-
Die
vorliegende Erfindung betrifft Imidazochinolinverbindungen mit Ether-
und Harnstoffunktionalität
in der 1-Stellung und Arzneimittel, die derartige Verbindungen enthalten.
Ein weiterer Aspekt der vorliegenden Erfindung betrifft die Verwendung
dieser Verbindungen zur Herstellung von Medikamenten zur Induktion
der Cytokin-Biosynthese in Tieren und zur Behandlung von Viruserkrankungen
und neoplastischen Erkrankungen. Die Erfindung stellt auch neue
Verbindungen bereit, die als Zwischenprodukte bei der Synthese der
obigen Verbindungen verwendet werden können.
-
Im
ersten zuverlässigen
Bericht über
das 1H-Imidazo[4,5-c]chinolin-Ringsystem
beschreiben Backman et al., J. Org. Chem. 15, 1278–1284 (1950),
die Synthese von 1-(6-Methoxy-8-chinolinyl)-2-methyl-1H-imidazo[4,5-c]chinolin
für eine
mögliche
Verwendung als Antimalariamittel. Danach wurde über Synthesen verschiedener
substituierter 1H-Imidazo[4,5-c]chinoline berichtet. Beispielsweise
synthetisierten Jain et al., J. Med. Chem. 11, S. 87–92 (1968),
die Verbindung 1-[2-(4-Piperidyl)ethyl]-1H-imidazo[4,5-c]chinolin
als mögliches
Antikonvulsivum und Herz-Kreislauf-Mittel. Außerdem berichteten Baranov
et al., Chem. Abs. 85, 94362 (1976), über einige 2-Oxoimidazo[4,5-c]chinoline
und Berenyi et al., J. Heterocyclic Chem. 18, 1537–1540 (1981), über bestimmte
2-Oxoimidazo[4,5-c]chinoline.
-
Später stellte
sich heraus, daß bestimmte
1H-Imidazo[4,5-c]chinolin-4-amine und 1- und 2-substituierte Derivate
davon zur Verwendung als Virustatika, Bronchodilatatoren und Immunmodulatoren
geeignet sind. Diese werden u.a. in den US-Patentschriften 4,689,338,
4,698,348, 4,929,624, 5,037,986, 5,268,376, 5,346,905 und 5,389,640,
auf die hiermit ausdrücklich
Bezug genommen wird, beschrieben.
-
Es
besteht nach wie vor Interesse am Imidazochinolin-Ringsystem.
-
In
der WO 92/15582 werden bestimmte 1-substituierte, 2-substituierte
1H-Imidazo[4,5-c]chinolin-4-amine beschrieben. In der WO 95/02598
werden bestimmte 6,7-propylen-,
-butylen- und -pentylenverbrückte
Imidazopyridin-4-amine beschrieben.
-
Bestimmte
1H-Imidazo[4,5-c]naphthyridin-4-amine, 1H-Imidazo[4,5-c]pyridin-4-amine und 1H-Imidazo[4,5-c]-chinolin-4-amine
mit einem etherhaltigen Substituenten in der 1-Stellung sind bekannt.
Diese werden in den US-Patentschriften
5,268,376, 5,389,640, 5,494,916 und WO 99/29693 beschrieben.
-
Trotz
dieser Versuche zur Auffindung von Verbindungen, die zur Verwendung
als die Immunantwort modifizierende Mittel geeignet sind, besteht
nach wie vor Bedarf an Verbindungen, die zur Modulierung der Immunantwort
durch Induktion der Cytokin-Biosynthese oder andere Mechanismen
befähigt
sind.
-
Es
wurde nun eine neue Klasse von Verbindungen gefunden, die zur Verwendung
bei der Induktion der Cytokin-Biosynthese in Tieren geeignet sind.
Gegenstand der vorliegenden Erfindung sind demgemäß Imidazochinolin-4-amin-
und Tetrahydroimidazochinolin-4-amin-Verbindungen mit einem ether-
und harnstoffhaltigen Substituenten in der 1-Stellung. Die Verbindungen
sind durch die Formeln (I) und (II), die nachstehend ausführlicher
definiert werden, definiert. Diese Verbindungen teilen die allgemeine
Strukturformel:
worin X, R
1,
R
2 und R die hier für jede Klasse von Verbindungen
mit den Formeln (I) und (II) angegebene Bedeutung besitzen.
-
Die
Verbindungen der Formeln (I) und (II) eignen sich zur Verwendung
als die Immunantwort modifizierende Mittel, da sie bei Verabreichung
an Tiere zur Induktion der Cytokin-Biosynthese und anderweitigen Modulierung
der Immunantwort befähigt
sind. Daher sind die Verbindungen zur Behandlung von verschiedenen
Leiden, z.B. Viruserkrankungen und Tumoren, die auf derartige Änderungen
der Immunantwort ansprechen, geeignet.
-
Gegenstand
der Erfindung sind ferner Arzneimittel, die die die Immunantwort
modifizierenden Verbindungen enthalten, und die Verwendung einer
Verbindung der Formel (I) oder (II) zur Herstellung von Medikamenten
zur Induktion der Cytokin-Biosynthese in einem Tier, zur Behandlung
einer Virusinfektion und/oder zur Behandlung einer neoplastischen
Erkrankung.
-
Darüber hinaus
werden Verfahren zur Synthese der erfindungsgemäßen Verbindungen beschrieben. Gegenstand
der Erfindung sind auch neue Zwischenprodukte, die bei der Synthese
dieser Verbindungen verwendet werden können.
-
Wie
oben erwähnt,
wurden bestimmte Verbindungen gefunden, die in Tieren die Cytokin-Biosynthese induzieren
und die Immunantwort modifizieren. Derartige Verbindungen werden
durch die nachstehend gezeigten Formeln (I) und (II) wiedergegeben.
-
Erfindungsgemäße Imidazochinolinverbindungen
mit Ether- und Harnstoffunktionalität in der
1-Stellung werden durch Formel (I) wiedergegeben:
wobei:
X für -CHR
5-, -CHR
5-Alkylen-
oder -CHR
5-Alkenylen- steht;
R
1 aus der Gruppe bestehend aus
-R
4-NR
8– CR
3-NR
5-Z-R
6-Alkyl;
-R
4-NR
8-CR
3-NR
5-Z-R
6-Alkenyl;
-R
4-NR
8-CR
3-NR
5-Z-R
6-Aryl;
-R
4-NR
8-CR
3-NR
5-Z-R
6-Heteroaryl;
-R
4-NR
8-CR
3-NR
5-Z-R
6-Heterocyclyl;
-R
4-NR
8-CR
3-NR
5R
7;
-R
4-NR
8-CR
3-NR
9-Z-R
6-Alkyl;
-R
4-NR
8-CR
3-NR
9-Z-R
6-Alkenyl;
-R
4-NR
8-CR
3-NR
9-Z-R
6-Aryl;
-R
4-NR
8-CR
3-NR
9-Z-R
6-Heteroaryl und
-R
4-NR
8-CR
3-NR
9-Z-R
6-Heterocyclyl;
ausgewählt ist;
R
2 aus der Gruppe bestehend aus
-Wasserstoff;
-Alkyl;
-Alkenyl;
-Aryl;
-Heteroaryl;
-Heterocyclyl;
-Alkylen-Y-alkyl;
-Alkylen-Y-alkenyl;
-Alkylen-Y-aryl;
und
-Alkyl oder -Alkenyl, das durch einen oder mehrere Substituenten
aus der Gruppe bestehend aus
-OH;
-Halogen;
-N(R
5)
2;
-CO-N(R
5)
2;
-CO-C
1-10-Alkyl;
-CO-O-C
1-10-Alkyl;
-N
3;
-Aryl;
-Heteroaryl;
-Heterocyclyl;
-CO-Aryl;
und
-CO-Heteroaryl
substituiert ist,
ausgewählt ist;
R
3 jeweils für =O oder =S steht;
R
4 jeweils unabhängig voneinander für Alkylen
oder Alkenylen, das durch eine oder mehrere Gruppen -O- unterbrochen
sein kann, steht;
R
5 jeweils unabhängig voneinander
für H oder
C
1-10-Alkyl
steht;
R
6 für eine Bindung, Alkylen oder
Alkenylen, das durch eine oder mehrere Gruppen -O- unterbrochen
sein kann, steht;
R
7 für H oder
C
1-10-Alkyl, das durch ein Heteroatom unterbrochen
sein kann, steht, oder R
7 mit R
5 verbunden sein
kann, um einen Ring zu bilden;
R
8 für H, C
1-10-Alkyl oder Arylalkyl steht; oder R
4 und R
8 miteinander
verbunden sein können,
um einen Ring zu bilden;
R
9 für C
1-10-Alkyl steht, das mit R
8 verbunden
sein kann, um einen Ring zu bilden;
Y jeweils unabhängig voneinander
für -O-
oder -S(O)
0-2- steht;
Z für eine Bindung,
-CO- oder -SO
2- steht;
n für 0 bis
4 steht; und
jedes vorhandene R unabhängig voneinander aus der Gruppe
bestehend aus C
1-10-Alkyl, C
1-10-Alkoxy,
Hydroxy, Halogen und Trifluormethyl ausgewählt ist;
oder ein pharmazeutisch
verträgliches
Salz davon.
-
Die
Erfindung umfaßt
auch Tetrahydroimidazochinolinverbindungen mit einem ether- und
harnstoffhaltigen Substituenten in der 1-Stellung. Derartige Tetrahydroimidazochinolinverbindungen
werden durch Formel (II) wiedergegeben:
wobei:
X für -OHR
5-, -OHR
5-Alkylen-
oder -CHR
5-Alkenylen- steht;
R
1 aus der Gruppe bestehend aus
-R
4-NR
8-CR
3-NR
5-Z-R
6-Alkyl;
-R
4-NR
8-CR
3-NR
5-Z-R
6-Alkenyl;
-R
4-NR
8-CR
3-NR
5-Z-R
6-Aryl;
-R
4-NR
8-CR
3-NR
5-Z-R
6-Heteroaryl;
-R
4-NR
8-CR
3-NR
5-Z-R
6-Heterocyclyl;
-R
4-NR
8-CR
3-NR
5R
7;
-R
4-NR
8-CR
3-NR
9-Z-R
6-Alkyl;
-R
4-NR
8-CR
3-NR
9-Z-R
6-Alkenyl;
-R
9-NR
8-CR
3-NR
9-Z-R
6-Aryl;
-R
4-NR
8-CR
3-NR
9-Z-R
6-Heteroaryl
und
-R
4-NR
8-CR
3-NR
9-Z-R
6-Heterocyclyl;
ausgewählt ist;
R
2 aus der Gruppe bestehend aus
-Wasserstoff;
-Alkyl;
-Alkenyl;
-Aryl;
-Heteroaryl;
-Heterocyclyl;
-Alkylen-Y-alkyl;
-Alkylen-Y-alkenyl;
-Alkylen-Y-aryl
und
-Alkyl oder -Alkenyl, das durch einen oder mehrere Substituenten
aus der Gruppe bestehend aus
-OH;
-Halogen;
-N(R
5)
2; -CO-N(R
5)
2;
-CO-C
1-10-Alkyl;
-CO-O-C
1-10-Alkyl;
-Na;
-Aryl;
-Heteroaryl;
-Heterocyclyl;
-CO-Aryl;
und
-CO-Heteroaryl
substituiert ist,
ausgewählt ist;
R
3 jeweils für =O oder =S steht;
R
4 jeweils unabhängig voneinander für Alkylen
oder Alkenylen, das durch eine oder mehrere Gruppen -O- unterbrochen
sein kann, steht;
R
5 jeweils unabhängig voneinander
für H oder
C
1-10-Alkyl
steht;
R
6 für eine Bindung, Alkylen oder
Alkenylen, das durch eine oder mehrere Gruppen -O- unterbrochen
sein kann, steht;
R
7 für H oder
C
1-10-Alkyl, das durch ein Heteroatom unterbrochen
sein kann, steht, oder R
7 mit R
5 verbunden sein
kann, um einen Ring zu bilden;
R
8 für H, C
1-10-Alkyl oder Arylalkyl steht; oder R
4 und R
8 miteinander
verbunden sein können,
um einen Ring zu bilden;
R
9 für C
1-10-Alkyl steht, das mit R
8 verbunden
sein kann, um einen Ring zu bilden;
Y jeweils unabhängig voneinander
für -O-
oder -S(O)
0-2- steht;
Z für eine Bindung,
-CO- oder -SO
2- steht;
n für 0 bis
4 steht; und
jedes vorhandene R unabhängig voneinander aus der Gruppe
bestehend aus C
1-10-Alkyl, C
1-10-Alkoxy,
Hydroxy, Halogen und Trifluormethyl ausgewählt ist;
oder ein pharmazeutisch
verträgliches
Salz davon.
-
Herstellung der Verbindungen
-
Erfindungsgemäße Verbindungen
können
gemäß Reaktionsschema
I hergestellt werden, wobei R, R2, R3, R4, R5,
R8, X und n die oben angegebene Bedeutung
besitzen, BOC tert-Butoxycarbonyl ist und R11-Z-R6-Alkyl, -Z-R6-Alkenyl,
-Z-R6-Aryl, -Z-R6-Heteroaryl,
-Z-R6-Heterocyclyl ist oder R11 R7 ist, wobei R6,
R7 und Z die oben angegebene Bedeutung besitzen.
-
In
Schritt (1) von Reaktionsschema I wird die Aminogruppe eines Aminoalkohols
der Formel X mit einer tert-Butoxycarbonylgruppe
geschützt.
Eine Lösung
des Aminoalkohols in Tetrahydrofuran wird in Gegenwart einer Base,
wie Natriumhydroxid, mit Di-tert-butyldicarbonat behandelt. Zahlreiche
Aminoalkohole der Formel X sind im Handel erhältlich; andere sind über bekannte
Synthesemethoden zugänglich.
-
In
Schritt (2) von Reaktionsschema I wird ein geschützter Aminoalkohol der Formel
XI in ein Iodid der Formel XII ungewandelt. Eine Lösung von
Triphenylphosphin und Imidazol in Dichlormethan wird mit Iod versetzt,
wonach eine Lösung
eines geschützten
Aminoalkohols der Formel XI in Dichlormethan zugegeben wird. Die
Umsetzung wird bei Umgebungstemperatur durchgeführt.
-
In
Schritt (3) von Reaktionsschema I wird ein 1H-Imidazo[4,5-c]chinolin-1-ylalkohol der
Formel XIII mit einem Iodid der Formel XII zu einem 1H-Imidazo[4,5-c]chinolin-1-ylether
der Formel XIV alkyliert. Der Alkohol der Formel XIII wird in einem
geeigneten Lösungsmittel,
wie N,N-Dimethylformamid, mit Natriumhydrid zu einem Alkoxid umgesetzt.
Das Iodid wird bei Umgebungstemperatur zu der Alkoxidlösung gegeben.
Nach beendeter Zugabe wird der Ansatz bei erhöhter Temperatur (~ 100°C) gerührt. Zahlreiche
Verbindungen der Formel XIII sind bekannt, siehe beispielsweise
Gerster, US-PS 4 689 338; andere sind über bekannte Syntheserouten leicht
zugänglich,
siehe beispielsweise Gerster et al., US-PS 5 605 899, und Gerster,
US-PS 5 175 296.
-
In
Schritt (4) von Reaktionsschema I wird ein 1H-Imidazo[4,5-c]chinolin-1-ylether der
Formel XIV mit einem herkömmlichen
Oxidationsmittel, das zur Bildung von N-Oxiden befähigt ist,
zu einem 1H-Imidazo[4,5-c]chinolin-5N-oxid
der Formel XV oxidiert. Vorzugsweise wird eine Lösung einer Verbindung der Formel XIV
in Chloroform bei Umgebungstemperatur mit 3-Chlorperoxybenzoesäure oxidiert.
-
In
Schritt (5) von Reaktionsschema I wird ein 1H-Imidazo[4,5-c]chinolin-5N-oxid der Formel
XV zu einem 1H-Imidazo[4,5-c]chinolin-4-amin der Formel XVI aminiert.
Hierbei wird (i) eine Verbindung der Formel XV mit einem Acylierungsmittel
umgesetzt und dann (ii) das Produkt mit einem Aminierungsmittel
umgesetzt. In Teil (i) von Schritt (5) wird ein N-Oxid der Formel
XV mit einem Acylierungsmittel umgesetzt. Geeignete Acylierungsmittel
sind u.a. Alkyl- oder Arylsulfonylchloride (z.B. Benzolsulfonylchlorid,
Methansulfonylchlorid, p-Toluolsulfonylchlorid).
Bevorzugt sind Arylsulfonyl chloride. Ganz besonders bevorzugt ist
para-Toluol-sulfonylchlorid.
In Teil (ii) von Schritt (5) wird das Produkt aus Teil (i) mit einem Überschuß eines
Aminierungsmittels umgesetzt. Geeignete Aminierungsmittel sind u.a.
Ammoniak (z.B. in Form von Ammoniumhydroxid) und Ammoniumsalze (z.B.
Ammoniumcarbonat, Ammoniumhydrogencarbonat, Ammoniumphosphat). Bevorzugt
ist Ammoniumhydroxid. Bei der Umsetzung geht man vorzugsweise so
vor, daß man
das N-Oxid der Formel XV in einem inerten Lösungsmittel, wie Dichlormethan
oder 1,2-Dichlorethan, löst,
gegebenenfalls unter Erhitzen, die Lösung mit dem Aminierungsmittel
versetzt und dann langsam das Acylierungsmittel zugibt. Gegebenenfalls
kann die Umsetzung in einem geschlossenen Druckbehälter bei
erhöhter
Temperatur (85–100°) durchgeführt werden.
-
In
Schritt (6) von Reaktionsschema I wird die Schutzgruppe durch Hydrolyse
unter sauren Bedingungen abgespalten, was ein 1H-Imidazo[4,5-c]chinolin-4-amin
der Formel XVII ergibt. Vorzugsweise wird die Verbindung der Formel
XVI bei Umgebungstemperatur oder unter gelindem Erwärmen in
Salzsäure/Ethanol
behandelt.
-
In
Schritt (7) von Reaktionsschema I wird ein 1H-Imidazo[4,5-c]chinolin-4-amin der Formel
XVII nach herkömmlichen
Synthesemethoden in einen Harnstoff oder Thioharnstoff der Formel
XVIII umgewandelt. So kann man beispielsweise eine Verbindung der
Formel XVII mit einem Isocyanat der Formel R12-N=C=O,
wobei R12 -R6-Alkyl,
-R6-Alkenyl, -R6-Aryl,
-R6-Heteroaryl oder -R6-Heterocyclyl
ist, umsetzen. Bei der Umsetzung kann man so vorgehen, daß man eine
Lösung
des Isocyanats in einem geeigneten Lösungsmittel, wie Dichlormethan
oder 1-Methyl-2-pyrrolidinon, bei Umgebungstemperatur zu einer Lösung einer
Verbindung der Formel XVII gibt. Alternativ dazu kann man eine Verbindung
der Formel XVII mit einem Thioisocyanat der Formel R12-N=C=S,
einem Acylisocyanat der Formel R12-C(O)-N=C=O,
einem Sulfonylisocyanat der Formel -R12-S(O2)-N=C=O oder einem Carbamoylchlorid der
Formel R13-N-C(O)Cl, wobei R13 R12 oder R7 ist, umsetzen.
Das Produkt oder ein pharmazeutisch verträgliches Salz davon kann nach
herkömmlichen
Methoden isoliert werden.
-
-
Erfindungsgemäße Verbindungen
können
gemäß Reaktionsschema
II hergestellt werden, wobei R, R2, R3, R4, R5,
R8, R11, X und n
die oben angegebene Bedeutung besitzen und BOC tert-Butoxycarbonyl
ist.
-
In
Schritt (1) von Reaktionsschema II wird die Aminogruppe eines Aminoalkohols
der Formel XIX mit einer tert-Butoxycarbonylgruppe geschützt. Eine
Lösung
des Aminoalkohols in Tetrahydrofuran wird in Gegenwart einer Base,
wie Natriumhydroxid, mit Di-tert-butyldicarbonat behandelt. Zahlreiche
Aminoalkohole der Formel XIX sind im Handel erhältlich; andere sind nach bekannten
Synthesemethoden zugänglich.
-
In
Schritt (2) von Reaktionsschema II wird ein geschützter Aminoalkohol
der Formel XX in ein Methansulfonat der Formel XXI umgewandelt.
Eine Lösung
einer Verbindung der Formel XX in einem geeigneten Lösungsmittel,
wie Dichlormethan, wird in Gegenwart einer Base, wie Triethylamin,
mit Methansulfonylchlorid behandelt. Die Umsetzung kann bei verminderter
Temperatur (0°C)
durchgeführt
werden.
-
In
Schritt (3a) von Reaktionsschema II wird ein Methansulfonat der
Formel XXI in ein Azid der Formel XXII umgewandelt. Eine Lösung einer
Verbindung der Formel XXI in einem geeigneten Lösungsmittel, wie N,N-Dimethylformamid,
wird mit Natriumazid versetzt. Die Umsetzung kann bei erhöhter Temperatur (80–100°C) durchgeführt werden.
-
In
Schritt (3b) von Reaktionsschema II wird eine Verbindung der Formel
XXII mit einem Halogenid der Formel Hal-R8 zu
einer Verbindung der Formel XXIII alkyliert. In Verbindungen, wobei
R8 Wasserstoff ist, wird dieser Schritt
weggelassen. Die Verbindung der Formel XXII wird in einem geeigneten
Lösungsmittel,
wie N,N-Dimethylformamid oder Tetrahydrofuran, mit Natriumhydrid
zu dem Anion umgesetzt und dann mit dem Halogenid vereinigt. Die
Umsetzung kann bei Umgebungstemperatur durchgeführt werden.
-
In
Schritt (4) von Reaktionsschema II wird ein Azid der Formel XXII
oder XXIII zu einem Amin der Formel XXIV reduziert. Vorzugsweise
wird die Reduktion mit einem herkömmlichen heterogenen Hydrierkatalysator,
wie Palladium auf Kohle, durchgeführt. Die Umsetzung kann zweckmäßigerweise
auf einer Parr-Apparatur in einem geeigneten Lösungsmittel, wie Methanol oder
Isopropanol, durchgeführt
werden.
-
In
Schritt (5) von Reaktionsschema II wird ein 4-Chlor-3-nitrochinolin der
Formel XXV mit einem Amin der Formel XXIV zu einem 3-Nitrochinolin
der Formel XXVI umgesetzt. Bei der Umsetzung kann man so vorgehen,
daß man
eine Lösung
einer Verbindung der Formel XXV in einem geeigneten Lösungsmittel,
wie Dichlormethan, in Gegenwart einer Base, wie Triethylamin, mit
einem Amin der Formel XXIV versetzt. Zahlreiche Chinoline der Formel
XXV sind bekannt oder nach bekannten Synthesemethoden zugänglich,
siehe beispielsweise Gerster, US-PS
4,689,338 und dort angegebene Literaturstellen.
-
In
Schritt (6) von Reaktionsschema II wird ein 3-Nitrochinolin der
Formel XXVI zu einem 3-Aminochinolin der Formel XXVII reduziert.
Vorzugsweise wird die Reduktion mit einem herkömmlichen heterogenen Hydrierkatalysator,
wie Platin auf Kohle, durchgeführt.
Die Umsetzung kann zweckmäßigerweise
auf einer Parr-Apparatur in einem geeigneten Lösungsmittel, wie Toluol, durchgeführt werden.
-
In
Schritt (7) von Reaktionsschema II wird eine Verbindung der Formel
XXVII mit einer Carbonsäure oder
einem Äquivalent
davon zu einem 1H-Imidazo[4,5-c]-chinolin
der Formel XIV umgesetzt. Geeignete Äquivalente einer Carbonsäure sind
u.a. Orthoester und Alkansäure-1,1-dialkoxyalkylester.
Die Carbonsäure
oder das Äquivalent
davon wird so gewählt,
daß sie
den gewünschten
Substituenten R2 in einer Verbindung der
Formel XIV liefert. So liefert beispielsweise Orthoameisensäuretriethylester
ein Verbindung, in der R2 Wasserstoff ist,
und Orthovaleriansäuretriethylester eine
Verbindung, in der R2 Butyl ist. Die Umsetzung
kann ohne Lösungsmittel
oder in einem inerten Lösungsmittel,
wie Toluol, durchgeführt
werden. Bei der Umsetzung wird so stark erhitzt, daß jeglicher
als Reaktionsnebenprodukt anfallende Alkohol oder jegliches als
Reaktionsnebenprodukt anfallendes Wasser ausgetrieben wird. Gegebenenfalls
kann man eine katalytisch wirksame Menge von Pyridinhydrochlorid
mitverwenden.
-
Alternativ
dazu kann man Schritt (7) durchführen,
indem man (i) eine Verbindung der Formel XXVII mit einem Acylhalogenid
der Formel R2C(O)Cl umsetzt und dann (ii)
cyclisiert. In Teil (i) wird das Acylhalogenid zu einer Lösung einer
Verbindung der Formel XXVII in einem inerten Lösungsmittel, wie Acetonitril
oder Dichlormethan, gegeben. Die Umsetzung kann bei Umgebungstemperatur
oder verminderter Temperatur durchgeführt werden. In Teil (ii) wird
das Produkt aus Teil (i) in einem alkoholischen Lösungsmittel
in Gegenwart einer Base erhitzt. Vorzugsweise wird das Produkt aus
Teil (i) in Gegenwart eines Überschusses
von Triethylamin in Ethanol refluxiert oder mit methanolischem Ammoniak
erhitzt.
-
Die
Schritte (8), (9), (10) und (11) werden auf die gleiche Art und
weise wie die Schritte (4), (5), (6) und (7) von Reaktionsschema
I durchgeführt.
-
-
Erfindungsgemäße Verbindungen
können
gemäß Reaktionsschema
III hergestellt werden, wobei R, R2, R3, R4, R5,
R8, R11, X und n
die oben angegebene Bedeutung besitzen.
-
In
Schritt (1) von Reaktionsschema III wird ein 1H-Imidazo[4,5-c]chinolin-4-amin
der Formel XVII zu einem 6,7,8,9-Tetrahydro-1H-imidazo[4,5-c]chinolin-4-amin
der Formel XXVIII reduziert. Bei der Reduktion geht man vorzugsweise
so vor, daß man
eine Verbindung der Formel XVII in Trifluoressigsäure suspendiert oder
löst, eine
katalytisch wirksame Menge von Platin(IV)-oxid zugibt und dann hydriert.
Die Umsetzung kann zweckmäßigerweise
in einer Parr-Apparatur durchgeführt
werden.
-
Der
Schritt (2) wird auf die gleiche Art und weise wie Schritt (7) von
Reaktionsschema I durchgeführt, was
ein 6,7,8,9-Tetrahydro-1H-imidazo[4,5-c]chinolin-4-amin der Formel
XXIX ergibt. Das Produkt oder ein pharmazeutisch verträgliches
Salz davon kann nach herkömmlichen
Methoden isoliert werden.
-
-
Erfindungsgemäße Verbindungen
können
auch gemäß Reaktionsschema
IV hergestellt werden, wobei R, R1, R2, X und n die oben angegebene Bedeutung
besitzen.
-
In
Schritt (1) von Reaktionsschema IV wird ein 4-Chlor-3-nitrochinolin der
Formel XXV mit einem Amin der Formel R1-O-X-NH2 zu einem 3-Nitrochinolin-4-amin der Formel
XXX umgesetzt. Die Umsetzung kann durch Zugabe des Amins zu einer
Lösung
einer Verbindung der Formel XXV in einem geeigneten Lösungsmittel,
wie Chloroform oder Dichlormethan, und gegebenenfalls Erhitzen durchgeführt werden.
Zahlreiche Chinoline der Formel XXV sind bekannt, siehe beispielsweise
Gerster, US-PS 4,689,338 und dort angegebene Literaturstellen.
-
In
Schritt (2) von Reaktionsschema IV wird ein 3-Nitrochinolin-4-amin
der Formel XXX nach der Methode von Schritt (6) von Reaktionsschema
II zu einem Chinolin-3,4-diamin
der Formel XXXI reduziert.
-
In
Schritt (3) von Reaktionsschema IV wird ein Chinolin-3,4-diamin
der Formel XXXI nach der Methode von Schritt (7) von Reaktionsschema
II zu einem 1H-Imidazo[4,5-c]chinolin der Formel XXXII cyclisiert.
-
In
Schritt (4) von Reaktionsschema IV wird ein 1H-Imidazo[4,5-c]chinolin
der Formel XXXII nach der Methode von Schritt (4) von Reaktionsschema
I zu einem 1H-Imidazo[4,5-c]chinolin-5N-oxid der Formel XXXIII oxidiert.
-
In
Schritt (5) von Reaktionsschema IV wird ein 1H-Imidazo[4,5-c]chinolin-5N-oxid
der Formel XXXIII nach der Methode von Schritt (5) von Reaktionsschema
I zu einem 1H-Imidazo[4,5-c]chinolin-4-amin der Formel I aminiert.
Das Produkt oder ein pharmazeutisch verträgliches Salz davon kann nach
herkömmlichen
Methoden isoliert werden.
-
Reaktionsschema
IV
-
-
Erfindungsgemäße Verbindungen
können
gemäß Reaktionsschema
V hergestellt werden, wobei R, R2, R3, R4, R5,
R8, R11, X und n
die oben angegebene Bedeutung besitzen.
-
In
Schritt (1) von Reaktionsschema V wird die BOC-Gruppe nach der Methode von Schritt
(6) von Reaktionsschema I aus einer Verbindung der Formel XIV abgespalten,
was ein 1H-Imidazo[4,5-c]chinolin der Formel XXXIV ergibt.
-
In
Schritt (2) von Reaktionsschema V wird ein 1H-Imidazo[4,5-c]chinolin
der Formel XXXIV nach der Methode von Schritt (7) von Reaktionsschema
I in einen Harnstoff oder Thioharnstoff der Formel XXXV umgewandelt.
-
In
Schritt (3) von Reaktionsschema V wird ein 1H-Imidazo[4,5-c]chinolin
der Formel XXXV nach der Methode von Schritt (4) von Reaktionsschema
I zu einem 1H-Imidazo(4,5-c]chinolin-5N-oxid der Formel XXXVI oxidiert.
-
In
Schritt (4) von Reaktionsschema V wird ein 1H-Imidazo[4,5-c]chinolin-5N-oxid
der Formel XXXVI nach der Methode von Schritt (5) von Reaktionsschema
I zu einem 1H-Imidazo[4,5-c]chinolin-4-amin der Formel XVIII aminiert.
Das Produkt oder ein pharmazeutisch verträgliches Salz davon kann nach
herkömmlichen Methoden
isoliert werden.
-
-
Gegenstand
der Erfindung sind auch neue Verbindungen, die als Zwischenprodukte
bei der Synthese der Verbindungen der Formeln (I) und (II) verwendet
werden können.
Diese Zwischenverbindungen haben die Strukturformeln (III) und (IV),
die nachstehend ausführlicher
beschrieben werden.
-
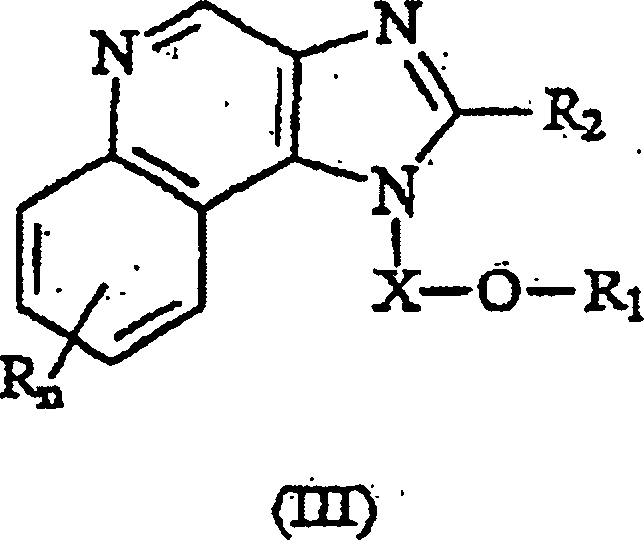
Eine
Klasse von Zwischenverbindungen hat die Formel III):
wobei:
X für -CHR
5-, -CHR
5-Alkylen-
oder -CHR
5-Alkenylen- steht;
R
1 aus der Gruppe bestehend aus
-R
4-NR
8-CR
3-NR
5-Z-R
6-Alkyl;
-R
4-NR
8-CR
3-NR
5-Z-R
6-Alkenyl;
-R
4-NR
8-CR
3-NR
5-Z-R
6-Aryl;
-R
4-NR
8-CR
3-NR
5-Z-R
6-Heteroaryl;
-R
4-NR
8-CR
3-NR
5-Z-R
6-Heterocyclyl;
und
-R
4-NR
8-CR
3-NR
5R
7;
-R
4-NR
8-CR
3-NR
9-Z-R
6-Alkyl;
-R
9-NR
8-CR
3-NR
9-Z-R
6-Alkenyl;
-R
4-NR
8-CR
3-NR
9-Z-R
6-Aryl;
-R
4-NR
8-CR
3-NR
9-Z-R
6-Heteroaryl;
und
-R
4-NR
8-CR
3-NR
9-Z-R
6-Heterocyclyl;
ausgewählt ist;
R
2 aus der Gruppe bestehend aus
-Wasserstoff;
-Alkyl;
-Alkenyl;
-Aryl;
-Heteroaryl;
-Heterocyclyl;
-Alkylen-Y-alkyl;
-Alkylen-Y-alkenyl;
-Alkylen-Y-aryl;
und
-Alkyl oder -Alkenyl, das durch einen oder mehrere Substituenten
aus der Gruppe bestehend aus
-OH;
-Halogen;
-N(R
5)
2;
-CO-N(R
5)
2;
-CO-C
1-10-Alkyl;
-CO-O-C
1-10-Alkyl;
-N
5;
-Aryl;
-Heteroaryl;
-Heterocyclyl;
-CO-Aryl
und
-CO-Heteroaryl
substituiert ist,
ausgewählt ist;
R
3 jeweils für =O oder =S steht;
R
4 jeweils unabhängig voneinander für Alkylen
oder Alkenylen, das durch eine oder mehrere Gruppen -O- unterbrochen
sein kann, steht;
R
5 jeweils unabhängig voneinander
für H oder
C
1-10-Alkyl
steht;
R
6 für eine Bindung, Alkyl oder
Alkenyl, das durch eine oder mehrere Gruppen -O- unterbrochen sein
kann, steht;
R
7 für H oder C
1-10-Alkyl,
das durch ein Heteroatom unterbrochen sein kann, steht, oder R
7 mit R
5 verbunden sein
kann, um einen Ring zu bilden;
R
8 für H, C
1-10-Alkyl oder Arylalkyl steht; oder R
4 und R
8 miteinander
verbunden sein können,
um einen Ring zu bilden;
R
9 für C
1-10-Alkyl steht, das mit R
8 verbunden
sein kann, um einen Ring zu bilden;
Y jeweils unabhängig voneinander
für -O-
oder -S(O)
0-2- steht;
Z für eine Bindung,
-CO- oder -SO
2- steht;
n für 0 bis
4 steht und
jedes vorhandene R unabhängig voneinander aus der Gruppe
bestehend aus C
1-10-Alkyl, C
1-10-Alkoxy,
Hydroxy, Halogen und Trifluormethyl ausgewählt ist;
oder ein pharmazeutisch
verträgliches
Salz davon.
-
Eine
andere Klasse von Zwischenverbindungen sind die Imidazochinolin-N-oxid-Verbindungen
der Formel (IV):
wobei:
X für -OHR
5-, -CHR
5-Alkylen-
oder -CHR
5-Alkenylen- steht;
R
1 aus der Gruppe bestehend aus
-R
4-NR
8-CR
3-NR
5-Z-R
6-Alkyl;
-R
4-NR
8-CR
3-NR
5-Z-R
6-Alkenyl;
-R
4-NR
8-CR
3-NR
5-Z-R
6-Aryl;
-R
4-NR
8-CR
3-NR
5-Z-R
6-Heteroaryl;
-R
4-NR
8-CR
3-NR
5-Z-R
6-Heterocyclyl;
-R
4-NR
8-CR
3-NR
5R
7;
-R
4-NR
8-CR
3-NR
9-Z-R
6-Alkyl;
-R
4-NR
8-CR
3-NR
9-Z-R
6-Alkenyl;
-R
4-NR
8-CR
3-NR
9-Z-R
6-Aryl;
-R
4-NR
8-CR
3-NR
9-Z-R
6-Heteroaryl;
und
-R
4-NR
8-CR
3-NR
9-Z-R
6-Heterocyclyl;
ausgewählt ist;
Y
jeweils unabhängig
voneinander für
-O- oder -S(O)
0-2- steht;
Z für eine Bindung,
-CO- oder -SO
2- steht;
R
4 jeweils
unabhängig
voneinander für
Alkylen oder Alkenylen, das durch eine oder mehrere Gruppen -O-
unterbrochen sein kann, steht;
R
5 jeweils
unabhängig
voneinander für
H oder C
1-10-Alkyl steht;
R
6 für eine Bindung,
Alkylen oder Alkenylen, das durch eine oder mehrere Gruppen -O-
unterbrochen sein kann, steht;
R
7 für H oder
C
1-10-Alkyl, das durch ein Heteroatom unterbrochen
sein kann, steht, oder R
7 mit R
5 verbunden sein
kann, um einen Ring zu bilden;
R
8 für H, C
1-10-Alkyl oder Arylalkyl steht; oder R
4 und R
8 miteinander
verbunden sein können,
um einen Ring zu bilden;
R
9 für C
1-10-Alkyl steht, das mit R
8 verbunden
sein kann, um einen Ring zu bilden;
n für 0 bis 4 steht; und
jedes
vorhandene R unabhängig
voneinander aus der Gruppe bestehend aus C
1-10-Alkyl,
C
1-10-Alkoxy, Halogen und Trifluormethyl
ausgewählt
ist;
oder ein pharmazeutisch verträgliches Salz davon.
-
Im
Rahmen der vorliegenden Erfindung schließen die Begriffe "Alkyl" und "Alkenyl" und das Präfix "Alk-" sowohl geradkettige
als auch verzweigtkettige Gruppen und cyclische Gruppen, d.h. Cycloalkyl
und Cycloalkenyl, ein. Sofern nicht anders vermerkt, enthalten diese
Gruppen 1 bis 20 Kohlenstoffatome, wobei Alkenylgruppen 2 bis 20
Kohlenstoffatome enthalten. Bevorzugte Gruppen enthalten insgesamt
bis zu 10 Kohlenstoffatome. Cyclische Gruppen können monocyclisch oder polycyclisch
sein und weisen vorzugsweise 3 bis 10 Ringkohlenstoffatome auf.
Beispiele für
cyclische Gruppen sind Cyclopropyl, Cyclopropylmethyl, Cyclopentyl,
Cyclohexyl und Adamantyl.
-
Außerdem können die
Alkylen- und Alkenylenteile von Gruppen -X- gegebenenfalls durch
einen oder mehrere Substituenten aus der Gruppe bestehend aus Alkyl,
Alkenyl, Aryl, Heteroaryl, Heterocyclyl, Arylalkyl, Heteroarylalkyl
und Heterocyclylalkyl substituiert sein.
-
Der
Begriff "Halogenalkyl" schließt Gruppen
ein, die durch ein oder mehrere Halogenatome substituiert sind,
einschließlich
perfluorierter Gruppen. Dies gilt auch für Gruppen mit dem Präfix "Halogen-". Beispiele für geeignete
Halogenalkylgruppen sind Chlormethyl, Trifluormethyl und dergleichen.
-
Im
Rahmen der vorliegenden Erfindung schließt der Begriff "Aryl" carbocyclische aromatische
Ringe oder Ringsysteme ein. Beispiele für Arylgruppen sind Phenyl,
Naphthyl, Biphenyl, Fluorenyl und Indenyl. Der Begriff "Heteroaryl" schließt aromatische
Ringe oder Ringsysteme ein, die mindestens ein Ringheteroatom (z.B.
O, S, N) enthalten. Geeignete Heteroarylgruppen sind u.a. Furyl,
Thienyl, Pyridyl, Chinolinyl, Isochinolinyl, Indolyl, Isoindolyl,
Triazolyl, Pyrrolyl, Tetrazolyl, Imidazolyl, Pyrazolyl, Oxazolyl,
Thiazolyl, Benzofuranyl, Benzothiophenyl, Carbazolyl, Benzoxazolyl,
Pyrimidinyl, Benzimidazolyl, Chinoxalinyl, Benzothiazolyl, Naphthyridinyl,
Isoxazolyl, Isothiazolyl, Purinyl, Chinazolinyl und so weiter.
-
"Heterocyclyl" schließt nichtaromatische
Ringe oder Ringsysteme ein, die mindestens ein Ringheteroatom (z.B.
O, S, N) enthalten, und schließt
alle vollständig
gesättigten
und teilweise ungesättigten
Derivate der oben aufgeführten
Heteroarylgruppen ein. Beispiele für heterocyclische Gruppen sind
Pyrrolidinyl, Tetrahydrofuranyl, Morpholinyl, Thiomorpholinyl, Piperidinyl,
Piperazinyl, Thiazolidinyl, Imidazolidinyl, Isothiazolidinyl und dergleichen.
-
Die
Aryl-, Heteroaryl- und Heterocyclylgruppen können gegebenenfalls durch einen
oder mehrere unabhängig
voneinander aus der Gruppe bestehend aus Alkyl, Alkoxy, Alkylthio,
Halogenalkyl, Halogenalkoxy, Halogenalkylthio, Halogen, Nitro, Hydroxy,
Mercapto, Cyano, Carboxy, Formyl, Aryl, Aryloxy, Arylthio, Arylalkoxy,
Arylalkylthio, Heteroaryl, Heteroaryloxy, Heteroarylthio, Heteroarylalkoxy,
Heteroarylalkylthio, Amino, Alkylamino, Dialkylamino, Heterocyclyl,
Heterocycloalkyl, Alkylcarbonyl, Alkenylcarbonyl, Alkoxycarbonyl,
Halogenalkylcarbonyl, Halogenalkoxycarbonyl, Alkylthio carbonyl,
Arylcarbonyl, Heteroarylcarbonyl, Aryloxycarbonyl, Heteroaryloxycarbonyl,
Arylthiocarbonyl, Heteroarylthiocarbonyl, Alkanoyloxy, Alkanoylthio,
Alkanoylamino, Arylcarbonyloxy, Arylcarbonylthio, Alkylaminosulfonyl,
Alkylsulfonyl, Arylsulfonyl, Heteroarylsulfonyl, Aryldiazinyl, Alkylsulfonylamino,
Arylsulfonylamino, Arylalkylsulfonylamino, Alkylcarbonylamino, Alkenylcarbonylamino,
Arylcarbonylamino, Arylalkylcarbonylamino, Heteroarylcarbonylamino,
Heteroarylalkylcarbonylamino, Alkylsulfonylamino, Alkenylsulfonylamino,
Arylsulfonylamino, Arylalkylsulfonylamino, Heteroarylsulfonylamino, Heteroarylalkylsulfonylamino,
Alkylaminocarbonylamino, Alkenylaminocarbonylamino, Arylaminocarbonylamino,
Arylalkylaminocarbonylamino, Heteroarylaminocarbonylamino, Heteroarylalkylaminocarbonylamino
und, im Fall von Heterocycyl, Oxo ausgewählte Substituenten substituiert
sein. Bei Bezeichnung von anderen Gruppen als „substituiert" oder „gegebenenfalls
substituiert" können diese
Gruppen auch durch einen oder mehrere der oben aufgelisteten Substituenten
substituiert sein.
-
Bestimmte
Substituenten sind im allgemeinen bevorzugt. Beispielsweise gehören zu den
bevorzugten Gruppen R1 -R4-NR8-CR3-NR5-Z-R6-Alkyl und -R4-NR8-CR3-NR5-Z-R6-Aryl, wobei die Alkyl- und Arylgruppen unsubstituiert
oder substituiert sein können;
und R4 ist vorzugsweise Ethylen oder n-Butylen,
oder R4 kann mit R8 verbunden
sein, um einen Ring zu bilden. Vorzugsweise sind keine Substituenten
R vorhanden (d.h. n ist 0). Bevorzugte Gruppen R2 sind
u.a. Wasserstoff, Alkylgruppen mit 1 bis 4 Kohlenstoffatomen (d.h.
Methyl, Ethyl, Propyl, Isopropyl, n-Butyl, sec-Butyl, Isobutyl und
Cyclopropylmethyl), Methoxyethyl und Ethoxymethyl. Für substituierte
Gruppen, wie substituierte Alkylgruppen oder substituierte Arylgruppen,
sind bevorzugte Substituenten u.a. Halogen, Nitril, Methoxy, Methylthio,
Trifluormethyl und Trifluormethoxy. Sofern vorhanden, können einer
oder mehrere dieser bevorzugten Substi tuenten in den erfindungsgemäßen Verbindungen
in beliebiger Kombination vorliegen.
-
Die
Erfindung schließt
die hier beschriebenen Verbindungen in allen ihren pharmazeutisch
verträglichen
Formen einschließlich
von Isomeren (z.B. Diastereomeren und Enantiomeren), Salzen, Solvaten,
Polymorphen und dergleichen ein. Insbesondere schließt die Erfindung
für den
Fall, daß eine
Verbindung optisch aktiv ist, speziell jedes Enantiomer der Verbindung
sowie racemische Gemische der Enantiomeren ein.
-
Arzneimittel
und biologische Wirkung Erfindungsgemäße Arzneimittel enthalten eine
therapeutisch wirksame Menge einer erfindungsgemäßen Verbindung gemäß obiger
Beschreibung in Kombination mit einem pharmazeutisch verträglichen
Träger.
-
Unter
dem Begriff „eine
therapeutisch wirksame Menge" versteht
man eine Menge der Verbindung, die zur Hervorrufung einer therapeutischen
Wirkung, wie Cytokin-Induktion,
Antitumorwirkung und/oder Antiviruswirkung, ausreicht. Die genaue
Menge der in einem erfindungsgemäßen Arzneimittel
verwendeten aktiven Verbindung variiert zwar gemäß dem Fachmann bekannten Faktoren,
wie der physikalischen und chemischen Beschaffenheit der Verbindung,
der Beschaffenheit des Trägers
und dem vorgesehenen Dosierungsschema, jedoch werden aller Voraussicht
nach die erfindungsgemäßen Zusammensetzungen
eine zur Bereitstellung einer Dosis von etwa 100 ng/kg bis etwa
50 mg/kg und vorzugsweise von etwa 10 μg/kg bis etwa 5 mg/kg der Verbindung
an den Patienten ausreichende Wirkstoffmenge enthalten. Es kommen
alle herkömmlichen
Dosierungsformen in Betracht, wie Tabletten, Pastillen, parenterale
Formulierungen, Sirupe, Cremes, Salben, Aerosolformulierungen, Transdermalpflaster,
Transmukosalpflaster und dergleichen.
-
Die
erfindungsgemäßen Verbindungen
können
als einziges Therapeutikum bei dem Behandlungsschema oder in Kombination
miteinander oder mit anderen Wirkstoffen einschließlich zusätzlichen
die Immunantwort modifizierenden Mitteln, Virustatika, Antibiotika
usw. verabreicht werden.
-
Es
hat sich gezeigt, daß die
erfindungsgemäßen Verbindungen
bei Versuchen, die gemäß den nachstehend
aufgeführten
Tests durchgeführt
wurden, die Produktion bestimmter Cytokine induzieren. Diese Ergebnisse
deuten darauf hin, daß die
Verbindungen zur Verwendung als die Immunantwort modifizierende
Mittel, die die Immunantwort auf einer Reihe von verschiedenen Wegen
modulieren können,
und somit zur Behandlung verschiedener Erkrankungen geeignet sind.
-
Zu
den Cytokinen, deren Produktion durch die Verabreichung von erfindungsgemäßen Verbindungen induziert
werden kann, gehören
im allgemeinen Interferon-α (IFN-α) und/oder Tumornekrosefaktor-α (TNF-α) sowie bestimmte
Interleukine (IL). Cytokine, deren Biosynthese durch erfindungsgemäße Verbindungen
induziert werden kann, sind u.a. INF-α, TNF-α, IL-1, IL-6, IL-10 und IL-12
sowie verschiedene andere Cytokine. Unter anderem können diese
und andere Cytokine die Produktion von Viren und das Wachstum von
Tumorzellen inhibieren, so daß die
Verbindungen zur Verwendung bei der Behandlung von Viruserkrankungen
und Tumoren geeignet sind. Gegenstand der Erfindung ist demgemäß die Verwendung
einer erfindungsgemäßen Verbindung
zur Herstellung eines Medikaments zur Induktion der Cytokin-Biosynthese.
-
Es
wurde gefunden, daß bestimmte
erfindungsgemäße Verbindungen
bevorzugt die Expression von IFN-α in
einer Population von hämatopoetischen
Zellen wie PBMCs (periphere mononukleare Blutzellen) mit pDC2-Zellen
(precursor dendritic cell-type 2) induzieren, ohne daß gleichzeitig
wesentliche Mengen inflammatorischer Cytokine gebildet werden.
-
Neben
der Fähigkeit
zur Induktion der Produktion von Cytokinen beeinflussen die erfindungsgemäßen Verbindungen
auch andere Aspekte der angeborenen Immunantwort. So kann beispielsweise
die Aktivität
natürlicher
Killerzellen stimuliert werden, was möglicherweise auf Cytokin-Induktion
zurückzuführen ist.
Die Verbindungen können
auch Makrophagen aktivieren, was wiederum die Sekretion von Stickstoffmonoxid
und die Produktion zusätzlicher
Cytokine stimuliert. Des weiteren können die Verbindungen Proliferation
und Differenzierung von B-Lymphozyten bewirken.
-
Erfindungsgemäße Verbindungen
haben auch eine Wirkung auf die erworbene Immunantwort. Beispielsweise
wird, obwohl nicht angenommen wird, daß eine direkte Wirkung auf
T-Zellen oder eine direkte Induktion von T-Zell-Cytokinen vorliegt, bei Verabreichung
der Verbindungen die Produktion des T-Helfer-Typ-1-Cytokins (Th1-Cytokins) IFN-γ indirekt
induziert und die Produktion der T-Helfer-Typ-2-Cytokine (Th2-Cytokine)
IL-4, IL-5 und IL-13 inhibiert. Aufgrund dieser Wirkung eignen sich
die Verbindungen zur Verwendung bei der Behandlung von Erkrankungen,
bei denen die Heraufregulierung der Th1-Antwort und/oder die Herabregulierung
der Th2-Antwort
erwünscht
ist. Angesichts der Fähigkeit
von erfindungsgemäßen Verbindungen
zur Inhibierung der Th2-Immunantwort
wird erwartet, daß die
Verbindungen zur Verwendung bei der Behandlung von atopischen Erkrankungen,
z.B. atopischer Dermatitis, Asthma, Allergie, allergischer Rhinitis;
systemischem Lupus erythematodes; als Impfhilfsstoff für zellvermittelte
Immunität
und möglicherweise als
Behandlung für
rezidivierende Pilzerkrankungen und Chlamydia geeignet sind.
-
Aufgrund
ihrer die Immunantwort modifizierenden Wirkungen sind die Verbindungen
zur Verwendung bei der Behandlung verschiedenster Leiden geeignet.
Aufgrund ihrer Fähigkeit
zur Induktion der Produktion von Cytokinen wie INF-α und/oder
TNF-α eignen
sich die Verbindungen besonders gut zur Verwendung bei der Behandlung
von Viruserkrankungen und Tumoren. Diese immunmodulierende Wirkung
legt nahe, daß erfindungsgemäße Verbindungen
zur Verwendung bei der Behandlung von Erkrankungen geeignet sind,
wie u.a. Viruserkrankungen einschließlich Feigwarzen; gemeinen
Warzen; Sohlenwarzen; Hepatitis B; Hepatitis C; Herpessimplexvirus
Typ I und Typ II; Molluscum contagiosum; Variola, insbesondere Variola
major; HIV; CMV; VZV; Rhinovirus; Adenovirus; Influenza und Parainfluenza;
intraepithelialer Neoplasien, wie zervikaler intraepithelialer Neoplasien;
Humanpapillomavirus (HPV) und damit einhergehenden Neoplasien; Pilzerkrankungen, z.B.
Candida-, Aspergillus- und Cryptococcenmeningitis; neoplastische
Erkrankungen, z.B. Basalzellenkarzinom, Haarzellenleukämie, Kaposi-Sarkom,
Nierenzellenkarzinom, Plattenepithelkarzinom, myelogischer Leukämie, multiplem
Myelom, Melanom, Non-Hodgkin-Lymphom,
kutanem T-Zellenlymphom und anderen Krebsarten; parasitischen Erkrankungen,
d.h. Pneumocystis carnii, Kryptosporidiose, Histoplasmose, Toxoplasmose,
Trypanosominfektion und Leishmaniase; und bakteriellen Infektionen,
z.B. Tuberkulose und Mycobacterium avium. Weitere Erkrankungen oder
Leiden, die mit den erfindungsgemäßen Verbindungen behandelt
werden können,
sind u.a. aktinische Keratose; Ekzem; Eosinophilie; essentielle
Thrombozythämie;
Lepra; multiple Sklerose; Ommen-Syndrom; diskoider Lupus; Bowen-Krankheit;
Bowenoide Papulose; Alopecia areata; die Inhibierung der Keloidbildung
nach Chirurgie und anderen Arten von postchirurgischen Narben. Außerdem könnten diese
Verbindungen die Heilung von Wunden einschließlich chronischer Wunden verbessern
oder stimulieren. Die Verbindungen können zur Verwendung bei der
Behandlung der opportunistischen Infektionen und Tumore, die nach
Suppression der zellvermittelten Immunität beispielsweise bei Transplantatpatienten,
Krebspatienten und HIV-Patienten auftreten, geeignet sein.
-
Eine
zur Induktion der Cytokin-Biosynthese wirksame Menge einer Verbindung
ist eine Menge, die dazu ausreicht, einen oder mehrere Zelltypen,
wie z.B. Monocyten, Makrophagen, dendritische Zellen und B-Zellen, zur Produktion
einer Menge eines oder mehrerer Cytokine, wie beispielsweise IFN-α, TNF-α, IL-1, IL-6,
IL-10 und IL-12, die gegenüber
dem Hintergrundniveau derartiger Cytokine erhöht ist, zu veranlassen. Die
genaue Menge variiert gemäß an sich
bekannten Faktoren, jedoch wird erwartet, daß es sich um eine Dosis von
etwa 100 ng/kg bis etwa 50 mg/kg und vorzugsweise etwa 10 μg/kg bis
etwa 5 mg/kg handelt. Gegenstand der Erfindung ist auch die Verwendung
einer erfindungsgemäßen Verbindung
oder Zusammensetzung zur Herstellung eines Medikaments zur Behandlung
einer Virusinfektion oder zur Behandlung einer neoplastischen Erkrankung.
Eine zur Behandlung oder Inhibierung einer Virusinfektion wirksame
Menge ist eine Menge, die einen Rückgang einer oder mehrerer
der Manifestationen der Virusinfektion, wie viralen Läsionen,
Virusbelastung, Geschwindigkeit der Virusproduktion und Mortalität im Vergleich
zu unbehandelten Kontrolltieren bewirkt. Die genaue Menge variiert
gemäß an sich
bekannten Faktoren, jedoch wird erwartet, daß es sich um eine Dosis von
etwa 100 ng/kg bis etwa 50 mg/kg und vorzugsweise etwa 10 μg/kg bis
etwa 5 mg/kg handelt. Eine zur Behandlung eines neoplastischen Leidens
wirksame Menge ist eine Menge, die eine Verringerung der Tumorgröße oder
der Zahl der Tumorfoki bewirkt. Wiederum variiert die genaue Menge
gemäß an sich
bekannten Faktoren, jedoch wird erwartet, daß es sich um eine Dosis von
etwa 100 ng/kg bis etwa 50 mg/kg und vorzugsweise etwa 10 μg/kg bis
etwa 5 mg/kg handelt.
-
Die
Erfindung wird anhand der folgenden Beispiele näher erläutert, die lediglich zur Illustration
dienen und die Erfindung in keiner Weise einschränken sollen.
-
In
den nachstehenden Beispielen wurden einige der Verbindungen mittels
halbpräparativer
HPLC gereinigt. Es wurde ein automatisiertes Reinigungssystem des
Typs Fraction Lynx von Waters verwendet. Die bei der halbpräparativen
HPLC erhaltenen Fraktionen wurden mit einem LC-TOFMS von Micromass
analysiert, und die entsprechenden Fraktionen wurden vereinigt und
in der Zentrifuge verdampft, was das Trifluoracetatsalz der gewünschten
Verbindung ergab. Die Strukturen wurden durch 1H-NMR
bestätigt.
-
Säule: Phenomenex
Luna C18(2), 10 × 50
mm, Teilchengröße 5 Mikron,
100 Å-Pore;
Durchflußrate: 25
ml/min; Gradientenelution von 5–65
% B in 4 min, dann 65 bis 95 % B in 0,1 min, dann 0,4 min Halten
bei 95 % B, wobei A = 0,05 % Trifluoressigsäure/Wasser und B = 0,05 % Trifluoressigsäure/Acetonitril;
Fraktionensammlung durch massenselektive Auslösung.
-
BEISPIEL
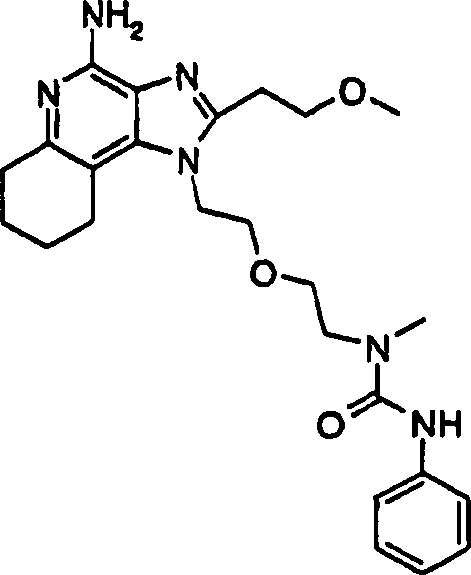
1 N-(2-{2-[4-Amino-2-(2-methoxyethyl)-1H-imidazo[4,5-c]-chinolin-1-yl]ethoxy}ethyl)-N'-phenylharnstoff
-
Teil A
-
Eine
Lösung
von 2-(2-Aminoethoxy)ethanol (29,0 g, 0,276 mol) in 180 ml Tetrahydrofuran
(THF) wurde unter N2 auf 0°C abgekühlt und
mit 140 ml 2 N NaOH-Lösung
behandelt. Dann wurde zu der schnell gerührten Lösung über einen Zeitraum von 1 h
eine Lösung
von Di-tert-butyldicarbonat (60,2 g, 0,276 mol) in 180 ml THF getropft.
Dann wurde die Reaktionsmischung auf Raumtemperatur kommen gelassen
und noch 18 h gerührt. Dann
wurde das THF unter vermindertem Druck abgezogen und die verbleibende
wäßrige Aufschlämmung durch
Zugabe von 150 ml 1 M H2SO4-Lösung auf
pH 3 gebracht. Nach Extraktion mit Essigsäureethylester (300 ml, 100
ml), wurden die vereinigten organischen Schichten mit H2O
(2×) und
Kochsalzlösung
gewaschen. Der organische Teil wurde über Na2SO4 getrocknet und aufkonzentriert, was 2-(2-Hydroxyethoxy)ethylcarbamidsäure-tert-butylester
in Form eines farblosen Öls
(47,1 g) ergab.
-
Teil B
-
Eine
schnell gerührte
Lösung
von 2-(2-Hydroxyethoxy) ethylcarbamidsäure-tert-butylester (47,1 g, 0,230
mol) in 1 l wasserfreiem CH2Cl2 wurde
unter N2 auf 0°C abgekühlt und mit Triethylamin (48,0
ml, 0,345 mol) behandelt. Dann wurde über einen Zeitraum von 30 min
Methansulfonylchlorid (19,6 ml, 0,253 mol) zugetropft. Die Reaktionsmischung
wurde dann auf Raumtemperatur kommen gelassen und dann noch 22 h
gerührt.
Dann wurde der Ansatz durch Zugabe von 500 ml gesättigter
NaHCO3-Lösung gequencht
und die organische Schicht abgetrennt. Dann wurde die organische
Phase mit H2O (3 × 500 ml) und Kochsalzlösung gewaschen.
Der organische Teil wurde über
Na2SO4 getrocknet
und aufkonzentriert, was 2-{2-[(tert-Butoxycarbonyl)amino]ethoxy}
ethylmethansulfonat in Form eines braunen Öls (63,5 g) ergab.
-
Teil C
-
Eine
gerührte
Lösung
von 2-{2-[(tert-Butoxycarbonyl)amino]ethoxy}ethylmethansulfonat
(63,5 g, 0,224 mol) in 400 ml N,N-Dimethylformamid (DMF) wurde mit
NaN3 (16,1 g, 0,247 mol) behandelt, wonach
die Reaktionsmischung unter N2 auf 90°C erhitzt
wurde. Nach 5 h wurde die Lösung
auf Raumtemperatur abgekühlt und
mit 500 ml kaltem H2O behandelt. Dann wurde
die Reaktionsmischung mit Et2O (3 × 3 00 ml)
extrahiert . Die vereinigten organischen Extrakte wurden mit H2O (4 × 100
ml) und Kochsalzlösung
(2 × 100
ml) gewaschen. Der organische Teil wurde über MgSO4 getrocknet
und aufkonzentriert, was 52,0 g 2-(2-Azidoethoxy)ethylcarbamidsäure-tert-butylester in Form
eines hellbraunen Öls
ergab.
-
Teil D
-
Eine
Lösung
von 2-(2-Azidoethoxy)ethylcarbamidsäure-tert-butylester (47,0 g, 0,204 mol)
in MeOH wurde mit 4 g 10% Pd auf Kohle behandelt und 24 h unter
H2 (3 kg/cm2) geschüttelt. Dann
wurde die Lösung über eine
Celiteschicht filtriert und aufkonzentriert, was 35,3 g rohen 2-(2-Aminoethoxy)ethylcarbamidsäure-tert-butylester
in Form einer farblosen Flüssigkeit
ergab, die ohne weitere Reinigung verwendet wurde.
-
Teil E
-
Eine
gerührte
Lösung
von 4-Chlor-3-nitrochinolin (31,4 g, 0,151 mol) in 500 ml wasserfreiem
CH2Cl2 wurde unter
N2 mit Triethylamin (43 ml, 0,308 mol) und
2-(2-Aminoethoxy)ethylcarbamidsäure-tert-butylester (0,151
mol) behandelt. Nach Rühren über Nacht
wurde die Reaktionsmischung mit H2O (2 × 300 ml)
und Kochsalzlösung
(300 ml) gewaschen. Der organische Teil wurde über Na2SO4 getrocknet und zu einem leuchtend gelben
Feststoff aufkonzentriert. Durch Umkristallisation aus Essigsäureethylester/Hexangemisch
wurden 43,6 g 2-{2-[(3-Nitrochinolin-4-yl)amino]ethoxy}ethylcarbamidsäure-tert-butylester in
Form von leuchtend gelben Kristallen erhalten.
-
Teil F
-
Eine
Lösung
von 2-{2-[(3-Nitrochinolin-4-yl)amino]ethoxy}ethylcarbamidsäure-tert-butylester
(7,52 g, 20,0 mmol) in Toluol wurde mit 1,5 g 5% Pt auf Kohle behandelt
und 24 h unter H2 (3 kg/cm2)
geschüttelt.
Dann wurde die Lösung über eine
Celiteschicht filtriert und aufkonzentriert, was 6,92 g rohen 2-{2-[(3-Aminochinolin-4-yl)amino]ethoxy}ethylcarbamidsäure-tert-butylester
in Form eines gelben Sirups ergab.
-
Teil G
-
Eine
Lösung
von 2-{2-[(3-Aminochinolin-4-yl)amino] ethoxy}ethylcarbamidsäure-tert-butylester
(10,2 g, 29,5 mmol) in 250 ml wasserfreiem CH2Cl2 wurde auf 0°C abgekühlt und mit Triethylamin (4,18
ml, 30,0 mmol) behandelt. Dann wurde über einen Zeitraum von 5 min
Methoxypropionylchlorid (3,30 ml, 30,3 mmol) zugetropft. Dann wurde
der Ansatz auf Raumtemperatur erwärmt und noch 1 h gerührt. Dann
wurde die Reaktionsmischung unter vermindertem Druck zu einem orangefarbenen
Feststoff aufkonzentriert. Dieser wurde in 250 ml EtOH gelöst und mit
12,5 ml Triethylamin versetzt. Die Mischung wurde zum Rückfluß erhitzt
und über
Nacht unter N2 gerührt. Dann wurde der Ansatz
unter vermindertem Druck bis zur Trockne aufkonzentriert und mit
300 ml Et2O behandelt. Dann wurde die Mischung
filtriert und das Filtrat unter vermindertem Druck aufkonzentriert,
was einen braunen Feststoff ergab. Der Feststoff wurde in 200 ml
heißem
MeOH gelöst
und mit Aktivkohle behandelt. Die heiße Lösung wurde filtriert und aufkonzentriert,
was 11,1 g 2-{2-[2-(2-Methoxyethyl)-1H-imidazo[4,5-c]chinolin-1-yl]ethoxy}ethylcarbamidsäure-tert-butylester
in Form eines gelben Sirups ergab.
-
Teil H
-
Eine
Lösung
von 2-{2-[2-(2-Methoxyethyl)-1H-imidazo[4,5-c]chinolin-1-yl]ethoxy}ethylcarbamidsäure-tert- butylester (10,22
g, 24,7 mmol) in 250 ml CHCl3 wurde mit
3-Chlorperoxybenzoesäure
(MCPBA, 77 %, 9,12 g, 40,8 mmol) behandelt. Nach 30 min Rühren wurde
die Reaktionsmischung mit 1%iger Na2CO3-Lösung (2 × 75 ml)
und Kochsalzlösung
gewaschen. Dann wurde die organische Schicht über Na2SO4 getrocknet und aufkonzentriert, was 10,6
g 2-{2-[2-(2-Methoxyethyl)-5-oxido-1H-imidazo[4,5-c]chinolin-1-yl]ethoxy}ethylcarbamidsäure-tert-butylester
in Form eines orangefarbenen Schaums ergab, der ohne weitere Reinigung
verwendet wurde.
-
Teil I
-
Eine
Lösung
von 2-{2-[2-(2-Methoxyethyl)-5-oxido-1H-imidazo[4,5-c]chinolin-1-yl]ethoxy}ethylcarbamidsäure-tert-butylester (10,6
g, 24,6 mmol) in 100 ml 1,2-Dichlorethan wurde auf 60°C erhitzt
und mit 10 ml konzentrierter NH4OH-Lösung behandlt.
Die schnell gerührt
Lösung
wurde über
einen Zeitraum von 10 min mit festem p-Toluolsulfonnylchlorid (7,05
g, 37,0 mmol) versetzt. Die Reaktionsmischung wurde mit noch 1 ml
konzentrierter NH4OH-Lösung behandelt und dann in
einem Druckbehälter
verschlossen und weitere 2 h erhitzt. Dann wurde die Reaktionsmischung
abgekühlt
und mit 100 ml CHCl3 behandelt. Die Reaktionsmischung
wurde dann mit H2O, 1%iger Na2CO3-Lösung
(2×) und
Kochsalzlösung
gewaschen. Der organische Teil wurde über Na2SO4 getrocknet und aufkonzentriert, was 10,6
g 2-{2-[4-Amino-2-(2-methoxyethyl)-1H-imidazo[4,5-c]-chinolin-1-yl]ethoxy}ethylcarbamidsäure-tert-butylester
in Form eines braunen Schaums ergab.
-
Teil J
-
2-{2-[4-Amino-2-(2-methoxyethyl)-1H-imidazo[4,5-c]-chinolin-1-yl]ethoxy}ethylcarbamidsäure-tert-butylester
(10,6 g, 24,6 mmol) wurde mit 75 ml 2 M HCl in EtOH behandelt, wonach
die Mischung unter Rühren zum
Rückfluß erhitzt
wurde. Nach 1,5 h wurde die Reaktionsmischung abgekühlt und
filtriert, was einen gummiartigen Feststoff ergab. Der Feststoff
wurde mit EtOH und Et2O gewaschen und unter
Vakuum getrocknet, was das Hydrochloridsalz in Form eines hellbraunen
Feststoffs ergab. Zur Herstellung der freien Base wurde das Hydrochloridsalz
in 50 ml H2O gelöst und mit 10%iger NaOH-Lösung behandelt.
Die wäßrige Suspension wurde
dann bis zur Trocknung aufkonzentriert, wonach der Rückstand
mit CHCl3 behandelt wurde. Die erhaltenen
Salze wurden abfiltriert, wonach das Filtrat aufkonzentriert wurde,
was 3,82 g 1-[2-(2-Aminoethoxy)ethyl]-2-(2-methoxyethyl)-1H-imidazo[4,5-c]chinolin-4-amin
in Form eines hellbraunen Pulvers ergab.
MS 330 (M + H)+;
1H-NMR (300
MHz, DMSO-d6) δ 8,10 (d, J = 8,1 Hz, 1H); 7,66
(d, J = 8,2 Hz, 1H); 7,40 (m, 1H); 7,25 (m, 1H); 6,88 (br s, 2H);
4,78 (t, J = 5,4 Hz, 2H); 3,89 (t, J = 4,8 Hz, 2H); 3,84 (t, J =
6,9 Hz, 2H); 3,54 (t, J = 5,4 Hz, 2H); 3,31 (s, 3H); 3,23 (t, J
= 6,6 Hz, 2H); 2,88 (t, J = 5,3 Hz, 2H).
-
Teil K
-
1-[2-(2-Aminoethoxy)ethyl]-2-(2-methoxyethyl)-1H-imidazo[4,5-c]chinolin-4-amin
(750 mg, 2,28 mmol) wurde in 30 ml wasserfreiem CH2Cl2 gelöst
und unter N2 auf 0°C abgekühlt. Dann wurde die Reaktionsmischung
mit Phenylisocyanat (247 μl,
2,28 mmol) und Et3N (0,64 ml, 4,56 mmol)
behandelt und langsam auf Raumtemperatur kommen gelassen. Nach 2
h Rühren
wurde die Reaktionsmischung unter vermindertem Druck zu einem gelben
Feststoff aufkonzentriert. Der gelbe Feststoff wurde in einer minimalen
Menge CH2Cl2 gelöst und mit
EtOAc versetzt, bis die Lösung
trübe wurde.
Die Mischung wurde über
Nacht in den Gefrierschrank gestellt, wobei sich weiße Kristalle
bildeten. Die Kristalle wurden abfiltriert und unter Vakuum getrocknet,
was 126 mg N-(2-{2-[4-Amino-2-(2-methoxyethyl)-1H-imidazo[4,5-c]chinolin-1-yl]ethoxy}ethyl)-N'-phenylharnstoff ergab. Fp. 171,0–174,0°C;
MS
449 (M + H)+;
1H-NMR
(300 MHz, DMSO-d6) δ 8,50 (s, 1H); 8,05 (d, J =
7,7 Hz, 1H); 7,62 (d, J = 8,8 Hz, 1H); 7,44–7,18 (m, 3H); 7,27–7,18 (m,
3H); 6,88 (t, J = 7,3 Hz, 1H); 6,54 (s, 2H); 6,12 (t, J = 5,5 Hz,
2H); 4,76 (t, J = 4,8 Hz, 2H); 3,88 (t, J = 5,3 Hz, 2H); 3,81 (t,
J = 6,7 Hz, 2H); 3,40 (t, J = 6,0 Hz, 2H); 3,28 (s, 3H); 3,25–3,14 (m,
4H);
13C (75 MHz, DMSO-d6) δ 155, 5,
152, 0, 149, 9, 140, 8, 132,7, 129,0, 126,8, 126,5, 121,5, 121,4,
120,5, 117,9, 115,1, 70,5, 69,4, 58,4, 45,5, 27,6;
Analyse:
berechnet für
C24H28N6O3·0,21H2O: %C, 63,73, %H, 6,33, %N, 18,58. Gefunden:
%C, 63,33, %H, 6,28, %N, 18,67.
-
BEISPIEL
2 N-(2-{2-[4-Amino-2-(2-methoxyethyl)-6,7,8,9-tetrahydro-1H-imidazo[4,5-c]chinolin-1-yl]ethoxy}ethyl)-N'-phenylharnstoff
-
Teil A
-
1-[2-(2-Aminoethoxy)ethyl]-2-(2-methoxyethyl)-1H-imidazo[4,5-c]chinolin-4-amin
(10,0 g, 27,3 mmol) wurde in 50 ml Trifluoressigsäure gelöst und mit
PtO2 (1,0 g) behandelt. Die Reaktionsmischung
wurde unter H2 (3 kg/cm2)
geschüttelt.
Nach 4 d wurden weitere 0,5 g PtO2 zugege ben,
wonach die Hydrierung noch 3 d fortgesetzt wurde. Dann wurde der
Ansatz über
Celite filtriert und unter vermindertem Druck aufkonzentriert, was
ein braunes Öl
ergab. Das Öl
wurde in 200 ml H2O gelöst und dann durch Zugabe von
10%iger NaOH-Lösung
basisch gestellt (pH ~ 11). Nach Extraktion mit CHCl3 (5 × 75 ml)
wurden die vereinigten organischen Schichten über Na2SO4 getrocknet und aufkonzentriert, was 5,
17 g 1-[2-(2-Aminoethoxy)ethyl]-2-(2-methoxyethyl)-6,7,8,9-tetrahydro-1H-imidazo[4,5-c]chinolin-4-amin
in Form eines hellbraunen Feststoffs ergab.
MS 334 (M + H)+;
1H-NMR (300
MHz, CDCl3) δ 5,19 (s, 2H); 4,49 (t, J =
5,4 Hz, 2H); 3,84 (t, J = 6,6 Hz, 2H); 3,71 (t, J = 5,4 Hz, 2H),
3,36 (t, J = 5,2 Hz, 2H); 3,51 (s, 3H); 3,15 (t, J = 6,6 Hz, 2H);
2,95 (m, 2H); 2,82 (m, 2H); 2,76 (t, J = 5,1 Hz, 2H); 1,84 (m, 4H),
1,47 (br s, 2H).
-
Teil B
-
1-[2-(2-Aminoethoxy)ethyl]-2-(2-methoxyethyl)-6,7,8,9-tetrahydro-1H-imidazo[4,5-c]chinolin-4-amin (919
mg, 2,76 mmol) wurde in 30 ml wasserfreiem CH2Cl2 gelöst
und unter N2 auf 0°C abgekühlt. Dann wurde die Reaktionsmischung
mit Phenylisocyanat (300 μl,
2,76 mmol) und Et3N (0,77 ml, 5,51 mmol)
behandelt und langsam auf Raumtemperatur kommen gelassen. Nach Rühren über Nacht
wurde die Reaktionsmischung dann durch Zugabe von gesättigter
NaHCO3-Lösung
(30 ml) gequencht. Die organische Schicht wurde abgetrennt und mit
H2O und Kochsalzlösung gewaschen, über Na2SO4 getrocknet und
unter vermindertem Druck aufkonzentriert, was einen gelben Feststoff
ergab. Der Feststoff wurde mit Et2O (30
ml) und einigen Tropfen MeOH trituriert. Der Feststoff wurde abfiltriert
und unter Vakuum getrocknet, was 460 mg N-(2-{2-[4-Amino-2-(2-methoxyethyl)-6,7,8,9-tetrahydro-1H-imidazo[4,5-c]chinolin-1-yl] ethoxy}ethyl)-N'-phenylharnstoff
in Form eines weißen
Pulvers ergab. Fp. 180–182°C;
MS
453 (M + H)+;
1H-NMR
(300 MHz, DMSO-d6) δ 8, 51 (s, 1H); 7,37 (d, J =
7,7 Hz, 2H); 7,19 (t, J = 8,2 Hz, 2H); 6,86 (t, J = 7,7 Hz, 1H);
6,11 (t, J = 5,5 Hz, 2H); 5,70 (s, 2H); 4, 43 (t, J = 5, 1 Hz, 2H)
; 3,78–3,69
(m, 4H); 3,39 (t, J = 5,6 Hz, 2H); 3,25 (s, 3H); 3,19 (m, 2H); 3,10
(t, J = 6,8 Hz, 2H); 2,91 (m, 2H); 2,64 (m, 2H); 1,72 (m, 4H);
13C (75 MHz, DMSO-d6) δ 155,5, 151,3,
149,3, 146,3, 140,8, 138,5, 129,0, 125,0, 121,4, 118,0, 105,6, 70,6, 70,5,
70,4, 58,4, 44,6, 39,2, 32,7, 27,6, 23,8, 23,1, 23,0;
Analyse:
berechnet für
C24H32N6O3: %C, 63,70, %H, 7,13, %N, 18,57. Gefunden:
%C, 63,33, %H, 7,16, %N, 18,66.
-
BEISPIEL
3 N-(2-{2-[4-Amino-2-(2-methoxyethyl)-1H-imidazo[4,5-c]-chinolin-1-yl]ethoxy}ethyl)-N-methyl-N'-phenylharnstoff
-
Teil A
-
Natriumhydrid
(60%ige Dispersion in Öl,
9,1 g, 228 mmol) wurde in einen Rundkolben gegeben und unter N2 mit Hexangemisch (3×) gewaschen. Das getrocknete Natriumhydrid
wurde mit 800 ml wasserfreiem THF behandelt. Dann wurde über einen
Zeitraum von 40 min eine Lösung
von 2-(2-Azidoethoxy)ethylcarbamidsäure-tert-butylester (41,9 g, 182 mmol) in 200
ml THF zu der gerührten
Natriumhydridlösung
gegeben. Nach vollständiger
Zugabe wurde der Ansatz noch 20 min gerührt und dann mit Methyliodid
(13,6 ml, 218 mmol) versetzt. Nach Rühren über Nacht wurde der Ansatz
mit 300 ml gesättigter
NaHCO3-Lösung
gequencht. Dann wurde die Reaktionsmischung mit 200 ml H2O und 1 l Et2O behandelt.
Die organische Phase wurde abgetrennt und mit H2O
und Kochsalzlösung
gewaschen. Der organische Teil wurde dann über MgSO4 getrocknet
und unter vermindertem Druck aufkonzentriert, was 41,9 g 2-(2-Azidoethoxy)ethyl(methyl)carbamidsäure-tert-butylester
in Form einer gelben Flüssigkeit
ergab.
-
Teil B
-
Eine
Lösung
von 2-(2-Azidoethoxy)ethyl(methyl)carbamidsäure-tert-butylester (41,9 g,
170 mmol) in 600 ml MeOH wurde mit 2,5 g 10 % Pd auf Kohle behandelt
und 24 h unter H2 (3 kg/cm2)
geschüttelt.
Dann wurde die Lösung über eine
Celiteschicht filtriert und aufkonzentriert, was 37,2 g rohen 2-(2-Aminoethoxy)ethyl(methyl)carbamidsäure-tert-butylester
in Form einer hellgelben Flüssigkeit
ergab.
-
Teil C
-
Eine
gerührte
Lösung
von 4-Chlor-3-nitrochinolin (32,3 g, 155 mol) in 400 ml wasserfreiem
CH2Cl2 wurde unter
N2 mit Triethylamin (43,1 ml, 310 mmol)
und 2-(2-Aminoethoxy)ethyl(methyl)carbamidsäure-tert-butylester (37,2 g, 171 mmol) behandelt.
Nach Rühren über Nacht
wurde die Reaktionsmischung mit H2O (2 × 300 ml)
und Kochsalzlösung
(300 ml) gewaschen. Der organische Teil wurde über Na2SO4 getrocknet und aufkonzentriert, was ein
braunes Öl
ergab. Durch Säulenchromatographie
(SiO2, 33% Essigsäureethylester/Hexangemisch –67% Essigsäure ethylester/Hexangemisch
wurden 46,7 g Methyl(2-{2-[(3-nitrochinolin-4-yl)amino]ethoxy}ethyl)carbamidsäure-tert-butylester
in Form eines gelben Feststoffs erhalten.
-
Teil D
-
Eine
Lösung
von Methyl(2-{2-[(3-nitrochinolin-4-yl)amino]ethoxy}ethyl)carbamidsäure-tert-butylester (6,56
g, 16,8 mmol) in 75 ml Toluol wurde mit 0,5 g 5% Pt auf Kohle behandelt
und 24 h unter H2 (3 kg/cm2) geschüttelt. Dann
wurde die Lösung über eine
Celiteschicht filtriert und aufkonzentriert, was 6,8 g rohen (2-{2-[(3-Aminochinolin-4-yl)amino]ethoxy}ethyl(methyl)
carbamidsäure-tert-butylester
in Form eines orangefarbenen Sirups ergab, der ohne weitere Reinigung
verwendet wurde.
-
Teil E
-
Eine
Lösung
von (2-{2-[(3-Aminochinolin-4-yl)amino]ethoxy}ethyl(methyl)carbamidsäure-tert-butylester
(6,05 g, 16,8 mmol) in 200 ml wasserfreiem CH2Cl2 wurde auf 0°C abgekühlt und mit Triethylamin (2,40
ml, 17,2 mmol) behandelt. Dann wurde über einen Zeitraum von 5 min
Methoxypropionylchlorid (1,72 ml, 17,2 mmol) zugetropft. Dann wurde
der Ansatz auf Raumtemperatur erwärmt und noch 3 h gerührt. Dann
wurde die Reaktionsmischung unter vermindertem Druck zu einem orangefarbenen
Feststoff aufkonzentriert. Dieser wurde in 200 ml EtOH gelöst und mit
7,2 ml Triethylamin versetzt. Die Mischung wurde zum Rückfluß erhitzt
und über
Nacht unter N2 gerührt. Dann wurde der Ansatz
unter vermindertem Druck bis zur Trockne aufkonzentriert und mit
300 ml Et2O behandelt. Dann wurde die Mischung
filtriert und das Filtrat unter vermindertem Druck aufkonzentriert,
was einen braunen Feststoff ergab. Dieser wurde in 300 ml CH2Cl2 gelöst und mit
H2O und Kochsalzlösung gewaschen. Der organische
Teil wurde über
Na2SO4 getrocknet
und unter vermindertem Druck aufkonzentriert, was ein braunes Öl ergab.
Das Öl
wurde in 100 ml heißem
MeOH gelöst
und mit Aktivkohle behandelt. Die heiße Lösung wurde filtriert und aufkonzentriert,
was 7,20 g 2-{2-[2-(2-Methoxyethyl)-1H-imidazo[4,5-c]chinolin-1-yl]ethoxy}ethyl(methyl)carbamidsäure-tert-butylester
in Form eines gelben Sirups ergab.
-
Teil F
-
Eine
Lösung
von 2-{2-[2-(2-Methoxyethyl)-1H-imidazo[4,5-c]chinolin-1-yl]ethoxy}ethyl(methyl)carbamidsäure-tert-butylester (7,20
g, 16,8 mmol) in 200 ml CH2Cl2 wurde
mit MCPBA (77%, 4,32 g, 19,3 mmol) behandelt. Nach 6 h Rühren wurde
die Reaktionsmischung mit gesättigter
NaHCO3-Lösung
behandelt, wonach die Schichten getrennt wurden. Der organische
Teil wurde mit H2O und Kochsalzlösung gewaschen
und dann über
Na2SO4 getrocknet
und aufkonzentriert, was 7,05 g 2-{2-[2-(2-Methoxyethyl)-5-oxido-1H-imidazo[4,5-c]chinolin-1-yl]ethoxy}ethyl(methyl)carbamidsäure-tert-butylester
in Form eines hellbraunen Feststoffs ergab.
-
Teil G
-
Eine
Lösung
von 2-{2-[2-(2-Methoxyethyl)-5-oxido-1H-imidazo[4,5-c]chinolin-1-yl]ethoxy}ethyl(methyl)carbamidsäure-tert-butylester
(7,05 g, 15,9 mmol) in 100 ml 1,2-Dichlorethan wurde auf 80°C erhitzt
und mit 5 ml konzentrierter NH4OH-Lösung behandelt.
Die schnell gerührte
Lösung
wurde über
einen Zeitraum von 10 min mit festem p-Toluolsulfonylchlorid (3,33
g, 17,5 mmol) versetzt. Die Reaktionsmischung wurde mit weiteren
5 ml konzentrierter NH4OH-Lösung behandelt
und dann in einem Druckbehälter
verschlossen und noch 4 h erhitzt. Dann wurde die Reaktionsmischung
abgekühlt
und mit 100 ml CH2Cl2 behandelt.
Dann wurde die Reaktionsmischung mit H2O,
1%iger Na2CO3-Lösung (3×) und Kochsalzlösung gewaschen.
Der organische Teil wurde über
Na2SO4 getrocknet
und aufkonzentriert, was 6,50 g 2-{2-[4-Amino-2-(2-methoxyethyl)-1H-imidazo[4,5-c]chinolin-1-yl]ethoxy} ethyl(methyl)carbamidsäure-tert-butylester
in Form eines braunen Öls
ergab.
-
Teil H
-
Eine
Lösung
von 2-{2-[4-Amino-2-(2-methoxyethyl)-1H-imidazo[4,5-c]chinolin-1-yl]ethoxy}ethyl(methyl)carbamidsäure-tert-butylester
(6,50 g, 14,7 mmol) wurde in 100 ml EtOH gelöst und mit 20 ml 2 M HCl in EtOH
behandelt, wonach die Mischung unter Rühren zum Rückfluß erhitzt wurde. Nach 6 h wurde
die Reaktionsmischung abgekühlt
und filtriert, was einen gummiartigen Feststoff ergab. Der Feststoff
wurde mit EtOH und Et2O gewaschen und unter
Vakuum getrocknet, was das Hydrochloridsalz in Form eines helbraunen
Pulvers ergab. Zur Herstellung der freien Base wurde das Hydrochloridsalz
in 50 ml H2O gelöst und mit 5 ml konzentriertem
NH4OH behandelt. Die wäßrige Suspension wurde mit
CH2Cl2 (5 × 50 ml)
extrahiert. Die vereinigten organischen Schichten wurden über Na2SO4 getrocknet und
auf konzentriert, was 3,93 g 2-(2-Methoxyethyl)-1-{2-[2-(methylamino)ethoxy]ethyl}-1H-imidazo(4,5-c]chinolin-4-amin in Form eines
hellbraunen Pulvers ergab.
MS 344 (M + H)+;
1H-NMR (300 MHz, DMSO-d6) δ 8,07 (d,
J = 7,7 Hz, 1H); 7,62 (dd, J = 1,0, 8,3 Hz, 1H); 7,42 (ddd, J =
1,0, 7,1, 8,2 Hz, 1H); 7,22 (ddd, J = 1,1, 7,1, 8,2 Hz, 1H); 6,49
(s, 2H); 4,75 (t, J = 5,1 Hz, 2H); 3,83 (t, J = 6,8 Hz, 4H); 3,35
(t, J = 5,6 Hz, 2H); 3,30 (s, 3H); 3,21 (t, J = 6,9 Hz, 2H); 2,45
(t, J = 5,6 Hz, 2H); 2,12 (s, 3H).
-
Teil I
-
2-(2-Methoxyethyl)-1-{2-[2-(methylamino)ethoxy]ethyl}-1H-imidazo[4,5-c]chinolin-4-amin
(929 mg, 2,71 mmol) wurde in 30 ml wasserfreiem CH2Cl2 gelöst
und mit Phenylisocyanat (300 μl,
2,76 mmol) behandelt. Nach Rühren
unter N2 über Nacht wurde die Reaktionsmischung
unter vermindertem Druck aufkonzentriert. Durch Reinigung mittels
Säulenchromatographie
(SiO2, 3% MeOH/CHCl3 mit
wäßrigem NH4OH gesättigt)
wurde das Produkt in Form eines weißen Feststoffs erhalten. Durch
Kristallisation aus H2O und MeOH wurden
610 mg N-(2-{2-[4-Amino-2-(2-methoxyethyl)-1H-imidazo[4,5-c]chinolin-1-yl]ethoxy}ethyl)-N-methyl-N'-phenylharnstoff
in Form von schuppigen weißen
Kristallen erhalten. Fp. 184,8–185,8°C;
MS
463 (M + H)+;
1H-NMR
(300 MHz, DMSO-d6) δ 8,16 (s, 1H); 8,06 (d, J =
7,7 Hz, 1H); 7,61 (dd, J = 1,0, 8,3 Hz, 1H); 7,43–7,38 (m,
3H); 7,25–7,17
(m, 3H); 6,91 (t, J = 7,3 Hz, 1H); 6,47 (s, 2H); 4,76 (t, J = 5,0
Hz, 2H); 3,88 (t, J = 5,1 Hz, 2H); 3,78 (t, J = 6,8 Hz, 2H); 3,48
(t, J = 5,2 Hz, 2H); 3,39 (t, J = 5,4 Hz, 2H); 3,27 (s, 3H); 3,20
(t, J = 6,8 Hz, 2H); 2,82 (s, 3H); 13C-NMR
(75 MHz, DMSO-d6) δ 155,6, 152,0, 151,9, 145,1,
140,9, 132,7, 128,5, 126,7, 126,6, 122,0, 121,4, 120,5, 120,1, 115,1,
70,5, 69,6, 69,4, 58,4, 47,7, 45,5, 35,4, 27,6.
Analyse: berechnet
für C25H30N6O3·0,12H2O: %C, 64,62; %H, 6,56; %N, 18,08. Gefunden:
%C, 64,69; %H, 6,65; %N, 18,09.
-
BEISPIEL
4 N-(2-{2-[4-Amino-2-(2-methoxyethyl)-6,7,8,9-tetrahydro-1H-imidazo[4,5-c]chinolin-1-yl]ethoxy}ethyl)-N-methyl-N'-phenylharnstoff
-
Teil A
-
2-(2-Methoxyethyl)-1-{2-[2-(methylamino)ethoxy]ethyl}-1H-imidazo[4,5-c]chinolin-4-amin
(4,22 g, 12,3 mmol) wurde in 25 ml Trifluoressigsäure gelöst und mit
PtO2 (0,5 g) behandelt. Die Reaktionsmischung
wurde unter H2 (3 kg/cm2)
geschüttelt.
Nach 4 d wurden weitere 0,5 g PtO2 zugegeben,
wonach die Hydrierung noch 3 d fortgesetzt wurde. Dann wurde der
Ansatz über
Celite filtriert und unter vermindertem Druck zu einem gelben Öl aufkonzentriert.
Das gelbe Öl
wurde in 50 ml H2O gelöst und mit 50 ml CHCl3 extrahiert. Der organische Teil wurde abgetrennt
und verworfen. Der wäßrige Teil
wurde dann durch Zugabe von 10%iger NaOH-Lösung basisch gestellt (pH ~
12). Nach Extraktion mit CHCl3 (6 × 50 ml)
wurden die vereinigten organischen Schichten über Na2SO4 getrocknet und zu einem braunen Öl aufkonzentriert.
Das braune Öl
wurde in 100 ml heißem MeOH
gelöst
und mit 1 g Aktivkohle behandelt. Die heiße Lösung wurde über Celite filtriert und bis
zur Trockne aufkonzentriert. Der erhaltene gummiartige Feststoff
wurde mehrmals mit Et2O aufkonzentriert,
was 3,19 g 2-(2-Methoxyethyl)-1-{2-[2-(methylamino)ethoxy]ethyl}-6,7,8,9-tetrahydro-1H-imidazo[4,5-c]chinolin-4-amin
in Form eines gebrochen weißen
Pulvers ergab.
MS 348 (M + H)+;
1H-NMR (300 MHz, CDCl3) δ 4,84 (s,
2H); 4,48 (t, J = 5,7 Hz, 2H); 3,84 (t, J = 6,7 Hz, 2H); 3,70 (t,
J = 5,7 Hz, 2H); 3,46 (t, J = 5,1 Hz, 2H); 3,36 (s, 3H); 3,14 (t,
J = 6,7 Hz, 2H); 2,96 (m, 2H); 2,83 (m, 2H); 2,65 (t, J = 5,1 Hz,
2H); 2,36 (s, 3H); 1,85 (m, 4H).
-
Teil B
-
2-(2-Methoxyethyl)-1-{2-[2-(methylamino)ethoxy]ethyl}-6,7,8,9-tetrahydro-1H-imidazo[4,5-c]chinolin-4-amin
(750 mg, 2,16 mmol) wurden in 30 ml wasserfreiem CH2Cl2 gelöst
und mit Phenylisocyanat (239 μl, 2,20
mmol) behandelt. Nach Rühren
unter N2 über Nacht wurde die Reaktionsmischung
unter vermindertem Druck aufkonzentriert. Durch Umkristallisieren
aus EtOAc zu CH2Cl2 wurden
170 mg N-(2-{2-[4-Amino-2-(2-methoxyethyl)-6,7,8,9-tetrahydro-1H-imidazo[4,5-c]chinolin-1-yl]ethoxy}ethyl)-N-methyl-N'-phenylharnstoff
in Form von lockeren weißen
Kristallen erhalten. Fp. 167,7–170,0 °C;
MS
467 (M + H)+;
1H-NMR
(300 MHz, DMSO-d6) δ 8,17 (s, 1H); 7,43 (d, J =
7,6 Hz, 2H); 7,21 (t, J = 7,9 Hz, 2H); 6,91 (t, J = 7,3 Hz, 1H);
5,65 (s, 2H); 4,43 (t, J = 5,0 Hz, 2H); 3,72 (t, J = 7,0 Hz, 2H);
3,70 (t, J = 5,2 Hz, 2H); 3,46–3,41
(m, 4H); 3,24 (s, 3H); 3,07 (t, J = 6,9 Hz, 2H); 2,92 (m, 2H); 2,85
(s, 3H); 2,64 (m, 2H); 1,72 (m, 4H);
13C-NMR
(75 MHz, DMSO-d6) δ 155,6, 151,2, 149,3, 146,3,
140,9, 138,4, 128,5, 124,9, 122,0, 120,1, 105,5, 70,7, 70,5, 69,5,
58,4, 48,0, 44,6, 35,5, 32,8, 27,6, 23,8, 23,1, 23,0.
Analyse:
berechnet für
C25H34N6O3: %C, 64,36; %H, 7,35; %N, 18,01. Gefunden:
%C, 64,04; %H, 7,38; %N, 18,02.
-
BEISPIEL
5 N-(2-{2-[4-amino-2-(2-methoxyethyl)-1H-imidazo[4,5-c]-chinolin-1-yl]ethoxy}ethyl)morpholin-4-carboxamid
-
Unter
Stickstoffatmosphäre
wurde 1-[2-(2-Aminoethoxy)ethyl]-2-(2-methoxyethyl)-1H-imidazo[4,5-c]chinolin-4-amin (0,75 g, 2,3
mmol) unter gelindem Erwärmen
und kräftigem
Rühren
in Dichlormethan (30 ml) und Triethylamin (0,64 ml, 4,6 mmol) gelöst. Die
Lösung
wurde in einem Eis/Wasser-Bad abgekühlt und tropfenweise mit 4-Morpholincarbonylchlorid
(0,27 ml, 2,3 mmol) versetzt. Dann wurde das Kühlbad weggenommen und der Ansatz
noch 4 h gerührt.
Der Ansatz wurde durch Zugabe von gesättigtem Natriumhydrogencarbonat
(25 ml) gequencht. Nach Phasentrennung wurde die organische Schicht
mit Wasser (3 × 25
ml) und Kochsalzlösung
(25 ml) gewaschen, getrocknet (Na2SO4), filtriert und aufkonzentriert, was einen
gelben Schaum ergab. Das Produkt wurde aus Dichlormethan und Essigsäureethylester
umkristallisiert. Die Kristalle wurden zur Entfernung von Lösungsmittelresten
mit Ether (2 × 5
ml) trituriert. Das schließlich
erhaltene Produkt wurde im Vakuumofen getrocknet, was 200 mg N-(2-{2-[4-Amino-2-(2-methoxyethyl)-1H-imidazo[4,5-c]chinolin-1-yl]ethoxy}ethyl)morpholin-4-carboxamid
in Form eines hellbraunen kristallinen Feststoffs ergab, Fp. 164–166°C.
1H-NMR (300 MHz, DMSO-d6) δ 8,06 (d,
J = 8,1 Hz, 1H), 7,61 (d, J = 7,3 Hz, 1H), 7,42 (t, J = 7,2 Hz,
1H), 7,23 (t, J = 7,8 Hz, 1H), 6,51 (s, 2H), 6,33 (t, J = 5,0 Hz,
1H), 4,74 (t, J = 4,3 Hz, 2H), 3,85–3,81 (m, 4H), 3,49 (t, J =
4,3 Hz, 4H), 3,33 (t, J = 5,9 Hz, 2H), 3,30 (s, 3H), 3,21 (t, J
= 6,8 Hz, 2H), 3,14 (t, J = 4,5 Hz, 4H), 3,08 (t, J = 6,0 Hz, 2H);
13C-NMR (75 MHz, DMSO-d6) δ 157,8, 151,9,
145,0, 132,7, 126,7, 126,6, 121,4, 120,5, 115,1, 70,4, 70,2, 69,2, 58,4,
45,5, 44,0, 27,6;
Analyse: berechnet für C22H30N6O4:
%C, 59,71, %H, 6,83, %N, 18,99. Gefunden: %C, 59,71, %H, 6,80, %N, 18,78;
MS(Cl) m/e 443 (M + H).
-
BEISPIEL
6 N-(2-{2-[4-Amino-2-(2-methoxyethyl)-1H-imidazo[4,5-c]chinolin-1-yl]ethoxy}ethyl)-N-methylmorpholin-4-carboxamid
-
2-(2-Methoxyethyl)-1-{2-[2-(methylamino)ethoxy]ethyl}-1H-imidazo[4,5-c]chinolin-4-amin
(802 mg, 2,34 mmol) wurde in 30 ml wasserfreiem CH2Cl2 gelöst
und unter N2 auf 0°C abgekühlt. Die gerührte Lösung wurde
mit Et3N (0,65 ml, 4,68 mmol) und Morpholincarbonylchlorid
(273 μl,
2,34 mmol) versetzt, wonach der Ansatz über Nacht auf Raumtemperatur
kommen gelassen wurde. Dann wurde die Reaktionsmischung durch Zugabe
von gesättigter
NaHCO3-Lösung
(30 ml) und CH2Cl2 (30
ml) gequencht. Die organische Schicht wurde abgetrennt und mit H2O und Kochsalzlösung gewaschen, über Na2SO4 getrocknet und
unter vermindertem Druck aufkonzentriert. Durch Reinigung mittels
Säulenchromatographie
(SiO2, 2–5 % MeOH/CHCl3 gesättigt mit
wäßrigem NH4OH) wurde das Produkt in Form eines farblosen
Schaums erhalten. Durch Kristallisation aus EtOAc wurden 640 mg
N-(2-{2-[4-Amino- 2-(2-methoxyethyl)-1H-imidazo[4,5-c]chinolin-1-yl]ethoxy}ethyl)-N-methylmorpholin-4-carboxamid
in Form von weißen
Kristallen erhalten. Fp. = 121,8–122,3°C.
MS 457 (M + H)+;
1H-NMR (500
MHz, DMSO-d6) δ 8,06 (dd, J = 0,9, 8,3 Hz,
1H); 7,61 (dd, J = 1,1, 8,3 Hz, 1H); 7,41 (ddd, J = 1,2, 7,0, 8,3
Hz, 1H); 7,22 (ddd, J = 1,3, 7,0, 8,1 Hz, 1H); 6,44 (s, 2H); 4,74
(t, J = 5,2 Hz, 2H); 3,84 (t, J = 5,2 Hz, 2H); 3,82 (t, J = 6,9
Hz, 2H); 3,50–3,43
(m, 6H); 3,30 (s, 3H); 3,20 (t, J = 6,9 Hz, 2H); 3,16 (t, J = 5,5
Hz, 2H); 2,88 (t, J = 4,7 Hz, 4H) ; 2, 59 (s, 3H) ;
13C-NMR (75 MHz, DMSO-d6) δ 163,8, 152,
0,151,8, 145,2, 132,7, 126,7, 121,3, 120,6, 115,1, 70,4, 69,4, 68,9, 66,1,
58,5, 49,1, 47,3, 45,5, 36,9, 27,7.
Analyse: berechnet für C23H32N6O4: %C, 60,51; %H, 7,07; %N, 18,41. Gefunden:
%C, 60,56; %H, 6,85; %N, 18,19.
-
BEISPIELE 7–21
-
Teil A
-
Eine
Lösung
von 2-{2-[(3-Aminochinolin-4-yl)amino]ethoxy}ethylcarbamidsäure-tert-butylester
(3,46 g, 10,0 mmol) in 50 ml Toluol wurde mit Orthovaleriansäuretriethylester
(2,5 ml, 14,5 mmol) behandelt, wonach die Reaktionsmischung zum
Rückfluß erhitzt
wurde. Dann wurde eine 25-mg-Portion Pyridiniumhydrochloride zugegeben
und noch 4 h am Rückfluß erhitzt.
Dann wurde der Ansatz unter vermindertem Druck bis zur Trockne aufkonzentriert.
Der Rückstand
wurde mit 50 ml CH2Cl2 gelöst und mit
gesättigtem
NaHCO3, H2O und
Kochsalzlösung
gewaschen. Der organische Teil wurde über Na2SO4 getrocknet und aufkonzentriert, was ein
grünes Öl ergab.
Das grüne Öl wurde
in 50 ml heißem
MeOH gelöst
und mit Aktivkohle behandelt. Die heiße Lösung wurde filtriert und aufkonzentriert,
was 4,12 g 2-[2-(2-Butyl-1H-imidazo[4,5-c]chinolin-1-yl)ethoxy]-ethylcarbamidsäure-tert-butylester
in Form eines gelben Öls
ergab.
-
Teil B
-
Eine
Lösung
von 2-[2-(2-Butyl-1H-imidazo[4,5-c]chinolin-1-yl)ethoxy]ethylcarbamidsäure-tert-butylester
(4,12 g, 10,0 mmol) in 50 ml CH2Cl2 wurde mit 3-Chlorperoxybenzoesäure (MCPBA,
77 %, 2,5 g, 11,2 mmol) behandelt. Nach 5 h Rühren wurde die Reaktionsmischung
mit gesättigter
NaHCO3-Lösung
behandelt, wonach die Schichten getrennt wurden. Der organische
Teil wurde mit H2O und Kochsalzlösung gewaschen
und dann über
Na2SO4 getrocknet
und aufkonzentriert, was 3,68 g 2-[2-(2-Butyl-5-oxido-1H-imidazo[4,5-c]chinolin-1-yl)ethoxy]ethylcarbamidsäure-tert-butylester
in Form eines hellbraunen Schaums ergab.
-
Teil C
-
Eine
Lösung
von 2-[2-(2-Butyl-5-oxido-1H-imidazo[4,5-c]chinolin-1-yl)ethoxy]ethylcarbamidsäure-tert-butylester (3,68
g, 8,60 mmol) in 100 ml 1,2-Dichlorethan wurde auf 80°C erhitzt
und mit 10 ml konzentrierter NH4OH-Lösung behandelt.
Die schnell gerührte
Lösung
wurde über
einen Zeitraum von 10 min mit festem p-Toluolsulfonylchlorid (1,87
g, 9,81 mmol) versetzt. Dann wurde die Reaktionsmischung in einem
Druckbehälter
verschlossen und noch 2 h erhitzt. Dann wurde die Reaktionsmischung
abgekühlt
und mit 100 ml CH2Cl2 behandelt.
Dann wurde die Reaktionsmischung mit H2O,
1%iger Na2CO3-Lösung (3×) und Kochsalzlösung gewaschen.
Der organische Teil wurde über
Na2SO4 getrocknet
und aufkonzentriert, was 3,68 g 2-[2-(4-Amino-2-butyl-1H-imidazo[4,5-c]chinolin-1-yl)ethoxy]ethylcarbamidsäure-tert-butylester
in Form eines hellbraunen Schaums ergab.
-
Teil D
-
Eine
Lösung
von 2-[2-(4-Amino-2-butyl-1H-imidazo[4,5-c]chinolin-1-yl)ethoxy]ethylcarbamidsäure-tert-butylester (3,68
g, 8,60 mmol) wurde in 20 ml 2 M HCl in EtOH suspendiert, wonach
die Mischung unter Rühren
zum Rückfluß erhitzt
wurde. Nach 3 h wurde die Reaktionsmischung zu einem Feststoff aufkonzentriert.
Der Feststoff wurde mit heißem
EtOH (50 ml) trituriert und filtriert, was 2,90 g des Produks in
Form des Hydrochloridsalzes ergab. Zur Herstellung der freien Base
wurde das Hydrochloridsalz in 50 ml H2O
gelöst
und mit 5 ml konzentriertem NH4OH behandelt.
Die wäßrige Suspension
wurde mit CH2Cl2 (3 × 50 ml)
extrahiert. Die vereinigten organischen Schichten wurden über Na2SO4 getrocknet und
aufkonzentriert, was 1-[2-(2-Aminoethoxy)ethyl]-2-butyl-1H-imidazo[4,5-c]chinolin-4-amin
in Form eines hellbraunen Pulvers ergab.
MS 328 (M + H)+;
1H-NMR (300
MHz, CDCl3) δ 7,95 (d, J = 8,3 Hz, 1H); 7,83
(d, J = 8,4 Hz, 1H); 7,50 (m, 1H); 7,30 (m, 1H); 5,41 (s, 2H); 4,69
(t, J = 5,6 Hz, 2H); 3,93 (t, J = 5,6 Hz, 2H); 3,39 (t, J = 5,1
Hz, 2H); 2,97 (t, J = 7,9 Hz, 2H); 2,76 (t, J = 5,1 Hz, 2H); 1,89
(m, 2H); 1,52 (m, 2H); 1,26 (br s, 2H); 1,01 (t, J = 7,3 Hz, 3H).
-
Teil E
-
Die
Verbindungen in der nachstehenden Tabelle wurden gemäß der Synthesemethode
von Schritt (7) von Reaktionsschema I oben nach der folgenden allgemeinen
Methode hergestellt.
-
Das
Isocyanat (84 μmol)
wurde in ein Reagenzglas mit einer Lösung von 1-[2-(2-Aminoethoxy)ethyl]-2-butyl-1H-imidazo[4,5-c]chinolin-4-amin
(25 mg, 76 μmol)
in Dichlormethan (5 ml) gegeben. Das Reagenzglas wurde verschlossen
und dann 20 h bei Umgebungstemperatur auf einen Schüttler gestellt.
Das Lösungsmittel
wurde durch Vakuumzentrifugation entfernt. Der Rückstand wurde mittels halbpräparativer
HPLC nach der oben beschriebenen Methode gereinigt. Die Struktur
der freien Base und die beobachtete genaue Masse (M + H) sind in
der nachstehenden Tabelle angegeben.
-
-
-
-
-
BEISPIELE 22–26
-
Teil A
-
Nach
der allgemeinen Methode von Teil A der Beispiele 7–21 wurde
4-Piperidinethanol (10 g, 77,4 mmol) mit Di-tert-butyldicarbonat (17,7 g, 81,3 mmol)
umgesetzt, was 13,1 g 4-(2-Hydroxyethyl)piperidin-1-carbonsäure-tert-butylester in Form
eines klaren Öls
ergab.
-
Teil B
-
Eine
Lösung
von Imidazol (3,89 g, 57,1 mmol) und Triphenylphosphin (14,98 g,
57,1 mmol) in Dichlormethan (350 ml) wurde in drei Portionen mit
Iod (7,97 g) versetzt. Nach fünf
Minuten wurde eine Lösung
der Substanz aus Teil A in Dichlormethan (70 ml) zugegeben. Die
Reaktionsmischung wurde über
Nacht bei Umgebungstemperatur gerührt. Nach Zugabe von mehr Iod
(7,97 g) wurde der Ansatz 1 h bei Umgebungstemperatur gerührt. Dann
wurde die Reaktionsmischung mit gesättigtem Natriumthiosulfat (2×) und Kochsalzlösung gewaschen, über Natriumsulfat
getrocknet, filtriert und dann unter vermindertem Druck auf konzentriert,
was einen öligen
Rückstand
ergab. Der Rückstand
wurde mittels Säulenchromatographie
(Kieselgel unter Verwendung von 20% Essigsäureethylester in Hexangemisch
als Elutionsmittel) gereinigt, was 15,52 g 4-(2-Iodethyl)piperidin-1-carbonsäure-tert-butylester
in Form eines blaßgelben Öls ergab.
-
Teil C
-
Unter
Stickstoffatmosphäre
wurde 2-(1H-imidazo[4,5-c]chinolin-1-yl)butan-1-ol (6,5 g, 26,9
mmol) in drei Portionen zu einer Lösung von Natriumhydrid (1,4
g 60%iges Material, 35,0 mmol) in wasserfreiem N,N-Dimethylformamid
gegeben. Die Reaktionsmischung wurde 45 Minuten rühren gelassen,
wonach die Gasentwicklung aufgehört
hatte. Dann wurde über
einen Zeitraum von 15 Minuten 4-(2-Iodethyl)piperidin-1-carbonsäure-tert-butylester
(10,05 g, 29,6 mmol) zugetropft. Die Reaktionsmischung wurde 2,5
h bei Umgebungstemperatur rühren
gelassen und dann auf 100°C
erhitzt und über
Nacht gerührt.
Die Reaktion war gemäß HPLC-Analyse
etwa 35% vollständig.
Nach Zugabe von gesättigter
Ammoniumchloridlösung
wurde die erhaltene Mischung 20 Minuten rühren gelassen und dann mit
Essigsäureethylester
(2×) extrahiert.
Die Essigsäureethylesterextrakte
wurden mit Wasser (2×)
und dann mit Kochsalzlösung
gewaschen, vereinigt, über
Natriumsulfat getrocknet, filtriert und dann unter vermindertem
Druck zu einem braunen Öl
aufkonzentriert. Das Öl wurde
mittels Säulenchromatographie
(Kieselgel unter Verwendung von 30% Essigsäure ethylester in Hexangemisch,
50% Essigsäureethylester
in Hexangemisch und Essigsäureethylester
als Elutionsmittel) gereinigt, was 2,2 g 4-{2-[2-(1H-Imidazo[4,5-c]chinolin-1-yl)butoxy]ethyl}piperidin-1-carbonsäure-tert-butylester
ergab.
-
Teil D
-
Nach
der allgemeinen Methode der Beispiele 7–21, Teil H, wurde die Substanz
aus Teil C oxidiert, was 4-{2-[2-(5-Oxido-1H-imidazo[4,5-c]chinolin-1-yl)butoxy]ethyl}piperidin-1-carbonsäure-tert-butylester
in Form eines Öls
ergab.
-
Teil E
-
Eine
Lösung
der Substanz aus Teil D in Dichlormethan (20 ml) wurde mit Ammoniumhydroxidlösung (20
ml) versetzt. Dann wurde über
einen Zeitraum von fünf
Minuten eine Lösung
von Tosylchlorid (0,99 g, 5,2 mmol) in Dichlormethan (10 ml) zugegeben.
Die erhaltene zweiphasige Reaktionsmischung wurde über Nacht rühren gelassen.
Dann wurde die Reaktionsmischung mit Chloroform und gesättigter
Natriumhydrogencarbonatlösung
verdünnt.
Nach Trennung der Schichten wurde die organische Schicht über Natriumsulfat
getrocknet, filtriert und dann unter vermindertem Druck zu einem
braunen Glas auf konzentriert. Diese Substanz wurde mittels Säulenchromatographie
(Kieselgel unter Verwendung von 50% Essigsäureethylester in Hexangemisch
und dann Essigsäureethylester
als Elutionsmittel) gereinigt, was 1,0 g 4-{2-[2-(4-Amino-1H-imidazo[4,5-c]chinolin-1-yl)butoxy]ethyl}piperidin-1-carbonsäure-tert-butylester
in Form eines blaßgelben
glasartigen Schaums ergab.
-
Teil F
-
Unter
Stickstoffatmosphäre
wurden 4-{2-[2-(4-Amino-1H-imidazo[4,5-c]chinolin-1-yl)butoxy]ethyl}piperidin-1-carbonsäure-tert-butylester
(1,00 g, 2,1 mmol) und 2 N ethanolische Salzsäure (10 ml, 20 mmol) vereinigt,
wonach die Lösung
14 h bei Umgebungstemperatur gerührt wurde.
Der nach Abziehen des Lösungsmittels
im Vakuum erhaltene hellbraune Feststoff wurde in Wasser gelöst. Dann
wurde gesättigtes
wäßriges Natriumhydrogencarbonat
zugegeben, bis der pH-Wert 10 erreichte. Nach Extraktion mit Dichlormethan
(3×) wurden
die organischen Fraktionen vereinigt, mit Kochsalzlösung gewaschen,
getrocknet (Na2SO4),
filtriert und im Vakuum vom größten Teil
des Lösungsmittels
befreit. Durch Zugabe von Hexan bildete sich ein Niederschlag. Durch
Vakuumfiltration wurden 0,5 g 1-{1-[(2-Piperidin-4-ylethoxy)methyl]propyl}-1H-imidazo(4,5-c]chinolin-4-amin
in Form eines hellbraunen Pulvers erhalten.
1H-NMR
(300 MHz, DMSO-d6): δ 8,34 (bs, 1H), 8,19 (d, J =
8,49 Hz, 1H), 7,61 (dd, J = 8,31, 1,13 Hz, 1H), 7,45–7,39 (m,
1H), 7,25–7,19
(m, 1H), 6,55 (s, 2H), 5,25–5,15
(m, 1H), 4,00–3,80
(m, 2H), 3,5–3,3
(m, 2H), 2,8–2,64
(m, 2H), 2,22–2,11
(m, 2H), 2,09–1,99
(m, 2H), 1,8–1,63
(bs, 1H), 1,37–1,0
(m, 5H), 0,95–0,7
(m, 5H);
13C-NMR (75 MHz, DMSO-d6): δ 152,8,
145,8, 140,6, 133,0, 127,8, 127,0, 126,9, 121,3, 121,0, 115,5, 71,8, 68,1,
58,4, 46,1, 36,3, 33,1, 32,7, 24,5, 9,9; MS (CI) m/e 368,2459 (berechnet
für C21H30N5O:
368,2450).
-
Teil G
-
Die
Verbindungen in der nachstehenden Tabelle wurden gemäß der Synthesemethode
von Schritt (7) von Reaktionsschema I oben nach der folgenden allgemeinen
Methode hergestellt.
-
Das
Isocyanat bzw. Isothiocyanat (75 μmol)
wurde in ein Reagenzglas mit einer Lösung von 1-{1-[(2-Piperidin-4-ylethoxy)methyl]propyl}-1H-imidazo[4,5-c]chinolin-4-amin (25 mg, 68 μmol) in Dichlormethan
(5 ml) gegeben. Das Reagenzglas wurde verschlossen und dann 20 h
bei Umgebungstemperatur auf einen Schüttler gestellt. Das Lösungsmittel
wurde durch Vakuumzentrifugation entfernt. Der Rückstand wurde mittels halbpräparativer HPLC
nach der oben beschriebenen Methode gereinigt. Die Struktur der
freien Base und die beobachtete genaue Masse (M + H) sind in der
nachstehenden Tabelle aufgeführt.
-
-
-
-
-
BEISPIELE 37–44
-
Teil A
-
Eine
Lösung
von 2-{2-[(3-Aminochinolin-4-yl)amino]ethoxy}ethylcarbamid-tert-butylester
(6,92 g, 20,0 mmol) in 100 ml Toluol wurde mit Orthoameisensäuretriethylester
(4,65 ml, 28,0 mmol) behandelt, wonach die Reaktionsmischung zum
Rückfluß erhitzt
wurde. Nach Zugabe einer 100-mg-Portion Pyridiniumhydrochlorid wurde
noch 2 h am Rückfluß erhitzt.
Dann wurde der Ansatz unter vermindertem Druck bis zur Trocknung
auf konzentriert.
-
Der
Rückstand
wurde in 200 ml CH2Cl2 gelöst und mit
gesättigtem
NaHCO3, H2O und
Kochsalzlösung gewaschen.
Der organische Teil wurde über
Na2SO4 getrocknet
und aufkonzentriert, was ein grünes Öl ergab. Das
grüne Öl wurde
in 200 ml heißem
MeOH gelöst
und mit 10 g Aktivkohle behandelt. Die heiße Lösung wurde filtriert und aufkonzentriert,
was 5,25 g 2-[2-(1H-Imidazo[4,5-c]chinolin-1-yl)ethoxy]ethylcarbamidsäure-tert-butylester in Form
eines hellgelben Sirups ergab.
-
Teil B
-
Eine
Lösung
von 2-[2-(1H-Imidazo[4,5-c]chinolin-1-yl)ethoxy]ethylcarbamidsäure-tert-butylester
(5,25 g, 14,7 mmol) in 200 ml CH2Cl2 wurde mit MCPBA (77%, 3,63 g, 16,3 mmol)
behandelt. Nach Rühren über Nacht
wurde die Reaktionsmischung mit gesättigter NaHCO3-Lösung behandelt, wonach die
Schichten getrennt wurden. Der organische Teil wurde mit H2O und Kochsalzlösung gewaschen und dann über Na2SO4 getrocknet und
aufkonzentriert, was 4,60 g 2-[2-(5-Oxido-1H-imidazo[4,5-c]chinolin-1-yl)ethoxy]ethylcarbamidsäure-tert-butylester
in Form eines hellbraunen Schaums ergab.
-
Teil C
-
Eine
Lösung
von 2-[2-(5-Oxido-1H-imidazo[4,5-c]chinolin-1-yl)ethoxy]ethylcarbamidsäure-tert-butylester
(4,60 g, 12,4 mmol) in 150 ml 1,2-Dichlorethan wurde auf 80°C erhitzt
und mit 10 ml konzentrierter NH4OH-Lösung behandelt. Die schnell
gerührte
Lösung
wurde über
einen Zeitraum von 10 min mit festem p-Toluolsulfonylchlorid (2,71
g, 14,2 mmol) versetzt. Die Reaktionsmischung wurde mit weiteren
2 ml konzentrierter NH4OH-Lösung behandelt
und dann in einem Druckbehälter
verschlossen und noch 3 h erhitzt. Dann wurde die Reaktionsmischung
abgekühlt
und mit 100 ml CH2Cl2 behandelt.
Dann wurde die Reaktionsmischung mit H2O,
1%iger Na2Cl3-Lösung (3×) und Kochsalzlösung gewaschen.
Der organische Teil wurde über Na2SO4 getrocknet und
aufkonzentriert, was 4,56 g 2-[2-(4-Amino-1H-imidazo [4,5-c]chinolin-1-yl)ethoxy]ethylcarbamidsäure-tert-butylester in Form
eines hellbraunen Schaums ergab.
-
Teil D
-
2-[2-(4-Amino-1H-imidazo[4,5-c]chinolin-1-yl)ethoxy]ethylcarbamidsäure-tert-butylester
(4,56 g, 12,3 mmol) wurde in 100 ml EtOH gelöst und mit 30 ml 2 M HCl in
EtOH behandelt, wonach die Mischung unter Rühren zum Rückfluß erhitzt wurde. Nach 3 h wurde
die Reaktionsmischung zu einem Feststoff aufkonzentriert. Der Feststoff
wurde mit heißem
EtOH (100 ml) trituriert und filtriert, was das Produkt in Form
des Hydrochloridsalzes ergab. Zur Herstellung der freien Base wurde
das Hydrochloridsalz in 50 ml H2O gelöst und mit 5
ml konzentriertem NH4OH behandelt. Die wäßrige Suspension
wurde mit CH2Cl2 (5 × 50 ml)
extrahiert. Die vereinigten organischen Schichten wurden über Na2SO4 getrocknet und
aufkonzentriert, was 1,35 g 1-[2-(2-Aminoethoxy) ethyl]-1H-imidazo[4,5-c]chinolin-4-amin in Form eines
hellbraunen Pulvers ergab.
MS 272 (M + H)+;
1H-NMR (300 MHz, CDCl3) δ 7,98 (d,
J = 8, 2 Hz, 1H); 7,88 (s, 1H); 7,84 (d, J = 8,4 Hz, 1H); 7,54 (m,
1H); 7,32 (m, 1H); 5,43 (s, 2H); 4,74 (t, J = 5,2 Hz, 2H); 3,97
(t, J = 5,2 Hz, 2H); 3,42 (t, J = 5,1 Hz, 2H); 2,78 (t, J = 5, 1 Hz,
2H); 1,10 (br s, 2H).
-
Teil E
-
Die
Verbindungen in der nachstehenden Tabelle wurden gemäß der Synthesemethode
von Schritt (7) von Reaktionsschema I oben nach der folgenden allgemeinen
Methode hergestellt.
-
1-[2-(2-Aminoethoxy)ethyl]-1H-imidazo[4,5-c]chinolin-4-amin (20 mg, 74 μmol) und
1-Methyl-2-pyrrolidinon (5 ml) wurden in einem Reagenzglas vereinigt
und dann unter Erhitzen mit Ultraschall behandelt, was eine Lösung ergab.
Nach Zugabe des Isocyanats (81 μmol)
wurde das Reagenzglas verschlossen und dann 20 h bei Umgebungstemperatur
auf einen Schüttler
gestellt. Das Lösungsmittel
wurde durch Vakuumzentrifugation entfernt. Der Rückstand wurde mittels halbpräparativer
HPLC nach der oben beschriebenen Methode gereinigt. Die Struktur
der freien Base und die beobachtete genaue Masse (M + H) sind in
der nachstehenden Tabelle aufgeführt.
-
-
-
-
CYTOKININDUKTION IN HUMANEN
ZELLEN
-
Zur
Beurteilung der Cytokininduktion dient ein In-vitro-Humanblutzellensystem. Die Aktivität gründet sich
auf die Messung von in Kulturmedien abgegebenem Interferon (α) und Tumornekrosefaktor
(α) (IFN
bzw. TNF), wie von Testerman et al. in „Cytokine Induction by the
Immunomodulators Imiquimod and S-27609", Journal of Leukocyte Biology, 58,
365–372
(September, 1995), beschrieben.
-
Blutzellenpräparation
zur Kultur
-
Vollblut
von gesunden menschlichen Spendern wird durch Venenpunktion in EDTA-Vacutainer-Röhrchen gesammelt.
Aus Vollblut werden durch Dichtegradientenzentrifugation mit Histopaque®-1077
periphere mononukleare Blutzellen (PBMCs) abgetrennt. Die PBMCs
werden zweimal mit Hank's
Balanced Salts Solution gewaschen und dann in einer Konzentration
von 3–4 × 106 Zellen/ml in RPMI komplett suspendiert.
Die PBMC-Suspension wird zu sterilen Flachboden-Gewebekulturplatten
mit 48 Vertiefungen (Costar, Cambridge, MA, oder Becton Dickinson
Labware, Lincoln Park, NJ) mit einem gleichen Volumen an RPMI-Komplettmedium
mit Testverbindung gegeben.
-
Vorbereitung der Verbindungen
-
Die
Verbindungen werden in Dimethylsulfoxid (DMSO) solubilisiert. Die
DMSO-Konzentration sollte eine End konzentration von 1% für die Zugabe
zu den Kulturvertiefungen nicht überschreiten.
-
Inkubation
-
Die
Lösung
der Testverbindung wird zu der ersten Vertiefung mit RPMI-Komplett
gegeben, wonach in den Vertiefungen serielle Verdünnungen
hergestellt werden. Dann wird die PBMC-Suspension im gleichen Volumen
in die Vertiefungen gegeben, wobei die Testverbindungskonzentrationen
in den gewünschten
Bereich gebracht werden. Die Endkonzentration an PBMC-Suspension beträgt 1,5–2 × 106 Zellen/ml. Die Platten werden mit sterilen
Kunststoffdeckeln abgedeckt, vorsichtig vermischt und dann in einer
5% Kohlendioxid enthaltenden Atmosphäre 18 bis 24 Stunden bei 37°C inkubiert.
-
Trennung
-
Nach
der Inkubation werden die Platten 5–10 Minuten bei 1000 U/min
(200 × g)
bei 4°C
zentrifugiert. Der zellenfreie Kulturüberstand wird mit einer sterilen
Polypropylenpipette entnommen und in sterile Polypropylenröhrchen überführt. Die
Proben werden bis zur Analyse bei –30 bis –70°C gehalten. Die Proben werden mittels
ELISA auf Interferon (α)
und Tumornekrosefaktor (α)
analysiert.
-
Analyse von Interferon
(α) und
Tumornekrosefaktor (α)
mittels ELISA
-
Die
Bestimmung der Konzentration von Interferon (α) erfolgt mittels ELISA unter
Verwendung eines Human-Multi-Species-Kits
von PBL Biomedical Laboratories, New Brunswick, NJ. Die Ergebnisse
werden in pg/ml ausgedrückt.
-
Die
Konzentration von Tumornekrosefaktor (α) (TNF) wird mit ELISA-Kits
von Genzyme, Cambridge, MA; R&D
Systems, Minneapolis, MN, oder Pharmingen, San Diego, CA, bestimmt.
Die Ergebnisse werden in pg/ml ausgedrückt.
-
Die
niedrigste gefundene Konzentration zur Induktion von Interferon
und die niedrigste gefundene Konzentration zur Induktion von Tumornekrosefaktor
für jede
Verbindung sind in nachstehender Tabelle aufgeführt. Ein "*" gibt
an, daß bei
keiner der getesteten Konzentrationen Induktion beobachtet wurde.
Im allgemeinen lag die höchste
getestete Konzentration bei 10 oder 30 μM.
-
-