden. Beispielsweise können Peptide
durch Festphasenverfahren synthetisiert, vom Harz abgespaltet und
mittels präparativer
Hochleistungs-Flüssig-Chromatographie
gereinigt werden (z.B. Creighton (1983) Proteins Structures und
Molecular Principles, WH Freeman und Co, New York NY). Die Zusammensetzung
der synthetischen Peptide kann mittels Aminosäure-Analyse oder Sequenzierung
bestätigt
werden (z.B. Edman-Abbauverfahren; Creighton, oben).
-
Die
direkte Synthese des Wirkstoffes oder von Varianten, Homologa, Derivaten,
Fragmenten oder Mimetika davon kann unter Verwendung von verschiedenen
Festphasen-Verfahren durchgeführt
werden (Roberge JY et al (1995) Science 269: 202–204), und die automatisierte
Synthese kann beispielsweise unter Verwendung des ABI 43 1 A Peptide-Synthesizer (Perkin
Elmer) entsprechend den vom Hersteller gelieferten Anleitungen erreicht
werden. Ausserdem können
die den Wirkstoff oder irgend einen Teil desselben umfassenden Aminosäurensequenzen
während
der direkten Synthese verändert
und/oder unter Verwendung von chemischen Verfahren mit einer Sequenz
von anderen Untereinheiten oder irgend einem Teil davon kombiniert
werden, woraus sich eine Wirkstoff- oder Targetvariante wie beispielsweise
eine NEP-Variante
ergibt.
-
In
einer alternativen Ausgestaltung der Erfindung kann die kodierende
Sequenz des Wirkstoff-Targets oder von Varianten, Homologa, Derivaten,
Fragmenten oder Mimetika davon als ganzes oder als Teil unter Verwendung
von im Fachgebiet wohlbekannten chemischen Verfahren synthetisiert
werden (siehe Caruthers MH et al (1980) Nuc Acids Res Symp Ser 215-23, Horn T et al
(1980) Nuc Acids Res Symp Ser 225–232).
-
MIMETIKUM
-
Der
hier verwendete Begriff "Mimetikum" bezieht sich auf
irgend eine chemische Verbindung, was ohne darauf beschränkt zu sein
ein Peptid, ein Polypeptid, einen Antikörper oder eine andere organische
chemische Verbindung umfasst, welche dieselbe qualitative Aktivität oder Wirkung
am Target haben wie ein Referenzwirkstoff.
-
CHEMISCHE
DERIVATE
-
Der
hier verwendete Begriff "Derivat" oder "derivatisiert" umfasst chemische
Modifikationen eines Wirkstoffes. Ein Beispiel für solche chemischen Modifikationen
wäre das
Ersetzen von Wasserstoff durch einen Halogengruppe, eine Alkylgruppe,
eine Acylgruppe oder eine Aminogruppe.
-
CHEMISCHE
MODIFIKATION
-
In
einer Ausgestaltung der vorliegenden Erfindung kann der Wirkstoff
ein chemisch modifizierter Wirkstoff sein.
-
Die
chemische Modifikation eines Wirkstoffes kann die Wasserstoffbindungs-Interaktion,
die Ladungs-Interaktion, die hydrophobe Interaktion, die Van Der
Waals Interaktion oder die Dipol-Interaktion zwischen dem Wirkstoff
und dem Target entweder verstärken
oder reduzieren.
-
In
einem der Aspekte kann der identifizierte Wirkstoff als Modell (beispielsweise
als Matrize) für
die Entwicklung von anderen Verbindungen wirken.
-
REKOMBINANTE VERFAHREN
-
Typischerweise
kann das im Assay verwendete Target durch rekombinante DNS-Verfahren
hergestellt werden.
-
POTENZIERUNG
VON cGMP
-
Der
hier unter Bezugnahme auf cGMP verwendete Begriff "Potenzierung" umfasst irgend eines
oder mehrere von: Erhöhung
der Wirksamkeit von cGMP, Erhöhung
des Spiegels von cGMP, Erhöhung
der Aktivität von
cGMP, Verringerung des Ausmasses des cGMP-Abbaus, Verringerung des
Ausmasses der cGMP-Inhibition.
-
Die
potenzierende Wirkung kann eine direkte Wirkung sein. Alternativ
kann sie eine sekundäre
Wirkung und/oder eine Wirkung stromabwärts sein.
-
Hier
wirkt der Wirkstoff, der cGMP potenziert, vorzugsweise auf ein IcGMP und/oder ein AMcGMP,
wobei der Modulator von cGMP eine schädliche Wirkung auf cGMP hat,
so dass der Wirkstoff die schädliche
Wirkung auf das IcGMP und/oder das AMcGMP gegenüber cGMP reduziert und/oder
eliminiert und/oder maskiert und/oder abhält.
-
Demnach
umfasst die vorliegende Erfindung eine Kombination von einem oder
mehreren I:IcAMP und einem oder mehreren
I:IcGMP. In einem Aspekt ist das I:IcGMP ein I:PDEcGMP.
-
IcAMP UND/ODER
AMcAMP
-
Es
wurde gezeigt, dass cAMP den genitalen (z.B. vaginalen oder klitoralen)
Blutfluss vermittelt, und durch Erhöhung der cAMP-Signalisierung
kann im Tiermodell der genitale (z.B. vaginale oder klitorale) Blutfluss
erhöht
werden. Demnach wird ein Wirkstoff, welcher die cAMP-vermittelte
Vasorelaxation auf reguliert/erhöht,
bei der Behandlung von FSAD wirksam sein. Zur Vereinfachung werden
diese Substanzen als Inhibitoren von IcAMP und/oder
von AMcAMP bezeichnet. Hier haben das IcAMP und das AMcAMP eine
schädliche
Wirkung auf die cAMP-Spiegel oder -Aktivität.
-
Demnach
kann der Wirkstoff eines oder mehrere sein von: ein I:IcAMP und/oder
ein I:AMcAMP.
-
Der
Wirkstoff kann eine einzelne Einheit sein, die befähigt ist,
zwei oder mehrere dieser Eigenschaften zu entfalten. Alternativ
oder zusätzlich
kann der Wirkstoff eine Kombination von Wirkstoffen sein, die befähigt sind,
eine oder mehrere dieser Eigenschaften zu entfalten.
-
Beispiele
von IcAMP und AMcAMP umfassen
NEP und einer oder mehrere PDE(s) oder irgend eine assoziierte Komponente.
Die assoziierte Komponente kann beispielsweise ein Rezeptor und/oder
eim Kofaktor sein.
-
Demnach
kann der erfindungsgemässe
Wirkstoff (d.h. I:NEP) in Kombination mit einem oder mehreren von:
einem I:PDEcAMP, einem I:NPY (gelegentlich
als NPYi bezeichnet), ein I:NPY Yn (gelegentlich
als NPY Yni bezeichnet) verwendet werden.
-
Ähnlicherweise
kann der Wirkstoff eine einzelne Einheit sein, die befähigt ist,
zwei oder mehrere dieser Eigenschaften zu entfalten. Alternativ
oder zusätzlich
kann der Wirkstoff eine Kombination von Wirkstoffen sein, die befähigt sind,
eine oder mehrere dieser Eigenschaften zu entfalten.
-
I:IcAMP UND/ODER
I:AMcAMP
-
In Übereinstimmung
mit der vorliegenden Erfindung wurde gefunden, dass es möglich ist,
FSAD unter Verwendung eines Wirkstoffes, welcher die schädliche Wirkung
auf das IcAMP und/oder das AMcAMP gegenüber cAMP
reduziert und/oder eliminiert und/oder maskiert und/oder abhält und/oder
verhindert, zu behandeln und/oder zu verhindern. Der Wirkstoff kann
sogar die cAMP-Spiegel wiederherstellen, die durch ein IcAMP und/oder ein AMcAMP verringert
wurden. Der Einfachheit halber werden diese Substanzen als I:IcAMP und/oder als I:AMcAMP bezeichnet.
Hier verhindert oder verringert das I:IcAMP und
das I:AMcAMP die schädliche Wirkung auf die cAMP-Spiegel
oder -Aktivität.
-
Demnach
ist der Wirkstoff in einem bevorzugten Aspekt ein I:IcAMP und/oder
ein I:AMcAMP, wobei das AMcAMP eine
schädliche
Wirkung auf AMcAMP hat.
-
AcAMP
-
In Übereinstimmung
mit der vorliegenden Erfindung wurde gefunden, dass eine der wichtigen
Ursachen von FSAD niederige Spiegel oder eine niederige Aktivität von cAMP
in den weiblichen Genitalien sind.
-
Demnach
kann der Wirkstoff ein U:AcAMP sein.
-
Demnach
sollte der erfindungsgemässe
Wirkstoff (d.h. I:NEP) vorzugsweise auch befähigt sein, als A:AC, A:VIPr,
A:VIPn, I:I:VIPr oder I:IvIPn zu
wirken und/oder zusammen mit irgend einem oder mehreren derselben
verwendet zu werden.
-
Der
Wirkstoff kann eine einzelne Einheit sein, die befähigt ist,
zwei oder mehrere dieser Eigenschaften zu entfalten. Alternativ
oder zusätzlich
kann der Wirkstoff eine Kombination von Wirkstoffen sein, die befähigt sind,
eine oder mehrere dieser Eigenschaften zu entfalten.
-
U:AcAMP
-
In
einem anderen Aspekt kann ein zusätzliches Target eine Komponente
sein, welche den cAMP-Spiegel erhöht. Demnach kann der Wirkstoff
auch als ein U:AC wirken.
-
Demnach
kann der erfindungsgemässe
Wirkstoff (d.h. I:NEP) beispielsweise auch befähigt sein, als U:AcAMP,
ein A:AC, ein A:VIPr, ein A:VIPn, ein I:I:VIPr oder ein I:I:VIPn zu wirken und/oder zusammen mit irgend einem
oder mehreren derselben verwendet zu werden.
-
Beispielsweise
könnte
das Target cAMP selber oder AC oder VIP (oder Kombinationen davon)
sein.
-
KOMBINATION von I:IcAMP UND/ODER I:McAMP AND/OR
U:AcAMP
-
In
einem anderen Aspekt kann der erfindungsgemässe Wirkstoff (d.h. I:NEP)
zusammen mit cAMP-Potenziatoren verwendet werden. Beispielsweise
kann der erfindungsgemässe
Wirkstoff (d.h. I:NEP) zusammen mit einem oder mehreren von:
- I:PDEcAMP
- I:PDEncAMP
- I:NPY
- I:NPY Yn
- I:NEP
- U:AcAMP
- A:AC
- A:VIPr
- A:VIPn
- I:I:VIPr
- I:I:VIPn
- CAMP-Mimetikum
- verwendet werden.
-
INHIBITOREN
-
Der
hier im Zusammenhang mit dem erfindungsgemässen Wirkstoff (d.h. I:NEP)
verwendete Begriff "Inhibitor" bedeutet einen Wirkstoff,
welcher die schädliche
Wirkung von einem IcAMP und/oder einem schädliche McAMP gegenüber cAMP reduzieren und/oder
eliminieren und/oder maskieren und/oder verhindern kann.
-
AKTIVATOR
-
Der
hier im Zusammenhang mit dem erfindungsgemässen Wirkstoff (d.h. I:NEP)
verwendete Begriff "Aktivator" bedeutet einen Wirkstoff,
welcher cAMP und/oder AcAMP erhöhen
und/oder bilden und/oder demaskieren kann und/oder die Wirkung von
cAMP und/oder eines AcAMP verstärken
oder sicherstellen kann. Der Aktiviator kann als ein Agonist wirken.
-
ANDERE AKTIVEN
KOMPONENTEN
-
In
einem anderen Aspekt kann der erfindungsgemässe Wirkstoff sogar zusammen
mit einer oder mehreren anderen aktiven Komponenten – beispielsweise
mit einem oder mehreren Wirkstoffen, die zur Potenzierung von cGMP
befähigt
sind – vorliegen.
-
AMINOSÄURENSEQUENZ
-
Der
hier verwendete Begriff "Aminosäurensequenz" ist gleichbedeutend
mit dem Begriff "Polypeptid" und/oder dem Begriff "Protein". In gewisse Fällen ist
der Begriff "Aminosäurensequenz" gleichbedeutend
mit dem Begriff "Peptid". In gewisse Fällen ist
der Begriff "Aminosäurensequenz" gleichbedeutend
mit dem Begriff "Protein".
-
Die
Aminosäurensequenz
kann durch Isolieren aus einer geeigneten Quelle hergestellt werden,
oder sie kann synthetisch hergestellt werden, oder sie kann unter
Verwendung von rekombinanten DNS-Verfahren hergestellt werden.
-
Ferner
wird eine Aminosäurensequenz
beschrieben, die befähigt
ist, in einem Assay als Target zu dienen, wobei das Assay zur Identifikation
von einem oder mehreren Wirkstoffen und/oder Derivaten davon vorgesehen
ist, welche die Aminosäurensequenz
dahingehend beeinflussen können,
dass das cAMP zwecks Behandlung von FSAD potenziert wird,
-
NUKLEOTID-SEQUENZ
-
Der
hier verwendete Begriff "Nukleotid-Sequenz" ist gleichbedeutend
mit dem Begriff "Polynukleotid".
-
Die
Nukleotid-Sequenz kann DNS oder RNS von genomischem oder synthetischem
oder rekombinantem Ursprung sein. Die Nukleotid-Sequenz kann doppelsträngig oder
einzelsträngig
sein, unabhängig
davon, ob sie den Sense- oder Antisense-Strang oder Kombinationen davon repräsentiert.
-
Für gewisse
Anwendungen ist die Nukleotid-Sequenz vorzugsweise DNS.
-
Für gewisse
Anwendungen wird die Nukleotid-Sequenz vorzugsweise unter Verwendung
von rekombinanten DNS-Verfahren
(z.B. rekombinanter DNS) hergestellt.
-
Für gewisse
Anwendungen ist die Nukleotid-Sequenz vorzugsweise cDNS.
-
Für gewisse
Anwendungen kann die Nukleotid-Sequenz vorzugsweise dieselbe sein
wie die natürlich vorkommende
Form für
diesen Aspekt.
-
Ausserdem
wird eine Nukleotid-Sequenz bereitgestellt, die für eine Substanz
kodiert, welche befähigt ist,
in einem Assay (beispielsweise in einem Hefe-Zwei-Hybrid-Assay)
als Target zu dienen, wobei das Assay zur Identifikation von einem
oder mehreren Wirkstoffen und/oder Derivaten davon vorgesehen ist,
welche zwecks Behandlung von FSAD die Aminosäurensequenz dahingehend beeinflussen
können,
dass das cAMP potenziert wird.
-
Eine
Fachperson wird verstehen, dass infolge der Entartung des genetischen
Codes zahlreiche unterschiedliche Nukleotid-Sequenzen für die Targets kodieren können. Ferner
versteht sich, dass Fachpersonen unter Verwendung von Routineverfahren
Nukleotid-Substitutionen vornehmen können, welche – ohne die durch
die erfindungsgemässe
Nukleotid-Sequenz
kodierte Aktivität
wesentlich zu beeinflussen – die
Codon-Belegung von jedem besonderen Wirtsorganismus reflektieren,
in welchem das Target exprimiert werden soll. Demnach umfassen die
Begriffe "Variante", "Homolog" oder "Derivat" im Zusammenhang
mit der in den beigefügten
Nukleotid-Sequenzprotokollen angegebenen Nukleotid-Sequenz jegliche
Substitution, Variation, Modifikation, Ersetzung, Deletion oder
Addition von einer oder mehreren Nukleinsäuren aus oder in die Sequenz,
sofern die resultierende Nukleotid-Sequenz für ein funktionelles Target
gemäss
der vorliegenden Erfindung kodiert (oder sogar für einen erfindungsgemässen Wirkstoff
(d.h. I:NEP), falls besagter Wirkstoff eine Nukleotid-Sequenz oder
eine Aminosäurensequenz
umfasst).
-
Wie
oben im Zusammenhang mit der Sequenzhomologie aufgezeigt, besteht
bevorzugt eine mindestens 75%-ige, noch bevorzugter mindestens 85%-ige,
noch bevorzugter mindestens 90%-ige Homologie zu den im beiliegenden
Sequenzprotokoll angegebenen Sequenzen. Besonders bevorzugt besteht
eine mindestens 95%-ige, noch bevorzugter eine mindestens 98%-ige
Homologie. Nukleotid-Homologie-Vergleiche können wie oben beschrieben durchgeführt werden.
Ein bevorzugtes Sequenz-Vergleichs-Programm
ist das oben beschriebene GCG-Wisconsin-Bestfit-Programm. Die vorgegebene Treffermatrix
hat einen Übereinstimmungswert
von 10 für
jedes identische Nukleotid und –9
für jede
Nicht-Übereinstimmung.
Der vorgegebene Abzug für
eine Lückenbildung
ist –50
und der vorgegebene Abzug für
eine Lückenausdehnung
ist –3
für jedes Nukleotid.
-
Ausserdem
werden Nukleotid-Sequenzen beschrieben, die fähig sind, selektiv zu den hier
angegebenen Sequenzen oder zu irgend einer Variante, einem Fragment
oder Derivat davon oder zum Komplement eines der obigen zu hybridisieren.
Nukleotid-Sequenzen haben bevorzugt eine Länge von mindestens 15 Nukleotiden,
besonders bevorzugt eine Länge
von mindestens 20, 30, 40 oder 50 Nukleotiden. Diese Sequenzen können als
Sonden, beispielsweise in einem diagnostischen Kit, verwendet werden.
-
VARIANTEN/HOMOLOGA/DERIVATE
-
Zusätzlich zu
den hier erwähnten
spezifischen Aminosäurensequenzen
und Nukleotid-Sequenzen umfasst die vorliegende Erfindung auch die
Verwendung von Varianten, Homologa und Derivaten davon. Hier kann
der Begriff "Homologie" mit "Identität" gleichbedeutend
sein.
-
Im
vorliegenden Zusammenhang wird davon ausgegangen, dass eine homologe
Sequenz eine Aminosäurensequenz
enthält,
die zu mindestens 75, 85 oder 90% identisch, vorzugsweise zu mindestens
95 oder 98% identisch ist. Inbesondere sollte Homolgie typischerweise
im Bezug auf diejeinigen Regionen der Sequenz betrachtet werden,
welche erwiesenermassen für
eine Aktivität
essenziell sind. Obwohl Homologie auch in Bezug auf die Ähnlichkeit
(d.h. Aminosäurenreste
mit ähnlichen
chemischen Eigenschaften/Funktionen) betrachtet werden kann, wird
Homologie im Zusammenhang mit der vorliegenden Erfindung vorzugsweise
in Bezug auf Sequenzidentität
auszudrücken.
-
Homologie-Vergleiche
können
von Auge oder, was noch üblicher
ist, mit Hilfe von ohne weiteres verfügbaren Sequenz-Vergleichsprogrammen
ausgeführt
werden. Diese kommerziell verfügbaren
Computerprogramme können
die % Homolgie zwischen zwei oder mehreren Sequenzen berechnen.
-
Die
%-Homolgie kann über
benachbarte Sequenzen berechnet werden, d.h. die eine Sequenz wird
mit der anderen Sequenz ausgerichtet, und jede Aminosäure in der
einen Sequenz wird direkt mit der entsprechenden Aminosäure in der
anderen Sequenz verglichen, jeweils ein Rest aufs Mal. Dies wird
als ein "lückenloses" Ausrichten bezeichnet.
Typischerweise werden solche lückenlosen
Ausrichtungen nur über
eine relativ kurze Anzahl von Resten durchgeführt.
-
Obwohl
es sich um ein sehr einfaches und konsistentes Verfahren handelt,
berücksichtigt
dieses nicht, dass beispielsweise in einem ansonsten identischen
Paar von Sequenzen eine Insertion oder Deletion den nachfolgenden
Aminosäurenrest
aus der Reihe bringt, was zu einer grossen Reduktion in der % Homolgie
führen
kann, wenn eine globale Ausrichtung vorgenommen wird.
-
Demzufolge
sind die meisten Sequenz-Vergleichsverfahren dazu ausgelegt, optimale
Ausrichtungen zu bilden, welche mögliche Insertionen und Deletionen
ohne ungebührlichen
Abzug beim Gesamthomolgiewert berücksichtigen. Dies wird durch
Einfügen
von "Lücken" in der Sequenzausrichtung
erreicht, wobei versucht wird, eine maximale lokale Homolgie herbeizuführen.
-
Allerdings
wesien diese komplexeren Verfahren jeder vorkommenden Lücke in der
Ausrichtung einen "Lücken-Abzug" zu, so dass für dieselbe
Anzahl von identischen Aminosäuren
eine Sequenz-Ausrichtung mit möglichst
wenigen Lücken – was einer
höhere Übereinstimmung
zwischen den beiden verglichenen Sequenzen entspricht – einen
höheren
Wert erreichen wird als ein solche mit zahlreichen Lücken. Typischerweise
werden "affine Lücken-Kosten" verwendet, welche
für das
Vorkommen einer Lücke
einen vergleichsweise hohen Zuschlag auferlegen und für jeden
zusätzlichen
Rest in der Lücke
einen kleineren Zuschlag auferlegen. Dies ist das am häufigsten
verwendete Lücken-Bewertungssystem.
Hohe Lücken-Zuschläge werden
natürlich
optimierte Ausrichtungen mit weniger Lücken ergeben. Die meisten Ausrichtungsprogramme
erlauben es, die Lücken-Zuschläge zu modifizieren.
Allerdings ist es beim Verwenden solcher Software für Sequenzvergleiche
bevorzugt, die vorgegebene Werte zu verwenden. Falls beispielsweise
das GCG Wisconsin Bestfit Paket (siehe unten) verwendet wird, beträgt der vorgegebene
Lücken-Zuschlag
für Aminosäurensequenzen –12 für eine Lücke und –4 für jede Extension.
-
Die
Berechnung der maximalen % Homolgie erfordert denmach erstens die
Bildung einer optimalen Ausrichtung, welche die Lücken-Zuschläge berücksichtigt.
Ein geeignetes Computerprogramm für die Durchführung einer
solchen Ausrichtung ist das GCG Wisconsin Bestfit Paket (University
of Wisconsin, U.S.A.; Devereux et al., 1984, Nucleic Acids Research
12:387). Beispiele für
andere Software, welche Sequenzvergleiche durchführen kann, umfassen ohne darauf
beschränkt
zu sein, das BLAST Paket (siehe Ausubel et al., 1999 ibid – Chapter
18), FASTA (Atschul et al., 1990, J. Mol. Biol., 403–410) und
das GENEWORKS Vergleichsinstrumente-Paket. Sowohl BLAST als auch
FASTA sind für
offline und online Suchen verfügbar
(siehe Ausubel et al., 1999 ibid, pages 7–58 to 7–60). Allerdings ist es bevorzugt,
das GCG Bestfit-Programm zu verwenden. Es ist auch ein neues Instrument
für den
Vergleich von Protein- und Nukleotid-Sequenzen verfügbar, das
als BLAST 2 Sequences bezeichnet wird (siehe FEMS Microbiol Lett
1999 174(2): 247-50;
FEMS Microbiol Lett 1999 177(1): 187–8 und tatiana@ncbi.nlm.nih.gov).
-
Obwohl
die endgültige
%-Homolgie in Bezug auf Identität
bestimmt werden kann, beruht der Ausrichtungsprozess als solcher
typischerweise nicht auf einem alles-oder-nichts- Paarvergleich. Vielmehr wird im Allgemeinen
eine skalierte Ähnlichkeitswert-Matrix
verwendet, welche jedem paarweisen Vergleich einen auf chemischer Ähnlichkeit
oder evolutionärem
Abstand basierenden Wert zuweist. Ein Beispiel für eine solche üblicherweise
verwendete Matrix ist die BLOSUM62-Matrix – die Vorgabematrix für das Programmpaket
BLAST. Die GCG-Wisconsin-Programme verwenden im Allgemeinen entweder
die öffentlichen
Vorgabewerte oder falls verfügbar
eine zugeschnitte Symbolvergleichstabelle (siehe Anwendungshandbuch
für weitere
Detaile). Es wird bevorzugt, für
das GCG Paket die öffentlichen
Vorgabewerte und im Falle von anderer Software die Vorgabematrix
wie BLOSUM62 zu verwenden.
-
Nachdem
die Software eine optimale Ausrichtung gebildet hat, ist es möglich die
%-Homolgie, vorzugsweise die %-Sequenzidentität zu berechnen.
Die Software tut dies typischerweise im Zuge des Sequenzvergleichs
und erzeugt ein numerisches Resultat.
-
Die
Sequenzen können
auch Deletionen, Insertionen oder Substitutionen von Aminosäurenresten
haben, welche eine stille Veränderung
bilden und zu einer funktionell äquivalenten
Substanz führen.
Vorsätzliche Aminosäuren-Substitutionen können auf
der Basis von Ähnlichkeit
in der Polarität,
Ladung, Löslichkeit,
Hydrophobie, Hydrophilie und/oder der amphiphilen Natur der Reste
gemacht werden, so lange die sekundäre Bindungsaktivität der Substanz
erhalten bleibt. Beispielsweise umfassen negativ geladene Aminosäuren Aspartansäure und
Glutaminsäure;
positiv geladene Aminosäuren
umfassen Lysin und Arginin; und Aminosäuren mit ungeladenen polaren
Kopfgruppen, die ähnliche
Hydrophile- Werte
aufweisen, umfassen Leucin, Isoleucin, Valin, Glycin, Alanin, Asparagin,
Glutamin, Serin, Threonin, Phenylalanin und Tyrosin.
-
Konservative
Substitutionen können
beispielsweise gemäss
der unten aufgeführten
Tabelle vorgenommen werden. Aminosäuren im selben Block in der
zweiten Spalte und vorzugsweise in derselben Zeile in der dritten
Spalte können
für einander
substituiert werden:
-
-
Es
können
homologe Substitutionen (Substitution und Ersatz werden hier beide
als Bezeichnung für den
Austausch eines bestehenden Aminosäurerestes durch einen alternativen
Rest verwendet), d.h. gleich-für-gleich
Substitutionen wie basisch für
basisch, sauer für
sauer, polar für
polar usw. vorkommen. Es können
auch nicht-homologe Substitutionen, d.h. Substitutionen von einer
Klasse von Resten zu einer anderen oder alternativ der Einschluss
von unnatürlichen
Aminosäuren
wie Ornithin (hiernach als Z bezeichnet), Diaminobuttersäure-ornithin
(hiernach als B bezeichnet), Norleucin-ornithin (hiernach als O
bezeichnet), Pyriylalanin, Thienylalanin, Naphthylalanin und Phenylglycin
vorkommen.
-
Ersetzungen
können
auch durch unnatürliche
Aminosäuren
einschliesslich; alpha*- und alpha-disubstituierte* Aminosäuren, N-Alkyl-Aminosäuren*, Milchsäure*, Halogenderivate
von natürlichen
Aminosäuren wie
Trifluorotyrosin*, p-Cl-Phenylalanin*, p-Br-Phenylalanin*, p-I-Phenylalanin*,
L-Allyl-glycine*, β-Alanin*, L-α-Aminobuttersäure*, L-γ-Aminobuttersäure*, L-α-Aminoisobuttersäure*, L-ε-Aminocapronsäure#, 7-Aminoheptansäure*, L-Methioninsulfon#*, L-Norleucin*, L-Norvalin*, p-Nitro-L-phenylalanin*,
L-hydroxyprolin#, L-Thioprolin*, Methylderivate von Phenylalanin
(Phe) wie 4-Methyl-Phe*,
Pentamethyl-Phe*, L-Phe-(4-amino)#, L-Tyr-(methyl)*, L-Phe-(4-isopropyl)*,
L-Tic-(1,2,3,4-Tetrahydroisochinolin-3-carbonsäure)*, L-Diaminopropionsäure# und L-Phe-(4-benzyl)* erfolgen. Die Bezeichnung*
wurde zum Zweck der obigen Diskussion (im Bezug auf homologe oder
nicht-homologe Substitution) zur Bezeichnung der hydrophoben Natur
des Derivates verwendet, während
# zur Bezeichnung der hydrophilen Natur des Derivates verwendet
wurde, #* weist auf amphiphile Eigenschaften
hin.
-
Variante
Aminosäurensequenzen
können
geeignete Spacergruppen umfassen, welche zwischen zwei beliebigen
Aminosäurenresten
der Sequenz eingeführt
werden können
und Alkylgruppen wie Methyl-, Ethyl- oder Propylgruppen sowie Aminosäurespacer
wie Glycin oder β-alanin-Reste
umfassen. Eine weitere Art von Variation umfasst die Gegenwart von
einem oder mehreren Aminosäurenresten
in peptoider Form, was Fachpersonen wohl bekannt sein wird. Zur
Vermeidung von Unklarheiten wird "die peptoide Form" verwendet, um auf Varianten eines Aminosäurenrestes
hinzuweisen, worin die α-Kohlenstoff-Substituentgruppe
am Stickstoffatom des Restes statt am α- Kohlenstoff ist. Verfahren zur Herstellung
von Peptiden in der peptoiden Form sind im Fachgebiet wohlbekannt,
beispielsweise Simon RJ et al., PNAS (1992) 89(20), 9367-9371 und Horwell
DC, Trends Biotechnol. (1995) 13(4), 132-134.
-
HYBRIDISIERUNG
-
Die
vorliegende Erfindung umfasst auch die Verwendung von Sequenzen,
die zu den hier dargestellten Target-Sequenzen hybridisieren können – beispielsweise
wenn der Wirkstoff eine Antisense-Sequenz ist.
-
Der
hier verwendete Begriff "Hybridisierung" soll "das Verfahren, durch
das ein Strang von Nukleinsäuren über Basen-Paarung
mit einem komplementären
Strang verknüpft
wird" sowie das
Verfahren der durch Polymerase-Kettenreaktions-(PCR)-Technologie
ausgeführten
Amplifikation umfassen.
-
Die
erfindungsgemässen
Nukleotid-Sequenzen, die zur selektiven Hybridisierung an die hier
angegebenen Nukleotid-Sequenzen
oder an deren Komplement befähigt
sind, werden im Allgemeinen zu mindestens 75%, bevorzugt zu mindestens
85 oder 90% und besonders bevorzugt zu mindestens 95% oder 98% homolog zu
den hier angegebenen entsprechenden komplementären Nukleotid-Sequenzen sein,
und zwar über
eine Region von mindestens 20, vorzugsweise mindestens 25 oder 30,
beispielsweise mindestens 40, 60 oder 100 oder mehr benachbarten
Nukleotiden. Bevorzugte erfindungsgemässe Nukleotid-Sequenzen werden
Regionen umfassen, die zu der in SEQ ID No 2 der Sequenz-Liste der
vorliegenden Erfindung angegebenen Nukleotid-Sequenz bevorzugt mindestens
80 oder 90% und besonders bevorzugt mindestens 95% homolog sind.
-
Der
Begriff "selektiv
hybridisierbar" bedeutet,
dass die Nukleotid-Sequenz, wenn sie als Sonde verwendet wird, unter
Bedinungen eingesetzt wird, wo eine Target-Nukleotid-Sequenz mit
der Sonde in einem signifikant über
dem Hintergrund liegenden Ausmass hybridisiert. Die Hintergrund-Hybridisierung kann
wegen anderen Nukleotid-Sequenzen vorkommen, die beispielsweise
in der gescreenten cDNS- oder genomischen DNS-Bibliothek vorhanden
sind. In solchen Fällen
impliziert "Hintergund" ein durch Interaktion
zwischen der Sonde und einem nicht-spezifischen DNS-Mitglied der
Bibliothek erzeugten Signal, welches weniger als 10-fach, vorzugsweise
weniger als 100-fach so intensiv ist wie die mit der Target-DNS
beobachtete spezifische Interaktion. Die Intensität der Interaktion
kann beispielsweise durch Radiomarkierung der Sonde, z.B. mit 32P, gemessen werden.
-
Hybridisierungsbedingungen
beruhen auf der Schmelztemperatur (Tm) des Nukleinsäure-Bindungskomplexes
wie in Berger und Kimmel (1987, Guide to Molecular Cloning Techniques,
Methods in Enzymology, Vol 152, Academic Press, San Diego CA) angegeben,
und verleihen eine definierte "Stringenz" wie unten erklärt.
-
Eine
maximale Stringenz kommt typischerweise bei ungefähr Tm-5°C (5°C unter dem
Tm der Sonde) vor; eine hohe Stringenz bei ungefähr 5°C bis 10°C unter Tm; eine mittlere Stringenz
bei ungefähr
10°C bis 20°C unter Tm;
und eine niedrige Stringenz bei ungefähr 20°C bis 25°C unter Tm. Wie es Fachpersonen
wohlbekannt sein wird, kann eine Hybridisierung maximaler Stringenz
zur Identifikation und zum Nachweis von identischen Nukleotid-Sequenzen
verwendet werden, während
eine Hybridisierung mittlerer (oder niedriger) Stringenz zur Identifikation
oder zum Nachweis von ähnlichen
oder verwandten Polynukleotid-Sequenzen verwendet werden kann.
-
In
einem bevorzugten Aspekt umfasst die vorliegende Erfindung Nukleotid-Sequenzen,
welche unter stringenten Bedingungen (z.B. 65°C und 0.l × SSC {1 × SSC = 0.15 M NaCl, 0.015
M Na3-Citrat, pH 7.0) zur erfindungsgemässen Nukleotid-Sequenz hybridisieren
können.
Wenn die erfindungsgemässe
Nukleotid-Sequenz doppelsträngig
ist, gehören
beide Stänge
des Duplexes, entweder individuell oder in Kombination, zur vorliegenden
Erfindung. Es versteht sich, dass wenn die Nukleotid-Sequenz einsträngig ist,
die komplementäre
Sequenz dieser Nukleotid-Sequenz ebenfalls zum Umfang der vorliegenden
Erfindung gehört.
-
Nukleotid-Sequenzen,
die zu den erfindungsgemässen
Sequenzen nicht zu 100% homolog sind, aber zum Umfang der Erfindung
gehören,
können
auf eine Vielzahl von Wegen erhalten werden. Andere Varianten der
hier beschriebenen Sequenzen können
beispielsweise durch Absuchen von DNS-Bibliotheken gewonnen werden,
die von einer Reihe von Quellen erzeugt wurden. Ausserdem können andere
virale/bakterielle oder zelluläre
Homologa, insbesondere in Zellen von Säugern (z.B. Zellen von Ratten,
der Mäusen,
Rindern und von Primaten) gefundene zelluläre Homologa, gewonnen werden,
und solche Homologa und Fragmente davon sind im Allgemeinen zur
selektiven Hybridisierung zu den hier in den Sequenzprotokolllen
gezeigten Sequenzen befähigt.
Solche Sequenzen können
durch Absuchen von cDNS-Bibliotheken oder genomischen DNS Bibliotheken
von anderen Tierspezies und durch Absuchen solcher Bibliotheken
mit Sonden, welche die gesamte oder Teile der hier angegebenen Nukleotid-Sequenz
unter Bedingungen mittlerer bis hoher Stringenz umfassen, gewonnen
werden. Ähnliche Überlegungen
gelten für
das Gewinnen von Spezies-Homologa und allele Varianten der erfindungsgemässen Aminosäuren- und/oder
Nukleotid-Sequenzen.
-
Varianten
und Strang/Spezies-Homologa können
auch unter Verwendung von degenerierter PCR gewonnen werden, welche
Primer verwendet, die zur Ansteuerung von Sequenzen innerhalb der
Varianten und Homologa ausgelegt sind, welche für erhaltene Aminosäurensequenzen
innerhalb der erfindungsgemässen Sequenzen
kodieren. Erhaltene Sequenzen können
beispielsweise durch Ausrichtung der Aminosäurensequenzen aus mehreren
Varianten/Homologa vorhergesagt weden. Sequenz-Ausrichtungen können unter
Verwendung von im Fachgebiet bekannter Computer-Software vorgenommen
werden. Beispielsweise wird das GCG Wisconsin PileUp Programm häufig verwendet.
Die bei der degenerierten PCR verwendeten Primer werden eine oder
mehrere degenerierte Positionen enthalten und werden unter Stringenz-Bedingungen
verwendet, die niedriger sind als diejenigen, die zum Klonieren
von Sequenzen mit Einzelsequenz-Primern gegen bekannte Sequenzen
verwendet werden.
-
Alternativ
können
solche Nukleotid-Sequenzen durch ortsgerichtete Mutagenese von charakterisierten
Sequenzen wie beispielsweise der in SEQ ID No 2 des erfindungsgemässen Sequenzprotokolls
angegebenen Nukleotid-Sequenz gewonnen werden. Dies kann nützlich sein,
wenn beispielsweise stille Codonveränderungen von Sequenzen benötigt werden
zur Optimierung der Codonpräferenzen
für eine
bestimmte Wirtzelle, in welcher die Nukleotid-Sequenzen exprimiert
werden. Andere Sequenzveränderungen
können
erwünscht sein,
um Restriktionsenzym-Erkennungsstellen einzuführen oder um die Aktivität des durch
die Nukleotid-Sequenzen kodierten Proteins zu verändern.
-
Die
erfindungsgemässen
Nukleotid-Sequenzen können
zur Bildung eins Primers, z.B. eines PCR-Primers, eines Primers
für eine
alternative Amplifikationsreaktion, einer Sonde, die z.B. auf konventionelle
Art unter Verwendung von radioaktiven oder nicht-radioaktiven Markierungen
mit einer erkennbaren Markierung versehen ist, verwendet werden,
oder die Nukleotid-Sequenzen können
in Vektoren kloniert werden. Solche Primer, Sonden und andere Fragmente
werden eine Länge
von mindestens 15, vorzugsweise mindestens 20, beispielsweise mindestens
25, 30 oder 40 Nukleotide haben und sind im Begriff Nukleotid-Sequenz
wie er hier verwendet wird ebenfalls enthalten.
-
Die
Nukleotid-Sequenzen wie beispielsweise DNS-Polynukleotide und erfindungsgemässe Sonden können rekombinant,
synthetisch oder durch irgend ein den Fachpersonen verfügbares Mittel
hergestellt werden. Sie können
auch mittels standardmässigen
Verfahren kloniert werden.
-
Im
Allgemeinen werden Primer synthetisch hergestellt, was eine schrittweise
Herstellung der gewünschten
Nukleinsäure-Sequenz – ein Nukleotid
aufs Mal – beinhaltet.
Verfahren, um dies mittels automatisierter Verfahren zu erreichen,
sind im Fachgebiet bekannt.
-
Längere Nukleotid-Sequenzen
werden im Allgemeinen unter Verwendung von rekombinanten Mitteln, beispielsweise
unter Verwendung eines PCR- (Polymerase-Kettenreaktion) Klonierungsverfahrens,
hergestellt. Dies umfasst das Erzeugen eines Paares von Primern
(z.B. aus ungefähr
15 bis 30 Nukleotiden), welche eine Region der zu klonierenden Targeting-Sequenz
flankieren, das in Kontakt bringen der Primer mit mRNS oder cDNS,
die aus Zellen von einem Tier oder Menschen gewonnen wurde, das
Durchführen
einer Polymerase-Kettenreaktion (PCR) unter Bedingungen, welche
eine Amplifikation der gewünschten
Region ergeben, das Isolieren des amplifizierten Fragmentes (z.B.
durch Reinigen des Reaktionsgemisches auf einem Agarosegel) und
das Wiedergewinnen der amplifizierten DNS. Die Primer können dahingehend
ausgestaltet sein, dass sie geeignete Restriktionsenzym-Erkennungsstellen
enthalten, so dass die amplifizierte DNS in einen geeigneten Klonierungsvektor
kloniert werden kann.
-
Wegen
der inhärenten
Entartung des genetischen Codes können andere DNS-Sequenzen,
welche im Wesentlichen für
dieselbe oder eine funktionell äquivalente
Aminosäurensequenz
kodieren, zum Klonieren und Exprimieren der Target-Sequenzen verwendet
werden. Wie den Fachpersonen bekannt ist, kann es für gewisse
Expressionssysteme vorteilhaft sein, die Target-Sequenzen mit nicht-natürlich vorkommenden
Codons zu erzeugen. Codons, die von einem bestimmten prokaryotischen
oder eukaryotischen Wirt bevorzugt werden (Murray E et al (1989)
Nuc Acids Res 17:477–508),
können
ausgewählt
werden, um beispielsweise die Geschwindigkeit der Target-Expression
zu erhöhen
oder um rekombinante RNS-Transkripte mit gewünschten Eigenschaften zu erzeugen,
beispielsweise mit einer längeren
Halbwertszeit als diejenige von Transkripten aus einer natürlich vorkommenden
Sequenz.
-
EXPRESSION
VON VEKTOREN
-
Die
als Target oder zur Expression des Targets verwendete Nukleotid-Sequenz
kann in einen rekombinanten replizierbaren Vektor eingebaut werden.
Der Vektor kann zur Repliaktion und Expression der Nukleotid-Sequenz
in und/oder aus einer kompatiblen Wirtzelle verwendet werden. Die
Expression kann durch Verwendung von Kontroll-Sequenzen, welche
Promotoren/Verstärker
und andere expressionsregulierende Signale umfassen, kontrolliert
werden. Es können
Prokaryotische Promotoren und in eukaryotischen Zellen funktionelle
Promotoren verwendet werden. Es können gewebespezifische oder
stimulispezifische Promotoren verwendet werden. Es können auch
chimäre
Promotoren, welche Sequenzelemente aus zwei oder mehreren unterschiedlichen
Promotoren wie oben beschrieben umfassen, verwendet werden.
-
Das
von einer rekombinanten Wirtzelle durch Expression der Nukleotid-Sequenz
erzeugte Protein kann je nach verwendeter Sequenz und/oder Vektor
ausgeschieden werden oder intrazellulär enthalten bleiben. Die kodierenden
Sequenzen können
mit Signal-Sequenzen versehen werden, welche die Ausscheidung der
substanzkodierenden Sequenzen durch eine bestimmte prokaryotische
oder eukaryotische Zellmembran lenken.
-
FUSIONSPROTEINE
-
Die
Target-Aminosäurensequenz
kann als Fusionsprotein hergestellt werden, um beispielsweise die Extraktion
und Reinigung zu erleichtern. Beispiele von Fusionsproteinpartnern
umfassen Glutathion-S-Transferase (GST), 6xHis, GAL4 (DNS-Bindungs-
und/oder Transkriptions-Aktivierungsdomänen) und β-Galactosidase.
Es kann auch vorteilhaft sein, eine proteolytische Spaltungstelle
zwischen dem Fusionsproteinpartner und der interessierenden Proteinsequenz
einzubauen, um das Entfernen von Fusionsprotein-Sequenzen zu erlauben.
Vorzugsweise wird das Fusionsprotein die Aktivität des Targets nicht behindern.
-
Das
Fusionsprotein kann ein(e) mit der erfindungsgemässen Substanz fusionierte(s)
Antigen oder antigene Determinante umfassen. In dieser Ausgestaltung
kann das Fusionsprotein ein nicht-natürlich vorkommendes Fusionsprotein
sein, das eine Substanz umfasst, welche als ein Adjuvans im Sinne
der Bereitstellung einer generalisierten Stimulation des Immunsystems
wirken kann. Das Antigen oder die antigene Determinante kann entweder
an das Amino- oder das Carboxyende der Substanz gebunden sein.
-
In
einer anderen Ausgestaltung der Erfindung kann die Aminosäurensequenz
an einer heterologe Sequenz gebunden sein, um ein Fusionsprotein
zu kodieren. Beispielsweise kann es für das Absuchen von Peptid-Bibliotheken
nach Wirkstoffen, die zur Beeinflussung der Substanz-Aktivität befähigt sind,
nützlich
sein, eine chimäre
Substanz zu kodieren, die ein heterologes Epitop exprimiert, das
von einem kommerziell verfügbaren
Antikörper
erkannt wird.
-
ANTIKÖRPER
-
Es
wird ein Antikörper
beschrieben. Zusätzlich
oder alternativ kann das Target ein Antikörper sein. Zusätzlich oder
alternativ kann das Mittel zum Nachweis des Targets ein Antikörper sein.
-
Antikörper können durch
Standardverfahren wie der Immunisierung mit der erfindungsgemässen Substanz
oder durch Verwendung einer Phagen-Display-Bibliothek hergestellt
werden.
-
Im
Rahmen der vorliegenden Erfindung umfasst der Begriff "Antikörper", wenn nicht anders
angegeben und ohne darauf beschränkt
zu sein, polyklonale, monoklonale, chimäre, "single-chain" Fab-Fragmente sowie durch eine Fab-Expressions-Bibliothek
erzeugte Fragmente wie auch Mimetika davon. Solche Fragmente umfassen
Fragmente ganzer Antikörper,
die ihre Bindungsaktivität
für eine
Target-Substanz
bewahren, Fv-, F(ab')-
und F(ab')2-Fragmente, sowie single-chain Antikörper (scFv),
Fusionsproteine und andere synthetische Proteine, welche die Antigen-Bindungsstelle
des Antikörpers
umfassen. Ausserdem können
die Antikörper
und Fragmente davon humanisierte Antikörper sein. Neutralisierende
Antikörper,
d.h. solche, welche die biologische Aktivität des Substanz-Polypeptids
inhibieren, sind für
Diagnostika und Therapeutika besonders bevorzugt.
-
Falls
polyklonale Antikörper
gewünscht
werden, wird ein ausgewählter
Säuger
(z.B. Maus, Hase, Ziege, Pferd, usw.) mit einem immunogenen Polypeptid
immunisiert, das ein aus einem identifizierten Wirkstoff und/oder
einer erfindungsgemässen
Substanz erhältliches
Epitop trägt.
-
In
Abhängigkeit
der Wirtspezies können
verschiedenen Adjuvanzien zur Erhöhung der immunologischen Antwort
verwendet werden. Solche Adjuvanzien umfassen ohne darauf beschränkt zu sein
Freund's, Mineralgele
wie Aluminiumhydroxid und oberflächenaktive
Substanzen wie Lysolecithin, pluronische Polyole, Polyanionen, Peptide, Ölemulsionen,
Schlüsselloch-Limpet-Hämocyanin
und Dinitrophenol. BCG (Bacilli Calmette-Guerin) und Corynebacterium
parvum sind potenziell nützliche
menschliche Adjuvanzien, welche in gereinigter Form verwendbar sind.
Die Polypeptid-Substanz wird an immunologisch kompromittierte Individuen zwecks
Stimulierung der systemischen Abwehr verabreicht.
-
Das
Serum vom immunisierten Tier wird gesammelt und nach bekannten Verfahren
behandelt. Falls ein Serum, das polyklonale Antikörper zu
einem Epitop enthält,
das von einem identifizierten erfindungsgemässen Wirkstoff und/oder einer
erfindungsgemässen
Substanz erhältlich
ist, zudem Antikörper
gegenüber
anderen Antigenen enthält,
können
die polyklonalen Antikörper
durch Immunoaffinitäts-Chromatographie gereinigt werden.
Verfahren zum Erzeugen und Verarbeiten von polyklonalem Antisera
sind im Fachgebiet bekannt. Damit solche Antikörper erzeugt werden können, beschreibt
die Erfindung auch erfindungsgemässe
Polypeptide oder Fragmente davon, die zwecks Verwendung als Immunogene
bei Tieren oder Menschen an ein anderes Polypeptid haptenisiert
sind.
-
Monoklonale
Antikörper,
die gegen Epitope gerichtet sind, welche von einem(r) erfindungsgemässen identifizierten Wirkstoff
und/oder Substanz erhältlich
sind, können
von einer Fachperson ohne weiteres hergestellt werden. Die allgemeine
Methodik zur Herstellung von monoklonalen Antikörpern durch Hybridome ist wohlbekannt.
Unsterbliche antikörpererzeugende
Zell-Linien können
durch Zellfusion und auch durch andere Verfahren wie direkte Transformation
von B-Lymphozyten mit onkogener DNS oder Transfektion mit Epstein-Barr-Virus
erzeugt werden. Paneele von gegen Orbit-Epitope gebildeten monoklonalen Antikörpern können nach
verschiedene Eigenschaften gescreent werden; d.h. nach Isotypen-
und Epitopen-Affinität.
-
Monoklonale
Antikörper
zur Substanz und/oder zum identifiziertem Wirkstoff können unter
Verwendung irgend eines Verfahrens, welches die Bildung von Antikörper-Molekülen durch
kontinuierliche Zell-Linien in Kulturen erlaubt, hergestellt werden.
Diese umfassen – ohne
darauf beschränkt
zu sein – das
ursprünglich von
Koehler und Milstein beschriebene Hybridom-Verfahren (1975 Nature
256:495–497),
das menschliche B-Zellen-Hybridom-Verfahren (Kosbor et al (1983)
Immunol Today 4:72; Cote et al (1983) Proc Natl Acad Sci 80:2026–2030) und
das EBV-Hybridom-Verfahren
(Cole et al (1985) Monoclonale Antibodies und Cancer Therapy, Alan
R Liss Inc, pp 77–96).
Ausserdem können
die für
die Bildung von "chimären Antikörpern" entwickelten Verfahren,
das Verspleissen von Maus-Antikörpergenen
an menschliche Antikörpergene
unter Gewinnung eines Moleküls
mit geeigneter Antigenspezifität
und biologischer Aktivität,
verwendet werden (Morrison et al (1984) Proc Natl Acad Sci 81:6851–6855; Neuberger
et al (1984) Nature 312:604–608;
Takeda et al (1985) Nature 314:452–454). Alternativ können Verfahren,
die zur Bildung von single-chain Antikörpern beschrieben wurden (US
Patent No. 4,946,779), zur Bildung substanzspezifischer single-chain
Antikörper
adaptiert werden.
-
Sowohl
monoklonale wie auch polyklonale Antikörper, die sich gegen Epitope
richten, die aus einem(r) identifizierten Wirkstoff und/oder Substanz
erhältlich
sind, eignen sich besonders für
die Diagnose, und solche, die neutralisierend sind, eignen sich
für die
passive Immunotherapie. Monoklonale Antikörper können insbesondere zur Erhöhung der
anti-idiotypen Antikörper
verwendet werden. Anti-idiotype Antikörper sind Immunoglobuline,
welche ein "inneres
Bild" der Substanz
und/oder des Wirkstoffes tragen, gegen die der Schutz erwünscht ist.
Verfahren zur Erhöhung
von anti-idiotypen
Antikörpern
sind im Fachgebiet bekannt. Diese anti-idiotypen Antikörper können auch
für die
Therapie nützlich
sein.
-
Antikörper können auch
durch Induktion der in-vivo-Bildung in der Lymphozytenpopulation
oder durch Screening von rekombinanten Immunoglobulin-Bibliotheken
oder Paneelen mit hochspezifischen Bindingsreagenzien wie in Orlandi
et al (1989, Proc NatI Acad Sci 86: 3833–3837) und Winter G und Milstein
C (1991; Nature 349:293–299)
beschrieben gebildet werden.
-
Es
können
auch Antikörper-Fragmente
gebildet werden, welche spezifische Bindungsstellen für die Substanz
enthalten. Beispielsweise umfassen solche Fragmente, ohne darauf
beschränkt
zu sein, die F(ab')2-Fragmente, welche durch Pepsinverdauung
des Antikörper-Moleküls erzeugt
werden können,
und die Fab-Fragmente, welche durch Reduktion der Disulfidbrücken der
F(ab')2-Fragmente
gebildet werden können. Alternativ
können
Fab-Expressions-Bibliotheken konstruiert werden, um eine schnelle
und einfache Identifikation von monoklonalen Fab-Fragmenten mit
der gewünschten
Spezifizität
zu ermöglichen
(Huse WD et al (1989) Science 256:1275–1281).
-
REPORTER
-
Eine
grosse Vielzahl von Reportern kann in den Assay-Verfahren (wie auch in den Screenings)
der vorliegenden Erfindung verwendet werden, wobei bevorzugte Reporter
einfach nachweisbare Signale (z.B. mittels Spektroskopie) liefern.
Beispielsweise kann ein Reportergen ein Enzym kodieren, das eine
Reaktion katalysiert, welche die Lichtabsorptionseigenschaften verändert.
-
Beispiele
von Reporter-Molekülen
umfassen ohne darauf beschränkt
zu sein β-Galactosidase,
Invertase, grünes
fluoreszierendes Protein, Luciferase, Chloramphenicol, Acetyltransferase, β-Glucuronidase, Exo-Glucanase
und Glucoamylase. Alternativ können
radioaktiv markierte oder fluoreszierend markierte Nukleotide in
naszente Transcripte eingebaut werden, welche dann identifizierbar
sind, wenn sie an Oligonukleotid-Sonden gebunden sind.
-
In
einer bevorzugten Ausgestaltung wird die Bildung des Reporter-Moleküls anhand
der enzymatischen Aktivität
des Reportergen-Produktes, beispielsweise von β-Galactosidase, gemessen.
-
Eine
Vielzahl von Protokollen zum Nachweis und zur Messung der Expression
des Targets, wie die Verwendung von für das Protein spezifischen
polyklonalen oder monoklonalen Antikörpern, sind im Fachgebiet bekannt.
Beispiele umfassen den Enzyme-linked-immunosorbent Assay (ELISA),
den Radioimmunoassay (RIA) und die Fluoreszenz-aktivierte Zellensortierung
(FACS). Ein zwei-stelliger, monoklonalbasierter Immunoassay, welcher
monoklonale Antikörper
die gegenüber
zwei nicht-interferierenden Epitopen auf Polypeptiden reaktiv sind,
wird bevorzugt, aber es kann auch ein kompetitiver Bindungsassay
verwendet werden. Diese und andere Assays sind unter anderem in
Hampton R et al (1990, Serological Methods, A Laboratory Manual,
APS Press, St Paul MN) und Maddox DE et al (1983, J Exp Med 15 8:121
1) beschrieben.
-
Eine
grosse Vielzahl von Markierungs- und Konjugationsverfahren sind
den Fachpersonen bekannt und können
in verschiedenen Nuklein- und Aminosäure-Assays verwendet werden.
Die Mittel zur Bildung von markierter Hybridisierung oder PCR-Sonden
zum Nachweis der Target-Polynukleotid-Sequenzen umfassen die Oligomarkierung,
die Nicktranslation und die Endmarkierung oder PCR-Amplifikation
unter Verwendung eines markierten Nukleotides. Alternativ kann die
kodierende Sequenz oder irgend ein Teil davon in einen Vektor zur
Bildung einer mRNS-Sonde kloniert werden. Solche Vektoren sind im
Fachgebiet bekannt, sind kommerziell verfügbar und können in vitro durch Zugabe
einer geeigneten RNS-Polymerase wie T7, T3 oder SP6 und von markierten
Nukleotiden zur Synthese von RNS-Sonden verwendet werden.
-
Eine
Anzahl von Unternehmen wie Pharmacia Biotech (Piscataway, NJ), Promega
(Madison, WI) und US Biochemical Corp (Cleveland, OH) vertreiben
kommerzielle Kits und Protokolle für diese Verfahren. Geeignet
Reporter-Moleküle
oder Markierungen umfassen Radionuklide, Enzyme, sowie fluoreszente,
chemilumineszente oder chromogene Wirkstoffe sowie Substrate, Kofaktoren,
Inhibitoren, magnetische Partikel und dergleichen. Patente, welche
die Verwendung von solchen Markierungen angeben, umfassen US-A-3817837; US-A-3850752; US-A-3939350;
US-A-3996345; US-A-4277437; US-A-4275149
und US-A-4366241. Es können
auch rekombinante Immunoglobuline hergestellt werden wie in US-A-4816567
gezeigt.
-
Zusätzliche
Verfahren zur Quantifizierung der Expression eines bestimmten Moleküls umfassen
das radioaktive Markieren (Melby PC et al 1993 J Immunol Methods
159:235–44)
oder das Biozinnylieren (Duplaa C et al 1993 Anal Biochem 229–36) von
Nukleotiden, die Koamplifikation einer Kontroll-Nukleinsäure, sowie Standardkurven,
auf denen die experimentellen Resultate interpoliert werden. Die
Quantifizierung von mehrfachen Proben kann durch Durchführen des
Assays in einem ELISA-Format, wobei das interessierende Oligomer
in verschiedenen Verdünnungen
vorliegt, beschleunigt werden, und eine spektrophotometrische oder
kalorimetrische Antwort erlaubt eine rasche Quantifizierung.
-
Obwohl
das Vorliegen/Fehlen von Marker-Gen-Expression darauf hinweist,
dass das interessierende Gen vorhanden ist, sollte dessen Gegenwart
und Expression bestätigt
werden. Falls beispielsweise die Nukleotid-Sequenz in eine Marker-Gen-Sequenz eingeführt wird,
können
rekombinante Zellen, welche dieselbe enthalten, durch das Fehlen
von Markergen-Funktion identifiziert werden. Alternativ kann ein
Markergen im Tandem mit einer für
ein Target kodierenden Sequenz unter der Kontrolle eines einzelnen
Promotors plaziert werden. Die Expression des Markergens als Antwort
auf die Induktion oder Selektion weist üblicherweise ebenfalls auf
die Expression des Targets hin.
-
Alternativ
können
Wirtzellen, welche die kodierende Sequenz für das Target enthalten und
die für
das Target kodierenden Regionen exprimieren, durch verschiedene
den Fachpersonen bekannten Verfahren identifiziert werden. Diese
Verfahren umfassen ohne darauf beschränkt zu sein die DNS-DNS- oder
DNS-RNS- Hybridisierung sowie Protein-Bioassay- oder Immunoassay-Verfahren,
welche membranbasierte, lösungsbasierte
oder chipbasierte Technologien zum Nachweis und/oder zur Quantifizierung
der Nukleinsäure
oder des Proteins umfassen.
-
ALLGEMEINE ASSAYS FÜR cAMP-AKTIVITÄT/SPIEGEL
-
Die
Fähigkeit
eines Testwirkstoffes, cAMP zu potenzieren, kann durch Messung einer
relevanten Zunahme oder Abnahme eines Targetspiegels bestimmt werden.
Zusätzlich
oder alternativ kann die Fähigkeit
eines Testwirkstoffes, cAMP zu potenzieren, durch Messung einer
relevanten Zunahme der cAMP-Spiegel bestimmt werden. Beispielsweise
können
die Lehren von Smith et al 1993 (Appl. Biochem. Biotechnol. 41:189–218) adaptiert
werden. Es gibt auch kommerziell erhältliche Immunoassay-Kits zur
Messung von cAMP (z.B. Amersham International, Arlington Heights,
IL und DuPont, Boston, MA). Details zu einem geeigneten cAMP-Assay
werden im experimentellen Abschnitt besprochen.
-
SCREENINGS
-
Irgend
ein oder mehrere geignete Targets – wie eine Aminosäurensequenz
und/oder Nukleotid-Sequenz – können zur
Identifikation von PcAMP in irgend einem
von zahlreichen Wirkstoff-Screening-Verfahren verwendet werden.
Das in einem solchen Test verwendete Target kann frei in Lösung vorliegen,
an einen festen Träger
gebunden sein, auf der Zelloberfläche liegen oder intrazellulär lokalisiert
sein. Das Target kann sogar in einem Tiermodell sein, wobei das
besagte Target ein exogenes Target oder ein eingeführtes Target
sein kann. Das Tiermodell wird ein nicht-menscliches Tiermodell
sein. Die Aufhebung der Target-Aktivität oder die Bildung von Bindungs-Komplexen
zwischen dem Target und dem zu testenden Wirkstoff kann gemessen
werden.
-
Verfahren
für das
Wirkstoff-Screening können
auf dem in Geysen, Europäische
Patent-Anmeldung 84/03564, veröffentlicht
am 13. September 1984 beschriebenen Verfahren basieren. Kurz gesagt,
werden grosse Anzahlen von unterschiedlichen kleinen Peptid-Testverbindungen
auf einem festen Substrat, beispielsweise auf Plastiknadeln oder
einer anderen Oberfläche
synthetisiert. Die Peptid-Testverbindungen werden mit einem geeigneten
Target oder einem Fragment davon umgesetzt und gewaschen. Die gebundenen
Einheiten werden alsdann nachgewiesen – beispielsweise durch Adaption
geeigneter, im Fachgebiet bekannter Verfahren. Ein gereinigtes Target
kann zur Verwendung in einem Wirkstoff-Screening-Verfahren auch direkt auf
Platten geschichtet werden. Alternativ können nicht-neutralisierende
Antikörper
verwendet werden, um das Peptid einzufangen und es auf einem festen
Träger
zu fixieren.
-
Es
wird auch die Verwendung von kompetitiven Wirkstoff-Screening-Assays
beschrieben, in welchen neutralisierende Antikörper, die zur spezifischen
Bindung an ein Target befähigt
sind, mit einer Testverbindung um die Bindung an ein Target konkurrieren.
-
Ein
anderes Screening-Verfahren erlaubt ein Hochdurchsatz-Screening (HTS) von
Wirkstoffen, welche eine geeignete Bindungsaffintät an die
Substanzen aufweisen, und beruht auf dem ausführlich in WO 84/03564 beschriebenen
Verfahren.
-
Es
ist zu erwarten, dass die hier erwähnten Assay-Verfahren für das Screening
von Testverbindungen sowohl in kleinem wie auch in grossem Massstab
und auch für
quantitative Assays geeignet sein wird.
-
Demnach
wird ferner ein Verfahren zur Identifikation von Wirkstoffen, die
cAMP potenzieren, beschrieben, wobei das Verfahren das Zusammenbringen
eines geeigneten Targets mit dem Wirkstoff und die anschliessende
Messung der Aktivität
und/oder der Spiegel von cAMP umfasst.
-
Es
wird ferner ein Verfahren zur Identifikation von Wirkstoffen, die
cAMP in den weiblichen sexuellen Genitalien selektiv potenzieren,
beschrieben, wobei das Verfahren das Zusammenbringen eines geeigneten Targets
aus weiblichen sexuellen Genitalien und die anschliessende Messung
der Aktivität
und/oder der Spiegel von cAMP umfasst.
-
Es
wird ferner ein Verfahren zur Identifikation von Wirkstoffen, die
cAMP potenzieren, beschrieben, wobei das Verfahren das Zusammenbringen
eines geeigneten Targets mit dem Wirkstoff und die anschliessende
Messung der Aktivität
und/oder der Spiegel des Targets umfasst.
-
Es
wird ferner ein Verfahren zur Identifikation von Wirkstoffen, die
cAMP in den weiblichen sexuellen Genitalien selektiv potenzieren,
beschrieben, wobei das Verfahren das Zusammenbringen eines geeigneten Targets
aus weiblichen sexuellen Genitalien und die anschliessende Messung
der Aktivität
und/oder der Spiegel des Targets umfasst.
-
In
einem bevorzugten Aspekt umfasst das erfindungsgemässe Screening
mindestens die foglenden Schritte (welche nicht notwendigerweise
in derselben Reihenfolge vorliegen müssen): (a) Durchführung eines in
vitro Screenings zur Bestimmung, ob ein Kandidaten-Wirkstoff die
relevante Aktivität
(beispielsweise eine Modulation von NEP, wie NEP aus Hundenieren)
besitzt; (b) Durchführung
eines oder mehrerer Selektivitäts-Screenings
zur Bestimmung der Selektivität
des besagten Kandidaten-Wirkstoffs (z.B. um festzustellen, ob besagter
Wirkstoff auch ein ACE-Inhibitor ist – beispielsweise unter Verwendung
des hier beschriebenen Assay-Protokolls); und (c) Durchführung eines
in vivo Screenings mit besagtem Kandidaten-Wirkstoff (z.B. unter
Verwendung eines funktionellen Tiermodelles). Typischerweise wird,
falls besagter Kandidaten-Wirkstoff das Screening (a) und das Screening
(b) besteht, daraufhin das Screening (c) durchgeführt.
-
DIAGNOSTIKA
-
Es
wird ferner eine diagnostische Zusammensetzung oder ein Kit zum
Nachweis einer Prädisposition für FSAD beschrieben.
In diesem Zusammenhang wird die Zusammensetzung oder der Kit eine
Einheit umfassen, die befähigt
ist, die Gegenwart von einem oder mehreren – oder sogar die Abwesenheit
von einem oder mehreren – der
Targets in einer Testprobe anzuzeigen. Vorzugsweise wird die Testprobe
aus den weiblichen sexuellen Genitalien oder aus einer Aussonderung
davon oder daraus gewonnen.
-
Beispielsweise
kann die diagnostische Zusammensetzung irgend eine der hier erwähnten Nukleotid-Sequenzen
oder eine Variante, ein Homologon, ein Fragment oder ein Derivat
davon, oder eine Sequenz, die zur Hybridisierung an die gesamte
oder an einen Teil von irgend einer Nukleotid-Sequenz befähigt ist, umfassen.
-
Um
eine Basis zur Diagnose einer Erkrankung bereitzustellen, sollten
Normal- oder Standardwerte für ein
Target bereitgestellt werden. Dies kann durch Zusammenbringen von
Körperflüssigkeiten
oder Zellextrakten, die von normalen tierischen oder menschlichen
Subjekten gewonnen wurden, mit einem Antikörper für ein Target unter einschlägig bekannten
Bedingungen, welche für
die Kompexbildung geeignet sind, erreicht werden. Die Menge der
standardmässigen
Kompexbildung kann durch deren Vergleich mit einer Verdünnungsreihe
von positiven Kontrollen, bei denen eine bekannte Menge von Antikörper mit
bekannten Konzentrationen eines gereinigten Targets zusammengegeben
wurde, quantifiziert werden. Danach können die aus normalen Proben erhaltenen
Standardwerte mit den aus Proben von möglicherweise von FSAD betroffenen
Subjekten erhaltenen Werten verglichen werden. Die Abweichung zwischen
dem Standardwert und dem Wert des Subjektes weist das Vorliegen
des Krankheitszustandes nach.
-
Ein
Target selbst oder irgend ein Teil davon kann die Basis für eine diagnostische
und/oder eine therapeutische Verbindung bereitstellen. Für diagnostische
Zwecke können
Target-Polynukleotid-Sequenzen verwendet werden, um die Genexpression
bei Krankheitszuständen,
Störungen
oder Erkrankungen, an welchen FSAD beteiligt sein kann, nachzuweisen
und zu quantifizieren.
-
Die
für das
Target kodierende Polynukleotid-Sequenz kann zur Diagnose von FSAD
verwendet werden, die von der Expression des Targets herrührt. Beispielsweise
können
die für
ein Target kodierenden Polynukleotid-Sequenzen in Hybridisierungs-
oder PCR-Assays von Geweben aus Biopsien oder Autopsien oder biologischen
Flüssigkeiten
verwendet werden, um Abnormitäten
bei der Target-Expression nachzeuweisen. Die Form von solchen qualitativen
oder quantitativen Verfahren kann Southern- oder Northern-Analyse, Dot-Blot-
oder andere membranbasierte Technologien; PCR-Technologien; Dipstick-,
Pin- oder Chip-Technologien; sowie ELISA- oder andere formale Mehrfachproben-Technologien
umfassen. Sämtliche
Verfahren sind im Fachgebiet bekannt und bilden in der Tat die Basis
für manche
kommerziell verfügbare
diagnostische Kits.
-
Solche
Assays können
auf die Evaluation der Wirksamkeit eines bestimmten therapeutischen
Behandlungsregimes zugeschnitten sein und können in Tierstudien, in klinischen
Studien oder zur Überwachung
der Behandlung eines einzelnen Patienten verwendet werden. Um eine
Basis zur Diagnose einer Erkrankung bereitzustellen, sollte ein
Normal- oder Standardprofil für
die Target-Expression bereitgestellt werden. Dies kann durch Zusammenbringen
von Körperflüssigkeiten
oder Zellextrakten, die von normalen tierischen oder menschlichen
Subjekten gewonnen wurden, mit dem Target oder einem Teil davon
unter Bedingungen, die für eine
Hybridisierung oder Amplifikation geeignet sind, erreicht werden.
Die standardmässige
Hybridisierung kann durch Vergleich der Werte von normalen Subjekten
mit denjenigen einer Verdünnungsreihe
von positiven Kontrollen in demselben Experiment, wobei eine bekannte
Menge von gereinigtem Target verwendet wird, quantifiziert werden.
Die von normalen Proben erhaltenen Standardwerte können mit
denjenigen aus Proben von Subjekten, die möglicherweise von einer mit
der Expression der für
das Target kodierenden Sequenz zusammenhängenden Störung oder Erkrankung betroffenen
sind, verglichen werden. Die Abweichung zwischen dem Standardwert
und dem Wert des Subjektes weist das Vorliegen des Krankheitszustandes
nach. Falls die Erkrankung bestätigt
ist, kann ein vorhandener therapeutischer Wirkstoff verabreicht
werden, und ein Behandlungsprofil oder -werte können erzeugt werden. Schliesslich
kann der Assay regelmässig
wiederholt werden, um zu untersuchen, ob die Werte gegen das Norm-
oder Standardmuster zurückstreben
oder dieses erreichen. Sukzessive Behandlungsprofile können verwendet
werden, um die Wirksamkeit einer Behandlung über eine Periode von mehreren
Tagen oder mehreren Monaten zu zeigen.
-
Demnach
wird die Verwendung eines Target-Polypeptids oder einer Variante,
eines Homologons, eines Fragmentes oder Derivates davon zur Bildung
von anti-Target-Antikörpern
beschrieben, welche beispielsweise zum Nachweis und zur Quantifizierung
der Target-Spiegel in einem FSAD-Zustand diagnostisch verwendet
werden kann.
-
Weiterhin
werden diagnostische Assays und Kits zum Nachweis eines Targets
in Zellen und Geweben beschrieben, wobei dieses ein als positive
Kontrolle verwendbares gereinigtes Target und anti-target Antikörper umfassen.
Solche Antikörper
können
in lösungsbasierten,
membranbasierten oder gewebebasierten Technologien verwendet werden,
um irgend einen Krankheitszustand oder eine Störung, die mit der Expression
von Target-Protein oder mit der Expression von Deletionen oder einer
Variante, eines Homologons, eines Fragmentes oder eines Derivates
davon zusammenhängen,
nachzuweisen.
-
ASSAY-VERFAHREN
-
Die
diagnostischen Zusammensetzungen und/oder Verfahren und/oder Kits
können
in den folgenden Verfahren verwendet werden, welche – ohne darauf
beschränkt
zu sein – umfassen:
kompetitive und nicht-kompetitive Assays, Radioimmunoassays, Biolumineszenz-
und Chemilumineszenz-Assays, fluorometrische Assays, Sandwich-Assays,
immunoradiometrische Assays, Dot-Blots,
Enzyme-linked-Assays einschliesslich ELISA, Mikrotiter Platten,
mit Antikörpern
belegte Streifen oder Stäbchen
zur schnellen Überwachung
von Urin oder Blut, Immunohistochemie und Immunozytochemie.
-
Beispielsweise
kann ein Immunohistochemie-Kit auch zur Lokalisation von NEP-Aktivität in genitalem Gewebe
verwendet werden. Dieser Immunohistochemie-Kit erlaubt unter Verwendung
sowohl von Licht- als auch von Elektronenmikroskopie die Lokalisation
von NEP in Gewebeschnitten und kultivierten Zellen, was sowohl für Forschungs-
als auch für
klinische Zwecke verwendet werden kann. Solche Informationen können für diagnostische
und möglicherweise
für therapeutische
Zwecke zum Nachweis und/oder zur Prävention und/oder zur Behandlung
einer FSD wie FSAD nützlich
sein. Für
jedes Kit sind der Bereich, die Sensitivität, die Genauigkeit, die Zuverlässigkeit,
die Spezifizität
und die Reproduzierbarkeit des Assays belegt. Die Intraassay- und
Interassay-Variation wird bei 20%, 50% und 80% Punkten auf den Standardkurven
der Verschiebung oder Aktivität
festgelegt.
-
SONDEN
-
Es
werden ferner Nukleinsäure-Hybridisierungs-
oder PCR-Sonden
angegeben, welche (insbesondere diejenigen, die zur selektiven Selektion
befähigt
sind) zum Nachweis von Polynukleotid-Sequenzen, einschliesslich
genomischen Sequenzen, die für
eine Target-Kodierungsregion oder eng verwandten Molekülen wie
Allelen kodieren, befähigt
sind. Die Spezifität
der Sonde, d.h. ob sie von einer hochgradig erhaltenen, erhaltenen
oder nicht-erhaltenen Region oder Domäne abgeleitet ist, und die
Stringenz der Hybridisierung oder Amplifikation (hoch, mittel oder
niedrig) wird bestimmen, ob die Sonde nur natürlich vorkommende Target-Kodierungssequenz
oder auch verwandte Sequenzen identifiziert. Sonden zum Nachweis
von verwandten Nukleinsäure-Sequenzen
werden aus erhaltenen oder hochgradig erhaltenen Nukleotid-Regionen
von Target-Familienmitgliedern ausgewählt, und solche Sonden können in
einem Pool von entarteten Sonden verwendet werden. Für den Nachweis
von identischen Nukleinsäure-Sequenzen,
oder wo eine maximale Spezifität
gewünscht
wird, werden die Nukleinsäure-Sonden
aus den nicht-erhaltenen Nukleotid-Regionen oder aus den einzigartigen
Regionen der Target-Polynukleotide ausgewählt. Der hier verwendete Begriff "nicht-erhaltene Nukleotid-Region" bezieht sich auf
eine Nukleotid-Region, die für
eine hier beschriebene target-kodierende Sequenz einzigartig ist
und bei verwandten Familienmitgliedern nicht vorkommt.
-
Die
in US-A-4683195, US-A-4800195 und US-A-4965188 beschriebene PCR
ergibt zusätzliche
Verwendungen für
Oligonukleotide, die auf Target-Sequenzen beruhen. Solche Oligomere
werden im Allgemeinen chemisch synthetisiert, aber sie können auch
enzymatisch erzeugt werden oder aus einer rekombinanten Quelle gebildet
werden. Oligomere umfassen im Allgemeinen zwei Nukleotid-Sequenzen,
und zwar eine mit Sense-Orientierung (5'->3') und eine mit Antisense
(3'<-5'), die unter optimierten
Bedingungen zur Identifizierung eines(r) spezifischen Gens oder
Bedingung verwendet werden. Die selben zwei Oligomere, verschachtelte
Sätze von
Oligomeren oder sogar ein entarteter Pool von Oligomeren können unter
weniger stringenten Bedingungen zum Nachweis und/oder zur Quantifizierung
von nahe verwandten DNS- oder RNS-Sequenzen verwendet werden.
-
Die
Nukleinsäure-Sequenz
für ein
Target kann auch verwendet werden, um wie vorgängig beschrieben Hybridisierungs-Sonden
zur Kartierung der endogenen genomischen Sequenz zu erzeugen. Die
Sequenz kann auf ein bestimmtes Chromosom oder auf eine spezifische
Region des Chromosoms kartiert werden, wobei wohlbekannte Verfahren
verwendet werden. Diese umfassen die in-situ-Hybridisierung an chromosomalen Ausstrichen
(Verma et al (1988) Human Chromosomes: A Manual of Basic Techniques,
Pergamon Press, New York City), flusssortierte chromosomale Präparationen
oder artifizielle Chromosomenkonstrukte wie YACs, bakterielle artifizielle
Chromosomen (BACs), bakterielle PI-Konstrukte oder Einzelchromosom
cDNS Bibliotheken.
-
Die
in-situ-Hybridisierung von chromosomalen Präparationen und physikalische
Kartierungsverfahren wie die Linkage-Analyse unter Verwendung von bewährten chromosomalen
Markern sind zur Ausdehnung von genetischen Karten von unschätzbarem
Wert. Beispiele von genetischen Karten können in Science (1995; 270:410f
und 1994; 265:1981f) gefunden werden. Das Platzieren eines Gens
auf das Chromosom einer anderen Säugerspezies kann oftmals assozierte
Marker zutage bringen, obwohl die Nummer oder der Arm eines bestimmten
mesnchlichen Chromosoms nicht bekannt ist. Neue Sequenzen können durch
physkialische Kartierung an Chromosomenarme oder Teile davon zugeordnet
werden. Dies liefert wertvolle Informationen für Wissenschaftler, die unter
Verwendung von Positions-Klonierung
oder anderen Gen-Erkennungsverfahren nach Krankheitsgenen suchen.
Nachdem eine Erkrankung oder ein Syndrom durch genetische Verknüpfung an
eine bestimmte Genomregion grob lokalisiert wurde, können jegliche
Sequenzen, die auf die besagte Region lokalisiert wurden, assozierte
oder regulatorische Gene für
die weitere Untersuchung darstellen. Die erfindungsgemässe Nukleotid-Sequenz kann auch
verwendet werden, um Unterschiede in der chromosomalen Lage infolge
Translokation, Inversion, usw. zwischen normalen Individuen, Trägern oder
betroffenen Individuen festzustellen.
-
WIRTZELLEN
-
Der
Begriff "Wirtzelle" – umfasst im Zusammenhang mit
der vorliegenden Erfindung jegliche Zellen, welche das Target für den Wirkstoff
enthalten könnten.
-
Demnach
werden Wirtzellen bereitgestellt, die mit einem Polynukleotid transformiert
oder transfiziert sind, welches das Target ist oder das Target exprimiert.
Vorzugsweise wird das besagte Polynukleotid in einem Vektor für die Replikation
und Expression von Polynukleotiden, die das Target sein oder das
Target exprimieren sollen, transportiert. Die Zellen werden so ausgewählt, dass
sie mit dem besagten Vektor kompatibel sind, und sie können beispielsweise
prokaryotische (beispielsweise bakterielle), Pilz-, Hefe- oder Pflanzenzellen
sein.
-
Das
gram-negative Bakterium E. coli wird häufig als Wirt für die heterologe
Genexpression verwendet. Allerdings haben grosse Mengen von heterologem
Protein die Tendenz, sich im Inneren der Zelle anzureichern. Die
anschliessende Reinigung des gewünschten
Proteins aus der Masse intrazellulärer Proteine von E. coli kann
gelegentlich schwierig sein.
-
Im
Gegensatz zu E.coli sind Bakterien des Genus Bacillus wegen ihrer
Fähigkeit,
Proteine in das Kulturmedium auszuschütten, als heterologe Wirte
sehr geeignet. Andere als Wirte geeignete Bakterien sind diejenigen
der Genera Streptomyces und Pseudomonas.
-
In
Abhängigkeit
von der Natur des Polynukleotides, welches für das erfindungsgemässe Polypeptid kodiert
und/oder von der Wünschbarkeit
einer Weiterverarbeitung des exprimierten Proteins, können eukaryotische
Wirte wie Hefe oder andere Pilze bevorzugt sein. Im Allgemeinen
werden Hefezellen gegenüber
Pilzzellen bevorzugt, weil sie einfacher zu manipulieren sind. Allerdings
werden gewisse Proteine entweder von den Hefezellen schlecht ausgeschüttet oder
in einigen Fällen
nicht richtig umgesetzt (z.B. Hyperglykosylierung in Hefe). In diesen
Fällen
sollte ein anderer Pilzwirt-Organismus ausgewählt werden.
-
Beispiele
von geeigneten Expressionswirten im Rahmen der vorliegenden Erfindung
sind Pilze wie Aspergillus-Spezies (wie die in EP-A-0184438 und
EP-A-0284603 beschriebenen) und Trichoderma-Spezies; Bakterien wie
Bacillus-Spezies (wie die in EP-A-0134048 und EP-A-0253455 beschrieben),
Streptomyces-Spezies
und Pseudomonas-Spezies; und Hefen wie Kluyveromyces-Spezies (wie
die in EP-A-0096430 und EP-A-0301670
beschrieben) und Saccharomyces-Spezies. Beispielsweise können typische
Expressionswirte aus Aspergillus niger, Aspergillus niger var. tubigenis,
Aspergillus niger var. awamori, Aspergillus aculeatis, Aspergillus
nidulans, Aspergillus orvzae, Trichoderma reesei, Bacillus subtilis,
Bacillus licheniformis, Bacillus amyloliquefaciens, Kluyveromyces
lactis und Saccharomyces cerevisiae ausgewählt werden.
-
Die
Verwendung von geeigneten Wirtzellen – wie Hefe-, Pilz- und Pflanzen-Wirtzellen – kann post-translationale
Modifikationen (z.B. Myristoylation, Glycosylation, Truncation,
Lapidation und Tyrosin-, Serin- oder Threonin-Phosphorylierung) ermöglichen,
die benötigt
werden, um den erfindungsgemässen
Produkten der rekombinanten Expression eine optimale biologische
Aktivität
zu verleihen.
-
ORGANISMUS
-
Der
Begriff "Organismus" umfasst im Zusammenhang
mit der vorliegenden Erfindung irgend einen Organismus, welcher
das Target und/oder die daraus erhaltenen Produkte umfassen kann.
Beispiele von Organismen können
einen Pilz, Hefe oder eine Pflanze umfassen.
-
Der
Begriff "transgener
Organismus" umfasst
im Zusammenhang mit der vorliegenden Erfindung irgend ein Organismus,
welcher das Target und/oder die erhaltenen Produkte enthält.
-
TRANSFORMATION DER WIRTZELLEN/WIRTORGANISMEN
-
Wie
bereits früher
erwähnt,
kann der Wirtorganismus ein prokaryotischer oder ein eukaryotischer
Organismus sein. Beispiele von geeigneten prokaryotischen Wirten
umfassen E. coli und Bacillus subtilis. Lehren über die Transformation von
prokaryotisch Wirten sind im Fachgebiet gut dokumentiert, siehe
beispielsweise Sambrook et al (Molecular Cloning: A Laboratory Manual,
2nd edition, 1989, Cold Spring Harbor Laboratory Press) und Ausubel
et al., Current Protocols in Molecular Biology (1995), John Wiley & Sons, Inc.
-
Falls
ein prokaryotischer Wirt verwendet wird, dann kann es nötig sein,
die Nukleotid-Sequenz vor der Transformation geeignet zu modifizieren – beispielsweise
durch Entfernen von Introns.
-
In
einer anderen Ausgestaltung kann der transgene Organismus eine Hefe
sein. In diesem Zusammenhang wurde Hefe häufig auch als Vehikel für die heterologe
Genexpression verwendet. Die Spezies Saccharomyces cerevisiae hat
eine lange Geschichte der industriellen Verwendung, wobei diese
die Verwendung für
die heterologe Genexpression einschliesst. Die Expression von heterologen
Genen in Saccharomyces cerevisiae ist in Goodey et al (1987, Yeast
Biotechnology, D R Berry et al, eds, pp 401–429, Allen und Unwin, London)
und in King et al (1989, Molecular und Cell Biology of Yeasts, E
F Walton und G T Yarronton, eds, pp 107–133, Blackie, Glasgow) diskutiert
worden.
-
Aus
verschiedenen Gründen
ist Saccharomyces cerevisiae für
die heterologe Genexpression gut geeignet. Erstens ist es für Menschen
nicht-pathogen und es ist nicht zur Bildung gewisser Endotoxine
fähig. Zweitens
hat es eine lange Geschichte einer sicheren Verwendung nach Jahrhunderten
der kommerziellen Nutzung für
verschiedene Zwecke. Dies hat zu einer breiten öffentlichen Akzeptanz geführt. Drittens
hat die dem Organismus gewidmete extensive kommerzielle Verwendung
und Forschung zu einer Menge von Wissen über die Genetik und Physiologie
sowie zu Charakteristika der Fermentation von Saccharomyces cerevisiae in
grossem Massstab geführt.
-
Eine Übersicht
der Prinzipien der heterologen Genexpression in Saccharomyces cerevisiae
und der Ausschüttung
von Genprodukten ist in E Hinchcliffe E Kenny (1993, "Yeast as a Vehicle
for the expression of heterologous genes", Yeasts, Vol 5, Anthony H Rose und
J Stuart Harrison, eds, 2nd edition, Academic Press Ltd.) gegeben.
-
Es
sind mehrere Arten von Hefevektoren, einschliesslich integrativer
Vektoren, welche für
deren Erhalt die Rekombination mit dem Wirtgenom benötigen, sowie
autonom replizierende Plasmidvektoren, verfügbar.
-
Um
das transgene Saccharomyces herzustellen, werden Expressionskonstrukte
durch Insertion der erfindungsgemässen Nukleotid-Sequenz in ein
für die
Expression in Hefe gestaltetes Konstrukt hergestellt. Es wurden
mehrere Arten von für
die heterologe Expression verwendeten Konstrukten entwickelt. Die
Konstrukte enthalten einen in Hefe aktiven Promotor, der mit der
erfindungsgemässen
Nukleotid-Sequenz fusioniert ist, wobei es sich gewöhnlich um
einen aus Hefe stammenden Promotor wie der GAL1-Promotor handelt.
Gewöhnlich
wird eine aus Hefe stammende Signalsequenz wie die für das SUC2-Signalpeptid
kodierende Sequenz verwendet. Ein in Hefe aktiver Terminator beendet
das Expressionssystem.
-
Für die Transformation
von Hefe wurden mehrere Transformationsprotokolle entwickelt. Beispielsweise
kann ein erfindungsgemässes
transgenes Saccharomyces durch Befolgen der Lehren von Hinnen et
al (1978, Proceedings of the National Academy of Sciences of the
USA 75, 1929); Beggs, J D (1978, Nature, London, 275, 104); und
Ito, H et al (1983, J Bacteriology 153, 163–168) hergestellt werden.
-
Die
transformierten Hefezellen werden unter Verwendung von verschiedenen
selektiven Markern selektioniert. Zu den für die Transformation verwendeten
Markern gehören
verschiedene auxotrophische Marker wie LEU2, HIS4 und TRP1 sowie
dominante Antibiotikaresistenzmarker wie Aminoglycosid-Antibiotikamarker, z.B.
G418.
-
Ein
anderer Wirtorganismus ist eine Pflanze. Das Basisprinzip bei der
Konstruktion von genetisch modifizierten Pflanzen ist das Einsetzen
von genetischer Information in das Pflanzengenom, so dass eine stabile Erhaltung
des eingeführten
genetischen Materials erreicht wird. Es existieren mehrere Verfahren
zum Einsetzen der genetischen Information, wobei die zwei Hauptprinzipien
das direkte Einsetzen der genetischen Information und das Einsetzen
der genetischen Information unter Verwendung eines Vektorsystems
sind. Eine Übersicht über die
allgemeinen Verfahren kann in Artikeln von Potrykus (Annu Rev Plant
Physiol Plant Mol Biol [1991] 42:205–225) und Christou (Agro-Food-Industry
Hi-Tech March/April 1994 17–27)
gefunden werden. Weitere Lehren zur Pflanzentransformation können in
EP-A-0449375 gefunden werden.
-
Demnach
wird ein Verfahren zum Transformieren einer Wirtzelle mit einer
Nukleotid-Sequenz beschrieben, welche das Target ist oder das Target
exprimiert. Wirtzellen, die mit der Nukleotid-Sequenz transformiert
werden, können
unter Bedingungen, die für
die Expression und die Rückgewinnung
des kodierenden Proteins aus der Zellkultur geeignet sind, kultiviert
werden. Das durch eine rekombinanten Zelle erzeugte Protein kann
in Abhängigkeit
von der verwendeten Sequenz und/oder dem Vektor ausgeschüttet werden,
oder es kann intrazellulär
lokalisiert sein. Wie von den Fachpersonen verständen wird, können Expressionsvektoren, welche
die kodierende Sequenzen enthalten, mit Signalsequenzen ausgestattet
werden, welche die Sekretion der kodierenden Sequenzen durch eine
bestimmte prokaryotische oder eukaryotische Zellmembran leiten.
Andere rekombinante Konstruktionen können die kodierende Sequenz
mit einer Nukleotid-Sequenz verknüpfen, die für eine Polypeptid-Domäne kodiert,
was die Reinigung von löslichen
Proteinen erleichtern wird (Kroll DJ et al (1993) DNS Cell Biol
12:441–53).
-
PHARMAZEUTISCHE
ZUSAMMENSETZUNGEN
-
Ferner
wird eine pharmazeutische Zusammensetzung angegeben, welche eine
therapeutisch wirksame Menge des erfindungsgemässen Wirkstoffes (d.h. I:NEP)
und ein(en) pharmazeutisch annehmbare(n/s) Träger, Lösungsmittel oder Hilfsstoffe
(einschliesslich Kombinationen davon) umfasst.
-
Die
pharmazeutischen Zusammensetzungen können zur Verwendung bei Menschen
oder Tieren in der Human- bzw. Veterinärmedizin vorgesehen sein und
werden typischerweise eines oder mehrere aus einem pharmazeutisch
annehmbaren Lösungsmittel,
Träger
oder Hilfsstoff umfassen. Annehmbare Träger oder Lösungsmittel für die therapeutische
Verwendung sind im Fachgebiet der Pharmazie wohlbekannt, und sind
beispielsweise, in Remington's
Pharmaceutical Sciences, Mack Publishing Co. (A. R. Gennaro edit.
1985) beschrieben. Die Wahl des pharmazeutischen Trägers, Hilfsstoffes
oder Lösungsmittels
kann im Hinblick auf den vorgesehenen Verabreichungsweg und gemäss der pharmazeutischen
Standardpraxis erfolgen. Die pharmazeutischen Zusammensetzungen
können
als oder zusätzlich
zum Träger,
Hilfsstoff oder Lösungsmittel
irgend ein(en) geeignetes(n) Bindemittel, Schmiermittel, Suspendierungsmittel,
Beschichtungsmittel, Lösungsvermittler
umfassen.
-
Es
können
Konservierungsmittel, Stabilisierungsmittel, Farbstoffe und sogar
Geschmacksmittel in der pharmazeutischen Zusammensetzung enthalten
sein. Beispiele von Konservierungsmitteln umfassen Natriumbenzoat,
Sorbinsäure
und Ester des p-Hydroxybenzoesäure.
Antioxidanzien und Suspendierungsmittel können ebenfalls verwendet werden.
-
Es
können
in Abhängigkeit
der unterschiedlichen Verabreichungssysteme unterschiedliche Zusammensetzungs-/Formulierungs-Erfordernisse
bestehen. Beispielsweise kann die erfindungsgemässe Zusammensetzung zur Verabreichung
unter Verwendung einer Mini-Pumpe oder über den Mukosaweg, beispielsweise
als ein Nasenspray oder Aerosol zur Inhalation oder als einnehmbare
Lösung,
oder zur parenteralen Verabreichung, bei welcher die Zusammensetzung
durch eine injziierbare Form zur Verabreichung über beispielsweise einen intravenösen, intramuskulären oder
subkutanen Weg formuliert sein. Alternativ kann die Formulierung
zur Verabreichung über
beide Wege ausgelegt sein.
-
Wenn
der Wirkstoff mukosal über
die gastrointestinale Mukosa verabreicht wird, sollte er fähig sein, während der
Passage durch den Magen-Darm-Trakt stabil zu bleiben; beispielsweise
sollte er gegenüber
dem proteolytischen Abbau resistent, bei saurem pH stabil und gegenüber der
zersetzenden Wirkung von Galle resistent sein.
-
Falls
angemessen, können
die pharmazeutischen Zusammensetzungen durch Inhalation, in der
Form eines Suppositorium oder Pessars, topisch in der Form einer
Lotion, Lösung,
Creme, Salbe oder Talkpuder, durch Verwendung eines Hautpflasters,
oral in der Form von Tabletten, die Hilfsstoffe wie Stärke oder
Laktose enthalten, oder in Kapseln oder Ovuli, entweder alleine
oder in Vermischung mit Hilfsstoffen, oder in der Form von Elixieren,
Lösungen
oder Suspensionen, die Geschmacksmittel oder Färbemittel enthalten, verabreicht werden,
oder sie können
parenteral, beispielsweise intravenös, intramuskulär oder subkutan
injiziiert werden. Für
die parenterale Verabreichung können
die Zusammensetzungen am besten in der Form einer sterilen wässrigen
Lösung,
welche andere Substanzen enthalten kann, beispielsweise genügend Salze
oder Monosaccharide, um die Lösung
mit dem Blut isotonisch zu machen, verwendet werden. Für die buccale
oder sublinguale Verabreichung können
die Zusammensetzungen in der Form von Tabletten oder Pastillen,
welche auf herkömmliche
Weise formuliert sein können,
verabreicht werden.
-
Für gewisse
Ausgestaltungen können
die Wirkstoffe auch in Kombination mit einem Cyclodextrin verwendet
werden. Von Cyclodextrinen ist bekannt, dass sie Einschluss- und
nicht- Einschluss-Komplexe
mit den Wirkstoffmolekülen
bilden. Die Bildung eines Wirkstoff-Cyclodextrin-Komplexes kann
die Löslichkeit,
die Auflösungsgeschwindigkeit,
die Bioverfügbarkeit
und/oder die Stabilitätseigenschaften
eines Wirkstoffmoleküls modifizieren.
Wirkstoff-Cyclodextrin-Komplexe
sind im Allgemeinen für
die meisten Dosierungsformen und Verabreichungswege nützlich.
Als Alternative zur direkten Komplexierung mit dem Wirkstoff kann
das Cyclodextrin als ein Hilfszusatz, z.B. als ein Träger, Lösungsmittel
oder Lösungsvermittler
verwendet werden. Alpha-, beta- und gamma-Cyclodextrine werden am
häufigsten
verwendet und sind geeignet. Beispiele sind in WO-A-91/11172, WO-A-94/02518
und WO-A-98/55148 beschrieben.
-
In
einer bevorzugten Ausgestaltung werden die erfindungsgemässen Wirkstoffe
systemisch (wie oral, buccal, sublingual), vorzugsweise oral verabreicht.
-
Demnach
liegt der Wirkstoff vorzugsweise in einer Form vor, die für die orale
Verabreichung geeignet ist.
-
Für gewisse
Ausgestaltungen wirkt der Wirkstoff – wenn im Gebrauch – vorzugsweise
nicht auf das Zentralnervensystem.
-
Für gewisse
Ausgestaltungen ist der Wirkstoff – wenn in Gebrauch – vorzugsweise
peripher wirksam.
-
VERABREICHUNG
-
Der
Begriff "verabreicht" umfasst die Verabreichung
durch virale oder nicht-virale Verfahren. Die viralen Verabreichungsmechanismen
umfassen ohne darauf beschränkt
zu sein adenovirale Vektoren, adeno-assoziierte virale (AAV) Vektoren,
herpes-virale Vektoren, retrovirale Vektoren, lentivirale Vektoren
und baculovirale Vektoren. Nicht-virale Verabreichungsmechanismen
umfassen die lipid-vermittelte Transfektion, Liposomen, Immunoliposomen,
Lipofektin, kationische faziale Amphiphile (CFAs) und Kombinationen
davon.
-
Der
erfindungsgemässe
Wirkstoff kann alleine verabreicht werden, aber er wird im Allgemeinen
als pharmazeutische Zusammensetzung verabreicht werden – z.B. wenn
der Wirkstoff in Vermischung mit einem geeigneten pharmazeutischen
Hilfsstoff, Lösungsmittel
oder Träger
vorliegt, welche im Hinblick auf den beabsichtigten Verabreichungsweg
und gemäss
der pharmazeutischen Standardpraxis ausgewählt sind.
-
Beispielsweise
kann der Wirkstoff (z.B. oral oder topisch) in der Form von Tabletten,
Kapseln, Ovuli, Elixieren, Lösungen
oder Suspensionen, welche Geschmacksmittel oder Färbemittel
enthalten können,
für Anwendungen
mit unmittelbarer, verzögerter,
modifizierter, anhaltender, gepulster oder kontrollierter Freisetzung verabreicht
werden.
-
Die
Tabletten können
Hilfsstoffe wie mikrokristalline Cellulose, Laktose, Natriumcitrat,
Kalziumcarbonat, dibasisches Kalziumphosphat und Glycin, Aufschlussmittel
wie Stärke
(bevorzugt Mais-, Kartoffel- oder Tapiokastärke), Natriumstärke-Glykollat,
Croscarmellose-Natrium- und gewisse komplexen Silikate und Granulations-Bindemittel
wie Polyvinylpyrrolidon, Hydroxypropylmethylcellulose (HPMC), Hydroxypropylcellulose (HPC),
Sucrose, Gelatine und Akazie enthalten. Ausserdem können Schmiermittel
wie Magnesiumstearat, Stearinsäure,
Glycerylbehenat und Talk eingeschlossen werden.
-
Feste
Zusammensetzungen ähnlichen
Typs können
auch als Füllmittel
in Gelatinekapseln verwendet werden. Bevorzugte Hilfsstoffe umfassen
in diesem Zusammenhang Laktose, Stärke, eine Cellulose, Milchzucker
oder Polyethylenglykole hohen Molekulargewichtes. Für wässrige Suspensionen
und/oder Elixiere kann der Wirkstoff mit verschiedenen Süssmitteln
oder Geschmacksmitteln, Färbemitteln
oder Farbstoffen, mit Emulgierungssmitteln und/oder Suspendierungsmitteln
und mit Verdünnungsmitteln
wie Wasser, Ethanol, Propylenglycol und Glycerin und Kombinationen
davon kombiniert werden.
-
Die
Verabreichungswege (Abgabe) umfassen ohne darauf beschränkt zu sein
eines oder mehrere von: oral (z.B. als eine Tablette, Kapsel oder
als eine einnehmbare Lösung),
topisch, mukosal (z.B. als ein Nasenspray oder Aerosol zur Inhalation),
nasal, parenteral (z.B. über
eine injizierbare Form), gastrointestinal, intraspinal, intraperitoneal,
intramuskulär,
intravenös,
intrauterin, intraocular, intradermal, intrakranial, intratracheal,
intravaginal, intrazerebroventrikulär, intrazerebral, subkutan,
ophthalmisch (einschliesslich intravitreal oder intracameral), transdermal,
rektal, buccal, vaginal, epidural, sublingual.
-
Es
versteht sich, dass nicht der gesamte Wirkstoff über denselben Weg verabreicht
werden muss. Ebenso gilt, dass falls die Zusammensetzung mehr als
eine aktive Komponente umfasst, dann diese Komponenten über unterschiedliche
Wege verabreicht werden können.
-
Falls
der erfindungsgemässe
Wirkstoff (d.h. I:NEP) parenteral verabreicht wird, dann umfassen
Beispiele von solcher Verabreichung eines oder mehrere von: intravenöse, intra-arterielle,
intraperitoneale, intrathecale, intraventrikuläre, intraurethrale, intrasternale,
intrakraniale, intramuskuläre
oder subkutane Verabreichung des Wirkstoffes; und/oder unter Verwendung
von Infusionsverfahren.
-
Für die parenterale
Verabreichung wird der Wirkstoff am besten in der Form einer sterilen
wässrigen Lösung verwendet,
welche andere Substanzen, beispielsweise genügend Salze oder Glucose, um
die Lösung mit
dem Blut isotonisch zu machen, enthalten kann. Die wässrigen
Lösungen
sollten falls nötig
geeignet gepuffert sein (vorzugsweise auf einen pH von 3 bis 9).
Die Herstellung von geeigneten parenteralen Formulierungen unter
sterile Bedingungen kann mittels standardmässigen pharmazeutischen Verfahren,
die den Fachpersonen wohl bekannt sind, ohne weiteres bewerkstelligt
werden.
-
Wie
angegeben, kann der erfindungsgemässe Wirkstoff (d.h. I:NEP)
intranasal oder durch Inhalation verabreicht werden und wird zweckmässigerweise
in der Form eines trockenen Puder-Inhalators oder eines Aerosolspray-Zubereitung
aus einem unter Druck stehenden Behälter, Pumpe, Spray oder Vernebler
unter Verwendung eines geeigneten Treibgases, z.B. Dichlorodifluoromethan,
Trichlorofluoromethan, Dichlorotetrafluoroethan, ein Hydrofluoroalkan
wie 1,1,1,2- Tetrafluoroethan
(HFA 134ATM) oder 1,1,1,2,3,3,3-Heptafluoropropan
(HFA 227EATM), Kohlenstoffdioxid oder andere
geeignete Gase verabreicht. Im Falle eines unter Druck stehenden
Aerosols kann die Dosierungseinheit durch Einsetzen eines Ventils
zur Verabreichung einer abgemessenen Menge bestimmt werden. Die
unter Druck stehenden Behälter,
Pumpen, Sprays oder Vernebler können
eine Lösung
oder Suspension der aktiven Verbindung enthalten, z.B. durch Verwendung
eines Gemisches von Ethanol und dem Treibgas als Lösungsmittel,
welches zusätzlich
ein Schmiermittel, z.B. Sorbitantrioleat, enthalten kann. Kapseln
und Patronen (beispielsweise aus Gelatine hergestellt) zur Verwendung
in einem Inhalator oder Insufflator können so formuliert werden,
dass sie ein Pudergemisch des Wirkstoffes und einer geeigneten Puderbasis
wie Laktose oder Stärke
enthalten.
-
Alternativ
kann der Wirkstoff in der Form eines Suppositoriums oder Pessars
verabreicht werden, oder er kann topisch in der Form eines Gels,
eines Hydrogels, einer Lotion, einer Lösung, einer Creme, einer Salbe oder
eines Staub-puders angewendet werden. Der Wirkstoff kann auch dermal
oder transdermal, beispielsweise unter Verwendung eines Hautpflasters,
verabreicht werden. Sie können
auch über
die pulmonalen oder rektalen Wege verabreicht werden. Sie können auch über den
okularen Weg verabreicht werden. Für die ophthalmische Verwendung
können
die Verbindungen als mikronisierte Suspensionen in isotonischer,
pH-angepasster, steriler Kochsalzlösung, oder bevorzugt als Lösungen in
isotonischer, pH-angepasster, steriler Kochsalzlösung, wahlweise in Kombination
mit einem Konservierungsmittel wie einem Benzylalkoniumchlorid,
formuliert sein. Alternativ können
sie als eine Salbe wie Petrolatum formuliert sein.
-
Für die topische
Applikation auf die Haut kann der Wirkstoff als eine geeignete Salbe
formuliert sein, welche die aktive Verbindung in suspendierter oder
gelöster
Form enthält,
beispielsweise ein Gemisch mit einem oder mehreren der Folgenden:
Mineralöl,
flüssiges
Petrolatum, weisses Petrolatum, Propylenglykol, Polyoxyethylen-polyoxypropylen-Verbindung, emulgierender
Wachs und Wasser. Alternativ kann er als eine geeignete Lotion oder
Creme formuliert sein, die beispielsweise in einem Gemisch mit einem
oder mehreren der Folgenden suspendiert oder aufgelöst ist:
Mineralöl,
Sorbitanmonostearat, ein Polyethylenglycol, flüssiges Paraffin, Polysorbat-60,
Cetylesterwachse, Cetearylalkohol, 2-Octyldodecanol, Benzylalksohol
und Wasser.
-
Die
erfindungsgemässen
Zusammensetzungen können
durch direkte Injektion verabreicht werden.
-
Für gewisse
Anwendungen wird der Wirkstoff vorzugsweise oral verabreicht.
-
Für gewisse
Anwendungen wird der Wirkstoff vorzugsweise topisch verabreicht.
-
DOSISSPIEGEL
-
Typischerweise
wird ein Arzt die aktuelle Dosierung bestimmen, welche für ein einzelnes
Subjekt am geeignetesten ist. Der spezifische Dosisspiegel und die
Häufigkeit
der Dosierung für
irgend einen bestimmten Patient können variiert werden und hängen von
einer Vielzahl von Faktoren ab, welche die Aktivität der verwendeten
spezifischen Verbindung, die metabolische Stabilität und die
Wirkungsdauer dieser Verbindung, das Alter, das Körpergewicht,
den allgemeinen Gesundheitszustand, das Geschlecht, die Ernährung, die
Art und die Zeit der Verabreichung, die Ausscheidungsgeschwindigkeit,
die Wirkstoffkombinationen, den Schweregrad des bestimmten Krankheitszustandes
und das sich der Therapie unterziehende Individuum umfassen. Der Wirkstoff
und/oder die erfindungsgemässe
Zusammensetzung können
in Übereinstimmung
mit einem Regimen von 1- bis 10-mal pro Tag, wie einmal oder zweimal
pro Tag, verabreicht werden.
-
Für die orale
und parenterale Verabreichung an menschliche Patienten kann der
tägliche
Dosierungsspiegel des Wirkstoffs in einer einzelnen oder in aufgeteilten
Dosen vorliegen.
-
In
Abhängigkeit
des Bedarfs kann der Wirkstoff mit einer Dosis von 0.01 bis 30 mg/kg
Körpergewicht, beispielsweise
von 0.1 to 10 mg/kg, vorzugsweise von 0.1 to 1 mg/kg Körpergewicht
verabreicht werden. Natürlich
sind die hier erwähnten
Dosierungen Beispiele für
den durchschnittlichen Fall. Es kann natürlich Einzelbeispiele geben,
wo höhere
oder tiefere Dosierungsbereiche bevorzugt werden.
-
FORMULIERUNG
-
Der
Wirkstoff kann zu einer pharmazeutischen Zusammensetzung formuliert
sein, beispielsweise durch Vermischen mit einem oder mehreren geeigneten
Trägern,
Lösungsmitteln
oder Hilfsstoffen unter Verwendung von im Fachgebiet bekannten Verfahren.
-
Nachfolgend
sind einige nicht-einschränkende
Beispiele von Formulierungen angegeben.
-
Formulierung
1: Eine Tablette wird unter Verwendung der folgenden Ingredienzien
hergestellt:
| | Gewicht/mg |
| Wirkstoff | 250 |
| Cellulose,
mikrokristallin | 400 |
| Kieselsäure, hochdispers | 10 |
| Stearinsäure | 5 |
| Total | 665 |
die Komponenten werden vermischt und zur Bildung
von Tabletten komprimiert, wovon jede 665 mg wiegt.
-
Formulierung
2: Eine intravenöse
Formulierung kann wie folgt hergestellt werden:
| Wirkstoff | 100
mg |
| Isotonische
Kochsalzlösung | 1.000
ml |
-
PHARMAZEUTISCH
AKTIVES SALZ
-
Der
Wirkstoff kann als ein pharmazeutisch annehmbares Salz verabreicht
werden. Typischerweise kann ein pharmazeutisch annehmbares Salz
ohne weiteres durch Verwendung einer gewünschten Säure oder ggf. Base hergestellt
werden. Das Salz kann aus der Lösung
ausgefällt
und durch Filtration gesammelt werden oder es kann durch Abdampfen
des Lösungsmittels
gewonnen werden.
-
TIERISCHE TESTMODELLE
-
Zur
Untersuchung und/oder zur Entwicklung von Therapien oder therapeutischen
Wirkstoffen für
die Behandlung von FSAD können
in-vivo-Modelle verwendet werden. Die Modelle könnten zur Untersuchung der Wirkung
von verschiedenen Werkzeugen/Leitverbindungen auf eine Vielzahl
von Parametern verwendet werden, die auf die sexuelle Erregungsantwort
hinweisen. Diese tierischen Testmodelle können als oder im erfindungsgemässen Assay
verwendet werden. Das tierische Testmodell wird ein nicht-menschliches
tierisches Testmodell sein.
-
Es
gibt eine Anzahl von Tiermodellen für die vaskulogene weibliche
Sexualstörung
(FSAD), die verwendet werden könnten.
-
Beispielsweise
kann auf invasive Tiermodelle verwiesen werden (siehe z.B. Park
et al., 1997). Dabei können
die vaginale und klitorale hämodynamische
Antwort bei normalen und atherosklerotischen weiblichen Hasen nach
Pelvisnervstimulation direkt aufgezeichnet werden. Die in-vivo-Wirkungen von
cAMP-Potenziatoren können
entweder bei normalen oder bei FSAD-Tieren untersucht werden.
-
Als
weiteres Beispiel kann auf nicht-invasive Tiermodelle verwiesen
werden (siehe z.B. die Übersicht von
Goldstein et al., 1998; Laan et al., 1998). Hier liefert die Pulswellen-Doppler-Ultrasonographie
ein Mittel zum Nachweis der Blutflussveränderungen in den vaginalen
und klitoralen Arterien. Dieses Modell kann zur Untersuchung von
vaskulogenen Wirkungen während
der pharmakologischen Verabreichung von Vasodilatatoren verwendet
werden.
-
Andere
nicht-invasive Verfahren, die verwendet werden können, umfassen die vaginale
Photoplethysmographie, welche ein quantitatives Mass für die Anschwellung
der Vaginalmukosa liefert, sowie Verfahren der vaginalen thermischen
Clearance, welche auf dem Prinzip beruhen, dass Veränderungen
des vaginalen Blutflusses durch Messung des Wärmeabflusses von einer auf
konstanter Temperatur gehaltenen intravaginalen Sonde gemessen werden
können.
-
EIN TIERMODELL
DER SEXUELLEN ERREGUNG
-
In
unseren Studien wurde ein robustes reproduzierbares Modell der Physiologie
der sexuellen Erregung entwickelt. Dieses Modell verwendet einen
anästhesierten
Hasen und benutzt zur Aufzeichnung des genitalen Blutflusses Laser-Doppler-Verfahren,
während
die kardiovaskulären
Parameter routinemässig
aufgezeichnet werden. Es ist möglich,
kleine Veränderungen
im vaginalen (und sogar im klitoralen) Blutfluss, die durch Stimulation
des Pelvisnervs oder durch Infusion von VIP in der Abwesenheit und
in Gegenwart von Testwirkstoffen induziert werden, zu messen.
-
Wir
glauben, dass unser Tiermodell die klinischen Daten direkt widerspiegelt.
Demnach kann dieses Modell für
die Untersuchung von Kandidaten-Wirkstoffen zur Behandlung von FSAD,
beispielsweise durch Messung der Erhöhung des vaginalen oder klitoralen
Blutflusses, verwendet werden.
-
PHYSIOLOGISCHE
MESSUNGEN DER WEIBLICHEN SEXUELLEN ERREGUNG
-
Es
kann eine Reihe von unterschiedlichen Verfahren zur Messung des
klitoralen und vaginalen Blutflusses verwendet werden. Beispielsweise
kann die vaginale Photoplethysmographie, die vaginale Wärmeauswasch-Technik,
die klitorale und vaginale kontrastverstärkte MRI, die klitorale/vulvale
gepulste Laser-Doppler-Bildgebung und die klitorale Ultrasonographie
eingesetzt werden.
-
Die
Quantifizierung der vaginalen Lubrikation kann ebenfalls gemessen
werden, wobei im Fachgebiet bekannte Verfahren – wie (a) das Wägen von
vaginalen Tampons prä-
und poststimulatiorisch und (b) die Messung des pH der vaginalen
Flüssigkeit – zum Einsatz
kommen. In Bezug auf den letzten Aspekt gilt, dass das im normalen
Ruhezustand saure Medium in der Vagina durch Annäherung an den pH-Wert des Blutes
akalischer wird, wenn bei der sexuellen Stimulation eine Transsudation
von Flüssigkeit
stattfindet.
-
NEP (neutrale Endopeptidase)
-
Gemäss der vorliegenden
Erfindung ist das Target ein PcAMP-Target, welches PcAMP-Target NEP ist.
-
Nukleotid-Sequenzen
und Aminosäurensequenzen
für NEP
sind aus der Literatur entnehmbar. Einige Sequenzen sind in den
hier aufgeführten
Sequenzprotokollen vorgestellt.
-
NEP
ist NEP (EC 3.4.24.11) (auch als Enkephalinase oder Endopeptidase-2
bekannt). Hier wurde in der Vagina von Menschen und Hasen NEP EC
3.4.24.11 mRNS und exprimiertes Protein gefunden.
-
Hier
wird angenommen, dass bei Frauen, einschliesslich solcher die an
FSAD leiden, VIP durch NEP EC3.4.24.11 abgebaut wird. Demnach werden
NEP-Inhibitoren die endogene vasorelaxierende Wirkung des während der
Erregung freigesetzten VIP potenzieren. Dies wird zu einer Behandlung
von FSAD führen,
beispielsweise durch Erhöhung
der vaginalen Anschwellung. Es wurde gezeigt, dass selektive Inhibitoren
von NEP EC 3.4.24.11 die durch Pelvisnervstimulation und durch VIP
induzierte Erhöhung
im genitalen (z.B. vaginalen oder klitoralen) Blutfluss verstärken. Ausserdem
verstärken
diese selektiven NEP-Inhibitoren die durch VIP und über Nerven
vermittelte Relaxationen von isolierter Vaginawand.
-
Hintergrundslehren über NEP
sind von Victor A. McKusick et al auf http://www3.ncbi.nlm.nih.gov/Omim/searchomim.htm
angegeben. Die folgenden Informationen betreffend NEP wurden von jener
Quelle entnommen.
-
Das
Antigen der gemeinen akuten lymphozytischen Leukämie ist ein wichtiger Zelloberflächenmarker bei
der Diagnose von akuter lymphozytischen Leukämie (ALL) beim Menschen. Es
ist auf den leukämischen Zellen
vom prä-B-Phänotyp vorhanden,
welche 85% der ALL-Fälle
ausmachen. CALLA ist jedoch nicht auf leukämische Zellen beschränkt und
wird auf einer Vielzahl von normalen Geweben gefunden. CALLA ist
ein Glycoprotein, das besonders in den Nieren vorkommt, wo es auf
dem Bürstenrand
der proximalen Tubuli und auf dem glomerulären Epithel vorliegt. Letarte
et al. (1988) haben eine für
CALLA kodierende cDNS kloniert und gezeigt, dass die aus der cDNS-Sequenz
abgeleitete Aminosäurensequenz
zu derjenigen von menschlicher membran-assoziierter neutraler Endopeptidase
(NEP; EC 3.4.24.11), auch als Enkephalinase bekannt, identisch ist.
NEP spaltet Peptide an der Aminostelle von hydrophoben Resten und
inaktiviert mehrere Peptidhormone, einschliesslich Glucagon, Enkephaline,
Sunstanz-P, Neurotensin, Oxytocin, und Bradykinin. Mittels cDNS-Transfektionsanalyse
bestätigten
Shipp et al. (1989), dass CALLA eine funktionell neutrale Endopeptidase
von dem Typ ist, der vorgängig
als Enkephalinase bezeichnet wurde. Barker et al. (1989) zeigten,
dass das CALLA-Gen, welches für
ein 100-kD Typ-II-Transmembran-Glycoprotein kodiert, als eine einzelne
Kopie mit einer Grösse
von mehr als 45 kb vorliegt, welches in malignem Gewebe, das Zelloberflächen-CALLA exprimiert,
nicht umgelagert wird. Das Gen wurde aufgrund der Untersuchung von
somatischen Zellhybriden auf das menschlichen Chromosom 3 lokalisiert,
und durch in-situ-Hybridisierung
wurde die Lokalisation auf 3q21-q27 regionalisiert. Tran-Paterson
et al. (1989) ordneten das Gen anhand von Southern blot Analyse
von DNS aus somatischen Mensch-Nagetier-Zellhybriden ebenfalls dem
Chromosom 3 zu. D'Adamio
et al. (1989) zeigten, dass das CALLA-Gen mehr als 80 kb umfasst
und aus 24 Exons zusammengesetzt ist."
-
I:NEP
-
Wie
oben angegeben, kann der Wirkstoff irgend ein geeigneter Wirkstoff
sein, der als I:NEP wirken kann.
-
Details über ein
geeignetes Assay-System zur Identifizierung und/oder Untersuchung
eines I:NEP sind im folgenden Abschnitt angegeben.
-
I:NEPs
werden in den folgenden Übersichtsartikeln
diskutiert:
- Pathol. Biol., 46(3), 1998, 191.
- Current Pharm. Design, 2(5), 1996, 443.
- Biochem. Soc. Trans., 21(3), 1993, 678.
- Handbook Exp. Pharmacol., 104/1, 1993, 547.
- TiPS, 11, 1990, 245.
- Pharmacol. Rev., 45(1), 1993, 87.
- Curr. Opin. Inves. Drugs, 2(11), 1993, 1175.
- Antihypertens. Drugs, (1997), 113.
- Chemtracts, (1997), 10(11), 804.
- Zinc Metalloproteases Health Dis. (1996), 105.
- Cardiovasc. Drug Rev., (1996), 14(2), 166.
- Gen. Pharmacol., (1996), 27(4), 581.
- Cardiovasc. Drug Rev., (1994), 12(4), 271.
- Clin. Exp. Pharmacol. Physiol., (1995), 22(1), 63.
- Cardiovasc. Drug Rev., (1991), 9(3), 285.
- Exp. Opin. Ther. Patents (1996), 6(11), 1147.
-
I:NEPs
sind in den folgenden Dokumenten beschrieben:
- EP-509442A
- US-192435
- US-4929641
- EP-599444B
- US-884664
- EP-544620A
- US-798684
- J. Med. Chem. 1993, 3821.
- Circulation 1993, 88(4), 1.
- EP-136883
- JP-851 36554
- US-4722810
- Curr. Pharm. Design, 1996, 2, 443.
- EP-640594
- J. Med. Chem. 1993, 36(1), 87.
- EP-738711-A
- JP-270957
- CAS # 115406-23-0
- DE-19510566
- DE-19638020
- EP-830863
- JP-98101565
- EP-733642
- WO9614293
- JP-08245609
- JP-96245609
- WO9415908
- JP05092948
- WO-9309101
- WO-9109840
- EP-519738
- EP-690070
- J. Med. Chem. (1993), 36, 2420.
- JP-95157459
- Bioorg. Med. Chem. Letts., 1996, 6(1), 65.
-
Bevorzugte
I:NEPs sind in den folgenden Dokumenten beschrieben:
- EP-A-0274234
- JP-88165353
- Biochem. Biophys. Res. Comm., 1989, 164, 58
- EP-629627-A
- US-77978
- Perspect. Med. Chem. (1993), 45.
- EP-358398-B
-
Bevorzugte
Beispiele von I:NEPs sind aus den folgenden Strukturen ausgewählt:
-
-
-
-
Besonders
bevorzugte I:NEPs sind aus den folgenden Strukturen ausgewählt:
-
-
-
Besonders
bevorzugte I:NEPs sind aus den folgenden Strukturen ausgewählt:
-
-
-
-
Diese
Verbindungen werden gemäss
den im experimentellen Abschnitt beschriebenen Lehren hergestellt
(unten). Diese Verbindungen wurden als Wirkstoffe getestet und erwiesen
sich zur Potenzierung von cAMP als nützlich und sind demnach für die Behandlung
von FSAD nützlich.
Einige der diese Verbindungen betreffenden experimentellen Daten
sind im experimentellen Abschnitt dargestellt (unten).
-
NEP-ASSAY
-
DIE
HERSTELLUNG UND DER ASSAY VON LÖSLICHER
(NEP) NEUTRALER ENDOPEPTIDASE AUS DEM NIERENKORTEX VON HUNDEN, RATTEN,
HASEN UND MENSCHEN.
-
Lösliche NEP
wird aus dem Nierenkortex gewonnen, und die Aktivität wird durch
Messung der Geschwindigkeit der Spaltung des NEP-Substrates Abz-D-Arg-Arg-Leu-EDDnp
unter Bildung von dessen fluoreszierendem Produkt Abz-D-Arg-Arg
bestimmt.
-
EXPERIMENTELLES VERFAHREN
-
1. MATERIALIEN
-
Sämtliches
Wasser ist zweifach entionisiert.
- 1.1 Gewebe
Menschliche
Niere IIAM (Pennsylvania. U.S.A.
Rattenniere
Hasenniere
Hundeniere
- 1.2 Homogenisationsmedium
100 mM Mannitol und 20 mM Tris
bei pH 7.1 2.42 g Tris (Fisher T/P630/60) wird mit 1 Liter Wasser
verdünnt, und
der pH wird unter Verwendung von 6M HCl bei Raumtemperatur auf 7.1
eingestellt. Dazu werden 18.22 g Mannitol (Sigma M-9546) zugegeben.
- 1.3 Tris-Puffer (NEP Puffer).
50 ml 50 mM Tris pH 7.4 (Sigma
T2663) werden in 950 ml Wasser verdünnt.
- 1.4 Substrat (Abz-D-Arg-Arg-Leu-EDDnp)
Wird von SNPE auf
Auftrag hergestellt und wird als Pulver bei –20°C gelagert. Eine 2 mM Stammlösung wird
durch sorgfältiges
Resuspendieren des Substrats in Tris Puffer hergestellt, wobei dieser
nicht verwirbelt oder beschallt werden darf. 600 μl Aliquote
der 2 mM-Stammlösung werden
während
bis zu einem Monat bei –20°C gelagert.
(Medeiros, M.A.S., Franca, M.S.F. et al., (1997), Brazilian Journal
of Medical und Biological Research, 30, 1157–1162).
- 1.5 Gesamtprodukt
Proben, die einer 100%igen Substrat-zu-Produkt
Umwandlung entsprechen, werden mit auf die Platte gegeben, um den
%-Substratumsatz zu bestimmen. Das Gesamtprodukt wird durch Inkubieren
von 1 ml des 2 mM Substrates mit 20 μl der Enzym-Stammlösung während 24
Stunden bei 37°C
erzeugt.
- 1.6 Stopplösung.
Eine
300 μM Stammlösung von
Phosphoramidon (Sigma R7385) wird in NEP-Puffer hergestellt und
in 50 μl
Aliquoten bei –20
gelagert.
- 1.7 Dimethylsulfoxid (DMSO).
- 1.8 Magnesiumchlorid – MgCl2.6H2O (Fisher MO600/53).
- 1.9 Black-Assay-Platten mit 96 Vertiefungen mit flachem Boden
(Costar 3915 oder Packard).
- 1.10 Topseal A (Packard 6005185).
- 1.11 1 Zentrifugenröhrchen
-
2. SPEZIFISCHE AUSSTATTUNG
-
- 2.1 Sorvall RC-5B Zentrifuge (SS34 GSA Rotor,
vorgekühlt
auf 4°C).
- 2.2 Braun Miniprimer Mixer.
- 2.3 Beckman CS-6R Zentrifuge.
- 2.4 Fluostar Galaxy.
- 2.5 Wesbart 1589 Schüttelinkubator.
-
3. METHODEN
-
- 3.1 GEWEBE-PRÄPARATION
- 3.2 NEP von Hunden, Ratten, Hasen und Menschen wird aus dem
Nierenkortex unter Verwendung einer von Booth, A.G. & Kenny, A.J. (1974)
Biochem. J. 142, 575–581
angepassten Methode erhalten.
- 3.3 Gefrorene Nieren werden auf Raumtemperatur auftauen gelassen
und der Kortex wird von der Medulla abgetrennt.
- 3.4 Der Kortex wird fein zerschnitten und in ungefähr 10 Volumina
des Homogenisationspuffers (1.2) unter Verwendung eines Braun Miniprimer
(2.2) homogenisiert.
- 3.5 Magnesiumchlorid (1.8) (20,3 mg/gm Gewebe) wird zum Homogenisat
zugegeben und während
15 Minuten in einem Eiswasserbad gerührt.
- 3.6 Das Homogenisat wird während
12 Minuten mit 1.500 g (3.820 rpm) in einer Beckman Zentrifuge (2.3) zentrifugiert,
und anschliessend wird der Überstand
in ein frisches Zentrifugenrohr gebracht und das Pellet verworfen.
- 3.7 Der Überstand
wird während
12 Minuten mit 15.000 g (12.100 rpm) in der Sovall Zentrifuge (2.1)
zentrifugiert, und der Überstand
wird entfernt.
- 3.8 Die hellrosa Phase am oberen Ende des verbleibenden Pellets
wird entfernt und erneut in Homogenisationspuffer, der Magnesiumchlorid
(9 mg MgCl in 5 ml Puffer pro 1 g Gewebe) enthält, suspendiert.
- 3.9 Die Suspension wird während
12 Minuten bei 2.200 g (4,630 rpm) in einer Beckman Zentrifuge zentrifugiert,
und anschliessend wird das Pellet verworfen.
- 3.10 Der Überstand
wird während
12 Minuten bei 15.000 g (12.100 rpm) unter Verwendung der Sorvall
Zentrifuge zentrifugiert, und der Überstand wird entfernt.
- 3.11 Das übrigbleibende
Pellet wird erneut in Homogenisationspuffer, welcher Magnesiumchlorid
(0,9 mg MgCl in 0,5 ml Puffer pro 1 g Gewebe) enthält, suspendiert.
Eine homogene Suspension wird unter Verwendung eines Braun Miniprimer
(2.2) erhalten. Diese wird dann in 100 μl Aliquoten eingefroren, um
die NEP-Aktivität zu bestimmen.
-
4.0 BESTIMMUNG DER NEP-AKTIVITÄT
-
Die
Aktivität
der vorgängig
aliquotierten NEP wird aufgrund ihrer Fähigkeit, das NEP-spezifische
Peptid-Substrat zu spalten, gemessen.
- 4.1 Eine
4%-ige DMSO/NEP-Pufferlösung
(4 ml DMSO in 96 ml NEP-Puffer) wird hergestellt.
- 4.2 Substrat, Gesamtprodukt, Enzym und Phosphoramidon-Stammlösung werden
zum Auftauen auf Eis belassen.
- 4.3 50 μl
4%-ige DMSO/NEP-Pufferlösung
werden in jede Vertiefung gegeben.
- 4.4 Die 2 mM Substrat-Stammlösung
wird 1:40 verdünnt,
woraus sich eine 50 μM
Lösung
ergibt. 100 μl
von 50 μM
Substrat wird in jede Vertiefung zugegeben (Endkonzentration im
Assay 25 μM).
- 4.5 50 μl
einer Reihe von Enzymverdünnungen
werden zugegeben, um die Reaktion auszulösen (gewöhnlich werden 1:100, 1:200,
1:400, 1:800, 1:1600 und 1:3200 verwendet). 50 μl des NEP-Puffers werden in die
leeren Vertiefungen zugegeben.
- 4.6 Das 2 mM Gesamtprodukt wird 1:80 verdünnt, woraus sich eine 25 μM Lösung ergibt.
200 μl des
25 μM Produkt
werden in die ersten vier Vertiefungen einer neuen Platte zugegeben.
- 4.7 Die Platten werden während
60 Minuten bei 37°C
in einem Schüttelinkubator
inkubiert.
- 4.8 Die 300 μM
Phosphoramidon-Stammlösung
wird 1:100 auf 300 nM verdünnt.
Die Reaktion wird durch Zugabe von 100 μl 300 nM Phosphoramidon gestoppt
und während
20 Minuten bei 37°C
in einem Schüttelinkubator
inkubiert, und anschliessend auf dem BMG Fluostar Galaxy (ex320/em420)
ausgemessen.
-
5. NEP-INHIBITIONS-ASSAYS
-
- 5.1 Substrat, Gesamtprodukt, Enzym und Phosphoramin-Stammlösungen werden
zum Auftauen auf Eis belassen.
- 5.2 Verbindungs-Stammlösungen
werden in 100% DMSO hergestellt und 1:25 in NEP-Puffer verdünnt, woraus
sich eine 4% DMSO-Lösung
ergibt. Alle weiteren Verdünnungen
werden in einer 4% DMSO-Lösung (4
ml DMSO in 96 ml NEP-Puffer) durchgeführt.
- 5.3 50 μl
der Verbindung werden zweifach in die Platte mit 96 Vertiefungen
gegeben, und 50 μl
des 4% DMSO/NEP-Puffers
werden in die Kontroll- und in die leeren Vertiefungen gegeben.
- 5.4 Die 2 mM Substrat-Stammlösung
wird mit NEP Puffer 1:40 verdünnt,
woraus sich eine 50 μM
Lösung ergibt
(275 μl
des 2 mM Substrats zu 10.73 ml Puffer zugegeben ist für 1 Platte
ausreichend).
- 5.5 Die Enzym-Stammlösung
wird mit NEP-Puffer verdünnt
(aus Aktivitäts-Tests
bestimmt).
- 5.6 Die 2 mM Gesamtprodukt-Stammlösung wird 1:80 mit NEP-Puffer verdünnt, woraus
sich eine 25 μM Lösung ergibt.
200 μl werden
zu den ersten vier Vertiefungen einer separaten Platte gegeben.
- 5.7 Die 300 μM
Phosphoramidon-Stammlösung
wird 1:1000 verdünnt,
woraus sich eine 300 μM
Stammlösung
(11 μM Phosphoramidon
zu 10.99 ml NEP-Puffer) ergibt.
- 5.8 In jede Vertiefung der Platte mit 96 Vertiefungen wird das
Folgende zugegeben:
- Tabelle Reagenzien, die in die Platte mit 96 Vertiefungen zuzugeben
sind.
- 5.9 Die Reaktion wird durch Zugabe des NEP-Enzyms ausgelöst, und
anschliessend wird während
1 Stunde bei 37°C
in einem Schüttelinkubator
inkubiert.
- 5.10 Die Reaktion wird durch Zugabe von 100 μl 300 nM Phosphoramidon gestoppt,
und es wird während 20
Minuten bei 37°C
in einem Schüttelinkubator
inkubiert, wonach auf dem BMG Fluostar Galaxy (ex320/em420) abgelesen
wird.
-
6. BERECHNUNGEN
-
Die
Aktivität
des NEP-Enzyms wird in Gegenwart und in Abwesenheit der Verbindung
bestimmt und in Prozentwerten ausgedrückt.
-
%-Kontroll-Aktivität (Umsatz
des Enzyms):
-
%-Aktivität mit Inhibitor:
-
Als
% der Kontrollen ausgedrückte
Aktivität:
-
Eine
sigmoidale Dosis-Antwort-Kurve wird an die %-Aktivitäten (% Kontrolle) vs. Verbindungskonzentration
angepasst und IC50-Werte werden mittels
der LabStats Ausgleichskurve in Excel berechnet.
-
PDE (Phosphodiesterase)
-
Gemäss einem
Aspekt der vorliegenden Erfindung kann ein zusätzliches Target ein anderes
PcAMP-Target wie PDE (Phosphodiesterase)
sein, insbesondere eine PDE, welche eine cAMP-hydrolysierende (und wahlweise
eine cGMP-hydrolysierende)
PDE ist.
-
Es
ist bekannt, dass cyclische Nukleotide wie cAMP und cGMP wichtige
intrazelluläre
second Messengers sind. Zyklische Nukleotid-Phosphodiesterasen – anderweitig
als PDEs bekannt – sind
eine Familie von Enzymen, welche den Abbau von cyclischen Nukleotiden
katalysieren und dabei eine der zellulären Komponenten darstellen,
welche die Konzentration von cyclischen Nukleotide regulieren.
-
In
den vergangenen Jahren sind, beruhend auf Substrataffinität und Kofaktorenerfordernissen
mindestens sieben PDE-Enzyme (wie PDEI – PDEVII) sowie viele Subtypen
dieser Enzyme identifiziert worden (Beavo JA und Reifsnyder DH,
Trends Pharmacol. Sci. 11:150 [1990]; Beavo J, In: Cyclic Nucleotide
Phosphodiesterases: Structure, Regulation und Drug Action., Beavo
J und Housley MD (Eds.). Wiley:Chichester, pp. 3–15 [1990]).
-
Beispiele
von PDEs umfassen: PDEI, welche eine Ca2+/Calmodulin-abhängige PDE
ist; PDEII, welche eine cAMP- und
cGMP-stimulierende PDE ist; PDEIII, welche eine cGMP-inhibierende PDE
ist; PDEIV, welche eine mit hoher Affinität cAMP-spezifische PDE ist;
und PDEV, welche eine cGMP spezifische PDE ist. PDEI usw. werden
manchmal als PDE-Typ I usw. oder PDE-Typ 1 usw bezeichnet.
-
Jede
PDE-Familie kann zwei oder mehrere Isoformen enthalten (d.h. es
können
zwei oder mehrere PDE-Isoenzyme sein). Beispielsweise ist von der
Säuger-PDE-IV,
dem Homologon des Drosophila Dunce Gens (Chen CN et al., Proc. Nat.
Acad. Sci. (USA) 83:9313 [1986]) bekannt, dass sie bei der Ratte
vier Isoformen hat (Swinnen JV et al., Proc. Nat. Acad. Sci. (USA)
86:5325 [1989]). Auch von menschlichen PDEs ist bekannt, dass sie
als Isoformen vorkommen und Splice-Varianten haben. Beispielsweise wurde
1990 über
das Klonieren einer menschlichen Isoform von PDEIV aus Monozyten
berichtet (Livi GP et al., Mol. Cell. Bio., 10:2678 [1990]).
-
Als
weiteres Beispiel haben andere Mitarbeiter unabhängig drei Splice-Varianten
von PDEIV kloniert, welche inzwischen als hPDEIV-B1, hPDEIV-B2,
und hPDEIV-B3 bezeichnet werden.
-
Lehren über cyclische
Nukleotid-Phosphodiesterasen können
auch in US-A-5932423 und US-A-5932465 gefunden werden.
-
Lehren
zu einer weiteren cyclischen Nukleotid-Phosphodiesterase – namentlich CN PCDE8 – können in
WO-A-97/35989 gefunden
werden. Gemäss
WO-A-97/35989 hat CN PCDE8 zwei Isozyme – welche als CN PCDE8A und
CN PCDE8B bezeichnet wurden. Der Begriff "Isozym" wird im Fachgebiet gelegentlich als "Isoform" angegeben.
-
Gemäss WO-A-97/35989
sind zahlreiche Inhibitoren von unterschiedlichen PDEs identifiziert
worden, und einige wurden der klinischen Evaluation unterzogen.
Beispielsweise werden PDEIII-Inhibitoren als antithrombotische Wirkstoffe,
als antihypertensive Wirkstoffe und als kardiotonische Wirkstoffe
entwickelt, die für die
Behandlung des kongestiven Herzversagens nützlich sind. Rolipram, ein
PDEIII-Inhibitor, ist zur Behandlung von Depression verwendet worden,
und anderen Inhibitoren von PDEIII werden als anti-inflammatorische Wirkstoffe
evaluiert. Von Rolipram wurde auch gezeigt, dass es den durch Lipopolysaccharid
(LPS) induzierten TNF-alpha inhibiert, von welchem gezeigt wurde,
dass er in vitro die Replikation von HIV-1 verstärkt. Demnach kann Rolipram
die HIV-1-Replikation inhibieren (Angel et al 1995 AIDS 9:1137–44). Ausserdem
wurde von Rolipram gezeigt, dass es beruhend auf seiner Fähigkeit,
die Bildung von TNF-alpha und -beta sowie von Interferon-gamma zu
unterdrücken,
bei der Behandlung der Enzephalomyelitis, dem experimentellen Tiermodell
für Multiple
Sklerose, wirksam ist (Sommer et al, 1995 Nat Med 1:244–248) und
dass es zu Behandlung der tardiven Dyskinesie wirksam sein kann
(Sasaki et al, 1995 Eur J Phamacol 282:71–76).
-
Gemäss WO-A-97/35989
gibt es auch nicht-spezifische PDE-Inhibitoren wie das zur Behandlung von Bronchialasthma
und anderen respiratorischen Erkrankungen verwendete Theophyllin
und das zur Behandlung von intermittierender Claudicatio und von
diabetesinduzierten peripheren vaskulären Erkrankungen verwendete
Pentoxiphyllin. Von Theophyllin wird angenommen, dass es bei der
Behandlung von respiratorischen Erkrankungen auf die Funktion der
glatten Muskeln in den Luftwegen sowie durch eine anti-inflammatorische oder
immunmodulatorische Kapazität
wirkt (Banner et al 1995 Respir J 8:996–1000), wobei angenommen wird, dass
es durch Inhibition der Hydrolyse sowohl von CN PDE cAMP- als auch
von cGMP wirkt (Banner et al 1995 Monaldi Arch Chest Dis 50:286–292). Pentoxiphyllin,
von dem auch bekannt ist, dass es die TNF-alpha-Bildung blockiert, kann die HIV-1-Replikation
inhibieren (Angel et al unten). Eine Liste von CN PDE-Inhibitoren ist in
Beavo 1995 oben gegeben.
-
Es
wurde vorgeschlagen, dass selektive Inhibitoren der PDEs samt ihren
Isozymen und ihren Subtypen zu einer wirksameren Therapie mit weniger
Nebenwirkungen führen
werden. Siehe beispielsweise die Angaben in den Übersichten von Wieshaar RE
et al, (J. Med. Chem., 28:537 [1985]), Giembycz MA (Biochem. Pharm.,
43:2041 [1992]) und Lowe JA und Cheng JB (Drugs of the Future, 17:799–807 [1992]).
-
Demnach
ist es bei gewissen Anwendungen wünschenswert, eine selektive
Inhibition eines einzelnen Typs von PDE zu haben.
-
Hintergrundlehren über PDEs
sind in Victor A. McKusick et al auf http://www3.ncbi.nlm.nih.gov/Omim/searchomim.htm
angegeben. Die folgenden Informationen betreffend PDE2 oder cGMP-stimulierte
PDE wurden dieser Quelle entnommen.
-
"Zyklische Nukleotide
dienen als second Messengers, welche eine Vielzahl von zellulären Antworten auf
extrazelluläre
Signale wie Hormone, Licht und Neurotransmitter vermitteln. Zyklische
Nukleotid-Phosphodiesterasen
(PDEs) spielen durch Regulierung der zellulären Konzentrationen von cyclischen
Nukleotiden eine Rolle bei der Signalübertragung. Säugerzellen
enthalten multiple PDEs, welche aufgrund ihrer Substrataffinität und ihrer
selektiven Sensitivität
auf Kofaktoren und inhibitorische Wirkstoffe in mindestens 7 Familien
unterteilt werden. Diese Familien sind: (I) Ca(2+)/calmodulin-abhängige PDEs;
(II) cGMP-stimulierte PDEs; (III) cGMP-inhibierte PDEs; (IV) cAMP-spezifische
PDEs; (V) cGMP-spezifische PDEs; (VI) Photorezeptoren-PDEs; und (VII) hohe
Affinität,
cAMP-spezifische. Aus den Aminosäurensequenzen
ist es ersichtlich, dass all diese PDE-Familien eine verwandte Domäne enthalten,
von der angenommen wird, dass sie die katalytische Domäne ist,
wobei eine ungefähr
30%-ige Sequenzidentität
zwischen den Familien vorliegt. Mitglieder derselben Familien sind
näher verwandt;
sie teilen sich 60 bis 80% Sequenz-Identität über die gesamte kodierende
Region. Michaeli et al. (1993) entwickelten ein hochsensitives funktionelles
Screening für
die Isolierung von cDNSs kodierenden cAMP-Phosphodiesterasen durch
Komplementierung von Defekten im Saccharomyces cerevisiae Strang,
dem beide endogenen cAMP-PDE-Gene, PDE1 und PDE2, fehlen. Drei Gruppen
von cDNSs, welche den 3 unterschiedlichen, für die cAMP-spezifischen PDEs
kodierenden humanen Gene entsprechen, wurden unter Verwendung dieses
funktionellen Screenings aus einer humanen Glioblastoma-cDNS-Bibliothekx
isoliert. Zwei der Gene waren mit der Drosophila 'dunce' cAMP-spezifischen
PDE nahe verwandt. Das dritte Gen, welches Michaeli et al. (1993)
als HCP1 bezeichneten, kodiert für
eine neue, cAMP-spezifische PDE. HCP1 hat eine mit den Sequenzen
der katalytisch Domänen
von allen cyclischen Nukleotid-PDEs verwandte Aminosäurensequenz.
Es ist eine cAMP-spezifische
PDE mit hoher Affinität,
welcher jedoch keine anderen Eigenschaften mit der cAMP-spezifischen
PDE-Familie teilt.
Die PDE-Aktivität
von HCP1 war gegenüber
cGMP oder anderen Inhibitoren der durch cGMP inhibierbaren PDEs
nicht sensitiv. Die biochemischen und pharmakologischen Eigenschaften
von HCP1 wiesen Michaeli et al. (1993) darauf hin, dass es sich
um ein Mitglied einer vorher nicht entdeckten cyclischen Nukleotid-PDE-Familie handelt,
welche sie als Familie VII bezeichneten. Die Northern Blot Analyse
wies auf die Gegenwart von hohen Spiegeln einer HCP1-RNS in menschlichen
Skelettmuskeln hin.
Mittels Southern Blot Analyse von somatischen
Zellhybrid-Linien
lokalisierten Milatovich et al. (1994) den HCP1-Lokus auf Chromosom 8; durch die Untersuchung
von somatischen Zellhybrid-Linien, welche unterschiedliche Regionen
von Chromosom 8 enthielten, regionalisierten sie die Zuordnung auf
8q13-q22. Han et al. (1998) lokalisierten das PDE7A Gen mittels
Fluoreszenz in situ Hybridisirung auf 8q13. Mittels interspezifischer
Rückkreuzungs-Analyse
lokalisierten sie das Maus-Pde7A-Gen
auf die proximale Region von Chromosom 3."
-
Hintergrundlehren über PDE2
wurden von Jennifer P. Macke et al in http://www3.ncbi.nlm.nih.gov/Omim/searchomim.htm
angegeben. Die folgenden Informationen betreffend cGMP-stimulierte PDE wurden
dieser Quelle entnommen.
-
"Rosman et al. (1997)
klonten eine dem menschlichem PDE2A entsprechende cDNS. Das PDE2A-Gen
kodiert für
ein 941-Aminosäuren-Polypeptid
mit einer vorhergesagten Molekülmasse
von 106 kD. Die Proteinsequenz ist mit den PDE2A-Sequenzen vom Rind
und der Ratte zu 90% identisch. Die Northern Blot Analyse zeigte,
dass PDE2A als eine 4.2-kb mRNS in variierendem Ausmass in allen
geprüften
menschlichen Geweben exprimiert wurde, wobei die stärkste Expression
im Gehirn erfolgte. Expressionsstudien ergaben, dass PDE2A cAMP
und cGMP hydrolysiert und durch den PDE2A-spezifischen Inhibitor
EHNA inhibiert wird."
-
Nukleotid-Sequenzen
und Aminosäurensequenzen
für PDEs
sind in der Literatur verfügbar.
Einige Sequenzen sind in den hier aufgeführten Sequenz-Listen angegeben.
-
In
einem Aspekt wird das PDE-Target aus irgend einem oder mehreren
der folgenden PDE-Enzyme ausgewählt:
PDEcAMP 1, PDEcAMP 2,
PDEcAMP 3, PDEcAMP 4,
PDEcAMP 7 und PDEcAMP 8.
-
In
einem besonders bevorzugten Aspekt wird das PDE-Target aus irgend
einem oder mehreren der folgenden PDE-Enzyme ausgewählt: PDEcAMP 1, PDEcAMP 2,
PDEcAMP 3, und PDEcAMP 4.
-
Vorzugsweise
ist erfindungsgemäss
das Verhältnis
von PDE zu Target mindestens 2.
-
I:PDE
-
Wie
oben angegeben, kann der zusätzliche
Wirkstoff irgend ein geeigneter Wirkstoff sein, welcher als ein
I:PDE wirken kann. Zusätzlich
oder alternativ kann der erfindungsgemässe Wirkstoff auch als ein
I:PDE wirken.
-
Beispiele
von I:PDE sind in EP-A-091133 und EP-A-0771799 angegeben.
-
Vorzugsweise
ist der I:PDE ein I:PDE2. Demnach sind bevorzugte Beispielverbindungen
diejenigen, die in EP-A-0771799
vorgestellt sind.
-
Der
Einfachheit halber wird nun der Anspruch 1 von EP-A-0771799 wiederholt:
-
Purin-6-on-Derivat
der allgemeinen Formel (I):
wobei:
- R1 für Wasserstoff
oder für
ein geradkettiges oder verzweigtes Alkyl mit bis zu 8 Kohlenstoffatomen
steht;
- R2 für
ein geradkettiges oder verzweigtes Acyl mit bis zu 4 Kohlenstoffatomen,
oder für
ein geradkettiges oder verzweigtes Alkyl mit bis zu 8 Kohlenstoffatomen,
das gegebenenfalls durch Hydroxyl, Azido oder eine Gruppe der Formel
-NR3R4 oder – OSO2R5substituiert ist,
steht, worin
- R3 und R4 gleich
oder verschieden sind und ein Cycloalkyl mit 3 bis 6 Kohlenstoffatomen,
Wasserstoff, Formyl, oder ein geradkettiges oder verzweigtes Alkyl
mit bis zu 6 Kohlenstoffatomen, das gegebenenfalls durch ein Hydroxy,
Carboxy oder ein geradkettiges oder verzweigtes Alkoxy oder Alkoxycarbonyl
mit jeweils bis zu 6 Kohlenstoffatomen oder durch eine Gruppe der
Formel -(CO)a-NR6R7 substituiert ist, bedeuten, worin
- a die Zahl 0 oder 1 bedeutet;
- R6 und R7 gleich
oder verschieden sind und Wasserstoff, Formyl, Hydroxy, Phenyl oder
ein geradkettiges oder verzweigtes Alkyl mit bis zu 6 Kohlenstoffatomen,
das gegebenenfalls durch Hydroxy oder ein geradkettiges oder verzweigtes
Alkoxy mit bis zu 5 Kohlenstoffatomen substituiert ist, bedeuten;
oder
- R3 und/oder R4 ein
geradkettiges oder verzweigtes Alkoxycarbonyl mit bis zu 6 Kohlenstoffatomen,
Carboxy oder ein geradkettiges oder verzweigtes Acyl mit bis zu
6 Kohlenstoffatomen, das gegebenenfalls durch Hydroxy oder ein geradkettiges
oder verzweigtes Alkoxy mit bis zu 4 Kohlenstoffatomen substituiert
ist, bedeuten; oder
- R3 und/oder R4 einen
Rest der Formel -(CO)b-T-NR8R9, -CO- R10, -SO2R11 oder -SO2NR12R13 bedeuten,
worin
- b die oben angegebene Bedeutung von a hat und mit diesem gleich
oder verschieden ist;
- T ein geradkettiges oder verzweigtes Alkyl mit bis zu 5 Kohlenstoffatomen
darstellen kann oder im Fall b ≠ 0 auch
eine Bindung darstellen kann;
- R8 und R9 die
oben angegebene Bedeutung von R6 und R7 haben und mit diesen gleich oder verschieden
sind;
- R10 einen gesättigten, partiell ungesättigten
oder ungesättigten
5- bis 7-gliedrigen Heterocyclus mit bis zu 3 Heteroatomen aus der
Reihe S, N und/oder O bedeutet, der gegebenenfalls auch über die
N-Funktion durch ein geradkettiges oder verzweigtes Alkyl, Alkoxy
oder Alkoxycarbonyl mit jeweils bis zu 4 Kohlenstoffatomen, Carboxyl,
Benzyloxycarbonyl oder Hydroxy substituiert sein kann;
- R11 ein geradkettiges oder verzweigtes
Alkyl mit bis zu 5 Kohlenstoffatomen, Benzyl oder Phenyl bedeutet;
- R12 und R13 gleich
oder verschieden sind und Wasserstoff, Phenyl oder ein geradkettiges
oder verzweigtes Alkyl mit bis zu 6 Kohlenstoffatomen bedeuten;
oder
- R3 und R4 gemeinsam
mit dem Stickstoffatom einen 5- oder
6-gliedrigen gesättigten,
partiell ungesättigten
oder ungesättigten
Heterocyclus bilden, der bis zu 3 Heteroatome aus der Reihe N, S
und/oder O oder einen Rest -NR14 enthalten
kann, und der gegebenenfalls durch Carbonyl, ein geradkettiges oder
verzweigtes Alkoxycarbonyl mit bis zu 5 Kohlenstoffatomen oder ein
geradkettiges oder verzweigtes Alkyl mit bis zu 5 Kohlenstoffatomen
substituiert ist, das seinerseits durch Hydroxy, Carboxy oder durch
ein geradkettiges oder verzweigtes Acyl, Alkoxy oder Alkoxycarbonyl
mit jeweils bis zu 6 Kohlenstoffatomen substituiert sein kann; worin
- R14 Wasserstoff, Carbonyl oder ein geradkettiges
oder verzweigtes Alkyl oder Alkoxycarbonyl mit jeweils bis zu 5
Kohlenstoffatomen bedeutet; und
- R5 Phenyl oder ein geradkettiges oder
verzweigtes Alkyl mit bis zu 5 Kohlenstoffatomen bedeutet;
- A für
eine geradkettige oder verzweigte Alkylen- oder Alkenylenkette mit
jeweils bis zu 6 Kohlenstoffatomen steht;
- D und L gleich oder verschieden sind und für ein Aryl mit 6 bis 10 Kohlenstoffatomen
oder für
einen 5- bis 7-gliedrigen aromatischen, gegebenenfalls benzokondensierten
Heterocyclus mit bis zu 3 Heteroatomen aus der Reihe S, N und/oder
O stehen, die gegebenenfalls bis zu 3-fach gleich oder verschieden
durch Halogen, Hydroxy, Nitro, Trifluormethyl, Carboxy, geradkettiges
oder verzweigtes Alkyl, Alkoxy oder Alkoxycarbonyl mit jeweils bis
zu 6 Kohlenstoffatomen oder durch eine Gruppe der Formel -(V)c-NR15R16 und/oder – OR17 substituiert sind; worin
- c die Zahl 0 oder 1 bedeutet;
- V einen Rest der Formel -CO oder -SO2 bedeutet;
- R15 und R16 gleich
oder verschieden sind und die oben angegebene Bedeutung von R3 und R4 haben;
- R17 Wasserstoff, ein geradkettiges oder
verzweigtes Alkenyl mit bis zu 8 Kohlenstoffatomen oder ein geradkettiges
oder verzweigtes Alkyl mit bis zu 8 Kohlenstoffatomen bedeutet,
das gegebenenfalls bis zu 3-fach gleich oder verschieden durch Hydroxy,
Carboxyl oder geradkettiges oder verzweigtes Alkoxycarbonyl mit
bis zu 5 Kohlenstoffatomen substituiert ist;
und/oder die Zyklen
gegebenenfalls durch ein Aryl mit 6 bis 10 Kohlenstoffatomen oder
durch einen 5- bis 7-gliedrigen aromatischen, gegebenenfalls benzokondensierten
Heterocyclus mit bis zu 3 Heteroatomen aus der Reihe S, N und/oder
O substituiert sind, die ihrerseits gegebenenfalls bis zu 2-fach
gleich oder verschieden durch Halogen, Hydroxy, Nitro, Carboxyl,
Trifluormethyl oder durch ein geradkettiges oder verzweigtes Alkyl, Alkoxy
oder Alkoxycarbonyl mit jeweils bis zu 5 Kohlenstoffatomen oder
durch eine Gruppe der Formel (V')d-NR18R19 substituiert
sind; worin
- d die oben angegebene Bedeutung von a hat und mit diesem gleich
oder verschieden ist;
- R18 und R19 die
oben angegebene Bedeutung von R3 und R4 haben und mit diesen gleich oder verschieden sind;
- V' die oben
angegebene Bedeutung von V hat und mit diesem gleich oder verschieden
ist; und/oder
das unten für
D angegebene Ringsystem darstellen, das gegebenenfalls durch ein
geradkettiges oder verzweigtes Acyl mit bis zu 5 Kohlenstoffatomen
substituiert ist, das gegebenenfalls durch Hydroxy, ein geradkettiges
oder verzweigtes Alkoxy mit bis zu 5 Kohlenstoffatomen oder durch
eine Gruppe der Formel -NR20R21 substituiert
ist; worin
- R20 und R21 gleich
oder verschieden und die oben angegebene Bedeutung von R3 und R4 haben; oder
- E für
einen Rest der Formel -CH2-Y-Z- steht; worin
- Y eine Bindung oder ein Sauerstoff- oder Schwefelatom oder die
-NH-Gruppe bedeutet;
- Z eine geradkettige oder verzweigte Alkylkette mit bis zu 5
Kohlenstoffatomen bedeutet;
- D für
einen Rest der Formel steht;
und
deren Tautomere und Salze.
-
Bevorzugte
I:PDEs werden aus den folgenden Strukturen ausgewählt:
-
-
-
NPY (Neuropeptid Y)
-
Gemäss einem
Aspekt der vorliegenden Erfindung ist das zusätzliche Target ein PcAMP-Target, wobei das PcAMP-Target
NPY oder einer seiner assoziierten Rezeptoren ist.
-
Nukleotid-Sequenzen
und Aminosäure-Sequenzen
für NPY
und seine Rezeptoren sind in der Literatur verfügbar. Einige Sequenzen sind
in den hier aufgeführten
Sequenzprotokollen angegeben.
-
Es
wurde hier gefunden, dass das Neuropeptid Y (NPY) einen inhibitorisch
regulierenden Einfluss auf die durch das vasoaktive intestinale
Peptid (VIP) vermittelte Vasorelaxation ausübt. Demnach wird die Inhibition
von NPY- Rezeptoren
zu einer verstärkten
Pelvisnerv- und VIP-vermittelten
Erhöhung
des genitalen (z.B. vaginalen oder klitoralen) Blutflusses führen. Dies
wird klinisch zu einer erhöhten
vaginalen und/oder klitoralen Anschwellung führen, welche letztlich über Plasma-Transsudation
zu einer erhöhten
Lubrikation und erhöhten vaginalen
Dehnbarkeit führen
wird. Demnach ist für
die Behandlung von FSAD das NPY oder einer seiner assoziierten Rezeptoren
ein geeignetes Target.
-
Demnach
ist der zusätzliche
Wirkstoff in einem bevorzugten Aspekt ein NPY Y1-,
Y2- oder Y5-Antagonist,
vorzugsweise ein oraler NPY Y1-, Y2- oder Y5-Antagonist.
Dieser Wirkstoff wird FSAD durch Erhöhung des genitalen (z.B. vaginalen
oder klitoralen) Blutflusses und durch Erhöhung der Lubrikation behandeln.
-
Der
NPY-vermittelte Antagonismus von VIP-induzierten Erhöhungen des
Blutflusses stellt demnach ein mögliches
therapeutisches Ziel dar, über
welches der Blutfluss im weiblichen Genitaltrakt beeinflusst werden
kann. Der Mechanismus, über
welchen dieser Antagonismus abläuft,
ist höchstwahrscheinlich über eine NPY
Y1-Rezeptor-induzierte Gi/o-Aktivierung.
In anderen physiologischen Systemen haben NPY Y1-Rezeptoren
mit der Vermittlung der Vasokonstriktion und Inhibierung der sympathetischen
Transmitter-Freisetzung in Zusammenhang gebracht (Lundberg et al.,
1996; eine NPY Y2 Wirkung kann nicht ausgeschlossen werden). Es
wird vermutet, dass NPY im weiblichen Genitaltrakt die Vasorelaxation
inhibiert, und zwar durch direkte Inhibition der Adenylatcyclase,
direkte Inhibition der VIP-Freisetzung oder sympathische Neurotransmission.
-
Wie
angegeben, ist ein zusätzliches
PcAMP-Target einer der NPY-Rezeptoren.
-
Die
neuronale Freisetzung von NPY reguliert die VIP-induzierte Vasorelaxation des vaginalen
vaskulären
Bettes. Dies erfolgt am wahrscheinlichsten über einen auf NPY Y1-Rezeptoren
beruhenden präsynaptischen
Mechanismus, obwohl eine postsynaptische Wirkungsart nicht ausgeschlossen
werden kann. Ein NPY-Antagonist wird die VIP-induzierte Vasodilation
des vaginalen vaskulären
Bettes potenzieren. Dies wird klinisch über die Anschwellung der Vaginawand
zu einer erhöhten
vaginalen Lubrikation und Dehnbarkeit führen.
-
Von
uns durchgeführte
Studien zur Expression von NPY-Rezeptoren
haben NPY Y1-, Y2-
und Y5-Rezeptor-Subtypen innerhalb der menschlichen
Vagina identifiziert.
-
Demnach
ist das zusätzliche
PcAMP-Target in einem Aspekt einer oder
mehrere der NPY Y1-, Y2-
und Y5-Rezeptor-Subtypen.
-
Hintergrundlehren über NPY
und die damit zusammenhängenden
Rezeptoren sind von Victor A. McKusick et al auf http://www3.ncbi.nlm.nih.gov/Omim/searchomim.htm
zusammengestellt worden. Der folgende Text betreffend NPY wurde
dieser Quelle entnommen.
-
"Das Neuropeptid Y
(NPY) ist ein häufiges
und weit verbreitetes Peptid im Nervensystem von Säugern. Es
zeigt eine Sequenz-Homolgie zum Peptid YY und über 50% Homolgie in der Aminosäuren- und
Nukleotid-Sequenz zum pankreatischen Polypeptid (PNP; 167780). NPY
ist ein 36-Aminosäuren-Peptid.
Minth et al. (1984) klonten das NPY- Gen ausgehend von der mRNS eines Pheochromocytoms.
Takeuchi et al. (1985, 1986) isolierten cDNS-Klone von NPY- und
PNP-Genen aus einem Pheochromocytom beziehungsweise einem pankreatischen
endokrinen Tumor. Unter Verwendung dieser cDNS-Sonden zur Analyse
der Genom-DNS aus Chromosomen-Zuordnungspaneelen von menschmaus-somatischen
Zellhybriden wurde dann die Frage untersucht, ob die Gene syntenisch
sind. Die Studien ergaben mit NPY auf 7pter-7q22 und PNP auf 17p11.1-17qter
eine Nichtsyntenie. Mittels Studien einer Rückkreuzung mit Mus spretus
lokalisierten Bahary et al. (1991) das homologe NPY-Gen zum Mauschromosom
6. Da das Mauschromosom 6 eine Homolgie zum menschlichen 7q hat,
ist es wahrscheinlich, dass das NPY-Gen beim Menschen in der Region
7cen-q22 lokalisiert ist. Meisler et al. (1987) schlossen eine nahe
Verknüpfung
zwischen den Loki für
zystische Fibrose (219700) und Neuropeptid Y aus. Terenghi et al.
(1987) bestimmten die Verteilung von mRNS kodierendem NPY in Neuronen
des zerebralen Kortex in chirurgischen Biopsieproben und in postmortem
Gehirn mittels in-situ-Hybridisierung-Verfahren. Sie zeigten eine
konsistente Lokalisation der NPY-Gen-Transkription und Expression
in normalen reifen kortikalen Neuronen. Baker et al. (1995) zeigten
mittels Fluoreszenz-in-situ-Hybridisierung,
dass das NPY-Gen auf 7p15.1 lokalisiert ist und als einzelne Kopie
vorliegt. Sie bemerkten, dass NPY eines der am höchsten konservierten bekannten
Peptide ist, das sich beispielsweise zwischen Menschen und Haien
nur in 3 Aminosäuren
unterscheidet. Das Neuropeptid Y ist ein an der Kontrolle des Energiegleichgewichtes
beteiligter Neuromodulator und wird im Hypothalamus von ob/ob-Mäusen überproduziert.
Um die Rolle von NPY bei der Antwort auf Leptin-(164160)-Mangel
zu bestimmen, erzeugten Erickson et al. (1996) ob/ob-Mäuse mit
NPY-Mangel. In der
Abwesenheit von NPY waren die ob/ob-Mäuse wegen verringerter Futteraufnahme
und erhöhtem
Energieverbrauch weniger übergewichtig
und weniger schwerwiegend von Diabetes, Sterilität und somatotropen Defekten
betroffen. Diese Resultate wurden dahingehend interpretiert, dass NPY
ein zentraler Effektor des Leptinmangels ist. Eine genetische Linkage-Analyse
von Ratten, welche selektiv auf Alkoholaffinität gezüchtet wurden, identifizierte
eine chromosomale Region, welche das NPY-Gen umfasst (Carr et al.,
1998). Alkoholaffine Ratten hatten im Vergleich zu nicht alkoholaffinen
Ratten in mehreren Hirnregionen niedriegere NPY-Spiegel. Thiele
et al. (1998) untersuchten daher den Alkoholkonsum von Mäusen, denen
infolge einer gezielten Gen-Disruption das NPY vollständig fehlte
(Erickson et al., 1996). Sie fanden, dass Mäuse mit NPY-Mangel im Vergleich
zu Mäusen
vom Wildtyp einen erhöhten
Konsum von Lösungen mit
6%, 10% und 20% (nach Volumen) Ethanol zeigten. Mäuse mit
NPY-Mangel waren auch weniger empfindlich auf die sedierenden/hypnotischen
Wirkungen von Ethanol, was durch eine schnellere Erholung aus dem ethanolinduzierten
Schlaf gezeigt wurde, die sogar dann eintrat, wenn sich die Ethanol-Plasmakonzentrationen
von denjenigen der Kontrollen nicht signifikant unterschieden. Im
Gegensatz dazu hatten transgene Mäuse mit einer Überexpression
eines markierten NPY-Gens in Neuronen, die dieses üblicherweise
exprimieren, eine geringere Präferenz
für Ethanol
und waren gegenüber
den sedierenden/hypnotischen Wirkungen von Ethanol empfindlicher
als die Kontrollen. Diese Daten lieferten direkte Hinweise, dass Alkoholkonsum
und -resistenz mit dem NPY Spiegel im Gehirn invers zusammenhängen. Als
Teil einer laufenden Studie über
die genetische Basis der Adipositas identifizierten Karvonen et
al. (1998) einen 1128T-C-Polymorphismus,
welcher im einzelnen Signalpeptidteil von pre-pro-NPY zur Substitution
von Leucin durch Prolin am Rest 7 führt. Dieser Polymorphismus
war nicht mit Adipositas oder dem Energiemetabolismus vergesellschaftet,
war aber sowohl bei normalgewichtigen als auch bei übergewichtigen
Finnen sowie bei übergewichtigen
holländischen
Subjekten signifikant und konsistent mit hohen Gesamtcholesterin-
und LDL-Cholesterin-Serumspiegeln
vergesellschaftet. Uusitupa et al. (1998) fanden den pro7-Polymorphismus
bei 14% der Finnen, aber nur bei 6% der Holländer. Subjekte mit pro7 in
NPY hatten im Durchschnitt um 0.6 bis 1.4 mMol/L höhere Gesamtcholesterin-Serumspiegel
als diejenigen ohne diese Genvariante. Da der Einfluss von pro7
NPY auf den Serumcholesterinspiegel bei normalgewichtigen Holländern nicht
gefunden werden konnte, kann angenommen werden, dass übergewichtige
Personen auf die Wirkung der Genvariante anfälliger sein können. Es
wurde berechnet, dass die Wahrscheinlichkeit, das pro7 in NPY zu
haben, bei übergewichtigen
Subjekten mit einem Gesamtserumcholesterin von oder 8 mMol/L oder
mehr 50 bis 60% betragen könnte.
Zumindest unter den Finnen ist die pro7-Form von NPY einer der stärksten genetischen
Faktoren, welche die Serumcholesterinspiegel beeinflussen. SIEHE
AUCH Allen und Bloom (1986); Dockray (1986); Maccarrone und Jarrott
(1986); Minth et al. (1986)."
-
Wie
angegeben, sind Hintergrundlehren über NPY und seine assoziierten
Rezeptoren von Victor A. McKusick et al zusammengestellt worden
(ibid). Der folgende Text betreffend NPYR1 wurde dieser Quelle entnommen.
-
"Das Neuropeptid Y
(NPY; 162640) ist eines der am häufigsten
vorkommenden Neuropeptide im Nervensystem von Säugern und entfaltet einen breiten
Bereich von wichtigen physiologischen Aktivitäten, welche Wirkungen auf die
psychomotorische Aktivität,
die Nahrungsaufnahme, die Regulierung der zentralen endokrinen Sekretion
sowie potente vasoaktive Wirkungen auf das kardiovaskuläre System
umfassen. Zwei wichtige Subtypen von NPY (Y1 und Y2) sind mittels
pharmakologischer Kriterien definiert worden. Die NPY Y1-Rezeptoren
wurden in eine Vielzahl von Geweben, namentlich in Gehirn, Milz,
Dünndarm,
Nieren, Hoden, Plazenta und glatten Muskeln in der Aorta identifiziert.
Der Y2-Rezeptor wird hauptsächlich
im zentralen Nervensystem gefunden. Herzog et al. (1992) berichteten über das
Klonieren einer für
den menschlichen NPY-Rezeptor
kodierenden cDNS, die sie als Mitglied der G-Protein-gekoppelten Rezeptor-Superfamilie
bestätigten.
Wenn der Rezeptor in den Ovarialzellen von Chinesischen Hamstern
(CHO) oder in menschlichen embryonalen Nierenzellen exprimiert wird,
entfaltet er eine charakteristische Ligandspezifität. In der
Nierenzell-Linie
war der Rezeptor an ein auf Keuchhustentoxin sensitives G-Protein
gekoppelt, das die Inhibition der Anreicherung von cyclischer AMP
vermittelt. Andererseits war der Rezeptor in der CHO-Zell-Linie
nicht mit der Inhibition der Adenylatcyclase, sondern vielmeher
mit der Erhöhung
des intrazellulären
Kalziums gekoppelt. Demnach war die second Messenger Kopplung des
NPY-Rezeptors zelltyp-spezifisch
und hing vom spezifischen, im Zelltyp vorhandenen Repertoire an
G-Proteinen und Effektorsystemen ab. Larhammar et al. (1992) klonten
und charakterisierten unabhängig
den Neuropeptid-Y-Rezeptor. Herzog et al. (1993) bestimmten die
molekulare Organisation und Regulierung des menschlichen NPY-Y1-Rezeptor-Genes. Im
Gegensatz zur zusammenhängenden Struktur
der meisten G-Protein-gekoppelten Rezeptor-Gene fanden sie, dass
das NPY-Y1-Rezeptor-Gen 3 Exons hat. Sie identifizierten zudem einen
gewöhnlichen
Pst1-Polymorphismus
im ersten Intron des Gens. Mittels hochauflösender Fluoreszenz-in-situ-Hybridisierung
lokalisierten sie das Gen auf 4q31.3-q32. Herzog et al. (1997) fanden,
dass die NPY1R- und NPY5R-(602001)-Gene auf dem Chromosom 4q31-q32
kolokalisiert sind. Die beiden Gene werden aus einer gewöhnlichen
Promoterregion in entgegengesetzter Richtung transkribiert. Eines
der alternierend gespleissten 5-Strich Exons des Y1-Rezeptor-Gens ist
ein Teil der kodierenden Sequenz des Y5-Rezeptors. Diese unübliche Anordnung
war für
Herzog et al. (1997) ein Hinweis, dass die beiden Gene durch ein
Gen-Duplikationsereignis entstanden und dass sie koordiniert exprimiert
werden könnten.
Mittels interspezifischer Rückkreuzungsanalyse
lokalisierten Lutz et al. (1997) die Npy1r- und Npy2r-Gene auf konservierte
Verknüpfungsgruppen
auf den Maus-Chromosomen
8 beziehungsweise 3, welche der distalen Region des menschlichen
Chromosoms 4q entsprechen." Wie
angegeben, sind Hintergrundlehren über NPY und seine assoziierten
Rezeptoren von Victor A. McKusick et al zusammengestellt worden
(ibid). Der folgende Text betreffend NPYR2 wurde dieser Quelle entnommen.
-
"Das Neuropeptid Y(NPY)
signalisiert über
eine Familie von im Gehirn und in sympathischen Neuronen vorhandenen
G-Protein-gekoppelten Rezeptoren. Mindestens 3 Arten von Neuropeptid-Y-Rezeptor
sind aufgrund von pharmakologischen Kriterien, Gewebeverteilung
und Struktur des kodierenden Gens definiert worden; siehe 162641
und 162643. Rose et al. (1995) berichteten über die Expressionsklonierung
in COS Zellen einer cDNS für
den menschlichen Typ-2-Rezeptor, NPY2R. Transfizierte Zellen zeigten
eine hohe Affinität für NPY (162640),
Peptid YY (PYY; 600781) und ein Fragment von NPY einschliesslich
Aminosäuren
13 bis 36. Das vorhergesagte 381-Aminosäuren-Protein hat 7 Transmembrandomänen, welche
für G-Protein-gekoppelte
Rezeptoren charakteristisch sind, und ist zum menschlichen Y1-Rezeptor
nur um 31% identisch (NPY1R; 162641). Eine 4-kb mRNS wurde auf Northern
Blots von Gewebeproben aus mehreren Regionen des Nervensystems nachgewiesen.
Gerald et al. (1995) klonierten die dem menschlichen Y2-Rezeptor
entsprechende cDNS aus einer cDNS-Expressions-Bibliothek des menschlichen
Hippocampus durch Verwendung eines radioaktiv markierten PYY-Bindungsassays.
Sie exprimierten das Y2-Gen in COS-7 Zellen und führten einen
Hormon-Bindungsassay durch, welcher zeigte, dass der Y2-Rezeptor (von höchster bis
niedrigster Affinität)
PYY, NPY und pankreatische Polypeptidhormone (PP; 167780) bindet.
Ammar et al. (1996) klonierten und charakterisierten das menschliche
Gen, welches für
den Typ 2 NPY-Rezeptor kodiert. Das Transkript umspannt 9 kb der
Genomsequenz und ist in 2 Exons kodiert. Wie im NPY-Rezeptor-Gen vom
Typ 1 wird die nichttranslatierte 5-Strich Region von NPY2R durch eine intervenierende
4.5-kb Sequenz unterbrochen. Ammar et al. (1996) zeigten mittels
Southern Analyse von Nager-Mensch Zellhybriden, gefolgt von Fluoreszenz-in-situ-Hybridisierung
(FISH), dass das NPY2R-Gen auf 4q31 lokalisiert ist, wobei diese
Region auch das NPY1R-Gen enthält, was
darauf hinweist, dass diese Subtypen trotz ihrer strukturellen Unterschiede
durch Gen-Duplikation entstanden sein könnten. Mittels interspezifischer
Rückkreuzungsanalyse
lokalisierten Lutz et al. (1997) die Npy1r- und Npy2r- Gene auf
konservierte Verknüpfungsgruppen
auf den Maus-Chromosomen 8 beziehungsweise 3, welche der distalen
Region des menschlichem Chromosoms 4q entsprechen."
-
Ein
Assay zur Bestimmung, ob ein mutmasslicher oder aktueller Wirkstoff
an NPY binden kann, ist in WO-A-98/52890 (siehe Seite 96 davon,
Zeilen 2 bis 28) beschrieben.
-
I:NPY
-
Wie
oben angegeben, kann der zusätzliches
Wirkstoff irgend ein geeigneter Wirkstoff sein, der als I:NPY (gelegentlich
als NPY-Antagonist bezeichnet) wirken kann. Zusätzlich oder alternativ kann
der erfindungsgemässe
Wirkstoff auch als I:NPY wirken.
-
I:NPYs
(insbesondere NPY-Antagonisten) werden in den folgenden Übersichtsartikeln
besprochen:
- Dunlop J, Rosenzweig-Lipson S: Therapeutic approaches
to obestity Exp Opin Ther Pat 1999 8 12 1683–1694
- Wang S, Ferguson KC, Burris TP, Dhurandhar NV: 8th annual international
conference on obesity and non-insulin dependent Diabetes mellitus:
novel drug developments. Exp Opin Invest Drugs 1999 8 7 1117–1125
- Ling AL: Neuropeptide Y receptor antagonists Exp Opin Ther Pat
1999 9 4 375–384
Adham N, Tamm J, Du P, Hou C, et al: Identification of residues
involved in the binding of the antagonist SNAP 6608 to the Y5 receptor Soc
Neurosci Abstr 1998 24 part 2 626.9
- Shu YZ, Cutrone JQ, Klohr SE, Huang S: BMS-192548, a tetracyclic
binding Inhibitor of Neuropeptid Y receptors, from Aspergillus niger
WB2346. II. Physico-chemical propeties and structural characterization
J Antibiot 1995 48 10 1060–1065
- Rigollier P, Rueger H, Whitebread S, Yamaguchi Y, Chiesi M,
Schilling W, Criscione L: Synthesis and SAR of CGP 71683A, a potent
and selective antagonist of the neuropeptid Y Y5 receptor. Int Symp
Med Chem 1998 15th Edinburgh 239
- Criscione L, Rigollier P, Batzl-Hartmann C, Rueger H, Stricker-Krongrad
A, et al: Food intake in free-feeding and energy-deprived lean rats
is mediated by the neuropeptide Y5 receptor. J Clin Invest 1998
102 12 2136–2145
- Neurogen Corp : NGD 95-1 Clin Trials Monitor 1996 5 10 Ab 19244
- Buttle LA: Anti-obesity drugs: on target for huge market sales.
Exp Opin Invest Drugs 1996 5 12 1583–1587
- Gehlert DR, Hipskind PA: Neuropeptide Y receptor antagonists
in obesity. Exp Opin Invest Drugs 1996 7 9 1827–1838
- Goldstein DJ, Trautmann ME: Treatments for obesity. Emerging
Drugs 1997 2 – 1–27
- Hipskind P A, Lobb K L, Nixon J A, Britton T C, Bruns R F, Catlow
J, Dieckman McGinty D K, Gackenheimer S L, Gitter B D, Iyengar S,
Schober D A, et al.: Potent and selective 1,2,3-trisubstituted indole
NPY Y-1 antagonists. J Med Chem 1997 40 3712–3714
- Zimmerman DM, Cantrall BE, Smith ECR, Nixon JA, Bruns RF, Gitter
B, Hipskind PA, Ornstein PL, Zarrinmayeh H, Britton TC, Schober
DA, Gehlert DR: Structure-activity relationships of a series of
1-substituted-4-methylbenzimidazole neuropeptide Y-1 receptor antagonists
Bioorganic Med Chem Lett 1998 8 5 473–476
- Zarrinmayeh H, Nunes A, Ornstein P, Zimmerman D, Arnold MB,
et al: Synthesis and evaluation of a series of novel 2-[(4-chlorophenoxy)methy]benzimidazoles
as selective neuropeptid Y Y1 receptor antagonists J Med Chem 1998
41 15 2709–2719
- Britton TC, Spinazze PG, Hipskind PA, Zimmerman DM, Zarrinmayeh
H, Schober DA, Gehlert DR, Bruns RF: Structure activity relationships
of a series of benzothiophene-dervied NPY-Y1 antagonists: optimization
of the C2 side chain Bioorganic Med Chem Lett 1999 9 3 475–480
- Zarrinmayeh H, Zimmerman DM, Cantrell BE, Schober DA, Bruns
RF, Gackenheimer SL, Ornstein PL, Hipskind PA, Britton TC, Gehlert
DR : Structure-activity relationship of a series of diaminoalkyl
substituted benzimidazole as neuropeptide Y Y1 receptor antagonists
Bioorganic Med Chem Lett 1999 9 5 647-652
- Murakami Y, Hara H, Okada T, Hashizume H, Kii M, Ishihara Y,
Ishikawa M, Mihara S-I, Kato G, Hanasaki K, Hagishita S, Fujimoto
M: 1,3-disubstituted benzazepines as novel, potent, selective neurpeptide
Y Y1 receptor antagonists J Med Chem 1999 42 14 2621–2632
- Rudolf K, Eberlein W, Engel W, Wieland HA, Willim KD, Entzeroth
M, Wienen W, Beck Sickinger AG, Doods HN : The first highly potent
and selective non-peptide neuropeptide Y Y1 receptor antagonist:
BIBP3226 Eur J Pharmacol 1994 271 2-3 R11–R13
- Wieland HA, Willim KD, Entzeroth M, Wienen W, Rudolf K, Eberlein
W, Engel W, Doods HN: Subtype selectivity and antagonist profile
of the nonpeptide neuropeptide Y1 receptor antagonist BIBP 3226
J Pharmacol Exp Ther 1995 275 1 143–149.
- Wright J, Bolton G, Creswell M, Downing D, Georgic L, Heffner
T, Hodges J, MacKenzie R, Wise L: 8-amino-6-(arylsulphonyl)-5-nitroquinolones: novel
nonpeptide neuropeptide Y1 receptor antagonists Bioorganic Med Chem
Lett 1996 6 15 1809–1814
- Capurro D, Huidobro-Toro JP: The involvement of neyropeptide
Y Y1 receptors in the blood pressure baroreflex:studies with BIBP
3226 and BIB 3304. Eur J Pharmacol 1999 376 3 251–255
- Dumont Y, Cadieux A, Doods H, Quirion R : New tools to investigate
neuropeptide Y receptors in the central and peripheral nervous systems:
BIBO-3304 (Y1), BIIE-246 (Y2) and [1251]-GR-231118 (Y1/Y4). Soc
Neurosci Abstr 1999 25 Part 1 Abs 74.11
- Hegde SS, Bonhaus DW, Stanley W, Eglen RM, Moy TM, Loeb M, et
al: Pharmacological evaluation of 1229U91, a high affinity and selective
neuropeptide Y(NPY) – Y1
rezeptor antagonist Pharmacol Res 1995 31 190
- Matthews JE, Chance WT, Grizzle MK, Heyer D, Daniels AJ: Food
intake inhibition and body weight loss in rats treated with GI 264879A,
an NPY-Y1 receptor. Soc Neurosci Abstr 1997 23 Pt 2 1346
- Doods HN, Willim K-D, Smith SJ: BIBP 3226: a selective and highly
potent NPY-Y1 antagonist Proc Br Pharmacol Soc 1994 13–16 Dec.
C47
- Rudolf K, Eberlein W, Engel W, Wieland HA, Willim KD, Entzeroth
M, Wienen W, Beck Sickinger AG, Doods HN: The first highly potent
and selective non-peptide neuropeptide Yy1 receptor antagonist:
BIBP3226 Eur J Pharmacol 1994 271 2-3 R11–R13
- Serradelil-Le-Gal C, Valette G, Rouby PE, Pellet A, Villanova
G, Foulon L, Lespy L, Neliat G, Chambon JP, Maffrand JP, Le-Fur
G: SR 120107A and SR 120819A: Two potent and selective, orally-effective
antagonists for NPY Y1 receptors Soc Neurosci Abstr 1994 20 Pt 1
907 – Abs
376.14
- Hong Y, Gregor V, Ling AL, Tompkins EV, Porter J, Chou TS, Paderes
G, Peng Z, Hagaman C, Anderes K, Luthin D, May J: Synthesis and
biological evaluation of novel guanylurea compounds as potent NPY
Y1 receptor antagonist Acs 1999 217 Anaheim MEDI 108
-
I:NPYs
(insbesondere NPY antagonists) werden in den folgenden Dokumenten
beschrieben:
- WO-98/07420
- WO-94/00486
- WO-96/22305
- WO-97/20821
- WO-97/20822
- WO-96/14307
- JP-07267988
- WO-96/12489
- US-5552422
- WO-98/35957
- WO-96/14307
- WO-94/17035
- EP-0614911
- WO-98/40356
- EP-0448765
- EP-0747356
- WO-98/35941
- WO-97/46250
- EP-0747357
-
Bevorzugte
Beispiele von I:NPYs sind aus den folgenden Strukturen ausgewählt. Diese
Verbindungen wurden geprüft
und zur Potenzierung von cAMP als nützlich befunden und demnach
zur Behandlung von FSAD als nützlich
gefunden. Einige der diese Verbindungen betreffenden experimentellen
Daten werden im experimentellen Abschnitt beschrieben (unten).
-
-
-
-
-
-
VIP (vasoaktives intestinales
Peptid)
-
Gemäss einem
Aspekt der vorliegenden Erfindung ist ein zusätzliches Target ein PcAMP-Target, wobei das PcAMP-Target
VIP oder einer seiner assoziierten Rezeptoren ist. Die gegenwärtige Klassifikation/Nomenklatur
bezeichnet diese als VPAC1, VPAC2 und PACAP.
-
Nukleotid-Sequenzen
und Aminosäuren-Sequenzen
für VIP
und seine Rezeptoren sind in der Literatur verfügbar. Einige Sequenzen sind
in den hier aufgeführten
Sequenzprotokollen angegeben.
-
Es
wurde gezeigt, dass VPAC1 und VPAC2 in der Vagina von Menschen wie
auch von Hasen vorliegt. PACAP war in der Vagina von Hasen wie auch
von Menschen nicht vorhanden.
-
VIP
ist ein wichtiger endogener Neurotransmitter, der während der
sexuellen Erregung freigesetzt wird und der für die nerveninduzierte vaginale
Vasodilatation des in der Vaginalwand lokalisierten vaskulären Betts verantwortlich
ist. Diese vasodilatorischen Wirkungen werden durch die Aktivierung
der Adenylatcyclase und die Bildung von cAMP vermittelt. Ohne sich
durch Therorie beschränken
zu wollen, kann diese Wirkung über die
VIP Rezeptorsubtypen VPAC1-, VPAC2- oder PACAP- (Hypophysen-Adenylatcyclase-aktivierendes
Peptid)-Rezeptoren vermittelt werden. Die VPAC2-
und PACAP-Rezeptoren
sind im ZNS am stärksten
exprimiert, und die Rezeptoren scheinen, obwohl sie in der Hypophyse
exprimiert werden, keine weit verbreitete biologische Funktion zu
haben.
-
Der
Wirkstoff könnte
VIP potenzieren und/oder als VIP-Mimetikum
oder Analogon davon wirken. Der Wirkstoff würde dann die vasorelaxatorischen
Wirkungen des während
der sexuelle Erregung freigesetzten endogenen VIP potenzieren und/oder
nachahmen. Der Wirkstoff kann auch eine additive Wirkung auf die
VIP-induzierten Relaxationen der vaginalen glatten Muskulatur haben.
Dies wird klinisch durch erhöhte
vaginale Lubrikation über
die Anschwellung der Vaginalwand und -dehnbarkeit zur FSAD-Behandlung
führen.
In dieser Ausgestaltung würde
allerdings das Mimetikum oder Analogon nicht die nachteiligen Eigenschaften
von VIP wie oben besprochen haben.
-
Hintergrundlehren über VIP
und seine assoziierten Rezeptoren sind von Victor A. McKusick et
al auf http://www3.ncbi.nlm.nih.gov/Omim/searchomim.htm angegeben.
Der folgende, VIP betreffende Text wurde dieser Quelle entnommen.
-
"Das vasoaktive intestinale
Peptid (VIP), ein ursprünglich
aus dem Duodenum von Schweinen isoliertes 28-Aminosäuren-Peptid,
ist nicht nur in gastrointestinalen Geweben, sondern auch in neuralen
Geweben, möglicherweise
als Neurotransmitter, vorhanden und entfaltet eine grosse Vielzahl
von biologischen Wirkungen. Weil VIP Ähnlichkeiten zu Glucagon, Secretin
und gastrischem inhibitorischem Peptid (GIP) zeigt, wird es als
Mitglied der Glucagon-Secretin Familie betrachtet. Das primäre Translationsprodukt
der für
VIP kodierenden mRNS (präpro-VIP)
hat ein Molekulargewicht von 20 Dalton. Durch Klonieren der zu der
für menschliches VIP
kodierenden mRNS komplementären
DNS-Sequenz fanden Itoh et al. (1983), dass der VIP-Vorläufer nicht nur
VIP, sondern auch ein neues Peptid aus 27 Aminosäuren enthält, das als PHM27 bezeichnet
wird und das aminoterminales Histidin und carboxyterminales Methionin
aufweist. Es unterscheidet sich von dem aus Schweinemagen isolierten
PHI17 durch 2 Aminosäuren;
PHI27 weist, wie seine Bezeichnung aufzeigt, ein carboxyterminales
Isoleucin auf. Linder et al. (1987) isolierten das menschliche Gen
für VIP
und PHM27 und studierten dessen Expression in verschiedenen Geweben
von Ratten. Heinz-Erian et al. (1985) schlugen vor, dass eine mangelhafte
Erneuerung von Schweissdrüsen
bei Patienten mit zystischer Fibrose durch das VIP-Neuropeptid ein
grundlegender Mechanismus für
den verminderten Wassergehalt und für die relative Undurchlässigkeit
des Epithels für
Chlorid und andere Ionen sein könnte,
welche die zystische Fibrose charakterisiert. Zur Überprüfung dieser
Hypothese verwendeten Gozes et al. (1987) den "Kandidaten-Gen"-Ansatz. Bodner
et al. (1985) hatten gezeigt, dass VIP und PHM-27 durch benachbarte
Exons kodiert werden. Gozes et al. (1987) verwendeten das für PHM-27-kodierende
Genomfragment zum Nachweis der Anwesenheit des VIP-Gens bei 6q21-qter.
Demnach schlossen sie ein defektes VIP-Gen als Kandidat für die primäre Ursache der
zystischen Fibrose aus (welche durch das Chromosom 7 kodiert ist).
-
Mittels
in-situ-Hybridisierungs-Verfahren ordneten Gozes et al. (1987) das
VIP-Gen dem 6q24 zu. Damit wurde VIP in die Region von MYB (189990)
platziert, welches auf 6q22 lokalisiert wurde. Gozes et al. (1987)
untersuchten eine funktionelle Beziehung zwischen den beiden Genen
in neuronalem Gewebe. Sie beobachteten einen scharfen Signal-Peak
von MYB mRNS im Hippocampus von 3 Tage alten Ratten, welcher dem
Signal von VIP mRNS voranging, das in diesem Bereich bei einem Alter
von 8 Tagen auftritt. Omary und Kagnoff (1987) fanden nukleäre Rezeptoren
für VIP
in einer menschlichen Kolonadenokarzinom-Zelllinie. Gotoh et al.
(1988) ordneten VIP dem Chromosom 6 zu mittels Spot Blot Hybridisierung
eines molekular klonierten Fragmentes des Gens an sortierte Chromosomen.
Die Lokalisation wurde mittels in-situ-Hybridisierung auf 6q26-q27
verfeinert."
-
Wie
angegeben, sind Hintergrundlehren über VIP und seine assoziierten
Rezeptoren in Victor A. McKusick et al angegeben (ibid). Der folgende
Text betreffend VIPR1 oder VPAC1 wurde dieser Quelle entnommen.
-
"Das vasoaktive intestinale
Peptid (VIP; 192320) ist ein octacosamerer neuroendokriner Mediator,
der vornehmlich in cholinergen präsynaptischen Neuronen des Zentralnervensystems
und in peripheren peptidergen Neuronen, die verschiedene Gewebe
innervieren, gefunden wird. Von den vielen neuroendokrinen Peptiden
mit immunologischen Funktionen unterscheidet sich VIP durch seine
Fähigkeit,
sowohl B- als auch T-Zellen direkt zu beeinflussen. Bestimmte unterschiedliche
Teilsätze
von neuralen, respiratorischen, gastrointestinalen und Immun- Zellen tragen spezifische
Rezeptoren mit hoher Affinität
für VIP,
welche mit einem zur Aktivierung der Adenylatcyclase befähigten Guanin-Nukleotid-Bindungs-(G)-Protein zusammenhängen. Libert
et al. (1991) fanden durch selektives) Amplifikation und Klonieren
aus Schilddrüsen-cDNS
vier neue Rezeptoren der G-Protein-gekoppelten Rezeptor-Familie. Eines
davon, als RDC1 bezeichnet, wurde von Sreedharan et al. (1991) als
der VIP-Rezeptor identifiziert. Libert et al. (1991) lokalisierten
das VIPR-Gen auf 2q37 mittels in-situ-Hybridisierung. Spätere Informationen liessen
Zweifel aufkommen, dass das auf 2q37 lokalisierte Gen tatsächlich das
VIP-Rezeptor-Gen war (Vassart, 1992). Die Sequenz, welche von Sreedharan
et al. (1991) als GPRN1 bezeichnet wurde und auf 2q37 lokalisiert
wurde, bindet gemäss
Wenger (1993) nicht an VIP. Sreedharan et al. (1995) isolierten
ein authentisches Typ I VIP-Rezeptor-Gen und lokalisierten es mittels
Fluoreszenz-in-situ-Hybridisierung
an der 3p22-Bande in einer mit nichtkleinzelligem Lungenkrebs zusammenhängenden
Region. Mittels interspezifischer Rückkreuzungs-Analyse lokalisierten
Hashimoto et al. (1999) das Maus-Vipr1-Gen auf die distale Region
des Chromosoms 9, eine Region, die eine Syntenie-Homolgie mit dem menschlichem
Chromosom 3p zeigt. Sreedharan et al. (1993) klonierten ein menschliches
intestinales VIP Rezeptor Gen; die ermittelte Aminosäuren-Sequenz
teilt 84% Identität
mit dem VIP-Rezeptor der Rattenlunge. Couvineau et al. (1994) isolierten
zwei VIPR cDNS Klone aus einer menschlichen jeunalen Epithelzellen-cDNS-Bibliothek.
Einer kodiert für
einen VIP-Rezeptor, der aus 460 Aminosäuren besteht und 7 mutmassliche
Transmembrandomänen
aufweist, wie dies auch andere G-Protein-gekoppelte Rezeptoren haben.
Der andere kodiert für
ein 495-Aminosäure-VIP-Rezeptor-verwandtes
Protein, das 100% Homolgie mit dem funktionellen VIP-Rezeptor über die
428 Aminosäuren
an der C-terminalen Region aufweist, aber eine vollständig divergente
67-Aminosäure-N-terminale
Domäne
enthält.
Bei Expression in COS-7 Zellen band das zweite Protein nicht an
radioiodiniertes VIP, obwohl es normalerweise gemäss Immunofluoreszenz-Studien
an der Plasmamembran adressiert wird. Für den Typ I VIP-Rezeptor, der
auch als Typ II PACAP-Rezeptor bezeichnet wird (siehe 102981 für einen
anderen Typ von PACAP-Rezeptor), wurde von Sreedharan et al. (1995)
gefunden, dass er ungefähr
22 kb umspannt und 13 Exons (im Bereich von 42 bis 1,400 bp) sowie
12 Introns (im Bereich von 0.3 bis 6.1 kb) enthält. Sreedharan et al. (1995)
charakterisierten auch den Promoter und die 5-prime Flankenregion
des Gens."
-
Wie
angegeben, sind Hintergrundlehren über VIP und seine assoziierten
Rezeptoren in Victor A. McKusick et al angegeben (ibid). Der folgende
Text betreffend VIPR2 oder VPAC2 wurde dieser Quelle entnommen.
-
"Das vasoaktive intestinale
Peptid (VIP; 192320) und das Hypophysen-Adenylatcyclase-aktivierende Polypeptid
(PACAP; 102980) sind homologe Peptide, welche als Neurotransmitter
und neuroendokrine Hormone wirken. Während die Rezeptoren für VIP und
PACAP Homolgie teilen, unterscheiden sie sich in ihren Substratspezifizitäten und
Expressionsmustern. Siehe VIPR1 (192321) und ADCYAPIRI(102981).
Svoboda et al. (1994) führten
eine PCR mit niedriger Stringenz unter Verwendung von Primern durch,
die auf Sequenzen beruhten, welche zwischen den VIP-Rezeptoren erhalten
bleiben. Sie klonierten das menschliche VIP2-Rezeptor Gen aus einer
Lymphoblasten-cDNS-Bibliothek.
Dieses Gen kodiert für
ein 438-Aminosäuren-Polypeptid,
das 86% Identität
mit dem Ratten-VIP2-Rezeptor
teilt. Sie exprimierten den menschlichen VIP2-Rezeptor in Ovarialzellen
des chinesischen Hamsters und fanden, dass es an PACAP-38, PACAP-27,
VIP und Helodermin bindet und dass die Bindung des Rezeptors an
irgend eines dieser Peptide die Adenylatcyclase aktiviert. Es wurde
gefunden, dass die Peptidbindung durch GTP inhibiert wird. Adamou
et al. (1995) klonierten das VIP2-Rezeptor-Gen aus einer menschlichen
Plazenta-cDNS-Bibliothek.
Northern Blotting ergab, dass VIPR2 als zwei Transkripte von 4.6
kb und 2.3 kb mit hohen Spiegeln in Skelettmuskeln und mit tiefen
Spiegeln in Herzen, Gehirn, Plazenta und Pankreas exprimiert wird.
Mackay et al. (1996) verwendeten Fluoreszenz-in-situ-Hybridisierung
um das VIPR2-Gen auf das menschliche Chromosom 7q36.3 zu lokalisieren.
Gemäss
weiteren Kartierungen mit Zelllinien, die von Patienten mit Holoprosenzephalie
vom Typ 3 (HPE3; 142945) abgeleitet wurden, liegt das VIPR2-Gen innerhalb der
minimalen kritischen HPE3-Region. Mackay et al. (1996) sagten aus,
dass obwohl VIPR2 zum HPE3-Phenotyp beitragen kann, es nicht der
einzige verantwortliche Faktor ist."
-
AC (Adenylatcyclase)
-
Gemäss einem
Aspekt der vorliegenden Erfindung ist ein zusätzliches Target ein PcAMP-Target, wobei das PcAMP-Target
AC ist.
-
Nukleotid-Sequenzen
und Aminosäuren-Sequenzen
für AC
sind in der Literatur verfügbar.
-
Zur
Bestätigung,
dass VIP die Vasorelaxation über
die Erhöhung
der intrazellulären
cAMP-Spiegel und die anschliessende Aktivierung der Adenylatcyclase
induziert, wurden während
der VIP-Stimulation die vaginalen cAMP-Konzentrationen gemessen und es wurde
Forskolin, ein Adenylatcyclase-Aktivator, verwendet, um die Wirkungen
der Aktivierung des cAMP/Adenylatcyclase-Pfads nachzuahmen.
-
In
diesen Studien wurde gefunden, dass die VIP-Behandlung und die Forskolin-Behandlung
die intrazellulären
cAMP-Konzentrationen
in isoliertem vaginalen Gewebe erhöhen.
-
Es
wurde zudem gefunden, dass Forskolin in einem Tiermodell der sexuellen
Erregung den vaginalen Blutfluss erhöht.
-
Ausserdem
wurde gefunden, dass Forskolin die Relaxation in isolierter Vagina
induziert.
-
Hintergrundlehren über AC sind
in Victor A. McKusick et al unter http://www3.ncbi.nlm.nih.gov/Omim/searchomim.htm
angegeben. Der folgende Text betreffend AC wurde dieser Quelle entnommen.
-
"Die Adenylylcyclase
(EC 4.6.1.1) katalysiert die Transformation von ATP zu cyclischem
AMP. Die enzymatische Aktivität
steht unter der Kontrolle von mehreren Hormonen, und verschiedene
Polypeptide nehmen an der Transduktion des Signals vom Rezeptor
zur katalytischen Einheit teil. Stimulatorische oder inhibitorische
Rezeptoren (Rs und Ri) interagieren mit G-Proteinen (Gs und Gi), welche eine GTPase-Aktivität entfalten, und
sie modulieren die Aktivität
der katalytischen Untereinheit der Adenylylcyclase. Parma et al.
(1991) klonierten eine der Adenylylcyclase des menschlichen Gehirns
entsprechende cDNS, die sie als HBAC1 bezeichnteten. Mittels in-situ-Hybridisierung
an metaphasigen chromosomalen Ausstrichen unter Verwendung von cDNS
für menschliches
Gehirn als Sonde zeigten Stengel et al. (1992), dass das Gen auf
8q24.2 lokalisiert ist. Ein hochgradig homologes Gen, ADCY2 (103071),
wurde mit Hilfe desselben Verfahrens 5p15.3 zugeordnet."
-
ALLGEMEINE
VERFAHREN DER REKOMBINANTEN DNS-METHODOLOGIE
-
Obwohl
die hier erwähnten
Verfahren im Allgemeinen im Fachgebiet wohlbekannt sind, kann insbesondere
auf Sambrook et al., Molecular Cloning, A Laboratory Manual (1989)
und Ausubel et al., Short Protocols in Molecular Biology (1999)
4th Ed, John Wiley & Sons, Inc. verwiesen werden. Die
PCR ist in US-A-4683195, US-A-4800195 und US-A-4965188 beschrieben.
-
ZUSAMMENFASSUNG
-
Zusammenfassend
bezieht sich die vorliegende Erfindung auf die Verwendung eines
I:NEP zur Behandlung von FSD, insbesondere FSAD.
-
BEISPIELE
-
Die
vorliegende Erfindung wird nachfolgend lediglich beispielhaft beschrieben,
wobei auf folgende Figuren verwiesen wird:
-
1,
welche eine Grafik ist;
-
2,
welche eine Grafik ist;
-
3,
welche eine Grafik ist;
-
4,
welche eine Grafik ist;
-
5,
welche eine Grafik ist;
-
6,
welche eine Grafik ist;
-
7,
welche eine Grafik ist;
-
8,
welche eine Grafik ist;
-
9,
welche eine Grafik ist;
-
10, welche eine Grafik ist;
-
11, welche eine Grafik ist; und
-
12, welche eine Grafik ist.
-
Es
versteht sich, dass der erfindungsgemässe Wirkstoff ein I:NEP ist.
Lehren über
I:PDE und I:NPY werden ebenfalls angegeben, da diese eine Fachperson
klar darauf hinweisen, dass der erfindungsgemässe Wirkstoff in Kombination
mit einem oder beiden dieser Typen von Wirkstoffen verwendet werden
kann, um die oben erwähnte
günstige
Wirkung zu erreichen. Diese zusätzlichen
Lehren sind ebenfalls angegeben, um unsere wegweisenden Befunde
weiter zu stützen.
-
Die
Figuren werden nun detaillierter besprochen.
-
1: – Die elektrische
Stimulation des Pelvisnervs induziert eine frequenzabhängige Erhöhung des vaginalen
Blutflusses im Modell der sexuellen Erregung beim anästhesierten
Hasen. Eine Erhöhung
der Stimulationsfrequenz induziert stärkere Erhöhungen des Blutflusses. Die
Veränderungen
wurden unter Verwendung von Laser-Doppler-Verfahren bestimmt.
-
2: – Vasoaktives
intestinales Peptid (VIP) induziert eine Erhöhung des vaginalen Blutflusses
im Modell der sexuellen Erregung beim anästhesierten Hasen. 2a zeigt wie der vaginale Blutfluss durch Infusionen
von VIP (intravenöser
Bolus) in einer konzentrationsabhängigen Art und Weise erhöht wird. 2b zeigt, dass zwei wiederholte Infusionen von
VIP ähnliche
Erhöhungen
des Blutflusses erzeugen. Man bemerke, dass die Dauer der Antwort
ebenfalls ähnlich
ist. Alle Veränderungen
wurden unter Verwendung von Laser-Doppler-Verfahren bestimmt.
-
3: – Vasoaktives
intestinales Peptid (VIP) reduziert den mittleren arteriellen Blutdruck
im Modell der sexuellen Erregung beim anästhesierten Hasen. Diese Grafik
zeigt die typischen Wirkungen der vasoaktiven Wirkstoffe und die
zur Untersuchung des vaginalen Blutflusses auf den mittleren arteriellen
Druck bei einem anästhesierten
Hasen verwendeten Stimulationsparameter. Diese beobachteten Wirkungen
sind typisch für
die bei allen geprüften
Tieren festgestellten Trends. VIP induzierte eine signifikante Abnahme
des mittleren arteriellen Druckes, während die Pelvisnerven-Stimulation, Kontroll-Infusionen
von Hepsalin oder Inhibitoren von PDEcAMP oder
NEP keine Wirkung auf den Blutdruck haben. Man bemerke, dass die
mit den VIP Infusionen zusammenhängende
Reduktion des Blutdrucks auch mit einer starken Erhöhung der
Herzfrequenz einhergeht.
-
4: – Die Aktivierung
des cAMP/Adenylatcyclase Pfads ahmt die VIP-vermittelte Vasorelaxation und
Relaxation der glatten Muskeln im vaginalen Gewebe nach. 4a zeigt, dass eine Infusion von Forskolin (40
nmol/kg iv Bolus, ein cAMP-Mimetikum) signifikante Erhöhungen des
vaginalen Blutflusses induziert. Man bemerke, dass die Amplitude
und Dauer der Antwort zu der durch VIP (20.0 μg/kg, iv Bolus) induzierten ähnlich ist.
Interessanterweise haben die Wirkungen auf den Blutfluss eine längere Wirkungsdauer
auf die äussere
Vaginalwand. Alle Veränderungen
wurden unter Verwendung von Laser-Doppler-Verfahren bestimmt. 4b zeigt, dass sowohl VIP (0.1 μM) als auch
Forskolin (10 μM)
die intrazellulären
Konzentrationen von cAMP signifikant über die basalen Spiegel in
der Hasenvagina erhöht. 4c zeigt, dass Forskolin potente Relaxationen
von präkontrahierten
(1 μM Phenylephrin)
Hasenvaginastreifen induziert mit einem IC50 ~
300 nM. Alle Veränderungen
wurden unter Verwendung von in-vivo-Laser-Doppler-Verfahren, biochemischen
cAMP-Enzym-Immunoassays oder durch in-vitro-Gewebe-Relaxation quantifiziert.
-
5: – Die Infusion
von VIP erhöht
den klitoralen Blutfluss, und die Aktivierung des cAMP/Adenylatcyclase
Pfads ahmt die VIP-vermittelte klitorale Vasorelaxation im Modell
der sexuellen Erregung beim anästhesierten
Hasen nach. Die Infusion von VIP (60–200 μg/kg) induziert eine konzentrationsabhängige Erhöhung des
klitoralen Blutflusses. Eine 115%-ige Erhöhung des klitoralen Blutflusses
wurde nach einer iv-Infusion von 200 μg/kg VIP beobachtet. Diese Wirkungen
von VIP auf den klitoralen Blutfluss können durch eine Infusion des
cAMP-Mimetikums Forskolin (FSK, 40 nmol/kg iv-Bolus) nachgeahmt
werden. Eine 156%-ige Erhöhung des klitoralen
Blutflusses wurde nach einer iv-Infusion von 40 nmol/kg Forskolin
beobachtet. Alle Erhöhungen waren
gegenüber
den Kontroll-Infusionen (Hepsalin) signifikant erhöht. Man
bemerke, dass die Amplitude der Antwort ähnlich zu der durch VIP (200 μg/kg, iv-Bolus)
induzierten ist und mit den beim vaginalen Blutfluss in 2 und 4 beobachteten
vergleichbar ist. Alle Veränderungen
wurden unter Verwendung von in-vivo-Laser-Doppler-Verfahren quantifiziert
und waren im Vergleich zu Vehikel-Infusionen (Hepsalin) signifikant
erhöht.
-
6: – Ein selektiver
Inhibitor von NEP EC 3.4.24.11 erhöht die unterleibsnervenstimulierten
(PNS) Erhöhungen
des vaginalen Blutflusses im Modell der sexuellen Erregung beim
anästhesierten
Hasen. Wiederholte PNS in Intervallen von 15 Minuten induzieren
reproduzierbare Erhöhungen
des vaginalen Blutflusses (weisser Balken). Die Verabreichung eines
NEP-Inhibitors (grauer
Balken) verstärkte
die durch submaximale Stimulationsfrequenzen (z.B. 4Hz) maximale
Erhöhung
des vaginalen Blutflusses im Vergleich zu den während zeitlich übereinstimmenden
Kontrollstimulationen oder mit Vehikel-Kontrollen beobachteten Erhöhungen (schraffierter
Balken). Die folgenden dosisabhängigen
Verstärkungen
wurden beobachtet: 0.3 mg/kg iv induzierte eine 40%-ige Erhöhung und
1.0 mg/kg iv induzierte eine 91%-ige Erhöhung (Mittelwert n=3). Der NEP-Inhibitor
hatte keine Wirkung auf den basalen (nicht stimulierten) vaginalen
Blutfluss (Daten nicht dargestellt). Alle Veränderungen wurden unter Verwendung
von Laser-Doppler-Verfahren bestimmt.
-
7: – Selektive
Inhibitoren von NEP EC 3.4.24.11 verstärken die VIP-induzierten Erhöhungen des vaginalen Blutflusses
im Modell der sexuellen Erregung beim anästhesierten Hasen. Wiederholte
Infusionen von VIP in Intervallen von 30 Minuten induzieren reproduzierbare
Erhöhungen
des vaginalen Blutflusses (siehe 2b).
Ein NEP-Inhibitor potenziert sowohl die Amplitude und verlängert auch
die Dauer des verstärkten Blutflusses
wenn diese Erhöhungen
durch submaximale Dosen von VIP, z.B. 6.0 μg/kg, induziert werden. Bei Dosierungen
von VIP, welche maximale Erhöhungen
im vaginalen Blutfluss induzieren, z.B. 60 μg/kg, potenzieren NEP-Inhibitoren
nur die Dauer des verstärkten
vaginalen Blutflusses. Die VIP-induzierte Erhöhungen in der Gegenwart eines
NEP-Inhibitors sind als geschlossene Dreiecke dargestellt, während die
Kontroll-VIP-Antworten als offene Dreiecke dargestellt sind. Eine
Kontroll-Infusion von Hepsalin hat keine Wirkung auf die Amplitude
der Antworten. Alle Veränderungen
wurden unter Verwendung von Laser-Doppler-Verfahren bestimmt.
-
8: – Ein selektiver
Inhibitor von PDEcAMP vom Typ 2 verstärkt die
unterleibsnervenstimulierten (PNS) Erhöhungen des vaginalen Blutflusses
im Modell der sexuellen Erregung beim anästhesierten Hasen. Wiederholte
PNS in Intervallen von 15 Minuten induzieren reproduzierbare Erhöhungen des
vaginalen Blutflusses (weisse Quadrate). Die Verabreichung eines
PDEcAMP Inhibitors vom Typ 2 verstärkte die
durch submaximale Stimulationsfrequenzen induzierte maximale Erhöhung des
vaginalen Blutflusses (schwarze Quadrate; bei 4Hz) im Vergleich
zu den während
zeitlich übereinstimmenden
Kontrollstimulationen beobachteten Erhöhungen (offene Quadrate). Eine
Infusion des PDE2-Inhibitors (500 μg/kg) induzierte eine 86.8±21.9%-ige
Verstärkung
des vaginalen Blutflusses (Mittelwert ± sem n=2). Alle Veränderungen wurden
unter Verwendung von Laser-Doppler-Verfahren bestimmt.
-
9: – Selektive
Inhibitoren von PDEcAMP vom Typ 2 verstärken die
VIP-induzierten Erhöhungen
des vaginalen Blutflusses im Modell der sexuellen Erregung beim
anästhesierten
Hasen. Wiederholte Infusionen von VIP in Intervallen von 30 Minuten
induzieren reproduzierbare Erhöhungen
des vaginalen Blutflusses (siehe 2b).
Ein selektiver PDEcAMP Inhibitor vom Typ
2 (25 μg/kg
iv-Bolus) potenziert die Dauer des durch VIP (60 μg/kg iv-Bolus)
induzierten erhöhten
vaginalen Blutflusses. VIP-induzierte Erhöhungen in der Gegenwart eines PDEcAMP-Inhibitors sind als geschlossene Dreiecke
dargestellt, während
die Kontroll-VIP-Antworten
als offene Dreiecke dargestellt sind. Eine Kontroll-Infusion von
Hepsalin hatte keine Wirkung auf die Amplitude der Antworten. Alle
Veränderungen
wurden unter Verwendung von Laser-Doppler-Verfahren bestimmt.
-
10: – Ein
selektiver Antagonist von NPY-Y1-Rezeptoren verstärkt die
unterleibsnervenstimulierten (PNS) Erhöhungen des vaginalen Blutflusses
im Modell der sexuellen Erregung beim anästhesierten Hasen. Wiederholte
PNS in Intervallen von 15 Minuten induzieren reproduzierbare Erhöhungen des
vaginalen Blutflusses (Daten nicht dargestellt). Die Verabreichung
eines NPY-Y1-Antagonisten (grauer Balken) verstärkte die durch submaximale
Stimulationsfrequenzen (z.B. 4Hz) induzierte maximale Erhöhung des
vaginalen Blutflusses im Vergleich zu den während den zeitlich übereinstimmenden
Kontrollstimulationen oder mit Vehikelkontrollen beobachteten Erhöhungen (schraffierter
Balken). Die folgenden dosisabhängigen
Verstärkungen
wurden beobachtet: 0.01 mg/kg iv induzierte eine 15.8±19.6%-ige
Erhöhung,
0.03 mg/kg iv induzierte eine 35.1±17.17%-ige Erhöhung, 0.10
mg/kg iv induzierte eine 60.1±16.9%-ige
Erhöhung
und 0.3 mg/kg iv induzierte eine 91.9±27.4%-ige Erhöhung (Mittelwert ± sem n=3).
Der NPY-Y1-Antagonist hatte keine Wirkung auf den basalen (nicht
stimulierten) vaginalen Blutfluss (Daten nicht dargestellt). Alle
Veränderungen
wurden unter Verwendung von Laser-Doppler-Verfahren bestimmt.
-
11 liefert eine Übersichtsgrafik für einige
der hier angegebenen Daten, wobei gezeigt wird, dass die Wirkstoffe
aufgrund der Potenzierung der endogenen cAMP-Spiegel sehr nützlich zur
Erhöhung
des vaginalen Blutflusses sind.
-
12: – Ein
selektiver Inhibitor von NEP EC 3.4.24.11 verstärkt die unterleibsnervenstimulierten (PNS)
Erhöhungen
des klitoralen Blutflusses im Modell der sexuellen Erregung beim
anästhesierten
Hasen. Die Verabreichung eines NEP-Inhibitors (graue Balken) verstärkte die
durch submaximale Stimulationsfrequenzen (z.B. 4Hz) induzierte maximale
Erhöhung
des klitoralen Blutflusses im Vergleich zu den während der zeitlich übereinstimmenden
Kontrollstimulationen oder mit Vehikelkontrollen beobachteten Erhöhungen (schraffierter
Balken). Die folgenden dosisabhängigen
Verstärkungen
wurden beobachtet: 1.0 mg/kg iv induzierte eine 1131%-ige Erhöhung (Mittelwert
n=3). Der NEP-Inhibitor hatte keine Wirkung auf den basalen (nicht stimulierten)
klitoralen Blutfluss. Alle Veränderungen
wurden unter Verwendung von Laser-Doppler-Verfahren ermittelt.
-
EIN ASSAY ZUR MESSUNG
DER cAMP AKTIVITÄT/SPIEGEL
-
Messung
von cAMP aus Proben von vaginalem Gewebe unter Verwendung eines
Biotrak cAMP Enzym-Immunoassay (EIA) Kits (Amersham Life Sciences
RPN 225).
-
Die
cAMP-Spiegel werden mittels EIA in Proben von vaginalem Gewebe gemessen.
Der EIA beruht auf der Kompetition zwischen nicht markiertem cAMP
und einer fixen Menge von mit Peroxidase markiertem cAMP für eine beschränkte Menge
von cAMP-spezifischem Antikörper.
-
1. MATERIALIEN
-
Alle
Materialien wurden, sofern nicht anders angegeben, von Amersham
Life Science cAMP EIA Kit (RPN 225) bezogen.
- 1.1
Mikrotiter-Platte: Platte mit 96 Vertiefungen, bedeckt mit Esel-anti-Hasen-IgG.
- 1.2 Assay-Puffer: 0.05 M Natriumacetat-Puffer pH 5.8, der nach
Rekonstitution 0.02% Rinderserumalbumin und 0.5% Konservierungsmittel
enthält.
Die Inhalte der Flaschen werden in einen Messzylinder unter Verwendung
von 3 × 15
ml destilliertem Wasser übergeführt. Das
endgültige
Volumen wird dann auf 500 ml eingestellt.
- 1.3 cAMP-Standard (für
Acetylierungsverfahren). cAMP bei 10.24 pmol/ml in 0.05 M Acetat-Puffer
pH 5.8, der nach Rekonstitution 0.02% Rinderserumalbumin und 0.5%
Konservierungsmittel enthält.
Standard wird für
die Verwendung in 2.5 ml des Assay-Puffers gelöst.
- 1.4 Antiserum. Anti-cAMP-Antikörper in 0.05 M Acetat-Puffer
pH 5.8. der nach Rekonstitution 0.02% Rinderserumalbumin und 0.5%
Konservierungsmittel enthält.
Vor der Verwendung wird der Antikörper mit 11 ml Assay-Puffer
verdünnt
und durch sanftes Umkippen zum Auflösen der Inhalte gemischt.
- 1.5 cAMP-Konjugat. cAMP Meerrettich Peroxidase in 0.05 M Acetat-Puffer
pH 5.8, der nach Rekonstitution 0.02% Rinderserumalbumin und 0.5%
Konservierungsmittel enthält.
Vor der Verwendung wird die Lösung mit
11 ml Assay-Puffer verdünnt
und durch sanftes Umkippen zum Auflösen der Inhalte gemischt.
- 1.6 Wasch-Puffer. 0.01 M Phosphat-Puffer pH 7.5, der nach Rekonstitution
0.05% (v/v) TweenTM 20 enthält. Die
Inhalte der Flaschen werden in einen Messzylinder unter Verwendung
von 3 × 15
ml destilliertem Wasser übergeführt. Das
endgültige
Volumen wird dann auf 500 ml eingestellt.
- 1.7 TMB-Substrat. 3,3',5,5'-Tetramethylbenzidin
(TMB)/Wasserstoffperoxid in 20% (v/v) Dimethylformamid.
-
Gebrauchsfertig.
- 1.8 Acetylierungsreagens. 2 ml Essigsäureanhydrid,
4 ml Triethylamin, hergestellt nach Bedarf.
- 1.9 Schwefelsäure
(1 M). 1 M Schwefelsäure
wird aus einer 18 M Stammlösung
(BDH) hergestellt. 1.11 ml der Säure
werden zu 18.8 ml destilliertem Wasser zugegeben.
-
2. SPEZIFISCHE AUSRÜSTUNG
-
- 2.1 Wegwerfbare 5 ml Reagenzgläser
- 2.2 Spektrophotometrischer Plattenleser (Spectra max 190)
- 2.3 Mikrotiter Plattenschüttler
(Luckham R100)
-
3. METHODEN
-
- – Gewebeproben-Herstellung.
Die Gewebe wurden in 5 ml Proben einer physiologischen Salzlösung mit der
relevanten Vorbehandlung, z.B. Agonisten, cAMP-Mimetika etc., behandelt.
Nach der Behandlung wurden die Proben in flüssigem Stickstoff schnellgefroren
und dann unter Verwendung eines Hammers zertrümmert. Das Pulver wurde in
ein Zentrifugenrohr gegeben und es wurde 1 ml 0.5M eiskalter Perchlorsäure (PCA)
zugegeben. Die Probe wurde durch Verwirbeln gemischt und während 1
Std. auf Eis belassen.
- – cAMP-Extraktion
aus Gewebeproben. Die Proben wurden während 5 Min. bei 10000 g bei
4°C zentrifugiert.
Der Überstand
wurde entfernt und in anderen Zentrifugenrohre gegeben. Das Pellet
wurde für
die Protein-Analyse bei –80°C aufbewahrt.
Die Überstand-Proben
wurden dann auf pH ~ 6 unter Verwendung von K3PO4 neutralisiert. Zentrifugieren während 5
Min bei 10000 g bei 4°C.
Der Überstand
wird wiedergewonnen und 4 Mal mit 5 Volumina (5 ml) wassergesättigtem
Diethylether gewaschen. Die obere Etherphase sollte nach jedem Waschen
verworfen werden. Die wässrige
Phase wird in ein kurzes dünnes
Glasrohr übergeführt und
unter einem Stickstoffstrom bei 60°C getrocknet. Der getrocknete
Extrakt wird in 1 ml des Assay-Puffers gelöst und bis zum Gebrauch im
Kühlschrank
gelagert (oder er kann tiefgefroren werden).
- – Stammreagenzien
werden auf Raumtemperatur äquilibriert,
und dann werden Arbeitslösungen
hergestellt.
- – cAMP-Standards
werden in Glasrohren, die als 2, 4, 8, 16, 32, 64, 128, 256 und
512 fmol markiert sind, hergestellt. Dies wird durch Zugabe von
1 ml des Assay-Puffers zu allen Rohren mit Ausnahme des 512 fmol
Standards erreicht. 1 ml Acetylierungsstandard (10.24 pmol/ml) wird
dann zu den beiden obersten Standards (256 und 512 fmol) zugegeben.
Der 256 fmol Standard wird verwirbelt und 1 ml wird zum 128 fmol Standard übergeführt. Dies
wird fortgesetzt bis zum 2 fmol Standard, wo 1 ml Lösung entsorgt
wird. Es wird noch ein 1 ml Assay-Puffer enthaltendes Nullstandardrohr
bereitgestellt.
- – Gewebeextraktproben
werden auf Eis aufgetaut (falls nötig) und 1 zu 100 (10 μl Proben
auf 990 μl
Assay-Puffer) in beschrifteten Glasrohren verdünnt.
- – Das
cAMP wird für
alle Standards und Proben in einer Kapelle durch Zugabe von 100 μl Acetylierungsreagens
acetyliert, welches entlang der Seite des Rohres zugegeben wird,
woraufhin umgehend verwirbelt wird.
- – 50 μl von allen
Standards und Proben werden in die geeigneten Vertiefungen der Platte
mit 96 Vertiefungen gegeben, und 150 μl Assay-Puffer werden in Vertiefungen
für die
nichtspezifische Bindung(NSB) gegeben.
- – 100 μl Antiserum
werden in alle Vertiefungen mit Ausnahme derjenigen für Blindproproben
(B) und NSB gegeben und anschliessend während 2 Stunden bei 3–5°C inkubiert.
- – Nach
der Inkubation werden 100 μl
cAMP-Peroxidase-Konjugat
in alle Vertiefungen mit Ausnahme derjenigen für B zugegeben und anschliessend
während
1 weiteren Stunde bei 3-5°C inkubiert.
- – Die
Platten werden durch kopfüber
stellen geleert und mit Löschpapier
betupft, und anschliessend wird jede Vertiefung viermal mit 400 μl Wasch-Puffer
gewaschen. Nach jedem Waschen werden die Platten wieder betupft,
um sicherzustellen, dass sämtlicher
Wasch-Puffer entfernt ist. 200 μl
TMB werden dann sofort in alle Vertiefungen verteilt.
- – Die
Platten werden vor Zugabe von 100 μl einer 1 M Schwefelsäure in alle
Vertiefungen während
30 Minuten bei Raumtemperatur auf einen Plattenschüttler gestellt.
Die optische Dichte wird auf dem Spectra max 190 bei 450 nm innerhalb
von 30 Minuten gemessen.
-
4. STANDARDS
-
Bei
jedem Assay werden die folgenden Standardrohre bereitgestellt:
-
4.1 Zugabe eines Standards
in Assay-Puffer
-
Eine
bekannte Menge von cAMP wird zum Assay-Puffer gegeben, um die Effizienz
des Assays zu bestimmen. 70 pmol/ml cAMP werden zum Assay-Puffer
zugegeben, welcher zu 35 fmol/Vertiefung im Assay äquivalent
ist, was in der Mitte der Dosis-Antwort-Kurve liegt.
| Bildung
von 1 ml Standard: | 68.4 μl 521 fmol/Vertiefung
Standard |
| | 931.6 μl Assay-Puffer |
-
4. Wirkungen
der Verbindungen auf die Platte
-
Standards
werden bereitgestellt, um zu bestimmen, ob die in den funktionellen
Studien verwendete Verbindung irgend eine Wirkung auf die Platte
mit 96 Vertiefungen hat oder das Bindungsverhalten von cAMP beeinflusst.
Dies umfasst:
- – Zugabe der Verbindung in
Assay-Puffer alleine, um die Wirkungen der Verbindung direkt auf
die Platte zu bestimmen.
- – Zugabe
der Verbindung in Plasma mit basalem Spiegel von cAMP, um die Wirkungen
der Verbindung auf das Bindungsverhalten von cAMP auf die Platte
zu bestimmen.
-
5
nM Konzentrationen der Verbindung werden in jeden Standard gegeben.
Es wurden 5 nM gewählt, weil
in der Vergangehnheit die Gesamtwirkstoffspiegel am Ende der Infusion
jeweils ungefähr
150–300
nM betrugen. Die Proben werden vor dem Testen um 1:100 verdünnt, sodass
5 nM auch höhere
als erwartete Gesamtwirkstoffkonzentrationen am Ende der Infusion
abdeckt sind.
-
5. BERECHNUNGEN
-
Der
Spectra max Plattenleser misst die optische Dichte (OD) bei 450
nm.
-
Die
Standardkurve wird durch Auftragen von %B/Bo (y-Achse) gegen cAMP
fmol/Vertiefung (x-Achse) auf dem Spectra max erzeugt.
%B/BO
(% gebunden) für
jede Probe und Standard werden wie folgt berechnet:
Bo = Nullstandard
(siehe Methoden 3.2)
-
-
Der
Wert in fmol/Vertiefungsvolumen kann dann für jede Probe direkt aus der
Standardkurve abgelesen werden. Die Werte werden dann auf pmol/ml
umgerechnet, und anschliessend wird der Mittelwert für jedes Proben-Paar
genommen.
-
Umrechnung
der Werte aus fmol/Vertiefung zu pmol/ml:
fmol auf pmol = man
teile durch 1000
Volumen in der Vertiefung = 50 μl .... So
(× 1000)/50
50
-
Die
Probe wird 1/100 verdünnt,
somit gilt gesamthaft = 1 × 1000/1000 × 100/50
= 2
-
Demnach
werden alle fmol/Vertiefungs-Werte mit 2 multipliziert um daraus
pmol/ml zu erhalten.
-
TIERTESTMODELL
-
DIE
POTENZIERUNG DER WIRKUNGEN VON ZYKLISCHEM ADENOSIN-3',5'-MONOPHOSPHAT (cAMP)
FÜHRT ZU
EINER ERHÖHUNG
DES VAGINALEN BLUTFLUSSES IM MODELL DER SEXUELLEN ERREGUNG BEI ANÄSTHESIERTEN
HASEN
-
1.0 Ziele
-
- 1. Entwicklung und Validierung eines Tiermodells
der weiblichen sexuellen Erregung.
- 2. Identifikation des (der) für die Regulierung des genitalen
Blutflusses im anästhesierten
Hasen verantwortlichen Mechanismus(en).
- 3. Identifikation von möglichen
Ansätzen
zur Verstärkung
des vaginalen und klitoralen Blutflusses.
- 4. Untersuchung des(der) Mechanismus(en), welche der Relaxation
der glatten Muskeln in der Vagina zugrunde liegen und Identifikation
von möglichen
Ansätzen
zur Verstärkung
der vaginalen Relaxation.
-
2.0 Einführung
-
Die
normale sexuelle Erregungsantwort besteht aus einer Anzahl von physiologischen
Antworten, die während
der sexuellen Erregung beobachtet werden. Diese Veränderungen,
wie das vaginale, labiale und klitorale Anschwellen ergeben sich
aus Erhöhungen
des genitalen Blutflusses. Das Anschwellen führt über die Plasma-Transsudation
zu einer erhöhten
vaginalen Lubrikation, zu einer erhöhten vaginalen Dehnbarkeit
(Relaxation der glatten Muskeln der Vagina) und zu Erhöhungen in
der vaginalen und klitoralen Sensibilität.
-
Die
weibliche sexuelle Erregungsstörung
(FSAD) ist eine weit verbreitete sexuelle Störung, die bis zu 40% der prä-, peri- und postmenopausalen
(±HRT)
Frauen betrifft. Die primären
Folgen der FSAD sind ein verringertes genitales Anschwellen, die
sich als Mangel der vaginalen Lubrikation und als Mangel an lustvollen genitalen
Empfindungen manifestieren. Sekundäre Folgen umfassen ein verringertes
sexuelles Begehren, Schmerzen während
des Geschlechtsverkehrs und Schwierigkeiten, den Orgasmus zu erreichen.
Die häufigste
Ursache von FSAD ist ein verringerter genitaler Blutfluss, welcher
zu einem verringerten vaginalen, labialen und klitoralen Anschwellen
führt (Park,
1997; Goldstein, 1998; Berman, 1999a, Werbin, 1999).
-
Wie
hier ausgeführt,
bezieht sich die vorliegende Erfindung auf ein Mittel zur Wiederherstellung
oder Potenzierung der normalen sexuellen Erregungsantwort bei an
FSAD leidenden Frauen, durch Erhöhung
des genitalen Blutflusses.
-
In
unseren Studien wurde cAMP (cyclisches Adenosin-3',5'-Monophosphat) als Mediator der vaginalen
Vasorelaxation zur Messung von kleinen Veränderungen im genitalen Blutfluss unter
Verwendung von Laser-Doppler-Verfahren identifiziert. Durch Verwendung
eines Inhibitors des VIP-Metabolismus (ein NEP EC3.4.24.11 Inhibitor)
wurde auch gezeigt, dass die während
der Pelvisnervenstimulation beobachteten Erhöhungen im genitalen Blutfluss
(d.h. die sexuelle Erregung) durch VIP vermittelt werden. Dies beruhte
auf der Entwicklung eines Tiermodells der sexuellen Erregung und
auf dem Nachweis, dass die Daten die während der weiblichen sexuellen
Erregung beobachteten physiologischen Veränderungen widerspiegeln. Das
Modell wurde dann zur Identifikation und Validierung von Mechanismen
verwendet, die den genitalen Blutfluss erhöhen, z.B. die direkte oder
indirekte Potenzierung der cAMP-vermittelten Vasorelaxation.
-
3.0 Methoden
-
3.1 Anästhesie-Protokoll
-
Weibliche
Neuseeland Hasen (~ 2.5 kg) wurden mit einer Kombination von Medetomidin
(DomitorTM) 0.5 ml/kg i.m. und Ketamin (VetalarTM) 0.25 ml/kg i.m. prämediziert, während die
Sauerstoffaufnahme über
eine Gesichtsmaske aufrechtgehalten wurde. Die Hasen wurden unter
Verwendung eines PortexTM kragenlosen Endotrachealtubus
3 ID tracheotomisiert, mit dem Ventiliergerät verbunden und bei einer Ventilationsrate
von 30–40
Atemzügen
pro Minute mit einem ungefähren
Atemvolumen von 18–20
ml gehalten, wobei ein maximaler Druck in den Atemwegen von 10 cm
H2O eingehalten wurde. Die Anästhesie
wurde dann auf Isofluran gewechselt und die O2-Beatmung
mit 2 l/Min. fortgesetzt. Die rechte marginale Ohrvene wurde unter
Verwendung eines 23G oder 24G Katheters kanüliert und eine Lactated Ringer-Lösung mit
0.5 ml/Min. perfundiert. Der Hase wurde während der invasiven Chirurgie
auf 3% Isofluran gehalten, wogegen zur Aufrechterhaltung der Anästhesie
auf 2% reduziert wurde.
-
3.2 Kanülierung
der Gefässe
-
Die
linke Leistenregion des Hasens wurde rasiert, und es wurde ein vertikaler
Schnitt mit einer Länge von
ungefähr
5 cm entlang des Schenkels vorgenommen. Die Femoralisvene und -arterie
wurden exponiert, isoliert und dann mit einem PVC-Katheter (17G) für die Infusion
der Wirkstoffe und Verbindungen kanüliert. Die Kanülierung
wurde für
die Femoralisarterie wiederholt, wobei der Katheter zu einer Tiefe
von 10 cm eingeführt wurde,
um sicherzustellen, dass der Katheter die Abdominalaorta erreichte.
Dieser arterielle Katheter wurde mit einem Gould-System verbunden,
um den Blutdruck aufzuzeichnen. Proben für die Blutgasanalyse wurden ebenfalls über den
arteriellen Katheter entnommen. Es wurden systolische und diastolische
Blutdruckwerte gemessen und der mittlere arterielle Druck unter
Verwendung der Formel (diastolisch × 2 + systolisch)/3 berechnet.
Die Herzfrequenz wurde über
das Pulsoxymeter und das Po-ne-mah-Datenerfassungs-Softwaresystem (Ponemah
Physiology Platform, Gould Instrument Systems Inc) gemessen.
-
3.3 Stimulation des Pelvisnervs
-
Es
wurde ein ventraler mittiger Schnitt in die abdominale Höhle gemacht.
Der Schnitt hatte eine Länge von
ungefähr
5 cm, genau oberhalb der Schamgegend. Das Fett und die Muskeln wurden
stumpf abgetrennt, um den hypogastrischen Nerv, welcher die Körperhöhle hinunter
verläuft,
freizulegen. Es war entscheidend, nahe bei der Seitenkurve der Schamwand
zu bleiben, um eine Beschädigung
der oberhalb der Schamgegend liegenden Femoralisvene und -arterie
zu vermeiden. Der Ischiasnerv und die Pelvisnerven liegen tiefer
und sind nach der weiteren Auftrennung auf der dorsalen Seite des
Hasens lokalisiert. Nachdem der Ischiasnerv identifiziert ist, wurde
der Pelvisnerv ohne weiteres lokalisiert. Der Begriff Pelvisnerv
wird hier lose verwendet; entsprechende Anatomiebücher identifizieren
die Nerven nicht genügend
detailliert. Jedenfalls verursacht die Stimulation des Nervs eine
Erhöhung
des vaginalen und klitoralen Blutflusses und eine Innervierung der
Pelvisregion. Der Pelvisnerv wurde vom umgebenden Gewebe freigelegt
und eine Harvard bipolare Stimulationselektrode wurde um den Nerv
platziert. Der Nerv wurde leicht angehoben um eine gewisse Spannung
zu geben, dann wurde die Elektrode in der Stellung gesichert. Ungefähr 1 ml
leichtes Paraffinöl
wurde um den Nerv und die Elektrode gegeben. Dieses wirkt als schützendes
Schmiermittel für
den Nerv und verhindert die Kontamination der Elektrode mit Blut.
Die Elektrode wurde mit einem Grass S88 Stimulator verbunden. Der
Pelvisnerv wurde unter Verwendung der folgenden Parameter stimuliert:
5V, Pulsbreite 0.5 ms, Dauer des Stimulus 10 Sekunden und Frequenzbereich
von 2 bis 16 Hz. Reproduzierbare Antworten wurden erhalten, wenn der
Nerv alle 15–20
Minuten stimuliert wurde.
-
Zu
Beginn von jedem Experiment wurde eine Frequenz-Antwort-Kurve bestimmt, um
die zur Verwendung als submaximale Antwort am besten geeignete Frequenz,
normalerweise 4Hz, zu bestimmen. Die zu prüfende(n) Verbindung(en) wurde(n) über die
Femoralisvene unter Verwendung einer Harvard 22 Infusionspumpe infundiert,
was einen kontinuierlichen 15-minütigen Stimulationszyklus
ermöglichte.
-
3.4 Positionierung der
Laser-Doppler-Sonden
-
Es
wurde ein ventraler mittiger Schnitt am kaudalen Ende der Schamgegend
gemacht, um die Schamgegend freizulegen. Bindegewebe wurde entfernt,
um die Tunica der Klitoris freizulegen und um sicherzustellen, dass
die Wand frei von kleinen Blutgefässen war. Die äussere Vaginalwand
wurde durch Entfernen von sämtlichem
Bindegewebe ebenfalls freigelegt. Eine Laser-Doppler-Flusssonde
wurde 3 cm in die Vagina eingeführt,
so dass die Hälfte
des Sondenschaftes immer noch sichtbar war. Eine zweite Sonde wurde
so positioniert, dass sie gerade über der äusseren Klitoriswand lag. Die
Position dieser Sonden wurde dann verstellt, bis ein Signal erhalten
wurde. Eine zweite Sonde wurde gerade über der Oberfläche eines
Blutgefässes
auf der äusseren
Vaginawand platziert. Beide Sonden wurden in ihrer Position festgeklemmt.
-
Die
vaginalen und klitoralen Blutflüsse
wurde entweder als Zahlenwerte direkt aus dem Flussmeter unter Verwendung
der Po-ne-mah-Datenerfassungs-Software (Ponemah Physiology Platform,
Gould Instrument Systems Inc) oder indirekt aus der Aufzeichnung
des Gould Kartenschreibers erfasst. Eine Kalibrierung wurde zu Beginn
des Experiments (0–125
ml/Min./100 g Gewebe) durchgeführt.
-
3.5 Infusion des vasoaktiven
intestinalen Peptides (VIP)
-
Die
infundierten Dosierungen von VIP (Bachem, H-3775) waren 2.0, 6.0,
20.0, 60.0 μg/kg
iv und wurden in einem Volumen von 0.5 ml physiologischer Kochsalzlösung infundiert.
VIP wurde unter Verwendung einer Harvard 22 Pumpe infundiert, wobei
mit 500 μl/Min. über ein
3-Wege-Ventil in die Femoralisvene infundiert wurde. Nach der VIP-Infusion
wurde der Katheter mit heparinisierter physiologischer Kochsalzlösung (Hepsalin)
gespült,
so dass kein VIP mehr im Katheter verblieb.
-
Für Experimente
mit Verwendung von VIP-Infusionen wurde eine initiale sensitivierende
Dosis-Antworts-Kurve (2–60 μg/kg) benötigt, um
reproduzierbare Antworten erhalten zu können. Als negative Kontrolle wurde
eine initiale Infusion von Hepsalin (50 Ul/ml) infundiert.
-
3.6 Infusion von Inhibitoren
-
NEP-(Neutrale
Endopeptidase EC3.4.24.11)-Inhibitoren, Phosphodiesterase-Typ 5-(PDE5)-Inhibitoren
und NPY-Y1-Antagonisten
wurden in physiologischer Kochsalzlösung oder 5%-iger Glucose-Lösung (200 μl 50% Glucose
in 1.8 ml Wasser für
Injektionszwecke) hergestellt. PDEcAMP-Inhibitoren
wurden in einer 40%-igen Ethanol-Lösung (200 μl 50% Glucose in 1.8 ml Wasser/Ethanol
zur Injektion) gelöst.
Die Inhibitoren und Vehikelkontrollen wurden mit derselben Geschwindigkeit
wie VIP infundiert. Die NEP-Inhibitoren wurden während 30 Minuten vor einer
VIP-Dosis-Antworts-Kurve belassen, während die NEP-Inhibitoren,
NPY-Y1-Rezeptor-Antagonisten und PDEcAMP-Inhibitoren
während
15 Minuten vor der Pelvisnerv-Stimulation
belassen wurden.
-
3.7 Messung der Relaxation
der glatten Muskeln in isolierter Hasenvagina
-
- 3.7 (a). Hasenvagina in-vitro-Herstellung:
Weibliche weisse Neuseeland Hasen (2.0–3.0 kg) wurden durch Genickbruch
getötet.
Die abdominale Höhle
wurde eröffnet
und die Vagina exzidiert. Gewebestreifen wurden longitudinal in
5 ml silanisierten Organkammern von Welsey Co. mittels geflochtener
Seidennähte (6/0-Mass)
mit einer initialen Ruhespannung von 1.5 g befestigt und in einem
bei 37°C
gehaltenem und mit 95%O2/5%CO2 begastem
Krebs-Bicarbonat-Puffer
gelagert. Die obere Ligatur von jedem Gewebestreifen wurde an einen
Kraft-Verschiebungs-Transducer mit einem 10 g Messbereich angebracht,
und so wurden Veränderungen
der isometrischen Kraft gemessen und unter Verwendung eines DART
In-vitro-Datenerfassungssystems aufgenommen. Die Gewebe wurden während 1.5
Stunden äquilibriert
und regelmässig
mit Krebs-Puffer gewaschen.
- 3.7 (b). Durch vasoaktives intestinales Peptid induzierte Relaxation
von Hasenvagina: Jedes Gewebe wurde unter Verwendung einer 1 μM Badkonzentration
von Phenylephrin kontrahiert. Nachdem die kontraktile Antwort ein
stabiles Plateau (~ 15 Minuten) erreichte, wurde VIP kumulativ in
log-Einheiten zur
Organkammer zugegeben, so dass sich Konzentrationen von 0.1 – 100 nM
ergeben. Die Relaxationsantworten wurden 5 Minuten nach der Zugabe
jeder Konzentration von VIP gemessen; die maximale Relaxation war
zu diesem Zeitpunkt erreicht worden. Die Gewebe erhielten dann entweder
einen Testwirkstoff (z.B. NEP- oder PDE-Inhibitor) oder DMSO-Vehikel (zeitlich übereinstimmende
Kontrolle).
- 3.7 (c). Analyse der Daten von VIP-Relaxations-Experimenten:
Für jede VIP-Konzentrations-Relaxations-Antworts-Kurve
wurden die durch VIP induzierten Relaxationsantworten als prozentualer
Anteil der maximalen durch Phenylephrin induzierten Kontraktion
ausgedrückt. Diese
Werte wurden dann gegen die log VIP-Konzentration aufgezeichnet,
und es wurden sigmoidale Kurven angepasst. Zum Zweck der Kurvenanpassung
wurde die minimale Relaxationsantwort auf 0% beschränkt, während die
maximale Relaxationsantwort frei angepasst werden konnte. Es wurde
die zur Erzeugung einer 50% Relaxation der Phenylephrin-Kontraktion
(EC50 PE) benötigte Konzentration
von VIP bestimmt.
- 3.7 (d). Durch elektrisches Feld stimulierte Relaxation von
Hasenvagina: Hasenvaginastreifen wurden wie im Abschnitt 3.7 (a)
beschrieben hergestellt. Die Gewebestreifen wurden zwischen zwei
Platinelektroden befestigt, die am oberen und unteren Ende der Organkammer
ungefähr
4 cm auseinander platziert waren. Jedes Gewebe wurde unter Verwendung
einer 1 μM
Badkonzentration von Phenylephrin kontrahiert. Nachdem die kontraktile
Antwort ein stabiles Plateau (~ 15 Minuten) erreichte, wurden die
Gewebe einer der Behandlung vorangehenden, durch ein elektrisches
Feld stimulierten (EFS) induzierten Relaxationskurve unterzogen.
Dies wurde bei 40 – 60
Volt durchgeführt
unter Verwendung von sequentiellen Frequenzen von 2, 4, 8 und 16
Hz, die als 10 Sekunden Züge
von Pulsen mit einer Breite von 0.5 Millisekunden abgegeben wurden.
Die Gewebe wurden zwischen jeder Frequenz (5 Minuten) auf die anfängliche,
präkontraktile Spannung
zurückkehren
gelassen, und es wurde das Ausmass der Relaxationsantwort ermittelt.
-
Nach
Vervollständigung
der EFS-Antworts-Kurve vor der Behandlung wurden alle Gewebe während 15
Minuten gewaschen, wobei die Gewebe auf die anfängliche Spannung zurückkehren
konnten. Die Gewebe erhielten dann entweder einen Testwirkstoff
(z.B. NEP- oder PDE-Inhibitor, Stickstoffmonoxid-Synthase-[NOS]-Inhibitor)
oder DMSO-Vehikel
(zeitlich übereinstimmende
Kontrolle). Die Gewebe wurden 15 Minuten nach der Zugabe der Verbindung
oder des Vehikels erneut mit Phenylephrin (1 μM) kontrahiert, und es wurde
eine EFS-induzierte Relaxations-Antworts-Kurve wie oben beschrieben
bestimmt.
-
Für die EFS-Experimente
wurde der Krebs-Puffer mit Atropin (10 μM) und Guanethidin (150 μM) supplementiert,
um jegliche cholinerge oder adrenerge neuronale Innervation der
Vagina zu vermeiden.
-
3.8 Messung der cAMP-Spiegel
in isolierter Hasenvagina
-
Messungen
von cAMP-Konzentrationen wurden aus Vaginalgewebe-Extrakten unter Verwendung
eines Biotrak-cAMP Enzym-Immunoassay
(EIA)-Kits (Amersham Life Sciences RPN 225) durchgeführt.
-
Isolierte
Vaginalgewebe-Proben wurden mit Testwirkstoffen (z.B. Forskolin
oder VIP) behandelt. Nach 5 Minuten wurden die Proben unter Verwendung
von flüssigem
Stickstoff schnellgefroren, homogenisiert, und cAMP wurde extrahiert.
Die cAMP-Spiegel wurden mittels EIA gemessen. EIA beruht auf einer
Kompetition zwischen nicht markiertem cAMP und einer fixen Menge
an mit Peroxidase markiertem cAMP für eine beschränkte Menge
von cAMP-spezifischem Antikörper.
-
3.9 Messung der Phosphodiesterase-(PDE)-Aktivität in isolierter
Hasenvagina
-
Zytosol-Extrakte
von menschlicher Vaginawand wurden von ABS Inc., Delaware (Alter
der Donatorinnen 41 und 60 Jahre alt) bezogen. Die PDE-Isoenzyme
wurden mittels Mono-Q-Anionenaustausch-Chromatographie
aufgetrennt und beruhend auf ihrer Substrat-Selektivität, Sensitivität auf allosterische
Modulatoren und selektive Inhibitoren charakterisiert. Es wurde
zudem eine Western Analyse unter Verwendung von spezifischen PDE-Isoenzym-Antikörpern zum
Nachweis der PDE-Expression in der menschlichen Vagina durchgeführt.
-
Sämtliche
Daten werden als Mittelwert ± s.e.m.
angegeben. Signifikante Veränderungen
wurden unter Verwendung von Student's t-Tests identifiziert.
-
4.0 Resultate und Diskussion
-
4.1 Tiermodell der sexuellen
Erregung
-
In
unseren Studien wurde ein robustes reproduzierbares Modell der Physiologie
der sexuellen Erregung entwickelt. Unter Verwendung dieses Modells
mit anästhesierten
Hasen ist es möglich,
mit Hilfe von Laser-Doppler-Verfahren kleine Veränderungen im genitalen Blutfluss
zu messen. Es wird die Stimulation des Pelvisnervs verwendet, um
die neuronalen Wirkungen der sexuellen Erregung zu simulieren.
-
Es
wurde gefunden, dass die Stimulation des Pelvisnervs frequenzabhängige Erhöhungen des
vaginalen und klitoralen Blutflusses induziert (siehe 1).
Diese Erhöhungen
des basalen Blutflusses sind signifikant, und zwar sowohl bei Messung
an der intravaginalen wie auch an der extravaginalen Wand. Die Stimulation
des Pelvisnervs bei 2Hz induzierte eine mittlere maximale vaginale
Blutflusserhöhung
von 10.3±1.8,
bei 4Hz 20.0±4.6,
8Hz 36.3±4.8
und 16Hz 46.6+4.7 ml/Min./100 g Gewebe (n=4); 15–20V, 0.5ms, 10s) und Erhöhungen des
klitoralen Blutflusses von 14.7±3.6 bei 2Hz, 29.4±1.4 bei
4Hz und 69.7±2.1
bei 8Hz. Diese Werte sind von ähnlicher
Amplitude wie die vorgängig
in Studien am Menschen und in Tiermodellen der Erregung beobachteten
(Berman, 1999a; Park, 1997).
-
Es
wurde gefunden, dass die submaximale Stimulation des Pelvisnervs
zu reproduzierbaren Erhöhungen
des genitalen Blutflusses führt
(z.B. ergab eine Stimulation mit 4Hz alle 15 Minuten eine mittlere
Erhöhung des
vaginalen Blutflusses von 8.50±0.10
ml/Min./100 g Gewebe n=8 und eine mittlere Erhöhung des klitoralen Blutflusses
von 13.65±0.86
ml/Min./100 g Gewebe n=11). Diese Reproduzierbarkeit bleibt bis
zu 5 Stunden erhalten. Die Reproduzierbarkeit dieser Antworten kann
verwendet werden zur Untersuchung von a.) der Identität von endogenen
vasoaktiven Wirkstoffen/Mechanismen, welche die genitale Anschwellung
vermitteln, und b.) des Einflusses von Wirkstoffen, welche zur Erhöhung des
vaginalen und/oder klitoralen Blutflusses wirksam sein könnten.
-
Es
wurde gefunden, dass die Pelvisnervstimulation im anästhesierten
Hasen keine nachteiligen kardiovaskulären Wirkungen hervorruft (siehe 3).
-
Der
genitale Blutfluss wird während
der sexuellen Erregung über
eine erhöhte
arterielle Blutzufuhr erhöht
(Berman, 1999) – wobei
die vaginale Arterie, der vaginale Zweig der Uterusarterie, die
interne Pudendalarterie und die mittleren Zweige der mittleren rektalen
Arterie alle an der Blutversorgung der Vagina und Klitoris beteiligt
sind. Der Pelvisnerv, welcher aus den spinalen Regionen S2/S4 stammt,
innerviert die weiblichen Genitalien und hat Zweige, die in der
unteren Vagina, Klitoris und den damit zusammenhängenden Blutgefässen enden.
Durch Stimulation des Pelvisnervs können die während der sexuellen Erregung
beobachteten Wirkungen des Blutfluss stimuliert werden, d.h. eine
Erhöhung
des arteriellen genitalen Blutflusses. Interessanterweise widerspiegelt
sich der erhöhte
arterielle Blutfluss nicht in einer entsprechenden venösen Entleerung, so
dass sich das Kapillarennetzwerk mit Blut anreichern kann. Das vaginale
Anschwellen führt über die
erhöhte Plasma-Transsudation
zur vaginalen Lubrikation, und dies ist eine der ersten während der
sexuellen Stimulation beobachteten Antworten im Pelvisbereich. Die
Neurotransmitter, welche nach der Pelvisnerv-Stimulation oder während der
sexuellen Erregung freigesetzt werden, sind zur Zeit noch nicht
identifiziert. Nerven, welche Neuropeptide und andere Neurotransmitter-Kandidaten enthalten
und die Vaskulatur und Mikrovaskulatur der Vagina und Klitoris innervieren,
sind immunhistochemisch identifziert worden. Diese Studien weisen
darauf hin, dass Calcitonin-Gen-assoziiertes Peptid (CGRP), Neuropeptid
Y (NPY), Stickstoffmonoxid-Synthase (NOS), Substanz P und vasoaktives
intestinales Peptid (VIP) alle in den Nerven vorliegen, welche die
menschliche Vagina und Klitoris innervieren (Hoyle, 1996; Burnett,
1997; Hauser-Kronberger, 1999).
-
4.2 Validierung des Modells
der sexuellen Erregung bei anästhesierten
Hasen
-
Um
die unter Verwendung dieses Modells erzeugten Blutfluss-Daten auf diejenigen
zu übertragen,
die in einem menschlichen Modell der sexuellen Erregung beobachtet
wurden, wurden unsere Daten direkt mit dem vaginalen Blutfluss und
den in präklinischen
Studien erhaltenen kardiovaskulären
Daten verglichen.
-
Es
wurde gefunden, dass die VIP-Infusion die folgenden Wirkungen im
Hasen-Modell der sexuellen Erregung hat:
- – Exogenes
VIP (iv-Bolus) induziert signifikante konzentrationsabhängige Erhöhungen des
vaginalen Blutflusses (siehe 2a).
Diese Erhöhungen
liegen signifikant über
den basalen Blutflusswerten, und zwar sowohl bei Messung an der
intravaginalen wie auch an der extravaginalen Wand. Der vaginale
Blutfluss wurde mit einer intravenösen Verabreichung von VIP (60 μg/kg) um
24.7±3.6
ml/Min./100 g Gewebe signifikant erhöht. Der Blutfluss blieb während ungefähr 11 Minuten
nach der Infusion über
den Basalwerten erhöht.
Niedrigere Dosierungen induzierten kleinere Erhöhungen, z.b. 6.0 μg/kg erhöhte den
Blutfluss um 7.5±1.3
ml/Min./1.00 g Gewebe, und der Blutfluss blieb während 7 Minuten nach der Infusion
erhöht.
- – Wiederholte
Infusionen mit ähnlichen
Dosierungen von VIP (iv in Intervallen von 30 Minuten) induzieren signifikante
reproduzierbare Erhöhungen
des vaginalen Blutflusses (siehe 2b).
- – VIP
(iv) erhöht
die Herzfrequenz signifikant und vermindert den mittleren arteriellen
Blutdruck (siehe 3). Bei 6.0 μg/kg verursachte
VIP (iv) eine signifikante Reduktion des mittleren arteriellen Blutdrucks um
13.2±0.7
mm Hg und eine signifikante Erhöhung
der Herzfrequenz um 16±4
Schläge
pro Minute.
- – Dieses
Tiermodell widerspiegelt direkt die nach der Infusion von VIP bei
gesunden Freiwilligen beobachteten klinischen Daten, z.B. erhöhter vaginaler
Blutfluss, verminderter Blutdruck und erhöhte Herzfrequenz. Demnach kann
dieses Modell verwendet werden, um den(die) Mechanismus(en) zu untersuchen,
die den während
der sexuellen Erregung auftretenden physiologischen Veränderungen
zugrunde liegen, und zusätzlich
um neue Ansätze
zur Erhöhung
des vaginalen Blutflusses und demzufolge die Behandlung von FSAD
zu validieren.
-
4.3 VIP-induziert Veränderungen
im vaginalen Blutfluss über
die Stimulation des cAMP/Adenvlatcyclase-Pfads
-
Ottesen
und Mitarbeiter zeigten, dass VIP bei gesunden Freiwilligen Erhöhungen des
vaginalen Blutflusses und der Lubrikation induziert. Allerdings
ist der Mechanismus, über
den VIP seine Wirkungen entfaltet, unklar. In der Literatur gibt
es zahlreiche Beispiele der VIP-Signalisierung über unterschiedliche second
Messenger Systeme, wobei diese cGMP/Guanylatcyclase (Ashur-Fabian,
1999), Kohlenstoffmonoxid/Häm-Oxygenase
(Fan, 1998) und cAMP/Adenylatcyclase (Schoeffter, 1985; Gu, 1992;
Foda, 1995) umfassen. Dies wird durch einen kürzlichen Bericht verdeutlicht,
welcher beschreibt, wie die relaxativen Wirkungen von VIP in der Uterusarterie
durch die Freisetzung von Stickstoffmonoxid erklärt werden kann (Jovanovic,
1998). Interessanterweise gibt es auch Hinweise für VIP-modulierende NO/cGMP
bei der männlichen
urogenitalen Funktion (Kim, 1994), und es gibt direkte Hinweise,
dass die Behandlung von menschlichen vaginalen glatten Muskeln-Zellkulturen mit
VIP (0.5 μM)
die cAMP-Spiegel nicht zu erhöhen
vermag (Traish, 1999 ibid).
-
In
dieser Studie wurde gezeigt, dass VIP über die Erhöhung der intrazellulären cAMP-Spiegel
die Vasorelaxation induziert. Anhand einer Serie von funktionellen
Experimenten wurde zusätzlich
zur biochemischen Messung der intrazellulären cAMP-Konzentrationen der
Blutfluss und die Relaxation der glatten Muskeln gemessen. Es wurde
Forskolin, ein Aktivator der Adenylatcyclase oder cAMP-Mimetikum,
verwendet, um die Wirkungen der Aktivierung des cAMP/Adenylatcyclase-Pfads
nachzuahmen. VIP und Forskolin haben identische Wirkungen auf die
physiologischen Wirkungen der Erregung auf den vaginalen Blutfluss
und die Relaxation.
-
VIP
(20 μg/kg)
und Forskolin (40 nmol/kg) induzieren signifikante Erhöhungen des
vaginalen Blutflusses um 13.2 beziehungsweise 12.7 ml/Min./100 mg
Gewebe (siehe 2a und 4a).
Diese durch VIP und Forskolin induzierten Veränderungen in der Amplitude
waren nicht signifikant unterschiedlich. Diese Erhöhungen liegen
signifikant über den
basalen Blutflusswerten, und zwar sowohl bei Messung an der intravaginalen wie
auch an der extravaginalen Wand.
-
Sowohl
VIP (0.1 μM)
als auch Forskolin (10 μM)
erhöhten
die intrazellulären
Konzentrationen von cAMP signifikant über die basalen Spiegel in
isoliertem Vaginagewebe (siehe 4b).
-
VIP
(0.1 μM)
und Forskolin (10 μM)
erhöhen
die basalen Konzentrationen von 276 nM um 156% beziehungsweise 238%.
Die Unterschiede in diesen prozentualen Anteilen widerspiegeln die
Unterschiede der verwendeten Konzentrationen von VIP und Forskolin,
z.B. relaxierte VIP bei einer Konzentration von 0.1 μM eine präkontrahierte
isolierte Vagina um zirka 80%, während
10 μM Forskolin
ausreichend ist, um das isolierte Gewebe vollständig zu relaxieren.
-
Ausserdem
konnte gezeigt werden, dass VIP und Forskolin in isoliertem vaginalen
Gewebe eine Relaxation mit EC50-Werten von
18.8±0.6
nM beziehungsweise 320±20
nM induziert (siehe 4c).
-
Diese
Daten belegen, dass VIP die vaginale Vasorelaxation über den
cAMP/Adenylatcyclase-Pfad induziert, und demzufolge kann dieses
Modell verwendet werden, um zu untersuchen, ob die Pelvisnerv-Stimulation,
d.h. die sexuelle Erregung, zur Freisetzung von VIP/Aktivierung
des cAMP/Adenylatcyclase-Pfads
führt. Ausserdem
können
auch Ansätze
zur Erhöhung
des vaginalen Blutflusses während
der sexuellen Erregung, z.B. durch direkte oder indirekte Verstärkung der
cAMP-Signalisierung,
untersucht werden.
-
4.4 cAMP ist der Mediator
der vaginalen Vasorelaxation
-
Der
Neurotransmitter und die second Messenger Kandidaten, welche für die Erhöhung des
vaginalen Blutflusses während
der sexuellen Erregung verantwortlich sind, sind derzeit nicht identifiziert.
Zur Zeit haben sich die Forscher auf den Stickstoffmonoxid (NO)/cGMP-Pfad
konzentriert. In Übereinstimmung
mit der vorliegenden Erfindung wurde gezeigt, dass: 1.) der cAMP/Adenylatcyclase-Pfad
die VIP-induzierten
Erhöhungen des
vaginalen Blutfluss vermittelt; 2.) VIP der während der sexuellen Erregung
freigesetzte Neurotransmitter ist und 3.) dass endogen freigesetztes
VIP seine vasorelaxatorischen Wirkungen über die Erhöhung von cAMP induziert.
-
Der
für die
Relaxation der Vaginawand verantwortliche Neurotransmitter ist derzeit
nicht identifiziert. Es wurde gezeigt, dass VIP der nach der Stimulation
des Pelvisnervs freigesetzte Neurotransmitter ist und dass cAMP
die VIP-vermittelte
Vasorelaxation vermittelt. Wirkstoffe, welche den Metabolismus von
VIP verhindern oder direkt die cAMP-Signalisierung erhöhen, verstärken die Pelvisnervstimulierten
Erhöhungen
des vaginalen Blutflusses, z.B. NEP-Inhibitoren beziehungsweise PDEcAMP-Inhibitoren (siehe folgende Abschnitte).
-
In
unseren Studien wurde gefunden, dass eine Rolle von NO bei der VIP-induzierten
vaginalen Relaxation ausgeschlossen werden kann. Ein wirksamer und
selektiver PDE-Typ-5-Inhibitor
hat eine minimale Wirkung auf VIP-induzierte Relaxationen von isolierten
glatten Muskeln der Vagina (30% Erhöhung der VIP-induzierten Relaxationen;
siehe Tablle 1).
-
Tabelle
1: Erhöhung
der VIP-vermittelten Relaxation von isolierter Hasenvagina. Diese
Tabelle zeigt die prozentuale Erhöhung des EC50 für die VIP-induzierten
Relaxationen von präkontrahierten
glatten Muskeln der Vagina (1 μM
Phenylephrin). Selektive Inhibitoren von PDEcAMP Typ
1, 2, 3 und 4 potenzierten alle die VIP-vermittelten Relaxationen
signifikant, während
ein selektiver Inhibitor von PDEcGMP Typ
5 oder Vehikelkontrolle keine Wirkung auf die VIP-vermittelten Relaxationen
hatten.
-
-
Es
wurde gezeigt, dass VIP auch der endogene NANC-(nicht-adrenerger, nicht-cholinerger)
Neurotransmitter ist, der insbesondere für EFS-induzierte Relaxationen
von isolierten glatten Muskeln der Vagina verantwortlich ist. Eine
hohe Dosis eines Stickstoffmonoxid-Synthase-Inhibitors (L-NOARG,
300 μM)
inhibiert nur 50% der EFS-induzierten Relaxationen. Ein NEP-Inhibitor
(1 μM),
welcher den NEP-induzierten Metabolismus von VIP verhindern wird
und demzufolge die VIP-Signalisierung
verstärkt,
verstärkt
die durch EFS induzierte nicht-Stickstoffmonoxid-NANC-Relaxation.
Es wurde gezeigt, dass sowohl NO als VIP den Tonus der glatten Muskeln
in der Vaginawand regulieren. Therapeutisch wird es möglich sein,
die Relaxationen der glatten Muskeln der Vagina mit Wirkstoffen,
welche die NO/cGMP- und/oder VIP/cAMP-vermittelte Signalisierung verstärken, zu
erhöhen.
-
4.5 VIP-induzierte klitorale
Vasorelaxation über
den cAMP-Pathway
-
Der
Neurotransmitter und die second Messenger-Kandidaten, welche für die Erhöhung des
klitoralen Blutflusses während
der sexuellen Erregung verantwortlich sind, sind derzeit nicht identifiziert.
In Übereinstimmung
mit laufenden Forschungen des vaginalen Blutflusses, haben Arbeiten
auf den Stickstoffmonoxid-(NO)/cGMP-Pathway spekuliert und sich
darauf fokussiert. Es gibt keine Berichte, dass VIP bei der Vermittlung
des klitoralen Blutflusses/Anschwellens eine Rolle spielt obwohl,
VIP enthaltende Neuronen im klitoralen Gewebe visualisert wurden
(Hauser-Kronberger et al., 1999).
-
In
dieser Studie wurde gezeigt, dass:
- 1. Die Infusion
von VIP den klitoralen Blutfluss erhöht
- 2. Der cAMP/Adenylatcyclase-Pfad die VIP-induzierte Erhöhung des
klitoralen Blutflusses vermittelt
- 3. VIP ein endogener klitoraler Neurotransmitter ist, welcher
während
der sexuellen Erregung freigesetzt wird:
- 1. Die Infusion von VIP (60–200 μg/kg, iv-Bolus)
induziert eine konzentrationsabhängige
Erhöhung
des klitoralen Blutflusses (5). Eine
115%-ige Erhöhung
des klitoralen Blutflusses wurde nach einer iv-Infusion von 200 μg/kg VIP
beobachtet. Dies war gegenüber
den Kontrollinfusionen (Hepsalin) signifikant erhöht.
- 2. Die Wirkungen von VIP auf den klitoralen Blutfluss können durch
eine Infusion des cAMP-Mimetikums Forskolin (40 nmol/kg iv-Bolus, 5)
nachgeahmt werden. Eine 156%-ige
Erhöhung
des klitoralen Blutfluss wurde nach einer iv-Infusion von 40 nmol/kg Forskolin beobachtet.
Dies war gegenüber
Kontrollinfusionen (Hepsalin) signifikant erhöht. Man bemerke, dass die Amplitude
der Antwort ähnlich
zu der durch VIP (200 μg/kg,
iv-Bolus) induzierten ist und mit denjenigen bezüglich des vaginalen Blutflusses
in 2 und 4 vergleichbar ist.
- 3. Selektive Inhibitoren von NEP EC 3.4.24.11 in klinisch relevanten
Dosen verstärken
die pelvisnervstimulierten Erhöhungen
des klitoralen Blutflusses signifikant (siehe 12). Ein NEP-Inhibitor verstärkt die maximale Erhöhung des
klitoralen Blutflusses bis zu 131 im Vergleich zu den Vehikel-Kontroll-Erhöhungen.
-
Diese
Daten belegen, dass VIP zur Erhöhung
des klitoralen Blutflusses/Vasorelaxation befähigt ist und dass dies durch
die Aktivierung des cAMP/Adenylatcyclase-Pfads nachgeahmt werden
kann. Der Befund, dass ein Inhibitor von NEP EC3.4.24.11 1 (das
für den
VIP-Metabolismus verantwortlich ist) die pelvisnervstimulierte Erhöhung des
klitoralen Blutflusses verstärkt,
zeigt, dass VIP ein Neurotransmitter ist, welcher während der
Pelvisnervstimulation/sexuellen Erregung freigesetzt wird.
-
4.6 Der genitale Blutfluss
wird durch pharmakologische Wirkstoffe welche direkt oder indirekt
die cAMP-Spiegel erhöhen,
verstärkt
-
FSAD
steht geht mit einem verringerten genitalen Blutfluss einher und
kann sich daraus ergeben. Mögliche
Ansätze
zur Behandlung dieser Störung
drehen sich um die Erhöhung
des genitalen Blutflusses. Nachdem nun feststeht, dass cAMP der
Mediator der genitalen Vasorelaxation ist und dass sich Erhöhungen von cAMP
aus neuronal freigesetztem VIP ergeben, wird angenommen, dass durch
Verstärkung
der cAMP-Signalisierung
als Folge der genitale Blutfluss erhöht sein wird, wodurch der genitale
Blutfluss auf normale Spiegel eingestellt und die FSAD behandelt
wird.
-
In
einem besonders bevorzugten Aspekt wurden drei Targets ausgewählt, um
die cAMP-vermittelte Vasorelaxation direkt oder indirekt zu erhöhen: PDEcAMP-Inhibitoren, z.B. PDEcAMP-Typ-2-Inhibitoren,
NEP-(EC 3.4.24.11)-Inhibitoren und Neuropeptid-Y-Y1-(NPY Y1)-Rezeptorantagonisten.
-
4.6.1 Neutrale Endopeptidase-(NEP
EC 3.4.24.11)-Inhibitoren
-
NEP
EC 3.4.24.11 metabolisiert VIP und beendet demzufolge die VIP-vermittelte
biologische Aktivität. NEP-Inhibitoren
werden die endogene vasorelaxatorische Wirkung des während der
Erregung freigesetzten VIP potenzieren. Dies wird die klinische
Wirkung einer Verstärkung
des genitalen Anschwellens haben.
-
Es
gibt keine vorgängigen
Literaturberichte über
die NEP EC3.4.24.11 Lokalisation oder über dessen funktionelle Rolle
im vaginalen Gewebe oder über
eine Rolle bei der sexuellen Erregung.
-
Die
selektiven Inhibitoren von NEP EC 3.4.24.11 in klinisch relevanten
Dosen verstärken
die pelvisnervstimulierten Erhöhungen
des vaginalen Blutflusses signifikant (siehe 6).
-
Ein
NEP-Inhibitor verstärkt
die maximale Erhöhung
des vaginalen Blutfluss bis zu 53% im Vergleich zu zeitlich übereinstimmenden
Kontrollerhöhungen.
Diese Erhöhung
der submaximalen Stimulationsfrequenzen (z.B. 4Hz) war dosisabhängig, z.B.
0.1 mg/kg iv induziert eine 35.0±7.6%-ige Erhöhung; 0.3 mg/kg iv induziert
eine 42.6.0±27.7%-ige
Erhöhung
und 1.0 mg/kg iv induziert eine 52.8±32.5%-ige Erhöhung. NEP-Inhibitoren
hatten keine Wirkung auf den basalen (nicht-stimulierten) vaginalen
Blutfluss. Demnach verstärken
die erfindungsgemässen
Wirkstoffe die Erregung durch Potenzieren der cAMP-Signalisierung,
statt dass sie die Erregung bei Abwesenheit von sexuellem Begehren,
d.h. durch direkte Erhöhung
der cAMP-Signalisierung, induzieren würden.
-
Die
selektiven Inhibitoren von NEP EC 3.4.24.11 in klinisch relevanten
Dosen verstärken
die pelvisnervstimulierten Erhöhungen
des klitoralen Blutflusses signifikant (siehe 12). Ein NEP-Inhibitor verstärkt die maximale Erhöhung des
klitoralen Blutflusses um bis zu 131% im Vergleich zu den Erhöhungen der
Vehikel-Kontrollen. NEP-Inhibitoren hatten keine Wirkung auf den
basalen (nicht stimulierten) vaginalen Blutfluss. Dies stützt unsere
Annahmen, dass die erfindungsgemässen
Wirkstoffe die Erregung durch Potenzieren der cAMP-Signalisierung,
statt dass sie die Erregung bei Abwesenheit von sexuellem Begehren,
d.h. durch direkte Erhöhung
der cAMP-Signalisierung, induzieren würden.
-
Die
selektiven Inhibitoren von NEP EC 3.4.24.11 in klinisch relevanten
Dosen verstärken
die VIP-induzierten Erhöhungen
des vaginalen Blutflusses im Vergleich zu zeitlich übereinstimmenden
Kontrollerhöhungen.
Bei submaximalen Dosierungen von VIP (z.B. 6.0 μg/kg) ergibt sich sowohl eine
signifikante Potenzierung der maximalen Erhöhung (95±6%) als auch eine Verlängerung
der Dauer der Erhöhung
(zirka 140% - von 7 bis zu über
17 Minuten; siehe 7). Die NEP-Inhibitoren verlängern die Dauer der VIP-induzierten
Erhöhung des
vaginalen Blutflussses signifikant, wenn sie in Kombination mit
einer Dosis von VIP verabreicht werden, welche eine maximale Flusserhöhung (zirka
80% Verlängerung
der Dauer – 11
bis 20 Minuten) erzeugt.
-
NEP-Inhibitoren
in klinisch relevanten Dosen verstärken die VIP-induzierten und
nervenvermittelten Relaxationen im isolierten Gewebe signifikant.
Der EC50 für VIP wird in der Gegenwart
eines selektiven NEP-Inhibitors (1 μM) signifikant von 18.8±0.6 nM
auf 2.9±0.3
nM verringert. Die Wirkung des NEP-Inhibitors ist konzentrationsabhängig.
-
NEP
EC 3.4.24.11 mRNS Signal und Protein wird exprimiert und wurde mittels
Northern und Western Analysen in der Vagina von Menschen und Hasen
identifiziert.
-
4.6.2 Phosphodiesterase-(PDE)-Inhibitoren
-
cAMP
wird durch cAMP-hydrolysierende PDEs, d.h. PDEcAMP abgebaut.
Die PDEcAMP-Inhibitoren werden die endogene
vasorelaxatorische Wirkung des während
der Erregung freigesetzten cAMP potenzieren. Dies sollte die klinische
Wirkung haben, das vaginale Anschwellen zu verstärken.
-
Es
gibt keine Literaturberichte über
die PDEcAMP-Lokalisation oder über eine
funktionelle Rolle dieser Isozyme im vaginalen Gewebe oder über eine
Rolle bei der sexuellen Erregung. Wir haben mittels PDE-Profiling
der Vagina von Mensch und Hasen gezeigt, dass die folgenden PDEcAMP 1, 2, 3, 4, 7 & 8 Isozyme vorhanden sind. Die Inhibitoren
dieser PDEcAMP stellen mögliche Wirkstoffe dar, um den
vaginalen Blutfluss zu verstärken
und/oder die glatten Muskeln der Vagina zu relaxieren.
-
Ein
selektiver Inhibitor des PDEcAMP Typ 2-Inhibitors
in klinisch relevanten Dosen verstärkt die pelvisnervstimulierten
Erhöhungen
des vaginalen Blutflusses signifikant (siehe 8). Ein
PDEcAMP-Typ-2-Inhibitor (500 μg/kg; iv)
verstärkt
die maximale Erhöhung
des vaginalen Blutflusses um 86.8±21.9% im Vergleich zu den während den
zeitlich übereinstimmenden
Kontrollen beobachteten Erhöhungen
(bei 4Hz).
-
Ein
selektiver PDEcAMP-Typ-2-Inhibitor verstärkte die
Dauer der VIP-(60 μg/kg)-induzierten
Erhöhungen
des maximalen vaginalen Blutflusses signifikant um über 100%
(gemessen bei einer 50% Amplitude; siehe 9). Der
selektive PDEcAMP-Typ-2-Inhibitor verstärkt die
durch die VIP-Stimulation induzierte maximale Erhöhung des
Blutflusses signifikant (zirka 15±3% [200 μg/kg]).
-
Verstärkte die
Dauer der VIP-induzierten Erhöhungen
des maximalen vaginalen Blutflusses signifikant um über 100% (gemessen
bei einer 50% Amplitude; siehe 8). Der
selektive PDEcAMP-Typ-2-Inhibitor verstärkt die
durch die Pelvisnervstimulation induzierte maximale Erhöhung des
Blutflusses signifikant (zirka 15±3% [200 μg/kg] bei 4Hz).
-
PDEcAMP-Inhibitoren verstärken VIP-induzierte Relaxationen
von präkontrahierten
isolierten glatten Muskeln der Vagina (1 μM Phenylephrin; siehe Tabelle
1). Die selektiven Inhibitoren der PDEcAMP Typ
1, 2, 3 und 4 potenzierten alle die VIP-vermittelten Relaxationen
signifikant. (210% bei 7 6 nM, 130% bei 8 nM, 220% bei 3.4 μM und 160%
bei 68 6 nM Potenzierung der VIP-EC50-Werte)
Diese Inhibitoren wurden in Dosen verabreicht, von denen bekannt
war, dass sie für
den jeweils interessierenden PDEcAMP selektiv sind. Ein selektiver
Inhibitor des PDEcGMP-Typ 5 oder der Vehikelkontrolle
hatte keine erkennbare Wirkung auf die VIP-vermittelten Relaxationen.
-
4.6.3 NPY-Y1-Rezeptorantagonisten
-
NPY
entfaltet einen inhibitorischen Einfluss über die VIP-vermittelte Vasorelaxation, und die NPY-Y1-Rezeptorantagonisten
werden die vasorelaxatorische Wirkung des während der Erregung freigesetzten
endogenen VIP erleichtern. Dies wird die klinisch Wirkung haben,
die vaginale Anschwellung zu verstärken.
-
Es
gibt keine Literaturberichte über
die NPY-Rezeptor-Lokalisation
oder über
eine funktionelle Rolle für
diese Rezeptoren im vaginalen Gewebe oder über eine Rolle bei der sexuellen
Erregung.
-
In
Studien über
die NPY-Rezeptor-Expression haben wir mittels Northern und Western
Analysen festgestellt, dass NPY Y1 Y2 und Y5 Rezeptor-Subtypen in
der Vagina von Menschen und Hasen vorhanden sind.
-
Die
selektiven Inhibitoren von NPY Y1 in klinisch relevanten Dosen verstärken die
pelvisnervstimulierten Erhöhungen
des vaginalen Blutflusses signifikant (siehe 10).
Ein NPY-Y1-Antagonist
verstärkte
die maximale Erhöhung
des vaginalen Blutflusses um bis zu 92% im Vergleich zu den Erhöhungen bei
zeitlich übereinstimmenden
Kontrollen. Diese Verstärkung
der submaximalen Stimulationsfrequenzen (z.B. 4Hz) war dosisabhängig, z.B.
0.01 mg/kg iv induziert eine 15.8±19.6%-ige Erhöhung; 0.03 mg/kg iv induziert
eine 35.1±17.17%-ige
Erhöhung;
0.10 mg/kg iv induziert eine 60.1±16.9%-ige Erhöhung und
0.3 mg/kg iv induziert eine 91.9±27.4%-ige Erhöhung (Mittel±sem n=3).
Die NPY-Y1-Antagonisten hatten keine Wirkung auf den basalen (nicht
stimulierten) vaginalen Blutfluss. Dies bestärkt unsere Ansicht, dass sie
die Erregung durch Potenzieren der cAMP-Signalisierung verstärken werden,
statt dass sie die Erregung bei Abwesenheit von sexuellem Begehren,
d.h. durch direkte Erhöhung
der cAMP-Signalisierung, induzieren würden.
-
4.7 Wirkungen von Wirkstoffen,
welche die cAMP verstärken
oder den vaginalen Blutfluss auf den mittleren arteriellen Blutdruck
im anästhesierten
Hasen verstärken.
-
Auf
der Suche nach einer oralen Therapie für FSAD ist es wünschenswert,
dass keine nachteiligen kardiovaskulären Wirkungen wie z.B. eine
Wirkung auf den Blutdruck oder auf die Herzfrequenz auftreten. In unseren
Studien wurde gefunden, dass Infusionen von VIP den mittleren arteriellen
Blutdruck signifikant verringern (siehe 3) und
die Herzfrequenz signifikant erhöhen.
Demnach ist der Wirkstoff in einem besonders bevorzugtem Aspekt
nicht VIP. Die Pelvisnervstimulation und Inhibitoren der PDEcAMP und NEP hatten jedoch keine Wirkung
auf den Blutdruck. Bei 6.0 μg/kg
verursachte VIP (iv) eine signifikante Reduktion des mittleren arteriellen
Blutdruckes um 13.2±0.7
mm Hg und eine signifikante Erhöhung
der Herzfrequenz um 16±4
Schläge
pro Minute. Bei höheren
Dosierungen wie 60.0 μg/kg
verursachte VIP (iv) eine signifikante Reduktion des mittleren arteriellen
Blutdruckes um 14.7±1.37
mm Hg, und dies war mit einer signifikanten Erhöhung der Herzfrequenz um 111±30 Schläge pro Minute
vergesellschaftet, welche dann den mittleren arteriellen Blutdruck
um 8.5±1.4
mmHg erhöhte.
-
GEPRÜFTE VERBINDUNGEN
-
Mehrere
der oben erwähnten
Verbindungen wurden entsprechend der vorliegenden Erfindung geprüft und erwiesen
sich erfindungsgemäss
als wirksam – d.h.
sie können
als PcAMP zur Behandlung von FSD, insbesondere
FSAD, wirken.
-
Diese
Verbindungen umfassen:
- Verbindung der Formel Ia ("FIa") – nämlich 5-[4-(Diethylamino)benzyl]-1-methyl-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-7-on.
FIa kann gemäss
den Lehren von EP-A-0911333 (Beispiel 50 davon) hergestellt werden.
- Verbindung der Formel II ("FII") – nämlich 9-(1-Acetyl-4-phenylbutyl)-2-[(3,4-dimethoxyphenyl)methyl]-1,9-dihydro-6H-purin-6-on. FII kann
gemäss
den Lehren von EP-A-0771799 (Beispiel 100 davon) hergestellt werden.
- Verbindung der Formel III ("FIII") – nämlich Milrinon.
FIII ist ein kommerziell verfügbares
Produkt.
- Verbindung der Formel IV ("FIV") – nämlich Rolipram.
FIV ist ein kommerziell verfügbares
Produkt.
- Verbindung der Formel V ("FV") – nämlich Cyclohexancarbonsäure, 3-[[[1-(2-Carboxy-4-pentenyl)cyclopentyl]carbonyl]amino]-,1-ethyl-ester.
FV kann gemäss
den Lehren von EP-A-0274234 (Beispiel 300 davon) hergestellt werden.
- Verbindung der Formel VI ("FVI") – nämlich Cyclohexancarbonsäure, 3-[[[1-(2-Carboxy-4-pentenyl)cyclopentyl]carbonyl]amino]-.
FVI kann gemäss
den Lehren von EP-A-0274234 (Beispiel 379 davon) hergestellt werden.
-
Insbesondere
sind FIa, FII, FIII und FIV PDEcAMP-Inhibitoren.
FIa ist ein I:PDEI, FII ist ein I:PDEII, FIII ist ein I:PDEIII und
FIV ist ein I:PDEIV.
-
Die
Daten dieser Verbindungen sind oben in den vorangehenden Abschnitten
mit den Beispielen vorgestellt worden – siehe beispielsweise Tabelle
I.
-
Es
ist offensichtlich, dass diese PDEcAMP-Inhibitoren
die VIP-induzierten Relaxationen von isoliertem Gewebe verstärken.
-
FII – welcher
ein selektiver I:PDEII ist – verstärkt in klinisch
relevanten Dosen die VIP-induzierte Erhöhung des vaginalen Blutflusses.
-
FII
verstärkt
in klinisch relevanten Dosen auch die durch den Pelvisnerv stimulierte
Erhöhung
des vaginalen Blutflusses.
-
FV
und FVI sind selektive Inhibitoren von NEP EC 3.4.24.11.
-
Die
oben in den vorgehenden Abschnitten mit den Beispielen präsentierten
Daten sind für
FVI. Allerdings wurden für
FV ähnlichen
Resultate erhalten.
-
Es
ist offensichtlich, dass FV und FVI in klinisch relevanten Dosen
die VIP-induzierte Erhöhung
des vaginalen Blutflusses verstärken.
-
FV
und FVI verstärken
in klinisch relevanten Dosen auch die durch den Pelvisnerv stimulierte
Erhöhung
des vaginalen Blutflusses.
-
FV
und FVI verstärken
in klinisch relevanten Dosen auch die VIP-induzierten und nervenvermittelten Relaxationen
von isoliertem Gewebe.
-
Zusätzliche
Verbindungen, die geprüft
wurden und die sich als wirksam erwiesen haben, umfassten:
- 2-[(1-{[(1-Benzyl-6-oxo-1,6-dihydro-3-pyridinyl)amino]carbonyl}cyclopentyl)methyl]-4-methoxybutansäure (F57)
- 2-{[1-({[3-(2-Oxo-1-pyrrolidinyl)propyl]amino}carbonylcyclopentyl]-methyl}-4-phenylbutansäure (F58)
- (+)-2-{[1-({[2-(Hydroxymethyl)-2,3-dihydro-1H-inden-2-yl]amino}carbonyl)cyclopentyl]-methyl}-4-phenylbutansäure (F59)
- 2-[(1-{[(5-Methyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}cyclopentyl)methyl]-4-phenylbutansäure (F60)
- cis-3-(2-Methoxyethoxy)-2-[(1-{[(4-{[(phenylsulfonyl)amino]carbonyl}cyclohexyl)-amino]carbonyl}cyclopentyl)methyl]propansäure (F61)
- (+)-2-{[1-({[2-(Hydroxymethyl)-2,3-dihydro-1H-inden-2-yl]amino}carbonyl)cyclopentyl]-methyl}pentansäure (F62)
- (+)-2-[(1-{[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}cyclopentyl)methyl]pentansäure (F63)
- 2-({1-[(3-Benzylanilino)carbonyl]cyclopentyl}methyl)-pentansäure (F64)
- 2-[(1-{[(1-Benzyl-6-oxo-1,6-dihydro-3-pyridinyl)amino]carbonyl}cyclopentyl)methyl]-pentansäure (F65)
- 2-{[1-({[(1R,3S,4R)-4-(Aminocarbonyl)-3-butylcyclohexyl]amino}carbonyl)cyclopentyl]methyl}pentansäure (F66)
- Jede der Verbindungen F57–66
ist ein I:NEP.
-
SYNTHESEN DER VERBINDUNGEN
F57–66
-
Im
folgenden Kommentar beziehen sich die Vorschriften auf die Synthese
von Zwischenprodukten, während
sich die Beispiele auf die Synthese der jeweiligen erfindungsgemässen Verbindungen
beziehen.
-
Beispiel
1 2-[(1-{[(1-Benzyl-6-oxo-1,6-dihydro-3-pyridinyl)amino]carbonyl}cyclopentyl)methyl]-4-methoxybutansäure (F57)
-
Ein
Gemisch des Benzylesters aus Vorschrift 1 (1/62) (850 mg, 1.64 mMol)
und 5% Palladium auf Aktivkohle (250 mg) in 40% wässrigem
Ethanol (21 ml) wurde während
30 Minuten bei 30 psi und Raumtemperatur hydriert. Das Reaktionsgemisch
wurde durch Hyflo® filtriert, und das Filtrat
wurde im Vakuum auf konzentriert. Der verbleibende Schaum wurde
mittels Säulenchromatographie
auf Silikagel unter Verwendung von Dichlormethan:Methanol (97:3)
als Laufmittel gereinigt, woraus sich die Titelverbindung als ein
weisser Schaum ergab, 550 mg, 79%; 1H NMR
(DMSO-d6, 300 MHz) d: 1.24–2.17 (m,
12H), 2.18–2.31
(m, 1H), 3.07 (s, 3H), 3.21 (t, 2H), 5.08 (s, 2H), 6.63 (d, 1H),
7.23–7.41
(m, 5H), 7.72 (d, 1H), 8.24 (s, 1H).
Anal. gefunden: C, 67.46;
H, 7.18; N, 6.24. C24H30N2O5 erfordert C,
67.58; H, 7.09; N, 6.57%.
-
Beispiel
2 2-{[1-({[3-(2-Oxo-1-pyrrolidinyl)propyl]amino}carbonylcyclopentyl]-methyl}-4-phenylbutansäure (F58)
-
Ein
Gemisch des Benzylesters aus Vorschrift 3 (3/67) (780 mg, 1.55 mMol)
und 10% Palladium auf Aktivkohle (100 mg) in Ethanol:Wasser (90:10
nach Volumen) (30 ml) wurde während
1.5 Stunden bei Raumtemperatur unter 60 psi H2-Druck
hydriert. Der Katalysator wurde abfiltriert, und das Filtrat wurde
im Vakuum aufkonzentriert, woraus sich die Titelverbindung als ein
weisser Schaum ergab, 473 mg, 74%; 1H NMR
(CDCl3, 300 MHz) d : 1.26–1.77 (m,
10H), 1.78–2.46
(m, 11H), 2.49–2.70
(m, 2H), 2.95–3.36
(m, 4H), 6.92–7.38
(m, 5H); Anal. gefunden: C, 64.05; H, 7.73; N, 6.22. C24H34N2O4;0.75H2O erfordert C, 65.88; H, 7.83; N, 6.40%.
-
Beispiel
3 (+)-2-{[1-({[2-(Hydroxymethyl)-2,3-dihydro-1H-inden-2-yl]amino}carbonyl)cyclopentyl]-methyl}-4-phenylbutansäure (F59)
-
2-{[1-({[2-(Hydroxymethyl)-2,3-dihydro-1H-inden-2-yl]amino}carbonyl)cyclopentyl]-methyl}-4-phenylbutansäure (WO
9110644) kann mittels standardmässigen
HPLC-Verfahren unter Verwendung einer AD-Säule und Hexan:Isopropanol:
Trifluoressigsäure
(70:30:0.2) als Laufmittel gereinigt werden, woraus sich die Titelverbindung
von Beispiel 3 ergab, 99.5% ee; [α]D = +9.1° (c
= 1.76 in Ethanol)
-
Beispiel
4 2-[(1-{[(5-Methyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}cyclopentyl)methyl]-4-phenylbutansäure (F60)
-
Ein
Gemisch des Benzylesters aus Vorschrift 4 (4/70) (187 mg, 0.39 mMol)
und 10% Palladium auf Aktivkohle (80 mg) in Ethanol (20 ml) wurde
während
18 Stunden bei 60 psi hydriert. Die DC-Analyse zeigte verbleibendes
Ausgangsmaterial, weshalb nochmals 10% Palladium auf Aktivkohle
(100 mg) zugegeben und die Reaktion während weiteren 5 Stunden fortgesetzt
wurde. Die DC-Analyse zeigte wiederum verbleibendes Ausgangsmaterial,
weshalb nochmals Katalysator (100 mg) zugegeben und die Hydrierung
während
18 Stunden fortgesetzt wurde. Das Gemisch wurde durch Arbocel® filtriert,
und das Filtrat wurde im Vakuum auf konzentriert und mit Dichlormethan
azeotropiert. Das Rohprodukt wurde mittels Chromatographie auf Silikagel
unter Verwendung einer Biotage®-Säule und Dichlormethan:Methanol
(95:5) als Laufmittel gereinigt, woraus sich die Titelverbindung
als ein klares Öl
ergab, 80 mg, 53%; 1H NMR (CDCl3,
300 MHz) d: 1.51–1.89
(m, 9H), 2.03 (m, 1H), 2.20 (m, 1H), 2.40 (m, 2H), 2.60 (m, 5H),
7.15–7.30
(m, 5H); LRMS : m/z 387.8 (MH+).
-
Beispiel
5 Cis-3-(2-Methoxyethoxy)-2-[(1-{[(4-{[(phenylsulfonyl)amino]carbonyl}cyclohexyl)amino]carbonyl}cyclopentyl)methyl]propansäure (F61)
-
Eine
Lösung
des tert-Butylesters aus Vorschrift 8 (8/66) (446 mg, 0.75 mMol)
in Dichlormethan (5 ml) und Trifluoressigsäure (5 ml) wurde während 18
Stunden bei Raumtemperatur gerührt.
Das Reaktionsgemisch wurde im Vakuum auf konzentriert, und der Rückstand
wurde mit Dichlormethan, dann Toluol und schliesslich Ether azeotropiert,
woraus sich die Titelverbindung als ein weisser Schaum ergab, 385
mg, 95%; 1H NMR (CDCl3,
400 MHz) d: 1.48–2.17
(m, 18H), 2.40 (s, 1H), 2.66 (s, 1H), 3.37 (s, 3H), 3.50–3.70 (m,
6H), 3.94 (s, 1H), 6.10 (d, 1H), 6.59 (s, 1H), 7.55 (t, 2H), 7.61
(m, 1H), 8.02 (d, 2H), 9.11 (s, 1H); Anal. gefunden: C, 54.88; H,
6.90; N, 5.04. C26H38N2O8S;1.7H2O erfordert C, 57.97; H, 7.11; N, 5.20%.
-
Beispiel
6 (+)-2-{[1-({([2-(Hydroxymethyl)-2,3-dihydro-1H-inden-2-yl]amino}carbonyl)cyclopentyl]-methyl}pentansäure (F62)
-
2-{[1-({[2-(Hydroxymethyl)-2,3-dihydro-1H-inden-2-yl]amino}carbonyl)cyclopentyl]-methyl}pentansäure (WO
9110644) wurde mittels HPLC unter Verwendung einer AD-Säule und
Hexan:Isopropanol:Trifluoressigsäure
(90:10:0.1) als Laufmittel weiter gereinigt, woraus sich die Titelverbindung
von Beispiel 6 ergab, 99% ee, [α]D = +10.4° (c
= 0.067, Ethanol).
-
Beispiel
7 (+)-2-[(1-{[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}cyclopentyl)methyl]pentansäure (F63)
-
Die
Säure aus
Vorschrift 18 (18/Bsp. 4) (824 mg) wurde mittels HPLC unter Verwendung
einer AD-Säule
und unter Verwendung von Hexan:Isopropanol:Trifluoressigsäure (85:15:0.2)
als Laufmittel weiter gereinigt, woraus sich die Titelverbindung
von Beispiel 7 als ein weisser Schaum ergab, 386 mg, 99% ee, 1H NMR (CDCl3, 400
MHz) δ:
0.90 (t, 3H), 1.38 (m, 6H), 1.50–1.79 (m, 9H), 2.19 (m, 1H),
2.30 (m, 1H), 2.44 (m, 1H), 2.60 (m, 1H), 2.98 (q, 2H), 12.10–12.27 (bs,
1H); LRMS: m/z 338 (MH); und [α]D = +3.8° (c
= 0.1, Methanol)
-
Beispiel
8 2-({1-[(3-Benzylanilino)carbonyl)cyclopentyl]methyl)pentansäure (F64)
-
Ein
Gemisch des Benzylesters aus Vorschrift 10 (10/53) (1.3 mg, 2.47
mMol) und 5% Palladium auf Aktivkohle (130 mg) in Wasser (10 ml)
und Ethanol (40 ml) wurde während
2 Stunden bei 30 psi und Raumtemperatur hydriert. Das Reaktionsgemisch
wurde durch Arbocel® filtriert, das Filtrat
wurde im Vakuum aufkonzentriert, und der Rückstand wurde mit Dichlormethan
zerstossen. Der erhaltene Gummi wurde mit Ether, dann Hexan zerstossen,
und bei 50°C
getrocknet, woraus sich die Titelverbindung als ein Feststoff ergab,
0.79g, 81%; 1H NMR (CDCl3,
300 MHz) d: 0.95 (t, 3H), 1.24–1.51
(m, 3H), 1.58-1.80
(m, 7H), 1.88 (dd, 1H), 2.15 (m, 2H), 2.24 (m, 1H), 2.48 (m, 1H),
4.00 (s, 2H), 6.98 (d, 1H), 7.24 (m, 6H), 7.40 (m, 3H); Anal. gefunden:
C, 75.48; H, 7.76; N, 3.59. C25H31NO3;0.25H2O erfordert C, 75.44; H, 7.98; N, 3.51%.
-
Beispiel
9 2-[(1-{[(1-Benzyl-6-oxo-1,6-dihydro-3-pyridinyl)amino]carbonyl}-cyclopentyl)methyl]-pentansäure (F65)
-
Die
Titelverbindung wurde als ein weisser Schaum mit einer Ausbeute
von 51% aus dem Benzylester aus Vorschrift 13 (13/56) nach einem ähnlichen
Verfahren wie dem in der Vorschrift 19 (19/Bsp. 21) beschriebenen
gewonnen mit der Ausnahme, dass das Produkt mittels Säulenchromatographie
auf Silikagel unter Verwendung von Ethylacetat als Laufmittel gereinigt
wurde; 1H NMR (CDCl3,
300 MHz) d: 0.96 (t, 3H), 1.28–1.80 (m,
12H), 2.01 (m, 1H), 2.30–2.52
(m, 2H), 5.02 (dd, 2H), 6.60 (d, 1H), 7.27 (m, 5H), 7.70 (s, 1H),
8.34 (s, 1H); Anal. gefunden: C, 69.52; H, 7.41; N, 6.51. C24H30N2O4;0.25H2O erfordert
C, 69.45; H, 7.41; N, 6.75.
-
Beispiel
10 2-{[1-({[(1R,3S,4R)-4-(aminocarbonyl)-3-butylcyclohexyl]amino}carbonyl)cyclopentyl]methyl}pentansäure (F66)
-
Verbindungen
der Formel ic, d.h. Verbindungen der allgemeinen Formel i, wobei
r1 Propyl ist, werden aus dem entsprechenden
tert-Butylester nach einem ähnlichen
Verfahren wie dem in Vorschrift 14 (14/Bsp. 1) beschriebenen hergestellt.
-
Vorschrift
1 (1/62) Benzyl-2-[(1-{[(1-benzyl-6-oxo-1,6-dihydro-3-pyridinyl)amino]carbonyl}cyclopentyl)methyl]-4-methoxybutanoat
-
Oxalylchlorid
(0.26 ml, 3.0 mMol) wurde zu einer eisgekühlten Lösung von 1-{2-[(Benzyloxy)carbonyl]-4-methoxybutyl}cyclopentancarbonsäure (
EP 274234 ) (1.0 g, 3.0 mMol)
und N,N-Dimethylformamid (2 Tropfen) in Dichlormethan (20 ml) zugegeben,
und das Reaktionsgemisch wurde während
2 Stunden bei Raumtemperatur gerührt.
Die Lösung
wurde im Vakuum auf konzentriert, und der Rückstand wurde mit Dichlormethan
(3 × 10
ml) azeotropiert. Das Produkt wurde in Dichlormethan (20 ml) gelöst, dann
in einem Eisbad abgekühlt.
Das Amin aus Vorschrift 2 (2/28) (600 mg, 3 mMol) und N-Methylmorpholin
(0.6 ml, 5.45 mMol) wurden zugegeben, und das Reaktionsgemisch wurde
während
18 Stunden bei Raumtemperatur gerührt. Das Reaktionsgemisch wurde
im Vakuum aufkonzentriert, und zwischen Wasser und Ether verteilt.
Die organische Phase wurde mit Chlorwasserstoffsäure (2N), Natriumbicarbonat-Lösung, dann
Wasser gewaschen, getrocknet (MgSO
4) und
im Vakuum eingedampft. Der erhaltene grüne Feststoff wurde mittels
Mitteldruck-Säulenchromatographie
auf Silikagel unter Verwendung von Ethylacetat:Hexan (90:10) als
Laufmittel gereinigt, woraus sich die Titelverbindung ergab, 880
mg, 57%;
1H NMR (CDCl
3,
300 MHz) d: 1.37–2.28
(m, 12H), 2.46–2.64
(m, 1H), 3.20 (s, 3H), 3.31 (m, 2H), 4.97 (dd, 2H), 5.08 (dd, 2H),
6.57 (d, 1H), 7.12 (m, 1H), 7.18-7.48
(m, 10H), 8.08 (d, 1H).
-
Vorschrift
2 (2/28) 5-Amino-1-benzyl-2(1H)-pyridinon
-
Ein
Gemisch von 1-Benzyl-5-nitro-1H-pyridin-2-on (Justus Liebigs Ann.
Chem. 484; 1930; 52) (1.0 g, 4.35 mMol) und granuliertem Zinn (3.5
g, 29.5 mMol) in konzentrierter Chlorwasserstoffsäure (14
ml) wurde während
1.5 Stunden bei 90°C
erhitzt. Die abgekühlte
Lösung
wurde mit Wasser verdünnt,
unter Verwendung von Natriumcarbonat-Lösung neutralisiert und mit
Ethylacetat (250 ml insgesamt) extrahiert. Die kombinierten organischen
Extrakte wurden filtriert, getrocknet (MgSO4)
und im Vakuum eingedampft, woraus sich die Titelverbindung als ein
hellgrüner
Feststoff ergab, (wurde mit der Zeit blau), 440 mg, 51%; 1H NMR (CDCl3, 250 MHz)
8: 4.12–4.47
(bs, 2H), 5.00 (s, 2H), 6.31 (d, 1H), 6.86 (s, 1H), 7.07 (m, 1H),
7.14–7.42
(m, 5H).
-
Vorschrift
3 (3/67) Benzyl-2-{[1-({[3-(2-Oxo-1-pyrrolidinyl)propyl]amino}carbonylcyclopentyl]-methyl}-4-phenylbutanoat
-
1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid-Hydrochlorid
(1.06 g, 5.53 mMol), 1-Hydroxybenzotriazol-Hydrat (0.60 g, 4.44
mMol) und 4-Methylmorpholin (0.56 g, 5.54 mMol) wurden nacheinander
zu einer gekühlten
Lösung
von 1-{2-[(Benzyloxy)carbonyl]-4-phenylbutyl}cyclopentan-carbosäure (
EP 274234 ) (1.5 g, 3.94 mMol)
in trockenem Dichlormethan (15 ml) bei Raumtemperatur, gefolgt von
N-(3-Aminopropyl)-2-pyrrolidinon
(0.56 g, 3.94 mMol) zugegeben, und das Reaktionsgemisch wurde während 18
Stunden bei Raumtemperatur gerührt.
Das Gemisch wurde mit Wasser, 2N Chlorwasserstoffsäure, gesättigter
wässriger
Natriumbicarbonat-Lösung
gewaschen, und dann getrocknet (MgSO
4) und
im Vakuum eingedampft. Das erhaltene gelbe Öl wurde mittels Säulenchromatographie
auf Silikagel unter Verwendung von Ethylacetat:Pentan (50:50) als
Laufmittel gereinigt, woraus sich die Titelverbindung als ein klarer
Gummi ergab, 800 mg, 40%;
1H NMR (CDCl
3, 300 MHz) d : 1.37-2.20 (m, 16H), 2.34–2.58 (m, 5H), 2.92–3.46 (m,
6H), 5.07 (d, 1H), 5.18 (d, 1H), 6.98–7.47 (m, 10H).
-
Vorschrift
4 (4/70) Benzyl-2-[(1-{[(5-methyl-1,3,4-thiadiazol-2-yl)aminolcarbonyl}cyclopentyl)methyl]-4-phenylbutanoat
-
Die
Titelverbindung wurde als ein klares Öl mit einer Ausbeute von 74%
aus 1-{2-[(Benzyloxy)carbonyl]-4-phenylbutyl}cyclopentan-carbonsäure (
EP 274234 ) und 2-Amino-5-methyl-1,3,4-thiadiazol
nach einem ähnlichen
Verfahren wie dem in Vorschrift 5 (5/68) beschriebenen erhalten;
1H NMR (CDCl
3, 400
MHz) d: 1.58–1.76
(m, 7H), 1.83–1.98
(m, 3H), 2.03 (m, 1H), 2.20 (m, 1H), 2.35 (m, 1H), 2.44 (m, 3H),
2.65 (s, 3H), 5.02 (dd, 2H), 7.00 (d, 2H), 7.15 (m, 1H), 7.19 (m,
2H), 7.35 (m, 5H); LRMS : m/z 478.7 (MH
+).
-
Vorschrift
5 (5/68) Benzyl-2-{[1-({[3-(methylamino)-3-oxopropyl]amino}carbonyl)cyclopentyl]methyl}-4-phenylbutanoat
-
1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid-Hydrochlorid
(122 mg, 0.64 mMol), 1-Hydroxybenzotriazol-Hydrat (86 mg, 0.64 mMol)
und 4-Methylmorpholin (173 μl,
1.59 mMol) wurden nacheinander zu einer gekühlten Lösung von 1-{2-[(Benzyloxy)carbonyl]-4-phenylbutyl}cyclopentan-carbonsäure (
EP 274234 ) (202 mg, 0.53
mMol) in N,N-Dimethylformamid (5 ml) bei Raumtemperatur, gefolgt
vom Amin-Hydrochlorid aus Vorschrift 6 (6/23) (146 mg, 1.06 mMol)
zugegeben, und das Reaktionsgemisch wurde während 18 Stunden bei 90°C gerührt. Die
abgekühlte
Lösung
wurde im Vakuum auf konzentriert und der Rückstand zwischen Wasser (20
ml) und Ethylacetat (100 ml) verteilt. Die Phasen wurden aufgetrennt,
die organische Phase wurde mit Wasser (3 × 30 ml), gesättigter
Kochsalzlösung
(25 ml) gewaschen, getrocknet (MgSO
4), und
im Vakuum eingedampft, woraus sich ein klares Öl ergab. Das Rohprodukt wurde
mittels Säulenchromatographie
auf Silikagel unter Verwendung von Dichlormethan:Methanol (98:2)
als Laufmittel gereinigt, woraus sich die Titelverbindung als ein
farbloses Öl
ergab, 162 mg, 67%;
1H NMR (CDCl
3, 400 MHz) δ: 1.38–1.53 (m, 2H), 1.53–1.96 (m, 8H),
2.02 (m, 2H), 2.27 (t, 2H), 2.46 (m, 3H), 2.76 (d, 3H), 3.44 (m,
2H), 5.13 (s, 2H), 5.79 (bs, 1H), 6.38 (m, 1H), 7.06 (d, 2H), 7.18
(m, 1H), 7.22 (m, 2H), 7.38 (m, 5H); LRMS : m/z 465.5 (MH
+).
-
Vorschrift
6 (6/23) 3-Amino-N-methylpropanamide-Hydrochlorid
-
Ein
Gemisch des Benzylcarbamates aus Vorschrift 7 (7/13) (7.92 g, 33.5
mMol) und 5% Palladium auf Aktivkohle (800 mg) in Ethanol (300 ml)
wurde während
4 Stunden bei 50 psi und Raumtemperatur hydriert. Das Reaktionsgemisch
wurde durch Arbocel® filtriert, mit Ethanol
durchgewaschen, und zu dem zusammengegebenen Filtraten wurde 1N
Chlorwasserstoffsäure
(36.9 ml, 36.9 mMol) zugegeben. Diese Lösung wurde im Vakuum eingedampft,
und der Rückstand
wurde mit Dichlormethan azeotropiert, woraus sich die Titelverbindung
als ein farbloser Schaum ergab, 4.66 g, 1H
NMR (DMSOd6, 300 MHz) δ: 2.46 (t, 2H), 2.60 (s, 3H),
2.95 (m, 2H), 7.98–8.16
(m, 2H).
-
Vorschrift
7 (7/13) Benzyl-3-(methylamino)-3-oxopropylcarbamat
-
Ein
Gemisch von N-[(Benzyloxy)carbonyl]-β-Alanin (10 g, 44.8 mMol), Methylamin-Hydrochlorid
(3.33 g, 49.28 mMol), 1-Hydroxybenzotriazol-Hydrat
(6.05 g, 44.8 mMol), 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid-Hydrochlorid
(10.3 g, 53.76 mMol) und N-Methylmorpholin (11.33 ml, 103 mMol)
in Dichlormethan (200 ml) wurde während 18 Stunden bei Raumtemperatur
gerührt.
Der erhaltene Niederschlag wurde abfiltriert, woraus sich das gewünschten
Produkt als ein farbloser Schaum ergab, und das Filtrat wurde im
Vakuum aufkonzentriert. Der Rückstand
wurde mittels Säulenchromatographie
auf Silikagel unter Verwendung eines Elutionsgradienten von Ethylacetat:Hexan
(90:10 bis 100:0) gereinigt, woraus sich zusätzliches Produkt ergab, 7.96
g, 75% insgesamt; 1H NMR (CDCl3, 300 MHz) δ: 2.42 (t,
2H), 2.80 (s, 3H), 3.50 (m, 2H), 5.21 (s, 2H), 5.49 (bs, 1H), 5.63
(bs, 1H), 7.36 (m, 5H); Anal. gefunden: C, 60.68; H, 7.00; N, 11.95.
C12H16N2O3 erfordert C, 61.00; H, 6.83; N, 11.86%.
-
Vorschrift
8 (8/66) Cis-tert-Butyl-3-(2-methoxyethoxy)-2-[(1-{[(4-{[(phenylsulfonyl)amino]carbonyl}-cyclohexyl)amino]carbonyl}cyclopentyl)methyl]propanoat
-
N,N'-Dicyclohexylcarbodiimid
(199 mg, 0.97 mMol), 4-Dimethylaminopyridin
(118 mg, 0.97 mMol) und Benzolsulfonamid (152 mg, 0.97 mMol) wurden
zu einer eisgekühlten
Lösung
des Säure
aus Vorschrift 9 (9/63) (400 mg, 0.878 mMol) in Dichlormethan (12
ml) und N,N-Dimethylformamid
(0.5 ml) zugegeben, und das Reaktionsgemisch wurde während 20
Stunden bei Raumtemperatur gerührt.
Das Gemisch wurde im Vakuum auf konzentriert und der Rückstand
in kaltem Ethylacetat suspendiert. Das erhaltene unlösliche Material
wurde abfiltriert, das Filtrat wurde mit Chlorwasserstoffsäure (1 N)
und Wasser gewaschen, dann getrocknet (MgSO4) und
im Vakuum eingedampft. Das Rohprodukt wurde mittels Säulenchromatographie
auf Silikagel unter Verwendung eines Elutionsgradienten von Dichlormethan:Methanol
(95:5 bis 90:10) gereinigt, woraus sich die Titelverbindung als
ein weisser Schaum ergab, 480 mg, 92%; 1H
NMR (CDCl3, 400 MHz) d: 1.44 (s, 9H), 1.63 (m,
13H), 1.80 (m, 2H), 1.88 (m, 1H), 1.98 (m, 2H), 2.36 (m, 1H), 2.57
(m, 1H), 3.38 (s, 3H), 3.40 (m, 1H), 3.51 (t, 2H), 3.58 (m, 3H),
3.95 (m, 1H), 5.92 (d, 1H), 7.56 (m, 2H), 7.62 (m, 1H), 8.05 (d,
2H), 8.75 (bs, 1H); LRMS : m/z 618 (MNa+).
-
Vorschrift
9 (9/63) 4-{[(1-{3-tert-Butoxy-2-[(2-methoxyethoxy)methyl]-3-oxopropyl}cyclopentyl)-carbonyl]amino}cyclohexancarbonsäure
-
Ein
Gemisch von Benzyl 4-{[(1-{3-tert-butoxy-2-[(2-methoxyethoxy)methyl]-3-oxopropyl}cyclopentyl)carbonyl]amino}cyclohexancarboxylat
(
EP 274234 ) und 10% Palladium
auf Aktivkohle (250 mg) in Wasser (10 ml) und Ethanol (50 ml) wurde
während
18 Stunden bei 50 psi und Raumtemperatur hydriert. Das Reaktionsgemisch
wurde durch Solkafloc
® filtriert, das Filtrat
wurde im Vakuum auf konzentriert, und der Rückstand wurde mit Toluol (3×) und dann
Dichlormethan (3×)
azeotropiert, woraus sich die Titelverbindung ergab, 2.0 g, 96%;
1H NMR (CDCl
3, 300
MHz) d: 1.48 (s, 9H), 1.53–1.84
(m, 14H), 1.94–2.10
(m, 5H), 2.60 (m, 2H), 3.40 (s, 3H), 3.41–3.63 (m, 5H), 3.96 (m, 1H),
5.90 (bd,
1H).
-
Vorschrift 10 (10/53)
-
Die
folgende Verbindung:
wobei:
wurde
aus dem Säurechlorid
aus Vorschrift 11 (11/3) und dem geeigneten Amin nach einem ähnlichen
Verfahren wie dem in Vorschrift 12 (12/52) beschriebenen hergestellt.
-
Vorschrift
11 (11/3) Benzyl-2-{[1-(chlorocarbonyl)cyclopentyl]methyl}pentanoat
-
Oxalylchlorid
(1.15 ml, 13.2 mMol) wurde zu einer eisgekühlten Lösung von 1-{2-[(Benzyloxy)carbonyl]pentyl}cyclopentancarbonsäure (
EP 274234 ) (2.0 g, 6.3 mMol)
in trockenem Dichlormethan (20 ml) zugegeben, und die Lösung wurde
während
2 Stunden bei Raumtemperatur gerührt.
Das Reaktionsgemisch wurde im Vakuum auf konzentriert, und der Rückstand
wurde mit Dichlormethan (3×)
azeotropiert, woraus sich die Titelverbindung als ein goldenes Öl ergab,
2.1 g;
1H NMR (CDCl
3,
300 MHz) d: 0.88 (t, 3H) , 1.28 (m, 2H) , 1.43 (m, 2H), 1.63 (m,
6H), 2.00 (m, 1H), 2.08–2.35
(m, 3H), 2.44 (m, 1H), 5.15 (s, 2H), 7.28 (m, 5H).
-
Vorschrift
12 (12/52) Benzyl-2-(11-[(3-pyridinylamino)carbonyl]cyclopentyl}methyl)pentanoat
-
Triethylamin
(0.11 ml, 0.78 mMol) wurde zu einem Gemisch des Säurechlorids
aus Vorschrift 11 (11/3) (200 mg, 0.60 mMol) und 2-Aminopyridin
(61 mg, 0.65 mMol) in Dichlormethan (3 ml) zugegeben, und das Reaktionsgemisch
wurde während
16 Stunden bei Raumtemperatur gerührt. Das Gemisch wurde im Vakuum
eingedampft, der Rückstand
wurde zwischen Natriumbicarbonat-Lösung (5 ml) und Ethylacetat
(20 ml) verteilt, und die Phasen wurden getrennt. Die organische
Phase wurde (MgSO4) getrocknet und im Vakuum
eingedampft, woraus sich ein Gummi ergab. Das Rohprodukt wurde mittels
Säulenchromatographie
auf Silikagel unter Verwendung von Ethylacetat als Laufmittel gereinigt,
woraus sich die Titelverbindung ergab, 130 mg; 1H NMR
(CDCl3, 400 MHz) d: 0.82 (t, 3H), 1.21 (m,
3H), 1.40 (m, 1H), 1.43–1.72
(m, 6H), 1.81 (d, 1H), 1.98 (m, 1H), 2.18 (m, 1H), 2.24 (m, 1H),
2.46 (m, 1H), 4.98 (m, 2H), 7.20–7.38 (m, 6H), 7.42 (s, 1H),
8.06 (d, 1H), 8.35 (d, 1H), 8.56 (s, 1H).
-
Vorschrift 13 (13/56)
-
Die
folgende Verbindung:
wobei:
wurde aus dem Säurechlorid
aus Vorschrift 11 (11/3) und dem geeigneten Amin nach einem ähnlichen
Verfahren wie dem in Vorschrift 12 (12/52) beschriebenen hergestellt.
-
Vorschrift
14 (14/Bsp. 1) 2-({1-[(1,3-Benzodioxol-5-ylamino)carbonyl]cyclopentyl}methyl)pentansäure
-
Trifluoressigsäure (5 ml)
wurde zu einer Lösung
des tert-Butylesters
aus Vorschrift 15 (15/34) (130 mg, 0.31 mMol) in Dichlormethan (5
ml) zugegeben, und die Lösung
wurde während
4 Stunden bei Raumtemperatur gerührt.
Das Reaktionsgemisch wurde im Vakuum aufkonzentriert, und der Rückstand
wurde mit Toluol und Dichlormethan azeotropiert, woraus sich die
Titelverbindung als ein klares Öl
ergab, 112 mg, 1H NMR (CDCl3,
400 MHz) δ0.83
(t, 3H), 1.22–1.40
(m, 3H), 1.50–1.72
(m, 8H), 1.95 (m, 1H), 2.10 (m, 2H), 2.19 (m, 1H), 4.30 (m, 2H),
5.93 (s, 2H), 5.99 (bs, 1H), 6.74 (m, 3H); LRMS: m/z 380 (MH-).
-
Vorschrift 15 (15/34)
-
Die
folgende Verbindung:
wobei:
wurde
aus der Säure
von Vorschrift 16 (16/1) und der geeigneten Aminverbindung nach
einem ähnlichen
Verfahren wie dem in Vorschrift 17 (17/33) beschriebenen hergestellt.
-
Vorschrift
16 (16/1) 1-[2-(tert-Butoxycarbonyl)-4-pentyl]-cyclopentan-carbonsäure
-
Ein
Gemisch von 1-[2-(tert-Butoxycarbonyl)-4-pentenyl]cyclopentan-carbonsäure (
EP 274234 ) (23 g, 81.5 mMol)
und 10% Palladium auf Aktivkohle (2 g) in trockenem Ethanol (200
ml) wurde während
18 Stunden bei 30 psi und Raumtemperatur hydriert. Das Reaktionsgemisch
wurde durch Arbocel
® filtriert, und das Filtrat wurde
im Vakuum auf konzentriert, woraus sich ein gelbes Öl ergab.
Das Rohprodukt wurde mittels Säulenchromatographie
auf Silikagel unter Verwendung von Ethylacetat:Pentan (40:60) als
Laufmittel gereinigt, woraus sich das gewünschte Produkt als ein klares Öl ergab,
21 g, 91%;
1H NMR (CDCl
3,
0.86 (t, 3H), 1.22–1.58 (m,
15H), 1.64 (m, 4H), 1.78 (dd, 1H), 2.00–2.18 (m, 3H), 2.24 (m, 1H);
LRMS : m/z 283 (M-H)
-.
-
Vorschrift
17 (17/33) tert-Butyl
2-{[1-({[1-(hydroxymethyl)cyclopentyl]amino}carbonyl)-cyclopentyl]methyl}pentanoat
-
1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid-Hydrochlorid
(41 mg, 0.21 mMol), 1-Hydroxybenzotriazol-Hydrat (27 mg, 0.2 mMol),
N-Methylmorpholin (35 μl,
0.31 mMol) und schliesslich 1-Amino-1-cyclopentanmethanol (25 mg,
0.22 mMol) wurden zu einer Lösung
der Säure
aus Vorschrift 16 (16/1) (150 mg, 0.53 mMol) in N,N-Dimethylformamid
(3 ml) zugegeben, und das Reaktionsgemisch wurde während 18
Stunden bei 90°C gerührt. Die
abgekühlte
Lösung
wurde mit Ethylacetat (90 ml) verdünnt, mit Wasser (3×25 ml)
und gesättigter Kochsalzlösung (25
ml) gewaschen, dann getrocknet (MgSO4) und
im Vakuum eingedampft. Das Rohprodukt wurde mittels Chromatographie
auf Silikagel, unter Verwendung von Ethylacetat:Pentan (30:70) als
Laufmittel gereinigt, woraus sich die Titelverbindung ergab, 38
mg, 57%; 1H NMR (CDCl3,
400 MHz) d: 0.88 (t, 3H), 1.29 (m, 3H), 1.41–1.78 (m, 26H), 1.78–1.98 (m,
4H), 2.04 (m, 1H), 2.26 (m, 1H), 3.59 (dd, 1H), 3.70 (dd, 1H), 4.80 (t,
1H), 5.81 (s, 1H); LRMS : m/z 380 (MH-).
-
Vorschrift 18 (18/Bsp.
4)
-
Eine
Verbindung der unten dargestellten Formel wurde aus dem entsprechenden
tert-Butylester nach einem ähnlichen
Verfahren wie dem in Vorschrift 14 (14/Bsp. 1) beschriebenen hergestellt.
-
-
-
Vorschrift
19 (19/Bsp. 21) 2-({1-[(3-Benzylanilino)carbonyl]cyclopentyl}methyl)pentansäure
-
Ein
Gemisch des Benzylesters aus Vorschrift 10 (10/53) (1.3 mg, 2.47
mMol) und 5% Palladium auf Aktivkohle (130 mg) in Wasser (10 ml)
und Ethanol (40 ml) wurde während
2 Stunden bei 30 psi und Raumtemperatur hydriert. Das Reaktionsgemisch
wurde durch Arbocel® filtriert, das Filtrat
wurde im Vakuum aufkonzentriert, und der Rückstand wurde mit Dichlormethan zerstossen,
und bei 50°C
getrocknet, woraus sich die Titelverbindung als ein Feststoff ergab,
0.79 g, 81%; 1H NMR (CDCl3,
300 MHz) δ:
0.95 (t, 3H), 1.24–1.51
(m, 3H), 1.58-1.80
(m, 7H), 1.88 (dd, 1H), 2.15 (m, 2H), 2.24 (m, 1H), 2.48 (m, 1H),
4.00 (s, 2H), 6.98 (d, 1H), 7.24 (m, 6H), 7.40 (m, 3H); Anal. gefunden:
C, 75.48; H, 7.76; N, 3.59. C25H31NO3;0.25H2O erfordert C, 75.44; H, 7.98; N, 3.51%.
-
ACE-ASSAY
-
DIE
HERSTELLUNG UND DER ASSAY VON LÖSLICHEM
ANGIOTENSIN-KONVERTIERENDEM-ENZYM
(ACE) AUS DEM NIERENKORTEX VON SCHWEINEN und MENSCHEN.
-
Die
Aktivität
von löslichem
ACE wird aus dem Nierenkortex gewonnen und durch Messung der Geschwindigkeit
der Spaltung des ACE-Substrats Abz-Gly-p-nitro-Phe-Pro-OH unter
Bildung seines fluoreszierenden Produktes Abz-Gly bestimmt.
-
1. MATERIALIEN
-
Sämtliches
Wasser ist doppelt entionisiert.
- 1.1 Menschliche
Niere IIAM (Pennsylvania. U.S.A.) oder UK Human Tissue Bank (UK
HTB)
- 1.2 Schweineniere ACE Sigma (A2580)
- 1.3 Homogenisationspuffer-1
100 mM Mannitol und 20 mM Tris
bei pH 7.1
2.42 g Tris (Fisher T/P630/60) werden mit 1 Liter
Wasser verdünnt,
und der pH wird unter Verwendung von 6M HCl bei Raumtemperatur auf
7.1 eingestellt. Dazu werden 18.22 g Mannitol (Sigma M-9546) zugegeben.
- 1.4 Homogenisationspuffer-2
100 mM Mannitol, 20 mM Tris
bei pH 7.1 und 10 mM MgCl2.6H2O
(Fisher MO600/53)
Zu 500 ml des Homogenisationspuffers 1 (1.4)
werden 1.017 g MgCl2 zugegeben.
- 1.5 Tris Puffer (ACE-Puffer).
50 mM Tris und 300 mM NaCl
bei pH 7.4
50 ml 50 mM Tris pH 7.4 (Sigma T2663) und 17.52
g NaCl (Fisher S/3160/60) werden in 1000 ml Wasser angesetzt.
- 1.6 Substrat (Abz-D-Gly-p-nitro-Phe-Pro-OH) (Bachem M-1100)
ACE-Substrat wird als Pulver bei –20°C gelagert. Eine 2 mM Stammlösung wird
durch sorgfältiges
Resuspendieren des Substrats in ACE-Puffer hergestellt, wobei dieses
weder verwirbelt noch beschallt werden darf. 400 μl Aliquote
der 2 mM Stammlösung
werden während
bis zu einem Monat bei –20°C gelagert.
- 1.7 Gesamtprodukt
Proben, die einer 100%-igen Substrat-zu-Produkt-Umwandlung
entsprechen, werden mit auf die Platte gegeben, um den %-Substratumsatz berechnen
zu können
(siehe Berechnungen). Das Gesamtprodukt wird durch Inkubieren von
1 ml des 2 mM Substrates mit 20 μl
der Enzym-Stammlösung
während
24 Stunden bei 37°C
erzeugt.
- 1.8 Stopplösung.
0.5M
EDTA (Promega CAS[6081/92/6]) wird mit ACE-Puffer 1:250 verdünnt, woraus
sich eine 2 mM Lösung ergibt.
- 1.9 Dimethylsulfoxid (DMSO).
- 1.10 Magnesiumchlorid MgCl2.6H2O (Fisher MO600/53).
- 1.11 Schwarze Platte mit 96 flachbodigen Vertiefungen (Costar
3915 oder Packard).
- 1.12 Topseal A (Packard 6005185).
- 1.13 Zentrifugenröhrchen
-
2. SPEZIFISCHE AUSÜSTUNG
-
- 2.1 Sorvall RC-5B Zentrifuge (SS34 GSA Rotor,
vorgekühlt
auf 4°C).
- 2.2 Braun Miniprimer Mixer.
- 2.3 Beckman CS-6R Zentrifuge.
- 2.4 BMG Fluostar Galaxy.
- 2.5 Wesbart 1589 Schüttelinkubator.
-
3. METHODEN
-
- 3.1 GEWEBE-PRÄPARATION
- 3.3 Menschliches ACE wird aus dem Nierenkortex unter Verwendung
einer aus Booth, A.G. & Kenny,
A.J. (1974) Biochem. J. 142, 575–581 angepassten Methode erhalten.
- 3.3 Gefrorene Nieren werden auf Raumtemperatur auftauen gelassen
und der Kortex wird von der Medulla abgetrennt.
- 3.4 Der Kortex wird fein zerschnitten und in ungefähr 10 Volumina
des Homogenisationspuffers 1 (1.4) unter Verwendung eines Braun
Miniprimer (2.2) homogenisiert.
- 3.5 Magnesiumchlorid (1.11) (20.3 mg/g Gewebe) wird zum Homogenisat
zugegeben und während
15 Minuten in einem Eiswasserbad gerührt.
- 3.6 Das Homogenisat wird während
12 Minuten mit 1,500 g (3,820 rpm) in einer Beckman Zentrifuge (2.3) zentrifugiert,
anschliessend wird der Überstand
in ein frisches Zentrifugenrohr transferiert und das Pellet verworfen.
- 3.7 Der Überstand
wird während
12 Minuten mit 15,000 g (12,100 rpm) in einer Sovall Zentrifuge
(2.1) zentrifugiert, und der Überstand
wird verworfen.
- 3.8 Die hellrosa Phase am oberen Ende des verbleibenden Pellets
wird entfernt und in Homogenisationspuffer 2 (1.5) (5 ml Puffer
pro 1 g Gewebe) resuspendiert.
- 3.9 Die Suspension wird während
12 Minuten bei 2,200 g (4,630 rpm) in einer Beckman Zentrifuge zentrifugiert,
und anschliessend wird das Pellet verworfen.
- 3.10 Der Überstand
wird während
12 Minuten bei 15,000 g (12,100 rpm) unter Verwendung der Sorvall
Zentrifuge zentrifugiert, und der Überstand wird verworfen.
- 3.11 Das finale Pellet wird in Homogenisationspuffer 2 (0.5
ml Puffer pro 1 g Gewebe) resuspendiert. Eine homogene Suspension
wird unter Verwendung eines Braun Miniprimer erhalten. Diese wird
dann in 100 μl Aliquoten
für die
Bestimmung der NEP-Aktivität
eingefroren.
-
4.0 BESTIMMUNG DER ACE-AKTIVITÄT
-
Die
Aktivität
des vorgängig
aliquotierten ACE wird aufgrund seiner Fähigkeit, das ACE-spezifische Peptid-Substrat
zu spalten, gemessen.
-
Schweine-ACE
(1.2) wird aufgetaut und in ACE-Puffer (1.6) bei 0.004U/μl resuspendiert,
und dies wird in 50 μl
Aliquoten eingefroren.
- 4.1 Es wird eine 4%-ige
DMSO/ACE-Pufferlösung
(4 ml DMSO in 96 ml ACE-Puffer) hergestellt.
- 4.2 Substrat (1.7), Gesamtprodukt (1.8) und Enzym (1.1, 1.2,
1.3) werden zum Auftauen auf Eis belassen.
- 4.3 50 μl
4%-ige DMSO/ACE-Pufferlösung
werden in jede Vertiefung gegeben.
- 4.4 Die 2 mM Substrat-Stammlösung
wird 1:100 verdünnt,
woraus sich eine 20 μM
Lösung
ergibt. 100 μl des
20 μM Substrats
werden in jede Vertiefung gegeben (Endkonzentration im Assay 10 μM).
- 4.5 50 μl
einer Reihe von Enzymverdünnungen
werden zugegeben, um die Reaktion auszulösen (gewöhnlich werden 1:100, 1:200,
1:400, 1:800, 1:1600 und 1:3200 verwendet). 50 μl des ACE-Puffers werden in die
leeren Vertiefungen gegeben.
- 4.6 Das 2 mM Gesamtprodukt wird 1:200 verdünnt, woraus sich eine 10 μM Lösung ergibt.
200 μl des
10 μM Produkt
werden in die ersten vier Vertiefungen einer neuen Platte gegeben.
- 4.7 Die Platten werden während
60 Minuten bei 37°C
in einem Schüttelinkubator
inkubiert.
- 4.8 Die Enzymreaktion wird durch Zugabe von 100 μl 2 mM EDTA
in ACE-Puffer gestoppt und während
20 Minuten bei 37°C
in einem Schüttelinkubator
inkubiert, und anschliessend auf dem BMG Fluostar Galaxy (ex320/em420)
vermessen.
-
5. ACE-INHIBITIONS-ASSAYS
-
- 5.1 Substrat, Gesamtprodukt und Enzym-Stammlösungen werden
zum Auftauen auf Eis belassen.
- 5.2 Verbindungs-Stammlösungen
werden in 100% DMSO hergestellt und 1:25 in ACE-Puffer verdünnt, woraus
sich eine 4% DMSO-Lösung
ergibt. Alle weiteren Verdünnungen
werden in einer 4% DMSO/ACE-Pufferlösung (4 ml DMSO in 96 ml ACE-Puffer)
durchgeführt.
- 5.3 50 μl
der Verbindung werden zweifach zur Platte mit 96 Vertiefungen gegeben,
und 50 μl
des 4% DMSO/ACE-Puffer werden in die Kontroll- und in die Blindvertiefungen
gegeben.
- 5.4 Die Schritte 5.2 und 5.3 können entweder manuell oder
unter Verwendung des Packard Multiprobenroboters durchgeführt werden.
- 5.5 Die 2 mM Substrat-Stammlösung
wird mit ACE-Puffer 1:100 verdünnt,
woraus sich eine 20 μM
Lösung ergibt
(10 μM Endkonzentration
im Assay) (110 μl
des 2 mM Substrats zu 10.89 ml Puffer zugegeben, ist für 1 Platte
ausreichend).
- 5.6 Die Enzym-Stammlösung
wird mit ACE-Puffer verdünnt,
gemäss
Bestimmung aus Aktivitätstests
(4.0).
- 5.7 Die 2 mM Gesamtprodukt-Stammlösung wird 1:200 mit ACE-Puffer verdünnt, woraus
sich eine 10 μM Lösung ergibt.
200 μl werden
in die ersten vier Vertiefungen einer separaten Platte gegeben.
- 5.8 Die 0.5 mM EDTA-Stammlösung
wird 1:250 verdünnt,
woraus sich eine 2 mM Stammlösung
(44 μl EDTA
zu 10.96 ml ACE-Puffer)
ergibt.
- 5.9 In jede Vertiefung der Platte mit 96 Vertiefungen werden
die folgenden Reagenzien zugegeben:
Tabelle 1: Reagenzien zur
Platte mit 96 Vertiefungen gegeben
- 5.10 50 μl
der höchsten
Konzentration von jeder im Assay verwendeten Verbindung werden zweifach
zur selben Platte mit 96 Vertiefungen gegeben wie das Gesamtprodukt
(5.7). 150 μl
des ACE-Puffers werden zugegeben, um etwaige Fluoreszenz der Verbindungen
zu bestimmen.
- 5.11 Die Reaktion wird durch Zugabe des ACE-Enzyms gestartet,
und anschliessend wird während
1 Stunde bei 37°C
in einem Schüttelinkubator
inkubiert.
- 5.12 Die Reaktion wird durch die Zugabe von 100 μl 2 mM EDTA
gestoppt, woraufhin während
20 Minuten bei 37°C
in einem Schüttelinkubator
inkubiert wird, und schliesslich erfolgt die Ablesung mit dem BMG
Fluostar Galaxy (ex320/em420).
-
6. BERECHNUNGEN
-
Die
Aktivität
des ACE-Enzyms wird in Gegenwart und in Abwesenheit der Verbindung
bestimmt und als prozentualer Anteil ausgedrückt.
-
FU
= Fluorezenz-Einheiten
- (i) %-Kontroll-Aktivität (Umsatz
des Enzyms):
- (ii) %-Aktivität
mit Inhibitor:
- (iii) Aktivität
ausgedrückt
als % der Kontrollen:
- (iv) %-Inhibition = 100 – %-Kontrolle
- (v) Für
fluoreszierende Verbindungen werden die mittleren FU von Blindproben,
welche die Verbindung (5.10) enthalten, von den zur Berechnung der
%-Aktivität
verwendeten mittleren FU der Verbindungswerte substrahiert.
-
Eine
sigmoidale Dosis-Antwort-Kurve wird an die %-Aktivitäten (% Kontrolle) vs. Verbindungskonzentration
angepasst und IC50-Werte werden mittels
der LabStats Ausgleichskurve in Excel berechnet.
-
SCHLUSSFOLGERUNGEN
-
Wir
haben ein Tiermodell entwickelt, das die während der weiblichen sexuellen
Erregung beobachtete physiologische Erregungsantwort widerspiegelt
und die bei menschlichen Probanden erhaltenen klinisch Daten direkt
widerspiegelt. Das Modell verwendet Laser-Doppler-Verfahren, um
kleine, durch eine Pelvisnervstimulation oder durch vasoaktive Neurotransmitter
induzierte Veränderungen
des vaginalen und klitoralen Blutfluss zu erfassen. Während der
sexuellen Erregung findet eine Erhöhung des genitalen Blutflusses
aufgrund erhöhter
Innervation des Pelvisnervs statt. Die im Tiermodell beobachtete
pelvisnervstimulierte Erhöhung
des vaginalen und klitoralen Blutflusses repräsentiert die während der
weiblichen sexuellen Erregung beobachteten endogenen vaskulären Wirkungen – d.h. das
Anschwellen. Demnach kann dieses Modell erstens verwendet werden,
um die an der Regulation des vaginalen und klitoralen Blutflusses
beteiligten Mechanismen zu identifizieren, und zweitens, um neue
Ansätze
für die
Verstärkung
des genitalen Blutflusses zu validieren.
-
In
dieser Studie wurde erfolgreich eine Kombination von in vivo, in
vitro und biochemischen Verfahren verwendet, um zu zeigen, dass
VIP den genitalen Blutfluss vermittelt und um cAMP als den die genitale
Vasorelaxation (und die Relaxation der Vaginawand) regulierenden
Mediator/second Messenger zu identifizieren. Unter Verwendung dieses
Tiermodells konnten wir zeigen, dass die Infusion von VIP Erhöhungen des
vaginalen und klitoralen Blutflusses induziert. Unter Verwendung
eines Inhibitor des VIP-Metabolismus (z.B. ein NEP-EC3.4.24.11-Inhibitor)
konnte wir zudem zeigen, dass die während der Pelvisnervstimulation
(d.h. während
der sexuellen Erregung) beobachteten Erhöhungen des genitalen Blutflusses
durch VIP vermittelt werden. Wir haben gezeigt, dass sich die VIP-vermittelten
Erhöhungen
des genitalen Blutflusses aus der Erhöhung von Gewebe-cAMP ergeben,
wogegen früher
für VIP
eine Erhöhung
des vaginalen Blutflusses bei gesunden Freiwilligen aufgezeigt aber
der zelluläre
Mechanismus nicht identifiziert wurde. Ausserdem haben wir gezeigt,
dass der genitale Blutfluss direkt mit einem cAMP-Mimetikum oder
indirekt durch Erhöhung
der cAMP-Konzentrationen
mit einem PDEcAMP-Typ-2-Inhibitor oder einem
NPY-Y1-Rezeptorantagonisten erhöht werden
kann.
-
Die
wichtigste Ursache der FSAD ist ein verminderter genitaler Blutfluss,
und dies manifestiert sich als ein verringertes vaginales, labiales
und klitorales Anschwellen. Die Behandlung von Frauen mit FSAD lässt sich
durch Wiederherstellung der normalen sexuellen Erregungsantwort
erreichen. Dies kann durch Erhöhung des
genitalen Blutflusses erreicht werden. Unser Ansatze zur Behandlung
von FSAD wird darin bestehen, den genitalen Blutfluss zu erhöhen und
dabei durch direkte oder indirekte Potenzierung der endogenen cAMP-Signalisierung,
z.B. mit einem Inhibitor der NEP (EC 3.4.24.11), einem cAMP-hydrolysierenden
PDE-Inhibitor oder
einem NPY-Rezeptorantagonisten die vaginale Anschwellung/Lubrikation
und die klitorale Anschwellung/Sensitivität zu potenzieren. Als Gesamtwirkung
wird dies die normale Erregungsantwort ohne kardiovaskuläre Nebenwirkungen
wiederherstellen oder potenzieren. Die sexuelle Erregung/Anschwellung
wird verstärkt,
nicht aber bei fehlendem sexuellen Begehren induziert wie dies mit
einigen exogen verabreichten vasoaktiven Wirkstoffen wie VIP der
Fall sein kann.
-
Zusammenfassend
bezieht sich die vorliegende Erfindung demnach unter anderem auf:
Verwendung
eines Wirkstoffes zur Herstellung eines Medikamentes für die Behandlung
von FSD, vorzugsweise FSAD; wobei der Wirkstoff zur Potenzierung
von cAMP in den sexuellen Genitalien von an FSD, vorzugsweise FSAD,
leidenden Frauen befähigt
ist; wobei der besagte Wirkstoff ein I:NEP ist.
-
In
einer besonders bevorzugten Ausgestaltung bezieht sich die vorliegende
Erfindung unter anderem auf:
Verwendung eines Wirkstoffes zur
Herstellung eines Medikamentes für
die Behandlung von FSD, vorzugsweise FSAD; wobei der Wirkstoff zur
Potenzierung von cAMP in den sexuellen Genitalien von an FSD, vorzugsweise
FSAD, leidenden Frauen befähigt
ist; und wobei der besagte Wirkstoff oral verabreicht wird; wobei
der besagte Wirkstoff ein I:NEP ist.
-
In
einer weiteren besonders bevorzugten Ausgestaltung bezieht sich
die vorliegende Erfindung unter anderem auf:
Verwendung eines
Wirkstoffes zur Herstellung eines Medikamentes für die Behandlung von FSD, vorzugsweise
FSAD; wobei der Wirkstoff zur Potenzierung von cAMP in den sexuellen
Genitalien von an FSD, vorzugsweise FSAD, leidenden Frauen befähigt ist;
und wobei der besagte Wirkstoff das endogene cAMP potenziert; wobei
der besagte Wirkstoff ein I:NEP ist.
-
In
einer weiteren besonders bevorzugten Ausgestaltung bezieht sich
die vorliegende Erfindung unter anderem auf:
Verwendung eines
Wirkstoffes zur Herstellung eines Medikamentes für die Behandlung von FSD, vorzugsweise
FSAD; wobei der Wirkstoff zur Potenzierung von cAMP in den sexuellen
Genitalien von an FSD, vorzugsweise FSAD, leidenden Frauen befähigt ist;
und wobei der besagte Wirkstoff oral verabreicht wird und wobei der
besagte Wirkstoff das endogene cAMP potenziert; wobei der besagte
Wirkstoff ein I:NEP ist.
-
REFERENZEN
FÜR DEN
ALLGEMEINEN TEXT
-
- Ashur-Fabian, O., Perl, O., Lilling, G., et al. (1999).
SNV, a lipophilic superactive VIP analog, acts through cGMP to promote
neuronal survival. Peptides, 20, 629–633.
- Berman, J.R., Berman, L. & Goldstein,
I. (1999). Female sexual dysfunction: Incidence, pathophysiology,
evaluation, and treatment options. Urology, 54, 385–391.
- Berman, J., Goldstein, I., Werbin, T. et al. (1999a). Double
blind placebo controlled study with crossover to assess effect of
sildenafil on physiological parameters of the female sexual response.
J. Urol., 161, 805.
- Burnett, A, Calvin, D., Silver, R. et al. (1997). Immunohistochemical
description of nitric oxide synthase isoforms in human clitoris.
J. Urol., 158, 75–78.
- Diagnostic and statistical manual of mental disorders-IV, American
Psychiatric Association: Washington, DC., 1987, pp 493–518.
- Fan, Y.P., Chakder, S. & Ratton,
S. (1998). Inhibitory effect of zinc protoporphyrin IX on lower
esophageal sphincter smooth muscle relaxation by vasoactive intestinal
polypeptide and other receptor agonists. J. Pharmacol. Exp. Ther.,
285, 468–474.
- Foda, H.D., Sharaf, H.H., Absood, A. et al. (1995). Pituitary
adenylate cyclase-activating peptide (PACAP), a VIP-like peptide,
has prolonged airway smooth muscle relaxant activity. Peptides,
16, 1057–1061.
- Frank, E., Anderson, C. & Rubinstein,
D. (1978). Frequency of sexual dysfunction in "normal" couples. N. Engl. J. Med., 229, 111–115.
- Goldstein, I. & Berman,
J.R. (1998). Vasculogenic female sexual dysfunction: vaginal engorgement
and clitoral erectile insufficiency syndromes. Int. J. Impot. Res.,
10, S84–S90.
- Gu, Z.F., Jensen, R.T. & Maton,
P.N. (1992). A primary role for protein kinase A in smooth muscle
relaxation induced by adrenergic agonists and neuropeptides. Am.
J. Physiol., 263, G360–G364.
- Hauser-Kronberger, C., Cheung, A., Hacker, G. et al. (1999).
Peptidergic innervation of the human clitoris. Peptides, 20, 539–543.
- Hoyle, C.H.V., Stones, R.W., Robson, T. et al. (1996). Innervation
of vasculature and microvasculature of the human vagina by NOS and
neuropeptide containing nerves. J. Anat., 188, 633–644.
- Ingenhoven, N. & Beck-Sickinger,
A.G. (1997). Flourescent labelled analogues of neuropeptide Y for
the characterisation of cells expressing NPY receptor subtypes.
J. Recept. Signal Transduct. Res., 17, 407–418.
- Jovanovic, A., Jovanovic, S., Tulic,. I. et al. (1998). Predominant
role for nitric oxide in the relaxation induced by vasoactive intestinal
polypeptide in human uterine artery. Mol. Human Reprod., 4, 71–76.
- Kaplan, H.S. (1974). The New Sex Therapy. London, Bailliere
Tindall.
- Kaplan, S.A., Reis, R.B., Kohm, I.J. et al. (1999). Safety and
efficacy of sildenafil in postmenopausal women with sexual dysfunction.
Urology, 53, 481–486.
- Kim, Y.C., Choi, H.K., Ahn, Y.S., et al. (1994). The effect
of vasoactive intestinal polypeptide (VIP) on rabbit cavernosal
smooth muscle contractility. J. Androl., 15, 392-739.
- Laan, E. & Everaerd,
W. (1998). Physiological measures of vaginal vasocongestion. Int.
J. Impot. Res., 10, S107–S110.
- Leiblum, S.R. (1998). Definition and classification of female
sexual disorders. Int. J. Impotence Res., 10, S104-S106.
- Levin, R.J. (1980). The physiology of sexual function in women.
Clin. Obstet. Gynecol., 7, 213–252.
- Levin, R.J. (1991). VIP, vagina, clitoral and preurethral glans:
An update on female genital arousal. Exp. Clin. Endocrinol., 98,
61–69.
- Levin, R.J. (1992). The mechanisms of human female sexual arousal.
Ann. Rev. Sex Res., 3,1–48.
- Levin, R.J. & Wagner,
G. (1986). TRH and vaginal blood flow-effects in concious women and anaesthetised sheep.
J. Physiol., 373, 83P.
- Lundberg, J.M., Modin, A. & Malmstrom,
R.E. (1996). Recent developments with neuropeptide Y receptor antagonists.
Trends. Pharmacol. Sci., 17, 301–304.
- Masters, W.H., Johnson, V.E. Human Sexual Response. Little,
Brown: Boston, 1996.
- Ottesen, B., Gerstenberg, T., Ulrichsen, H. et al. (1983). Vasoactive
intestinal polypeptide (VIP) increases vaginal blood flow and inhibits
smooth muscle activity in women. Eur. J. Clin. Invest., 13, 321–324.
- Ottesen, B., Wagner, G. & Fahrenkrug,
J. Peptide innervation of the sexual organs. In: Handbook of Sexology, Vol.
6, The Pharmacological and Endocrinology of Sexual Function, Sitsen,
J.M.A. (eds), Amsterdam: Elsevier Science Publishers (1988), Kapitel
4, S. 66–97.
- Ottensen, B., Pedersen, B., Nielsen, J. et al. (1987). Vasoactive
intestinal polypeptide (VIP) provokes vaginal lubrication in normal
women. Peptides, 8, 797–800.
- Park, K., Goldstein, I., Andry, C., et al. (1997). Vasculogenic
female sexual dysfunction: The hemodynamic basis for vaginal engorgement
insufficiency and clitoral erectile insufficiency. Int. J. Impotence
Res., 9, 27–37.
- Rosen, R., Taylor, J., Leiblum, S. et al. (1993). Prevalence
of sexual dysfunction in women: results of a survey of 329 women
in an outpatient gynecological clinic. J. Sex Marital Ther., 19,
171–188.
- Schiavi, R.C. & Seagraves,
R.T. (1995). The biology of sexual function. Psychiat. Clin. North.
Am., 18, 7–23.
- Schoeffter, P. & Stoclet,
J.C. (1985). Effect of vasoactive intestinal polypeptide (VIP) on
cyclic AMP level and relaxation in rat isolated aorta. Eur. J. Pharmacol.,
109, 275–279.
- Serradeil-Le Gal, C., Valette, G., Rouby, P.E. et al. (1995).
SR 120819A, an orally-active and selective neuropeptide Y Y1 receptor
antagonist. FEBS Letters, 3, 192-196.
- Sjoberg, I. (1992). The vagina: Morphological, functional and
ecological aspects. Acta Obst. Gynecol. Scand., 71, 84-85.
- Spector, I.P. & Carey,
M.P. (1990). Incidence and prevalence of sexual dysfunctions: a
critical review of the empirical literature. Arch. Sex. Behav.,
19, 389–408.
- Wagner, G. (1992). Aspects of genital physiology and pathology.
Sem. Neurol., 12, 87–97.
- Werbin, T., Salimpour, P., Berman, L., et al. (1999). Effect
of sexual stimulation and age on genital blood flow in women with
sexual stimulation. J. Urol., 161, 688
- Wincze, J.P., Albert, A. & Bansal,
S. (1993). Sexual arousal in diabetic females: Physiological and
self-report measures. Arch. Sex Behav., 22, 587–601.
- Wieland, H.A., Willim, K.D., Entzeroth, M. et al. (1995). Subtype
selectivity and antagonist profile of the nonpeptide Y1 receptor
antagonist BIBP 3226. J Pharmacol Exp Ther., 275, 143–9.
-
REFERENZEN
FÜR DEN
PDE-ABSCHNITT
-
- Han, P.; Fletcher, C. F.; Copeland, N. G.; Jenkins, N. A.;
Yaremko, L. M.; Michaeli, T.: Assignment of the mouse Pde7A gene
to the proximal region of chromosome 3 and of the human PDE7A gene
to chromosome 8q13. Genomics 48: 275–276, 1998.
- 2. Michaeli, T.; Bloom, T. J.; Martins, T.; Loughney, K.; Ferguson,
K.; Riggs, M.; Rodgers, L.; Beavo, J. A.; Wigler, M.: Isolation
and characterization of a previously undetected human cAMP phosphodiesterase
by complementation of cAMP phosphodiesterase-deficient Saccharomyces
cerevisiae. J. Biol. Chem. 268: 12925–12932, 1993.
- 3. Milatovich, A.; Bolger, G.; Michaeli, T.; Francke, U.: Chromosome
localizations of genes for five cAMP-specific phosphodiesterases
in man and mouse. Somat. Cell Molec. Genet. 20: 75–86, 1994.
- 4. Rosman, G. J.; Martins, T. J.; Sonnenburg, W. K.; Beavo,
J. A.; Ferguson, K.; Loughney, K.: Isolation and characterization
of human cDNAs encoding a cGMP-stimulated 3-prime,5-prime-cyclic
nucleotide phosphodiesterase. Gene 191: 89–95, 1997.
-
REFERENZEN FÜR DEN NEP-ABSCHNITT
-
- 1. Barker, P. E.; Shipp, M. A.; D'Adamio, L.; Masteller, E. L.; Reinherz,
E. L. The common acute lymphoblastic leukemia antigen gene maps
to chromosomal region 3(q21-q27). J. Immun. 142: 283–287, 1989.
- 2. D'Adamio,
L.; Shipp, M. A.; Masteller, E. L.; Reinherz, E. L.: Organization
of the gene encoding common acute lymphoblastic leukemia antigen
(neutral endopeptidase 24.11): multiple miniexons and separate 5-prime untranslated
regions. Proc. Nat. Acad. Sci. 86: 7103–7107, 1989.
- 3. Letarte, M.; Vera, S.; Tran, R.; Addis, J. B. L.; Onizuka,
R. J.; Quackenbush, E. J.; Jongeneel, C. V.; McInnes, R. R.: Common
acute lymphocytic leukemia antigen is identical to neutral endopeptidase.
J. Exp. Med. 168: 1247–1253,
1988.
- 4. Shipp, M. A.; Vijayaraghavan, J.; Schmidt, E. V.; Masteller,
E. L.; D'Adamio,
L.; Hersh, L. B.; Reinherz, E. L.: Common acute lymphoblastic leukemia
antigen (CALLA) is active neutral endopeptidase 24.11 ('enkephalinase'): direct evidence
by cDNA transfection analysis. Proc. Nat. Acad. Sci. 86: 297–301, 1989.
- 5. Tran-Paterson, R.; Willard, H. F.; Letarte, M.: The common
acute lymphoblastic leukemia antigen (neutral endopeptidase--3.4.24.11)
gene is located on human chromosome 3. Cancer Genet. Cytogenet.
42: 129–134, 1989.
-
REFERENZEN FÜR DEN NPY-ABSCHNITT
-
- 1. Allen, J. M.; Bloom, S. R.: Neuropeptide Y: a putative
neurotransmitter. Neurochem. Int. 8: 1–8, 1986.
- 2. Bahary, N.; Zorich, G.; Pachter, J. E.; Leibel, R. L.; Friedman,
J. M.: Molecular genetic linkage maps of mouse chromosomes 4 and
6. Genomics 11: 33–47,
1991.
- 3. Baker, E.; Hort, Y. J.; Ball, H.; Sutherland, G. R.; Shine,
J.; Herzog, H.: Assignment of the human neuropeptide Y gene to chromosome
7p15.1 by nonisotopic in situ hybridization. Genomics 26: 163–164, 1995.
- 5. Carr, L. G.; Foroud, T.; Bice, P.; Gobbett, T.; Ivashina,
J.; Edenberg, H.; Lumeng, L.; Li, T. K.: A quantitative trait locus
for alcohol consumption in selectively bred rat lines. Alcohol Clin.
Exp. Res. 22: 884–887,
1998.
- 5. Dockray, G. J.: Neuropeptide Y: in search of a function.
Neurochem. Int. 8: 9–11,
1986.
- 6. Erickson, J. C.; Clegg, K. E.; Palmiter, R. D.: Sensitivity
to leptin and susceptibility to seizures of mice lacking neuropeptide
Y. Nature 381: 415–421,
1996. PubMed ID 8632796
- 7. Erickson, J. C.; Hollopeter, G.; Palmiter, R. D.: Attenuation
of the obesity syndrome of ob/ob mice by the loss of neuropeptide
Y. Science 274: 1704–1706,
1996.
- 8. Karvonen, M. K.; Pesonen, U.; Koulu, M.; Niskanen, L.; Laakso,
M.; Rissanen, A.; Dekker, J. M.; Hart, L. M.; Valve, R.; Uusitupa,
M. I.: Association of a leucine(7)-toproline(7) polymorphism in
the signal peptide of neuropeptide Y with high serum cholesterol
and LDL cholesterol levels. Nature Med. 4: 1434–1437, 1998.
- 9. Maccarrone, C.; Jarrott, B.: Neuropeptide Y: a putative neurotransmitter.
Neurochem. Int. 8: 13–22,
1986.
- 10. Meisler, M. H.; Spence, J. E.; Dixon, J. E.; Caldwell, R.
M.; Minth, C. D.; Beaudet, A. L.: Exclusion of close linkage between
the loci for cystic fibrosis and neuropeptide Y on human chromosome
7. Cytogenet. Cell Genet. 44: 175–176, 1987.
- 11. Minth, C. D.; Andrews, P. C.; Dixon, J. E.: Characterization,
sequence, and expression of the cloned human neuropeptide Y gene.
J. Biol. Chem. 261: 11974-11979,
1986.
- 12. Minth, C. D.; Bloom, S. R.; Polak, J. M.; Dixon, J. E.:
Cloning, characterization, and DNA sequence of a human cDNA encoding
neuropeptide tyrosine. Proc. Nat. Acad. Sci. 81: 4577–4581, 1984.
- 13. Takeuchi, T.; Gumucio, D.; Eddy, R.; Meisler, M.; Minth,
C.; Dixon, J.; Yamada, T.; Shows, T.: Assignment of the related
pancreatic polypeptide (PPY) and neuropeptide Y (NPY) genes to regions
on human chromosomes 17 and 7. (Abstract) Cytogenet. Cell Genet.
40: 759 only, 1985,
- 14. Takeuchi, T.; Gumucio, D. L.; Yamada, T.; Meisler, M. H.;
Minth, C. D.; Dixon, J. E.; Eddy, R. E.; Shows, T. B.: Genes encoding
pancreatic polypeptide and neuropeptide Y are on human chromosomes
17 and 7. J. Clin. Invest. 77: 1038-1041, 1986.
- 15. Terenghi, G.; Polak, J. M.; Hamid, Q.; O'Brien, E.; Denny, P.; Legon, S.; Dixon,
J.; Minth, C. D.; Palay, S. L.; Yasargil, G.; Chan-Palay, V.: Localization
of neuropeptide Y mRNA in neurons of human cerebral cortex by means
of in situ hybridization with a complementary RNA probe. Proc. Nat.
Acad. Sci. 84: 7315–7318,
1987.
- 16. Thiele, T. E.; Marsh, D. J.; Ste. Marie, L.; Bernstein,
I. L.; Palmiter, R. D.: Ethanol consumption and resistance are inversely
related to neuropeptide Y levels. Nature 396: 366–369, 1998.
- 17. Uusitupa, M. I. J.; Karvonen, M. K.; Pesonen, U.; Koulu,
M.: Neuropeptide Y: a novel link between the neuroendocrine system
and cholesterol metabolism. Ann. Med. 30: 508–510, 1998.
-
REFERENZEN FÜR DEN NPYR1-ABSCHNITT
-
- 1. Herzog, H.; Baumgartner, M.; Vivero, C.; Selbie, L. A.;
Auer, B.; Shine, J.: Genomic organization, localization, and allelic
differences in the gene for the human neuropeptide Y Y1 receptor.
J. Biol. Chem. 268: 6703–6707,
1993.
- 2. Herzog, H.; Darby, K.; Ball, H.; Hort, Y.; Beck-Sickinger, A.; Shine,
J.: Overlapping gene structure of the human neuropeptide Y receptor
subtypes Y1 and Y5 suggests coordinate transcriptional regulation.
Genomics 41: 315–319,
1997.
- 3. Herzog, H.; Hort, Y. J.; Ball, H. J.; Hayes, G.; Shine, J.;
Selbie, L. A.: Cloned human neuropeptide Y receptor couples to two
different second messenger systems. Proc. Nat. Acad. Sci. 89: 5794–5798, 1992.
- 4. Larhammar, D.; Blomgvist, A. G.; Yee, F.; Jazin, E.; Yoo,
H.; Wahlestedt, C.: Cloning and functional expression of a human
neuropeptide Y/peptide YY receptor of the Y1 type. J. Biol. Chem.
267: 10935–10938,
1992.
- 5. Lutz, C. M.; Frankel, W. N.; Richards, J. E.; Thompson, D.
A.: Neuropeptide Y receptor genes on human chromosome 4q31-q32 map
to conserved linkage groups on mouse chromosomes 3 and 8. Genomics
41: 498–500,
1997.
-
REFERENZEN FÜR DEN NPYR2-ABSCHNITT
-
1.
Ammar, D. A.; Eadie, D. M.; Wong, D. J.; Ma, Y.-Y.; Kolakowski,
L. F., Jr.; Yang-Feng, T. L.; Thompson, D. A.: Characterization
of the human type 2 neuropeptide Y receptor gene (NPY2R) and localization
to the chromosome 4q region containing the type 1 neuropeptide Y
receptor gene. Genomics 38: 392–398,
1996.
-
2.
Gerald, C.; Walker, M. W.; Vaysse, P. J.-J.; He, C.; Branchek, T.
A.; Weinshank, R. L.: Expression cloning and pharmacological characterization
of a human hippocampal neuropeptide Y/peptide YY Y2 receptor subtype.
J. Biol. Chem. 270: 26758–26761,
1995.
-
3.
Lutz, C. M.; Frankel, W. N.; Richards, J. E.; Thompson, D. A.: Neuropeptide
Y receptor genes on human chromosome 4q31-q32 map to conserved linkage
groups on mouse chromosomes 3 and 8. Genomics 41: 498–500, 1997.
-
4.
Rose, P. M.; Fernandes, P.; Lynch, J. S.; Frazier, S. T.; Fisher,
S. M.; Kodukula, K.; Kienzle, B.; Seethala, R.: Cloning and functional
expression of a cDNA encoding a human type 2 neuropeptide Y receptor.
J. Biol. Chem. 270: 22661-22664,
1995.
-
REFERENZEN FÜR DEN VIP-ABSCHNITT
-
- 1. Bodner, M.; Fridkin, M.; Gozes, I.: Coding sequences
for vasoactive intestinal peptide and PHM-27 peptide are located
on two adjacent exons in the human genome. Proc. Nat. Acad. Sci.
82: 3548–3551,
1985.
- 2. Gotoh, E.; Yamagami, T.; Yamamoto, H.; Okamoto, H.: Chromosomal
assignment of human VIP/PHM-27 gene to 6q26-q27 region by spot blot
hybridization and in situ hybridization. Biochem. Int. 17: 555–562, 1988.
- 3. Gozes, I.; Avidor, R.; Yahav, Y.; Katznelson, D.; Croce,
C. M.; Huebner, K.: The gene encoding vasoactive intestinal peptide
is located on human chromosome 6p21-6qter. Hum. Genet. 75: 41–44, 1987.
- 4. Gozes, I.; Nakai, H.; Byers, M.; Avidor, R.; Weinstein, Y.;
Shani, Y.; Shows, T. B.: Sequential expression in the nervous system
of C-MYB and VIP genes, located in human chromosomal region 6q24.
Somat. Cell Molec. Genet. 13: 305-313, 1987.
- 5. Heinz-Erian, P.; Dey, R. D.; Flux, M.; Said, S. I.: Deficient
vasoactive intestinal peptide innervation in sweat glands of cystic
fibrosis patients. Science 229: 1407-1408, 1985.
- 6. Itoh, N.; Obata, K.; Yanaihara, N.; Okamoto, H.: Human preprovasoactive
intestinal polypeptide contains a novel PHI-27-like peptide, PHM-27.
Nature 304: 547–549,
1983.
- 7. Linder, S.; Barkhem, T.; Norberg, A.; Persson, H.; Schalling,
M.; Hokfelt, T.; Magnusson, G.: Structure and expression of the
gene encoding the vasoactive intestinal peptide precursor. Proc.
Nat. Acad. Sci. 84: 605–609, 1987.
- 8. Omary, M. B.; Kagnoff, M. F.: Identification of nuclear receptors
for VIP on a human colonic adenocarcinoma cell line. Science 238:
1578–1581,
1987.
-
REFERENZEN FÜR DEN AC-ABSCHNITT
-
1.
Parma, J.; Stengel, D.; Gannage, M.-H.; Poyard, M.; Barouki, R.;
Hanoune, J.: Sequence of a human brain adenylyl cyclase partial
cDNA: evidence for a consensus cyclase domain. Biochem. Biophys.
Res. Commun. 179: 455–462,
1991.
-
2.
Stengel, D.; Parma, J.; Gannage, M.-H.; Roeckel, N.; Mattei, M.-G.;
barouki, R.; Hanoune, J.: Different chromosomal localization of
two adenylyl cyclase genes expressed in human brain. Hum. Genet.
90: 126–130,
1992.
-
REFERENZEN FÜR DEN VPAC1-ABSCHNITT
-
- 1. Couvineau, A.; Rouyer-Fessard, C.; Darmoul, D.; Maoret,
J.-J.; Carrero, I.; Ogier-Denis, E.; Laburthe, M.: Human intestinal
VIP receptor: cloning and functional expression of two cDNA encoding
proteins with different N-terminal domains. Biochem. Biophys. Res.
Commun. 200: 769–776,
1994.
- 2. Hashimoto, H.; Nishino, A.; Shintani, N.; Hagihara, N.; Copeland,
N. G.; Jenkins, N. A.; Yamamoto, K.; Matsuda, T.; Ishihara, T.;
Nagata, S.; Baba, A.: Genomic organization and chromosomal location
of the mouse vasoactive intestinal polypeptide 1 (VPAC-1) receptor.
Genomics 58: 90–93,
1999.
- 3. Libert, F.; Passage, E.; Parmentier, M.; Simons, M.-J.; Vassart,
G.; Mattei, M.-G.: Chromosomal mapping of A1 and A2 adenosine receptors,
VIP receptor, and a new subtype of serotonin receptor. Genomics
11: 225–227,
1991.
- 4. Sreedharan, S. P.; Huang, J.-X.; Cheung, M.-C.; Goetzl, E.
J.: Structure, expression, and chromosomal localization of the type
I human vasoactive intestinal peptide receptor gene. Proc. Nat.
Acad. Sci. 92: 2939–2943, 1995.
- 5. Sreedharan, S. P.; Patel, D. R.; Huang, J.-X.; Goetzl, E.
J.: Cloning and functional expression of a human neuroendocrine
vasoactive intestinal peptide receptor. Biochem. Biophys. Res. Commun.
193; 546–553,
1993.
- 6. Sreedharan, S. P.; Robichon, A.; Peterson, K. E.; Goetzl,
E. J.: Cloning and expression of the human vasoactive intestinal
peptide receptor. Proc. Nat. Acad. Sci. 88: 4986-4990, 1991.
- 7. Vassart, G.: Personal Communication. Brussels, Belgium, 1/15/1992.
8. Wenger, G. D.: Personal Communication. Columbus, Ohio, 8/3/1993.
-
REFERENZEN FÜR DEN VPAC2-ABSCHNITT
-
1.
Adamou, J. E.; Aiyar, N.; Van Horn, S.; Elshourbagy, N. A.: Cloning
and functional characterization of the human vasoactive intestinal
peptide (VIP)-2 receptor. Biochem. Biophys. Res. Commmun. 209: 385–392, 1995.
-
2.
Mackay, M.; Fantes, J.; Scherer, S.; Boyle, S.; West, K.; Tsui,
L.-C.; Belloni, E.; Lutz, E.; Van Heyningen, V.; Harmar, A. J.:
Chromosomal localization in mouse and human of the vasoactive intestinal
peptide receptor type 2 gene: a possible contributor to the holoprosencephaly
3 phenotype. Genomics 37: 345–353, 1996.
-
3.
Svoboda, M.; Tastenoy, M.; Van Rampelbergh, J.; Goossens, J.-F.;
De Neef, P.; Waelbroeck, M.; Robberecht, P.: Molecular cloning and
functional characterization of a human VIP receptor from SUP-T1
lymphoblasts. Biochem. Biophy. Res. Commun. 205: 1617–1624, 1994.
-
ABKÜRZUNGEN
- FSD
- = weibliche Sexualstörung (female
sexual dysfunction)
- FSAD
- = weibliche sexuelle
Erregungsstörung
(female sexual arousal disorder)
- cAMP
- = cyclisches Adenosin-3',5'-monophosphat
- cGMP
- = cyclisches Guanosin-3',5'-monophosphat
- PcAMP
- = Potenziator von
cAMP
- PcGMP
- = Potenziator von
cGMP
- AcAMP
- = Aktivator von cAMP
- AcGMP
- = Aktivator von cGMP
- AMcAMP
- = Adversmodulator
von cAMP
- AMcGMP
- = Adversmodulator
von cGMP
- IcAMP
- = Inhibitor von cAMP
- IcGMP
- = Inhibitor von cGMP
- I:IcAMP
- = Inhibitor eines
Inhibitors von cAMP
- I:IcGMP
- = Inhibitor eines
Inhibitors von cGMP
- I:AMcAMP
- = Inhibitor eines
Adversmodulators von cAMP
- I:AMcGMP
- = Inhibitor eines
Adversmodulators von cGMP
- U:AcAMP
- = Aufregulator(upregulator)
eines Aktivators von cAMP
- U:AcGMP
- = Aufregulator(upregulator)
eines Aktivators von cGMP
- AC
- = Adenylatcyclase
- A:AC
- = Aktivator von AC
- NEP
- = neutrale Endopeptidase
- I:NEP
- = Inhibitor von NEP
- VIP
- = vasoaktives intestinales
Peptid
- VIPr
- = Rezeptor von VIP
(kann als VIPR bezeichnet sein)
- VIPn
- = Rezeptor-Subtyp
von VIP (wie VIPR1, VIPR2)
- A:VIPr
- = Aktivator von VIPr
- A:VIPn
- = Aktivator von VIPn
- I:VIPr
- = Inhibitor von VIPr
- I:VIPn
- = Inhibitor von VIPn
- I:I:VIPr
- = Inhibitor eines
Inhibitors von VIPr
- I:I:VIPn
- = Inhibitor eines
Inhibitors von VIPn
- PDE
- = Phosphodiesterase
- PDEn
- = PDE Familie (e.g.
PDE1, PDE2 etc.)
- PDEcAMP
- = cAMP hydrolysierende
PDE
- PDEcGMP
- = cGMP hydrolysierende
PDE
- I:PDE
- = Inhibitor einer
PDE
- I:PDEcAMP
- = Inhibitor einer
cAMP hydrolysierenden PDE
- I:PDEcAMP
- = Inhibitor einer
cAMP hydrolysierenden PDE- Familie
- NPY
- = Neuropeptid Y
- NPYr
- = Rezeptor von NPY
(kann als NPYR bezeichnet sein)
- NPY Yn
- = Yn Rezeptor-Subtyp
von NPY (z.B. NPY Y1) (z.B. NPYR1)
- I:NPY
- = Inhibitor von NPY
- I:NPY Yn
- = Inhibitor von NPY
Yn
- kDa
- = Kilodalton
- bp
- = Basenpaare
- kb
- = Kilobasenpaare
-
In
der wissenschaftlichen Literatur bis zu 1993 sind Beispiele für die Bestimmung
der oralen Bioverfügbarkeit
bei wachen Hunden aus den nachfolgenden Artikeln verfügbar:
- 1. Bristol Myers Squibb für das orale Antibiotikum Cefprozil:
Absolute
Bioavailability of Cefprozil after Oral Administration in Beagles
(Barbhaiya et al., 1992, Antimicrobial Agents and Chemotherapy,
vol 36, pp 687-689)
- 2. Carlo Erba für
das Zytostatikum Iododoxorubicin:
Pharmacokinetics of Iododoxorubicin
in the Rat, Dog, and Monkey (Edwards et al., 1991, Drug Metabolism
and Disposition, vol 19, pp 938–945)
- 3. Eli Lilly für
den experimentellen ZNS-antiischämischen
Wirkstoff LY256548:
Pharmacokinetics of a Novel Butylated Hydroxytoluene-Thiazolidinone CNS
Antiischemic Agent LY256548 in Rats, Mice, Dogs, and Monkeys (Ruterbories
and Lindstrom, 1990, Drug Metabolism and Disposition, vol 18, pp
674–679)
- 4. Wellcome für
den Folatantagonisten Piritrexim:
The Disposition and Metabolism
of [14C]Piritrexim in Dogs after Intravenous
and Oral Administration (Woolley et al., 1991, Drug Metabolism and
Disposition, vol 19, pp 1139–1146)
- 5. ICI für
das Antiandrogen Casodex:
The Pharmacokinetics of Casodex in
Laboratory Animals (Cockshott et al., 1991, Xenobiotica, vol 21,
pp 1347-1355)
- 6. Glaxo für
den Calciumantagonisten Lacidipin:
Absorption, Distribution
and Excretion of Lacidipine, a Dihydropyridine Calcium Antagonist,
in Rat and Dog (Pellegatti et al., 1990, Xenobiotica, 1990, vol
20, pp 765–777)
- 7. Fujisawa für
den Aldosereduktase-Inhibitor Zenarestat:
Absorption, Distribution
and Excretion of Zenarestat, a New Aldose Reductase Inhibitor, in
Rats and Dogs (Tanaka et al., 1992, Xenobiotica, vol 22, pp 57–64)
-
In
den obigen Beispielen wird für
die Bestimmung der oralen Bioverfügbarkeit auch die Verwendung von
wachen Mäusen,
Ratten und Affen zitiert.
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-






















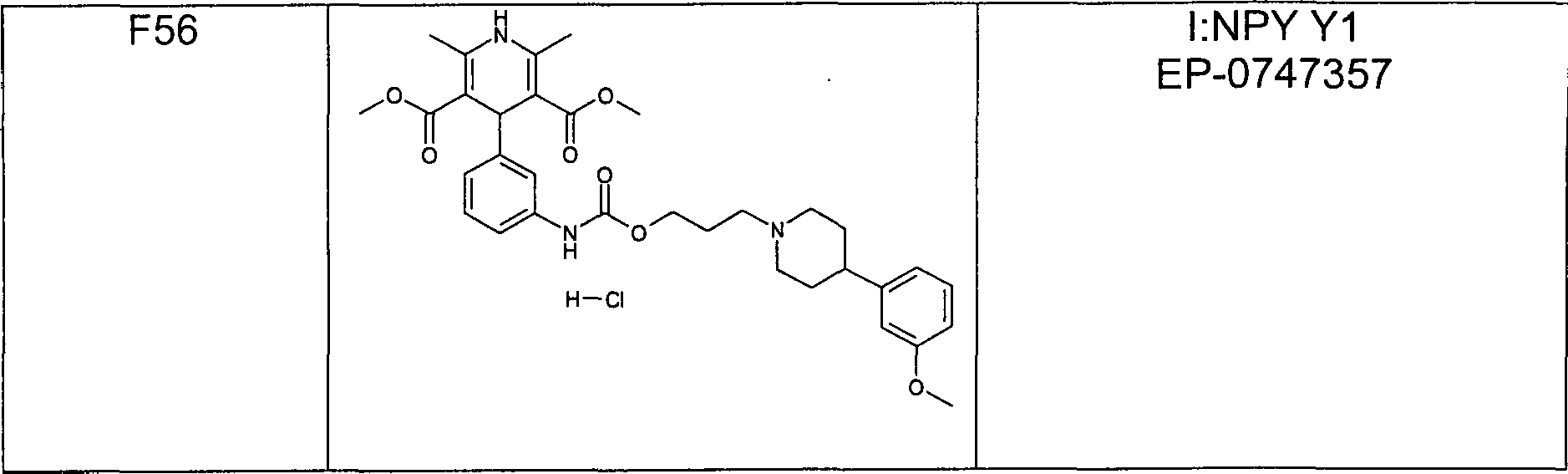





















 wurde aus dem Säurechlorid aus Vorschrift 11 (11/3) und dem geeigneten Amin nach einem ähnlichen Verfahren wie dem in Vorschrift 12 (12/52) beschriebenen hergestellt.
wurde aus dem Säurechlorid aus Vorschrift 11 (11/3) und dem geeigneten Amin nach einem ähnlichen Verfahren wie dem in Vorschrift 12 (12/52) beschriebenen hergestellt.


 wurde aus dem Säurechlorid aus Vorschrift 11 (11/3) und dem geeigneten Amin nach einem ähnlichen Verfahren wie dem in Vorschrift 12 (12/52) beschriebenen hergestellt.
wurde aus dem Säurechlorid aus Vorschrift 11 (11/3) und dem geeigneten Amin nach einem ähnlichen Verfahren wie dem in Vorschrift 12 (12/52) beschriebenen hergestellt.

 wurde aus der Säure von Vorschrift 16 (16/1) und der geeigneten Aminverbindung nach einem ähnlichen Verfahren wie dem in Vorschrift 17 (17/33) beschriebenen hergestellt.
wurde aus der Säure von Vorschrift 16 (16/1) und der geeigneten Aminverbindung nach einem ähnlichen Verfahren wie dem in Vorschrift 17 (17/33) beschriebenen hergestellt.


























