-
Die
vorliegende Erfindung betrifft ein Verfahren zur Herstellung von
Copolymeren von Ethylen mit alpha-Olefinen.
-
Metallocenverbindungen
mit zwei verbrückten
Cyclopentadienylgruppen sind als Katalysatorkomponenten für die Homo-
und Copolymerisationsreaktion von Olefinen bekannt.
-
So
wird beispielsweise in der US-PS 5,001,205 die Herstellung von Copolymeren
von Ethylen mit alpha-Olefinen in Gegenwart eines katalytischen
Systems, das eine Biscyclopentadienylverbindung von Zr, Ti und Hf
und Methylalumoxan (MAO) als Cokatalysator enthält, beschrieben. Von den Ausführungsbeispielen wird
die Copolymerisation von Ethylen mit Propylen in Gegenwart von verbrücktem oder
unverbrücktem
(Tetrahydroindenyl)zirconiumdichlorid beschrieben.
-
Die
Homogenität
der alpha-Olefinverteilung in der Kette ist zwar gegenüber aus
herkömmlichen
Katalysatoren vom Ziegler-Natta-Typ auf Titan- oder Vanadiumbasis
erhaltenen Copolymeren verbessert, jedoch immer noch nicht zufriedenstellend,
so daß eine
weitere Verbesserung höchst
wünschenswert
ist.
-
Metallocenverbindungen
mit zwei Cyclopentadienylgruppierungen, die über ein einziges Atom verbrückt sind,
sind ebenfalls bekannt.
-
So
wird beispielsweise in der PCT-Anmeldung WO 96/22995 eine Klasse
von über
ein einziges Kohlenstoffatom verbrückten Metallocenen und deren
Verwendung in Katalysatoren für
die Polymerisation von Olefinen, insbesondere von Propylen, beschrieben.
Bei der Klasse von Metallocenverbindungen, die zur Verwendung bei
Propylenpolymerisationen besonders gut geeignet sein soll, handelt
es sich um diejenige der über
ein einziges Kohlenstoffatom verbrückten Bisindenyle, bei denen
die Indenylgruppierungen in 3-Stellung mit Kohlenstoff-, Silicium-
oder Germaniumatomen mit drei Kohlenwasserstoffsubstituenten substituiert
sind. Es werden aber weder Beispiele für Copolymerisationen von Ethylen
mit einem alpha-Olefin noch irgendwelche Informationen über die
Eigenschaften der erhältlichen
Ethylen-Copolymere angegeben. Insbesondere finden sich keine Angaben über die
Comonomerverteilung entlang der Polymerkette.
-
Wünschenswert
wäre die
Wahl von Katalysatoren, mit denen Ethylen-Copolymere mit verbesserter Homogenität der Verteilung
der Comonomereinheiten entlang der Polymerkette erhältlich sind.
Es wurde nun unerwarteterweise gefunden, daß man auf Ethylen basierende
Copolymere mit hohem Molekulargewicht und extrem homogener Verteilung der
Comonomereinheiten in der Polymerkette bei Temperaturen von technischem
Interesse herstellen kann, indem man die Polymerisationsreaktion
von Ethylen in Gegenwart von Metallocenkatalysatoren durchführt, die
spezielle einatomig verbrückte
Bisindenylverbindungen enthalten, die in der 3-Stellung der Indenylgruppe
substituiert sind.
-
Gegenstand
der vorliegenden Erfindung ist daher nach einer ersten Ausgestaltung
ein Verfahren zur Herstellung von Ethylen-Copolymeren mit mehr als
50 Mol-% von Ethylen abgeleiteten Einheiten, bei dem man Ethylen
in Gegenwart eines Katalysators, der durch Inberührungbringen
- (A)
einer Metallocenverbindung der Formel (I): worin
die Substituenten
R1 für
Wasserstoffatome oder C1-C20-Alkylgruppen
stehen,
die Substituenten R2 für CHR10R11-, SiR12R13R14-
oder GeR15R16R17-Gruppen stehen, wobei R10,
R11, R12 und R15 für
Wasserstoffatome, C1-C20-Alkyl-,
C3-C20-Cycloalkyl-,
C2-C20-Alkenyl-, C6-C20-Aryl-, C7-C20-Alkylaryl- oder C7-C20-Arylalkylreste, die gegebenenfalls Silicium-
oder Germaniumatome enthalten, stehen,
R13,
R14, R16 und R17 für
C1-C20-Alkyl-, C3-C20-Cycloalkyl-,
C2-C20-Alkenyl-,
C6-C20-Aryl-, C7-C20-Alkylaryl- oder
C7-C20-Arylalkylreste,
die gegebenenfalls Silicium- oder Germaniumatome enthalten, stehen,
R3 und R4 gleich oder
verschieden sind und für
Wasserstoffatome oder -CHR5R6-Gruppen
stehen,
R3 und R4 einen
gegebenenfalls Heteroatome enthaltenden Ring mit 3 bis 8 Kohlenstoffatomen
bilden können,
R5 und R6 gleich oder
verschieden sind und für
Wasserstoffatome, C1-C20-Alkyl-,
C3-C20-Cycloalkyl-, C2-C20-Alkenyl-, C6-C20-Aryl-, C7-C20-Alkylaryl-
oder C7-C20-Arylalkylreste,
die einen gegebenenfalls Heteroatome enthaltenden Ring mit 3 bis
8 Kohlenstoffatomen bilden können,
stehen,
die Substituenten R7 gleich
oder verschieden sind und für
C1-C20-Alkyl-, C3-C20-Cycloalkyl-C2-C20-Alkenyl-, C6-C20-Aryl-, C7-C20-Alkylaryl- oder C7-C20-Arylalkylreste, die gegebenenfalls Silicium-
oder Germaniumatome enthalten, stehen und zwei benachbarte Substituenten
R7 einen Ring mit 5 bis 8 Kohlenstoffatomen
bilden können,
wobei n für
eine ganze Zahl von 0 bis 4 steht,
M für ein Übergangsmetall aus der Gruppe
3, 4, 5 oder 6 oder der Lanthaniden- oder Actinidengruppe des Periodensystems
der Elemente (neue IUPAC-Version) steht,
X gleich oder verschieden
ist und für
einen monoanionischen Liganden, wie ein Wasserstoffatom, ein Halogenatom
oder eine R8-, -OR8-,
-OSO2CF3-, -OCOR8-, -SR8-, -NR8 2- oder -PR8 2-Gruppe, worin
die Substituenten R8 für einen C1-C20-Alkyl-, C3-C20-Cycloalkyl-,
C2-C20-Alkenyl-,
C6-C20-Aryl-, C7-C20-Alkylaryl-
oder C7-C20-Arylalkylrest,
der gegebenenfalls Silicium- oder Germaniumatome enthält, stehen,
steht,
p für
eine ganze Zahl von 0 bis 3 steht, die gleich der Oxidationsstufe
des Metalls M minus zwei ist; mit
- (B) einem Alumoxan und/oder einer zur Bildung eines Alkylmetallocenkations
befähigten
Verbindung erhältlich
ist, mit mindestens einem unter alpha-Olefinen, Cycloolefinen und
Polyenen ausgewählten
Comonomer polymerisiert.
-
Das Übergangsmetall
M wird vorzugsweise unter Titan, Zirconium und Hafnium ausgewählt.
-
Die
Substituenten X stehen vorzugsweise für Chloratome oder Methylgruppen.
-
Der
Substituent R1 steht vorzugsweise für ein Wasserstoffatom.
-
Als
Beispiele für
Metallocenverbindungen, die zur Verwendung bei dem erfindungsgemäßen Verfahren
geeignet sind, seien genannt:
Methylenbis(3-methylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(3-methylindenyl)zirconiumdichlorid
und -dimethyl;
Methylenbis(3-ethylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(3-ethylindenyl)zirconiumdichlorid
und -dimethyl;
Methylenbis(3-dimethylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(3-dimethylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Methylenbis(3-dimethylgermylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(3-dimethylgermylindenyl)zirconiumdichlorid
und -dimethyl;
Methylenbis(3-trimethylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(3-trimethylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Methylenbis(3-triethylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(3-triethylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Methylenbis(3-trimethylgermylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(3-trimethylgermylindenyl)zirconiumdichlorid
und -dimethyl;
Methylenbis(3-diphenylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(3-diphenylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Methylenbis(3-diethylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(3-diethylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Methylenbis(2-methyl-3-trimethylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(2-methyl-3-trimethylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Methylenbis(2-methyl-3-diethylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(2-methyl-3-diethylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Methylenbis(3-benzylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(3-benzylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Methylenbis(3-cyclopentylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(3-cyclopentylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Methylenbis(2-ethyl-3-diethylsilylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(2-ethyl-3-diethylsilylindenyl)zirconiumdichlorid
und -dimethyl.
-
In
den Metallocenverbindungen der Formel (I), in der R2 für eine CHR10R11-Gruppe steht,
steht R10 vorzugsweise nicht für ein Wasserstoffatom.
Besonders bevorzugt stehen weder R10 noch
R11 für
ein Wasserstoffatom.
-
Für die Verwendung
bei dem erfindungsgemäßen Verfahren
sind diejenigen Metallocene der Formel (I) besonders interessant,
in denen R1 für ein Wasserstoffatom und R2 für
eine CHR10R11-Gruppe
steht.
-
Beispiele,
die zu dieser Klasse gehören,
sind u.a.:
Methylenbis(3-isopropylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(3-isopropylindenyl)zirconiumdichlorid
und -dimethyl;
Methylenbis(3-isobutylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(3-isobutylindenyl)zirconiumdichlorid
und -dimethyl;
Methylenbis(3-isopentylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(3-isopentylindenyl)zirconiumdichlorid
und -dimethyl;
Methylenbis(3-diphenylmethylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(3-diphenylmethylindenyl)zirconiumdichlorid
und -dimethyl;
Methylenbis(3-biscyclohexylmethylindenyl)zirconiumdichlorid
und -dimethyl;
Isopropylidenbis(3-biscyclohexylmethylindenyl)zirconiumdichlorid
und -dimethyl.
-
Ganz
besonders bevorzugt handelt es sich bei den Metallocenverbindungen
der Formel (I) um Methylenbis(3-isopropylindenyl)zirconiumdichlorid
und Isopropylidenbis(3-isopropylindenyl)zirconiumdichlorid.
-
Die
Metallocenverbindungen der Formel (I) können durch Umsetzen der entsprechenden
Indenylliganden mit einer zur Bildung eines delokalisierten Anions
am Cyclopentadienylring befähigten
Verbindung und mit einer Verbindung der Formel MXp+2,
worin M, X und p die oben angegebene Bedeutung besitzen, hergestellt werden.
-
Die
Liganden der Formel (I) können
nach verschiedenen Verfahren hergestellt werden. Ein besonders gut
geeignetes Verfahren zur Herstellung der Liganden der Formel (I),
worin R3 und R4 für Wasserstoffatome stehen,
wird in der eigenen europäischen
Patentanmeldung 97200933.6 beschrieben. Ein besonders gut geeignetes
Verfahren zur Herstellung der Liganden der Formel (I), worin die
Substituenten R3 und R4 nicht
für Wasserstoffatome
stehen, wird in der EP-A 0 722 949 beschrieben.
-
In
dem Fall, daß mindestens
ein Substituent X in der herzustellenden Metallocenverbindung der
Formel (I) nicht für
Halogen steht, muß man
mindestens einen Substituenten X in dem erhaltenen Metallocen durch
mindestens einen Substituenten X, bei dem es sich nicht um ein Halogen
handelt, substituieren. Die Reaktion zur Substitution von Substituenten
X durch Substituenten X, bei denen es sich nicht um Halogen handelt, wird
nach allgemein angewandten Verfahren durchgeführt. So kann man beispielsweise
dann, wenn es sich bei den gewünschten
Substituenten X um Alkylgruppen handelt, die Metallocene mit Alkylmagnesiumhalogeniden
(Grignard-Reagentien) oder mit Alkyllithiumverbindungen zur Reaktion
bringen.
-
In
dem bei dem erfindungsgemäßen Verfahren
verwendeten Katalysator können
sowohl die Metallocenverbindungen der Formel (I) als auch das Alumoxan
als Produkte der Umsetzung mit einer metallorganischen Aluminiumverbindung
der Formel AlR9 3 oder
Al2R9 6,
worin die Substituenten R9 gleich oder voneinander verschieden
sind und die gleiche Bedeutung wie die Substituenten R besitzen
oder für
Halogenatome stehen, vorliegen. Die bei dem erfindungsgemäßen Verfahren
verwendeten Alumoxane sind durch Inberührungbringen von Wasser mit
einer metallorganischen Aluminiumverbindung der Formel AlR9 3 oder Al2R9 6,
worin die Substituenten R9 gleich oder voneinander
verschieden sind und die gleiche Bedeutung wie oben besitzen, mit der
Maßgabe,
daß mindestens
ein Substituent R9 nicht für Halogen
steht, erhältlich.
Das Molverhältnis
zwischen dem Aluminium und dem Wasser liegt im Bereich von 1:1 bis
100:1.
-
Als
Beispiele für
Aluminiumverbindungen der Formel AlR9 3 oder Al2R9 6 seien genannt:
Al(Me)3, Al(Et)3, AlH(Et)2, Al(iBu)2, AlH(iBu)2, Al(iHex)3, Al(iOct)3, AlH(iOct)2, Al(C6H5)3,
Al(CH2C6H5)3, Al(CH2CMe3)3,
Al(CH2SiMe3)3, Al(Me)2iBu, Al(Me)2Et, AlMe(Et)2, AlMe(iBu)2, Al(CH2-CH(Me)CH(Me)2)3, Al(Me)2iBu, Al(Me)2Cl,
Al(Et)2Cl, AlEtCl2 und
Al2(Et)3Cl3, worin Me = Methyl, Et = Ethyl, iBu = Isobutyl,
iHex = Isohexyl, iOct = 2,4,4-Trimethylpentyl.
-
Unter
den obigen Aluminiumverbindungen sind Trimethylaluminium (TMA),
Triisobutylaluminium (TIBAL) und Tris(2,4,4-trimethylpentyl)pentyl)aluminium
(TIOA) bevorzugt.
-
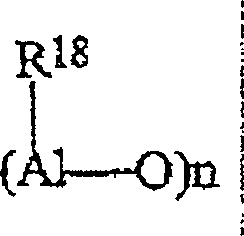
Die
in dem erfindungsgemäßen Katalysator
verwendeten Alumoxane werden als lineare, verzweigte oder cyclische
Verbindungen erachtet, die mindestens eine Gruppe des folgenden
Typs enthalten:
worin die Substituenten R
18 gleich oder verschieden sind und für gegebenenfalls
Wasserstoffatome, Silicium- oder Germaniumatome enthaltende C
1-C
20-Alkyl-, C
3-C
20-Cycloalkyl-,
C
2-C
20-Alkenyl-,
C
6-C
20-Aryl-, C
7-C
20-Alkylaryl-
oder C
7-C
20-Arylalkylreste
oder eine -O-Al(R
18)
2-Gruppe
stehen und gegebenenfalls einige Substituenten R
18 für Halogenatome
stehen können.
-
Insbesondere
kann man im Fall von linearen Verbindungen Alumoxane der Formel:
worin n für 0 oder eine ganze Zahl von
1 bis 40 steht und die Substituenten R
18 die
oben angegebene Bedeutung besitzen, oder im Fall von cyclischen
Verbindungen Alumoxane der Formel:
worin n für eine ganze Zahl von 2 bis
40 steht und die Substituenten R
18 die oben
angegebene Bedeutung besitzen, verwenden.
-
Die
Substituenten R18 stehen vorzugsweise für Ethyl-,
Isobutyl- oder 2,4,4-Trimethylpentylgruppen.
-
Als
Beispiele für
Alumoxane, die zur erfindungsgemäßen Verwendung
geeignet sind, seien Methylalumoxan (MAO), Isobutylalumoxan (TIBAO),
2,4,4-Trimethylpentylalumoxan (TIOAO) und 2,3-Dimethylbutylalumoxan
genannt.
-
Das
Molverhältnis
zwischen dem Aluminium und dem Metall der Metallocenverbindung liegt
im allgemeinen zwischen 10:1 und 20 000:1 und vorzugsweise zwischen
100:1 und 5000:1.
-
Beispiele
für zur
Bildung eines Alkylmetallocenkations befähigte Verbindungen sind u.a.
Verbindungen der Formel Y+Z–,
worin Y+ für eine zur Abgabe eines Protons
und zur irreversiblen Reaktion mit einem Substituenten X der Verbindung
der Formel (I) befähigte
Brönsted-Säure steht
und Z– für ein kompatibles
nichtkoordinierendes Anion, das die bei der Reaktion der beiden
Verbindungen entstehende aktive katalytische Spezies stabilisieren
kann, und so labil ist, daß es
durch ein Olefinsubstrat verdrängt
werden kann, steht. Vorzugsweise besteht das Anion Z– aus
einem oder mehreren Boratomen.
-
Besonders
bevorzugt handelt es sich bei dem Anion Z– um
ein Anion der Formel BAr4 (–),
worin die Substituenten Ar gleich oder verschieden sein können und
für Arylreste,
wie Phenyl, Pentafluorphenyl, oder Bis(trifluormethyl)phenyl, stehen.
Tetrakispentafluorphenylborat ist besonders bevorzugt. Außerdem können zweckmäßigerweise
Verbindungen der Formel BAr3 verwendet werden.
Verbindungen dieser Art werden beispielsweise in der veröffentlichten
internationalen Patentanmeldung WO 92/00333 beschrieben.
-
Die
erfindungsgemäßen Katalysatoren
können
auch geträgert
verwendet werden. Hierzu wird die Metallocenverbindung (A) bzw.
das Produkt der Umsetzung davon mit der Komponente (B) bzw. zunächst die Komponente
(B) und dann die Metallocenverbindung (A) auf Trägern, wie beispielsweise Siliciumoxid,
Aluminiumoxid, Magnesiumhalogeniden, Styrol/Divinylbenzol-Copolymeren,
Polyethylen oder Polypropylen, abgeschieden.
-
Eine
geeignete Klasse von verwendbaren Trägern bilden poröse organische
Träger,
die mit aktive Wasserstoffatome enthaltenden Gruppen funktionalisiert
sind. Besonders gut sind diejenigen geeignet, bei denen es sich
bei dem organischen Träger
um ein teilvernetztes Styrolpolymer handelt. Träger dieser Art werden in der
europäischen
Anmeldung EP-633 272 beschrieben.
-
Eine
andere Klasse von inerten Trägern,
die zur erfindungsgemäßen Verwendung
besonders gut geeignet sind, bilden die porösen Olefinpräpolymere
und insbesondere die porösen
Propylenpräpolymere
gemäß der internationalen
Patentanmeldung WO 95/26369.
-
Eine
weitere Klasse von inerten Trägern
zur erfindungsgemäßen Verwendung
bilden die porösen
Magnesiumhalogenide, wie z.B. diejenigen gemäß der internationalen Patentanmeldung
WO 95/32995.
-
Die
so erhaltene feste Verbindung kann in Kombination mit der weiteren
Zugabe der Alkylaluminiumverbindung, entweder als solche oder gegebenenfalls
nach vorheriger Umsetzung mit Wasser, bei der Gasphasenpolymerisation
eingesetzt werden.
-
Durch
Polymerisation von Ethylen mit alpha-Olefinen in Gegenwart der obigen
speziellen Metallocene kann man in hohen Ausbeuten bei Temperaturen
von technischem Interesse (d.h. über
50°C) Ethylen-Copolymere
mit extrem homogener Verteilung der Comonomere in der Polymerkette
erhalten, d.h. die Zahl der Sequenzen aus zwei oder mehr aufeinanderfolgenden
Einheiten der von dem alpha-Olefin abgeleiteten Einheiten ist sehr
klein. Die Analyse der Verteilung der alpha-Olefine in den erfindungsgemäßen Copolymeren
wurde mittels 13C-NMR-Spektroskopie durchgeführt. Die
Zuordnungen wurden im Fall von Ethylen/1-Hexen-Copolymeren gemäß J. C.
Randall, Macromol. Chem. Phys. (1989), 29, 201, vorgenommen.
-
Das
erfindungsgemäße Verfahren
zur Polymerisation von Olefinen kann in der Flüssigphase, gegebenenfalls in
Gegenwart von inerten Kohlenwasserstofflösungsmitteln, oder in der Gasphase
durchgeführt
werden. Das Kohlenwasserstofflösungsmittel
kann entweder aromatisch, wie Toluol, oder aliphatisch, wie Propan, Hexen,
Heptan, Isobutan oder Cyclohexan, sein.
-
Die
Polymerisationstemperatur liegt im allgemeinen zwischen –100°C und +100°C und insbesondere zwischen
10°C und
+90°C. Der
Polymerisationsdruck liegt im allgemeinen zwischen 0,5 und 100 bar.
-
Je
niedriger die Polymerisationstemperatur, desto höher sind die resultierenden
Molekulargewichte der erhaltenen Polymere.
-
Da
die Polymerisationsausbeuten von der Reinheit der Metallocenverbindung
des Katalysators abhängen,
können
die nach dem erfindungsgemäßen Verfahren
erhaltenen Metallocenverbindungen als solche verwendet oder Reinigungsbehandlungen
unterworfen werden.
-
Die
Komponenten des Katalysators können
schon vor der Polymerisation miteinander in Berührung gebracht werden. Die
Vorkontaktkonzentrationen für
die Metallocenkomponente (A) liegen im allgemeinen zwischen 10 und
10–8 mol/l
und für
die Komponente (B) im allgemeinen zwischen 10 und 10–8 mol/l.
Die Vorkontaktierung erfolgt im allgemeinen in Gegenwart eines Kohlenwasserstofflösungsmittels
und gegebenenfalls in Gegenwart von geringen Monomermengen. Bei
der Vorkontaktierung kann man auch ein nichtpolymerisierbares Olefin,
wie Isobuten, 2-Buten und dergleichen, verwenden.
-
Die
nach dem erfindungsgemäßen Verfahren
erhältlichen
Copolymere enthalten mehr als 50 mol-% und vorzugsweise zwischen
80 und 99 mol-% von Ethylen abgeleitete Einheiten.
-
Der
Gehalt an von alpha-Olefin abgeleiteten Einheiten liegt vorzugsweise
bei bis zu weniger als 50 mol-% und besonders bevorzugt zwischen
1 und 20 mol-%.
-
Der
Gehalt an von Polyen abgeleiteten Einheiten liegt vorzugsweise zwischen
0 und 4% und besonders bevorzugt zwischen 0 und 3%.
-
Beispiele
für alpha-Olefine,
die bei dem erfindungsgemäßen Verfahren
als Comonomere verwendet werden können, sind u.a. Propylen, 1-Buten,
1-Penten, 4-Methyl-1-penten, 1-Hexen,
1-Octen, 4,6-Dimethyl-1-hepten, 1-Decen, 1-Dodecen, 1-Tetradecen,
1-Hexadecen, 1-Octadecen,
1-Eicosen und Allylcyclohexan.
-
Beispiele
für cyclische
Olefine, die bei dem erfindungsgemäßen Verfahren als Comonomere
verwendet werden können,
sind u.a. Cyclopenten, Cyclohexen und Norbornen.
-
Die
erfindungsgemäßen Copolymere
können
auch von Polyenen abgeleitete Einheiten enthalten.
-
Die
Polyene, die in den erfindungsgemäßen Copolymeren als Comonomere
verwendet werden können,
gehören
zu den folgenden Klassen:
- – cyclopolymerisierbare nichtkonjugierte
Diolefine, wie beispielsweise 1,5-Hexadien, 1,6-Heptadien und 2-Methyl-1,5-hexadien;
- – zur
Lieferung ungesättigter
monomerer Einheiten befähigte
Diene, insbesondere konjugierte Diene, wie beispielsweise Butadien
und Isopren, und lineare nichtkonjugierte Diene, wie beispielsweise
trans-1,4-Hexadien, cis-1,4-Hexadien, 6-Methyl-1,5-heptadien, 3,7-Dimethyl-1,6-octadien
und 11-Methyl-1,10-dodecadien.
-
Polyene,
bei denen es sich nicht um nichtkonjugierte alpha-omega-Diolefine
mit 6 oder mehr Kohlenstoffatomen handelt, werden vorzugsweise in
Mengen zwischen 0 und 3 mol-%
als zweites alpha-Olefin-Comonomer verwendet.
-
Eine
besonders interessante Ausführungsform
der vorliegenden Erfindung besteht aus Copolymeren von Ethylen mit
1-Hexen oder höheren
alpha-Olefinen.
-
Die
erfindungsgemäßen Copolymere
sind durch eine extrem homogene Verteilung der Comonomere und genauer
gesagt dadurch, daß sie
eine extrem kleine Zahl von Sequenzen aus zwei oder mehr aufeinanderfolgenden
alpha-Olefin-Einheiten enthalten, gekennzeichnet.
-
Die
Analyse der Verteilung der Comonomereinheiten in den erfindungsgemäßen Copolymeren
wurde mittels 13C-NMR-Spektroskopie durchgeführt. Die
Zuordnungen wurden gemäß Randall,
Macromol. Chem. Phys. 1989, 29, 201, vorgenommen. Im Fall von Ethylen/1-Hexen
wird die Triadenverteilung mit Hilfe der folgenden Beziehung berechnet: HHH = Tββ EHE = Tδδ HHE
= Tβδ HEH
= Sββ HHE=Sβδ EEE = 0,5(Sδδ + 0,5Sγδ)worin
EHE, HHE und HHH die Sequenz Ethylen/1-Hexen/Ethylen, 1-Hexen/1-Hexen/Ethylen bzw.
1-Hexen/1-Hexen/1-Hexen im Copolymer bedeuten. Für die NMR-Nomenklatur siehe
H. Carmen, R. A. Harrington, C. E. Wilkes, Macromolecules, 10, 537
(1977). Die Werte sind normalisiert. Je höher die Zahl isolierter 1-Hexen-Einheiten
in der Polymerkette, desto näher
liegen die Werte des Verhältnisses
EHE/(EHE + HHE + HHH) an eins.
-
Die
Zahl der 1-Hexen-Sequenzen scheint eine Funktion der Menge der in
der Kette vorliegenden 1-Hexeneinheiten zu sein.
-
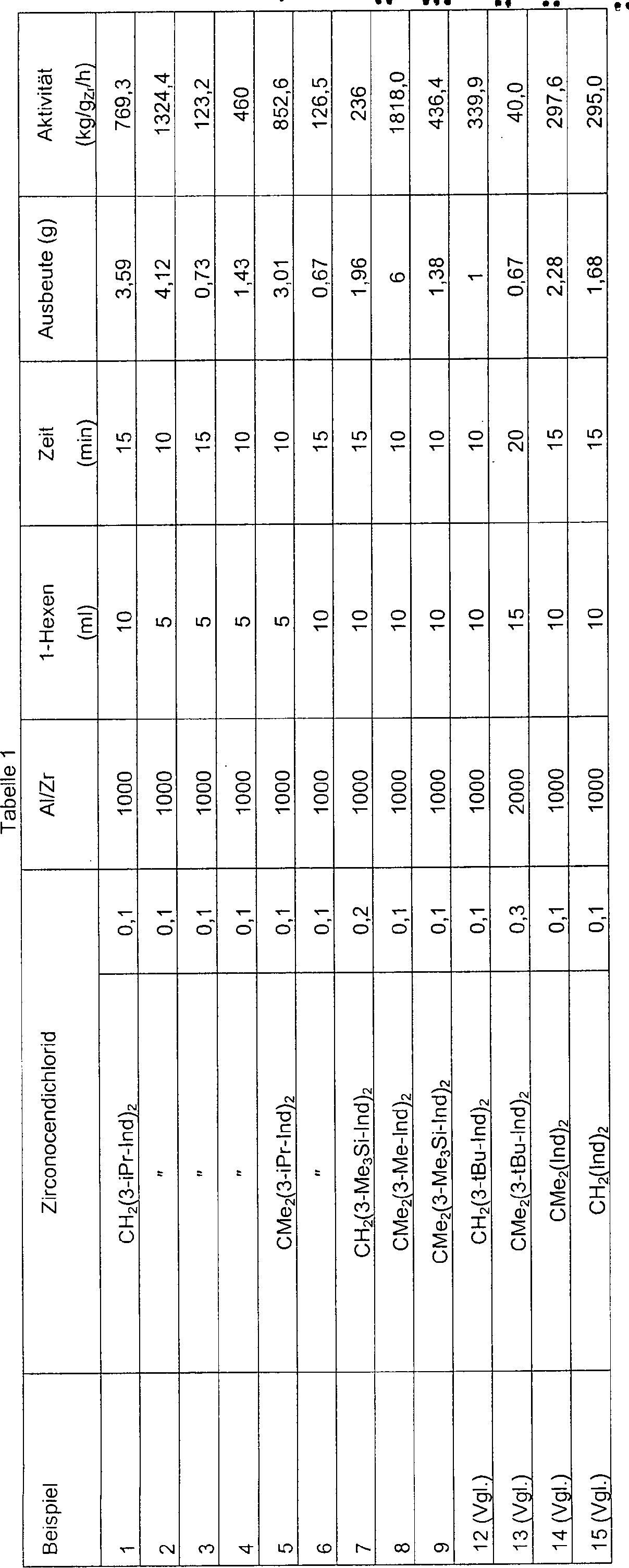
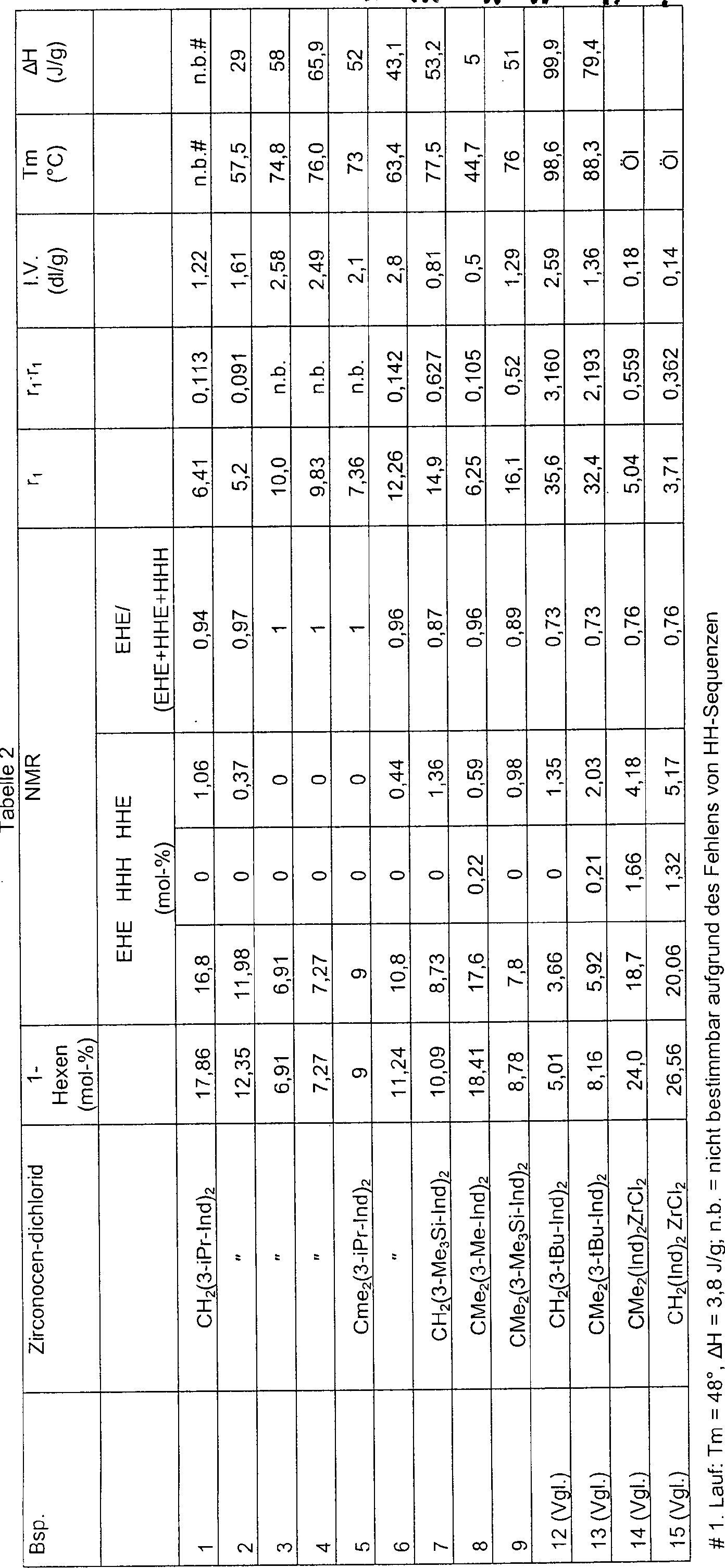
Die
Tabellen 2 und 3 beziehen sich auf nach einem erfindungsgemäßen Verfahren
erhaltene Ethylen/1-Hexen-Copolymere.
-
Insbesondere
sind in Tabelle 2 die Verhältnisse
EHE/(EHE + HHE + HHH) als Funktion des Molprozentanteils von 1-Hexen
in der Kette für
nach einem erfindungsgemäßen Verfahren
in Gegenwart der oben angegebenen Metallocenverbindungen erhaltene
Ethylen/1-Hexen-Copolymere angegeben. Bei gleichen Mengen von 1-Hexen-Einheiten
sind die Werte für
das Verhältnis
EHE/(EHE + HHE + HHH) für
die erfindungsgemäßen Copolymere
immer höher
als diejenigen für
die mit in den Vergleichsbeispielen verwendeten Metallocenen erhaltenen
Copolymere, was die verbesserte Verteilung von 1-Hexen-Einheiten
in der Kette reflektiert.
-
In
den erfindungsgemäßen Copolymeren
scheint das Produkt der Reaktivitätsverhältnisse r1·r2, worin r1 für die relative
Reaktivität
des Comonomers gegenüber
Ethylen und r2 für die relative Reaktivität von Ethylen gegenüber dem
Comonomer steht, sehr klein zu sein. Insbesondere ist es im allgemeinen
kleiner als 0,30, vorzugsweise kleiner als 0,20 und besonders bevorzugt
kleiner als 0,15. Die Diaden wurden aus der Triadenverteilung berechnet.
-
Im
Fall von Ethylen/1-Hexen wird das Produkt der Reaktivitätsverhältnisse
r1·r2 gemäß J. Uozomi,
K. Soga, Mak. Chemie, 193, 823, (1992), nach der folgenden Formel
berechnet: r1 = 2EE/(EH)X r1·r2 =
4(EE·HH)/EH2, worinX = Monomeren-Molverhältnis [E]/[H]
im Polymerisationsbad.
-
Insbesondere
erfüllt
das Verhältnis
EHE/(EHE + HHE + HHH) die folgenden Beziehung: EHE/(EHE
+ HHE + HHH) ≥ 0,75vorzugsweise: EHE/(EHE + HHE + HHH) ≥ 0,85besonders bevorzugt EHE/(EHE + HHE + HHH) ≥ 0,9.
-
Die
intrinsische Viskosität
(I.V.) der erfindungsgemäßen Copolymere
liegt im allgemeinen über
0,5 dl/g und vorzugsweise über
1,0 dl/g. Die intrinsische Viskosität kann Werte von 3,0 dl/g und
noch höher
erreichen.
-
Das
Molekulargewicht der Polymere kann auch durch Variation der Art
oder Konzentration der Katalysatorkomponenten oder durch Verwendung
von Molekulargewichtsreglern, wie beispielsweise Wasserstoff, variiert
werden.
-
Die
erfindungsgemäßen Copolymere
zeichnen sich im allgemeinen durch eine enge Molekulargewichtsverteilung
aus. Die Molekulargewichtsverteilung wird durch das Verhältnis Mw/Mn repräsentiert,
das für die
erfindungsgemäßen Copolymere
bei Verwendung des Metallocens in Form eines reinen Isomers im allgemeinen
unter 4, vorzugsweise unter 3,5 und besonders bevorzugt unter 3
liegt.
-
Die
Molekulargewichtsverteilung kann durch Verwendung von Mischungen
verschiedener Metallocenverbindungen oder durch Durchführung der
Polymerisation in mehreren Stufen bei verschiedenen Polymerisationstemperaturen
und/oder verschiedenen Konzentrationen der Molekulargewichtsregler
variiert werden.
-
Die
erfindungsgemäßen Copolymere
können
durch herkömmliche
Verarbeitung thermoplastischer Materialien (Abformen, Extrusion,
Spritzguß usw.)
in Formkörper
umgewandelt werden.
-
Die
folgenden Beispiele sollen die Erfindung erläutern, ohne ihren Schutzbereich
einzuschränken.
-
ALLGEMEINE
VORSCHRIFTEN UND CHARAKTERISIERUNGEN
-
Es
werden folgende Abkürzungen
verwendet:
- THF
- = Tetrahydrofuran
- Et2O
- = Ethylether
- NaOEt
- OEt = Natriumethoxid
- tBuOK
- = Kalium-tert.-butoxid
- DMSO
- = Dimethylsulfoxid
- DMF
- = N,N-Dimethylformamid
- BuLi
- = Butyllithium
-
Alle
Arbeiten wurden unter Stickstoff nach herkömmlichen Schlenktechniken durchgeführt. Lösungsmittel
wurden über
blauem Natriumbenzophenonketyl (Et2O), CaH2 (CH2Cl2)
bzw. AliBu3 (Kohlenwasserstoffe) destilliert
und unter Stickstoff aufbewahrt. BuLi (Aldrich) wurde in Lieferform
verwendet.
-
Die 1H-NMR-Analysen der Metallocene wurden auf
einem Spektrometer der Bauart DPX 200 von Bruker durchgeführt (CD2Cl2, bezogen auf
das mittlere Signal des Tripletts von restlichem CHDCl2 bei
5,35 ppm). Alle NMR-Lösungsmittel
wurde vor Gebrauch über
P2O5 getrocknet
und destilliert. Die Proben wurden unter Stickstoff nach standardmäßigen Inertatmosphärentechniken
hergestellt.
-
Die 13C-NMR- und 1H-NMR-Analysen
der Polymere wurden auf einem Spektrometer der Bauart DPX 400 von
Bruker mit einer Betriebsfrequenz von 400,13 MHz bzw. 100,61 MHz
durchgeführt.
Die Proben wurden als Lösungen
in Tetrachlordideuteroethan bei 120°C analysiert.
-
Die
intrinsische Viskosität
(I.V.) wurde bei 135°C
in Tetralin gemessen.
-
Die
Schmelzpunkte der Polymere (Tm) wurden mittels Differentialkalorimetrie
(D.S.C.) auf einem DSC-Instrument von Mettler folgendermaßen gemessen.
Etwa 10 mg einer aus der Polymerisation erhaltenen Probe wurden
auf –25°C abgekühlt und
danach mit einer Scangeschwindigkeit von 20°C/Minute auf 200°C erhitzt.
Die Probe wurde 5 Minuten bei 200°C
gehalten und danach mit einer Scangeschwindigkeit von 20°C/Minute
auf 0°C
abgekühlt.
Danach wurde ein zweiter Scan mit einer Scangeschwindigkeit von
10°C/Minute durchgeführt. Die
angegebenen Werte wurden beim zweiten Scan erhalten.
-
Die
Molekulargewichtsverteilung wurde mittels GPC auf einem Instrument
WATERS 150 in ortho-Dichlorbenzol bei 135°C bestimmt.
-
HERSTELLUNG
DER METALLOCENE
-
Die
Synthese von rac-Isopropylidenbis(3-isopropylindenyl)zirconiumdichlorid (rac-CMe2(3-iPr-Ind)ZrCl2), rac-Isopropylidenbis(3-trimethylsilylindenyl)zirconiumdichlorid (rac-CMe2(3-Me3Si-Ind)2ZrCl2), rac-Isopropylidenbis(3-methylindenyl)zirconiumdichlorid (rac-CMe2(3-Me-Ind)2ZrCl2), rac-Isopropylidenbis(3-tert.-butylindenyl)zirconiumdichlorid (rac-CMe2(3-tBu-Ind)2ZrCl2) wurde gemäß WO 96/22995
durchgeführt.
-
Synthese von rac-Methylenbis(3-t-butyl-1-indenyl)zirconiumdichlorid
-
(a) Synthese von t-Butylinden
-
42,0
g Inden (technisch, 94%ig gemäß GC, 39,5
g, 340 mmol), 50 gew.-%iges wäßriges KOH
(308 g in 308 ml) und 15,8 g Adogen (Aldrich, 34 mmol) als Lösung in
139,7 g tert.-Butylbromid
(1019,6 mmol) wurden bei Raumtemperatur in dieser Reihenfolge in
einen ummantelten 1-l-Glasreaktor mit mechanischem Rührer (Büchi) gegeben.
Die organische Phase färbt
sich grün.
Die Mischung wird auf 60°C
erhitzt und zwei Stunden kräftig
gerührt
(wobei ein Druckanstieg auf 2,5 bar Überdruck beobachtet wird) und
dann auf Raumtemperatur abgekühlt.
Die Gesamtreaktionszeit beträgt
3 h. Die organische Phase wird mit technischem Hexan (3 × 200 ml)
extrahiert und mittels GC analysiert. Umsatz: 74,5 Gew.-% 3-tert.-Butylinden
und 1,8% 1-tert.-Butylinden, nicht umgesetztes Inden 13,7 Gew.-%.
Nach Eindampfen der Lösung
unter vermindertem Druck (Rotationsverdampfer) wurde die erhaltene
dunkelbraune viskose Flüssigkeit
bei 1 mm Hg destilliert, wobei die zwischen 70 und 80°C siedende
Fraktion aufgefangen wurde (40 g, 76,8% 3-tert.-Butylinden und 19,5%
1-tert.-Butylinden, kein Inden.
-
(b) Synthese von Bis(3-t-butylindenyl)methan
-
In
einem 1-l-Dreihalskolben mit Rührstab
wurden in der angegebenen Reihenfolge 10,32 g BuOK (92 mmol), 400
ml DMF und 80,6 g tert.-Butylinden (98,2%ig gemäß GC, 460 mmol) vorgelegt wonach über einen Zeitraum
von 15 min 18,6 ml wäßrige Formaldehydlösung (37%ig,
6,9 g, 230 mmol) tropfenweise zugetropft wurden. Es wird eine leicht
exotherme Reaktion beobachtet, und die Lösung färbt sich rot. Die Mischung
wurde 2 Stunden bei Raumtemperatur gerührt, wonach die Reaktion durch
Gießen
der Mischung auf Eis und NH4Cl gequencht,
mit Et2O (2 × 250 ml) extrahiert und unter
vermindertem Druck aufkonzentriert wurde, was ein oranges öliges Produkt
mit der folgenden Zusammensetzung gemäß GC ergab: 0,3% 1-tBulnd; 2,8% 3-tBulnd; 78,3%; Zielprodukt
Rest Nebenprodukt. Rohproduktausbeute: 83,6 g, was einer Ausbeute
von 79,9% entspricht. Das orange ölige Produkt kristallisierte
beim Stehen (etwa 1 h). Dieses Produkt wurde durch Waschen mit Pentan
weiter gereinigt, wobei Bis(1-tert.-butyl-3-indenyl)methan in Form eines hellgelben
Pulvers mit einer GC-Reinheit von 99,8% zurückbleibt.
-
(c) Synthese von Methylenbis(3-t-butyl-1-indenyl)zirconiumdichlorid
-
In
einem 250-ml-Schlenkrohr wurden 11,0 g reines Bis(1-tert.-butyl-3-indenyl)methan
(30,9 mmol) in 200 ml Et2O gelöst, wonach
die Lösung
auf –15°C abgekühlt wurde.
Dann wurden über
einen Zeitraum von 15 min unter Rühren 40 ml 1,6 M BuLi in Hexan
(63,3 mmol) zugetropft. Die Lösung
wird auf Raumtemperatur kommen gelassen und 4,5 Stunden gerührt. Es
entwickelt sich eine zunehmende Trübung, und schließlich bildet
sich eine gelbe Suspension. 7,2 g ZrCl4 (30,9
mmol) wurden in 200 ml Pentan aufgeschlämmt. Die beiden Mischungen
wurden beide auf –80°C abgekühlt, wonach
die Lithiumsalzlösung
in Et2O schnell zu der ZrCl4-Aufschlämmung in
Pentan gegeben wurde. Das Kühlbad
wird weggenommen. Nach 20 min schlägt die Farbe der Aufschlämmung von
gelb nach rot um. Die Reaktionsmischung wurde über Nacht bei Raumtemperatur
gerührt
und dann unter vermindertem Druck bis zur Trockne eingedampft. Das
rote Pulver wurde in 200 ml Pentan aufgeschlämmt und in eine Filtrationsapparatur
mit das System ober- und unterhalb der Fritte verbindendem Seitenarm
(zur Ermöglichung
des Refluxierens von Lösungsmittel),
einem Auffangkolben am unteren Ende und einem Blasenkühler am
oberen Ende überführt. Der
rote Feststoff wurde etwa 3,5 Stunden mit refluxierendem Pentan
extrahiert. Das Filtrat wurde unter vermindertem Druck bis zur Trockne
eingedampft, was eine rote Paste ergab, die rac-CH2(3-tBu-Ind)2ZrCl2 enthielt, das von seinem meso-Isomer frei
war, aber polymere Nebenprodukte enthielt. Die Paste wurde zweimal
mit Et2O (20 + 10 ml) gewaschen, was 1 g
reines Produkt ergab. Der rote Feststoff auf der Fritte wurde mit
CH2Cl2 weiter extrahiert,
bis das Filtrat hellorange war (6 Stunden), und getrocknet. Gemäß 1H-NMR liegt reines rac-CH2(3-tBu-Ind)2ZrCl2 (7,25 g) vor. Die Gesamtausbeute (8,25
g rotes Pulver) von rac-CH2(3-tBu-Ind)2ZrCl2 beträgt 52%. 1H-NMR (CDCl3, d,
ppm): s, 1,41, tBu, 18H; s, 4,78, CH2, 2H; s, 5,79, 2H, Cp-H; m, 7,15, 2H, m,
7,36, 2H; m, 7,47, 2H; m, 7,78, 2H.
-
Synthese von Methylenbis(3-isopropyl-1-indenyl)ZrCl2
-
(a) Synthese von 3-Isopropyl-1-inden
-
In
einem 0,5-l-Kolben wurden 25 g Inden (Aldrich, 94,4%ig) in 140 ml
Et2O vorgelegt, auf –20°C abgekühlt und in etwa 30 min tropfenweise
mit 141 ml n-BuLi (1,6 M in Hexan, 226 mmol) versetzt. Die Reaktionsmischung
wurde auf Raumtemperatur kommen gelassen und dann 5 Stunden gerührt (braunorange
Lösung). Diese
Lösung
wurde dann langsam zu einer bei 0°C
gehaltenen Lösung
von 101 ml i-PrBr (Aldrich, MW 123 g/mol, d = 1,31 g/ml, 1,07 mol)
in 140 ml Et2O gegeben. Es wurde unter Rühren 72
Stunden bei Raumtemperatur umgesetzt. Proben für die GC-Analyse wurden nach
24 h (Inden = 12,1%, i-PrInd = 56,5%, (i-Pr)2Ind
= 18,8%), 48 h (Inden = 4,6%, i-PrInd = 66,8%, (i-Pr)2Ind
= 16,5%) und am Ende (Inden = 4,8%, i-PrInd = 65,3%, (i-Pr)2Ind = 16,8%) entnommen. Die Mischung wurde
auf 300 g Eis gegossen, wonach die Wasserschicht mit Et2O
(3 × 200
ml) extrahiert wurde und die Et2O-Waschlösung mit
der organischen Schicht vereinigt, über MgSO4 getrocknet
und nach Filtratrion unter Vakuum vom Lösungsmittel befreit wurde,
was 30,9 g eines gelben Öls
ergab (Ausbeute gemäß GC-Analyse
62%). 18 g dieses Öls
wurden (unter Zugabe von NaOH-Plätzchen
zur Verhinderung von Polymerisation mit einer 20-cm-Vigreux-Kolonne),
destilliert, wobei die bei 10 mmHg bei 95–105°C siedende Fraktion aufgefangen
wurde, 10 g, GC: i-Prlnd (2 Isomere) = 92,1%, (i-Pr)Ind = 6,7%. 1H-NMR (CDCl3, d,
ppm): d: 1,45, 1,47, 6H; m, 3,47, CH, 1H; s, 3,47, 2H, CH2; s, 6,35, 1H; m, 7,47, 2H; m, 7,3–7,74H.
Das Hauptisomer ist 3-i-Pr-Inden.
-
(b) Synthese von Bis(3-isopropylindenyl)methan
-
In
einem 500-ml-Dreihalskolben mit Rührstab wurden in der angegebenen
Reihenfolge 10 g i-Pr-Inden (92%ig, MG 158, 58,3 mmol) als Lösung in
250 ml DMSO und 1,42 g t-BuOK
(MG 112,82, 12,6 mmol) vorgelegt. Die gelbe Lösung wird grün. Dann
werden in 15 min 2,56 ml wäßrige Formaldehydlösung (37%ig,
MG 30,03, 31,6 mmol) in 70 ml DMSO zugegeben. Es wird eine schwach
exotherme Reaktion beobachtet, und die. Lösung färbt sich dunkelbraun. Nach
beendeter Zugabe wurde die Reaktionsmischung 16 h bei Raumtemperatur
gerührt.
Dann wurde die Reaktion durch Gießen der Mischung auf 200 g
Eis mit 0,3 g NH4Cl gequencht. Das organische
Produkt wurde mit Et2O extrahiert, wonach
die Wasserschicht mit Et2O (3 × 100 ml)
gewaschen wurde und die organischen Schichten vereinigt, über MgSO4 getrocknet, filtriert und aufkonzentriert
wurden, was 13,65 g gelbes Öl
ergab, das gemäß GC-Analyse
32% des gewünschten
Produkts enthielt.
-
(c) Synthese von Methylenbis(3-isopropylindenyl)ZrCl2
-
In
einem 250-ml-Schlenkrohr wurden 13,6 g rohes Bis(3-isopropyl-1-indenyl)methan
in 200 ml Et2O gelöst, wonach die Lösung auf –80°C abgekühlt wurde. Über einen
Zeitraum von 15 min wurden unter Rühren 33,3 ml 2,5 M BuLi in
Hexan (83,2 mmol) zugetropft. Die Lösung wurde auf Raumtemperatur
kommen gelassen und 5 Stunden gerührt. Es entwickelt sich eine
zunehmende Trübung,
und schließlich
bildet sich ein oranger Niederschlag. Nach Abziehen von Et2O unter Vakuum wurden 200 ml Toluol zugegeben.
9,7 g ZrCl4 (MG 233,03, 41,62 mmol) wurden
in 200 ml Toluol aufgeschlämmt.
Die beiden Mischungen wurden beide auf –80°C abgekühlt, wonach die ZrCl4-Aufschlämmung
in Toluol schnell zu der Lithiumsalzlösung in Toluol gegeben wurde.
Das Kühlbad
wird entfernt. Die Reaktionsmischung wird über Nacht bei Raumtemperatur
gerührt.
Filtration: Der Rückstand
war eine klebrige Masse (verworfen). Das Filtrat wurde unter vermindertem
Druck auf 25 ml eingeengt, wonach der ausgefallene Feststoff abfiltriert
wurde: 1H-NMR (CD2Cl2, d, ppm): 92% meso; ps-t, 1,31, i-Pr, 12H;
Quintett, 3,32, CH, 2H; Quartett, 4,84, 4,91, 5,01, 5,08, 2H, CH2-Brücke;
s, 5,81, 2H, Cp-H; t, 6,9–7,0,
2H; t, 7,06–7,15,
2H; m, 7,47–755,
4H.
-
Das
Filtrat wurde getrocknet, was einen roten klebrigen Feststoff (5,8
g) ergab, der in 30 ml Et2O und 2 ml CH2Cl2 dispergiert
und bei 0°C
filtriert wurde. Der Rückstand
wurde getrocknet, was 1 g rotes Pulver ergab. Gemäß 1H-NMR liegt chemisch reines CH2(3-i-PrInd)2ZrCl2 (80% racemisch,
20% meso) vor. 1H-NMR (CD2Cl2, d, ppm): 1,17, 1,21, CH3, 6H; d, 1,31,
1,34, CH3, 6H; Quintett, 3,13–3,20,
CH, 2H; s, 4,82, 2H, CH2-Brücke;
s, 5,78, 2H, Cp-H; t, 7,07–7,13,
2H; t, 7,25–7,30,
2H; d, 7,47–7,52,
2H; d, 7,60–7,65,
2H.
-
Synthese von Methylenbis(3-trimethylsilyl-1-indenyl)zirconiumdichlorid
-
(a) Synthese von Bis(1-trimethylsilyl-3-indenyl)methan
-
In
einem 250-ml-Schlenkrohr wurden 9,56 g wie in Synthese 10 beschrieben
erhaltenes Bis(1-indenyl)methan (39,1 mmol) in 70 ml Et2O
gelöst,
wonach die Lösung
auf –78°C abgekühlt wurde. Über einen
Zeitraum von 30 Minuten wurden unter Rühren 33,0 ml 2,5 M BuLi in
Hexan (82,5 mmol) zugetropft. Die erhaltene Lösung wurde auf Raumtemperatur
kommen gelassen und dann 3 Stunden gerührt, was eine dunkelbraune, leicht
trübe Lösung ergab.
10,5 ml Chlortrimethylsilan (82,7 mmol) wurden in 50 ml Et2O gelöst.
Die beiden Mischungen wurden beide auf –78°C abgekühlt, wonach die Lithiumsalzlösung in
Et2O über
einen Zeitraum von 20 Minuten zu der Chlortrimethylsilanlösung in
Et2O gegeben wurde; die Farbe der Lösung schlug
von braun nach rötlichbraun
um. Nach Wegnehmen des Kühlbads
wurde die Reaktionsmischung über
Nacht bei Raumtemperatur gerührt.
Nach 20 Stunden wurde die etwas klarere Lösung mit einigen ml MeOH gequencht, filtriert
und aufkonzentriert, was 11,28 g Bis(1-trimethylsilyl-3-indenyl)methan
in Form eines dunkelbraunen Öls ergab
(74,2% Ausbeute, meso/rac = 1/1).
1H-NMR
(CDCl3, δ,
ppm): –0,04
bis –0,03
(s, 18H, CH3); 3,35–3,45 (m, 2H, CH oder CH2-Brücke); 3,93–4,00 (bs,
2H, CH2-Brücke oder CH); 6,30–6,40 (m,
2H, Cp-H); 7,10–7,50
(m, 8H).
-
(b) Synthese von Methylenbis(3-trimethylsilyl-1-indenyl)zirconiumdichlorid
CH2(3-Me3Si-Ind)2ZrCl2
-
In
einem 250-ml-Schlenkrohr wurden 4,90 g wie oben angegeben erhaltenes
Bis(1-trimethylsilyl-3-indenyl)methan
(12,6 mmol) in 70 ml Et2O gelöst, wonach
die Lösung
auf –70°C abgekühlt wurde.
Dann wurden unter Rühren
10,6 ml 2,5 M BuLi in Hexan (26,5 mmol) zugetropft. Die Lösung wurde
auf Raumtemperatur kommen gelassen und 3 Stunden gerührt. Es
entwickelte sich eine zunehmende Trübung, und schließlich bildete
sich eine dunkelbraune Suspension. 2,94 g ZrCl4 (12,6
mmol) wurden in 50 ml Pentan aufgeschlämmt. Die beiden Mischungen
wurden beide auf –70°C abgekühlt, wonach
die Lithiumsalzlösung
in Et2O schnell zu der ZrCl4-Aufschlämmung in
Pentan gegeben wurde; dann wurde das Kühlbad weggenommen. Die Reaktionsmischung
wurde über
Nacht bei Raumtemperatur gerührt,
wobei die Farbe der Suspension nach rötlichbraun umschlug. Nach Filtrieren
wurde der Rückstand
aufkonzentriert und dann mit Toluol extrahiert, was ein rosarotes
Pulver ergab. Gemäß 1H-NMR-Analyse lag meso/rac-CH2(3-Me3Si-1-Ind)2ZrCl2 im Verhältnis 75/25
vor. Das Filtrat wurde getrocknet, was einen dunkelbraunen klebrigen
Feststoff ergab, und mit Pentan versetzt; die erhaltene Mischung
wurde 1 Stunde bei Raumtemperatur gerührt und dann filtriert. Der
Rückstand
wurde schließlich
getrocknet, was 1,87 g eines orangen Pulvers ergab. Gemäß 1H-NMR-Analyse lag rac/meso-CH2(3-Me3Si-1-Ind)2ZrCl2 in einem Verhältnis von 81/19 vor (27,0%
Ausbeute).
1H-NMR (CD2Cl2, δ,
ppm): 0,22 (s, 6H, CH3); 0,34 (s, 6H, CH3); 4,79 (s, CH2-Brücke, 2H);
4,93 (q, CH2-Brücke, 2H); 6,47 (s, Cp-H, 2H);
6,57 (s, Cp-H, 2H); 7,06–7,72
(m, 16H).
-
POLYMERISATION
-
Methylalumoxan (MAO)
-
Eine
im Handel (von Witco) erhältliche
10%ige Toluollösung
wurde im Vakuum zu einer festen, glasartigen Substanz getrocknet,
die fein zerstoßen
und im Vakuum von allen flüchtigen
Bestandteilen befreit wurde (4–6
Stunden, 0,1 mmHg, 50°C),
was ein weißes,
freifließendes
Pulver ergab.
-
Tris(2,4,4-trimethylgentyl)aluminium
(TIOA)
-
Eine
im Handel (von Witco) erhältliche
Probe wurde in dem angegebenen Lösungsmittel
zu einer 1 M Lösung
verdünnt.
-
BEISPIEL 1
-
Ethylen/1-hexencopolymerisation
-
Ein
200-ml-Glasautoklav mit Magnetrührer,
Temperaturanzeigeeinrichtung und Ethylenzufuhrleitung wurde gereinigt
und bei 35°C
mit Ethylen gespült.
Bei Raumtemperatur wurden 90 ml Heptan und 10 ml 1-Hexen eingetragen.
Das Katalysatorsystem wurde separat in 10 ml Heptan hergestellt,
indem nacheinander 0,22 ml 1 M Toluollösung von MAO und 0,1 mg (2,04 × 10–6 mol)
Methylenbis(3-isopropyl-1-indenyl)zirconiumdichlorid in der kleinstmöglichen
Menge Toluol gelöst
eingetragen wurden. Nach 5 Minuten Rühren wurde die Lösung unter
Ethylenstrom in den Autoklaven eingetragen, wonach der Reaktor geschlossen,
die Temperatur auf 70°C erhöht und der
Reaktor bis zu einem Druck von 4,5 bar mit Ethylen beaufschlagt
wurde. Der Gesamtdruck wurde durch Zufuhr von Ethylen konstant gehalten.
Nach 15 Minuten wurde die Polymerisation durch Abkühlen, Entgasen
des Reaktors und Eintragen von 1 ml Methanol beendet. Das Produkt
wurde mit saurem Methanol und dann mit Methanol gewaschen und schließlich im
Ofen bei 60°C
unter Vakuum getrocknet. Die Ausbeute betrug 3,59 g, was einer Aktivität von 769,3
kg/gZr.h entspricht. Die intrinsische Viskosität des Polymers betrug 1,22
dl/g.
-
Die
Charakterisierungsdaten des so erhaltenen Copolymers sind in Tabelle
2 aufgeführt.
-
BEISPIEL 2
-
Beispiel
1 wurde wiederholt, jedoch mit der Abwandlung, daß anstelle
von 10 ml 1-Hexen 5 ml 1-Hexen eingetragen wurden.
-
Die
Polymerisationsbedingungen sind in Tabelle 1 aufgeführt.
-
Die
Charakterisierungsdaten des so erhaltenen Copolymers sind in Tabelle
2 aufgeführt.
-
BEISPIEL 3
-
Beispiel
1 wurde wiederholt, jedoch mit dem Unterschied, daß anstelle
von MAO 0,27 mmol TIOA/H2O (Molverhältnis Al/H2O = 2,11) verwendet wurden. Die Ausbeute
betrug 0,73 g, was einer Aktivität
von 123,2 kg/gZr.h entspricht. Die intrinsische Viskosität des Polymers
betrug 2,58 dl/g.
-
Die
Polymerisationsbedingungen sind in Tabelle 1 aufgeführt.
-
Die
Charakterisierungsdaten des erhaltenen Copolymers sind in Tabelle
2 aufgeführt.
-
BEISPIEL 4
-
Beispiel
1 wurde wiederholt, jedoch unter Verwendung von 0,22 mmol einer
Mischung von TIOA-O/MAO im Verhältnis
9:1. TIOA-O wurde bei einem Molverhältnis Al/H2O
von 2,07 erhalten.
-
Die
Polymerisationsbedingungen sind in Tabelle 1 aufgeführt.
-
Die
Charakterisierungsdaten des erhaltenen Copolymers sind in Tabelle
2 aufgeführt.
-
BEISPIEL 5
-
Beispiel
1 wurde wiederholt, jedoch mit der Abwandlung, daß 0,12 mg
Me2C(3-iPrInd)2ZrCl2 und 0,24 mmol MAO verwendet wurden.
-
Die
Polymerisationsbedingungen sind in Tabelle 1 aufgeführt.
-
Die
Charakterisierungsdaten des erhaltenen Copolymers sind in Tabelle
2 aufgeführt.
-
BEISPIEL 6
-
Beispiel
1 wurde wiederholt, jedoch unter Verwendung von 0,12 mg CMe2(3-i-PrInd)2ZrCl2 und 0,24 mmol einer Mischung von TIOA-O/MAO
im Verhältnis
9:1. TIOA-O wurde bei einem Molverhältnis Al/H2O
von 2,07 erhalten.
-
Die
Polymerisationsbedingungen sind in Tabelle 1 aufgeführt.
-
Die
Charakterisierungsdaten des erhaltenen Copolymers sind in Tabelle
2 aufgeführt.
-
BEISPIEL 7
-
Beispiel
1 wurde wiederholt, jedoch mit der Abwandlung, daß 0,2 mg
H2C(3-Me3Si-Ind)2ZrCl2 verwendet wurden.
-
Die
Polymerisationsbedingungen sind in Tabelle 1 aufgeführt.
-
Die
Charakterisierungsdaten des erhaltenen Copolymers sind in Tabelle
2 aufgeführt.
-
BEISPIEL 8
-
Beispiel
1 wurde wiederholt, jedoch unter Verwendung von 0,1 mg CMe2(3-Me-Ind)2ZrCl2 und 0,23 mmol TIOA/H2O
(Molverhältnis
Al/H2O = 2,07) und ohne MAO.
-
Die
Polymerisationsbedingungen sind in Tabelle 1 aufgeführt.
-
Die
Charakterisierungsdaten des erhaltenen Copolymers sind in Tabelle
2 aufgeführt.
-
BEISPIEL 9
-
Beispiel
1 wurde wiederholt, jedoch mit der Abwandlung, daß 0,12 mg
Me2C(3-Me3Si-Ind)2ZrCl2 verwendet wurden.
-
Die
Polymerisationsbedingungen sind in Tabelle 1 aufgeführt.
-
Die
Charakterisierungsdaten des erhaltenen Copolymers sind in Tabelle
2 aufgeführt.
-
BEISPIEL 10
-
Ethylen/1-octen-Copolymerisation
-
Ein
260 ml Glasautoklav mit Magnetrührer,
Temperaturanzeigeeinrichtung und Ethylenzufuhrleitung wurde gereinigt
und bei 35°C
mit Ethylen gespült.
Bei Raumtemperatur wurden 86 ml Heptan und 4,1 ml über LiAlH4 destilliertes 1-Octen eingetragen. Das
Katalysatorsystem wurde separat hergestellt, indem nacheinander
MAO (0,21 mmol in Form einer 1 M Toluollösung) und 0,1 mg (0,000205
mg.at.Zr) des Metallocens gemäß Beispiel
1 in (der kleinstmöglichen
Menge) Toluol gelöst
eingetragen wurden. Nach 5 Minuten Rühren wurde die Lösung mit
Heptan auf 10 ml verdünnt
und unter Ethylen strom in den Autoklaven eingetragen, wonach der Reaktor
geschlossen, die Temperatur auf 70°C erhöht und mit einem Druck von
4 bar beaufschlagt wurde. Der Gesamtdruck wurde durch Zufuhr von
Ethylen 20 Minuten konstant gehalten. Die Polymerisation wurde durch Abkühlen, Entgasen
des Reaktors und Eintragen von 1 ml Methanol beendet. Das erhaltene
Polymer wurde mit saurem Methanol und dann mit Methanol gewaschen
und im Ofen bei 60°C
unter Vakuum getrocknet. Es wurden 1,68 g Polymer (270 kg/gZr/h)
mit den folgenden Eigenschaften erhalten: I.V. = 1,82 dl/g; 1-Octen
= 5,73 mol-%,
Tm = 92,5°C; ΔH = 63 J/g;
Triadenverteilung
in mol-%: [EXE] = 5,73; [XXX] = 0; [EXE]/Xmt =
1, wobei X für
1-Octen steht.
-
BEISPIEL 11
-
Ethylen/1-decen-Polymerisation
-
Ein
200 ml Glasautoklav mit Magnetrührer,
Temperaturanzeigeeinrichtung und Ethylenzufuhrleitung wurde gereinigt
und bei 35°C
mit Ethylen gespült.
Bei Raumtemperatur wurden 85 ml Heptan und 5 ml über LiAlH4 destilliertes
1-Octen eingetragen. Das Katalysatorsystem wurde hergestellt, indem
nacheinander MAO (0,22 mmol in Form einer 1 M Toluollösung und
0,1 mg (0,000205 mg.at.Zr) des Metallocens gemäß Beispiel 1 in (der kleinstmöglichen
Menge) Toluol gelöst
eingetragen wurden. Nach 5 Minuten Rühren wurde die Lösung mit
Heptan auf 10 ml verdünnt
und unter Ethylenstrom in den Autoklaven eingetragen, wonach der
Reaktor geschlossen, die Temperatur auf 70°C erhöht und mit einem Druck von
4 bar beaufschlagt wurde. Der Gesamtdruck wurde durch Zufuhr von
Ethylen 10 Minuten konstant gehalten. Die Polymerisation wurde durch
Abkühlen,
Entgasen des Reaktors und Eintragen von 1 ml Methanol beendet. Das
erhaltene Polymer wurde mit saurem Methanol und dann mit Methanol
gewaschen und im Ofen bei 60°C
unter Vakuum getrocknet. Es wurden 3,2 g Polymer (1045 kg/gZr/h)
mit den folgenden Eigenschaften erhalten: I.V. = 2,01 dl/g; 1-Decen
= 7,48 mol-%,
Tm = 72,8°C; ΔH = 63 J/g;
Triadenverteilung
in mol-%: [EXE] = 7,48; [XXX] = 0; [XXE] = 0; [EXE]/Xtot =
1, wobei X für
1-Decen steht.
-
BEISPIEL 12 (zum Vergleich)
-
Beispiel
1 wurde wiederholt, jedoch unter Verwendung von rac-CH2(3-tBu-Ind)2ZrCl2.
-
Die
Polymerisationsbedingungen sind in Tabelle 1 aufgeführt.
-
Die
Charakterisierungsdaten des erhaltenen Copolymers sind in Tabelle
2 aufgeführt.
-
BEISPIEL 13 (zum Vergleich)
-
Beispiel
1 wurde wiederholt, jedoch unter Verwendung von 0,3 mg rac-CMe2(3-tBu-Ind)2ZrCl2 und 1,15 mmol
TIOA/H2O (Molverhältnis Al/H2O
= 4,18) und 15 ml 1-Hexen.
-
Die
Polymerisationsbedingungen sind in Tabelle 1 aufgeführt.
-
Die
Charakterisierungsdaten des erhaltenen Copolymers sind in Tabelle
2 aufgeführt.
-
BEISPIEL 14 (zum Vergleich)
-
Beispiel
1 wurde wiederholt, jedoch unter Verwendung von rac-CMe2(Ind)2ZrCl2. Die Polymerisationsbedingungen
sind in Tabelle 1 aufgeführt.
-
Die
Charakterisierungsdaten des erhaltenen Copolymers sind in Tabelle
2 aufgeführt.
-
BEISPIEL 15 (zum Vergleich)
-
Beispiel
1 wurde wiederholt, jedoch unter Verwendung von rac-CH2(Ind)2ZrCl2.
-
Die
Polymerisationsbedingungen sind in Tabelle 1 aufgeführt.
-
Die
Charakterisierungsdaten des erhaltenen Copolymers sind in Tabelle
2 aufgeführt.
-
-

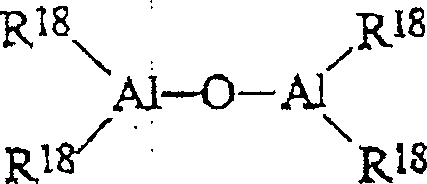
 worin n für eine ganze Zahl von 2 bis 40 steht und die Substituenten R18 die oben angegebene Bedeutung besitzen, verwenden.
worin n für eine ganze Zahl von 2 bis 40 steht und die Substituenten R18 die oben angegebene Bedeutung besitzen, verwenden.