DE69918107T2 - Ionomere und ionenleitende zusammensetzungen - Google Patents
Ionomere und ionenleitende zusammensetzungen Download PDFInfo
- Publication number
- DE69918107T2 DE69918107T2 DE69918107T DE69918107T DE69918107T2 DE 69918107 T2 DE69918107 T2 DE 69918107T2 DE 69918107 T DE69918107 T DE 69918107T DE 69918107 T DE69918107 T DE 69918107T DE 69918107 T2 DE69918107 T2 DE 69918107T2
- Authority
- DE
- Germany
- Prior art keywords
- ionomer
- liquid
- radicals
- polymer
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 239000000203 mixture Substances 0.000 title claims description 56
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 75
- 229920000554 ionomer Polymers 0.000 claims description 60
- 238000000034 method Methods 0.000 claims description 36
- 239000007788 liquid Substances 0.000 claims description 32
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 18
- 239000007772 electrode material Substances 0.000 claims description 15
- -1 alkali metal cation Chemical class 0.000 claims description 13
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 claims description 11
- 239000006229 carbon black Substances 0.000 claims description 10
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 9
- 229910052731 fluorine Inorganic materials 0.000 claims description 9
- 125000001153 fluoro group Chemical group F* 0.000 claims description 9
- 229920000098 polyolefin Polymers 0.000 claims description 9
- 229910052799 carbon Inorganic materials 0.000 claims description 8
- 150000002500 ions Chemical class 0.000 claims description 8
- 229910052783 alkali metal Inorganic materials 0.000 claims description 7
- 239000011888 foil Substances 0.000 claims description 6
- 150000005677 organic carbonates Chemical class 0.000 claims description 6
- 239000002245 particle Substances 0.000 claims description 6
- 230000008569 process Effects 0.000 claims description 6
- KMTRUDSVKNLOMY-UHFFFAOYSA-N Ethylene carbonate Chemical compound O=C1OCCO1 KMTRUDSVKNLOMY-UHFFFAOYSA-N 0.000 claims description 5
- HBBGRARXTFLTSG-UHFFFAOYSA-N Lithium ion Chemical compound [Li+] HBBGRARXTFLTSG-UHFFFAOYSA-N 0.000 claims description 5
- 239000002585 base Substances 0.000 claims description 5
- 150000001768 cations Chemical class 0.000 claims description 5
- 239000011737 fluorine Substances 0.000 claims description 5
- 125000001183 hydrocarbyl group Chemical group 0.000 claims description 5
- 150000001340 alkali metals Chemical class 0.000 claims description 4
- 229940006487 lithium cation Drugs 0.000 claims description 4
- 229910052751 metal Inorganic materials 0.000 claims description 4
- 239000002184 metal Substances 0.000 claims description 4
- 229910052757 nitrogen Inorganic materials 0.000 claims description 4
- 229910052760 oxygen Inorganic materials 0.000 claims description 4
- 125000005010 perfluoroalkyl group Chemical group 0.000 claims description 4
- IEJIGPNLZYLLBP-UHFFFAOYSA-N dimethyl carbonate Chemical compound COC(=O)OC IEJIGPNLZYLLBP-UHFFFAOYSA-N 0.000 claims description 3
- 125000001434 methanylylidene group Chemical group [H]C#[*] 0.000 claims description 3
- 239000000377 silicon dioxide Substances 0.000 claims description 3
- 239000010954 inorganic particle Substances 0.000 claims 2
- OIFBSDVPJOWBCH-UHFFFAOYSA-N Diethyl carbonate Chemical compound CCOC(=O)OCC OIFBSDVPJOWBCH-UHFFFAOYSA-N 0.000 claims 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 claims 1
- JBTWLSYIZRCDFO-UHFFFAOYSA-N ethyl methyl carbonate Chemical compound CCOC(=O)OC JBTWLSYIZRCDFO-UHFFFAOYSA-N 0.000 claims 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 claims 1
- 239000004071 soot Substances 0.000 claims 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 claims 1
- 229920000642 polymer Polymers 0.000 description 77
- 239000010408 film Substances 0.000 description 27
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 24
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 22
- 239000003054 catalyst Substances 0.000 description 21
- 239000002904 solvent Substances 0.000 description 18
- 239000012528 membrane Substances 0.000 description 14
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 14
- 239000000523 sample Substances 0.000 description 14
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 13
- 229910052744 lithium Inorganic materials 0.000 description 13
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 12
- 239000005977 Ethylene Substances 0.000 description 11
- 229920001577 copolymer Polymers 0.000 description 11
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 11
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 10
- 239000002243 precursor Substances 0.000 description 10
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 9
- 238000010348 incorporation Methods 0.000 description 8
- RUOJZAUFBMNUDX-UHFFFAOYSA-N propylene carbonate Chemical compound CC1COC(=O)O1 RUOJZAUFBMNUDX-UHFFFAOYSA-N 0.000 description 8
- 238000003756 stirring Methods 0.000 description 8
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 7
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 7
- 239000007787 solid Substances 0.000 description 7
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical class [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 6
- 239000004698 Polyethylene Substances 0.000 description 6
- 150000001336 alkenes Chemical class 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 238000001035 drying Methods 0.000 description 6
- 239000011572 manganese Substances 0.000 description 6
- 239000000178 monomer Substances 0.000 description 6
- 238000006243 chemical reaction Methods 0.000 description 5
- 239000011521 glass Substances 0.000 description 5
- 238000002844 melting Methods 0.000 description 5
- 230000008018 melting Effects 0.000 description 5
- 239000003921 oil Substances 0.000 description 5
- 229920000573 polyethylene Polymers 0.000 description 5
- 239000011541 reaction mixture Substances 0.000 description 5
- RFFLAFLAYFXFSW-UHFFFAOYSA-N 1,2-dichlorobenzene Chemical compound ClC1=CC=CC=C1Cl RFFLAFLAYFXFSW-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 239000004215 Carbon black (E152) Substances 0.000 description 4
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 4
- 238000005481 NMR spectroscopy Methods 0.000 description 4
- 229920000557 Nafion® Polymers 0.000 description 4
- 239000000654 additive Substances 0.000 description 4
- 238000007334 copolymerization reaction Methods 0.000 description 4
- 239000006185 dispersion Substances 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 229920002313 fluoropolymer Polymers 0.000 description 4
- 238000005227 gel permeation chromatography Methods 0.000 description 4
- 239000010439 graphite Substances 0.000 description 4
- 229910002804 graphite Inorganic materials 0.000 description 4
- 229930195733 hydrocarbon Natural products 0.000 description 4
- 238000005160 1H NMR spectroscopy Methods 0.000 description 3
- YEJRWHAVMIAJKC-UHFFFAOYSA-N 4-Butyrolactone Chemical compound O=C1CCCO1 YEJRWHAVMIAJKC-UHFFFAOYSA-N 0.000 description 3
- 229910013716 LiNi Inorganic materials 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- ATHHXGZTWNVVOU-UHFFFAOYSA-N N-methylformamide Chemical compound CNC=O ATHHXGZTWNVVOU-UHFFFAOYSA-N 0.000 description 3
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 239000000010 aprotic solvent Substances 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- 239000008367 deionised water Substances 0.000 description 3
- 229910021641 deionized water Inorganic materials 0.000 description 3
- 239000012530 fluid Substances 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- 238000005342 ion exchange Methods 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 229920000570 polyether Polymers 0.000 description 3
- 238000006116 polymerization reaction Methods 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 150000004763 sulfides Chemical class 0.000 description 3
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- BFKJFAAPBSQJPD-UHFFFAOYSA-N tetrafluoroethene Chemical group FC(F)=C(F)F BFKJFAAPBSQJPD-UHFFFAOYSA-N 0.000 description 3
- DHKHKXVYLBGOIT-UHFFFAOYSA-N 1,1-Diethoxyethane Chemical compound CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 2
- LSUPXLRKMCJALP-UHFFFAOYSA-L 1-[2-(2,3,3a,4,5,6,7,7a-octahydro-1H-inden-1-yl)ethyl]-2,3,3a,4,5,6,7,7a-octahydro-1H-indene zirconium(2+) dichloride Chemical compound [Cl-].[Cl-].[Zr++].C(CC1CCC2CCCCC12)C1CCC2CCCCC12 LSUPXLRKMCJALP-UHFFFAOYSA-L 0.000 description 2
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 2
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- 229920002799 BoPET Polymers 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- 229910000733 Li alloy Inorganic materials 0.000 description 2
- 239000005041 Mylar™ Substances 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 2
- 125000000217 alkyl group Chemical group 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- LSXWFXONGKSEMY-UHFFFAOYSA-N di-tert-butyl peroxide Chemical compound CC(C)(C)OOC(C)(C)C LSXWFXONGKSEMY-UHFFFAOYSA-N 0.000 description 2
- 238000000113 differential scanning calorimetry Methods 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 238000000921 elemental analysis Methods 0.000 description 2
- OIBMEBLCOQCFIT-UHFFFAOYSA-N ethanesulfonyl fluoride Chemical compound CCS(F)(=O)=O OIBMEBLCOQCFIT-UHFFFAOYSA-N 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 239000004811 fluoropolymer Substances 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- 229910002102 lithium manganese oxide Inorganic materials 0.000 description 2
- QEXMICRJPVUPSN-UHFFFAOYSA-N lithium manganese(2+) oxygen(2-) Chemical class [O-2].[Mn+2].[Li+] QEXMICRJPVUPSN-UHFFFAOYSA-N 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 229910052976 metal sulfide Inorganic materials 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 229910052759 nickel Inorganic materials 0.000 description 2
- 230000000379 polymerizing effect Effects 0.000 description 2
- FGIUAXJPYTZDNR-UHFFFAOYSA-N potassium nitrate Chemical compound [K+].[O-][N+]([O-])=O FGIUAXJPYTZDNR-UHFFFAOYSA-N 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052596 spinel Inorganic materials 0.000 description 2
- 239000011029 spinel Substances 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 125000001273 sulfonato group Chemical group [O-]S(*)(=O)=O 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 239000011135 tin Substances 0.000 description 2
- 229910052718 tin Inorganic materials 0.000 description 2
- 229910052723 transition metal Inorganic materials 0.000 description 2
- 229910000314 transition metal oxide Inorganic materials 0.000 description 2
- 229910052720 vanadium Inorganic materials 0.000 description 2
- XPHKPKDWWZNHGN-UHFFFAOYSA-N 1,1,2,2-tetrafluoro-2-(1,1,2,2,3,3,4,4-octafluorodec-9-enoxy)ethanesulfonyl fluoride Chemical compound C=CCCCCC(F)(F)C(F)(F)C(F)(F)C(F)(F)OC(F)(F)C(F)(F)S(F)(=O)=O XPHKPKDWWZNHGN-UHFFFAOYSA-N 0.000 description 1
- HFSKWPUHEMGYMQ-UHFFFAOYSA-N 1,3-dioxolan-2-one Chemical compound O=C1OCCO1.O=C1OCCO1 HFSKWPUHEMGYMQ-UHFFFAOYSA-N 0.000 description 1
- OCJBOOLMMGQPQU-UHFFFAOYSA-N 1,4-dichlorobenzene Chemical compound ClC1=CC=C(Cl)C=C1 OCJBOOLMMGQPQU-UHFFFAOYSA-N 0.000 description 1
- TUFKHKZLBZWCAW-UHFFFAOYSA-N 2-(1-ethenoxypropan-2-yloxy)ethanesulfonyl fluoride Chemical compound C=COCC(C)OCCS(F)(=O)=O TUFKHKZLBZWCAW-UHFFFAOYSA-N 0.000 description 1
- SVQOKUWDNBOKFD-UHFFFAOYSA-N 2-ethenoxyethanesulfonyl fluoride Chemical compound FS(=O)(=O)CCOC=C SVQOKUWDNBOKFD-UHFFFAOYSA-N 0.000 description 1
- YXYFONWVDGFHJH-UHFFFAOYSA-N 3-fluoro-4-fluorosulfonyloxybut-1-ene Chemical class C=CC(F)COS(F)(=O)=O YXYFONWVDGFHJH-UHFFFAOYSA-N 0.000 description 1
- 229920003026 Acene Polymers 0.000 description 1
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 description 1
- 0 C*c1cccc(*C)c1N(CCN1c2c(C(C)C)cccc2C(C)C)N1Br Chemical compound C*c1cccc(*C)c1N(CCN1c2c(C(C)C)cccc2C(C)C)N1Br 0.000 description 1
- GBQOEJASLZIOJB-UHFFFAOYSA-N CC(C(C)N1c2c(C)cccc2C)N(C2C(C)=CC=CC2C)[N+]1(Br)Br Chemical compound CC(C(C)N1c2c(C)cccc2C)N(C2C(C)=CC=CC2C)[N+]1(Br)Br GBQOEJASLZIOJB-UHFFFAOYSA-N 0.000 description 1
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 229910008163 Li1+x Mn2-x O4 Inorganic materials 0.000 description 1
- 229910011939 Li2.6 Co0.4 N Inorganic materials 0.000 description 1
- 229910012851 LiCoO 2 Inorganic materials 0.000 description 1
- 229910013290 LiNiO 2 Inorganic materials 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 1
- 241001282736 Oriens Species 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000004721 Polyphenylene oxide Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 229910004298 SiO 2 Inorganic materials 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- FKNQFGJONOIPTF-UHFFFAOYSA-N Sodium cation Chemical compound [Na+] FKNQFGJONOIPTF-UHFFFAOYSA-N 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- 239000011954 Ziegler–Natta catalyst Substances 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- XHCLAFWTIXFWPH-UHFFFAOYSA-N [O-2].[O-2].[O-2].[O-2].[O-2].[V+5].[V+5] Chemical class [O-2].[O-2].[O-2].[O-2].[O-2].[V+5].[V+5] XHCLAFWTIXFWPH-UHFFFAOYSA-N 0.000 description 1
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910001413 alkali metal ion Inorganic materials 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 239000006183 anode active material Substances 0.000 description 1
- 239000011260 aqueous acid Substances 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 239000003990 capacitor Substances 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 239000006182 cathode active material Substances 0.000 description 1
- 238000005341 cation exchange Methods 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 238000005345 coagulation Methods 0.000 description 1
- 230000015271 coagulation Effects 0.000 description 1
- IVMYJDGYRUAWML-UHFFFAOYSA-N cobalt(ii) oxide Chemical class [Co]=O IVMYJDGYRUAWML-UHFFFAOYSA-N 0.000 description 1
- 239000000571 coke Substances 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 238000005260 corrosion Methods 0.000 description 1
- 230000007797 corrosion Effects 0.000 description 1
- 150000004292 cyclic ethers Chemical class 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- RAABOESOVLLHRU-UHFFFAOYSA-N diazene Chemical compound N=N RAABOESOVLLHRU-UHFFFAOYSA-N 0.000 description 1
- 229910000071 diazene Inorganic materials 0.000 description 1
- 229940117389 dichlorobenzene Drugs 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 238000007598 dipping method Methods 0.000 description 1
- UZZWBUYVTBPQIV-UHFFFAOYSA-N dme dimethoxyethane Chemical compound COCCOC.COCCOC UZZWBUYVTBPQIV-UHFFFAOYSA-N 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 238000005868 electrolysis reaction Methods 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 125000006005 fluoroethoxy group Chemical group 0.000 description 1
- 239000000446 fuel Substances 0.000 description 1
- 229910021485 fumed silica Inorganic materials 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000007863 gel particle Substances 0.000 description 1
- 230000009477 glass transition Effects 0.000 description 1
- 239000011491 glass wool Substances 0.000 description 1
- 229920000578 graft copolymer Polymers 0.000 description 1
- 239000002946 graphitized mesocarbon microbead Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 238000007654 immersion Methods 0.000 description 1
- 239000003014 ion exchange membrane Substances 0.000 description 1
- 125000003010 ionic group Chemical group 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 238000010030 laminating Methods 0.000 description 1
- 239000001989 lithium alloy Substances 0.000 description 1
- 229910001416 lithium ion Inorganic materials 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- 229920001684 low density polyethylene Polymers 0.000 description 1
- 239000004702 low-density polyethylene Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- AMWRITDGCCNYAT-UHFFFAOYSA-L manganese oxide Inorganic materials [Mn].O[Mn]=O.O[Mn]=O AMWRITDGCCNYAT-UHFFFAOYSA-L 0.000 description 1
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 1
- PPNAOCWZXJOHFK-UHFFFAOYSA-N manganese(2+);oxygen(2-) Chemical class [O-2].[Mn+2] PPNAOCWZXJOHFK-UHFFFAOYSA-N 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- 239000002931 mesocarbon microbead Substances 0.000 description 1
- 239000012968 metallocene catalyst Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- 239000011325 microbead Substances 0.000 description 1
- 229910052750 molybdenum Inorganic materials 0.000 description 1
- 239000011733 molybdenum Substances 0.000 description 1
- 229910000476 molybdenum oxide Inorganic materials 0.000 description 1
- 229910052758 niobium Inorganic materials 0.000 description 1
- 239000010955 niobium Substances 0.000 description 1
- GUCVJGMIXFAOAE-UHFFFAOYSA-N niobium atom Chemical compound [Nb] GUCVJGMIXFAOAE-UHFFFAOYSA-N 0.000 description 1
- 150000004767 nitrides Chemical class 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- VWBWQOUWDOULQN-UHFFFAOYSA-N nmp n-methylpyrrolidone Chemical compound CN1CCCC1=O.CN1CCCC1=O VWBWQOUWDOULQN-UHFFFAOYSA-N 0.000 description 1
- 229920006120 non-fluorinated polymer Polymers 0.000 description 1
- 230000009022 nonlinear effect Effects 0.000 description 1
- 229920000620 organic polymer Polymers 0.000 description 1
- 150000002898 organic sulfur compounds Chemical class 0.000 description 1
- PQQKPALAQIIWST-UHFFFAOYSA-N oxomolybdenum Chemical class [Mo]=O PQQKPALAQIIWST-UHFFFAOYSA-N 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 229920003223 poly(pyromellitimide-1,4-diphenyl ether) Polymers 0.000 description 1
- 229920006267 polyester film Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920001721 polyimide Polymers 0.000 description 1
- 229920006254 polymer film Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 235000010333 potassium nitrate Nutrition 0.000 description 1
- 239000004323 potassium nitrate Substances 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 229910001415 sodium ion Inorganic materials 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- OBTWBSRJZRCYQV-UHFFFAOYSA-N sulfuryl difluoride Chemical group FS(F)(=O)=O OBTWBSRJZRCYQV-UHFFFAOYSA-N 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- 229920001169 thermoplastic Polymers 0.000 description 1
- 239000004416 thermosoftening plastic Substances 0.000 description 1
- 239000010409 thin film Substances 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 229910001935 vanadium oxide Inorganic materials 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
- C08F8/44—Preparation of metal salts or ammonium salts
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/02—Ethene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/18—Manufacture of films or sheets
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/34—Silicon-containing compounds
- C08K3/36—Silica
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L101/00—Compositions of unspecified macromolecular compounds
- C08L101/12—Compositions of unspecified macromolecular compounds characterised by physical features, e.g. anisotropy, viscosity or electrical conductivity
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L23/04—Homopolymers or copolymers of ethene
- C08L23/08—Copolymers of ethene
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01B—CABLES; CONDUCTORS; INSULATORS; SELECTION OF MATERIALS FOR THEIR CONDUCTIVE, INSULATING OR DIELECTRIC PROPERTIES
- H01B1/00—Conductors or conductive bodies characterised by the conductive materials; Selection of materials as conductors
- H01B1/06—Conductors or conductive bodies characterised by the conductive materials; Selection of materials as conductors mainly consisting of other non-metallic substances
- H01B1/12—Conductors or conductive bodies characterised by the conductive materials; Selection of materials as conductors mainly consisting of other non-metallic substances organic substances
- H01B1/122—Ionic conductors
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/056—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes
- H01M10/0564—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes the electrolyte being constituted of organic materials only
- H01M10/0565—Polymeric materials, e.g. gel-type or solid-type
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M50/00—Constructional details or processes of manufacture of the non-active parts of electrochemical cells other than fuel cells, e.g. hybrid cells
- H01M50/40—Separators; Membranes; Diaphragms; Spacing elements inside cells
- H01M50/409—Separators, membranes or diaphragms characterised by the material
- H01M50/411—Organic material
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M50/00—Constructional details or processes of manufacture of the non-active parts of electrochemical cells other than fuel cells, e.g. hybrid cells
- H01M50/40—Separators; Membranes; Diaphragms; Spacing elements inside cells
- H01M50/409—Separators, membranes or diaphragms characterised by the material
- H01M50/411—Organic material
- H01M50/414—Synthetic resins, e.g. thermoplastics or thermosetting resins
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/10—Fuel cells with solid electrolytes
- H01M8/1016—Fuel cells with solid electrolytes characterised by the electrolyte material
- H01M8/1018—Polymeric electrolyte materials
- H01M8/102—Polymeric electrolyte materials characterised by the chemical structure of the main chain of the ion-conducting polymer
- H01M8/1023—Polymeric electrolyte materials characterised by the chemical structure of the main chain of the ion-conducting polymer having only carbon, e.g. polyarylenes, polystyrenes or polybutadiene-styrenes
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/10—Fuel cells with solid electrolytes
- H01M8/1016—Fuel cells with solid electrolytes characterised by the electrolyte material
- H01M8/1018—Polymeric electrolyte materials
- H01M8/1039—Polymeric electrolyte materials halogenated, e.g. sulfonated polyvinylidene fluorides
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/10—Fuel cells with solid electrolytes
- H01M8/1016—Fuel cells with solid electrolytes characterised by the electrolyte material
- H01M8/1018—Polymeric electrolyte materials
- H01M8/1069—Polymeric electrolyte materials characterised by the manufacturing processes
- H01M8/1081—Polymeric electrolyte materials characterised by the manufacturing processes starting from solutions, dispersions or slurries exclusively of polymers
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M2300/00—Electrolytes
- H01M2300/0085—Immobilising or gelification of electrolyte
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/62—Selection of inactive substances as ingredients for active masses, e.g. binders, fillers
- H01M4/621—Binders
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/10—Fuel cells with solid electrolytes
- H01M8/1016—Fuel cells with solid electrolytes characterised by the electrolyte material
- H01M8/1018—Polymeric electrolyte materials
- H01M8/1058—Polymeric electrolyte materials characterised by a porous support having no ion-conducting properties
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/30—Hydrogen technology
- Y02E60/50—Fuel cells
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P70/00—Climate change mitigation technologies in the production process for final industrial or consumer products
- Y02P70/50—Manufacturing or production processes characterised by the final manufactured product
Description
- TECHNISCHES GEBIET DER ERFINDUNG
- Die Erfindung betrifft Ionomere, die funktionalisierte Polyolefine mit Fluoralkylsulfonat-Seitengruppen aufweisen, und ionenleitende Zusammensetzungen, die durch Zugabe von Lösungsmitteln daraus gebildet werden. Die erfindungsgemäßen ionenleitenden Zusammensetzungen sind in Batterien, Brennstoffzellen, Elektrolysezellen, Ionenaustauschmembranen, Sensoren, elektrochemischen Kondensatoren und modifizierten Elektroden verwendbar.
- TECHNISCHER HINTERGRUND DER ERFINDUNG
- Die Bildung ionenleitender Membranen und Gele aus organischen Polymeren, die ionenleitende Seitengruppen enthalten, ist dem Fachmann seit langem bekannt. Derartige Polymere werden als Ionomere bezeichnet. Besonders gut bekannte, weitverbreitet kommerziell eingesetzte Ionomermembranen sind Nafion®-Membranen, die von E. I. du Pont de Nemours and Company beziehbar sind. Nafion® entsteht durch Copolymerisation von Tetrafluorethylen (TFE) mit Perfluor(3,6-dioxa-4-methyl-7-octensulfonylfluorid), wie in US-A-3282875 offenbart. Gleichfalls bekannt sind Copolymere von TFE mit Perfluor(3-oxa-4-pentensulfonylfluorid), wie in US-A-4358545 offenbart. Die so gebildeten Copolymere werden durch Hydrolyse in die Ionomerform umgewandelt, typischerweise durch Einwirkung einer geeigneten wäßrigen Base, wie in US-A-3282875 offenbart. Als geeignete Kationen für die oben zitierten Ionomere sind Lithium, Natrium und Kalium dem Fachmann alle gut bekannt.
- In den oben zitierten Polymeren bieten die Fluoratome mehr als einen Vorteil. Die Fluorgruppen an den Kohlenstoffatomen in der Nähe der Sulfonylgruppe in der Seitenkette liefern die Elektronegativität, um die Kationen hinreichend instabil für die Bereitstellung einer hohen Ionenleitfähigkeit zu machen. Der Austausch dieser Fluoratome durch Wasserstoff führt zu einer erheblichen Verminderung der Ionenbeweglichkeit und einem daraus folgenden Leitfähigkeitsverlust.
- Die übrigen Fluoratome verleihen dem Polymer die normalerweise mit fluorierten Polymeren verbundene chemische und Wärmebeständigkeit. Diese hat sich in Anwendungen wie z. B. dem bekannten "Chlor-Alkali-Prozeß" als sehr wertvoll erwiesen. Hochfluorierte Polymere haben jedoch auch Nachteile, wo ein geringerer Bedarf für hohe chemische und Wärmebeständigkeit besteht. Die fluorierten Monomere sind teurer als ihre Olefin-Gegenstücke, erfordern höhere Verarbeitungstemperaturen und erfordern oft kostspielige korrosionsbeständige Verarbeitungsanlagen. Ferner ist die Herstellung von Lösungen und Dispersionen von Fluorpolymeren schwierig. Außerdem ist es schwierig, feste Klebeverbindungen mit Fluorpolymeren herzustellen. Bei Materialien, die in elektrochemischen Zellen eingesetzt werden, kann es beispielsweise vorteilhaft sein, bis zu einem gewissen Grade auf Kosten der chemischen und Wärmebeständigkeit eine bessere Verarbeitbarkeit zu erreichen. Daher gibt es einen Anreiz zur Entwicklung von Ionomeren mit hochinstabilen Kationen, die nichtfluorierte Polymerhauptketten aufweisen.
- Zahlreiche Veröffentlichungen offenbaren Polyether entweder mit proximalen Ionenspezies im Polymer oder in Kombination mit ionischen Salzen. Die Leitfähigkeiten liegen im Bereich von 10–5 S/cm und weniger. Le Nest et al., Polymer Communications 28, 303 (1987), offenbaren eine Zusammensetzung von Polyetherglycol-Oligomeren, die durch zu dem verwandten Lithium-Ionomer hydrolysierte Phosphat- oder Thiophosphat-Komponenten aneinander gebunden sind. In Kombination mit Propylencarbonat wurde eine Leitfähigkeit im Bereich von 1–10 × 10–4 S/cm realisiert. Eine Übersicht über die verwandte Technik ist in Fauteux et al., Electrochimica Acta 40, 2185 (1995) zu finden.
- Benrabah et al., Electrochimica Acta 40 2259 (1995) offenbaren Polyether, die durch Lithiumoxytetrafluorsulfonate und deren Derivate vernetzt sind. Es sind keine aprotischen Lösungsmittel beigemischt. Mit Zugabe von Lithiumsalzen wurde eine Leitfähigkeit von < 10–4 S/cm erzielt.
- Armand et al., US-A-5627292, offenbaren Copolymere, die aus Vinylfluorethoxysulfonylfluoriden oder cyclischen Ethern mit Fluorethoxysulfonylfluoridgruppen mit Polyethylenoxid, Acrylnitril, Pyridin und anderen Monomeren gebildet werden. Es werden Lithium-Sulfonat-Ionomere gebildet. Es werden keine aprotischen Lösungsmittel beigemischt. Die Leitfähigkeit war < 10–4 S/cm.
- Narang et al., US-A-5633098, offenbaren Acrylat-Copolymere mit einer funktionalisierten Polyolefin-Hauptkette und Seitengruppen, die Tetrafluorethoxylithiumsulfonatgruppen enthalten. Die Comonomere, welche die Sulfonatgruppen enthalten, sind in Molverhältnissen von 50–100% vorhanden. Es werden Zusammensetzungen offenbart, die das Polymer und ein aus Propylencarbonat, Ethylencarbonat und Dimethoxyethan bestehendes Lösungsmittelgemisch aufweisen. Die Ionenleitfähigkeit dieser Zusammensetzungen lag im Bereich von 10–4–10–3 S/cm.
- Brookhart et al., WO 9623010A2, offenbaren ein Copolymer, das aus Ethen und 1,1,2,2-Tetrafluor-2-[(1,1,2,2,3,3,4,4-octafluor-9-decenyl)oxy]ethansulfonylfluorid über eine katalysierte Reaktion mit Verwendung von Diimin-Übergangsmetall-Komplexen gebildet wird. Das so gebildete Polymer weist eine Polyethylen-Hauptkette mit statistisch verteilten Seitengruppen aus 1,1,2,2-Tetrafluor-2-[(1,1,2,2,3,3,4,4-octafluor-(meistens)octoxyl]ethansulfonylfluorid sowie Alkylzweige auf.
- ZUSAMMENFASSUNG DER ERFINDUNG
- Die Erfindung stellt ein Ionomer mit einer Hauptkette und Seitengruppen bereit, wobei die Hauptkette im wesentlichen aus Methyleneinheiten besteht und die Seitengruppen ionische Radikale mit der Formel
- Die vorliegende Erfindung stellt ferner eine ionenleitende Zusammensetzung bereit, die das oben beschriebene Ionomer und eine darin eingesaugte Flüssigkeit aufweist.
- Die vorliegende Erfindung offenbart ferner ein Verfahren zur Bildung eines Ionomers, wobei das Verfahren aufweist: Inkontaktbringen eines Polyolefins, das eine Hauptkette und Seitengruppen aufweist, mit einer Lösung einer Alkalimetallbase, wobei die Hauptkette im wesentlichen aus Methylen- und Methineinheiten besteht und die Seitengruppen ionische Radikale mit der Formel
- Ferner wird ein Verfahren zur Bildung einer leitfähigen Zusammensetzung offenbart, bei dem das obige Ionomer mit einer Flüssigkeit in Kontakt gebracht wird.
- Außerdem ist hier eine Elektrode eingeschlossen, die mindestens ein aktives Elektrodenmaterial, damit vermischt das hier offenbarte Ionomer und eine darin eingesaugte Flüssigkeit aufweist.
- Ferner wird eine elektrochemische Zelle offenbart, die eine positive Elektrode, eine negative Elektrode, ein zwischen der positiven und der negativen Elektrode angeordnetes Trennelement und eine Einrichtung zum Anschließen der Zelle an eine äußere Last oder Quelle aufweist, wobei mindestens eine Komponente aus der Gruppe, die aus dem Trennelement, der Kathode und der Anode besteht, das obige Ionomer aufweist.
- AUSFÜHRLICHE BESCHREIBUNG
- In einer bevorzugten Ausführungsform des erfindungsgemäßen Polyolefin-Ionomers besteht die Hauptkette im wesentlichen aus Olefinradikalen, von denen 1–20 Mol.-% Seitengruppen in Form eines Radikals mit der folgenden Formel aufweisen:
- Die Olefinradikale, welche die Hauptkette des erfindungsgemäßen Polyolefin-Ionomers bilden, sind im wesentlichen nichtsubstituiert, mit der Ausnahme, daß 1–20 Mol-% der Olefinradikale der Hauptkette in einer bevorzugten Ausführungsform der Erfindung eine Seitengruppe in Form des Radikals (I) aufweisen. In einer besonders bevorzugten Ausführungsform weisen 2–10 Mol-% der Olefinradikale der Hauptkette eine Seitengruppe in Form des Radikals (I) auf.
- Wie dem Fachmann bekannt, sind Grad und Typ der Verzweigung in einem Polyolefin von den bei der Polymerisation verwendeten Monomeren und dem Verfahren abhängig, durch das die Polymerisation erzielt wird. Ethylen, das durch verschiedene katalytische Verfahren polymerisiert wird, weist kurzkettige Verzweigungen mit einer Häufigkeit von < 1 bis ca. 150 pro 1000 Methylengruppen in der Hauptkette auf, in Abhängigkeit vom verwendeten Katalysator und den Reaktionsbedingungen. Die so gebildeten kurzkettigen Verzweigungen sind meist Methyl- oder Ethylgruppen.
- Wenn das polymerisierte Olefinmonomer höher als Ethylen ist, dann nimmt die Anzahl der Verzweigungen erheblich zu, da dann jeder Monomereinheit mindestens eine Seitenkette inhärent ist.
- In der Praxis zeigt sich, daß die Kettenverzweigung eine wesentliche Auswirkung auf die Ionenleitfähigkeit der erfindungsgemäßen leitfähigen Zusammensetzungen hat. Um die höchste Leitfähigkeit zu erzielen, wird eine Verzweigungshäufigkeit von 5–90 Methylverzweigungen pro 1000 Methylenen bevorzugt, wenn das erfindungsgemäße Ionomer aus Polymeren hergestellt wird, die auf den hierin beschriebenen katalytischen Wegen synthetisiert werden. Ein höherer Verzweigungsgrad erscheint tolerierbar, wenn das Ionomer auf dem nachstehend beschriebenen Weg über ein Propfpolymer hergestellt wird.
- Die bevorzugten erfindungsgemäßen Ionomere werden zweckmäßig nach dem Fachmann bekannten Verfahren erzeugt, indem ein nichtionischer Sulfonylhalogenid-Vorläufer mit einer Alkalimetallhydroxidlösung in Kontakt gebracht wird und dadurch das Polymer zu dem Alkalimetallsalz hydrolysiert wird. In der Praxis der Erfindung stellt man fest, daß sie Säureform des erfindungsgemäßen Ionomers am leichtesten erzeugt wird, indem zuerst der nichtionische Vorläufer einer Alkalimetallhydroxidlösung ausgesetzt wird und anschließend ein Ionenaustausch mit einer wäßrigen Säure erfolgt. Durch dem Fachmann bekannte Ionenaustauschverfahren kann das Alkalimetallion gegen weitere einwertige Metalle ausgetauscht werden, wie z. B. Kupfer oder Silber.
- Die für die praktische Ausführung der Erfindung bevorzugten Vorläuferpolymere können durch Copolymerisation eines oder mehrerer Olefine, vorzugsweise Ethylen, mit einem substituierten Olefin-Comonomer gebildet werden, vorzugsweise mit der Formel
- Diimin-Übergangsmetall-Komplexe, wie von Brookhart offenbart und nachstehend durch Beispiele erläutert, sind bevorzugte Katalysatoren zur Ausbildung der nichtionischen Vorläuferpolymere, die für das erfindungsgemäße Verfahren bevorzugt werden. In dem so gebildeten Polymer besteht die Hauptkette im wesentlichen aus Olefinradikalen, von denen 1–20 Mol-%, vorzugsweise 2–10 Mol-%, Seitengruppen aus 1,1,2,2-Tetrafluor-2-[1,1,2,2,3,3,4,4-octafluor-(meist)octoxyl]ethansulfonylfluorid aufweisen, wobei das Polymer weniger als 150, vorzugsweise 50–90 Alkylverzweigungen, meist Methyl- und Ethylverzweigungen pro 1000 Methylene aufweist. Die Katalysatorstruktur hat eine bestimmende Wirkung auf die Anzahl der Kettenverzweigungen. Andere Katalysatoren, die sich für die praktische Ausführung der Erfindung eignen, sind unter anderem Metallocen- und Ziegler-Natta-Katalysatoren. Die am stärksten bevorzugten Katalysatoren sind Nickeldiimin-Katalysatoren, die durch die Strukturen B und D in der untenstehenden Tabelle dargestellt werden, in Kombination mit PMAO. Diese Katalysatoren bieten eine wünschenswerte Kombination von guten Einbauraten des Comonomers, Verzweigungsgraden im bevorzugten Bereich, alles zusammen mit einer hohen Polymerausbeute.
- Im Fall der bevorzugten Diimin-Nickel-Katalysatoren hat sich bei der praktischen Ausführung der vorliegenden Erfindung gezeigt, daß sich aus der Verwendung voluminöserer Katalysatoren ein höherer Polymerverzweigungsgrad ergibt. Weniger voluminöse Katalysatoren sind mit einem stärkeren Einbau des sulfonylhaltigen substituierten Olefin-Comonomers verbunden.
- Der bei metallocen-katalysierten Copolymerisationen erzielte Verzweigungsgrad (siehe zum Beispiel Yang et al., J. Am. Chem. Soc. 116, S. 10015ff., 1994) der bevorzugten erfindungsgemäßen Comonomere ist im allgemeinen niedrig und von einer niedrigeren Leitfähigkeit begleitet. Die Verwendung eines Termonomers, vorzugsweise eines Olefins mit drei oder mehr Kohlenstoffatomen in der Kette, in Kombination mit einem Metallocen- oder Ziegler-Natta-Katalysator kann jedoch den Verzweigungsgrad im resultierenden Polymer erhöhen und zu einer höheren Ionenleitfähigkeit führen.
- Andere Mittel sind gleichfalls für die Bildung der erfindungsgemäßen Ionomere geeignet. Dazu gehören die Bildung des erfindungsgemäßen Ionomers durch Pfropfen bzw. Anpolymerisieren eines Radikals mit der Formel
- In einer anderen Ausführungsform wird ein Polymer mit einer Hauptkette, die im wesentlichen aus Olefinradikalen besteht, von denen 1–20 Mol-% Seitengruppen in Form eines Radikals mit der folgenden Formel aufweisen:wobei die Rf- und Rf'-Gruppen lineares oder verzweigtes Perfluoralkylen, O- oder Cl-haltiges Perfluoralkylen oder ein Perfluorarylradikal sind und nicht alle gleich zu sein brauchen, R ein Kohlenwasserstoffradikal mit n = 0 oder 1 ist. Das Natriumion kann durch einfache, dem Fachmann bekannte Kationenaustauschverfahren z. B. durch ein Lithiumion ausgetauscht werden.
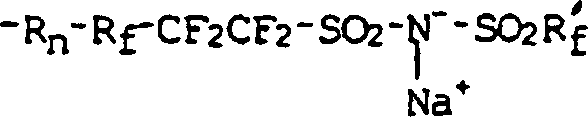
- In einer weiteren Ausführungsform wird ein Polymer mit einer Hauptkette, die im wesentlichen aus Olefinradikalen besteht, von denen 1–20 Mol-% Seitengruppen in Form eines Radikals mit der folgenden Formel aufweisen:wobei M+ ein Alkalimetall ist, die Rf und Rf'-Gruppen lineares oder verzweigtes Perfluoralkylen, O- oder Cl-haltiges Perfluoralkylen oder ein Perfluorarylradikal sind und nicht alle gleich zu sein brauchen, und wobei R ein Hydrocarbyl bzw. Kohlenwasserstoffradikal mit n = 1 ist.
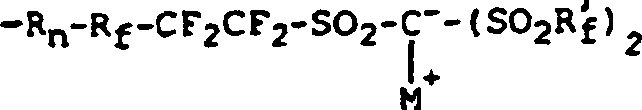
- Bei der praktischen Ausführung der Erfindung hat sich gezeigt, daß der Comonomer-Einbaugrad eine hochgradig nichtlineare Wirkung auf die Leitfähigkeit der erfindungsgemäßen leitfähigen Zusammensetzungen hat. Für Comonomer-Konzentrationen unterhalb etwa 2 Mol-% liegt die Leitfähigkeit im Bereich von 0 bis etwa 10–5 S/cm, nahezu unabhängig vom Verzweigungsgrad oder der verwendeten Flüssigkeit. Leitfähige Zusammensetzungen, die eine Leitfähigkeit von 10–5 S/cm aufweisen, sind von relativ begrenztem Nutzen.
- Bei einem Comonomer-Einbau von etwa 2–3 Mol-% steigt die Ionenleitfähigkeit beträchtlich an und weist eine starke Abhängigkeit von der Verzweigung und der Wahl der Flüssigkeiten auf, die für die Bildung der leitfähigen Zusammensetzung eingesetzt werden. Im Bereich von etwa 3–10 Mol-% werden Leitfähigkeiten im Bereich von 10–5 bis 10–2 S/cm erzielt, während eine mäßige Abhängigkeit von der Comonomer-Konzentration beobachtet wird. Einen geringen zusätzlichen Vorteil erzielt man bei Comonomer-Konzentrationen von etwa 10 Mol-% gegenüber etwa 6–7 Mol-%. Die Erfinder glauben, daß ein Grund für diesen Effekt der "abnehmenden Erträge" ist, daß die Katalysatoren, die zum Erzielen eines höheren Comonomer-Einbaus in das Polymer erforderlich sind, auch weniger verzweigtes Polymer erzeugen, so daß die beiden Effekte sich ein wenig selbst aufheben. Trotzdem wird in erfindungsgemäßen leitfähigen Zusammensetzungen, in denen Propylencarbonat als Flüssigkeit verwendet wird, eine Leitfähigkeit weit oberhalb 10–5 S/cm bei Comonomer-Konzentrationen im Polymer von weniger als 10 Mol-% beobachtet, im starken Gegensatz zu den Lehren des Standes der Technik.
- In einer bevorzugten Ausführungsform der vorliegenden Erfindung ist die Ionenfunktionalität in dem Ionomer vorzugsweise in einer Konzentration von 1–10 Mol-%, besonders bevorzugt von 3–7 Mol-%, vorhanden.
- Es gibt zwar keine Grenze für die Form oder die Proportionen eines Gegenstands, der aus den erfindungsgemäßen Ionomeren geformt wird, aber dünne Folien oder Membranen sind von besonderem Nutzen. Die erfindungsgemäßen Ionomere sind nicht vollständig thermoplastisch und sind nicht so leicht verarbeitbar wie die nichtionischen Vorläuferpolymere, von denen sie abgeleitet sind. Daher findet man es zweckmäßig, Membranen aus den Vorläuferpolymeren durch Verfahren zu formen, die dem Fachmann allgemein bekannt sind und nachstehend beschrieben werden. Besonders bequem ist das Extrudieren von Folien mit Hilfe eines Schneckenextruders und eines Breitschlitzmundstücks. Alternativ können Folien aus der Schmelze gepreßt werden. Als weitere Alternative können Folien aus Lösungen oder Dispersionen der Vorläuferpolymere durch Gießen auf ein Substrat und Koagulation gegossen werden. Kein spezielles Verfahren wird gegenüber einem anderen bevorzugt, und das konkrete Verfahren wird entsprechend den Bedürfnissen des jeweiligen Praktikers ausgewählt.
- Die erfindungsgemäßen Ionomere weisen im trockenen Zustand eine Ionenleitfähigkeit bei Raumtemperatur von etwa 10–7 bis 10–6 S/cm auf. Bei der praktischen Ausführung der Erfindung zeigt sich jedoch, daß zahlreiche Flüssigkeiten, wenn sie in das erfindungsgemäße Ionomer eingesaugt werden, die Leitfähigkeit um Größenordnungen verbessern. Um die brauchbarsten Ausführungsformen der vorliegenden Erfindung zu erzielen, hat es sich daher als wünschenswert erwiesen, leitfähige Zusammensetzungen herzustellen, in denen Flüssigkeiten in das erfindungsgemäße Ionomer eingesaugt werden.
- Die verwendete Flüssigkeit wird durch die Anwendung bestimmt. Allgemein gesagt, hat man bei der praktischen Ausführung festgestellt, daß die Leitfähigkeit des flüssigkeitshaltigen Ionomers mit zunehmender Flüssigkeitsaufnahme in Gew.-%, steigender Dielektrizitätskonstante der Flüssigkeit und zunehmender Lewis-Basizität der Flüssigkeit zunimmt, während beobachtet wurde, daß die Leitfähigkeit mit zunehmender Viskosität und zunehmender Molekülgröße der verwendeten Flüssigkeit abnimmt. Daher kann ein stark basisches Lösungsmittel von niedriger Viskosität und kleiner Molekülgröße, aber niedriger Dielektrizitätskonstante eine höhere Leitfähigkeit in einer gegebenen Membran liefern als ein größeres, stärker viskoses, weniger basisches Lösungsmittel mit sehr hoher Dielektrizitätskonstante. Natürlich kommen auch andere Überlegungen ins Spiel. Zum Beispiel kann eine zu hohe Löslichkeit des Ionomers in der Flüssigkeit unerwünscht sein. Oder die Flüssigkeit kann bei der vorgesehenen Verwendung elektrochemisch instabil sein.
- Eine besonders bevorzugte Ausführungsform weist das Lithium-Ionomer in Kombination mit aprotischen Lösungsmitteln auf, vorzugsweise organischen Carbonaten, die in Lithiumbatterien verwendbar sind.
- Die bevorzugte erfindungsgemäße Elektrode weist ein Gemisch aus einem oder mehreren aktiven Elektrodenmaterialien in Teilchenform, dem erfindungsgemäßen Ionomer, mindestens einem elektronenleitenden Zusatzstoff und mindestens einem organischen Carbonat auf. Beispiele verwendbarer aktiver Anodenmaterialien schließen ein, sind aber nicht beschränkt auf Kohlenstoff (graphitartige, koksartige Kohlenstoffe, Mesocarbone, Polyacene usw.) und Kohlenstoff mit Lithiumeinschlüssen, Lithium-Metall-Nitride, wie z. B. Li2,6Co0,4N, zinnoxidgrundierte Gläser, metallisches Lithium und Lithiumlegierungen, wie z. B. Legierungen von Lithium mit Aluminium, Zinn, Magnesium, Silicium, Mangan, Eisen und Zink. Anoden mit Lithiumeinschlüssen unter Verwendung von Kohlenstoff werden bevorzugt. Verwendbare aktive Kathodenmaterialien schließen ein, sind aber nicht beschränkt auf Übergangsmetalloxide und -sulfide, lithiierte Übergangsmetalloxide und -sulfide und schwefelorganische Verbindungen. Beispiele davon sind Cobaltoxide, Manganoxide, Molybdänoxide, Vanadiumoxide, Sulfide von Titan, Molybdän und Niobium, lithiierte Oxide, wie z. B. Spinell-Lithium-Mangan-Oxide, Li1+xMn2-xO4, chromdotierte Spinell-Lithium-Mangan-Oxide, LixCryMnzO4, LiCoO2, LiNiO2, LiNixCo1-xO2, mit 0 < x < 1 und einem bevorzugten Bereich von 0,5 < x < 0,95, LiCoVO4 und Gemische davon. LiNixCo1-xO2 wird bevorzugt. Ein stark bevorzugter elektronenleitender Hilfsstoff ist Ruß, vorzugsweise Super P-Ruß, beziehbar von MMM S. A. Carbon, Brüssel, Belgien, im Konzentrationsbereich von 1–10%. Vorzugsweise liegt der Volumenanteil des Lithium-Ionomers in der fertigen Elektrode zwischen 4 und 40%.
- Die erfindungsgemäße Elektrode kann zweckmäßig durch Auflösen aller Polymerbestandteile in einem gewöhnlichen Lösungsmittel und Vermischen mit den Rußteilchen und den aktiven Elektrodenteilchen hergestellt werden. Für Kathoden ist das bevorzugte aktive Elektrodenmaterial LiNixCo1-xO2, wobei 0 < x < 1 ist, während für Anoden das bevorzugte aktive Elektrodenmaterial graphitierte Mesocarbon-Mikroperlen sind. Zum Beispiel kann eine bevorzugte erfindungsgemäße Lithiumbatterie-Elektrode hergestellt werden, indem erfindungsgemäßes Ionomer in einem Gemisch aus Aceton und Dimethylformamid aufgelöst, anschließend Teilchen des aktiven Elektrodenmaterials und Ruß zugesetzt werden, gefolgt von der Abscheidung und dem Trocknen einer Schicht auf einem Substrat. Die entstehende bevorzugte Elektrode weist aktives Elektrodenmaterial, leitfähigen Ruß und erfindungsgemäßes Ionomer auf, wobei das Gewichtsverhältnis des Ionomers zu dem aktiven Elektrodenmaterial vorzugsweise zwischen 0,05 und 0,8 und das Gewichtsverhältnis von Ruß zum aktiven Elektrodenmaterial zwischen 0,01 und 0,2 liegt. Am stärksten bevorzugt liegt das Gewichtsverhältnis des Ionomers zum aktiven Elektrodenmaterial zwischen 0,1 und 0,25, und das Gewichtsverhältnis von Ruß zum aktiven Elektrodenmaterial liegt zwischen 0,02 und 0,1. Diese Elektrode kann dann aus Lösung auf einen geeigneten Träger gegossen werden, wie z. B. eine Glasplatte oder eine Stromkollektor-Metallfolie, und unter Anwendung von dem Fachmann bekannten Verfahren zu einer Schicht geformt werden. Die so erzeugte Elektrodenschicht kann dann durch Laminieren in eine mehrschichtige elektrochemische Zellenstruktur eingebaut werden, wie weiter unten beschrieben.
- Es kann wünschenswert sein, der erfindungsgemäßen Trennmittel-Zusammensetzung Zusatzstoffe beizumischen, die für Zwecke wie z. B. die Bindungsverbesserung ihrer Bestandteile oder die Bereitstellung einer verbesserten strukturellen Integrität eines daraus gefertigten Gegenstands nützlich sein können. Ein besonders bevorzugter Zusatzstoff ist SiO2, das einfach durch Dispergieren seiner Teilchen in die gleiche Lösung beigemischt werden kann, aus der das Trennmittel geformt wird, wie weiter oben beschrieben. Bevorzugt sind Siliciumdioxidteilchen mit einer mittleren Teilchengröße von weniger als 1,0 μm, wobei das Siliciumdioxid in der Beimischung in einem Anteil von bis zu 50 Gew.-% der Gesamtmenge vorhanden ist.
- In einem alternativen Verfahren kann die Dispersion aus dem aktiven Elektrodenmaterial und dem wahlfreien Ruß und anderen Zusatzstoffen zunächst auf eine Oberfläche gegossen und anschließend das erfindungsgemäße Ionomer in organischer Carbonatlösung zugesetzt werden.
- Die Erfindung wird in den folgenden konkreten Ausführungsformen näher erläutert.
- BEISPIELE
- Die nachstehend beschriebenen nichtionischen Vorläuferpolymere I–XIV wurden durch Copolymerisation von Ethylen mit einem Comonomer mit der Formel
- Wie aus Tabelle 1 ersichtlich, wurden die Polymere I–X und XIV synthetisiert, indem die angegebenen Mengen des angegebenen Katalysators, Comonomers und Lösungsmittels in einem Schlenk-Kolben in einem stickstoffgespülten Trockenschrank vereinigt wurden. Die Struktur des bezeichneten Katalysators ist in Tabelle 2 angegeben. Das Gemisch wurde dann aus dem Trockenschrank entnommen und unter einen Ethylen-Druck von 1 atm gesetzt. Das Gemisch wurde 15 Minuten mit Ethylen gespült, während es durch Eintauchen in ein Eiswasserbad gekühlt wurde. 2,2 ml einer Lösung von 7,1% Polymethylalumoxan (PMAO) in Toluol (4,4 ml für die Polymere VII und XIV) wurden dann eingeleitet, um die Reaktion auszulösen, und das Gemisch wurde während der angegebenen Zeit gerührt. Am Ende der angegebenen Zeit wurden dem Reaktionsgemisch langsam 5 ml Methanol zugesetzt, wonach das Gemisch in 150 ml Methanol dekantiert wurde und anschließend 1,5 ml konzentrierte wäßrige HCl zugesetzt wurde. Das entstehende Gemisch wurde etwa 30 Minuten lang gerührt. Das entstehende weiße feste Polymer wurde gefiltert, mit sechs 20 ml-Teilmengen Methanol gewaschen und im Vakuum getrocknet.
- Das Polymer XI wurde synthetisiert, indem die angegebenen Mengen des angegebenen Katalysators, Comonomers und Dichlormethan-Lösungsmittels in einem Schlenk-Kolben vereinigt wurden. Die Reaktion wurde ausgelöst, indem das Gemisch unter 1 atm Ethylen bei Raumtemperatur gebracht wurde; es wurde kein PMAO zugesetzt. Die Reaktion ging 4260 Minuten lang unter Rühren vonstatten. Das entstehende Polymer war eine ölige Flüssigkeit. Das Reaktionsprodukt wurde gefiltert. Dem Filtrat wurden 350 ml Methanol unter Rühren zugesetzt. Ein abgeschiedenes Öl wurde isoliert und in 100 ml Dichlormethan wiederaufgelöst, und anschließend wurden 350 ml Methanol zugesetzt. Ein hellgelbes Ölprodukt wurde isoliert und im Vakuum getrocknet. Das so erzeugte Material wies eine Glasübergangstemperatur von –66°C und keinen Schmelzpunkt auf.
- Die Polymere XII und XIII wurde erzeugt, indem die angegebenen Mengen des angegebenen Katalysators, Comonomers und Lösungsmittels in einem Schlenk-Kolben innerhalb eines Trockenschranks vereinigt wurden. Das Gemisch wurde unter 1 atm Ethylen gebracht und 15 Minuten mit Ethylen gespült. 10 ml einer Lösung von 7,1% PMAO in Toluol wurden zugesetzt um die Reaktion auszulösen, die unter Rühren während der angegebenen Zeit bei der angegebenen Temperatur weiterlief. 350 ml Methanol wurden langsam dem Reaktionsgemisch zugesetzt, gefolgt von 5 ml konzentrierter HCl. Das weiße feste Polymer wurde gefiltert, mit Methanol gewaschen und im Vakuum getrocknet.
- Das Molekulargewicht wurde durch Gelpermeationschromatographie unter Verwendung von Polyethylen-Standards bestimmt. Die Schmelzpunkte wurden unter Verwendung eines DuPont-Differentialscanningkalorimeters, Modell 912, durch Abkühlung auf –100°C und anschließende Erwärmung mit 10°C/min auf 150°C bestimmt. In den nachstehend beschriebenen Beispielen werden die Polymere mit römischen Zahlen I-XVII bezeichnet. Die verwendeten Katalysatoren werden in Tabelle 1 durch eine Buchstabenkennzeichnung bezeichnet, die den in Tabelle 2 aufgeführten Katalysatoren entspricht. Der prozentuale Anteil des Comonomers im Polymer wurde durch eine Kombination von Protonen- und 13C-magnetischer Kernresonanz bestimmt.
-

- TABELLE 2: Bei der Synthese der Polymere I–XIV eingesetzte Katalysatoren

-

- Die nachstehend beschriebenen nichtionischen Vorläuferpolymere XV und XVI wurden durch Copolymerisation von Propylen mit einem Comonomer der folgenden Formel gebildet:
- Das verwendete Lösungsmittel war Toluol.
- Das Polymer XV wurde synthetisiert, indem in einem Schlenk-Kolben innerhalb eines Trockenschranks 2,3 mg (0,0055 mMol) des Katalysators rac. Ethylenbis(indenyl)zirconium(IV)dichlorid, 2,72 g (7,11 mMol) CH2=CH(CH2)4(CF2)2O(CF2)2SO2F und 25 ml Toluol vereinigt wurden. Dieses Gemisch wurde in einem Eiswasserbad unter 3 psig (122 kPa) Propylen gesetzt und wurde 10 Minuten mit Propylen gespült. Dem Gemisch wurde PMAO (7,2 ml Toluollösung von 7,1 Gew.-%) zugesetzt. Nach einstündigem Rühren unter 3 psig (122 kPa) Propylen bei 0°C wurde dem Reaktionsgemisch langsam Methanol (150 ml) zugesetzt, gefolgt von 5 ml konzentrierter HCl. Das weiße feste Polymer wurde gefiltert, mit Methanol gewaschen und im Vakuum getrocknet. Man erhielt Copolymer (1,35 g). Gemäß 13C-NMR war das Polymer isotaktisch. 1H-NMR (TCE-d2) zeigte einen Comonomer-Einbau von 2,9 Mol-% an. Gemäß Differentialscanningkalorimetrie weist das Copolymer einen Schmelzpunkt von 133°C auf Die Gelpermeationschromatographie (TCB, 135°C, Polyethylen-Standard) ergab: Mw = 23200; Mn = 11000; Mw/Mn = 2,1.
- Das Polymer XVI wurde synthetisiert, indem in einem Schlenk-Kolben innerhalb eines Trockenschranks 2,3 mg (0,0055 mMol) des Katalysators rac. Ethylenbis(indenyl)zirconium(IV)dichlorid, 5,5 g (0,0144 Mol) CH2=CH(CH2)4(CF2)2O(CF2)2SO2F und 25 ml Toluol vereinigt wurden. Dieses Gemisch wurde in einem Eiswasserbad unter 3 psig (122 kPa) Propylen gesetzt und wurde 10 Minuten mit Propylen gespült. Dem Gemisch wurde PMAO zugesetzt (4,0 ml Toluollösung von 12,9 Gew.-%). Nach zweistündigem Rühren unter 3 psig (122 kPa) Propylen bei 0°C wurde dem Reaktionsgemisch langsam Methanol (5 ml) zugesetzt. Das Gemisch wurde dann in 150 ml Methanol gegossen, gefolgt von 5 ml konzentrierter HCl. Nach 20 Minuten Rühren bei Raumtemperatur wurde das weiße feste Polymer gefiltert, mit Methanol gewaschen und im Vakuum getrocknet. Man erhielt Copolymer (4,6 g). Gemäß 13C-NMR war das Polymer isotaktisch. 1H-NMR (TCE-d2) zeigte einen Comonomer-Einbau von 3,8 Mol-% an. Gemäß Differentialscanningkalorimetrie weist das Copolymer einen Schmelzpunkt von 124°C auf. Die Gelpermeationschromatographie (TCB, 135°C, Polyethylen-Standard) ergab: Mw = 39200; Mn = 20900; Mw/Mn = 1,9.
- Das nachstehend beschriebene nichtionische Vorläuferpolymer XVII wurde durch Anpolymerisieren von CH2=CH(CF2)2O(CF2)2SO2F an Niederdruckpolyethylen gebildet.
- Das Polymer XVII wurde synthetisiert, indem eine Lösung von 13,03 g Niederdruckpolyethylen (Aldrich, Mw = 125000) in 100 ml o-Dichlorbenzol bei 125°C mit 10,15 g CH2=CH(CF2)2O(CF2)2SO2F unter Stickstoff vereinigt wurde und anschließend eine Dichlorbenzollösung von tert-Butylperoxid (1,23 g tBuOOtBu in 20 ml o-Dichlorbenzol) langsam zugesetzt wurde. Die Zugabe war innerhalb 7 h beendet. Die Lösung wurde dann über Nacht gerührt. Die Lösung wurde in 500 ml Methanol gegossen und in einem Labormischer vermischt und anschließend filtriert, wobei die Schritte der Auflösung in Methanol, des Vermischens und der Filtration insgesamt viermal ausgeführt wurden. Das feste Polymer wurde dann dreimal mit Methanol gewaschen und im Vakuum getrocknet. Man erhielt weißes Polymer (21,3 g). Gemäß 1H-NMR betrug der Anteil des Comonomer-Einbaus 5,4 Mol-%. Gemäß NMR betrug die Verzweigungshäufigkeit 7 Me/1000CH2. GPC (TCB, 135°C, PE-Standard): Mw = 65700; Mn = 4820; P/D = 15.3. Gemäß DSC hat das Copolymer einen Schmelzpunkt von 118°C.
- Aus den erfindungsgemäßen Polymeren wurden erfindungsgemäße Folien gefertigt, die mit F1-F16, F19, F21–F23 bezeichnet wurden.
- Es wurde festgestellt, daß das Polymer VI zu spröde für eine Weiterbehandlung war, wahrscheinlich wegen des niedrigen Molekulargewichts, und nicht zu einer freistehenden Folie verarbeitet werden konnte. Das Polymer XI war ein Öl, das nicht bei Raumtemperatur zu einer freistehenden Folie verarbeitet werden konnte.
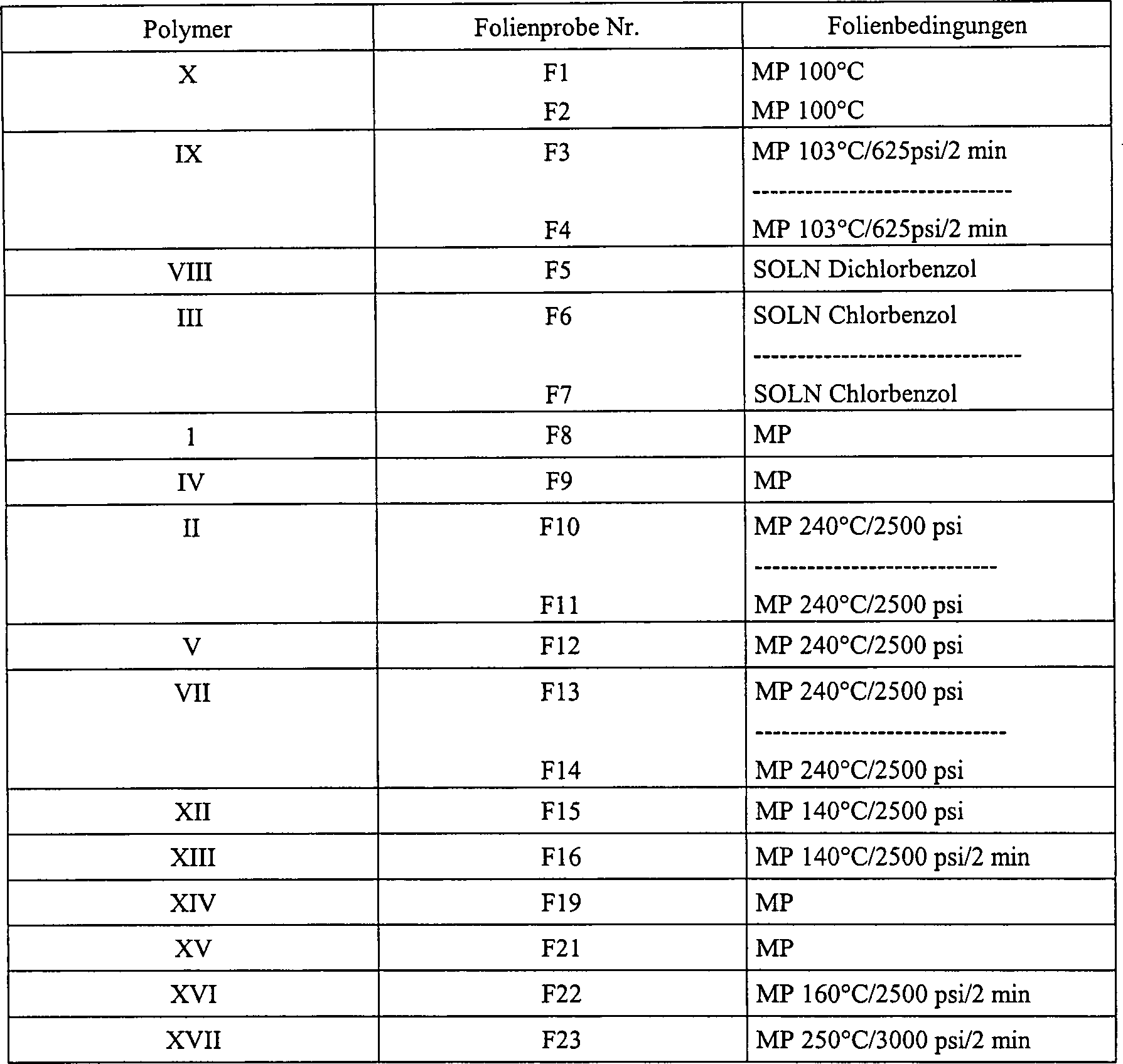
- Alle anderen oben angegebenen Polymere, die als Polymere I–V und als Polymere VII–X und XII-XVII bezeichnet wurden, wurden zu Folien verarbeitet. Aus der Schmelze gepreßte Folien, die in Tabelle 3 als "MP" bezeichnet werden, mit Abmessungen im Bereich von 3,75 cm × 3,75 cm bis 7,5 cm × 7,5 cm, wurden geformt, indem 0,25–5,0 g des getrockneten Polymers zwischen zwei Blätter Kapton®-Polyimidfolie, beziehbar von DuPont, Wilmington, Delaware, eingebracht und zwischen die Platten einer hydraulischen Presse (Modell P218C, Pasadena Hydraulic Industries, City of Industry, CA) eingelegt wurden, die mit ESCS-Temperaturreglern von Omron Electronics Inc. (Schaumburg, IL) ausgestattet war. Das Polymer wurde 2 Minuten vorgewärmt, anschließend 2 Minuten gepreßt und dann unter Druck abgekühlt. Die entstehenden Folien hatten Dicken im Bereich von etwa 63 bis 127 Mikrometer. Die angegebenen spezifischen Temperaturen und Drücke sind in Tabelle 3 dargestellt.
- Aus der Lösung gegossene Folien, in Tabelle 3 mit "soln" bezeichnet, wurden hergestellt, indem 0,25–5,0 g Polymer durch Erhitzen des Lösungsmittels bis zur Auflösung des Polymers in dem angegebenen Lösungsmittel aufgelöst und anschließend auf eine Glasgießschale mit Mulden von 2,5 cm × 2,5 cm gegossen wurden. Das Lösungsmittel wurde bei Raumtemperatur verdampft und ließ Polymerfolien mit Dicken im Bereich von etwa 25–127 Mikrometer zurück. TABELLE 3 – FORMEN VON POLYMERFOLIEN
- 1 psi
- 1 lb/in2 = 6,895 kPa
- BEISPIELE 1–22
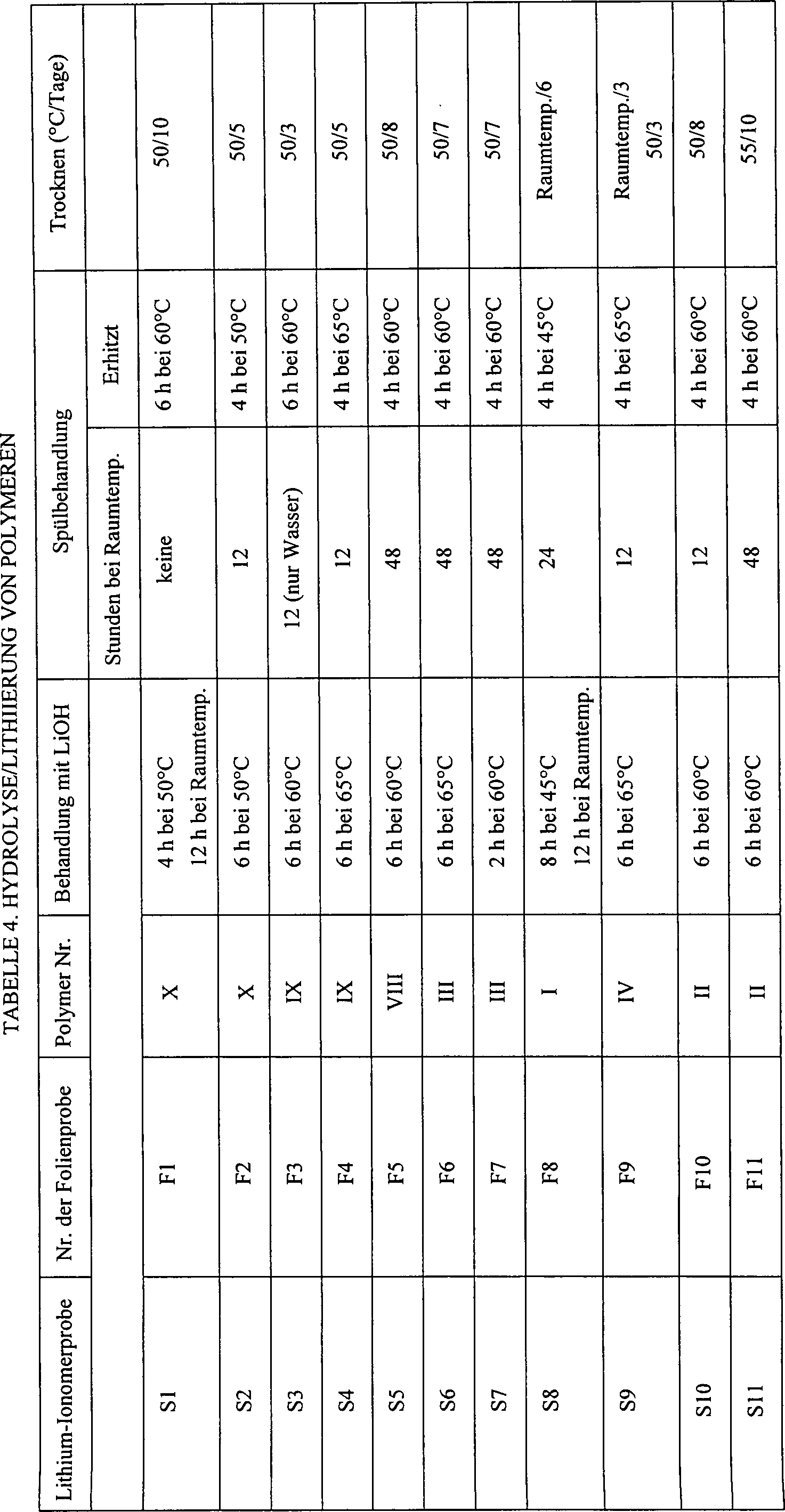
- Jede der in Tabelle 3 beschriebenen Folienproben F1–F16, F19, F21–F23 zuzüglich zwei Öl-Teilmengen des Polymers XI wurden durch Behandlung mit einer gesättigten LiOH-Lösung in 1 : 1 Wasser/Methanol hydrolysiert, anschließend in 1 : 1 Wasser/Methanol bei Raumtemperatur gespült und dann in einem frischen 1 : 1 Wasser/Methanol-Gemisch erhitzt. Wenn nicht anders angegeben, wurden die Proben dann in einem Vakuumofen, Modell 1430, beziehbar von VWR Scientific, West Chester, PA, bei einem Druck von etwa 220 Torr (29,33 kPa) getrocknet. Tabelle 4 zeigt die Dauer und Temperatur der Einwirkung der LiOH-Lösung auf die Folie, die Dauer der Spülung bei Raumtemperatur, die Temperatur und Dauer der Warmspülung und die Temperatur und Dauer der Trocknung der hydrolysierten Folie.
-
-

-

- BEISPIEL 23
- Eine Probe von 4,43 g Polymer XIV in Form eines Polymerbrockens im Polymerisationszustand, hergestellt wie in Beispiel XIV beschrieben, wurde einer Hydrolyse ausgesetzt, indem sie zwei Stunden in einen Überschoß einer gesättigten LiOH-Lösung in einem auf 70°C vorgewärmten 1 : 1 Wasser/Methanol-Gemisch eingetaucht wurde, anschließend auf 75°C erwärmt und weitere 2 Stunden gehalten wurde, anschließend der Reihe nach auf Raumtemperatur abgekühlt und 12 Stunden gehalten und danach wieder auf 75°C erwärmt und 4 Stunden lang gehalten wurde. Das resultierende hydrolysierte Polymer wurde dann 12 Stunden bei Raumtemperatur in einem 1 : 1 Wasser/Methanol-Gemisch gespült, anschließend 4 Stunden bei 80°C in einem frischen 1 : 1 Gemisch von Wasser/Methanol gespült, danach 12 Stunden bei Raumtemperatur in einem 1 : 1 Gemisch von Wasser/Methanol gespült.
- Das hydrolysierte Polymer wurde dann in THF aufgelöst und durch Gießen auf eine Glasplatte, anschließende Verdampfung des THF und Trennung der Folie von der Platte zu einer Folie gegossen. Die so geformte hydrolysierte Folie wird nachstehend als Probe S20 bezeichnet.
- BEISPIEL 24
- 0,2817 g lithiierte Polymerprobe S19, hergestellt wie vorstehend beschrieben, wurde in 10 ml THF eingebracht und bis zur Auflösung schwach erwärmt. 0,056 g Cabot Cab-o-sil® TS530 wurde zugesetzt und bis zur Dispersion gerührt. Die Dispersion wurde auf eine runde Petrischale aus Teflon® PFA von 50 mm Durchmesser gegossen, und man ließ das Lösungsmittel verdunsten, um die hydrolysierte Folie zu formen, die nachstehend als Probe S24 bezeichnet wird.
- BEISPIELE 25–181
- Die getrockneten hydrolysierten Folien der Beispiele 1–24, S1–S24, wurden im noch warmen Zustand in einen abgedichteten Behälter übertragen und in eine Manipulationskammer mit einer trockenen Stickstoffatmosphäre unter Überdruck befördert, in der die Membran aus dem abgedichteten Behälter entnommen und auf Raumtemperatur gebracht wurde. Noch in der Manipulationskammer wurde die Membran dann in mehrere Abschnitte von 1,0 cm × 1,5 cm Größe geschnitten. Typischerweise wurden die Proben im Präparationszustand dann 24–48 Stunden unter Vakuum auf 100°C erwärmt.
- Eine abgekühlte Membranprobe von 1,0 cm × 1,5 cm wurde dann in einem verschlossenen Glasfläschchen 24 Stunden bei Raumtemperatur in einem Überschuß einer oder mehrerer Flüssigkeiten getränkt. Die verwendeten Flüssigkeiten sind alle im Handel erhältlich und wurden im Anlieferungszustand verwendet. Nach dem Eintauchen wurde die Membranprobe aus dem Flüssigkeitsbad entnommen, mit einem Papierhandtuch abgetupft, um überschüssige Flüssigkeit zu entfernen, und getestet.
- Die Ionenleitfähigkeit wurde unter Anwendung eines sogenannten Vierpunktsondenverfahrens bestimmt, das in einem Artikel mit dem Titel "Proton Conductivity of Nafion® 117 As Measured by a Four-Electrode AC Impedance Method" (Protonenleitfähigkeit von Nafion® 117, gemessen nach einem Vier-Elektroden-Wechselstromimpedanzverfahren) von Y. Sone et al., J. Electrochem. Soc., 143, 1254 (1996) beschrieben wird. Das beschriebene Verfahren gilt für Membranen für wäßrige Elektrolyte. Das Verfahren wurde modifiziert, um die hierin angegebenen Meßwerte für nichtwäßrige Lösungsmittel zu erhalten, indem die beschriebene Vorrichtung in eine abgedichtete, mit trockenem Stickstoff gespülte Manipulationskammer gesetzt wurde, um eine etwaige Wassereinwirkung zu minimieren. Das Verfahren wurde außerdem modifiziert, indem die Punkt- bzw. Spitzensonden, die in dem veröffentlichten Verfahren eingesetzt wurden, durch parallele lineare Sonden ausgetauscht wurden, welche die volle Breite der Testprobe überspannten.
- Eine Folie von 1,0 cm × 1,5 cm wurde trockengetupft und in die Leitfähigkeitsmeßzelle eingesetzt. Die Zellenimpedanz wurde über den Bereich von 10 Hz bis 100000 Hz bestimmt, und der Wert mit dem Phasenwinkel null im höheren Frequenzbereich (gewöhnlich 500–5000 Hz) wurde dem Volumenwiderstand der Probe in Ohm zugeschrieben. Der unaufbereitete Widerstandswert wurde dann unter Verwendung der Zellenkonstante und der Dicke der durch Flüssigkeit gequollenen Folie in einen Leitfähigkeitswert in S/cm umgerechnet.
- Die folgenden Abkürzungen sind benutzt worden:
DEC Diethylcarbonat DEE Diethoxyethan DMC Dimethylcarbonat DME 1,2-Dimethoxyethan DMF N,N-Dimethylformamid DMSO Dimethylsulfoxid EC Ethylencarbonat (1,3-Dioxolan-2-on) GBL γ-Butyrolacton MA Methylacetat MeOH Methanol MG Methylglycolat NMF N-Methylformamid NMP N-Methylpynolidon PC Propylencarbonat PEG Poly(ethylenglycol) THF Tetrahydrofuran - BEISPIELE 25–138
- Bei den so ausgeführten Tests, in denen erfindungsgemäße hydrolysierte Folien S1–S24 mit den angegebenen Flüssigkeiten kombiniert wurden, um die beschriebenen erfindungsgemäßen leitfähigen Zusammensetzungen zu bilden, zeigte sich, daß die in Tabelle 5 dargestellten Zusammensetzungen eine höhere Ionenleitfähigkeit bei Raumtemperatur als 10–5 S/cm aufweisen.
- TABELLE 5

- Es wurde beobachtet, daß die in den oben beschriebenen Beispielen verwendeten Proben von S15 ihre körperliche Integrität in besonders hohem Grade beibehielten, während sie in die zahlreichen, in Tabelle 5 aufgeführten Lösungsmittel getaucht wurden.
- BEISPIELE 139–140
- Probekörper von jeder der hydrolysierten Folienproben S2, S4, S5, S6 und S7 wurden nach dem oben beschriebenen Verfahren in Propylencarbonat (PC) getaucht, wobei aber die Einwirkungsdauer entweder 2 Stunden oder 54 Stunden betrug, wie in Tabelle 6 angegeben.
- TABELLE 6 IONENLEITFÄHIGKEITEN VON HYDROLYSIERTEN FOLIEN IN PROPYLENCARBONAT BEI RAUMTEMPERATUR

- BEISPIELE 141–150
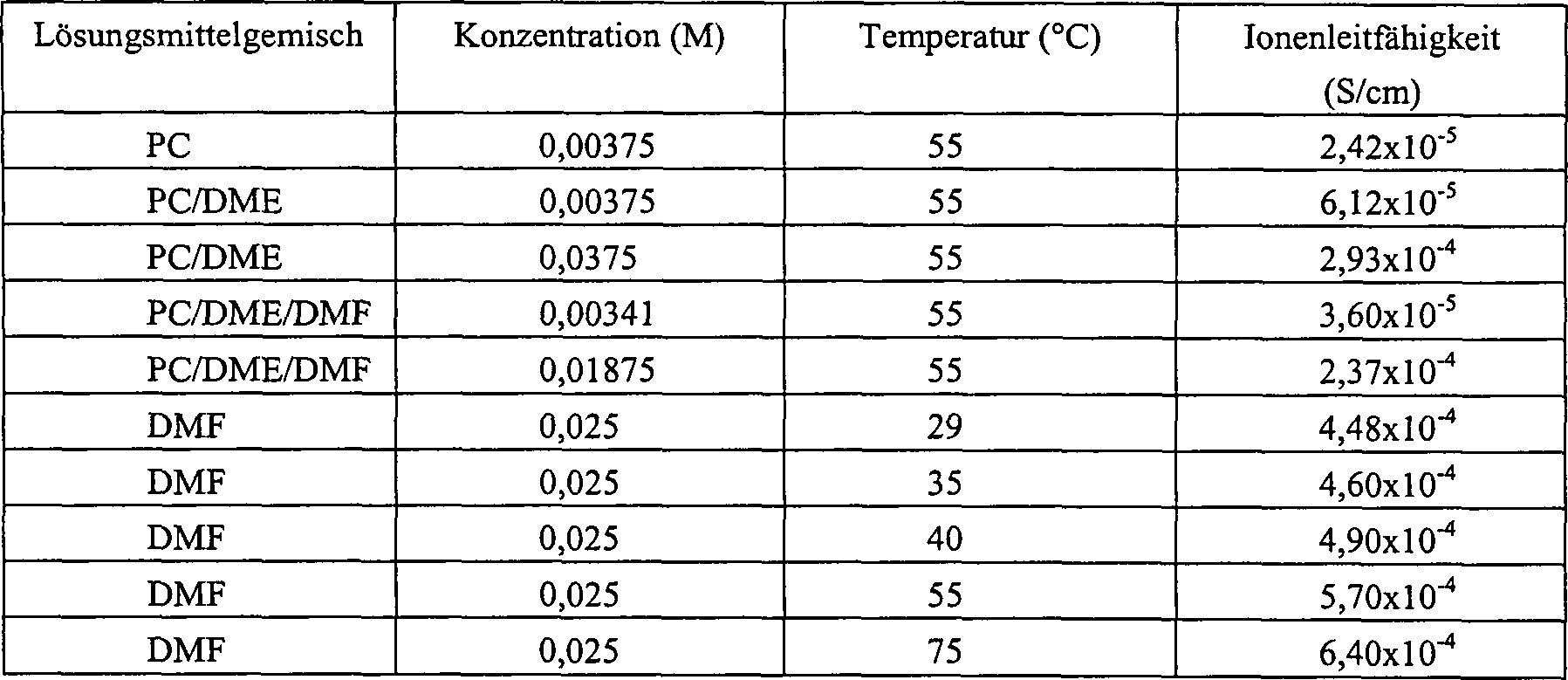
- Das durch Hydrolyse des Polymers XI gebildete polymere Öl wurde mit den in Tabelle 7 angegebenen Lösungsmitteln vermischt, um Gele bei den angegebenen Temperaturen zu bilden. Leitfähigkeitsmessungen wurden unter Verwendung einer Orien-Flüssigkeitstauchsonde zur Leitfähigkeitsmessung ausgeführt. Die Ergebnisse sind in Tabelle 7 dargestellt.
- TABELLE 7 IONENLEITFÄHIGKEITEN VON GELLÖSUNGEN DES POLYMERS XI
- BEISPIELE 150–173
- Für Proben der in Tabelle 8 angegebenen hydrolysierten Folien wurde die Leitfähigkeit sowohl in der lithiierten Form, die gemäß der obigen Beschreibung hergestellt wurde, als auch in der Säureform in einem wäßrigen Medium bestimmt. Die Membranen in Säureform wurden aus den Membranen in Lithium-Form hergestellt, indem die Membran eine Stunde lang in einen Überschuß an 1,0 M Salpetersäre (analysenrein, EM Science, Gibbstown, NJ) getaucht, anschließend eine Stunde in entionisiertem Wasser bei T = 80°C gespült und dann in entionisiertem Wasser abgekühlt wurde.
- Die Folien wurden dann durch zweistündiges Eintauchen in entionisiertes Wasser und Erwärmen auf T = 80°C und anschließendes Abkühlen auf Raumtemperatur behandelt. Die Leitfähigkeit wurde unter Anwendung der gleichen Verfahren wie oben gemessen, außer daß alle Messungen außerhalb der Manipulationskammer ausgeführt wurden.
- TABELLE 8 IONENLEITFÄHIGKEITEN VON COPOLYMEREN VON ETHYLEN- UND FLUORSULFONATMONOMEREN IN Li+- UND H+-FORM, ÄQUILIBRIERT MIT FLÜSSIGEM ENTIONISIERTEM WASSER BEI RAUMTEMPERATUR
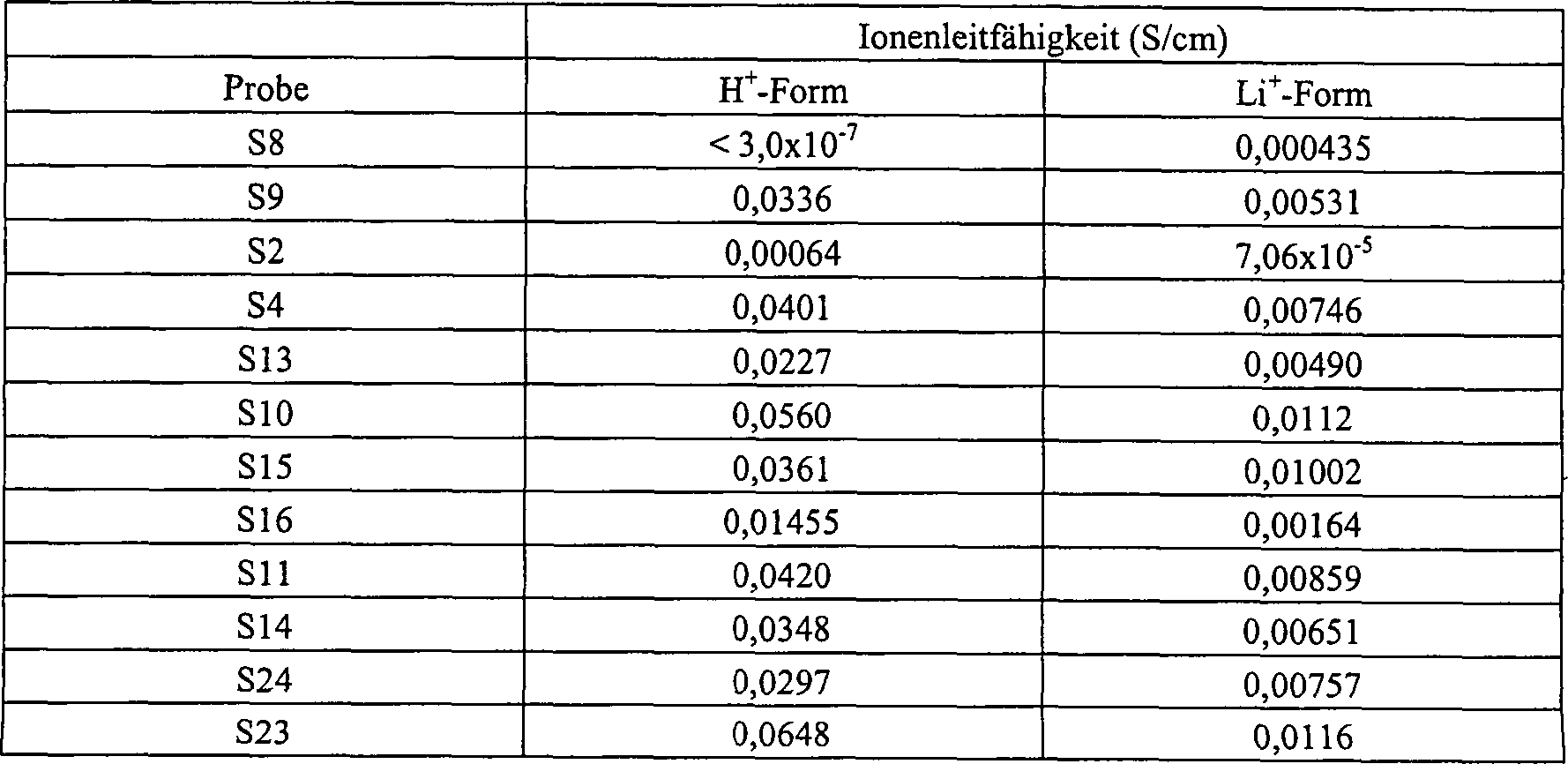
- BEISPIEL 174
- Das Polymer IX, das gemäß der obigen Offenbarung synthetisiert wurde, wird nach dem in Desmarteau, J. Fluorine Chem. 52, S. 7ff: (1991) gelehrten Verfahren, das hier durch Verweis einbezogen wird, mit CF3CF2SO2NNaSi(CH3)3 zur Reaktion gebracht. Das so gebildete Polymer wird 6 Stunden bei Raumtemperatur mit einem H2SO4-Überschuß von 30% behandelt, um das Polymer in Protonenform (H+-Form) zu bilden. Das so entstandene Polymer in Protonenform wird dann bei Raumtemperatur 5 Stunden lang mühelos einem Ionenaustausch in einem großen Überschuß von 0,1 M LiOH in einer 50/50-Wasser/Methanol-Lösung ausgesetzt. Das entstehende Ionomer ist ein Polymer mit einer im wesentlichen nichtsubstituierten Ethylen-Hauptkette und 3,9 Mol-% einer Seitengruppe, die ein Radikal mit der folgenden Formel aufweist:
-
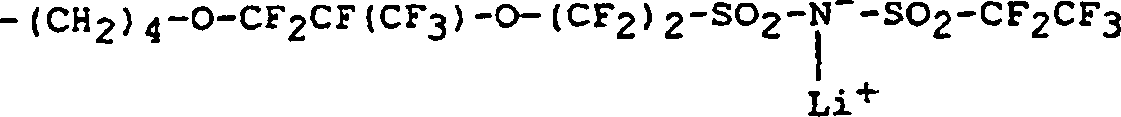
- Das Li-Imid-Ionomer wird durch NMR und Elementaranalyse bestätigt.
- BEISPIEL 175
- (CF3CF2'SO2)2C(BrMg)2 wird nach dem Verfahren von Seppelt, Inorg. Chem. 27, S. 2135ff. (1988) aus CH2(SO2CF2CF3)2 und CH3MgBr synthetisiert. Einer gerührten THF-Lösung des gemäß der obigen Beschreibung synthetisierten Polymers IX wird (CF3CF2'SO2)2C(BrMg)2 bei 0°C zugesetzt. Nach abgeschlossener Zugabe wird das Reaktionsgemisch über Nacht bei Raumtemperatur gerührt. Das THF wird dann abgepumpt, 3 M HCl wird zugesetzt, die Lösung wird mehrere Stunden gerührt und gefiltert. Der Feststoff wird mit Wasser gewaschen, um anorganische Salze zu entfernen, und dann mit 0,1 M LiCl in 50/50 Wasser/MeOH bei Raumtemperatur über Nacht behandelt. Man erhielt RfSO2C(Li)(SO2CF3)2.
- Da resultierende Ionomer ist ein Polymer mit einer im wesentlichen nichtsubstituierten Polyethylen-Hauptkette und 3,9 Mol-% einer Seitengruppe, die ein Radikal mit der folgenden Formel
-

- Das Li-Methid-Ionomer wird durch NMR und Elementaranalyse bestätigt.
- BEISPIEL 176
- Eine getrocknete 0,5 g-Probe des hydrolysierten Polymers von Beispiel 23 zusammen mit 12 g Tetrahydrofuran (THF) wurde in einen abgedichteten Kolben eingebracht und 4 Stunden mit 400 U/min und einer Temperatur von 65°C gerührt. Quarzstaub (125 g TS530, Cabot Corp., Boston, MA) wurde zugesetzt, und das Rühren wurde einige Minuten fortgesetzt, um das Siliciumdioxid zu dispergieren. Die Suspension wurde durch einen Glaswollepfropfen gefiltert, um etwaige Gelteilchen zu entfernen, und mit einer Rakel mit einer Rahmenhöhe von 0,050 Zoll auf Mylar®-Polyesterfolie, beziehbar von DuPont, gegossen. Das THF ließ man bei Umgebungstemperatur verdunsten, woraus sich eine Folie von 32 bis 40 μm Dicke ergab. Die so erzeugte Folie wurde als Trennfolie in der nachstehend beschriebenen Batteriekonstruktion verwendet.
- Eine zweite Probe des hydrolysierten Polymers von Beispiel 23 wurde bei der Ausbildung der positiven Elektrode eingesetzt, die in der nachstehend beschriebenen Batterie verwendet wird: 0,2 g des getrockneten Ionomers wurden in einen abgedichteten Kolben mit 7 g Tetrahydrofuran eingebracht und 2 Stunden bei einer Temperatur von 70°C mit 400 U/min gerührt. Ruß (0,05 g SP-Ruß, MMM S. A. Carbon, Brüssel, Belgien) und Graphit (0,75 g Mesophasenkohlenstoff-Mikrokügelchen MCMB 25–28, Osaka Gas, Japan) wurden zugesetzt, und das Gemisch wurde weitere 15 Minuten gerührt. Die Aufschwemmung wurde auf silanierte Mylar®-Folie, beziehbar von DuPont, gegossen, und das THF ließ man bei Umgebungstemperatur verdunsten.
- Aus der Graphitfolie wurde eine runde positive Elektrode von 12 mm Durchmesser ausgestanzt und unter Vakuum bei 100°C getrocknet und ergab eine Masse von 11,6 mg (8,7 mg Graphit) und eine Dicke von 160 μm. Aus der oben erwähnten Trennelementfolie wurde ein Trennelement ausgestanzt (11,5 mg Masse, 43 μm dick und rund mit 19 mm Durchmesser). Diese Elemente wurden 10 min in einem Überschuß von wasserfreiem Ethylencarbonat (EC)/Dimethylcarbonat (DMC) getränkt und saugten 21,8 mg Flüssigkeiten auf. Sie wurden zusammen mit einer negativen Lithiumfolienelektrode in eine Knopfzelle der Größe 2325 montiert. Die Zelle wurde mit konstantem Strom von 0,5 mA auf eine Spannung von 0,01 V entladen, an welchem Punkt die Spannung konstant gehalten wurde, bis der Strom auf weniger als 0,05 mA abfiel. Die Kapazität bei der ersten Entladung betrug 2,42 mAh, was 280 mAh pro g positivem Graphitelektrodenmaterial entspricht. Die Zelle wurde mit einer Geschwindigkeit von 0,5 mA auf 1,2 V aufgeladen, und dann wurde die Spannung auf 1,2 V konstant gehalten, bis der Ladestrom auf weniger als 0,05 mA abfiel. Die Ladekapazität betrug 1,97 mAh, woraus sich auf einen elektrochemischen Wirkungsgrad von 81% im ersten Entlade-/Ladezyklus schließen läßt. Die Zelle wurde auf ähnliche Weise wie oben wiederholt entladen und aufgeladen, wobei die Kapazität bei der 14. Entladung 1,85 mAh betrug.
Claims (28)
- Ionomer mit einer Hauptkette und Seitengruppen, wobei die Hauptkette im wesentlichen aus Methylen- und Methineinheiten besteht und die Seitengruppen ionische Radikale mit der Formel
- Ionomer nach Anspruch 1, wobei a = 0; X = 0 gilt; Rf durch die Formel
- Ionomer nach Anspruch 2, wobei M+ ein Alkalimetall-Kation, Rf' Fluor ist, x = 1 oder 2, y = 0, m = 1 oder 2, z = 4 gilt und R Ethenyl, Propenyl oder Butenyl ist.
- Ionomer nach Anspruch 3, wobei M+ ein Lithium-Kation ist.
- Ionomer nach Anspruch 1, wobei die Konzentration der ionischen Radikale 1–20 Mol-% beträgt.
- Ionomer nach Anspruch 1, wobei die Konzentration der ionischen Radikale 2–10 Mol-% beträgt.
- Ionomer nach Anspruch 1, das ferner bis zu 150 kurzkettige Verzweigungen pro 1000 Methylengruppen in der Hauptkette aufweist.
- Ionomer nach Anspruch 1, das 5–90 kurzkettige Verzweigungen pro 1000 Methylengruppen in der Hauptkette aufweist.
- Ionomer nach Anspruch 1 in Form eines Films oder einer Folie.
- Ionomer nach Anspruch 1, das ferner beigemischte anorganische Teilchen aufweist.
- Ionomer nach Anspruch 10, wobei die anorganischen Teilchen Siliciumdioxid-Teilchen mit einer mittleren Teilchengröße von weniger als 1,0 μm sind, wobei das Siliciumdioxid in der Beimischung in einem Anteil bis zu 50 Gew.-% der Gesamtmasse vorhanden ist.
- Ionenleitende Zusammensetzung, die ein Ionomer nach Anspruch 1 und eine darin eingesaugte Flüssigkeit aufweist.
- Ionenleitende Zusammensetzung nach Anspruch 12, wobei die Flüssigkeit Wasser oder Methanol ist.
- Ionenleitende Zusammensetzung nach Anspruch 12, wobei die Flüssigkeit aprotisch ist.
- Ionenleitende Zusammensetzung nach Anspruch 14, wobei die Flüssigkeit aus der Gruppe ausgewählt ist, die aus organischen Carbonaten und deren Gemischen besteht.
- Ionenleitende Zusammensetzung nach Anspruch 13, wobei die Flüssigkeit ein Gemisch aus Ethylencarbonat und mindestens einer Flüssigkeit ist, die aus der Gruppe ausgewählt ist, die aus Dimethylcarbonat, Methylethylcarbonat und Diethylcarbonat besteht.
- Ionenleitende Zusammensetzung nach Anspruch 12, wobei M+ ein Lithium-Kation ist, Rn = (CH2)4, Rf (CF2)x, X = O, a = 0 gilt, und die Konzentration der ionischen Radikale 2–10 Mol-% beträgt, wobei die Zusammensetzung ferner bis zu 150 kurzkettige Verzweigungen pro 1000 Methyleneinheiten in der Hauptkette aufweist und die Flüssigkeit aus der Gruppe ausgewählt ist, die aus organischen Carbonaten und deren Gemischen besteht.
- Ionenleitende Zusammensetzung nach Anspruch 12 in einer Form, die aus der Gruppe ausgewählt ist, die aus einem Film, einer Folie und einem Gel besteht.
- Ionenleitende Zusammensetzung nach Anspruch 17 in einer Form, die aus der Gruppe ausgewählt ist, die aus einem Film, einer Folie und einem Gel besteht.
- Ionenleitende Zusammensetzung nach Anspruch 18, die ferner eine(n) mikroporöse(n), elektrisch isolierende(n) Folie oder Film aufweist, in deren (dessen) Mikroporen das Gel eingesaugt ist.
- Ionenleitende Zusammensetzung nach Anspruch 19, die ferner eine(n) mikroporöse(n), elektrisch isolierende(n) Folie oder Film aufweist, in deren (dessen) Mikroporen das Gel eingesaugt ist.
- Verfahren zur Bildung eines Ionomers, wobei das Verfahren aufweist: Inkontaktbringen eines Polyolefins, das eine Hauptkette und Seitengruppen aufweist, mit einer Lösung einer Alkalimetallbase, wobei die Hauptkette im wesentlichen aus Methylen- und Methineinheiten besteht und die Seitengruppen Radikale mit der Formel
- Verfahren zur Bildung einer leitfähigen Zusammensetzung, wobei das Verfahren das Inkontaktbringen des Ionomers nach Anspruch 1 mit einer Flüssigkeit aufweist.
- Elektrode, die mindestens ein aktives Elektrodenmaterial, das damit vermischte Ionomer nach Anspruch 1 und eine darin eingesaugte Flüssigkeit aufweist.
- Elektrode nach Anspruch 24, wobei M+ ein Lithium-Kation ist, Rn = (CH2)4, Rf (CF2)x, X = O, a = 0 gilt und die Konzentration der ionischen Radikale 2–10 Mol-% beträgt, wobei die Zusammensetzung ferner bis zu 150 kurzkettige Verzweigungen pro 1000 Methyleneinheiten in der Hauptkette aufweist und die Flüssigkeit aus der Gruppe ausgewählt ist, die aus organischen Carbonaten und deren Gemischen besteht.
- Elektrode nach Anspruch 25, die ferner Ruß aufweist.
- Elektrode nach Anspruch 26, wobei das Masseverhältnis des Ionomers zum aktiven Elektrodenmaterial zwischen 0,05 und 0,8 und das Masseverhältnis des Rußes zum aktiven Elektrodenmaterial zwischen 0,01 und 0,2 liegt.
- Elektrochemische Zelle, die eine positive Elektrode, eine negative Elektrode, ein zwischen den positiven und negativen Elektroden angeordnetes Trennelement und ein Mittel zum Anschluß der Zelle an eine äußere Last oder Quelle aufweist, wobei mindestens ein Element der Gruppe, die aus dem Trennelement, der positiven Elektrode und der negativen Elektrode besteht, das Ionomer nach Anspruch 1 aufweist.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US61132 | 1993-05-13 | ||
| US09/061,132 US6100324A (en) | 1998-04-16 | 1998-04-16 | Ionomers and ionically conductive compositions |
| PCT/US1999/008255 WO1999052954A1 (en) | 1998-04-16 | 1999-04-15 | Ionomers and ionically conductive compositions |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE69918107D1 DE69918107D1 (de) | 2004-07-22 |
| DE69918107T2 true DE69918107T2 (de) | 2005-01-27 |
Family
ID=22033801
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69918107T Expired - Fee Related DE69918107T2 (de) | 1998-04-16 | 1999-04-15 | Ionomere und ionenleitende zusammensetzungen |
Country Status (9)
| Country | Link |
|---|---|
| US (2) | US6100324A (de) |
| EP (1) | EP1082358B1 (de) |
| JP (1) | JP2002511502A (de) |
| KR (1) | KR100541312B1 (de) |
| CN (1) | CN1297457A (de) |
| AU (1) | AU3645599A (de) |
| CA (1) | CA2326845A1 (de) |
| DE (1) | DE69918107T2 (de) |
| WO (1) | WO1999052954A1 (de) |
Families Citing this family (68)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6100324A (en) * | 1998-04-16 | 2000-08-08 | E. I. Du Pont De Nemours And Company | Ionomers and ionically conductive compositions |
| JP2003520872A (ja) * | 2000-01-19 | 2003-07-08 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー | グラフト共重合体の製造法 |
| BR0113004A (pt) | 2000-07-28 | 2003-06-24 | Du Pont | Processo para enxerto de radical livre |
| EP1220344B2 (de) * | 2000-12-26 | 2012-08-01 | Asahi Glass Company, Limited | Festpolymer-Elektrolyt Membran Festpolymer Brennstoffzelle und Fluorpolymer |
| US6727019B2 (en) * | 2001-03-22 | 2004-04-27 | Ilion Technology | Electrochemical cell having an ionomer binder of Li-AMPS and associated fabrication |
| CA2459357A1 (en) | 2001-10-15 | 2003-04-24 | E. I. Du Pont De Nemours And Company | Solid polymer membrane for fuel cell prepared by in situ polymerization |
| US7094851B2 (en) | 2001-12-06 | 2006-08-22 | Gore Enterprise Holdings, Inc. | Low equivalent weight ionomer |
| US6861489B2 (en) | 2001-12-06 | 2005-03-01 | Gore Enterprise Holdings, Inc. | Low equivalent weight ionomer |
| US6679979B2 (en) * | 2001-12-12 | 2004-01-20 | Ballard Power Systems Inc. | Aqueous ionomeric gels and products and methods related thereto |
| EP1474839B1 (de) * | 2002-02-06 | 2010-04-07 | Battelle Memorial Institute | Polymerelektrolytmembranen zur verwendung in brennstoffzellen |
| AU2003210939A1 (en) * | 2002-02-06 | 2003-09-02 | Battelle Memorial Institute | Polymer electrolyte membrane fuel cell system |
| US20050053818A1 (en) * | 2002-03-28 | 2005-03-10 | Marc St-Arnaud | Ion exchange composite material based on proton conductive functionalized inorganic support compounds in a polymer matrix |
| WO2004051782A1 (en) * | 2002-06-28 | 2004-06-17 | Dubitsky Yuri A | Fuel cell incorporating a polymer electrolyte membrane grafted by irradiation |
| US7317047B2 (en) | 2002-09-24 | 2008-01-08 | E.I. Du Pont De Nemours And Company | Electrically conducting organic polymer/nanoparticle composites and methods for use thereof |
| EP1546283B1 (de) | 2002-09-24 | 2012-06-20 | E.I. Du Pont De Nemours And Company | Elektrisch leitfähige kompositwerkstoffe basierend auf organischen polymeren und nanoteilchen sowie deren verwendung |
| ATE404609T1 (de) | 2002-09-24 | 2008-08-15 | Du Pont | Wasserdispergierbare polythiophene hergestellt unter verwendung von kolloiden auf basis von polymersäuren |
| JP2006500461A (ja) | 2002-09-24 | 2006-01-05 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー | 電子工学用途のための、ポリマー酸コロイドを用いて製造された水分散性ポリアニリン |
| US7390438B2 (en) | 2003-04-22 | 2008-06-24 | E.I. Du Pont De Nemours And Company | Water dispersible substituted polydioxythiophenes made with fluorinated polymeric sulfonic acid colloids |
| US7537682B2 (en) | 2004-03-17 | 2009-05-26 | California Institute Of Technology | Methods for purifying carbon materials |
| US7351358B2 (en) | 2004-03-17 | 2008-04-01 | E.I. Du Pont De Nemours And Company | Water dispersible polypyrroles made with polymeric acid colloids for electronics applications |
| US8147962B2 (en) | 2004-04-13 | 2012-04-03 | E. I. Du Pont De Nemours And Company | Conductive polymer composites |
| JP4978191B2 (ja) * | 2004-06-30 | 2012-07-18 | Tdk株式会社 | 直接アルコール型燃料電池及びその製造方法 |
| JP4506403B2 (ja) * | 2004-10-15 | 2010-07-21 | ダイキン工業株式会社 | イオン伝導体 |
| US7722785B2 (en) | 2005-06-27 | 2010-05-25 | E.I. Du Pont De Nemours And Company | Electrically conductive polymer compositions |
| US7727421B2 (en) | 2005-06-27 | 2010-06-01 | E. I. Du Pont De Nemours And Company Dupont Displays Inc | Electrically conductive polymer compositions |
| WO2007002740A2 (en) | 2005-06-28 | 2007-01-04 | E. I. Du Pont De Nemours And Company | Buffer compositions |
| KR101356296B1 (ko) | 2005-06-28 | 2014-02-06 | 이 아이 듀폰 디 네모아 앤드 캄파니 | 높은 일 함수의 투명한 도체 |
| WO2007041415A2 (en) | 2005-09-30 | 2007-04-12 | Battelle Memorial Institute | Polymers for use in fuel cell components |
| US20100221603A1 (en) * | 2006-03-03 | 2010-09-02 | Rachid Yazami | Lithium ion fluoride battery |
| US7563542B2 (en) * | 2005-10-05 | 2009-07-21 | California Institute Of Technology | Subfluorinated graphite fluorides as electrode materials |
| US8232007B2 (en) | 2005-10-05 | 2012-07-31 | California Institute Of Technology | Electrochemistry of carbon subfluorides |
| US8377586B2 (en) | 2005-10-05 | 2013-02-19 | California Institute Of Technology | Fluoride ion electrochemical cell |
| US7794880B2 (en) | 2005-11-16 | 2010-09-14 | California Institute Of Technology | Fluorination of multi-layered carbon nanomaterials |
| US20070218364A1 (en) * | 2005-10-05 | 2007-09-20 | Whitacre Jay F | Low temperature electrochemical cell |
| US7560497B2 (en) * | 2005-10-24 | 2009-07-14 | Yuan Ze University | Perfluorocarbon ionomer membrane with high proton conductivity and preparation thereof |
| US8216680B2 (en) | 2006-02-03 | 2012-07-10 | E I Du Pont De Nemours And Company | Transparent composite conductors having high work function |
| KR101398780B1 (ko) | 2006-06-01 | 2014-06-18 | 이 아이 듀폰 디 네모아 앤드 캄파니 | 전도성 중합체 조성물 |
| WO2007145975A2 (en) | 2006-06-05 | 2007-12-21 | E. I. Du Pont De Nemours And Company | Process for forming an organic light-emitting diode and devices made by the process |
| KR101387894B1 (ko) | 2006-06-30 | 2014-04-25 | 이 아이 듀폰 디 네모아 앤드 캄파니 | 전도성 중합체 및 부분적으로 플루오르화된 산 중합체의 안정화된 조성물 |
| US8658309B2 (en) * | 2006-08-11 | 2014-02-25 | California Institute Of Technology | Dissociating agents, formulations and methods providing enhanced solubility of fluorides |
| US7718319B2 (en) | 2006-09-25 | 2010-05-18 | Board Of Regents, The University Of Texas System | Cation-substituted spinel oxide and oxyfluoride cathodes for lithium ion batteries |
| US7456314B2 (en) * | 2006-12-21 | 2008-11-25 | E.I. Du Pont De Nemours And Company | Partially fluorinated ionic compounds |
| US20080191172A1 (en) | 2006-12-29 | 2008-08-14 | Che-Hsiung Hsu | High work-function and high conductivity compositions of electrically conducting polymers |
| CA2679635A1 (en) * | 2007-03-14 | 2008-09-18 | California Institute Of Technology | High discharge rate batteries |
| US20080232032A1 (en) * | 2007-03-20 | 2008-09-25 | Avx Corporation | Anode for use in electrolytic capacitors |
| US7649730B2 (en) | 2007-03-20 | 2010-01-19 | Avx Corporation | Wet electrolytic capacitor containing a plurality of thin powder-formed anodes |
| US20080251768A1 (en) | 2007-04-13 | 2008-10-16 | Che-Hsiung Hsu | Electrically conductive polymer compositions |
| US8241526B2 (en) | 2007-05-18 | 2012-08-14 | E I Du Pont De Nemours And Company | Aqueous dispersions of electrically conducting polymers containing high boiling solvent and additives |
| KR20100094475A (ko) | 2007-10-26 | 2010-08-26 | 이 아이 듀폰 디 네모아 앤드 캄파니 | 격납된 층을 제조하기 위한 방법 및 재료, 및 이를 사용하여 제조된 소자 |
| US8216685B2 (en) | 2008-05-16 | 2012-07-10 | E I Du Pont De Nemours And Company | Buffer bilayers for electronic devices |
| CN102203984A (zh) * | 2008-11-04 | 2011-09-28 | 加州理工学院 | 具有可溶性阳极的混合型电化学发生器 |
| TW201030086A (en) | 2008-12-09 | 2010-08-16 | Du Pont | Electrically conductive polymer compositions |
| WO2010077710A2 (en) | 2008-12-09 | 2010-07-08 | E. I. Du Pont De Nemours And Company | Electrically conductive polymer compositions |
| US8461758B2 (en) | 2008-12-19 | 2013-06-11 | E I Du Pont De Nemours And Company | Buffer bilayers for electronic devices |
| US8785913B2 (en) | 2008-12-27 | 2014-07-22 | E I Du Pont De Nemours And Company | Buffer bilayers for electronic devices |
| US8766239B2 (en) | 2008-12-27 | 2014-07-01 | E I Du Pont De Nemours And Company | Buffer bilayers for electronic devices |
| TW201100480A (en) | 2009-03-12 | 2011-01-01 | Du Pont | Electrically conductive polymer compositions for coating applications |
| US8223473B2 (en) | 2009-03-23 | 2012-07-17 | Avx Corporation | Electrolytic capacitor containing a liquid electrolyte |
| JP5587980B2 (ja) | 2009-04-21 | 2014-09-10 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー | 導電性ポリマー組成物およびそれから作製されたフィルム |
| CN102395627B (zh) | 2009-04-24 | 2015-04-01 | E.I.内穆尔杜邦公司 | 导电聚合物组合物和由其制得的膜 |
| WO2010142772A1 (en) * | 2009-06-12 | 2010-12-16 | Solvay Solexis S.P.A. | Fluoroionomers dispersions having low surface tension, low liquid viscosity and high solid content |
| EP2459379A4 (de) | 2009-07-27 | 2015-05-06 | Du Pont | Verfahren und materialien zur herstellung abgegrenzter schichten und damit hergestellte vorrichtungen |
| CN104136417A (zh) * | 2011-12-06 | 2014-11-05 | 3M创新有限公司 | 具有侧官能团的氟化低聚物 |
| CN104798244B (zh) | 2013-10-31 | 2017-07-28 | 株式会社Lg化学 | 锂二次电池 |
| KR102217106B1 (ko) | 2016-05-09 | 2021-02-22 | 주식회사 엘지화학 | 고분자 전해질용 조성물 및 이를 포함하는 리튬 이차전지 |
| WO2017196012A1 (ko) | 2016-05-09 | 2017-11-16 | 주식회사 엘지화학 | 고분자 전해질용 조성물 및 이를 포함하는 리튬 이차전지 |
| JP6843889B2 (ja) | 2016-05-27 | 2021-03-17 | エルジー・ケム・リミテッド | 電気導電性ポリマー組成物におけるジヘテロアミン |
| WO2021066544A1 (ko) * | 2019-09-30 | 2021-04-08 | 코오롱인더스트리 주식회사 | 높은 분산 안정성을 갖는 이오노머 분산액, 그 제조방법, 및 그것을 이용하여 제조된 고분자 전해질막 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3282875A (en) * | 1964-07-22 | 1966-11-01 | Du Pont | Fluorocarbon vinyl ether polymers |
| GB1034197A (en) * | 1963-09-13 | 1966-06-29 | Du Pont | Sulphonic acid derivatives of fluorocarbon vinyl ethers, and polymers thereof |
| JPS568047B2 (de) * | 1972-07-14 | 1981-02-21 | ||
| US4358545A (en) * | 1980-06-11 | 1982-11-09 | The Dow Chemical Company | Sulfonic acid electrolytic cell having flourinated polymer membrane with hydration product less than 22,000 |
| JPS5785826A (en) * | 1980-11-17 | 1982-05-28 | Japan Atom Energy Res Inst | Cation exchange membrane |
| JPS57131376A (en) * | 1981-02-06 | 1982-08-14 | Japan Atom Energy Res Inst | Electrolyzing method for water |
| US5627292A (en) * | 1992-02-21 | 1997-05-06 | Centre National De La Recherche Scientifique | Monomers derived from perhalogenated sultones and polymers obtained from these monomers |
| US5548055A (en) * | 1995-01-13 | 1996-08-20 | Sri International | Single-ion conducting solid polymer electrolytes |
| EP0805826B1 (de) * | 1995-01-24 | 2003-04-23 | E.I. Du Pont De Nemours And Company | Polyolefine |
| US5798417A (en) * | 1996-10-15 | 1998-08-25 | E. I. Du Pont De Nemours And Company | (Fluorovinyl ether)-grafted high-surface-area polyolefins and preparation thereof |
| US6100324A (en) * | 1998-04-16 | 2000-08-08 | E. I. Du Pont De Nemours And Company | Ionomers and ionically conductive compositions |
-
1998
- 1998-04-16 US US09/061,132 patent/US6100324A/en not_active Expired - Lifetime
-
1999
- 1999-04-15 CA CA002326845A patent/CA2326845A1/en not_active Abandoned
- 1999-04-15 CN CN99805082A patent/CN1297457A/zh active Pending
- 1999-04-15 EP EP99918576A patent/EP1082358B1/de not_active Expired - Lifetime
- 1999-04-15 KR KR1020007011363A patent/KR100541312B1/ko not_active IP Right Cessation
- 1999-04-15 JP JP2000543510A patent/JP2002511502A/ja not_active Withdrawn
- 1999-04-15 AU AU36455/99A patent/AU3645599A/en not_active Abandoned
- 1999-04-15 DE DE69918107T patent/DE69918107T2/de not_active Expired - Fee Related
- 1999-04-15 WO PCT/US1999/008255 patent/WO1999052954A1/en active IP Right Grant
-
2000
- 2000-06-07 US US09/588,885 patent/US6268430B1/en not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| JP2002511502A (ja) | 2002-04-16 |
| WO1999052954A1 (en) | 1999-10-21 |
| US6268430B1 (en) | 2001-07-31 |
| US6100324A (en) | 2000-08-08 |
| CA2326845A1 (en) | 1999-10-21 |
| EP1082358A1 (de) | 2001-03-14 |
| CN1297457A (zh) | 2001-05-30 |
| EP1082358B1 (de) | 2004-06-16 |
| AU3645599A (en) | 1999-11-01 |
| DE69918107D1 (de) | 2004-07-22 |
| KR20010042662A (ko) | 2001-05-25 |
| KR100541312B1 (ko) | 2006-01-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69918107T2 (de) | Ionomere und ionenleitende zusammensetzungen | |
| DE69911378T2 (de) | Fluorierte ionomere und verwendung davon | |
| DE69922827T2 (de) | Ionomere und polymere für elektrochemische anwendungen | |
| EP0574791B1 (de) | Polymerelektrolyt-Membran und Verfahren zu ihrer Herstellung | |
| DE60125012T2 (de) | Perfluoropolyether Additive für elektrochemische Anwendungen | |
| DE69918117T2 (de) | Aromatische polymere mit fluorierten ionischen gruppen | |
| DE69637433T2 (de) | Hybrider polymerverbundelektrolyt und nichtwässrige elektrochemische zelle | |
| US6287722B1 (en) | Continuous melt process for fabricating ionically conductive articles | |
| DE102009020181A1 (de) | Sulfonierte Perfluorcyclobutanblockcopolymere und protonenleitende Polymermembranen | |
| DE69433319T2 (de) | Vernetzbare Copolyether und ihre Verwendung als polymerische Elektrolyten | |
| DE60031022T2 (de) | Polymerelektrolyten für Lithium-Akkumulatoren | |
| CN111313083A (zh) | 一种复合固态电解质薄膜及其制备和应用 | |
| DE102004021074A1 (de) | Sulfonimid enthaltende Poly(Arylenether) und Poly(Arylenethersulfone), Verfahren zu deren Herstellung und Verwendung derselben | |
| DE60037392T2 (de) | Fester elektrolyt aus vernetztem polymer und dessen verwendung | |
| CN112038687A (zh) | 双层复合固态电解质膜及其制备方法 | |
| CN110224173B (zh) | 一种可自愈合的锂电池用固态聚合物电解质及其制备方法 | |
| CN110734517A (zh) | 一种聚碳酸酯基嵌段聚合物电解质制备及应用 | |
| JP3348513B2 (ja) | 高分子固体電解質電池 | |
| Ghahramani et al. | Novel Single-ion conducting gel polymer electrolyte with honeycomb-like morphology prepared using brush copolymer for lithium-ion battery application | |
| DE112018007588T5 (de) | Poly(pyrocarbonat)-basierte polymerelektrolyte für hochspannungs-lithiumionenbatterien | |
| JP3843505B2 (ja) | 高分子電解質及び電池 | |
| DE102014113833A1 (de) | Polymerelektrolyt-Membran und Verfahren zur Herstellung | |
| DE112007001464T5 (de) | Kondensierte Ringe enthaltender Polymerelektrolyt und Anwendung hierfür | |
| US20040024174A1 (en) | Copolymer of ethylene oxide and at least one substituted oxirane carrying a cross-linkable function, process for preparation thereof, and use thereof for producing ionically conductive materials | |
| US11942598B2 (en) | Ionic liquid softened polymer electrolyte for zinc ion batteries |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition | ||
| 8339 | Ceased/non-payment of the annual fee |