EP2431380A2 - Macrocyclic antagonist of the motilin receptor for treatment of gastrointestinal dysmotility disorders - Google Patents
Macrocyclic antagonist of the motilin receptor for treatment of gastrointestinal dysmotility disorders Download PDFInfo
- Publication number
- EP2431380A2 EP2431380A2 EP20110075225 EP11075225A EP2431380A2 EP 2431380 A2 EP2431380 A2 EP 2431380A2 EP 20110075225 EP20110075225 EP 20110075225 EP 11075225 A EP11075225 A EP 11075225A EP 2431380 A2 EP2431380 A2 EP 2431380A2
- Authority
- EP
- European Patent Office
- Prior art keywords
- diarrhea
- compound
- boc
- compounds
- induced
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- 0 *CCCc(cccc1)c1OCC(C*I)N=O Chemical compound *CCCc(cccc1)c1OCC(C*I)N=O 0.000 description 27
- ZUWVFUDWXWCWJQ-UHFFFAOYSA-N CC[n]1nnc(C)c1C Chemical compound CC[n]1nnc(C)c1C ZUWVFUDWXWCWJQ-UHFFFAOYSA-N 0.000 description 2
- HRJDEHQWXAPGBG-YFKPBYRVSA-N CC(C)(C)OC(N[C@@H](CO1)C1=O)=O Chemical compound CC(C)(C)OC(N[C@@H](CO1)C1=O)=O HRJDEHQWXAPGBG-YFKPBYRVSA-N 0.000 description 1
- XNHNRNGHLWOFLP-FULLSBAXSA-N CC(C)[C@H](C(N[C@@H](CC1CC1)C(NCCCc(cccc1)c1OCCN[C@@H]1Cc2cc(F)ccc2)=O)=O)NC1=O Chemical compound CC(C)[C@H](C(N[C@@H](CC1CC1)C(NCCCc(cccc1)c1OCCN[C@@H]1Cc2cc(F)ccc2)=O)=O)NC1=O XNHNRNGHLWOFLP-FULLSBAXSA-N 0.000 description 1
- MQQNXHJDLKZGTQ-UHFFFAOYSA-N CCCNc1ncc[s]1 Chemical compound CCCNc1ncc[s]1 MQQNXHJDLKZGTQ-UHFFFAOYSA-N 0.000 description 1
- UEOXMXATZKGJJK-UHFFFAOYSA-N CCN1N(C)C=C=C1 Chemical compound CCN1N(C)C=C=C1 UEOXMXATZKGJJK-UHFFFAOYSA-N 0.000 description 1
- QYFGFCYIHATFNM-UHFFFAOYSA-O CCN1[NH+]=NNC1C Chemical compound CCN1[NH+]=NNC1C QYFGFCYIHATFNM-UHFFFAOYSA-O 0.000 description 1
- ICFWLGYCUXWLGI-UHFFFAOYSA-N CCNc1ncccn1 Chemical compound CCNc1ncccn1 ICFWLGYCUXWLGI-UHFFFAOYSA-N 0.000 description 1
- LUFSLKYOKXBZES-UHFFFAOYSA-N CC[n]1[n-][nH+]cc1C Chemical compound CC[n]1[n-][nH+]cc1C LUFSLKYOKXBZES-UHFFFAOYSA-N 0.000 description 1
- FLNMQGISZVYIIK-UHFFFAOYSA-N CC[n]1nccc1 Chemical compound CC[n]1nccc1 FLNMQGISZVYIIK-UHFFFAOYSA-N 0.000 description 1
- MJIOJWXRXDWUBV-UHFFFAOYSA-N CC[n]1nnc(C)c1 Chemical compound CC[n]1nnc(C)c1 MJIOJWXRXDWUBV-UHFFFAOYSA-N 0.000 description 1
- HPZIIBYLAYWBSN-UHFFFAOYSA-N CC[n]1nnnc1C Chemical compound CC[n]1nnnc1C HPZIIBYLAYWBSN-UHFFFAOYSA-N 0.000 description 1
- WJKISRFVKIOBCQ-UHFFFAOYSA-N Cc(cc1)cc(F)c1O Chemical compound Cc(cc1)cc(F)c1O WJKISRFVKIOBCQ-UHFFFAOYSA-N 0.000 description 1
- YSCJTQOLJKEMCX-UHFFFAOYSA-N Cc1c[n](C[IH]C)nn1 Chemical compound Cc1c[n](C[IH]C)nn1 YSCJTQOLJKEMCX-UHFFFAOYSA-N 0.000 description 1
- VJYXZJGDFJJDGF-UHFFFAOYSA-N Cc1cc(C(F)(F)F)ccc1 Chemical compound Cc1cc(C(F)(F)F)ccc1 VJYXZJGDFJJDGF-UHFFFAOYSA-N 0.000 description 1
- ZUGXFNTVGYBMPE-UHFFFAOYSA-N ICc1c[s]cn1 Chemical compound ICc1c[s]cn1 ZUGXFNTVGYBMPE-UHFFFAOYSA-N 0.000 description 1
Images






















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0812—Tripeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/08—Drugs for disorders of the alimentary tract or the digestive system for nausea, cinetosis or vertigo; Antiemetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/12—Antidiarrhoeals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/16—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by singly-bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D273/00—Heterocyclic compounds containing rings having nitrogen and oxygen atoms as the only ring hetero atoms, not provided for by groups C07D261/00 - C07D271/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Definitions
- the present invention relates to novel conformationally-defined macrocyclic compounds that bind to and/or are functional modulators of the motilin receptor including subtypes, isoforms and/or variants thereof.
- These macrocyclic compounds possess appropriate pharmacological properties to be useful as therapeutics for a range of disease indications.
- these compounds are useful for treatment and prevention of disorders characterized by hypermotilenemia or gastrointestinal hypermotility, including, but not limited to diarrhea, cancer treatment-related diarrhea, cancer-induced diarrhea, chemotherapy-induced diarrhea, radiation enteritis, radiation-induced diarrhea, stress-induced diarrhea, chronic diarrhea, AIDS-related diarrhea, C .
- the compounds possess utility for the treatment of diseases and disorders characterized by poor stomach or intestinal absorption, such as short bowel syndrome, celiac disease and cachexia.
- the compounds may also be used to treat inflammatory diseases and disorders of the gastrointestinal tract, such as inflammatory bowel disease, ulcerative colitis, Crohn's disease and pancreatitis.
- GI gastrointestinal
- absorption, secretion, blood flow and motility Mulvihill, S.J.; et al. in Basic and Clinical Endocrinology, 4th edition, Greenspan, F.S.; Baxter, J.D., Eds., Appleton & Lange: Norwalk, CT, 1994, pp 551-570 ). Since interactions between the brain and GI system are critical to the proper modulation of these functions, these peptides can be produced locally in the GI tract or distally in the CNS.
- motilin a linear 22-amino acid peptide
- motilin plays a critical regulatory role in the GI physiological system through governing of fasting gastrointestinal motor activity.
- the peptide is periodically released from the duodenal mucosa during fasting in mammals, including humans. More precisely, motilin exerts a powerful effect on gastric motility through the contraction of gastrointestinal smooth muscle to stimulate gastric emptying, decrease intestinal transit time and initiate phase III of the migrating motor complex (MMC) in the small bowel.
- MMC migrating motor complex
- Motilin can exert these effects through receptors located predominantly on the human antrum and proximal duodenum, although its receptors are found to some degree along the entire GI tract.
- Motilin receptors in the brain have been suggested to play a regulatory role in a number of CNS functions, including feeding and drinking behavior, micturition reflex, central and brain stem neuronal modulation, and pituitary hormone secretion ( Itoh, Z. Peptides 1997, 18, 593-608 ; Asakawa, A.; Inui, A.; Momose, K.M.; et al. Peptides 1998, 19, 987-990 and Rosenfeld, D.J.; Garthwaite, T.L. Physiol. Behav.
- motilin agonists as therapeutic agents to enhance.motility.
- the first of these, peptidic agonists of the motilin receptor have clinical application for the treatment of hypomotility disorders, in particular gastroparesis, ( Haramura, M.; Tsuzuki, K.; Okamachi, A.; et al. Bioorg. Med. Chem. 2002, 10, 1805-1811 ; U.S. Pat. Nos.
- the macrolide antibiotic erythromycin has long been known to have stimulation of GI motility as a side effect and, hence, has been utilized as a treatment for gastroparesis. This effect has subsequently been shown to be mediated through interaction at the motilin receptor.
- ABT-229 (alemcinal, Talley, N.J.; Verlinden, M.; Snape, W.; et al. Aliment. Pharmacol. Ther. 2000, 14, 1653-1661 ; Talley, N.J.; Verlinden, M.; Geenan, D.J.; et al. Gut 2001, 49, 395-401 ; Chen, C.L.; Orr, W.C.; Verlinden, M.H.; et al. Aliment. Pharmacol.
- Gastroenterology 2005, 128, A464 ; Carreras, C.W.; Burlingame, M.; Carney, J.; et al. Can. J. Gastroenterol. 2005,19, 15 ) has been described and appears to circumvent many of these problems.
- a method useful for analyzing the therapeutic efficiency of these types of molecules has also been formulated ( U.S. Pat. No. 6,875,576 ; U.S. Pat. Appl. Publ. 2002/192709 ; Intl. Pat. Appl. Publ. WO 02/64092 ).
- non-peptide, non-motilide motilin agonists have been reported ( U.S. Pat. No. 7,262,195 ; U.S. Pat. Appl. Publ. No. 2004/152732 ; 2005/065156 ; Intl. Pat. Appl. Publ. WO 02/137127 ; WO 02/92592 ; WO 2005/027908 ; WO 2005/027637 ; Jap. Pat. Abstr. Publ. No. 09249620 ).
- BMS-591348 has been described as possessing a pharmacological profile that avoids the tachyphylaxis issues that plagued many of the previous motilin agonist efforts.
- antagonists of the motilin receptor are potentially useful as therapeutic treatments for diseases associated with hypermotilinemia and/or gastrointestinal hypermotility, including diarrhea, cancer treatment-related diarrhea, cancer-induced diarrhea, chemotherapy-induced diarrhea, radiation enteritis, radiation-induced diarrhea, stress-induced diarrhea, chronic diarrhea, AIDS-related diarrhea, C . difficile associated diarrhea, traveller's diarrhea, diarrhea induced by graph versus host disease, other types of diarrhea, dyspepsia, irritable bowel syndrome, functional gastrointestinal disorders, chemotherapy-induced nausea and vomiting (emesis) and post-operative nausea and vomiting. Current treatments for these conditions are ineffective in many cases. Loperamide, an opioid agonist, is useful for milder diarrhea and generally does not work in a high percentage of patients.
- Octreotide a somatostatin agonist
- Diarrhea is a common and serious side-effect experienced by cancer patients resulting from surgery, bone marrow transplantation, chemotherapy and radiation treatment.
- chemotherapeutic regimens particularly those including fluoropyrimidines and irinotecan, result in chemotherapy-induced diarrhea (CID) rates as high as 50-80%.
- CID chemotherapy-induced diarrhea
- Acute radiation enteritis (ARE) or radiation induced intestinal dysfunction occurs in 75% of patients undergoing radiation therapy, typically occurring in the second or third week of therapy. Characterized by abdominal cramping and diarrhea, this is a serious and feared side effect that results in increased overall treatment time as well as reduced quality of life and can even result in death. In 5-15% of patients, the condition becomes chronic. In addition to discomfort, this side effect decreases the therapeutic benefit from radiation treatment by increasing the overall treatment time.
- chronic diarrhea can arise as a result of numerous medical conditions.
- chronic diarrhea is a common problem for patients with human immunodeficiency virus infection, especially those with advanced disease. This is a debilitating side effect that occurs in 60-90% of AIDS patients.
- Traveller's diarrhea affects over 50% of travellers to some destinations, particularly tropical ones, and is estimated to afflict over 11 million individuals annually. Apart from the disruption to business, travel and vacation schedules, this condition is often accompanied by other clinical manifestations such as nausea, vomiting, abdominal pain, fecal urgency, bloody stools, and fever.
- Lima A.A.M. Curr. Opin. Infect. Dis. 2001, 14, 547-552 ; Al-Abri, S.S.; Beeching, N.J.; Nye, F.J. Lancet Infect. Dis. 2005, 5, 349-360 ; DuPont, H.L. Gastroenterol. Clin. North Am. 2006, 35, 337-353 .
- Clostridium difficile is the etiological agent responsible for about one-third of cases of antibiotic-associated diarrhea and is estimated to cause a $1 billion annual cost in the U.S. Antibiotic-associated diarrhea is more common in the hospital setting with up to 29% of patients developing the condition, resulting in increased length of stay, increased cost of care, and increased mortality.
- GVHD graft versus host disease
- IBS Irritable bowel syndrome
- IBS patients are sub-classified into diarrhea-predominant (IBS-d), constipation-predominant (IBS-c) or those alternating between these two patterns (IBS-m).
- Antispasmodics, tricyclic antidepressants, selective serotonin reuptake inhibitors, laxatives, antidiarrheals, and bulking agents have not proven to be widely effective and tend to treat symptoms, rather than underlying pathophysiology.
- dyspepsia Another extremely common GI disorder, dyspepsia, is characterized by chronic or recurrent upper GI distress with no obvious physical cause.
- Typical symptoms include gastric fullness, bloating, pain, nausea and vomiting. This disease has prevalence as high as 20% annually in Western countries. It accounts for up to 5% of all visits to primary care physicians and 30% of visits to GI specialists.
- the patient population can be categorized into various subsets based upon symptoms.
- the largest patient subset (up to 60%) suffers from dyspepsia with no known organic cause, otherwise known as "functional dyspepsia (FD)." FD has a major impact on quality of life and health care resources.
- FD functional dyspepsia
- Circulating plasma motilin levels are also seen to be raised in patients suffering from dyspepsia.
- Chemotherapy-induced nausea and vomiting is one of the most severe adverse effects resulting from cancer treatment and is often cited as the side effect most feared by patients. From 70-80% of patients receiving cancer chemotherapy experience CINV. In addition to a significant deterioration in quality of life, this condition often requires modification or delay of chemotherapeutic regimens with concomitant negative impact on the effectiveness of treatment.
- CINV Chemotherapy-induced nausea and vomiting
- PONV Post-operative nausea and vomiting
- motilin In addition to its actions on motility, motilin is involved in inducing secretion within the gastrointestinal tract. Motilin plays a role in gastric and pancreatic secretion in dogs.
- motilin antagonists In addition to treatment of disorders characterized by hypermotility, the use of motilin antagonists would also be useful in the treatment of diseases and disorders characterized by poor stomach or intestinal absorption. A motilin antagonist would slow gastrointestinal motility thereby permitting longer GI exposure time for absorption of necessary nutrients.
- diseases and disorders include celiac disease, a chronic disorder afflicting almost 1% of the population.
- Celiac disease is a GI disorder characterized by inflammation, leading to injury to the mucosal lining of the small intestine. The inflammation results when gliadin, a protein found in gluten-containing foods, is ingested by genetically susceptible individuals. The mucosal damage and subsequent malabsorption of nutrients can lead to numerous complications.
- Short bowel syndrome is a medical condition that occurs after resection of a substantial portion of small intestine and is characterized by malnutrition.
- cancer cachexia is a therapeutic condition characterized by weight loss and muscle wasting and afflicts approximately 50% of all cancer patients and is the main cause of death in more than 20% of patients. Additionally, this condition has been shown to be a strong independent risk factor for mortality.
- motilin has been implicated in inflammatory disorders of the GI system. Elevated motilin levels have been observed in patients with inflammatory bowel disease ( Besterman, H.S.; Mallinson, C.N.; et al. Scand. J. Gastroenterol. 1983, 18, 845-852 ), ulcerative colitis ( Greenberg, G.R.; Buchan, A.M.; McLeod, R.S.; Preston, P.; Cohen, Z. Gut 1989, 30, 1721-1730 ) and chronic pancreatitis ( Besterman, H.S.; Adrian, T.E.; Bloom, S.R. et al.
- motilin antagonists may be used as anti-inflammatory agents, in particular for use in the GI tract, with beneficial properties relative to existing treatments.
- motilin antagonists As a novel approach to treat hypermotility and malabsorption disorders, efforts have lagged those directed at agonists.
- a variety of peptidic compounds have been described as antagonists of the motilin receptor [(ANQ-11125: Peeters, T.L.; Depoortere, I.; Macielag, M.J.; Marvin, M.S.; Florance, J.R.; Galdes, A. Biochem. Biophys. Res. Comm.
- Cyclization of peptidic derivatives is a method that can be employed to improve the properties of a linear peptide both with respect to metabolic stability and conformational freedom. Cyclic molecules tend to be more resistant to metabolic enzymes. Such cyclic peptide motilin antagonists have been reported, highlighted by GM-109. ( Takanashi, H.; Yogo, K.; Ozaki, M.; Akima, M.; Koga, H.; Nabata, H. J. Pharm. Exp. Ther.
- Macrocyclic peptidomimetics have been previously described as antagonists of the motilin receptor and their uses for the treatment of a variety of GI disorders and to modulate the migrating motor complex summarized.
- peptidomimetic macrocyclic motilin antagonists are distinguished from the aforementioned cyclic peptide motilin antagonists in that it was found that such peptidic derivatives containing D-amino acids were devoid of activity.
- the D-stereochemistry is beneficial for two of the three building elements.
- the tether portion of the molecule provides a non-peptidic component and, hence, distinct structures.
- the peptidomimetic macrocycles of the present invention are demonstrated to have binding and functional activity at the motilin receptor.
- binding potency and target affinity are factors in drug discovery and development, also important for development of viable pharmaceutical agents are optimization of pharmacokinetic (PK) and pharmacodynamic (PD) parameters.
- PK pharmacokinetic
- PD pharmacodynamic
- the macrocyclic compounds of the present invention have been found to possess desirable pharmacological characteristics, while maintaining sufficient binding affinity and selectivity for the motilin receptor, as illustrated in the Examples. These combined characteristics make them more suitable for development as pharmaceutical agents.
- the present invention provides novel conformationally-defined macrocyclic compounds with improved pharmacological properties. These compounds can function as antagonists of the motilin receptor.
- the present invention is directed to compounds of formula I: and pharmaceutically acceptable salts, hydrates or solvates thereof wherein: Y is wherein (L 5 ) and (L 6 ) indicates the bonds to L 5 and L 6 of formula I, respectively;
- Ar of formula I is selected from:
- R 1 of formula I is selected from methyl, ethyl, isopropyl and cyclopropyl.
- R 2 of formula I is selected from: wherein M is selected from the group consisting of CH 2 , O, NH, and NCH 3 .
- L 1 , L 2 , L 3 and L 4 in formula I are each CH, L 5 is O and L 6 is CH 2 .
- R 3 , R 4 , R 5 and R 6 in formula I are each hydrogen; or R 3 is methyl and R 4 , R 5 and R 6 are each hydrogen; or R 3 is hydroxymethyl and R 4 , R 5 and R 6 are each hydrogen; or R 3 , R 4 and R 6 are each hydrogen and R 5 is methyl; or R 3 and R 5 are each methyl and R 4 and R 6 are each hydrogen.
- Y in formula I is selected from: wherein (L 5 ) and (L 6 ) indicate the bond to L 5 and L 6 , respectively.
- the present invention relates to compounds of formula II: wherein R 25 is selected from hydrogen, alkyl, aryl, cycloalkyl, heterocyclic and heteroaryl; R 26 is selected from hydrogen, alkyl, aryl, acyl, carboxyalkyl, carboxyaryl, sulfonyl and a standard protecting group used for amino acids; R 27 is selected from the group consisting of hydrogen and alkyl; and R 28 is selected from the group consisting of wherein M is selected from the group consisting of CH 2 , O, NH, and NCH 3 .
- the present invention relates to compounds of formula III: wherein X 10 and X 11 are independently selected from the group consisting of hydrogen, halogen, trifluoromethyl and lower alkyl; R 40 , R 41 , R 42 , and R 43 are independently selected from the group consisting of hydrogen, lower alkyl and substituted lower alkyl; R 44 is selected from the group consisting of hydrogen, alkyl, acyl, carboxyalkyl, carboxyaryl, sulfonyl and a protecting group for an amine functional group; R 45 is selected from the group consisting of hydrogen and alkyl; R 46 is selected from the group consisting of hydrogen, alkyl, acyl, sulfonyl and a protecting group for a hydroxy functional group; L 11 , L 12 , L 13 and L 14 are independently selected from the group consisting of CH and N; with the proviso that the total number of nitrogens in the ring must be 0, 1, 2 or 3; L 15 and L 16 are independently selected from the group
- Compounds of formula III can be used in another aspect of the invention to provide compounds of formula I.
- Compounds of formula III can further be used to provide macrocyclic compounds including a building block structure, such as an amino acid building block structure, and the compounds of formula III.
- the present invention also relates to methods of using the compounds as described herein and as used for the preparation of a medicament for prevention and/or treatment of the disorders described herein.
- alkyl refers to straight or branched chain saturated or partially unsaturated hydrocarbon groups having from 1 to 20 carbon atoms, in some instances 1 to 8 carbon atoms.
- lower alkyl refers to alkyl groups containing 1 to 6 carbon atoms. Examples of alkyl groups include, but are not limited to, methyl, ethyl, isopropyl, tert-butyl, 3-hexenyl, and 2-butynyl.
- unsaturated is meant the presence of 1, 2 or 3 double or triple bonds, or a combination of the two. Such alkyl groups may also be optionally substituted as described below.
- C 2 -C 4 alkyl indicates an alkyl group with 2, 3 or 4 carbon atoms.
- cycloalkyl refers to saturated or partially unsaturated cyclic hydrocarbon groups having from 3 to 15 carbon atoms in the ring, in some instances 3 to 7, and to alkyl groups containing said cyclic hydrocarbon groups.
- examples of cycloalkyl groups include, but are not limited to, cyclopropyl, cyclopropylmethyl, cyclopentyl, 2-(cyclohexyl)ethyl, cycloheptyl, and cyclohexenyl.
- Cycloalkyl as defined herein also includes groups with multiple carbon rings, each of which may be saturated or partially unsaturated, for example decalinyl, [2.2.1]-bicycloheptanyl or adamantanyl. All such cycloalkyl groups may also be optionally substituted as described below.
- aromatic refers to an unsaturated cyclic hydrocarbon group having a conjugated pi electron system that contains 4n+2 electrons where n is an integer greater than or equal to 1.
- Aromatic molecules are typically stable and are depicted as a planar ring of atoms with resonance structures that consist of alternating double and single bonds, for example benzene or naphthalene.
- aryl refers to an aromatic group in a single or fused carbocyclic ring system having from 6 to 15 ring atoms, in some instances 6 to 10, and to alkyl groups containing said aromatic groups.
- aryl groups include, but are not limited to, phenyl, 1-naphthyl, 2-naphthyl and benzyl.
- Aryl as defined herein also includes groups with multiple aryl rings which may be fused, as in naphthyl and anthracenyl, or unfused, as in biphenyl and terphenyl.
- Aryl also refers to bicyclic or tricyclic carbon rings, where one of the rings is aromatic and the others of which may be saturated, partially unsaturated or aromatic, for example, indanyl or tetrahydronaphthyl (tetralinyl). All such aryl groups may also be optionally substituted as described below.
- heterocycle refers to saturated or partially unsaturated monocyclic, bicyclic or tricyclic groups having from 3 to 15 atoms, in some instances 3 to 7, with at least one heteroatom in at least one of the rings, said heteroatom being selected from O, S or N.
- Each ring of the heterocyclic group can contain one or two O atoms, one or two S atoms, one to four N atoms, provided that the total number of heteroatoms in each ring is four or less and each ring contains at least one carbon atom.
- the fused rings completing the bicyclic or tricyclic heterocyclic groups may contain only carbon atoms and may be saturated or partially unsaturated.
- heterocyclic also refers to alkyl groups containing said monocyclic, bicyclic or tricyclic heterocyclic groups. Examples of heterocyclic rings include, but are not limited to, 2- or 3-piperidinyl, 2- or 3-piperazinyl, 2- or 3-morpholinyl. All such heterocyclic groups may also be optionally substituted as described below
- heteroaryl refers to an aromatic group in a single or fused ring system having from 5 to 15 ring atoms, in some instances 5 to 10, which have at least one heteroatom in at least one of the rings, said heteroatom being selected from O, S or N.
- Each ring of the heteroaryl group can contain one or two O atoms, one or two S atoms, one to four N atoms, provided that the total number of heteroatoms in each ring is four or less and each ring contains at least one carbon atom.
- the fused rings completing the bicyclic or tricyclic groups may contain only carbon atoms and may be saturated, partially unsaturated or aromatic.
- the N atoms may optionally be quaternized or oxidized to the N-oxide.
- Heteroaryl also refers to alkyl groups containing said cyclic groups.
- Examples of monocyclic heteroaryl groups include, but are not limited to pyrrolyl, pyrazolyl, pyrazolinyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, thiadiazolyl, isothiazolyl, furanyl, thienyl, oxadiazolyl, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, and triazinyl.
- bicyclic heteroaryl groups include, but are not limited to indolyl, benzothiazolyl, benzoxazolyl, benzothienyl, quinolinyl, tetrahydroisoquinolinyl, isoquinolinyl, benzimidazolyl, benzopyranyl, indolizinyl, benzofuranyl, isobenzofuranyl, chromonyl, coumarinyl, benzopyranyl, cinnolinyl, quinoxalinyl, indazolyl, purinyl, pyrrolopyridinyl, furopyridinyl, thienopyridinyl, dihydroisoindolyl, and tetrahydroquinolinyl.
- tricyclic heteroaryl groups include, but are not limited to carbazolyl, benzindolyl, phenanthrollinyl, acridinyl, phenanthridinyl, and xanthenyl. All such heteroaryl groups may also be optionally substituted as described below.
- hydroxy refers to the group -OH.
- alkoxy refers to the group -OR a , wherein R a is alkyl, cycloalkyl or heterocyclic. Examples include, but are not limited to methoxy, ethoxy, tert-butoxy, cyclohexyloxy and tetrahydropyranyloxy.
- aryloxy refers to the group -OR b wherein R b is aryl or heteroaryl. Examples include, but are not limited to phenoxy, benzyloxy and 2-naphthyloxy.
- amino acyl indicates an acyl group that is derived from an amino acid.
- amino refers to an -NR d R e group wherein R d and R e are independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, heterocyclic, aryl and heteroaryl.
- R d and R e together form a heterocyclic ring of 3 to 8 members, optionally substituted with unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl, unsubstituted heteroaryl, hydroxy, alkoxy, aryloxy, acyl, amino, amido, carboxy, carboxyalkyl, carboxyaryl, mercapto, sulfinyl, sulfonyl, sulfonamido, amidino, carbamoyl, guanidino or ureido, and optionally containing one to three additional heteroatoms selected from O, S or N.
- R f and R g together form a heterocyclic ring of 3 to 8 members, optionally substituted with unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl, unsubstituted heteroaryl, hydroxy, alkoxy, aryloxy, acyl, amino, amido, carboxy, carboxyalkyl, carboxyaryl, mercapto, sulfinyl, sulfonyl, sulfonamido, amidino, carbamoyl, guanidino or ureido, and optionally containing one to three additional heteroatoms selected from O, S or N.
- R i and R j together form a heterocyclic ring of 3 to 8 members, optionally substituted with unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl, unsubstituted heteroaryl, hydroxy, alkoxy, aryloxy, acyl, amino, amido, carboxy, carboxyalkyl, carboxyaryl, mercapto, sulfinyl, sulfonyl, sulfonamido, amidino, carbamoyl, guanidino or ureido, and optionally containing one to three additional heteroatoms selected from O, S or N.
- Carboxyalkyl refers to the group -CO 2 R k , wherein R k is alkyl, cycloalkyl or heterocyclic.
- Carboxyaryl refers to the group -CO 2 R m , wherein R m is aryl or heteroaryl.
- cyano refers to the group -CN.
- halo refers to fluoro, fluorine or fluoride, chloro, chlorine or chloride, bromo, bromine or bromide, and iodo, iodine or iodide, respectively.
- hydroxymethyl refers to the group -CH 2 OH.
- mercapto refers to the group -SR n wherein R n is hydrogen, alkyl, cycloalkyl, heterocyclic, aryl or heteroaryl.
- nitro refers to the group -NO 2 .
- trifluoromethyl refers to the group -CF 3 .
- R r and R s together form a heterocyclic ring of 3 to 8 members, optionally substituted with unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl, unsubstituted heteroaryl, hydroxy, alkoxy, aryloxy, acyl, amino, amido, carboxy, carboxyalkyl, carboxyaryl, mercapto, sulfinyl, sulfonyl, sulfonamido, amidino, carbamoyl, guanidino or ureido, and optionally containing one to three additional heteroatoms selected from O, S or N.
- R x and Ry together form a heterocyclic ring or 3 to 8 members, optionally substituted with unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl, unsubstituted heteroaryl, hydroxy, alkoxy, aryloxy, acyl, amino, amido, carboxy, carboxyalkyl, carboxyaryl, mercapto, sulfinyl, sulfonyl, sulfonamido, amidino, carbamoyl, guanidino or ureido, and optionally containing one to three additional heteroatoms selected from O, S or N.
- R aa and R bb together form a heterocyclic ring of 3 to 8 members, optionally substituted with unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl, unsubstituted heteroaryl, hydroxy, alkoxy, aryloxy, acyl, amino, amido, carboxy, carboxyalkyl, carboxyaryl, mercapto, sulfinyl, sulfonyl, sulfonamido, amidino, carbamoyl, guanidino or ureido, and optionally containing one to three additional heteroatoms selected from O, S or N.
- optionally substituted is intended to expressly indicate that the specified group is unsubstituted or substituted by one or more suitable substituents, unless the optional substituents are expressly specified, in which case the term indicates that the group is unsubstituted or substituted with the specified substituents.
- various groups may be unsubstituted or substituted (i.e., they are optionally substituted) unless indicated otherwise herein (e.g., by indicating that the specified group is unsubstituted).
- R cc , R dd , R ee , R ff , R gg , R hh , R ii , R jj , R mm , R pp , R qq and R rr are independently selected from hydrogen, unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl or unsubstituted heteroaryl; and wherein R kk and R nn are independently selected from unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl or unsubstituted heteroaryl.
- R gg and R hh , R jj and R kk or R pp and Rqq together form a heterocyclic ring of 3 to 8 members, optionally substituted with unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl, unsubstituted heteroaryl, hydroxy, alkoxy, aryloxy, acyl, amino, amido, carboxy, carboxyalkyl, carboxyaryl, mercapto, sulfinyl, sulfonyl, sulfonamido, amidino, carbamoyl, guanidino or ureido, and optionally containing one to three additional heteroatoms selected from O, S or N.
- substituted for aryl and heteroaryl groups includes as an option having one of the hydrogen atoms of the group replaced by cyano, nitro or tri
- substitution is made provided that any atom's normal valency is not exceeded and that the substitution results in a stable compound.
- such substituted group is preferably not further substituted or, if substituted, the substituent comprises only a limited number of substituted groups, in some instances 1, 2, 3 or 4 such substituents.
- stable compound or “stable structure” refers to a compound that is sufficiently robust to survive isolation to a useful degree of purity and formulation into an efficacious therapeutic agent.
- amino acid refers to the common natural (genetically encoded) or synthetic amino acids and common derivatives thereof, known to those skilled in the art.
- standard or “proteinogenic” refers to the genetically encoded 20 amino acids in their natural configuration.
- unnatural or “unusual” refers to the wide selection of non-natural, rare or synthetic amino acids such as those described by Hunt, S. in Chemistry and Biochemistry of the Amino Acids, Barrett, G.C., Ed., Chapman and Hall: New York, 1985 .
- fragment with respect to a dipeptide, tripeptide or higher order peptide derivative indicates a group that contains two, three or more, respectively, amino acid residues.
- amino acid side chain refers to any side chain from a standard or unnatural amino acid, and is denoted R AA .
- the side chain of alanine is methyl
- the side chain of valine is isopropyl
- the side chain of tryptophan is 3-indolylmethyl.
- agonist refers to a compound that duplicates at least some of the effect of the endogenous ligand of a protein, receptor, enzyme or the like.
- antagonist refers to a compound that inhibits at least some of the effect of the endogenous ligand of a protein, receptor, enzyme or the like.
- variable when applied to a receptor is meant to include dimers, trimers, tetramers, pentamers and other biological complexes containing multiple components. These components can be the same or different.
- peptide refers to a chemical compound comprised of two or more amino acids covalently bonded together.
- peptidomimetic refers to a chemical compound designed to mimic a peptide, but which contains structural differences through the addition or replacement of one of more functional groups of the peptide in order to modulate its activity or other properties, such as solubility, metabolic stability, oral bioavailability, lipophilicity, permeability, etc. This can include replacement of the peptide bond, side chain modifications, truncations, additions of functional groups, etc.
- non-peptide peptidomimetic When the chemical structure is not derived from the peptide, but mimics its activity, it is often referred to as a "non-peptide peptidomimetic.”
- protecting group refers to any chemical compound that may be used to prevent a potentially reactive functional group, such as an amine, a hydroxyl or a carboxyl, on a molecule from undergoing a chemical reaction while chemical change occurs elsewhere in the molecule.
- a potentially reactive functional group such as an amine, a hydroxyl or a carboxyl
- a number of such protecting groups are known to those skilled in the art and examples can be found in " Protective Groups in Organic Synthesis," Theodora W. Greene and Peter G. Wuts, editors, John Wiley & Sons, New York, 3rd edition, 1999 [ISBN 0471160199 ].
- amino protecting groups include, but are not limited to, phthalimido, trichloroacetyl, benzyloxycarbonyl, tert-butoxycarbonyl, and adamantyloxycarbonyl.
- amino protecting groups are carbamate amino protecting groups, which are defined as an amino protecting group that when bound to an amino group forms a carbamate.
- amino carbamate protecting groups are allyloxycarbonyl (Alloc), benzyloxycarbonyl (Cbz), 9-fluorenylmethoxycarbonyl (Fmoc), tert-butoxycarbonyl (Boc) and ⁇ , ⁇ -dimethyl-3,5-dimethoxybenzyloxycarbonyl (Ddz).
- hydroxyl protecting groups include, but are not limited to, acetyl, tert-butyldimethylsilyl (TBDMS), trityl (Trt), tert-butyl, and tetrahydropyranyl (THP).
- carboxyl protecting groups include, but are not limited to methyl ester, tert-butyl ester, benzyl ester, trimethylsilylethyl ester, and 2,2,2-trichloroethyl ester.
- solid phase chemistry refers to the conduct of chemical reactions where one component of the reaction is covalently bonded to a polymeric material (solid support as defined below). Reaction methods for performing chemistry on solid phase have become more widely known and established outside the traditional fields of peptide and oligonucleotide chemistry.
- solid support refers to a mechanically and chemically stable polymeric matrix utilized to conduct solid phase chemistry. This is denoted by "Resin,” “P-” or the following symbol:
- polystyrene polyethylene, polyethylene glycol, polyethylene glycol grafted or covalently bonded to polystyrene (also termed PEG-polystyrene, TentaGelTM, Rapp, W.; Zhang, L.; Bayer, E. In Innovations and Perspectives in Solid Phase Synthesis. Peptides, Polypeptides and Oligonucleotides; Epton, R., Ed.; SPCC Ltd.: Birmingham, UK; p 205 ); polyacrylate (CLEARTM), polyacrylamide, polyurethane, PEGA [polyethyleneglycol poly(N,N-dimethylacrylamide) co-polymer, Meldal, M.
- polystyrene polyethylene, polyethylene glycol, polyethylene glycol grafted or covalently bonded to polystyrene
- PEG-polystyrene TentaGelTM, Rapp, W.; Zhang, L.; Bayer, E. In Innovations and Perspectives
- This solid support can include as non-limiting examples aminomethyl polystyrene, hydroxymethyl polystyrene, benzhydrylamine polystyrene (BHA), methylbenzhydrylamine (MBHA) polystyrene, and other polymeric backbones containing free chemical functional groups, most typically, -NH 2 or -OH, for further derivatization or reaction.
- the materials used as resins are insoluble polymers, but certain polymers have differential solubility depending on solvent and can also be employed for solid phase chemistry.
- polyethylene glycol can be utilized in this manner since it is soluble in many organic solvents in which chemical reactions can be conducted, but it is insoluble in others, such as diethyl ether.
- reactions can be conducted homogeneously in solution, then the product on the polymer precipitated through the addition of diethyl ether and processed as a solid. This has been termed "liquid-phase" chemistry.
- linker when used in reference to solid phase chemistry refers to a chemical group that is bonded covalently to a solid support and is attached between the support and the substrate typically in order to permit the release (cleavage) of the substrate from the solid support. However, it can also be used to impart stability to the bond to the solid support or merely as a spacer element. Many solid supports are available commercially with linkers already attached.
- the term "effective amount” or “effective” is intended to designate a dose that causes a relief of symptoms of a disease or disorder as noted through clinical testing and evaluation, patient observation, and/or the like, and/or a dose that causes a detectable change in biological or chemical activity.
- the detectable changes may be detected and/or further quantified by one skilled in the art for the relevant mechanism or process.
- the dosage will vary depending on the administration routes, symptoms and body weight of the patient but also depending upon the compound being administered.
- Administration of two or more compounds "in combination” means that the two compounds are administered closely enough in time that the presence of one alters the biological effects of the other.
- the two compounds can be administered simultaneously (concurrently) or sequentially. Simultaneous administration can be carried out by mixing the compounds prior to administration, or by administering the compounds at the same point in time but at different anatomic sites or using different routes of administration.
- the phrases "concurrent administration”, “administration in combination”, “simultaneous administration” or “administered simultaneously” as used herein, means that the compounds are administered at the same point in time or immediately following one another. In the latter case, the two compounds are administered at times sufficiently close that the results observed are indistinguishable from those achieved when the compounds are administered at the same point in time.
- pharmaceutically active metabolite is intended to mean a pharmacologically active product produced through metabolism in the body of a specified compound.
- solvate is intended to mean a pharmaceutically acceptable solvate form of a specified compound that retains the biological effectiveness of such compound.
- examples of solvates include compounds of the invention in combination with water, isopropanol, ethanol, methanol, DMSO, ethyl acetate, acetic acid, or ethanolamine.
- Novel macrocyclic compounds of the present invention include macrocyclic compounds comprising a building block structure including a tether component that undergoes cyclization to form the macrocyclic compound.
- the building block structure can comprise amino acids (standard and unnatural) and a tether component as described herein.
- Macrocyclic compounds of the present invention further include those of formula I: and pharmaceutically acceptable salts, hydrates or solvates thereof wherein: Y is wherein (L 5 ) and (L 6 ) indicates the bonds to L 5 and L 6 of formula I, respectively;
- Ar is selected from the group consisting of: R 1 is selected from the group consisting of lower alkyl and cycloalkyl; R 2 is selected from the group consisting of lower alkyl, substituted lower alkyl, cycloalkyl and substituted cycloalkyl; R 3 , R 4 , R 5 , and R 6 are independently selected from the group consisting of hydrogen, lower alkyl and substituted lower alkyl; R 7 is selected from the group consisting of hydrogen, lower alkyl, hydroxy and amino; R 10a and R 10b are independently selected from the group consisting of hydrogen, lower alkyl and substituted lower alkyl; X 1 , X 2 , X 6 , X 7 , X 8 and X 9
- the present invention includes isolated compounds.
- An isolated compound refers to a compound that, in some embodiments, comprises at least 10%, at least 25%, at least 50% or at least 70% of the compounds of a mixture.
- the compound, pharmaceutically acceptable salt thereof or pharmaceutical composition containing the compound exhibits a statistically significant binding and/or antagonist activity when tested in biological assays at the human motilin receptor.
- the compounds of formula I disclosed herein have asymmetric centers.
- the inventive compounds may exist as single stereoisomers, racemates, and/or mixtures of enantiomers and/or diastereomers. All such single stereoisomers, racemates, and mixtures thereof are intended to be within the scope of the present invention. In particular embodiments, however, the inventive compounds are used in optically pure form.
- the terms "S” and "R” configuration as used herein are as defined by the IUPAC 1974 Recommendations for Section E, Fundamentals of Stereochemistry ( Pure Appl. Chem. 1976, 45, 13-30 .)
- the compounds may be prepared as a single stereoisomer or a mixture of stereoisomers.
- the non-racemic forms may be obtained by either synthesis or resolution.
- the compounds may, for example, be resolved into the component enantiomers by standard techniques, for example formation of diastereomeric pairs via salt formation.
- the compounds also may be resolved by covalently bonding to a chiral moiety.
- the diastereomers can then be resolved by chromatographic separation and/or crystallographic separation. In the case of a chiral auxiliary moiety, it can then be removed.
- the compounds can be resolved through the use of chiral chromatography. Enzymatic methods of resolution could also be used in certain cases.
- an “optically pure” compound is one that contains only a single enantiomer.
- the term “optically active” is intended to mean a compound comprising at least a sufficient excess of one enantiomer over the other such that the mixture rotates plane polarized light.
- Optically active compounds have the ability to rotate the plane of polarized light. The excess of one enantiomer over another is typically expressed as enantiomeric excess (e.e.).
- the prefixes D and L or R and S are used to denote the absolute configuration of the molecule about its chiral center(s).
- the prefixes "d” and “I” or (+) and (-) are used to denote the optical rotation of the compound ( i.e. , the direction in which a plane of polarized light is rotated by the optically active compound).
- the "I” or (-) prefix indicates that the compound is levorotatory ( i.e. , rotates the plane of polarized light to the left or counterclockwise) while the "d” or (+) prefix means that the compound is dextrarotatory ( i.e. , rotates the plane of polarized light to the right or clockwise).
- the sign of optical rotation, (-) and (+) is not related to the absolute configuration of the molecule, R and S.
- a compound of the invention having the desired pharmacological properties will be optically active and, can be comprised of at least 90% (80% e.e.), at least 95% (90% e.e.), at least 97.5% (95% e.e.) or at least 99% (98% e.e.) of a single isomer.
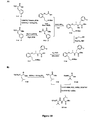
- the compounds of formula I can be synthesized using traditional solution synthesis techniques or solid phase chemistry methods. Synthetic methods for this general type of macrocyclic structure are described in Intl. Pat. Appls. WO 01/25257 , WO 2004/111077 , WO 2005/012331 , WO 2005/012332 , WO 2006/009645 and WO 2006/009674 , including purification procedures described in WO 2004/111077 and WO 2005/012331 . Representative procedures for compounds of the invention are presented in the Examples.
- Reagents and solvents were of reagent quality or better and were used as obtained from various commercial suppliers unless otherwise noted.
- DMF, DCM (CH 2 Cl 2 ), DME, CH 3 CN and THF used are of DriSolv ® (EMD Chemicals, Inc., part of Merck KGaA, Darmstadt, Germany) or synthesis grade quality except for (i) deprotection, (ii) resin capping reactions and (iii) washing.
- NMP used for the amino acid (AA) coupling reactions is of analytical grade.
- DMF was adequately degassed by placing under vacuum for a minimum of 30 min prior to use. Homogeneous catalysts were obtained from Strem Chemicals, Inc. (Newbury Port, MA, USA).
- Cbz-, Boc- and Fmoc-protected amino acids and side chain protected derivatives, including those of N-methyl and unnatural amino acids, were obtained from commercial suppliers or synthesized through standard methodologies known to those in the art.
- Ddz-amino acids were either synthesized by standard methods, or obtained commercially from Orpegen (Heidelberg, Germany) or Advanced ChemTech (Louisville, KY, USA). Bts-amino acids were synthesized by established procedures.
- Analytical TLC was performed on pre-coated plates of silica gel 60F254 (0.25 mm thickness) containing a fluorescent indicator and were visualized using the method(s) and reagent(s) indicated, for example using ultraviolet light (UV) and/or ceric-molybdic acid (CMA) solution (prepared by mixing 100 mL of sulfuric acid, 10 g ceric ammonium sulfate and 25 g ammonium molybdate).
- UV ultraviolet light
- CMA ceric-molybdic acid
- concentrated/evaporated/removed under reduced pressure indicates removal of solvent and volatile components utilizing a rotary evaporator under either water aspirator pressure (typically 10-30 torr) or the stronger vacuum provided by a mechanical oil vacuum pump ("high vacuum,” typically ⁇ 1 torr) as appropriate for the solvent being removed. Drying of a compound “ in vacuo " or under “high vacuum” refers to drying using an oil vacuum pump at low pressure ( ⁇ 1 torr).
- “Flash chromatography” was performed using silica gel 60 (230-400 mesh, EMD Chemicals, Still, W. C.; Kahn, M.; Mitra, A. J. Org. Chem.
- “Dry pack” indicates chromatography on silica gel that has not been pre-treated with solvent, generally applied on larger scales for purifications where a large difference in R f exists between the desired product and any impurities.
- “dried in the standard manner” is that the resin is dried first in air (1 h), and subsequently under vacuum (oil pump usually) until full dryness is attained ( ⁇ 30 min to O/N). Glassware used in air and water sensitive reactions were dried in an oven at least O/N and cooled in a desiccator prior to use.
- Amino acids, Boc- and Fmoc-protected amino acids and side chain protected derivatives, including those of N-methyl and unnatural amino acids were obtained from commercial suppliers [for example: Advanced ChemTech (Louisville, KY, USA), Bachem (Bubendorf, Switzerland), ChemImpex (Wood Dale, IL, USA), Novabiochem (subsidiary of Merck KGaA, Darmstadt, Germany), PepTech (Burlington, MA, USA), Synthetech (Albany, OR, USA), AstaTech (Bristol, PA, USA)] or synthesized through standard methodologies known to those in the art.
- Ddz-amino acids were either obtained commercially from Orpegen (Heidelberg, Germany) or Advanced ChemTech (Louisville, KY, USA) or synthesized using standard methods utilizing Ddz-OPh or Ddz-N 3 .
- Bts-amino acids were synthesized by known methods.
- N-Alkyl amino acids in particular N-methyl amino acids, are commercially available from multiple vendors (Bachem, Novabiochem, Advanced ChemTech, ChemImpex, Peptech).
- N-alkyl amino acid derivatives were accessed via literature methods. ( Hansen, D. W., Jr.; Pilipauskas, D. J. Org. Chem. 1985, 50, 945-950 .)
- Aziridine-2-carboxylic acid was constructed using reported methods from the literature ( Baldwin, J. E. et al.
- R 25 is selected from hydrogen, alkyl, aryl, cycloalkyl, heterocyclic and heteroaryl
- R 26 is selected from hydrogen, alkyl, aryl, acyl, carboxyalkyl, carboxyaryl, sulfonyl and a standard protecting group used for amino acids
- R 27 is selected from the group consisting of hydrogen and alkyl
- R 28 is selected from the group consisting of wherein M is selected from the group consisting of CH 2 , O, NH, and NCH 3 .
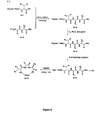
- Tethers were obtained from the methods previously described in Intl. Pat. Appl. Nos. WO 01/25257 , WO 2004/111077 , WO 2005/012331 , WO 2006/009645 and WO 2006/009674 . Procedures for the synthesis of representative tethers as described herein are also presented in the Examples below. Tethers useful for the compounds of the present invention can include suitable tethers as described in the references cited above as well as known and/or novel tethers described herein.
- Exemplary tethers include, but are not limited to, the following: and intermediates in the manufacture thereof, wherein PG 1 is selected from hydrogen and a protecting group for an amine functional group and PG 2 is selected from hydrogen and a protecting group for a hydroxy functional group.
- (R)-substituted tether building blocks result in compounds of formula I with an (S)-stereocenter.
- (S)-substituted tether building blocks result in compounds of formula I with an (R)-stereocenter.
- Tethers T90 and T91 are introduced along with AA 1 as fragments F2 and F1 respectively as described in Examples 6 and 5.
- the relevant deprotection and coupling protocols required for the assembly of the cyclization precursors are performed utilizing standard procedures and those described in WO 2004/111077 , WO 2006/009645 and WO 2006/009674 as appropriate for the nature of the building block.
- the final macrocycles are obtained after application of the appropriate deprotection sequences.
- HPLC analyses were performed on a Waters Alliance ® system 2695 running at 1 mL/min using an Xterra ® MS C18 column (or comparable) 4.6 x 50 mm (3.5 ⁇ m) and the indicated gradient method.
- a Waters 996 PDA provided UV data for purity assessment (Waters Corporation, Milford, MA).
- the second part (40%) went to an evaporative light scattering detector (ELSD, Polymer Laboratories, now part of Varian, Inc., Palo Alto, CA, PL-ELS-1000TM) for purity assessment and the last portion (10%) went to a chemiluminescence nitrogen detector (CLND, Antek ® Model 8060, Antek Instruments, Houston, TX, part of Roper Industries, Inc., Duluth, GA) for quantitation and purity assessment. Data was captured and processed utilizing the most recent version of the Waters Millennium ® software package.
- ELSD evaporative light scattering detector
- CLND chemiluminescence nitrogen detector
- Enantiomeric and diastereomeric purity were assessed using appropriate chiral HPLC columns using a Waters Breeze system (or comparable). Although other packing materials can be utilized, particularly useful columns for these analyses are: Chiralpak AS-RH and Chiralcel OD-RH (Chiral Technologies, West Chester, PA, USA).
- Preparative HPLC purifications were performed on final deprotected macrocycles using the Waters FractionLynx ® system, on an XTerra ® MS C18 column (or comparable) 19 x 100 mm (5 ⁇ m). The injections were done using an At-Column-Dilution configuration with a Waters 2767 injector/collector and a Waters 515 pump running at 2 mL/min. The mass spectrometer, HPLC, and mass-directed fraction collection are controlled via MassLynx ® software version 3.5 with FractionLynx ® .
- the compounds of the present invention were evaluated for their ability to interact at the human motilin receptor utilizing a competitive radioligand binding assay, fluorescence assay or Aequorin functional assay as described below. Such methods can be conducted in a high throughput manner to permit the simultaneous evaluation of many compounds.
- the competitive binding assay at the human motilin receptor was carried out analogously to assays described in the literature.
- the reaction is initiated by addition of 10 ⁇ L of [ 125 I]-motilin (final conc. 0.04 - 0.06 nM) to each well. Plates are sealed with TopSeal-A, vortexed gently and incubated at room temperature for 2 hours.
- the reaction is arrested by filtering samples through pre-soaked (0.3% polyethyleneimine, 2 h) Multiscreen Harvest plates using a Tomtec Harvester, washed 9 times with 500 ⁇ L of cold 50 mM Tris-HCl (pH 7.4), and than plates are air-dried in a fumehood for 30 minutes. A bottom seal is applied to the plates prior to the addition of 25 ⁇ L of MicroScint-0 to each well. Plates are than sealed with TopSeal-A and counted for 30 sec per well on a TopCount Microplate Scintillation and Luminescence Counter (PerkinElmer) where results are expressed as counts per minute (cpm).
- TopSeal-A TopCount Microplate Scintillation and Luminescence Counter
- K i values were calculated using a K d value of 0.16 nM for [ 125 I]-motilin (previously determined during membrane characterization).
- D max 1 - test concentration with maximal displacement - non - specific binding total binding - non - specific binding x 100 where total and non-specific binding represent the cpm obtained in the absence or presence of 1 ⁇ M motilin, respectively.
- Binding activity at the motilin receptor for representative compounds of the present invention is shown in Table 3. Compound structures for Table 3 are presented with the various groups as defined for the general structure of formula I. Table 3: Binding Activity at the Human Motilin Receptor for Representative Compounds of the Invention Compound No. K i (nM) Compound No.
- Compounds were provided as dry films at a quantity of approximately 1.2 ⁇ mol in pre-formatted 96-well plates. Compounds were dissolved in 100% DMSO at a concentration of 10 mM and stored at -20°C until further use. Daughter plates were prepared at a concentration of 500 ⁇ M in 30% DMSO with 0.1% BSA and stored at -20°C until testing. On the test day, compounds were allowed to thaw at room temperature and than diluted in assay buffer according to the desired test concentrations. Under these conditions, the maximum final DMSO concentration in the assay was 0.6%.
- Cells are collected from culture plates with Ca 2+ and Mg 2+ -free phosphate buffered saline (PBS) supplemented with 5 mM EDTA, pelleted for 2 minutes at 1000 x g, resuspended in assay buffer (see above) at a density of 5 x 10 6 cells/mL and incubated overnight in the presence of 5 ⁇ M coelenterazine. After loading, cells were diluted with assay buffer to a concentration of 5 x 10 5 cells/mL.
- PBS Ca 2+ and Mg 2+ -free phosphate buffered saline
- assay buffer see above
- test compound or motilin reference agonist
- 50 ⁇ L of the cell suspension was mixed with 50 ⁇ L of the appropriate concentration of test compound or motilin (reference agonist) in 96-well plates (duplicate samples).
- the emission of light resulting from receptor activation was recorded using the Functional Drug Screening System 6000 'FDSS 6000' (Hamamatsu Photonics K.K., Japan).
- an approximate EC 80 concentration of motilin i.e. 0.5 nM; 100 ⁇ L was injected onto the cell suspension containing the test compounds (duplicate samples) 15-30 minutes after the end of agonist testing and the consequent emission of light resulting from receptor activation was measured as described in the paragraph above.
- E the measured RLU value at a given agonist concentration (C)
- E max the maximal response
- EC 50 the concentration producing 50% stimulation
- n is the slope index.
- results for agonist testing results for each concentration of test compound were expressed as percent activation relative to the signal induced by motilin at a concentration equal to the EC 80 (i.e. 0.5 nM). For antagonist testing, results for each concentration of test compound were expressed as percent inhibition relative to the signal induced by motilin at a concentration equal to the EC 80
- ExpresSProfile a broad target screen offered commercially by CEREP (Poitiers, France).
- CEREP CEREP
- a single-point (10 ⁇ M) binding assay is performed across 50 individual receptors from four distinct target classes: non-peptide G-Protein Coupled Receptors (GPCRs), peptide GPCRs, ion channels and amine transporters.
- GPCRs non-peptide G-Protein Coupled Receptors
- peptide GPCRs peptide GPCRs
- ion channels amine transporters
- Binding activity at the motilin receptor for representative compounds of the present invention is shown in Table 4.
- Compound structures for Table 4 are presented in Table 3 with the various groups as defined for the general structure of formula I.
- Table 4 Functional Activity at the Motilin Receptor for Representative Compounds of the Invention Compound No. IC 50 (nM) a Compound No.
- IC 50 (nM) a 502 B 543 C 503 C 544 A 504 C 545 A 505 B 546 A 511 C 547 B 512 C 548 B 513 C 550 A 515 B 552 A 516 C 555 A 529 B 557 B 530 C 563 B 532 C 564 A 533 B 565 B 534 B 568 B 535 B 569 B 536 C 570 A 537 A 571 B 538 A 572 A 539 A 573 B 540 A 574 D 541 B 585 B 542 B 586 C a Functional activity determined using standard method, IC 50 values are expressed as follows: A ⁇ 1 nM, B ⁇ 10 nM, C ⁇ 100 nM, D ⁇ 1000 nM
- CHO membranes were immobilized into 96-well FlashPlate microplates.
- Test compound, GTP ⁇ S, motilin and [ 35 S]-GTP ⁇ S were combined in each well according to the Assay Volumes described above.
- the reaction is initiated by addition of 100 mL of [ 35 S]-GTP ⁇ S to each well. Each plate is sealed (TopSeal-A) and incubated in the dark at room temperature for 150 min. Then, plates are counted for 30 seconds per well on the TopCount NXT.
- Top and Bottom correspond to the top and bottom values of the dose-response curve calculated by GraphPad Prism.
- Duodenal segments were vertically suspended in organ chambers of 10 mL filled with Krebs buffer and connected to an isotonic force transducer, with a preload of 1 g. After a stabilization period, the muscle strips were challenged with 10 -4 M acetylcholine and washed. This was repeated until a stable maximal contraction was obtained (2-3 times), with an interval of at least 20 minutes.
- test compounds were added to the bath. After a 15 minute incubation, a dose response to motilin was recorded by adding logarithmically increasing concentrations of motilin to the bath (final concentration 10 -9 to 10 -6 M). A blank experiment (no test compound present) was also performed. At the end of the dose response curve, a supramaximal dose of acetylcholine (10 -4 M) was given and this response was used as a reference (100% contraction).
- Rat models have been utilized for evaluation of compounds in chemotherapy-induced diarrhea (CID).
- CID chemotherapy-induced diarrhea
- mice have been used to test agents for amelioration of gastrointestinal toxicity of chemotherapeutic agents.
- Boushey RP, Yusta B, Drucker DJ. Cancer Res 2001, 61, 687-93 Zhao, J.; Huang, L.; Belmar, N.; Buelow, R.; Fong, T. Clin. Canc. Res. 2004, 10, 2851-2859 .
- dogs are known to suffer from diarrhea during treatment with common chemotherapeutic agents.
- rats and mice do not possess a functional motilin receptor and would not be appropriate animal models for a motilin antagonist, but could be used to differentiate effects at the motilin receptor from non-specific effects.
- the dog is an appropriate model species as it possesses a functional motilin receptor and has been used previously for studies involving motilin modulation.
- Other species appropriate for such an evaluation include hamster, pig, rabbit, shrew, guinea pig and opossum.
- methods analogous to those described for rats and mice could be adapted to canine or other models to test the compounds of the invention.
- the effect of treatment with compounds of the invention on the frequency and severity of incidences of diarrhea in a dog or other animal model can be used as a measure of the effectiveness of the compounds of the invention.
- the objective of this study was to determine the efficacy of representative macrocyclic compounds of the present invention in chemotherapy-induced diarrhea (CID) following intravenous administration to the male beagle dog ( Canis familiaris ) for up to 32 days. Comparison with existing agents utilized for this condition, octreotide and loperamide, was an additional objective.
- CID chemotherapy-induced diarrhea
- TA1 irinotecan solution or irinotecan hydrochloride
- TA1 irinotecan solution or irinotecan hydrochloride
- Cycle 1 irinotecan solution was administered at a dose level of 4 mg/kg/day by one hour infusion via implanted catheter for 5 consecutive days followed by a 9-day rest period.
- irinotecan solution was administered by one hour infusion via implanted catheter for four consecutive days at a dose level of 4 mg/kg/day and by bolus injection (over approximately one minute) at a dose level of 8 mg/kg/day via implanted catheter for the fifth day followed by a 5-day rest period; for Cycle 3, one spare animal was added to Group 1.
- Irinotecan formulation was prepared with irinotecan hydrochloride powder and administered to the four animals by bolus injection via implanted catheter at a dose level of 4 mg/kg/day for five consecutive days. These four animals were observed for 9 days and subjected to necropsy following an overnight period without food.
- irinotecan formulation was prepared with irinotecan hydrochloride powder and administered by bolus injection via implanted catheter up to two treatment cycles for a 28-day study period. In each treatment cycle, animals were treated with a single daily dose at 6 mg/kg for 5 consecutive days followed by a 9-day rest period without treatment. Before commencing treatment with irinotecan (Group 4) or upon onset of irinotecan-induced diarrhea (Group 2, 3, 6 and 7), these animals were treated with TA2 (compound 552) (Group 2 to 4), octreotide (Group 6) or loperamide (Group 7) as detailed below.
- b TA2 (compound 552) was administered by infusion to all the animals in the group via an implanted catheter over 45 minutes.
- the treatment with compound 552 started on the day of onset of diarrhea, if diarrhea was observed in any of the three animals from the group in the morning or on the following day if diarrhea was observed in the afternoon. Treatment with compound 552 then continued throughout the remainder of the 28-day study period.
- the daily dosage i.e.
- 5.0 and 15.0 mg/kg/day for Groups 2 and 3 respectively was divided into two equal doses (ie 2.5 and 7.5 mg/kg/occasion for Groups 2 and 3 respectively) and infused at least 6 hours apart on each day.
- the animals received 0.9% Sodium Chloride for Injection USP at a rate of 2.5 mL/hour between each infusion of compound 552.
- c TA2 (compound 552) was administered to all three animals in the group following the same procedures for Groups 2 and 3, starting four days before the start of TA 1 (irinotecan) treatment and throughout the 28-day study period.
- the daily dose (5.0 mg/kg/day) was divided into two equal doses (2.5 mg/kg/occasion) and infused at least 6 hours apart on each day.
- the animals in the Vehicle Control group (5% dextrose for injection, USP) were treated with the vehicles for irinotecan and compound 552.
- the vehicle for irinotecan was administered by a slow bolus injection (approximately over one minute) via infusion line for up to two treatment cycles. In each treatment cycle, animals were treated with single daily doses for 5 consecutive days followed by a 9-day rest period.
- the vehicle for compound 552 was administered by following the dosing procedures for compound 552 administration or 23 days (from Day 6 to Day 28.
- mice in the group were dosed with octreotide by subcutaneous injection on the day of onset of irinotecan-induced diarrhea, if diarrhea was observed in any of the three animals from the group in the morning or on the following day if diarrhea was observed in the afternoon.
- the treatment was maintained throughout the remainder of the 28-day study period.
- the daily dosage (0.021 mg/kg/day) was divided into three equal doses (0.007 mg/kg/occasion) and administered by subcutaneous injection early in the morning, noon and late in the afternoon, respectively. Each administration was separated by at least 4 hours ( ⁇ 30 minutes).
- test compounds, controls and comparators were prepared at appropriate intervals. All formulations intended for administration to the test animals were stored refrigerated at 2-8°C. Irinotecan was obtained as a sterile solution for intravenous injection containing irinotecan hydrochloride trihydrate at a concentration of 20 mg/mL, ready to use. The irinotecan formulation for dosing the animals (0.5 mg/mL) in Group 1 (Cycles 1 and 2 only) was achieved by dilution of 20 mg/mL formulation with 5% dextrose for injection, USP. The dosing formulation was stored refrigerated (2 to 8°C), protected from light and used within 48 hours.
- irinotecan For the remaining treatment with irinotecan (i.e. Cycle 3 for Group 1; Groups 2 to 4; Groups 6 and 7), a stock irinotecan formulation was prepared from irinotecan solid (irinotecan hydrochloride) and used for preparation of a dose formulation as described below:
- Octreotide was provided as a sterile lyophilisate and reconstituted with sterile water for injection to yield a stock solution (10 mg/mL).
- the stock solution was stored refrigerated (2 to 8°C). On each day of dosing, the stock solution is diluted with sterile water for injection to a final concentration of 0.1 mg/mL.
- Compound 552 was formulated as a solution with 10% hydroxypropyl-13-cyclodextrin (97%) in water.
- representative samples 1.0 mL in duplicate, except for octreotide, where only 0.1 mL each will be collected) were collected at appropriate intervals during the study.
- a total of 21 males and 2 spare males were obtained from a commercial supplier (for example Marshall BioResources, North Rose, New York) with ages 5 to 7 months old at onset of treatment and weights 6-9 kg at onset of treatment.
- the animals were acclimated between receipt of the animals and the start of treatment to accustom the dogs to the laboratory environment prior to the start of the study approximately 3 weeks for Group 1, 5 weeks for Group 2 to 4, and 3 weeks for Group 5 to 7.
- Each dog was housed in a stainless steel cage equipped with an automatic watering system supplemented by water bottles as appropriate. Each cage was clearly labeled with a color-coded cage card indicating the study, group and animal numbers, sex and dose level.
- Each animal was uniquely identified by the supplier by means of a tattoo on the ventral aspect of one pinna.
- the animal room environment was controlled (targeted ranges: temperature 21 ⁇ 3°C, relative humidity 50 ⁇ 20%, 12 hours light, 12 hours dark, 10-15 air changes per hour) except during designated procedures. Temperature and relative humidity were monitored continuously and recorded 4 times daily.
- a standard certified commercial dog chow (for example 400 g of Teklad Certified 25% Lab Dog Diet #8727C) was available to each dog once daily during a minimum two-hour feeding period but up to a maximum of 4 hours. During acclimation, the animals were slowly acclimated to the two-hour feeding regimen. Concentrations of the constituents of the diet and contaminants (e.g., heavy metals, aflatoxin, organophosphates, pesticides and chlorinated hydrocarbons) were controlled and routinely measured by the manufacturers. Municipal tap water (which has been purified by reverse osmosis, ultraviolet light and further filtered with a 0.2 ⁇ m filter) was provided to the animals ad libitum except during designated procedures.
- Municipal tap water (which has been purified by reverse osmosis, ultraviolet light and further filtered with a 0.2 ⁇ m filter) was provided to the animals ad libitum except during designated procedures.
- the dogs were prepared for surgery approximately 2-3 weeks before starting treatment. These animals were food deprived overnight prior to the surgical procedure and water was removed on the morning of surgery to insert an intravenous catheter.
- the dogs were injected intramuscularly with a pre-anesthetic cocktail (e.g. butorphanol, acepromazine and glycopyrrolate) for preparation of the surgical sites.
- a pre-anesthetic cocktail e.g. butorphanol, acepromazine and glycopyrrolate
- animals were anesthetized by isoflurane inhalation.
- the intravenous catheter consisted of a length of medical-grade tubing inserted into the femoral vein and advanced into the vena cava.
- the catheter was exteriorized at the nape of the neck using a trocar which created a subcutaneous tunnel from the inguinal area to the dorso-cervical area. During the recovery from anesthesia, each animal was dressed in a clean jacket.
- the catheter was attached to a swivel device, via a jacket and tether system, and connected to a syringe by another section of medical-grade tubing.
- the pumps and reservoirs were contained in a specially designed box positioned on the exterior of the cage.
- antibiotic for example benzanthine penicillin G and procaine penicillin G
- a topical antiseptic was applied to the surgical site once daily, as needed.
- the syringes/bags containing the saline solution were changed with appropriate frequency during the pretreatment period.
- cycle 1 For the animals in Group 1, treatment with test article No. 1 (irinotecan) consisted of three (3) cycles: Cycle 1, irinotecan was administered at a dose level of 4 mg/kg/day by one hour infusion via implanted catheter for 5 consecutive days followed by a 9-day rest period. Cycle 2, irinotecan was administered by one hour infusion via implanted catheter for four consecutive days at a dose level of 4 mg/kg/day and by bolus injection (approximately over one minute) at a dose level of 8 mg/kg/day via implanted catheter for the fifth day followed by a 5-day rest period; Cycle 3, one spare animal was added to Group 1.
- Irinotecan formulation was prepared with irinotecan hydrochloride powder and administered to the four animals by bolus injection via implanted catheter at a dose level of 4 mg/kg/day for five consecutive days.
- irinotecan formulation that was prepared with irinotecan hydrochloride powder was administered by bolus injection via implanted catheter up to two treatment cycles for a 28-day study period. In each treatment cycle, animals were treated with a single daily dose at 6 mg/kg for 5 consecutive days followed by a 9-day rest period without treatment.
- the control/vehicle article for irinotecan was administered to all dogs in Group 5 by following the same dosing procedures for irinotecan administration.
- the dose volume was 8 mL/kg for Cycle 1 and the first 4 days of Cycle 2 for Group 1, 2 mL/kg for the last day of Cycle 2 for Group 1, and 3 mL/kg for Groups 2 to 4, 6 and 7.
- the actual volume infused to each dog was calculated and adjusted based on the most recent practical body weight of each animal.
- the test article No. 2 (compound 552) was intravenously infused via an implanted catheter over 45 minutes to all the animals in Group 4 four days before the start of TA1 (irinotecan) treatment and throughout the 28-day study period.
- TA2 (compound 552) was administered to all the animals in Groups 2 and 3 on the same day of onset of irinotecan-induced diarrhea, if diarrhea was observed in any one of the three animals from the group in the morning, or on the following day of diarrhea onset if diarrhea was observed in anyone of the three animals in the group in the afternoon, and throughout the remainder of the 28-day study period.
- the daily dosage i.e.
- 5.0, 15.0 and 5.0 mg/kg/day for Groups 2 to 4 respectively was administered by two equal doses (i.e. 2.5, 7.5 and 2.5 mg/kg/occasion for Groups 2 to 4 respectively) which were separate by at least 6 h.
- Each dose was infused at a dose volume of 10 mL/kg/h.
- the vehicle for compound 552 was administered to all dogs in Group 5 by following the same dosing procedures and regimen for compound 552 administration for 23 days (from Day 6 to Day 28).
- the actual volume infused was calculated and adjusted based on the most recent practical body weight of each animal.
- the syringes/bags were changed at appropriate intervals as necessary and the weights recorded prior to the start and at the end of the infusion.
- Octreotide was administered by subcutaneous injection to all animals in Group 6 starting on the same day of diarrhea onset, if diarrhea was observed in any one of the three animals from the group in the morning, or on the following day of diarrhea onset, if diarrhea was observed in any one of the three animals in the goup in the afternoon.
- the treatment was maintained throughout the remainder of the 28-day study period.
- the daily dosage (0.021 mg/kg/day) was divided into three equal doses (0.007 mg/kg/occasion) and administered by subcutaneous injection approximately early in the morning, noon and late in the afternoon, respectively. Each administration was separated by at least 4 hours ( ⁇ 30 minutes).
- the dose volume for each administration was 0.07 mL/kg. The actual dose volume was calculated and adjusted based on the most recent practical body weight of each animal.
- Loperamide was administered by oral gavage to all animals in Group 7 starting on the same day of diarrhea onset, if diarrhea was observed in any one of the three animals of the group in the morning, or on the following day of diarrhea onset, if diarrhea was observed in anyone of the three animals in the group in the afternoon.
- the treatment was maintained throughout the remainder of the 28-day study period.
- the daily dosage (0.18 mg/kg/day) was divided into three equal doses (0.06 mg/kg/occasion) and administered by oral gavage approximately early in the morning, noon and late in the afternoon, respectively. Each administration was separated at least 4 h ( ⁇ 30 min).
- the dose volume for each administration was 0.6 mL/kg. The actual dose volume was calculated and adjusted based on the most recent practical body weight of each animal.
- the infusion line in each animal was maintained by infusion of 0.9% Sodium Chloride for Injection USP at a rate of 2.5 mL/h between each treatment of irinotecan, compound 552 or vehicles.
- Cage-side clinical signs (ill health, behavioral changes, stool changes etc.) were recorded once daily during the acclimation period except on detailed clinical examination days. Cage-side clinical signs were recorded twice a day (am and pm) during the treatment/observation period except on the days of a detailed clinical examination where they were recorded once (PM). A detailed clinical examination of each dog was performed once pretreatment, weekly during the treatment/observation periods and before necropsy. Particular attention was paid to the surgical sites.
- Body weights were recorded for all animals once prior to group assignment, and approximately one week prior to initiation of treatment. Body weights were recorded for all animals up to 1 day prior to dosing and twice weekly thereafter during the treatment/observation periods, as well as terminally prior to necropsy (fasted). Food consumption was measured daily during the week prior to treatment and throughout the treatment/observation periods.
- a series of blood samples (approximately 2.0 mL each) were removed from each dog in Groups 2 and 3 after the first (am) infusion on the first day of treatment with compound 552 and after the second (pm) infusion on the last day of treatment with compound 552. (Note: the second (pm) infusion on the first day of treatment with compound 552 was performed after completion of the last blood sampling).
- each dog was bled by venipuncture and the samples collected into tubes containing the anticoagulant, K 2 EDTA. Tubes were placed on wet ice pending processing. On each occasion, samples were collected prior to the start of infusion, 5, 15, 30, 45, 50, 65, 95, 135 minutes, 3 and 6 hours after the start of infusion of compound 552. Following collection, the samples were centrifuged at approximately 1500xg, approximately 4°C, for at least 10 minutes and the resulting plasma recovered and dispensed into two aliquots and stored frozen (at approximately -80°C) pending shipment.
- the samples were centrifuged at approximately 1500xg, approximately 4°C, for at least 10 minutes and the resulting plasma recovered and dispensed into two aliquots and stored frozen (at approximately -80°C) pending shipment.
- the data was analyzed for homogeneity of variance using Levene Median and for normality using Kolmogorov-Smimov tests.
- Homogeneous data was analyzed using the Analysis of Variance and the significance of intergroup differences was analyzed using Dunnett's test.
- Heterogeneous data was analyzed using Kruskal-Wallis test and the significance of intergroup differences between the control and treated groups assessed using Dunn's test. A significance level of p ⁇ 0.05 was reported.
- compound 552 a potent and selective motilin antagonist, demonstrated superior efficacy in the treatment of irinotecan-induced CID in dogs versus the current standard of care.
- the compound proved to be more effective, with a quicker onset and longer duration of action as well.
- Plasma concentrations of compound 552 in male beagle dogs receiving low and high intravenous doses of compound 552 as part of the above experiment were determined.
- Plasma samples were prepared for analysis by solid phase extraction and analyzed by LC-MS/MS.
- the average C max observed at 30 min after the start of the 45-min infusion was about 1.5 and 1.8 ⁇ g/mL on the first and last day of treatment with compound 552, respectively, for the animals receiving 2.5 mg/kg.
- the average C max observed for the high dose group administered with 7.5 mg/kg occurred between 15 and 45 min after the start of the 45-min infusion and was about 3.4 and 5.0 ⁇ g/mL on the first and last day of treatment with compound 552, respectively.
- the average AUC 0-t was similar on the first (78,233 ng.min/mL) and last day (83,047 ng.min/mL) of dosing for the low dose group (2.5 mg/kg/occasion) and was proportionally higher on the first day (246,073 ng.min/mL) of dosing for the high dose group (7.5 mg/kg/occasion).
- the average AUC 0-t was higher (380,758 ng.min/mL) on the last day of dosing for the high dose group, however the higher average value-resulted from one animal exhibiting particularly high AUC 0-t .
- the plasma concentration data demonstrated dose-related exposure for compound 552 in beagle dogs during the CID efficacy study.
- Cytochrome P450 enzymes are implicated in the phase I metabolism of drugs. The majority of drug-drug interactions are metabolism-based and, moreover, these interactions typically involve inhibition of cytochrome P450s. Six CYP450 enzymes (CYPIA2, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4) appear to be commonly responsible for the metabolism of most drugs and the associated drug-drug interactions. Assays to determine the binding of compounds of the invention to the various metabolically important isoforms of cytochrome P450 metabolizing enzymes are commercially available, for example NoAb BioDiscoveries (Mississaugua, ON, Canada) and Absorption Systems (Exton, PA, USA).
- the pharmacokinetic and pharmacodynamic properties of drugs are largely a function of the reversible binding of drugs to plasma or serum proteins such as albumin and ⁇ 1 -acid glycoprotein.
- plasma or serum proteins such as albumin and ⁇ 1 -acid glycoprotein.
- drugs with low plasma protein binding generally have large volumes of distribution and rapid clearance since only unbound drug is available for glomerular filtration and, in some cases, hepatic clearance.
- the ideal range for plasma protein binding is in the range of 87-98% for most drug products.
- Protein binding studies were performed using human plasma. Briefly, 96-well microplates were used to incubate various concentrations of the test article for 60 min at 37°C. Bound and unbound fractions are separated by equilibrium dialysis, where the concentration remaining in the unbound fraction is quantified by LC-MS or LC-MS-MS analysis. Drugs with known plasma protein binding values such as quinine ( ⁇ 35%), warfarin ( ⁇ 98%) and naproxen ( ⁇ 99.7%) were used as reference controls.
- Table 6 Results for representative compounds of the invention are summarized in Table 6.
- Table 8 Inhibition of CYP P450 Isozymes by Representative Compounds of the Invention Compound IC 50 ( ⁇ M) a 3A4 3A4 1A2 2A6 2B6 2C8 2C9 2C19 2D6 2E1 (BQ) (BFC) 551 10.4 2.37 n/a n/a n/a 26.9 37.0 30.1 >100 553 3.48 0.0192 n/a n/a n/a >100 44.5 n/a 80.8 >100 a n/a indicates IC 50 value much above 100 ⁇ M
- the Caco-2 cell line derived from a human colorectal carcinoma, has become an established in vitro model for the prediction of drug absorption across the human intestine.
- Assays to determine the permeability of compounds of the invention utilizing Caco-2 cells are commercially available, for example NoAb BioDiscoveries (Mississaugua, ON, Canada) and Absorption Systems (Exton, PA, USA).
- PAMPA parallel artificial membrane permeability assays
- Permeability across the Caco-2 cell layer was determined by growing the cells on a membrane placed between two (donor and acceptor) chambers. Drug candidates are typically added to the apical (A) side of the cell layer and their appearance in the basolateral (B) side is measured over incubation time. Permeability in this direction represents intestinal absorption. Permeability may also be determined from the basolateral to the apical side of the Caco-2 cell. A higher apical to basolateral P app , compared to the basolateral to apical P app , is indicative of carrier-mediated transport. P-gp mediated transport is suggested when a higher basolateral to apical P app is observed relative to the apical to basolateral P app .
- Permeability (10 ⁇ M) for compounds of the invention in the apical to basolateral and basolateral to apical direction were tested in duplicate. Samples will be collected from the donor and acceptor chambers at the beginning (0 min) and following 60 min of incubation at 37°C and stored frozen at -70°C until bioanalysis. Samples for each test compound generated from the Caco-2 permeability assay were further analyzed by LC-MS-MS. The permeability of [ 3 H]-mannitol and [ 3 H]-propranolol were determined in parallel as controls.
- the pharmacokinetic behavior of compound of the invention can be ascertained by methods well known to those skilled in the art. ( Wilkinson, G. R. "Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination” in Goodman & Gilman's The Pharmacological Basis of Therapeutics, Tenth Edition, Hardman, J.G.; Limbird, L.E., Eds., McGraw Hill, Columbus, OH, 2001, Chapter 1 .) The following method was used to investigate the pharmacokinetic parameters (elimination half-life, total plasma clearance, etc.) for intravenous, subcutaneous and oral administration of compounds of the present invention.
- Rats male, Sprague-Dawley ( ⁇ 250 g)
- Rats/Treatment Group 6 (2 subsets of 3 rats each, alternate bleeds)
- test compound was sent in solution in a formulation (such as with cyclodextrin) appropriate for dosing. It will be appreciated by one skilled in the art that appropriate modifications to this protocol can be made as required to adequately test the properties of the compound under analysis.
- 0.7 mL of blood were collected from each animal. It is expected that this volume of blood will yield a sample of at least 0.3 mL of plasma.
- EDTA was used as an anti-coagulant for whole blood collection. Whole blood samples were chilled and immediately processed by centrifugation to obtain plasma.
- Plasma samples were stored frozen (-70°C) until analysis.
- Analytical detection of parent compound in plasma samples performed by LC-MS after an appropriate preparation protocol: extraction using solid phase extraction (SPE) cartridges (Oasis MCX, Oasis HLB) or liquid-liquid extraction.
- SPE solid phase extraction
- na not applicable c Done in the presence of 10% beta-cyclodextrin d Done without cyclodextrin
- motilin stimulates the contractile activity in smooth muscle strips isolated from the rabbit gastric antrum ( Van Assche, G.; Depoortere, I.; Thijs, T.; Janssens, J.J.; Peeters, T.L. Concentration-dependent stimulation of cholinergic motor nerves or smooth muscle by [Nle13]-motilin in the isolated rabbit gastric antrum. Eur. J. Pharmacol. 1997, 337, 267-274 .) or from the circular muscle of-the duodenum as described in Method 3-D above.
- Motilin (threshold concentration of 10 -8 M) enhances contractions induced by electrical field stimulation (4 Hz) by a post-ganglionic interaction with the cholinergic neurotransmission. At higher concentrations, motilin causes a tonic contraction interacting directly with the antral smooth muscle. Analogously, the effects of motilin and [Nle 13 ]-motilin (Calbiochem), as selective agonists of the motilin receptor, on smooth muscle strips isolated from the gastric antrum, the circular muscle of the duodenum and the colon of the shrew were investigated.
- test compounds were tested on different regions of the GI tract of the shrew (gastric antrum, ileum and colon) and to evaluate the antagonist affinity of test compounds in this model system.
- the ability of varying concentrations of test compound to antagonize the motilin or [Nle 13 ]-motilin-induced contractions was measured in organ baths as described above.
- Compound 552 (0.01 ⁇ M-10 ⁇ M) caused a dose-dependent inhibition of the contractile response to 0.3 ⁇ M motilin or to 0.3 ⁇ M [Nle 13 ]-motilin.
- data are mean ⁇ SEM and the number of animals was as indicated for each treatment.
- Compound 552 did not cause a significant change in basal contractile activity (data not shown).
- Animal models can be used to investigate the effect of treatment with representative compounds of the present invention on amelioration of the symptoms or prevention of the development of diarrhea induced in different manners, in particular by stress or by PGE 2 .
- rats and mice do not express motilin receptors
- the musk shrew ( Suncus murinus ) is one appropriate animal model expressing the motilin receptor and suitable for this work.
- opossum, rabbit or the pig are also suitable animal models for this type of evaluation.
- Food and water was provided ad libitum in accordance with the species requirements: Food: 3 part Purina Cat Chow and 1 part Mink Chow (for example Wisconsin "Mink Complete Pellets-Grow-Fur”: crude protein ⁇ 34%, crude fat >20%, crude fiber >4%).
- the food was placed in a small plastic dish within the cage.
- Water Slightly acidic (pH 5.5) distilled water, to reduce contamination of tap water supply.
- the cages and water bottles were changed weekly. All animals were acclimated to the animal facility for at least 1 week prior to initiating the study.
- the shrews were placed in individual restraint cages for 1 h at room temperature. Fecal output (number of pellets) was measured during 1 h of immobilization and within 1 h after the restraint stress. The consistency was evaluated using a 3 level scoring system: 0, normal; 1, soft; 2 unformed. ( Saito, T.; Mizutani, F.; Iwanaga, Y.; Morikawa, K.; Kato, H. Laxative and anti-diarrheal activity of polycarbophil in mice and rats. Jpn. J.
- test compound 0.1, 1 and 10 mg/kg s.c. or other appropriate doses and mode of administration
- vehicle Animals were placed in individual restraint cages for 1 h at room temperature and fecal output was measured as described above. The animals had free access to food and water prior to the experiment.
- test compound 0.1, 1 and 10 mg/kg s.c. or other appropriate doses and mode of administration
- vehicle 0.1, 1 and 10 mg/kg s.c. or other appropriate doses and mode of administration
- Diarrhea was induced by administration of PGE 2 (0.3 mg/kg, i.p.) and changes in stool consistency were evaluated as described above.
- the animals were acclimated with free access to food and water.
- the macrocyclic compounds of the present invention or pharmacologically acceptable salts thereof according to the invention may be formulated into pharmaceutical compositions of various dosage forms.
- one or more compounds, including optical isomers, enantiomers, diastereomers, racemates or stereochemical mixtures thereof, or pharmaceutically acceptable salts thereof as the active ingredient is intimately mixed with appropriate carriers and additives according to techniques known to those skilled in the art of pharmaceutical formulations.
- a pharmaceutically acceptable salt refers to a salt form of the compounds of the present invention in order to permit their use or formulation as pharmaceuticals and which retains the biological effectiveness of the free acids and bases of the specified compound and that is not biologically or otherwise undesirable. Examples of such salts are described in Handbook of Pharmaceutical Salts: Properties, Selection, and Use, Wermuth, C.G. and Stahl, P.H. (eds.), Wiley-Verlag Helvetica Acta, Zurich, 2002 [ISBN 3-906390-26-8 ].
- salts include alkali metal salts and addition salts of free acids and bases:
- pharmaceutically acceptable salts include sulfates, pyrosulfates, bisulfates, sulfites, bisulfites, phosphates, monohydrogenphosphates, dihydrogenphosphates, metaphosphates, pyrophosphates, chlorides, bromides, iodides, acetates, propionates, decanoates, caprylates, acrylates, formates, isobutyrates, caproates ; heptanoates, propiolates, oxalates, malonates, succinates, suberates, sebacates, fumarates, maleates, butyne-1,4-dioates, hexyne-1,6-dioates, benzoates, chlorobenzoates, methylbenzoates, dinitrobenzoates, hydroxybenzoates, methoxybenzoates, phthalates,
- a desired salt may be prepared by any suitable method known to those skilled in the art, including treatment of the free base with an inorganic acid, such as, without limitation, hydrochloric acid, hydrobromic acid, hydroiodic, carbonic acid, sulfuric acid, nitric acid, phosphoric acid, and the like, or with an organic acid, including, without limitation, formic acid, acetic acid, propionic acid, maleic acid, succinic acid, mandelic acid, fumaric acid, malonic acid, pyruvic acid, oxalic acid, stearic acid, ascorbic acid, glycolic acid, salicylic acid, pyranosidyl acid, such as glucuronic acid or galacturonic acid, alpha-hydroxy acid, such as citric acid or tartaric acid, amino acid, such as aspartic acid or glutamic acid, aromatic acid, such as benzoic acid or cinnamic acid, sulfonic acid, such as p-tol
- an inorganic acid such
- an inventive compound is an acid
- a desired salt may be prepared by any suitable method known to the art, including treatment of the free acid with an inorganic or organic base, such as an amine (primary, secondary, or tertiary); an alkali metal or alkaline earth metal hydroxide; or the like.
- an inorganic or organic base such as an amine (primary, secondary, or tertiary); an alkali metal or alkaline earth metal hydroxide; or the like.
- suitable salts include organic salts derived from amino acids such as glycine, lysine and arginine; ammonia; primary, secondary, and tertiary amines such as ethylenediamine, N,N'-dibenzylethylenediamine, diethanolamine, choline, and procaine, and cyclic amines, such as piperidine, morpholine, and piperazine; as well as inorganic salts derived from sodium, calcium, potassium, magnesium, manganese, iron, copper, zinc, aluminum, and lithium.
- compositions for oral administration may be, for example, solid preparations such as tablets, sugar-coated tablets, hard capsules, soft capsules, granules, powders and the like, with suitable carriers and additives being starches, sugars, binders, diluents, granulating agents, lubricants, disintegrating agents and the like. Because of their ease of use and higher patient compliance, tablets and capsules represent the most advantageous oral dosage forms for many medical conditions.
- compositions for liquid preparations include solutions, emulsions, dispersions, suspensions, syrups, elixirs, and the like with suitable carriers and additives being water, alcohols, oils, glycols, preservatives, flavoring agents, coloring agents, suspending agents, and the like.
- suitable carriers and additives being water, alcohols, oils, glycols, preservatives, flavoring agents, coloring agents, suspending agents, and the like.
- Typical preparations for parenteral administration comprise the active ingredient with a carrier such as sterile water or parenterally acceptable oil including polyethylene glycol, polyvinyl pyrrolidone, lecithin, arachis oil or sesame oil, with other additives for aiding solubility or preservation may also be included.
- a carrier such as sterile water or parenterally acceptable oil including polyethylene glycol, polyvinyl pyrrolidone, lecithin, arachis oil or sesame oil, with other additives for aiding solubility or preservation may
- compositions according to embodiments of the present invention include those suitable for oral, rectal, topical, inhalation (e.g., via an aerosol) buccal (e.g., sub-lingual), vaginal, topical (i.e., both skin and mucosal surfaces, including airway surfaces), transdermal administration and parenteral (e.g., subcutaneous, intramuscular, intradermal, intraarticular, intrapleural, intraperitoneal, intrathecal, intracerebral, intracranially, intraarterial, or intravenous), although the most suitable route in any given case will depend on the nature and severity of the condition being treated and on the nature of the particular active agent which is being used.
- compositions for injection will include the active ingredient together with suitable carriers including propylene glycol-alcohol-water, isotonic water, sterile water for injection (USP), emulPhorTM-alcohol-water, cremophor-ELTM or other suitable carriers known to those skilled in the art.
- suitable carriers including propylene glycol-alcohol-water, isotonic water, sterile water for injection (USP), emulPhorTM-alcohol-water, cremophor-ELTM or other suitable carriers known to those skilled in the art.
- carriers may be used alone or in combination with other conventional solubilizing agents such as ethanol, propylene glycol, or other agents known to those skilled in the art.
- the compounds may be used by dissolving or suspending in any conventional diluent.
- the diluents may include, for example, physiological saline, Ringer's solution, an aqueous glucose solution, an aqueous dextrose solution, an alcohol, a fatty acid ester, glycerol, a glycol, an oil derived from plant or animal sources, a paraffin and the like. These preparations may be prepared according to any conventional method known to those skilled in the art.
- compositions for nasal administration may be formulated as aerosols, drops, powders and gels.
- Aerosol formulations typically comprise a solution or fine suspension of the active ingredient in a physiologically acceptable aqueous or non-aqueous solvent.
- Such formulations are typically presented in single or multidose quantities in a sterile form in a sealed container.
- the sealed container can be a cartridge or refill for use with an atomizing device.
- the sealed container may be a unitary dispensing device such as a single use nasal inhaler, pump atomizer or an aerosol dispenser fitted with a metering valve set to deliver a therapeutically effective amount, which is intended for disposal once the contents have been completely used.
- the dosage form comprises an aerosol dispenser, it will contain a propellant such as a compressed gas, air as an example, or an organic propellant including a fluorochlorohydrocarbon or fluorohydrocarbon.
- compositions suitable for buccal or sublingual administration include tablets, lozenges and pastilles, wherein the active ingredient is formulated with a carrier such as sugar and acacia, tragacanth or gelatin and glycerin.
- a carrier such as sugar and acacia, tragacanth or gelatin and glycerin.
- compositions for rectal administration include suppositories containing a conventional suppository base such as cocoa butter.
- compositions suitable for transdermal administration include ointments, gels and patches.
- compositions known to those skilled in the art can also be applied for percutaneous or subcutaneous administration, such as plasters.
- compositions comprising the active ingredient or ingredients in admixture with components necessary for the formulation of the compositions
- other conventional pharmacologically acceptable additives may be incorporated, for example, excipients, stabilizers, antiseptics, wetting agents, emulsifying agents, lubricants, sweetening agents, coloring agents, flavoring agents, isotonicity agents, buffering agents, antioxidants and the like.
- additives there may be mentioned, for example, starch, sucrose, fructose, dextrose, lactose, glucose, mannitol, sorbitol, precipitated calcium carbonate, crystalline cellulose, carboxymethylcellulose, dextrin, gelatin, acacia, EDTA, magnesium stearate, talc, hydroxypropylmethylcellulose, sodium metabisulfite, and the like.
- the composition is provided in a unit dosage form such as a tablet or capsule.
- kits including one or more containers comprising pharmaceutical dosage units comprising an effective amount of one or more compounds of the present invention.
- the present invention further provides prodrugs comprising the compounds described herein.
- prodrug is intended to mean a compound that is converted under physiological conditions or by solvolysis or metabolically to a specified compound that is pharmaceutically active.
- the "prodrug” can be a compound of the present invention that has been chemically derivatized such that, (i) it retains some, all or none of the bioactivity of its parent drug compound, and (ii) it is metabolized in a subject to yield the parent drug compound.
- the prodrug of the present invention may also be a "partial prodrug" in that the compound has been chemically derivatized such that, (i) it retains some, all or none of the bioactivity of its parent drug compound, and (ii) it is metabolized in a subject to yield a biologically active derivative of the compound.
- Known techniques for derivatizing compounds to provide prodrugs can be employed. Such methods may utilize formation of a hydrolyzable coupling to the compound.
- the present invention further provides that the compounds of the present invention may be administered in combination with a therapeutic agent used to prevent and/or treat metabolic and/or endocrine disorders, gastrointestinal disorders, cardiovascular disorders, obesity and obesity-associated disorders, central nervous system disorders, genetic disorders, hyperproliferative disorders and inflammatory disorders.
- a therapeutic agent used to prevent and/or treat metabolic and/or endocrine disorders, gastrointestinal disorders, cardiovascular disorders, obesity and obesity-associated disorders, central nervous system disorders, genetic disorders, hyperproliferative disorders and inflammatory disorders.
- agents include analgesics (including opioid analgesics), anesthetics, antifungals, antibiotics, antiinflammatories (including nonsteroidal anti-inflammatory agents), anthelmintics, antiemetics, antihistamines, antihypertensives, antipsychotics, antiarthritics, antitussives, antivirals, cardioactive drugs, cathartics, chemotherapeutic agents (such as DNA-interactive agents, antimetabolites, tubulin-interactive agents, hormonal agents, and agents such as asparaginase or hydroxyurea), corticoids (steroids), antidepressants, depressants, diuretics, hypnotics, minerals, nutritional supplements, parasympathomimetics, hormones (such as corticotrophin releasing hormone, adrenocorticotropin, growth hormone releasing hormone, growth hormone, thyrptropin-releasing hormone and thyroid stimulating hormone), sedatives, sulfonamides, stimulants, sympathomimetics, tranquilizer
- Subjects suitable to be treated according to the present invention include, but are not limited to, avian and mammalian subjects, and are preferably mammalian.
- Mammals of the present invention include, but are not limited to, canines, felines, bovines, caprines, equines, ovines, porcines, rodents ( e.g . rats and mice), lagomorphs, primates, humans, and the like, and mammals in utero. Any mammalian subject in need of being treated according to the present invention is suitable.
- Human subjects are preferred. Human subjects of both genders and at any stage of development ( i.e ., neonate, infant, juvenile, adolescent, adult) can be treated according to the present invention.
- Illustrative avians according to the present invention include chickens, ducks, turkeys, geese, quail, pheasant, ratites ( e.g., ostrich) and domesticated birds ( e.g., parrots and canaries), and birds in ovo.
- ratites e.g., ostrich
- domesticated birds e.g., parrots and canaries
- the present invention is primarily concerned with the treatment of human subjects, but the invention can also be carried out on animal subjects, particularly mammalian subjects such as mice, rats, dogs, cats, livestock and horses for veterinary purposes.
- animal subjects particularly mammalian subjects such as mice, rats, dogs, cats, livestock and horses for veterinary purposes.
- the invention can be further carried out for drug screening and drug development purposes.
- the compounds of the present invention or an appropriate pharmaceutical composition thereof may be administered in an effective amount. Since the activity of the compounds and the degree of the therapeutic effect vary, the actual dosage administered will be determined based upon generally recognized factors such as age, condition of the subject, route of delivery and body weight of the subject.
- the dosage can be from about 0.1 to about 100 mg/kg, administered orally 1-4 times per day.
- compounds can be administered by injection at approximately 0.01 - 20 mg/kg per dose, with administration 1-4 times per day. Treatment could continue for weeks, months or longer. Determination of optimal dosages for a particular situation is within the capabilities of those skilled in the art.
- the compounds of formula I of the present invention can be used for the prevention and treatment of a range of gastrointestinal disorders characterized by hypermotility or hypermotilinemia.
- the macrocyclic compounds of the present invention can be used to treat diarrhea, cancer treatment-related diarrhea, chemotherapy-induced diarrhea, radiation enteritis, radiation-induced diarrhea, stress-induced diarrhea, chronic diarrhea, AIDS-related diarrhea, C . difficile associated diarrhea, traveller's diarrhea, diarrhea induced by graph versus host disease and other types of diarrhea.
- the present invention is directed to a method of treating irritable bowel syndrome, inflammatory bowel disease, dyspepsia, functional gastrointestinal disorders, chemotherapy-induced nausea and vomiting (emesis), post-operative nausea and vomiting, Crohn's disease, gastroesophogeal reflux disorders, ulcerative colitis, pancreatitis, infantile hypertrophic pyloric stenosis, carcinoid syndrome, malabsorption syndrome, diabetes mellitus, obesity, postgastroenterectomy syndrome, atrophic colitis or gastritis, gastric stasis, gastrointestinal dumping syndrome, celiac disease, short bowel syndrome, cachexia and eating disorders leading to obesity in humans and other mammals comprising administering a therapeutically effective amount of a compound of formula I.
- treatment is not necessarily meant to imply cure or complete abolition of the disorder or symptoms associated therewith.
- the compounds of the present invention can further be utilized for the preparation of a medicament for the treatment of a range of medical conditions involving gastrointestinal motility disorders.
- Step 1-2 Thiazol-2-yl-carbamic acid benzyl ester (AA1-2).
- 2- aminothiazole AA1-A , 3.0 g, 30.0 mmol
- triethylamine 6.30 mL, 45.0 mmol
- benzyl chloroformate 5 mL, 36 mmol
- the reaction was stirred at RT O/N.
- the reaction mixture was washed with a saturated aqueous solution of NaHCO 3 , then water and concentrated to dryness under reduced pressure. The resulting yellow solid was crystallized from ethanol to afford AA1-2 in 70% yield.
- Step 1-3 Boc-Dap(Z-thiazol-2-yl) (Boc-AA1) This procedure is based on that presented for other substrates in Gautam Panda et al. SynLett 2004, 4, 714-716 .
- a mixture of Boc-Ser-Weinreb amide AA1-1 (3.0 g, 12.0 mmol) and triphenylphosphine (4.75 g, 18 mmol) in anhydrous THF (100 mL) was cooled in an ice bath.
- a solution of DIAD (3.63 g, 18 mmol) in THF (20 mL) was added to this mixture dropwise.
- Step 2-1 Boc-Serine- ⁇ -lactone (AA2-1). This procedure is based on that found in the literature ( Vederas, J.C.; et al. J. Am. Chem. Soc. 1987, 109, 4649-4659 ). Into a dry 250 mL 3-neck flask equipped with a mechanical stirrer under a nitrogen atmosphere was added triphenylphosphine (4.5 g, 17.1 mmol, 1.1 eq), followed by 100 mL of an anhydrous THF:CH 3 CN (1:9) mixture.
- triphenylphosphine 4.5 g, 17.1 mmol, 1.1 eq
- Step 2-2 1-Trimethylsilylimidazole (AA2-2).
- HMDS 11.46 mL, 0.055 mol, 1.5 eq
- imidazole 5.0 g, 0.074 mol, 2 eq
- the reaction mixture heated to reflux for 3 h.
- the reaction mixture was then concentrated to dryness under reduced pressure. Distillation of the residue gave AA2-2 as a colorless oil in 85%.
- this product should be stored at 0°C under argon.
- Step 2-3 Boc-imidazol-1-yl-Ala (AA2). Similar to the method described in Vederas, J.C.; et al. J. Am. Chem. Soc. 1985, 107, 7105-7109 , trimethylsilylimidazole AA2-2 (0.488 g, 3.50 mmol, 1.3 eq) in dry CH 3 CN (10 mL) was treated dropwise over 5 min with AA2-1 (0.5 g, 2.67 mmol, 1 eq) in dry CH 3 CN (10 mL) under argon, then the mixture stirred for 28 h.
- Z-Dap(Me)-OH hydrochloride salt (AA4-3) .
- a dry 500 mL round bottom flask under nitrogen atmosphere was charged with 60 ml of anhydrous CH 3 CN followed by N-methyl trimethylsilylamine ( AA4-0 , synthesized as described in Step 4-6, 1.9 mL, 13.3 mmol, 1.5 eq.).
- Step 4-3 Z-Dap(Boc-Me)-OH (AA4-4).
- AA4-3 To a flask containing the crude AA4-3 (8.9 mmol, 1.0 eq based on theoretical yield) was added 90 mL of anhydrous DCM. The mixture was cooled to 0°C and diisopropylethylamine (DIPEA, 7.8 mL, 44.5 mmol, 5.0 eq) added dropwise, which helps to solubilize the crude substrate.
- DIPEA diisopropylethylamine
- Di-tert-butyl dicarbonate 2.1 g, 9.8 mmol, 1.1 eq
- Step 4-4 H-Dap(Boc-Me)-OH (AA4-5).
- a solution of AA4-4 (2.5 g, 7.1 mmol, 1.0 eq.) in 75 mL of MeOH was added to 10% palladium on activated carbon (250 mg, 10% w/w) under a nitrogen atmosphere. Hydrogen was then bubbled into the mixture until completion of the reaction. [TLC BuOH:AcOH:H 2 O (4:1:1), UV, ninhydrin].
- the mixture was then purged with nitrogen, filtered on a Celite ® (World Minerals Inc., Santa Barbara, CA) pad and rinsed well with MeOH (3 x 25 mL). The filtrate was evaporated under reduced pressure and dried under vacuum to give 1.5 g (93%) of AA5-5 as a pale yellow solid.
- Step 4-5 Ddz-Dap(Boc-Me)-OH (Ddz-AA4).
- AA4-5 1.9 g, 8.7 mmol, 1.0 eq
- MeOH MeOH
- Triton B 4.3 mL, 9.5 mmol, 1.1 eq
- the mixture was stirred 30 min at room temperature, then concentrated under reduced pressure.
- the residue was azeotroped with toluene (2x) to remove excess H 2 O and MeOH.
- the resulting slurry was dissolved in 20 mL of anhydrous DMF and Ddz-OPh (3.1 g, 9.5 mmol, 1.1 eq) added in one portion.
- the mixture was stirred 36-48 h at 50°C under nitrogen. DMF was then removed under high vacuum and the residue diluted in H 2 O (50 mL). The resulting aqueous phase (checked to ensure pH was 9 and adjusted if necessary) was washed with Et 2 O until TLC indicates no phenol in the aqueous phase.
- the basic aqueous phase was cooled to 0°C, Et 2 O (50 mL) added, and the aqueous phase acidified with citrate buffer (1.0 M, pH 3.5). The separated aqueous phase was extracted with Et 2 O (2 x 50 mL).
- Step 4-6 Synthesis of N-methyl trimethylsilylamine (AA4-0).
- Methylamine (25.0 mL, 0.56 mol, 2.5 eq) was condensed at -20°C, then added to 250 mL of anhydrous Et 2 O at - 20°C under a nitrogen atmosphere.
- Freshly distilled (from LAH) chlorotrimethylsilane (28.1 mL, 0.22 mol, 1.0 eq) was added dropwise to the solution at -20°C. A white precipitate was formed.
- the reaction was warmed to room temperature and stirred for 4 h. The mixture was then cooled to 0°C and filtered cold.
- Step 5-1 3-(3-Fluorophenyl)-2-hydroxy-propionic acid benzyl ester (F1-2).
- Sodium nitrite (3.77 g, 54.6 mmol) was added in portions to a solution of L-3-fluorophenylalanine (F1-1, 5.0 g, 27.3 mmol) in acetic acid (82 mL) which is maintained at room temperature by immersion in an ice-water bath bearing a thermocouple. The solution is stirred at room temperature for 1 hr, then concentrated under reduced pressure. Water was then added to the residue and the product extracted with Et 2 O, dried over MgSO 4 , filtrated and concentrated under reduced pressure to yield 2.57g (42%) of the corresponding acid.
- Step 5-2 ⁇ 3-[2-(2-Amino-3-methyl-butoxy)-phenyl]-propyl ⁇ -carbamic acid tert-butyl ester (F1-5).
- Diisopropyl azodicarboxylate (DIAD, 3.13 mL, 15.9 mmol) is added dropwise to a well-stirred solution of triphenylphosphine (4.17 g, 15.9 mmol) in THF (150 mL) at 0°C under nitrogen. The mixture is stirred at 0°C for 30 min, then warmed to room temperature.
- the PPh 3 -DIAD adduct should precipitate as a white solid.
- Step 5-3 2- ⁇ 1-[2-(3- tert -Butoxycarbonylaminopropyl)-phenoxymethyl]-2-methyl-propyl-amino ⁇ -3-(3-fluorophenyl)-propionic acid benzyl ester F1.
- Freshly distilled (CF 3 SO 2 ) 2 O (29.5 ⁇ L, 175 ⁇ mol) was added to a solution of the alcohol F1-2 (45 mg, 164 ⁇ mol) in CH 2 Cl 2 at 0°C over 5 min, followed by 2,6-lutidine (22 ⁇ L, 189 ⁇ mol).
- the related fragment F2 was made following the same synthetic route as described for fragment F1 as illustrated in Figure 4 .
- Step 7-2 Ddz-amino-alkyne [T38-4(S)].
- T38-2(S) (23.9 g, 86.0 mmol, 1.0 eq)
- Distilled (after being stirred O/N with CaH 2 ) triethylamine (45 mL) was then added to the solution and the mixture purged by bubbling with argon for 5 min.
- Step 7-3 Ddz-Amino-alcohol [Ddz-T38(S)].
- T38-4(S) 35.0 g, 81.9 mmol, 1.0 eq
- EtOH 400 mL
- platinum oxide 1.86 g, 8.2 mmol, 0.1 eq
- Hydrogen was bubbled into the mixture O/N while stirring. Reaction was monitored by 1 H NMR until completion. When the reaction was complete, nitrogen was bubbled for 10 min to remove the excess of hydrogen.
- T38(S) is used to make compounds of formula I with an (R)-stereocenter.
- T38(R) is used to synthesize compounds of formula I with an (S)-stereocenter as described further in Examples 10 and 11.
- protected derivatives of tethers T92 and T93 can be obtained using analogous methods to that of Example 7 starting from 3-fluoro-2-iodophenol and 6-fluoro-2-iodophenol, respectively, or the corresponding 2-bromophenols.
- Step 8-2 3-(2-Methoxy-phenyl)-2-methyl-propionic acid ethyl ester (T81-3).
- T81-2 7.3 g, 23 mmol
- ethanol 100 ml
- Pd/C 730 mg
- Hydrogen was bubbled through the solution, then the solution stirred under hydrogen atmosphere for O/N.
- the suspension was filtered through a pad of silica gel and washed with ethyl acetate.
- the combined filtrate and washes were concentrated under reduced pressure to give T81-3 which was suitable for use in the next step without any further purification (yield: 86-90%).
- Step 8-3 3-(2-Methoxy-phenyl)-2-methyl-propan-1-ol (T81-4).
- An addition of water (18.8 mL) was effected very slowly (under a nitrogen flow) followed by a solution of 15% sodium hydroxide (18.8 mL), then water (56 mL).
- the residual salt was filtered and the filtrate concentrated under reduced pressure to give alcohol T81-4 (99%) which was used without any further purification in the next step.
- Step 8-4 Acetic acid 3-(2-methoxy-phenyl)-2-methyl-propyl ester [T81-5(S)] and 3-(2-Methoxy-phenyl)-2-methyl-propan-1-ol [T81-5(R)] .
- Enzyme Amano PS (1.7 g) and racemic alcohol T81-4 (30.7, 0.67 mol) were mixed with TBME (292 mL) in a reaction flask containing a magnetic stirring bar with no cap.
- TBME was placed in a dessicator and used to restore the volume of reaction after 24 h (due to loss by evaporation).
- the mixture was stirred for 24 h in a dessicator in the presence of saturated MgCl 2 (5-10 mL of a saturated solution of MgCl 2 in water was prepared and stored in the dessicator without any cap). After 24 h, the pre-equilibrated solvent was added to the reaction flask to compensate for loss by evaporation. Vinyl acetate (56 mL, 3.7 equiv) was added to the reaction flask, which was immediately sealed with a septum. The mixture was stirred at room temperature until a conversion of 42% was obtained as determined by LC-MS (aliquot starting after 2 h up to 5.5 h at which time the reaction was typically complete). The reaction was filtered through a medium fritted glass-sintered funnel.
- the alcohol T81-5(R) was retreated with the Amano PS with the same procedure used for the racemic alcohol except that the reaction was permitted to proceed until a conversion of 20% as determined by LC-MS (aliquots starting at 2 h, usually reached this level after 8.5 h). Then the mixture was treated as described for T81-5(S) to give the alcohol T81-6(R) (14.1 g) and the acetate T81-5(S). ( J. Chem. Soc., Perkin Trans. 1 2000, 367 .) This alcohol can be substituted for the T81-6(S) isomer in Step 8-6 below and transformed into T81 containing an 8R-stereocenter.
- Step 8-5 3-(2-Methoxy-phenyl)-2-methyl-propan-1-ol [T81-6(S)].
- Methanol (135 mL, 4x the volume of BBr 3 ) was added slowly at 0°C, followed by water.
- Step 8-6 3-[2-(2-Hydroxy-propoxy)-phenyl]-2-methyl-propan-1-ol [T81-7(1R,8S)].
- DMF 16 mL
- potassium carbonate 1.6 g, 8.42 mmol, 1 eq
- Step 8-7 Toluene-4-sulfonic acid 3-[2-(2-hydroxy-propoxy)-phenyl]-2-methyl-propyl ester [T81-8(1R,8S)].
- Step 8-8 1-[2-(3-Azido-2-methyl-propyl)-phenoxy]-propan-2-ol [T81-9(1R,8S)].
- T81-8(1R,8S) 2.2 g, 5.85 mmol, 1.0 eq
- NaN 3 1.85 g, 0.03 mol, 5 eq
- Step 8-9 1-[2-(3-Amino-2-methyl-propylpphenoxy]-propan-2-ol [T81-10 (1R,8S)].
- T81-9((1R,8S) (1.20 g, 4.8 mmol, 1.0 eq) in ethyl acetate (25 mL) was carefully added 10% Pd/C (0.12 g). Hydrogen was bubbled continuously through the solution and the resulting mixture stirred at room temperature O/N.
- Step 8-10 13-[2-(2-Hydroxy-propoxy)-phenyl]-2-methyl-propyl ⁇ -carbamic acid tert-butyl ester [Boc-T81 (1R,8S)].
- T81-10(1R,8S) (1.12 g, 5.01 mmol, 1.0 eq) in THF:H 2 O (1:1, 20 mL) was added K 2 CO 3 (1.43 g, 10.0 mmol, 2.0 eq) and Boc 2 O (3.30 g, 15.0 mmol, 3 eq).
- the mixture was stirred at room temperature O/N.
- the reaction was monitored by TLC [(ethyl acetate/hexanes, 4/6).
- T81(1R;8S) and T81(1R,8R) are used to make compounds of formula I with a (1S)-stereocenter.
- T81(1S,8S) and T81(1S,8R) are used to synthesize compounds of formula I with a (1R)-stereocenter.
- Step 9-1 In a round bottom flask equipped with a Dean-Stark apparatus, was suspended amino acid G-1 (0.2 mol, 1.0 eq) in toluene (1 L per 0.2 mol, solvent/wash amounts in the procedure are scaled for smaller or larger scales).
- p-Toluenesulfonic acid (pTSA, 1.2 eq) and benzyl alcohol (5.0 eq) were added and the mixture was stirred at reflux 16-18 h. The mixture was cooled down to room temperature and a white precipitate was formed. The precipitate was diluted with MTBE (500 mL), filtered and triturated with MTBE (3 x 500 mL). The solid was dried under high vacuum for 16-18 h to give the intermediate tosylate salt.
- the tosylate salt was taken up in an aqueous solution of 1 M Na 2 CO 3 (200 mL).
- the basic aqueous phase was extracted with EtOAc (4 x 150 mL) and the combined organic phases were washed with brine (1 x 100 mL), dried over MgSO 4 , filtered, concentrated under reduced pressure and dried in vacuo to give the free amino-ester G2.
- esters to the benzyl ester can also be employed for the procedure with modification of the deprotection protocol.
- Step 9-2 To a solution of freshly free based G-2 (1.0 eq.) in DCM (90-100 mL) was added triethylamine (TEA, 1.1 eq), pyridine (3.4 eq) and Bts-Cl (2.0 eq). The reaction was stirred O/N at room temperature. The reaction mixture was diluted with DCM (100 mL), washed sequentially with aqueous citrate buffer (2x), brine (1x), dried over MgSO 4 , filtered and concentrated under reduced pressure. The resulting residue was purified by flash chromatography to give Bts protected amino-ester G-3.
- TEA triethylamine
- pyridine 3.4 eq
- Bts-Cl 2.0 eq
- Step 9-3 Protected tether G-4 (1.5 eq.), triphenylphosphine (8.5g, 32.4 mmol, 1.5 eq) and G-3 (1.0 eq) were dissolved in anhydrous THF (100 mL). The solution was cooled to 0°C in an ice-water bath and diisopropylazodicarboxylate (DIAD, 1.45 eq) was added dropwise. The preformed DIAD:PPh 3 adduct (Example 38) could be used for this step as well. After addition, the mixture was stirred 1 h at 0°C, then warmed up slowly to room temperature and stirred for 16-18 h under nitrogen. THF was removed under reduced pressure and the crude residue was purified by flash chromatography to give alkylated amino acid ester G-5.
- DIAD diisopropylazodicarboxylate
- Step 9-4 To a solution of G-5 (1.0 eq) in DMF (75 mL), was added solid sodium carbonate (5.0 eq), followed by 2-mercaptoethanol (5.0 eq). The mixture was stirred for 16-18 h at room temperature. Reaction was monitored by HPLC (Grad_A4). Water (150 mL) was added and the aqueous phase extracted with EtOAc (3 x 75 mL). The combined organic phases were washed with brine (1 x 100mL), dried over MgSO 4 , filtered and concentrated under reduced pressure. The crude residue was purified by flash chromatography to give debetsylated amino acid ester G-6. It is important to make sure no benzothiazole:mercaptoethanol adduct is left as determined by LC-MS after the reaction as this side product has been shown to inhibit the subsequent hydrogenolysis reaction.
- Step 9-5 This step is described for approximately 15 mmol scale.
- a round bottom flask under nitrogen atmosphere was charged with 10% Pd/C (50% wet, 10% by weight) and 95% EtOH (70 mL), followed by a solution of G-6 (15 mmol, 1.0 eq) in EtOH 95% (70 mL).
- the mixture was stirred under hydrogen bubbling using a gas dispersion tube for 4 hours. A precipitate formed in the mixture after 2 h. EtOAc or THF (100 mL) was added to obtain a stirring suspension.
- Step 9-6 For a salt of the amino acid benzyl ester ( G-8, 1 eq, described for 15 mmol scale), the solid was taken up in an aqueous solution of 1 M Na 2 CO 3 (50 mL). The basic aqueous phase was extracted with EtOAc (4 x 50 mL) and the combined organic phases were washed with brine (1 x 600 mL), dried over MgSO 4 , filtered, concentrated under reduced pressure and dried under high vacuum 4-6 h to give the free amino-ester G-9. Step 9-7.
- the organic phase was washed sequentially with an aqueous solution of citrate buffer (1 M, pH 3.5) (2 x 50 mL), H 2 O (1 x 50 mL), an aqueous solution of saturated NaHCO 3 (2 x 50 mL) and brine (1 x 50 mL).
- the organic phase was dried over MgSO 4 , filtered, concentrated under reduced pressure and dried under high vacuum 16-18 h to give crude dipeptide.
- Step 9-8 To a solution of acid fragment G-7 (1.0 eq) and dipeptide hydrochloride salt G-11 (1.05 eq) in anhydrous THF/DCM (1:1) (10 mL, described for 1.50 mmol) at 0°C was added DIPEA (5.0 eq), followed by HATU (1.05 eq). The mixture was stirred 1 h at 0°C, allowed to warm to room temperature and stirred 16-18 h under nitrogen. Solvent was removed under reduced pressure and the residue dissolved in EtOAc (50 mL).
- the organic phase was washed sequentially with an aqueous solution of citrate buffer (1 M, pH 3.5) (2 x 25 mL), an aqueous solution of saturated NaHCO 3 (2 x 25 mL) and brine (1 x 25 mL).
- the organic phase was dried over MgSO 4 , filtered and concentrated under reduced pressure.
- the crude residue was purified by flash chromatography to give the desired protected alkylated tripeptide G12.
- Step 9-9 A round bottom flask under a nitrogen atmosphere was charged with 10% Pd/C (50% wet, 10% weight) and 95% EtOH 95% (5 mL, described for 1.30 mmol), followed by a solution of G-12 (1.0 eq) in 95% EtOH (8 mL). The mixture was stirred under hydrogen bubbling for 4 h. The reaction mixture turned very thick after 2-3 h. THF was added to dissolve, at least partially, the suspension. The mixture was purged with nitrogen for 5 min to remove the excess of hydrogen, filtered on a Celite ® (World Minerals Inc., Santa Barbara, CA) pad and washed with THF until there was no further material eluting. The solvent was evaporated under reduced pressure and the resulting solid dried under high vacuum to give acid G-13.
- Step 9-10 Boc protected G-13 (1.0 eq, described for 1.30 mmol) was dissolved in TFA/TES/DCM (33/3/64) (9.8 eq, approximately 3 mL). The mixture was stirred 30 min at room temperature and evaporated under reduced pressure. The residue was co-evaporated twice with THF, then dried under high vacuum for 16-18 h to give macrocyclic precursor G-14.
- Step 9-11 To a solution of G-14 (1.0 eq, described for 1.30 mmol) in anhydrous THF (sufficient to form a 25 mM solution, although solutions of 10 mM concentration are also acceptable) were added DIPEA (5.0 eq) and DEPBT (1.1 eq). The mixture was stirred at room temperature 16-18 h. THF was evaporated under reduced pressure and the residue was taken up in 1 M Na 2 CO 3 (aq):EtOAc (1:1) (50 mL). The separated basic aqueous phase was washed with brine (2 x 25 mL), dried over MgSO 4 , filtered and evaporated under reduced pressure. The crude residue was purified by flash chromatography or reverse phase HPLC to give the macrocycle G-15.
- Table 13 below lists the yields of this final macrocyclization step for other representative compounds of the invention that have been synthesized according to this standard procedure.
- Table 13 Representative Yields for Step 9-11 Compound Macrocyclization Yield a Concentration 513 73% 25 mM 554 75% 25 mM 555 78% 25 mM 557 76% 25 mM 558 72% 25 mM 559 83% 10 mM 560 73% 10 mM 561 55% 10 mM 562 87% 10 mM a Yield determined after silica gel purification, based on linear precursor prior Boc deprotection.
- Step 10-1 Chlorination of H-(D)Tyr-OMe.
- Step 10-2 Double Bts protection. To a solution of 10-2, HCl salt in DCM (50 mL) was added pyridine (5.8 mL, 68 mmol, 6.8 eq) and Et 3 N (3.0 mL, 22 mmol, 2.2 eq), followed by solid Bts-Cl (9.3 g, 40 mmol, 4 eq). The mixture was then stirred for 16 h at rt. It was diluted with DCM (100 mL), washed sequentially with citrate buffer (2 x 50 mL), saturated aqueous sodium bicarbonate (2 x 50 mL), brine once then dried on MgSO 4 and concentrated.
- Step 10-3 Fukuyama-Mitsunobu.
- Boc-T38(R) 2.2 g, 7.2 mmol, 1.4 eq
- PPh 3 1.9 g, 7.2 mmol, 1.4 eq
- DIAD 1.5 mL, 7.2 mmol, 1.4 eq
- Step 10-4 Debetsylation.
- sodium carbonate 3.3 g, 31 mmol, 5 eq
- 2-mercaptoethanol 2.2 mL, 31 mmol, 5 eq
- the mixture was then stirred for 16 h under N 2 , then DMF was evaporated and the mixture was diluted with water (200 mL) and pH was adjusted to 8.
- the aqueous layer was extracted with EtOAc (3 x 100 mL). The combined organic layers were washed with brine, then dried with MgSO 4 and concentrated under reduced pressure.
- Step 10-5 Ester hydrolysis.
- Step 10-6 Fragment coupling.
- 10-6 (1.94 g, 3.8 mmol, 1.0 eq) and 10-7 (synthesized as described in Step 10-9, 1.24 g, 4.6 mmol, 1.2 eq) in DMF (20 mL) were added HATU (1.75 g, 4.6 mmol, 1.2 eq) and DIPEA (4.0 ml, 23 mmol, 5 eq.).
- the mixture was stirred for 16 h at rt under N 2 , then the solvent evaporated under reduced pressure.
- the residue was taken up in water (20 mL), the pH adjusted to 8-9 and the product extracted with EtOAc (2 x 20 mL) and DCM (2 x 20 mL).
- Step 10-7 Deprotections. To 10-8 (1.5 g, 2.0 mmol) was added a mixture of TFA (70 mL), DCM (30 mL) and Et 3 SiH (3 mL). The mixture was stirred at rt for 2 h, then concentrated under reduced pressure. It was evaporated twice with DCM, then dried in vacuo. The crude residue, 10-9, was azeotroped with THF (2x) prior to macrocyclization in Step X-8.
- Step 10-8 Macrocyclization.
- DEPBT 1.2 g, 4.0 mmol, 2 eq
- DIPEA 4.2 mL, 24 mmol, 6 eq
- THF was removed and the product purified by flash chromatography (100% EtOAc) to give compound 552 as a white foam (0.80 g, 68.3% for two steps).
- Similar yields (70.9%) and purity (89.3/91.1/100, UV, CLND, ELSD) were obtained by modifying the above procedure to replace the flash chromatography with passage through a silica gel pad.
- LC-MS UV, CLND, ELSD
- 92.9/100/98.6; t R 5.84 min; (M+H) + 586.
- Compound 552 was dissolved in acetonitrile (60 mg.mL) and 1.5 eq. 0.5 M HCl (aq) added to immediately form a solid precipitate. The solid was collected by filtration and washed with acetonitrile. The resulting HCl salt of compound 552 can be recrystallized one or more times from 95% EtOH using heptane to induce solid formation.
- Step 11-1 In a round bottom flask equipped with a Dean-Stark apparatus, was suspended H-(D)Phe(3F)-OH ( 11-1 , 35.6 g, 194 mmol, 1.0 eq) in toluene (1 L). para Toluenesulfonic acid (44.3 g, 233 mmol, 1.2 eq), benzyl alcohol (101 mL, 970 mmol, 5.0 eq) were added and the mixture was stirred at reflux 16-18 hours. The mixture was cooled down to room temperature and a white precipitate formed. The precipitate was diluted with MTBE (500 mL), filtered and triturated with MTBE (3 x 500 mL). The solid was dried under high vacuum for 16-18 h to give the tosylate salt as a white solid (85.1 g, 98.6%).
- the tosylate salt (85.1 g) was taken up in an aqueous solution of 1 M Na 2 CO 3 (210 mL). The basic aqueous phase was extracted with EtOAc (4 x 150 mL), then the combined organic phases were washed with brine (1 x 100 mL), dried over MgSO 4 , filtered, concentrated under reduced pressure and dried under high vacuum to give the free amino-ester 11-2 as a clear oil (49.5 g, 95%).
- Step 11-2 To a solution of freshly free based 11-2 (49.5 g, 181 mmol, 1.0 eq) in DCM (90 mL) was added triethylamine (27.8 mL, 616 mmol, 1.1 eq), pyridine (49.8 mL, 616 mmol, 3.4 eq) and Bts-Cl (84.7 g, 363 mmol, 2.0 eq). The reaction was stirred O/N at room temperature during which time the solution turned orange. The reaction mixture was diluted with DCM (100mL), washed sequentially with citrate buffer (2x) and brine (1x), dried over MgSO 4 , filtered and concentrated under reduced pressure.
- Step 11-3 Boc-T38(R) tether ( 11-4, 10.0 g, 32.4 mmol, 1.5 eq), triphenylphosphine (8.5 g, 32.4 mmol, 1.5 eq) and Bts-(D)Phe(3F)-OBn ( 11-3, 10.2 g, 21.6 mmol, 1.0 eq) were dissolved in anhydrous THF (108 mL). The solution was cooled to 0°C in an ice-water bath and diisopropylazodicarboxylate (DIAD, 6.2 mL, 31.3 mmol, 1.45 eq) was added dropwise.
- DIAD diisopropylazodicarboxylate
- Step 11-4 To a solution of alkylated amino acid 11-5 (14.0 g, 18.4 mmol, 1.0 eq) in DMF (74 mL), was added sodium carbonate (9.8 g, 92.0 mmol, 5.0 eq), followed by 2-mercaptoethanol (6.5 mL, 92.0 mmol, 5.0 eq). The mixture was stirred for 16-18 h at room temperature. Reaction was monitored by HPLC [(Grad_A4): 9.69 min (product), 15.37 min (starting material)]. Water (150 mL) was added and the aqueous phase extracted with EtOAc (3 x 75mL).
- Step 11-5 A round bottom flask under nitrogen atmosphere was charged with 10% Pd/C (50% wet, 785 mg, 10% by weight) and 95% EtOH (70 mL), followed by a solution of amino acid ester 11-6 (7.85 g, 13.9 mmol, 1.0 eq) in 95% EtOH (70 mL). The mixture was stirred under hydrogen bubbling using a gas dispersion tube for 4 h. A precipitate formed after 2 h. EtOAc (100 mL) was added to obtain a stirring suspension.
- Step 11-6 Commercially available H-Leu-OBn tosylate salt (11-8, 6.1 g) was taken up in an aqueous solution of 1 M Na 2 CO 3 (50 mL). The basic aqueous phase was extracted with EtOAc (4 x 50 mL) and the combined organic phases were washed with brine (1 x 580 mL), dried over MgSO 4 , filtered, concentrated under reduced pressure and dried under vacuum 4-6 h to give the free amino-ester 11-9 as a pale yellow oil (3.44 g, 100%).
- Step 11-7 Freshly free based H-Leu-OBn ( 11-9, 3.44 g, 15.5 mmol, 1.0 eq) and Boc-(D)Val-OH (11-10, 3.54 g, 16.3 mmol, 1.05 eq) were dissolved in anhydrous THF:CH 2 Cl 2 (1:1, 81 mL). 6-Cl-HOBt (2.63 g, 15.5 mmol, 1.0 eq) and DIPEA (14.2 mL, 81.5 mmol, 5.0 eq) were added. The mixture was cooled to 0°C in an ice-water bath and EDC (3.28 g, 17.1 mmol, 1.1 eq) was added.
- Step 11-8 To a solution of 11-7 (750 mg, 1.58 mmol, 1.0 eq) and 11-11 (591 g, 1.66 mmol, 1.05 eq) in anhydrous THF:DCM (1:1, 10.5 mL) at 0°C was added DIPEA (1.4 mL, 7.9 mmol, 5.0 eq), followed by HATU (631 mg, 1.66 mmol, 1.05 eq). The mixture was stirred 1 h at 0°C, allowed to warm to room temperature and stirred 16-18 h under nitrogen. Solvent was removed under reduced pressure and the residue was dissolved in EtOAc (50 mL).
- Step 11-10 Boc protected alkylated tripeptide 11-13 (911 mg, 1.33 mmol, 1.0 eq) was dissolve in a TFA/TES/DCM (33/3/64, 3.0 mL, 12.9 mmol, 9.8 eq). The mixture was stirred 30 min at room temperature and evaporated under reduced pressure. The oily residue was co-evaporated twice with THF, then dried under high vacuum for 16-18 h to give macrocyclic precursor 11-14 as a pale yellow gummy solid (1.27 g, > 100%).
- TLC: [EtOAc:MeOH (9:1)]: R f baseline (UV, ninhydrin).
- Step 11-11 To a solution of macrocyclic precursor 11-14 [1.27 g, 1.33 mmol (based on quantity obtained after hydrogenolysis), 1.0 eq) in anhydrous THF (53.6 mL, for a 25 mM solution) were added DIPEA (1.17 mL, 6.79 mmol, 5.0 eq) and DEPBT (440 mg, 1.47 mmol, 1.1 eq). The mixture was stirred at room temperature 16-18 h. THF was evaporated under reduced pressure and the residue was taken up in 1 M Na 2 CO 3 :EtOAc (1:1, 50 mL).
- Step 15-1 (Panda, G.; Rao, N. V. Synlett 2004 , 714.) To a stirred suspension of Boc-L-Ser-OH ( 15-1 , 5.0 g, 24.5 mmol, 1.0 eq) in 160 mL of anhydrous DMF at room temperature was added N,N-diisopropylethylamine (DIPEA, 4.25 mL, 24.5 mmol, 1.0 eq).
- DIPEA N,N-diisopropylethylamine
- Step 15-2 To a mixture of the alcohol 15-2 (5.0 g, 20 mmol, 1 eq) and triethylamine (5.60 mL, 40.3 mmol, 2 eq) in 100 mL of anhydrous dichloromethane under N 2 , was added freshly distilled methanesulfonyl chloride (2.35 mL, 30 mmol, 1.5 eq) dropwise over 5 min. The reaction was allowed to warm to room temperature and stirred for 3 h. The mixture was washed sequentially with cold water, 1 M citrate buffer, saturated aqueous sodium bicarbonate, and brine.
- Step 15-3 To a solution of 15-3 (5.0 g, 15.3 mmol, 1 eq) in 60 ml of anhydrous DMF was added sodium azide (3.0 g, 46 mmol, 3 eq) under an argon atmosphere and the mixture stirred at 50°C for 12 h. ( CAUTION! Such quantities of NaN 3 should be handled carefully . ). The reaction mixture was cooled to 0°C and water (100 mL) added very slowly. The reaction mixture was extracted with ethyl acetate (3 x 100 mL).
- Such amino macrocycles can also be synthesized by assembly with appropriately protected Dap derivatives using Example 9 or the solid phase procedure illustrated in Figure 7 .
- the reaction was cooled to -78°C and an excess of propylene gas was bubbled (over 10 eq) into the solution and the flask sealed. The mixture was stirred at room temperature O/N. The reaction was cooled to 0°C, propylene was released slowly, and Cu was filtered through Celite ® (World Minerals Inc., Santa Barbara, CA) and washed with acetonitrile. All solvents were removed under reduced pressure. The resulting residue was purified by purified by flash chromatography (ethyl acetate:methanol, 95:05) to give 526 (76% yield).
- Step 25-1 To a solution of 17-2 (1.0 g, 1.73 mmol, 1 eq) in DCM (10 mL) at room temperature was added Bts-Cl (0.41 g, 1.73 mmol, 1 eq), and triethylamine (0.175g, 1.73 mmol, 1 eq). The mixture was stirred at room temperature O/N. The reaction mixture was washed with 0.1 N HCl, dried over magnesium sulfate and concentrated to dryness under reduced pressure. The resulting residue was purified by flash chromatography (EtOAc:hexanes, 1:1) to give 25-1 (95% yield).
- Step 25-2 To a solution of 25-1 (50 mg, 64.5 ⁇ mol, 1 eq) in THF (10 mL) at room temperature was added methanol (26 ⁇ L, 0.645 mmol, 10 eq), the pre-formed triphenylphosphine-DIAD adduct (synthesized as described in Example 38, 32 mg, 0.071 mmol, 1.1 eq). The mixture was stirred at room temperature O/N. The reaction mixture was concentrated to dryness under reduced pressure. The residue was purified by flash chromatography (ethyl acetate:hexanes, 1:1) to give 25-2 (98% yield).
- Step 25-3 25-2 (50 mg) was dissolved in ethanol:THF (1:1,2 mL) at room temperature, treated with thiophenol resin (1 g) and agitated on an orbital shaker for 2 h. The resin was filtered off and washed several times with ethanol:THF. The filtrate and washings were removed under reduced pressure and the crude residue subjected to HPLC purification to give 25-3 (50% yield).
- Compound 546 was synthesized from 17-20 using the standard procedure of Example 25 in ⁇ 44% overall yield for the three step sequence.
- Compound 548 was synthesized from 17-13 using the standard procedure of Example 25 in 40% overall yield for the three step sequence.
- Monomethylamino-containing macrocycles are also synthesized using building block Boc-AA4 in solid phase methods as illustrated in Figure 7 or in solution phase procedures as described in Example 9.
- Compound 550 was synthesized from X-13 using the standard procedure of Example 26 in 80% yield.
- Step 27-1 To a solution of 27-1 (synthesized from the corresponding macrocycle containing a free secondary amine and a protected primary amine by treatment with Bts-Cl similarly to as described in Step 11-2 followed by deprotection of the Boc under standard conditions, 3.3 g, 3.63 mmol, 1 eq) in TFA (50 mL, 0.653 mol, 180 eq) at room temperature was added polystyrene (PS) sulfonamide resin (Argonaut Technologies, now part of Biotage AB, Uppsala, Sweden, 1.1 mmol/g,, 16.8 g, 18.2 mmol, 5 eq). The mixture was stirred at 70°C for 2 h. The resin was filtered off and washed with TFA and DCM several times. The filtrate and washings were removed under reduced pressure. The resulting residue was purified by flash chromatography (100% ethyl acetate) to give 27-2 (70% yield).
- PS polysty
- Step 27-2 To a solution of 27-2 (1.2 g, 1.55 mmol, 1 eq) in acetonitrile/TMOF (6 mL/2 mL) at room temperature was added formaldehyde (0.426 ml, 15.5 mmol, 10 eq), NaBH 3 CN (97 mg, 15.5 mmol, 10 eq) and 1 drop of acetic acid. The reaction was stirred at room temperature for 2 h. The solution was cooled to 0°C, then 1 mL of a solution of 25% NaOH slowly added and the reaction stirred for 5 min.
- Step 27-3 To a solution of 27-3 (1.33 g, 1.66 mmol, 1 eq) in DMF (10 mL) at room temperature was added mercaptopropanoic acid (0.9 g, 8.30 mmol, 5 eq), and K 2 CO 3 (1.15 g, 8.30 mmol, 5 eq). The mixture was stirred at room temperature for 12 h. The reaction mixture was diluted with ethyl acetate and water, then extracted with ethyl acetate. The combined organic layers were washed with brine, dried over magnesium sulfate and concentrated to dryness under reduced pressure. The resulting residue was purified by flash chromatography (ethyl acetate:methanol, 9:1) to give 27-4 (80% yield).
- Step 28-1 To a solution of 538 (0.200 g, 0.352 mmol, 1 eq) in DCM (2 mL) at room temperature were added trifluoroacetic anhydride (TFAA, 122 ⁇ L, 0.882 mmol, 2.5 eq) and triethylamine (TEA, 250 ⁇ L, 1.76 mmol, 5 eq). The reaction was stirred at 0°C for 1 h. The mixture was then diluted with 10 mL of dichloromethane and washed with 0.1 N HCl and brine, dried over magnesium sulfate and concentrated to dryness under reduced pressure. The residue was purified by flash chromatography (ethyl acetate:hexanes, 1:1) to give 28-1 (35% yield)
- Step 28-2 To a solution of 28-1 (66 mg, 0.0994 mmol, 1 eq) in ethyl acetate (5 mL) at room temperature was added Pd/C (15 mg, 20% by weight), and 1 drop of acetic acid. The mixture was stirred at room temperature under a hydrogen atmosphere using a balloon O/N. The Pd/C was filtered through Celite ® (World Minerals Inc., Santa Barbara, CA) and washed with ethyl acetate. The filtrate and washings were removed under reduced pressure to provide 28-2.
- Pd/C Celite ® (World Minerals Inc., Santa Barbara, CA)
- Step 28-3 The crude 28-2 was dissolved in acetonitrile/TMOF (6 mL/2 mL) at room temperature and formaldehyde (22 ⁇ L, 0.0995 mmol, 3 eq), NaBH 3 CN (6.15 mg, 0.0298 mmol, 2 eq) and 1 drop of acetic acid were added. The mixture was stirred at room temperature for 2 h. The solution was cooled to 0°C and 1 mL of a solution of 25% NaOH was slowly added and the mixture stirred for 5 min. The aqueous phase was extracted with ethyl acetate (3x), washed with brine, dried over MgSO 4 , filtered and concentrated under reduced pressure.
- Nitrile 33-1 obtained from solid phase synthesis with appropriate amino acid building blocks according to the procedure outlined in Figure 7 , 218 mg crude
- Nitrile 33-1 was dissolved in a solution of 1.25 M HCl in EtOH (25 mL) at 0°C and gaseous HCl was bubbled into the solution for 30 min. The mixture was then stirred at room temperature for 2 h. The solution was evaporated under reduced pressure, then dried under high vacuum. The crude residue was dissolved in a solution of 2 M ammonia in ethanol (25 mL) and the mixture stirred at room temperature O/N. The solution was evaporated under reduced pressure and purified by reverse phase HPLC to give 33-3 (17.4 mg). Removal of the Bts group was achieved as described for Example 31.
- Step 34-1 3-(2-(R) Hydroxyethyl-1-oxy)-2-bromopyridine [T94-2(R)].
- 2-Bromo-3-pyridinol T94-1 2.0 g, 12 mmol, 1 eq
- DMF anhydrous, 50 mL
- Potassium carbonate 1.6 g, 12 mmol, 1 eq
- (R)-methyl carbonate (1 mL, 12 mmol, 1 eq.
- the reaction was then heated to 85°C using an oil-bath for 72 h.
- Step 34-2 3-(2-(R) Hydroxyethyl-1-oxy)-2-(1- tert -butoxycarbonylamino-prop-2-yn-3-yl)pyridine [T94-3(R)].
- T94-2(R) (1 g, 4.3 mmol, 1 eq) and Boc-propargylamine derivative (1.1 g, 6.46 mmol, 1.5 eq) were dissolved in Et 3 N (distilled over CaH 2 ):DMF (1:3, 15 mL) and the reaction mixture was degassed by bubbling argon through the solution.
- Step 34-3 3-(2-(R)Hydroxyethyl-1-oxy)-2-(1- tert -butoxycarbonylaminoprop-3-yl)pyridine [Boc-T94(R)].
- the acetylenic compound T94-3(R) (5.0 g, 16 mmol) was dissolved in EtOH (80 mL), then PtO 2 (4 mol%, 150 mg) was added and the mixture stirred under a hydrogen atmosphere (a balloon full of hydrogen gas was used) O/N.
- Step 35-1 A solution of 3-hydroxypyridine ( T95-1 , 14.0 g, 0.147 mmol) in THF/DMF (315 mL/315 mL) was cooled to 0°C, then potassium tert -butoxide (24.7 g, 221 mmol) added in small portions. The mixture was stirred for 5 min, then MOM-Cl (13.0 mL, 162 mmol) added dropwise over 10 min. A saturated aqueous solution of ammonium chloride was added and the THF removed under reduced pressure. The aqueous phase was then extracted with Et 2 O (3x). The combined organic phases were dried over MgSO 4 , filtered and concentrated under reduced pressure. The crude T95-2 (orange oil) was used as obtained in the next step.
- Step 35-2 A solution of T95-2 (6.0 g, 43 mmol) and TMEDA (7.8 mL, 52 mmol) in THF (175 mL) was cooled to -78°C and agitated with a mechanical stirrer. nBuLi (1.56 M in hexane, 31.4 mL, 49 mmol) was added dropwise. A sticky orange thick syrup was formed and dissolved over time. The solution was stirred for 1 h then I 2 (14.2 g, 56 mmol) was added and the solution stirred another 1 h at -78°C then warmed up to rt O/N. Water was added to the reaction and the THF removed under reduced pressure.
- Step 35-3 To a solution of T95-3 (6.5 g, 24.6 mmol) in DCM (49 mL) at rt was added TFA (49 mL). The mixture was stirred for 2 h at rt. The solvent was removed under reduced pressure and re-treated with the same reagents under the same conditions for an additional hour. The reaction was monitored by 1 H NMR spectroscopy on a worked-up reaction aliquot. A saturated aqueous solution of sodium bicarbonate was added and the aqueous phase extracted with EtOAc (3x). The combined organic phases were dried over MgSO 4 , filtered and concentrated under reduced pressure to give 5.1 g (94%) of T95-4 as a yellowish solid, which was used as such for the following step.
- Step 35-4 To a solution of 3-hydroxy-4-iodopyridine (5.09 g, 23.0 mmol) in DMF (95 mL) were added (R)-propylene carbonate ( T95-A , 2.35 g, 23.0 mmol)) and potassium carbonate (3.18 g, 23.0 mmol). The solution was stirred at 85°C for 72 h. Water was added and the aqueous phase extracted with EtOAc (3x). The combined organic phases were washed with brine (1x) and water (1x), dried over MgSO 4 , filtered and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (100% EtOAc) to give 1.4 g (24%) of T95-4(R) .
- Step 35-5 A solution of iodide T95-4(R) (1.27 g, 4.56 mmol) and Boc-propargylamine (1.06 g, 6.83 mmol) in CH 3 CN/TEA (13.2 mL/5.4 mL) was degassed for 5 min with argon, then Pd(Cl) 2 (PPh 3 ) 2 (95 mg) and Cul (25 mg) were added. The solution was heated at 50°C O/N. The solvent was removed under reduced pressure and the resulting residue was purified by flash chromatography (100% EtOAc) to give 1.23 g (89%) of T95-5(R) .
- T95(R) is used to make compounds of formula I with an (S)-stereocenter as described further in Example 37.
- T95(S) is used to synthesize compounds of formula I with an (R)-stereocenter.
- Step 36-1 To a solution of 4-hydroxypyridine ( T96-1 , 7.0 g, 74 mmol) in CCl 4 (360 mL) at rt was added NBS (26.2 g, 0.147 mol). The solution was stirred for 24 h in the dark (covered with aluminum foil). The mixture was concentrated under reduced pressure and the resulting residue triturated with MeOH, then with acetone to give 18.9 g (100%) of T96-2 .
- Step 36-2 To a solution of T96-2 (8.0 g, 31.3 mmol) in THF (307 mL) at -90°C was added slowly a solution of n-BuLi (1.15 M in hexanes, 71 mL). The mixture was stirred for 40 min, then water (10 eq) was added and the mixture warmed to room temperature. The solvent was removed under reduced pressure and the resulting residue purified by flash chromatography (acetone:MeOH, 10:1) to give 3.22 g (60%) of T 96-3 .
- Step 36-3 T 96-3 (3.22 g, 18.4 mmol), PPh 3 (4.82 g, 18.4 mmol) and the mono-PMB ether of (R)-glycerol ( T96-A , synthesized from the corresponding diol using standard methods, 1.8 g, 9.2 mmol) were dissolved in THF (14 mL). The mixture was cooled to 0°C, then DIAD (3.5 mL, 17.9 mmol) added dropwise maintaining the temperature at 0°C. The solution was stirred at 0°C for 1 h, then at rt O/N. The mixture was concentrated under reduced pressure and the residue purified by flash chromatography (50/50 EtOAc/Hex) to yield T96-4(R) (42%).
- Step 36-4 To a solution of T96-4(R) (1.25 g, 3.56 mmol) in DIPEA (20 mL) was added Boc-propargylamine and the mixture degassed with argon for 10 min. Triphenylphosphine (121 mg), CuI (29 mg) and Pd(Cl) 2 PPH 3 (160 mg) were then added. The solution was stirred at 70°C O/N. The reaction mixture was concentrated under reduced pressure and the resulting residue purified by flash chromatography (gradient, 40% to 90% EtOAc/Hexane) to give 1.3 g (80%) of T96-5(R) .
- Step 36-5 Pd/C (10%, 50% wet, 628 mg) was added to a solution of T96-5(R) (900 mg) and AcOH (1.5 eq) in 95% EtOH (20 mL). The solution was saturated with H 2 at 1300 psi. The mixture was filtered through Celite ® (World Minerals Inc., Santa Barbara, CA) and washed several times with EtOAc, then with MeOH. The combined filtrate and washings were concentrated under reduced pressure and the crude Boc-T96(R) obtained was used in the synthesis of macrocyclic compounds of the invention without any further purification.
- Celite ® World Minerals Inc., Santa Barbara, CA
- T96(R) is used to make compounds of formula I with an (S)-stereocenter.
- T96(S) is used to synthesize compounds of formula I with an (R)-stereocenter.
- Step 37-1 Boc-T95(R) (901 mg, 2.90 mmol), triphenylphosphine (762 mg, 2.90 mmol) and Bts-(D)Phe(3F)-OBn (976 mg, 2.07 mmol) were dissolved in anhydrous THF (10 mL). The solution was cooled to 0°C in an ice-water bath and diisopropylazodicarboxylate (DIAD, 551 ⁇ L) was added dropwise. Once the addition was completed, the mixture was stirred for 1 h at 0°C, then warmed up slowly to room temperature and stirred for an additional 16-18 h under nitrogen.
- DIAD diisopropylazodicarboxylate
- Step 37-2 To a solution of alkylated amino acid 37-1 (778 mg, 1.02 mmol) in DMF (6 mL) was added sodium carbonate (519 mg, 5 eq), followed by 2-mercaptoethanol (346 ⁇ L, 5 eq). The mixture was stirred for 16-18 hours at room temperature. Water was added and the aqueous phase was extracted with EtOAc (3x). The combined organic phases were washed with brine (1x), dried over MgSO 4 , filtered and concentrated under reduced pressure. The crude residue was purified by flash chromatography (gradient 6/3 EtOAc/Hexane to 100% EtOAc) to give 552 mg of 37-2 (96%).
- Step 37-3 Pd/C (10%, 50% wet, 628 mg) was carefully added to a solution of 37-2 (502 mg, 0.888 mmol) and AcOH (2 drops) in 95% EtOH (6 mL). The solution was saturated with hydrogen (H 2 gas bubbled into the reaction mixture for 2 h), then stirred O/N under H 2 atmosphere. The mixture was filtered through Celite ® (World Minerals Inc., Santa Barbara, CA) and washed several times with EtOAc, then with MeOH. The filtrate was concentrated under reduced pressure and the crude 37-3 (94%) thus obtained was used in the next step without any further purification.
- Celite ® World Minerals Inc., Santa Barbara, CA
- Step 37-4 To a solution of the amino acid zwitterion 37-3 (293 mg, 0.617 mmol) and H-D-Val-Nva-OBn hydrochloride salt (422 mg, 1.23 mmol) in anhydrous THF (3.0 mL) at 0°C was added DIPEA (1.1 mL, 6.17 mmol), followed by HATU (469 mg, 1.23 mmol). The mixture was stirred for 16-18 h under nitrogen. The solvent was removed under reduced pressure and the resulting residue dissolved in EtOAc. The organic phase was washed sequentially with an aqueous solution of citrate buffer (1 M, pH 3.5, 2x), a saturated aqueous of sodium bicarbonate (2x) and brine (2x). The organic phase was dried over MgSO 4 , filtered and concentrated under reduced pressure. The residue thus obtained was purified by flash chromatography (80/20 EtOAc /Hex) to give 290 mg of 37-4 (62%).
- Step 37-5 Pd/C (10%, 50% wet, 28 mg) was added to a solution of 37-4 (284 mg, 0.373 mmol) in 95% EtOH (4 mL). The solution was saturated with hydrogen (H 2 gas bubbled into the reaction mixture for 2 h), then stirred O/N under a H 2 atmosphere. The mixture was filtered through Celite ® (World Minerals Inc., Santa Barbara, CA) and washed several times with EtOAc, then with MeOH. The filtrate was concentrated under reduced pressure and the crude 37-5 obtained, 256 mg (100%), was used in the next step without any further purification.
- Celite ® World Minerals Inc., Santa Barbara, CA
- Step 37-6 Boc protected alkylated tripeptide 37-5 (256 mg) was dissolved in a TFA/TES/DCM (33/3/64, 17 mL) mixture. The reaction was stirred I h at room temperature, then concentrated under reduced pressure. The resulting oily residue was co-evaporated with DCM (3x), then THF (3x) and dried under reduced pressure to give macrocyclic precursor 37-6 .
- Diisopropyl azodicarboxylate (DIAD, 1 eq) is added dropwise to a well-stirred solution of triphenylphosphine (1 eq) in THF (0.4 M) at 0°C under nitrogen. After completion of the addition, the mixture is stirred at 0°C for an additional 30 min. The white solid obtained is collected by filtration (use medium sized fritted filters) and washed with cold anhydrous THF until the washes are colorless. Finally, the white precipitate is washed once with anhydrous Et 2 O. The adduct is then dried well in vacuo , and stored under nitrogen. (It is important to store this reagent under anhydrous conditions! The adduct can be used in place of the separately added reagents in Mitsunobu-type reactions.
- Formulations designed to enhance the solubility of drugs often result in both increasing oral bioavailability and clinical efficacy.
- the most popular approaches incorporate the active lipophilic component into inert lipid vehicles ( Christopher J. H. Porter & al. Lipid and lipid-based formulation; optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug. Disc. 2007, 6, 231-246 ; Aungst, B.J. Novel formulation strategies for improving oral bioavailability of drugs with poor membrane permeation or presystemic metabolism. J. Pharm. Sci. 1993, 81, 979-987 ), such as oils ( Burcham, D.L.; Maurin, M.B.; Hausner, E.A.; Huang, S.M.
Abstract
Description
- This application claims the benefit of United States Provisional Patent Application Serial No.
60/825,237, filed September 11, 2006 - The present invention relates to novel conformationally-defined macrocyclic compounds that bind to and/or are functional modulators of the motilin receptor including subtypes, isoforms and/or variants thereof. These macrocyclic compounds possess appropriate pharmacological properties to be useful as therapeutics for a range of disease indications. In particular, these compounds are useful for treatment and prevention of disorders characterized by hypermotilenemia or gastrointestinal hypermotility, including, but not limited to diarrhea, cancer treatment-related diarrhea, cancer-induced diarrhea, chemotherapy-induced diarrhea, radiation enteritis, radiation-induced diarrhea, stress-induced diarrhea, chronic diarrhea, AIDS-related diarrhea, C. difficile associated diarrhea, traveller's diarrhea, diarrhea induced by graph versus host disease, other types of diarrhea, dyspepsia, irritable bowel syndrome, functional gastrointestinal disorders, chemotherapy-induced nausea and vomiting (emesis) and post-operative nausea and vomiting. In addition, the compounds possess utility for the treatment of diseases and disorders characterized by poor stomach or intestinal absorption, such as short bowel syndrome, celiac disease and cachexia. The compounds may also be used to treat inflammatory diseases and disorders of the gastrointestinal tract, such as inflammatory bowel disease, ulcerative colitis, Crohn's disease and pancreatitis.
- A number of peptide hormones are involved in the control of the different functions in the gastrointestinal (GI) tract, including absorption, secretion, blood flow and motility (Mulvihill, S.J.; et al. in Basic and Clinical Endocrinology, 4th edition, Greenspan, F.S.; Baxter, J.D., Eds., Appleton & Lange: Norwalk, CT, 1994, pp 551-570). Since interactions between the brain and GI system are critical to the proper modulation of these functions, these peptides can be produced locally in the GI tract or distally in the CNS.
- One of these peptide hormones, motilin, a linear 22-amino acid peptide, plays a critical regulatory role in the GI physiological system through governing of fasting gastrointestinal motor activity. As such, the peptide is periodically released from the duodenal mucosa during fasting in mammals, including humans. More precisely, motilin exerts a powerful effect on gastric motility through the contraction of gastrointestinal smooth muscle to stimulate gastric emptying, decrease intestinal transit time and initiate phase III of the migrating motor complex (MMC) in the small bowel. (Itoh, Z., Ed., Motilin, Academic Press: San Diego, CA, 1990, ASIN: 0123757304; Itoh, Z. Peptides 1997, 18, 593-608; Nelson, D.K. Dig. Dis. Sci. 1996, 41, 2006-2015; Peeters, T.L.; Vantrappen, G.; Janssens, J. Gastroenterology 1980, 79, 716-719; Itoh, Z.; Sekiguchi, T. Scand. J. Gastroenterol. Suppl. 1983, 82, 121-134; Itoh, Z.; Aizawa, I.; Sekiguchi, T. Clin. Gastroenterol. 1982, 11, 497-521; Luiking, Y.C.; Peeters, T.L.; Stolk, M.F.; Nieuwenhuijs, V.B.; Portincasa, P.; Depoortere, I.; Van Berge Henegouwen, G.P.; Akkermans, L.M.A. Gut 1998, 42, 830-835.)
- Motilin can exert these effects through receptors located predominantly on the human antrum and proximal duodenum, although its receptors are found to some degree along the entire GI tract. (Peeters, T.L.; Bormans, V.; Vantrappen, G. Regul. Pept. 1988, 23, 171-182; Poitras, P.; Miller, P.; Dickner, M.; Mao, Y.K.; Daniel, E.E.; St-Pierre, S.; Trudel, L. Peptides 1996, 17, 701-707; Miller, P.; Trudel, L.; St-Pierre, S.; Takanashi, H.; Poitras, P. Peptides 2000, 21, 283-287; Takeshita E, Matsuura B, Dong M, Miller LJ, Matsui H, Onji M. J. Gastroenterol. 2006, 41, 223-230.) Therefore, motilin hormone is involved in motility of both the upper and lower parts of the GI system. In addition, motilin and its receptors have been found in the CNS and periphery, suggesting a physiological role in the nervous system that has not yet been definitively elucidated. (Peeters, T.L.; Tang, M. Peptides 2007, 28, 625-631; Liu, M.; Dong, L.; Duan, Z.; Zhu, W.-y.; Cui, Y.; Lei, L. J. Med. Colleges PLA 2005, 20, 321-326; Thielemans, L.; Depoortere, I.; Van Assche, G.; Bender, E.; Peeters, T.L. Brain Res. 2001, 895, 119-128; Depoortere, I.; Peeters, T.L. Am. J. Physiol. 1997, 272, G994-G999 and O'Donohue, T.L.; et al. Peptides 1981, 2, 467-477.) Recently, motilin receptors were found to be expressed in Purkinje cells of both human and rat cerebellum. (Chen, H.; Chen, L.; Wang, J.J.; Wei, H.J.; Yung, W.H. NeuroReport 2007, 18, 1345-1349.) Motilin receptors in the brain have been suggested to play a regulatory role in a number of CNS functions, including feeding and drinking behavior, micturition reflex, central and brain stem neuronal modulation, and pituitary hormone secretion (Itoh, Z. Peptides 1997, 18, 593-608; Asakawa, A.; Inui, A.; Momose, K.M.; et al. Peptides 1998, 19, 987-990 and Rosenfeld, D.J.; Garthwaite, T.L. Physiol. Behav. 1987, 39, 753-756). Studies in infants have also indicated a role for motilin in the long-term regulation of energy balance. (Savino, R.; Grassino, E.C.; Fissore, M.F.; et al. Clin. Endocrinol. 2006, 65, 158-162.)
- The recent identification and cloning of the human motilin receptor (Intl. Pat. Appl. Publ.
WO 99/64436 - Two primary avenues have been pursued to discover and develop motilin agonists as therapeutic agents to enhance.motility. (Peeters, T.L. Neurogastroenterol. Motil. 2006, 18, 1-5; Sandham, D.A.; Plannkuche, H.-J. Ann. Rep. Med. Chem. 2006, 41, 211-219.) The first of these, peptidic agonists of the motilin receptor, have clinical application for the treatment of hypomotility disorders, in particular gastroparesis, (Haramura, M.; Tsuzuki, K.; Okamachi, A.; et al. Bioorg. Med. Chem. 2002, 10, 1805-1811;
U.S. Pat. Nos. 5,422,341 ;5,432,261 ;5,459,049 ;5,695,952 ;5,721,353 ;5,734,012 ;6,018,037 ;6,380,158 ;6,420,521 ,6,838,438 ;U.S. Pat. Appl. Publ. 2001/041791 ;2003/176640 ;2004/254345 ;2005/065156 ;2005/080116 ,2005/106146 ;2005/208626 ; Intl. Pat. Appl. Publ.WO 98/42840 WO 01/00830 WO 02/059141 terminal 14 residues of motilin, has shown promising results in early human studies. (Park, M.I.; Ferber, I.; Camilleri, M.; et al. Neurogastroenterol. Motil. 2006, 18, 28-36; Intl. Pat. Appl. Publ.WO 2006/138023 ;WO 2006/138026 ;U.S. Pat. Appl. Publ. 2006/287243 ;2006/293243 .) - The macrolide antibiotic erythromycin has long been known to have stimulation of GI motility as a side effect and, hence, has been utilized as a treatment for gastroparesis. This effect has subsequently been shown to be mediated through interaction at the motilin receptor. (Hasler, W.L.; Heldsinger, A.; Chungal, O.Y. Am. J. Physiol. 1992, 262, G50-G55; Peeters, T.L. ; Weber, F.H., Jr.; Richards, R.D.; McCallum, R.W. Am. J. Gastroenterol. 1993, 88, 485-490.) However, use of erythromycin therapy can be associated with nausea, diarrhea, cramping and abdominal pain and, further, must be limited in duration to avoid development of bacterial resistance. Therefore, as the second major strategy aimed at motilin agonist therapeutics, the development of derivatives of erythromycin (commonly referred to as motilides) which have little or no antibiotic activity, yet maintain the GI stimulatory effects, has been the subject of a considerable number of research efforts. (Faghih, R.; Nellans, H.N.; Platter, J.J. Drugs of the Future 1998, 23, 861-872; Salat, P.; Parikh, V. Ind. J. Pharmacol. 1999, 31, 333-339; Wu, Y.J. Curr. Pharm. Des. 2000, 6, 181-223; Inatomi, N.; Sato, F.; Itoh, Z.; Omura, S. Mode of action of macrolides with motilin agonistic activity - motilides. Macrolide Antibiotics, 2nd edition, Omura, S., ed., Academic Press: San Diego, CA, 2002, pp 501-531;
U.S. Pat. Nos. 4,677,097 ;4,920,102 ;5,008,249 ;5,175,150 ;5,418,224 ;5,470,961 ;5,523,401 ;5,523,418 ;5,538,961 ;5,554,605 ;5,578,579 ;5,658,888 ;5,712,253 ;5,854,407 ;5,912,235 ;5,922,849 ;6,077,943 ;6,084,079 ;6,100,239 ;6,165,985 ;6,403,775 ;6,562,795 ;6,750,205 ;6,939,861 ;6,946,482 ;7,211,568 ;U.S. Pat. Appl. Publ. 2002/025936 ;2002/094962 ;2003/220271 ;2004/138150 ;2004/147461 ;2005/119195 ;2006/270616 ; Intl. Pat. Appl. Publ.WO 01/60833 WO 02/051855 WO 2004/19879 WO 2005/18576 WO 2006/070937 ;WO 2006/127252 .) Generally disappointing results in clinical trials have been observed for such motilides as EM-574 (Satoh, M.; Sakai, T.; Sano, I.; et al. J. Pharmacol. Exp. Ther. 1994, 271, 574-579; Choi, M.G.; Camilleri, M.; Burton, D.D.; Johnson, S.; Edmonds, A. J. Pharmacol. Exp. Ther. 1998, 285, 37-40), ABT-229 (alemcinal, Talley, N.J.; Verlinden, M.; Snape, W.; et al. Aliment. Pharmacol. Ther. 2000, 14, 1653-1661; Talley, N.J.; Verlinden, M.; Geenan, D.J.; et al. Gut 2001, 49, 395-401; Chen, C.L.; Orr, W.C.; Verlinden, M.H.; et al. Aliment. Pharmacol. Ther. 2002, 16, 749-757; Netzer, P.; Schmitt, B.; Inauen, W. Aliment. Pharmacol. Ther. 2002, 16, 1481-1490) and GM-611 (mitemcinal, Peeters, T.L. Curr. Opin. Investig. Drugs. 2001, 2, 555-557; Koga, H.; Takanashi, H.; Itoh, Z.; Omura, S. Drugs of the Future 2002, 27, 255-272; Takanashi, H.; Yogo, K.; Ozaki, K.; Koga, H.; Itoh, Z.; Omura, S. Pharmacology 2007, 79, 137-148; Ozaki, K.I.; Yogo, K.; Sudo, H.; Onoma, M.; Kamei, K.; Akima, M.; Koga, H.; Itoh, Z.; Omura, S.; Takanashi, H. Pharmacology 2007, 79, 223-235; Ozaki, K.; Sudo, H.; Muramatsu, H.; Yogo, K.; Kamei, K.; Koga, H.; Itoh, Z.; Omura, S.; Takanashi, ; McCallum, R.W.; Cynshi, O. Aliment. Pharmacol. Ther. 2007, 26, 107-116), primarily due to issues such as poor bioavailability, chemical instability and tachyphylaxis. (Thielemans, L.; Depoortere, I.; Perret, J.; et al. J. Pharmacol. Exp. Ther. 2005, 313, 1397-1405; Mitselos, A.; Depoortere, I.; Peeters, T.L. Biochem. Pharmacol. 2007, 73, 115-124.) Nonetheless, due to the therapeutic potential of such agents, the search for motilin agonists in this class has continued and, recently, KOS-2187 (Carreras, C.W.; Liu, Y.; Chen, Y.; et al. Gastroenterology 2005, 128, A464; Carreras, C.W.; Burlingame, M.; Carney, J.; et al. Can. J. Gastroenterol. 2005,19, 15) has been described and appears to circumvent many of these problems. A method useful for analyzing the therapeutic efficiency of these types of molecules has also been formulated (U.S. Pat. No. 6,875,576 ;U.S. Pat. Appl. Publ. 2002/192709 ; Intl. Pat. Appl. Publ.WO 02/64092 - Similarly, non-peptide, non-motilide motilin agonists have been reported (
U.S. Pat. No. 7,262,195 ;U.S. Pat. Appl. Publ. No. 2004/152732 ;2005/065156 ; Intl. Pat. Appl. Publ.WO 02/137127 WO 02/92592 WO 2005/027908 ;WO 2005/027637 ; Jap. Pat. Abstr. Publ. No.09249620 - On the other hand, antagonists of the motilin receptor are potentially useful as therapeutic treatments for diseases associated with hypermotilinemia and/or gastrointestinal hypermotility, including diarrhea, cancer treatment-related diarrhea, cancer-induced diarrhea, chemotherapy-induced diarrhea, radiation enteritis, radiation-induced diarrhea, stress-induced diarrhea, chronic diarrhea, AIDS-related diarrhea, C. difficile associated diarrhea, traveller's diarrhea, diarrhea induced by graph versus host disease, other types of diarrhea, dyspepsia, irritable bowel syndrome, functional gastrointestinal disorders, chemotherapy-induced nausea and vomiting (emesis) and post-operative nausea and vomiting. Current treatments for these conditions are ineffective in many cases. Loperamide, an opioid agonist, is useful for milder diarrhea and generally does not work in a high percentage of patients. Octreotide, a somatostatin agonist, is used off-label as a diarrheal treatment, but is relatively expensive, given by injection, and also not effective in many instances. Further, motilin levels have been observed to be elevated in patients with acute diarrhea (Besterman, H.S.; Christofides, N.D.; Welsby, P.D.; et al. Gut 1983, 24, 665-671) and traveler's diarrhea (Besterman, H.S.; Cook, G.C.; Sarson, D.L.; et al. Br. Med. J. 1979,17, 1252-1255).
- Diarrhea is a common and serious side-effect experienced by cancer patients resulting from surgery, bone marrow transplantation, chemotherapy and radiation treatment. (Stem, J.; Ippoliti, C. Sem. Oncol. Nurs. 2003, 19, 11-16; Benson, A.B.,III; Ajani, J.A.; Catalano, R.B.; et al. J. Clin. Oncol. 2004, 22, 2918-2926; O'Brien, B.E.; Kaklamani, V.G.; Benson, A.B.III Clin. Colorectal Canc. 2005, 4, 375-381.) Certain chemotherapeutic regimens, particularly those including fluoropyrimidines and irinotecan, result in chemotherapy-induced diarrhea (CID) rates as high as 50-80%. (Arbuckle, R.B.; Huber, S.L.; Zacker, C. The ; Saltz, L.B. J. Support. Oncol. 2003, 1, 35-46; Goldberg-Arnold, R.J.; Gabrail, N.; Raut, M.; Kim, R.; Sung, J.C.Y.; Zhou, Y. J. Support. Oncol. 2005, 3, 227-232; Sharma, R.; Tobin, P.; Clarke, S.J. Lancet Oncol. 2005, 6, 93-102; Gibson, R.J.; Keefe, D.M.K. Support. .) The implications of CID include increased morbidity and mortality. This presents a significant problem as, in 2001, over 1.4 million individuals in the U.S. were undergoing cancer chemotherapy. A large heterogeneous study of cancer patients at all stages of treatment placed the prevalence of diarrhea at 14%. (M.D. Anderson Symptom Inventory, ). However, for certain types of cancer, the occurrence is higher. In colorectal cancer, for example, more than half of patients experienced diarrhea rated serious (grade 3) or higher. Resulting from tissue damage in the intestine caused by drugs designed to thwart the rapid growth of tumor cells, it also affects the cells lining the intestinal wall. No effective therapy exists for this damage nor for the associated diarrhea.
- In general, from 10-20% of patients experience CID, although for some chemotherapeutic agents the incidence can be as high as 90%. In approximately 20% of patients, the adverse effects are so severe, it requires a halt to or reduction of the treatment regimen and, often, hospitalization. In addition, parenteral nutrition often must be taken due to the inability of patients to take nourishment normally. Hence, this has an effect on the efficacy of the chemotherapy. Indeed, a review of clinical trials in colorectal cancer revealed higher death rates primarily due to gastrointestinal toxicity. (Rothenberg, M.L.; Meropol, N.J.; Poplin, E.A.; VanCutsem, E.; Wadler, S. J. Clin. Onco/. 2001, 19, 3801-3807.) Current pharmacological treatments only work in some patients and are much less effective against the more serious grades of diarrhea. (MacNaughton, W.K. Aliment. Pharmacol. Ther. 2000,14, 523-528).
- Acute radiation enteritis (ARE) or radiation induced intestinal dysfunction occurs in 75% of patients undergoing radiation therapy, typically occurring in the second or third week of therapy. Characterized by abdominal cramping and diarrhea, this is a serious and feared side effect that results in increased overall treatment time as well as reduced quality of life and can even result in death. In 5-15% of patients, the condition becomes chronic. In addition to discomfort, this side effect decreases the therapeutic benefit from radiation treatment by increasing the overall treatment time. (MacNaughton, W.K. Aliment. Pharmacol. Ther. 2000, 14, 523-528; Nguyen, N.P.; Antoine, J.E.; Dutta, S.; Karlsson, U.; Sallah, ; Gwede, C.K. Sem. Nursing Oncol. 2003, 19, 6-10.)
- Indeed, chronic diarrhea can arise as a result of numerous medical conditions. (Schiller, L.R. Curr. Treat. Options Gastroenterol. 2005, 8, 259-266; Spiller, R. Neurogastroenterol. Motil. 2006, 18, 1045-1055.) For example, chronic diarrhea is a common problem for patients with human immunodeficiency virus infection, especially those with advanced disease. This is a debilitating side effect that occurs in 60-90% of AIDS patients. (Cohen, J.; West, A.B.; Bini, E.J. Gastroenterol. Clin. North Am. 2001, 30, 637-664; Oldfield, E.C.,III Rev. Gastroenterol. Disord. 2002, 2, 176-88; Sestak, K.; Curr. HIV Res. 2005, 3, 199-205; Thom, K.; Forrest, G. Curr. Opin. Gastroenterol. 2006, 22, 18-23.) Additionally, psychological factors, such as stress, are known to play a role in adversely affecting the proper functioning of the GI tract. (North, C.S.; Alpers, D.H.; Thompson, S.J.; Spitznagel, E.L. Dig. Dis. Sci. 1996, 41, 633-640; Kamm, M.A. Eur. J. Surg. Suppl. 1998, 583, 37-40 ; Botha, C.; Libby, G. Br. J. Hosp. Med. (Lond.) 2006, 67, 344-349.)
- Traveller's diarrhea affects over 50% of travellers to some destinations, particularly tropical ones, and is estimated to afflict over 11 million individuals annually. Apart from the disruption to business, travel and vacation schedules, this condition is often accompanied by other clinical manifestations such as nausea, vomiting, abdominal pain, fecal urgency, bloody stools, and fever. (Lima, A.A.M. Curr. Opin. Infect. Dis. 2001, 14, 547-552; Al-Abri, S.S.; Beeching, N.J.; Nye, F.J. Lancet Infect. Dis. 2005, 5, 349-360; DuPont, H.L. Gastroenterol. Clin. North Am. 2006, 35, 337-353.)
- Clostridium difficile is the etiological agent responsible for about one-third of cases of antibiotic-associated diarrhea and is estimated to cause a $1 billion annual cost in the U.S. Antibiotic-associated diarrhea is more common in the hospital setting with up to 29% of patients developing the condition, resulting in increased length of stay, increased cost of care, and increased mortality. (Bartlett, J.G. N. Engl. J. Med. 2002, 346, 334-339; Kelly, C.P.; Pothoulakis, C.; LaMont, J.T. N. Engl. J. Med. 1994, 330, 257-262; Kyne, L.; Farrell, R.J.; Kelly, C.P. Gastroenterol. Clin. N. Am. 2001, 30, 753-777; Malnick, S.D.H.; Zimhony, O. Ann. Pharmacother. 2002, 36, 1767-1775; Hull, M.W.; Beck, P.L. Can. Fam. Phys. 2004, 50, 1536-1540; Schroeder, M.S. Am. Fam. Phys. 2005, 71, 921-928; Voth, D.E.; Ballard, J.D. Clin. Microbiol. Rev. 2005, 18, 247-263.) It is a serious condition with a mortality rate as high as 25% in frail elderly patients. Recently, the incidence and severity of C. difficile-associated diarrhea (CADD) has begun to increase dramatically. (Frost. F:; Craun, G.F.; Calderon, R.L. Emerg. Infect. Dis. 1998, 4, 619-625; Olfield, E.C. Rev. Gastroenterol. Disord. 2006, 6, 79-96.)
- Diarrhea is also induced in patients with graft versus host disease (GVHD). GVHD is a common, potentially life-threatening complication of allogenic hematopoietic stem cell transplantation. Gastrointestinal GVHD frequently involves the colon and complicates management of these seriously ill patients. (Flowers, M.E.; Kansu, E.; Sullivan, K.M. Hematol Oncol Clin North Am. 1999, 13, 1091-1112; Ross, W.A.; Couriel, D. Curr. Opin. Gastroenterol. 2005, 21, 64-69.) In addition, diarrhea is a common side effect after other types of transplantation with an incidence ranging from 10% to 43%. Diarrhea is also a frequent side effect of immunosuppressive medications. (Ginsburg, P.M.; Thuluvath, P.J. Liver Transpl. 2005, 11, 881-890.)
- Irritable bowel syndrome (IBS) is the most common functional GI disorder with an estimated worldwide prevalence of 10-15%. (Saito, Y.A.; Schoenfeld, P.; Locke, G.R. Am. J. Gastroenterol. 2002, 97, 1910-1915; Gilkin, R.J., Jr. Clin. Ther. 2005, 27, 1696-1709; Lacy, B.E.; De Lee, R. J. Clin. Gastroenterol. 2005, 39, S230-S242; Talley, N.J. Intern. Med. J. 2006, 36, 724-728; Ohman, L.; Simren, M. Dig. Liver Dis. 2007, 39, 201-215.) The total annual cost attributable to IBS is estimated to be $30 billion, including $10 billion in direct costs from physician visits and prescription pharmaceuticals, as well as a significant cost from missed work days. (Talley, N.J.; Gabriel, S.E.; Harmsen, W.S.; et al. Gastroenterology 1995, 109, 1736-1741; Maxion-Bergemann, S.; Thielecke, F.; Abel, F.; Bergemann, R. Pharmacoeconomics 2006, 24, 21-37.) IBS patients are sub-classified into diarrhea-predominant (IBS-d), constipation-predominant (IBS-c) or those alternating between these two patterns (IBS-m). Treatments for these various subsets generally must be approached with separate and specific therapies. Antispasmodics, tricyclic antidepressants, selective serotonin reuptake inhibitors, laxatives, antidiarrheals, and bulking agents have not proven to be widely effective and tend to treat symptoms, rather than underlying pathophysiology. (Schoenfeld, P. Gastroenterol. Clin. North Am. 2005, 34, 319-335; Cremonini, F.; Talley, N.J. Nat. Clin. Pract. Gastroenterol. Hepatol. 2005, 2, 82-88; Andersen, V.; Camilleri, M. Drugs 2006, 66, 1073-1088.) The plasma levels of motilin have been shown to be elevated in patients with IBS. (Simren, M.; Bjornsson, E.S.; Abrahamsson, H. Neurogastroenterol. Motil. 2005, 17, 51-57.) Motilin antagonists, hence, would be a useful treatment for patients with IBS. They would likely be more suited to IBS-d and to a lesser extent, IBS-m. IBS-d is manifested by fecal urgency and frequent loose bowel movements (> 3 per day). Individuals suffering from IBS-d account for approximately one-third of the entire IBS patient population.
- Another extremely common GI disorder, dyspepsia, is characterized by chronic or recurrent upper GI distress with no obvious physical cause. (Tack, J.; Bisschops, R.; Sarnelli, G. Gastroenterology 2004, 127, 1239-1255; Kleibeuker, J.H.; Thijs, J.C. Curr.. Opin. Gastroenterol. 2004, 20, 546-550; Talley, N.J.; Vakil, N.; et al. Am. J. Gastroenterol. 2005, 100, 2324-2337; Talley, N.J.; Vakil, N.; Moayyedi, P. Gastroenterology 2005, 129, 1756-1780; Smith, M.L. Dig. Liver Dis. 2005, 37, 547-558; Saad, R.J.; Chey, W.D. Aliment. Pharmacol. Ther. 2006, 24, 475-492; Suzuki, H.; Nishizawa, T.; Hibi, T. J. Gastroenterol. 2006, 41, 513-523.; Mahadeva, S.; Goh, K.L. World J. Gastroenterol. 2006, 12, 2661-2666; Monkemuller K, Malfertheiner P. World J. Gastroenterol. 2006, 12, 2694-2700; Mizuta, Y.; Shikuwa, S.; Isomoto, H.; Mishima, R.; Akazawa, Y.; Masuda, J.; Omagari, K.; Takeshima, F.; Kohno, S. J. Gastroenterol. 2006, 41, 1025-1040; Chua, A.S. World J. Gastroenterol. 2006, 12, 2656-2659.) Typical symptoms include gastric fullness, bloating, pain, nausea and vomiting. This disease has prevalence as high as 20% annually in Western countries. It accounts for up to 5% of all visits to primary care physicians and 30% of visits to GI specialists. As with IBS, the patient population can be categorized into various subsets based upon symptoms. (Choung, R.S.; Locke, G.R. III; Schleck, C.D.; Zinsmetister, A.R.; Talley, N.J. Am. J. Gastroenterol. 2007, 102, 1983-1989.) However, the largest patient subset (up to 60%) suffers from dyspepsia with no known organic cause, otherwise known as "functional dyspepsia (FD)." FD has a major impact on quality of life and health care resources. In analogy with IBS, no widely-accepted therapy for the treatment of FD currently exists. (Stanghellini, V.; De Ponti, F.; De Giorgio, R.; et al. Drugs 2003, 63, 869-892; Cremonini, F.; Delgado-Aros, S.; Talley, N.J. Best Pract. Res. Clin. Gastroenterol. 2004, 18, 717-733; McNally, M.A.; Talley, N.J. Curr. Treatment Opt. Gastroenterol. 2007, 10, 157-168.) Circulating plasma motilin levels are also seen to be raised in patients suffering from dyspepsia. (Kusano, M.; Sekiguchi, T.; Kawamura, O.; Kikuchi, K.; Miyazaki, M.; Tsunoda, T.; Horikoshi, T.; Mori, M. Am. J. Gastroenterol. 1997, 92, 481-484; Kamerling, I.M.; Van Haarst, A.D.; Burggraaf, J.; Schoemaker, R.C.; Biemond, I.; Heinzerling, H.; Jones, R.; Cohen, A.F.; Masclee, A.A. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G776-G781.) As with IBS, motilin antagonists would mitigate the effects of motilin in such patients.
- Chemotherapy-induced nausea and vomiting (CINV), or emesis, is one of the most severe adverse effects resulting from cancer treatment and is often cited as the side effect most feared by patients. From 70-80% of patients receiving cancer chemotherapy experience CINV. In addition to a significant deterioration in quality of life, this condition often requires modification or delay of chemotherapeutic regimens with concomitant negative impact on the effectiveness of treatment. Despite recent progress in the development and availability of new approaches to mitigating the effects of CINV, there remains a compelling need for alternative strategies for patients for whom current treatments are inadequate. (Lindley, C.M.; Hirsch, J.D.; O'Neill, C.V.; Transau, M.D.; Gilbert, C.S.; Osterhaus, J.T. Qual. Life Res. 1992, 1, 331-340; Martin, M. Oncology 1996, 53, 26-31; Kovac, A.L. Drug Safety 2003, 26, 227-259; Grunberg, S.M. J. Support. Oncol. 2004, 2, 1-12; Jordan, K.; Kasper, C.; Schmoll, H.-J. Eur. J. Canc. 2005, 41, 199-205; Herrstedt, J.; Dombemowsky, P. Basic Clin. Pharmacol. Toxicol. 2007, 101, 143-150.)
- Post-operative nausea and vomiting (PONV) is a common complication from surgery, occurring in 30-50% of patients. PONV can lead to unintended or extended hospitalization, electrolyte abnormalities and strain on surgical sutures, plus a substantial negative effect on quality of life. As such, it increases health care costs and decreases patient satisfaction. The importance of dealing with PONV has become well-recognized in the medical community and there is a need for effective treatments. (Osoba, D.; Zee, B.; Warr, D.; et al. Support. ; Kovac, A.L. ; Gan, T.J. J. Am. Med. Assoc. 2002, 287, 1233-1236; Tramèr, M.R. Best Pract. Res. Clin. Anaesthesiol. 2004, 18, 693-701; Habib, A.S.; Gan, T.J. Can. J. Anesth. 2004, 51, 326-341; Golembiewski, J.; Chernin, E.; Chopra, T. Am. J. Health-Syst. Pharm. 2005, 62, 1247-1260.)
- In addition to its actions on motility, motilin is involved in inducing secretion within the gastrointestinal tract. Motilin plays a role in gastric and pancreatic secretion in dogs. (Konturek, S.J.; Dembinski, A.; Krol, R.; Wunsch, E. Scand. J. Gastroenterol. 1976, 11, 57-61; Magee, D.F.; Naruse, S. J. Physiol. 1984, 355, 441-447 ; Lee, K.L.; Shiratori, K.; Chen, Y.F.; Chang, T.-M.; Chey, W.Y. Am. J. Physiol. 1986, 14, G759-764.) Similarly, intestinal secretagogue activity in humans has been described for [Nle13]-motilin. (Kachel, G.W.; Frase, L.L.; Domschke, W.; Chey, W.Y.; Krejs, G.J. Gastroenterology 1984, 87, 550-556.) Hence, an antagonist of the motilin receptor may show anti-secretory effects and play a role as an anti-diarrheal agent. Anti-secretory agents have proven to be effective anti-diarrheal therapeutics. (Farthing, M.J.G. Exp. Opin. Invest. ; Farthing, M.J.G. Dig. Dis. 2006, 24, 47-58.) This dual role, as both an anti-motility and an anti-secretory agent would make motilin antagonists even more effective therapeutics for the treatment of diarrheal conditions.
- In addition to treatment of disorders characterized by hypermotility, the use of motilin antagonists would also be useful in the treatment of diseases and disorders characterized by poor stomach or intestinal absorption. A motilin antagonist would slow gastrointestinal motility thereby permitting longer GI exposure time for absorption of necessary nutrients. Such diseases and disorders include celiac disease, a chronic disorder afflicting almost 1% of the population. Celiac disease is a GI disorder characterized by inflammation, leading to injury to the mucosal lining of the small intestine. The inflammation results when gliadin, a protein found in gluten-containing foods, is ingested by genetically susceptible individuals. The mucosal damage and subsequent malabsorption of nutrients can lead to numerous complications. (Alaedini, A.; Green, P.H.R. Ann. Intern. Med. 2005, 142, 289-298; Koning, F. Gastroenterology 2005, 129, 1294-1301; Chand, N.; Mihas, A.A. J. Clin. Gastroenterol. 2006, 40, 3-14 ; Westerberg, D.P.; Gill, J.M.; Dave, B.; et al. J. Am. Osteopath. Assn. 2006, 106, 145-151; Jones, R.B.; Robins, G.G.; Howdle, P.D. Curr. Opin. Gastroenterol. 2006, 22, 117-123; Green, P.H.R.; Jabri, B. Ann. Rev. Med. 2006, 57, 207-221; Hill, I.D. Curr. Treat. Options Gastroenterol. 2006, 9, 399-408.) The only current treatment is modification to a gluten-free diet.
- Short bowel syndrome is a medical condition that occurs after resection of a substantial portion of small intestine and is characterized by malnutrition. (Misiakos, E.P.; Macheras, A.; Kapetanakis, T.; Liakakos, T. J. Clin. Gastroenterol. 2007, 41, 5-18.; Buchman, A.L. Gastroenterology. 2006, 130 (Suppl. 1), S5-S15; Jackson, C.; Buchman, A.L. Curr. Gastroenterol. Rep. 2005, 7, 373-378; Scolapio, J.S. Curr. Opin. Gastroenterol. 2004, 20, 143-145; Buchman, A.L.; Scolapio, J.; Fryer, J. Gastroenterology 2003, 124, 1111-1134; Westergaard, H. Sem. Gastrointest. Dis. 2002, 13, 210-220.) The syndrome is particularly distressing in children, where mortality and morbidity are very high. (Vanderhoof, J.A.; Young, R.J.; Thompson, J.S. ; Vanderhoof, J.A. J. Ped Gastroenterol. Nutri. 2004, 39, 5768-5771; Sukhotnik, I.; Coran, A.G.; et al. Pediatr. Surg. Int. 2005, 21, 947- 953.) No current pharmacological agents are currently approved for SBS, which is typically treated through intestinal adaptation or rehabilitation in order to improve nutritional status of SBS patients. (DiBaise, J.K.; Young, R.J.; Vanderhoof, J.A. Am. J. Gastroenterol. 2004, 99, 1823-1832.)
- Additionally, the potential for improving nutrient absorption through the use of motilin antagonists could be useful in the treatment of cachexia, a wasting disorder common in serious illnesses such as cancer, AIDS, chronic heart failure and other cardiovascular diseases, and renal disease, as well as in the aged. Cancer cachexia is a therapeutic condition characterized by weight loss and muscle wasting and afflicts approximately 50% of all cancer patients and is the main cause of death in more than 20% of patients. Additionally, this condition has been shown to be a strong independent risk factor for mortality. (Kern, K.A.; Norton, J.A. ; Tisdale, M.J. J. Natl. Cancer Inst. 1997, 89, 1763-1773; Gagnon, B.; Bruera, E. Drugs 1998, 55, 675-688; Inui, A. CA Cancer J. Clin. 2002, 52, 72-91.) Likewise, patients suffering from chronic heart failure are at serious risk from a similar wasting syndrome. (Springer ,J.; Filippatos, G.; Akashi, Y.J.; Anker, S.D. Curr. Opin. Cardiol. 2006, 21, 229-233; Akashi, Y.J.; Springer, J.; Anker, S.D. Curr. Heart Fail. Rep. 2005, 2, 198-203; Anker, S.D.; Steinborn, W.; Strassburg, S. Ann. Med. 2004, 36, 518-529.) This condition also affects an increasing proportion of the elderly. (Morley, J.E. J. Gerontology, Ser. A: Biol. Sci. Med. Sci. 2003, 58A, 131-137.)
- Additionally, the association between intestinal inflammation and altered intestinal motility is well-established. Further, motilin has been implicated in inflammatory disorders of the GI system. Elevated motilin levels have been observed in patients with inflammatory bowel disease (Besterman, H.S.; Mallinson, C.N.; et al. Scand. J. Gastroenterol. 1983, 18, 845-852), ulcerative colitis (Greenberg, G.R.; Buchan, A.M.; McLeod, R.S.; Preston, P.; Cohen, Z. ) and chronic pancreatitis (Besterman, H.S.; Adrian, T.E.; Bloom, S.R. et al. Digestion 1982, 24, 195-208).. In addition, increased motilin and mRNA expression have been found in a rabbit colitis model (Depoortere, I.; Van Assche, G.; Peeters, T.L. Neurogastroenterol. Motil. 2001, 13, 55-63.) Increased plasma motilin concentrations were also obtained in patients after intestinal resection (Besterman, H.S.; Adrian, T.E..; Mallinson, C.N. Gut 1982, 23, 854-861) and ileostomy (Kennedy, H.J.; Sarson, D.L.; Bloom, S.R.; et al. Digestion 1982, 24, 133-136). It has further been shown that non-steriodal anti-inflammatory drugs can induce hypermolitinemia, disturb the interdigestive migrating motor complex, and contribute to the formation of gastric ulcers. (Narita, T.; Okabe, N.; Hane, M.; et al. J. Vet. Pharmacol. Ther. 2006, 29, 569-577.) Therefore, motilin antagonists may be used as anti-inflammatory agents, in particular for use in the GI tract, with beneficial properties relative to existing treatments.
- Despite the potential offered by motilin antagonists as a novel approach to treat hypermotility and malabsorption disorders, efforts have lagged those directed at agonists. A variety of peptidic compounds have been described as antagonists of the motilin receptor [(ANQ-11125: Peeters, T.L.; Depoortere, I.; Macielag, M.J.; Marvin, M.S.; Florance, J.R.; Galdes, A. Biochem. Biophys. Res. Comm. 1994, 198, 411-416); (OHM-11526: Farrugia, G.; Macielag, M.J.; Peeters, T.L.; Sarr, M.G.; Galdes, A.; Szurszewski, J.H. Am. J. Physiol. 1997, 273, G404-G412; Depoortere, I.; Macielag, M.J.; Galdes, A.; Peeters, T.L. Eur. J. Pharmacol. 1995, 286, 241-247); (MA-2029: Mitselos, A.; Depoortere, I.; Peeters, T.L. Biochem. Pharmacol. 2007, 73, 115-124); Poitras, P.; Miller, P.; Gagnon, D.; St-Pierre, S. Biochem. Biophys. Res. Comm. 1994, 205, 449-454;
U.S. Pat. Nos. 5,470,830 ;6,255,285 ;6,586,630 ;6,720,433 ;U.S. Pat. Appl. Publ. 2003/176643 ; Intl. Pat. Appl. Publ.WO 99/09053 WO 00/17231 WO 00/44770 WO 02/64623 - Cyclization of peptidic derivatives is a method that can be employed to improve the properties of a linear peptide both with respect to metabolic stability and conformational freedom. Cyclic molecules tend to be more resistant to metabolic enzymes. Such cyclic peptide motilin antagonists have been reported, highlighted by GM-109. (Takanashi, H.; Yogo, K.; Ozaki, M.; Akima, M.; Koga, H.; Nabata, H. J. Pharm. Exp. Ther. 1995, 273, 624-628; Haramura, M.; Okamachi, A.; Tsuzuki, K.; Yogo, K.; Ikuta, M.; Kozono, T.; Takanashi, H.; Murayama, E. Chem. Pharm. Bull. 2001, 49, 40-43; Haramura, M.; Okamachi, A.; Tsuzuki, K.; Yogo, K.; Ikuta, M.; Kozono, T.; Takanashi, H.; Murayama, E. J. Med. Chem. 2002, 45, 670-675;
U.S. Patent Nos. 7,018,981 ;U.S. Pat. Appl. Publ. 2003/191053 ; Intl. Pat. Appl. Publ.WO 02/16404 07138284 - Macrocyclic peptidomimetics have been previously described as antagonists of the motilin receptor and their uses for the treatment of a variety of GI disorders and to modulate the migrating motor complex summarized. (Intl. Pat. Appl. Publ.
WO 2004/111077 ;U.S. Pat. Appl. Publ. 2005/054562 ;U.S. Prov. Pat. Appl. 60/938,655 ;U.S. Prov. Pat. Appl. 60/939,280 ; Marsault, E.; Hoveyda, H.R.; Peterson, M.L.; Saint-Louis, C.; Landry, A.; Vézina, M.; Ouellet, L.; Wang, Z.; Ramaseshan, M.; Beaubien, S.; Benakli, K.; Beauchemin, S.; Déziel, R.; Peeters, T.; Fraser, G.L. J. Med. Chem. 2006, 49, 7190-7197; Marsault, E.; Benakli, K.; Beaubein, S.; Saint-Louis, C.; Déziel, R.; Fraser, G. Bioorg Med. Chem. Lett. 2007, 17, 4187-4190.) These peptidomimetic macrocyclic motilin antagonists are distinguished from the aforementioned cyclic peptide motilin antagonists in that it was found that such peptidic derivatives containing D-amino acids were devoid of activity. In contrast, for the tripeptidomimetic compounds of the present invention, the D-stereochemistry is beneficial for two of the three building elements. Further, the tether portion of the molecule provides a non-peptidic component and, hence, distinct structures. - The peptidomimetic macrocycles of the present invention are demonstrated to have binding and functional activity at the motilin receptor. Although binding potency and target affinity are factors in drug discovery and development, also important for development of viable pharmaceutical agents are optimization of pharmacokinetic (PK) and pharmacodynamic (PD) parameters. A focus area for research in the pharmaceutical industry has been to better understand the underlying factors which determine the suitability of molecules in this manner, often colloquially termed its "drug-likeness." (Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Adv. Drug Delivery Rev. 1997, 23, 3-25; Muegge, I. Med. Res. Rev. 2003, 23, 302-321; Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. J. Med. Chem. 2002, 45, 2615-2623.) For example, molecular weight, log P, membrane permeability, the number of hydrogen bond donors and acceptors, total polar surface area (TPSA), and the number of rotatable bonds have all been correlated with compounds that have been successful in drug development. Additionally, experimental measurements of plasma protein binding, interaction with cytochrome P450 enzymes, and pharmacokinetic parameters are employed in the pharmaceutical industry to select and advance new drug candidates.
- However, these parameters have not been widely explored or reported within the macrocyclic structural class. This creates tremendous challenges in drug development for such molecules. The macrocyclic compounds of the present invention have been found to possess desirable pharmacological characteristics, while maintaining sufficient binding affinity and selectivity for the motilin receptor, as illustrated in the Examples. These combined characteristics make them more suitable for development as pharmaceutical agents.
- Other motilin antagonists, which are non-peptidic and non-macrocyclic in nature have also been reported. [(RWJ-68023: Beavers, M.P.; Gunnet, J.W.; Hageman, W.; Miller, W.; Moore, J.B.; Zhou, L.; Chen, R.H.K.; Xiang, A.; Urbanski, M.; Combs, D.W.; Mayo, K.H.; Demarest, K.T. Drug Design Disc. 2001, 17, 243-251); Johnson, S.G.; Gunnet, J.W.; Moore, J.B.; et al. Bioorg. Med. Chem. Lett. 2006, 16, 3362-3366;
U.S. Patent Nos. 5,972,939 ;6,384,031 ;6,392,040 ;6,423,714 ;6,511,980 ;6,624,165 ;6,667,309 ;6,967,199 ;U.S. Pat. Appl. Publ. 2001/041701 ;2001/056106 ,2002/002192 ;2002/013352 ;2002/103238 ;2002/111484 ;2003/203906 ;2005/148584 ;2007/054888 ; Intl. Pat. Appl. Publ.WO 99/21846 WO 01/68620 WO 01/68621 WO 01/68622 WO 01/85694 - Indeed, neither of the previously well-studied motilin antagonists, RWJ-68023 and GM-109, possessed the preferred full profile for a potential pharmaceutical product targeting this receptor including high binding affinity and functional activity at the motilin receptor, good Caco-2 membrane permeability, appropriate pharmacokinetic profile [reasonable plasma half-life (t1/2) and clearance values (ClT)], oral bioavailability, sufficient solubility to facilitate formulation and in vivo efficacy. In contrast, the macrocyclic motilin antagonists of the present invention have been found to possess at least some, if not all, of these favorable characteristics.
- The present invention provides novel conformationally-defined macrocyclic compounds with improved pharmacological properties. These compounds can function as antagonists of the motilin receptor.
- According to aspects of the present invention, the present invention is directed to compounds of formula I:


- Ar is selected from the group consisting of:

R2 is selected from the group consisting of lower alkyl, substituted lower alkyl, cycloalkyl and substituted cycloalkyl;
R3, R4, R5, and R6 are independently selected from the group consisting of hydrogen, lower alkyl and substituted lower alkyl;
R7 is selected from the group consisting of hydrogen, lower alkyl, hydroxy and amino;
R10a and R10b are independently selected from the group consisting of hydrogen, lower alkyl and substituted lower alkyl;
X1, X2, X6, X7, X8 and X9 are independently selected from the group consisting of hydrogen, halogen, trifluoromethyl and lower alkyl;
X3, X4 and X5 are independently selected from the group consisting of hydrogen, hydroxyl, alkoxy, halogen, trifluoromethyl and lower alkyl;
L1, L2, L3 and L4 are independently selected from the group consisting of CH and N; with the proviso that the total number of nitrogens in the ring must be 0, 1, 2 or 3;
L5 and L6 are independently selected from the group consisting of O, CR8aR8b and NR9; wherein R8a and R8b are independently selected from the group consisting of hydrogen and lower alkyl; and R9 is selected from the group consisting of hydrogen, lower alkyl, formyl, acyl and sulfonyl. - In particular aspects of the invention, Ar of formula I is selected from:



- In another aspect of the invention, R1 of formula I is selected from methyl, ethyl, isopropyl and cyclopropyl.
- In other aspects of the invention, R2 of formula I is selected from (CH2)mCH3, (CH2)n1R11, CH2N3, (CH2)n2NR12R13, (CH2)n3C(=NR14)NR15R16, (CH2)n4NR17C(=NR18)NR19R20, and (CH2)n5NHC(=O)NR21R22 wherein m is 0, 1, 2, or 3; n1, n2, n3, n4 and n5 are independently 1, 2, 3 or 4; R11, R13, R16, R17, R20, R21 and R22 are independently selected from hydrogen and lower alkyl; R12, R15 and R19 are independently selected from hydrogen, lower alkyl, carboxyalkyl, carboxyaryl and sulfonyl; R14 and R18 are independently selected from hydrogen, lower alkyl, carboxyalkyl, carboxyaryl sulfonyl and cyano.
- In additional aspects of the invention, R2 of formula I is selected from:



- In another aspect of the invention, L1, L2, L3 and L4 in formula I are each CH, L5 is O and L6 is CH2.
- In other aspects of the invention, R3, R4, R5 and R6 in formula I are each hydrogen; or R3 is methyl and R4, R5 and R6 are each hydrogen; or R3 is hydroxymethyl and R4, R5 and R6 are each hydrogen; or R3, R4 and R6 are each hydrogen and R5 is methyl; or R3 and R5 are each methyl and R4 and R6 are each hydrogen.
- In yet another aspect, Y in formula I is selected from:

- In an additional aspect, the present invention relates to compounds of formula II:





R40, R41, R42, and R43 are independently selected from the group consisting of hydrogen, lower alkyl and substituted lower alkyl;
R44 is selected from the group consisting of hydrogen, alkyl, acyl, carboxyalkyl, carboxyaryl, sulfonyl and a protecting group for an amine functional group;
R45 is selected from the group consisting of hydrogen and alkyl;
R46 is selected from the group consisting of hydrogen, alkyl, acyl, sulfonyl and a protecting group for a hydroxy functional group;
L11, L12, L13 and L14 are independently selected from the group consisting of CH and N; with the proviso that the total number of nitrogens in the ring must be 0, 1, 2 or 3;
L15 and L16 are independently selected from the group consisting of O, CR47R48 and NR49; wherein R47 and R48 are independently selected from the group consisting of hydrogen and lower alkyl; and R49 is selected from the group consisting of hydrogen, lower alkyl, formyl, acyl and sulfonyl. - Compounds of formula III can be used in another aspect of the invention to provide compounds of formula I. Compounds of formula III can further be used to provide macrocyclic compounds including a building block structure, such as an amino acid building block structure, and the compounds of formula III.
- In another aspect, the present invention also relates to methods of using the compounds as described herein and as used for the preparation of a medicament for prevention and/or treatment of the disorders described herein.
- The foregoing and other aspects of the present invention are explained in greater detail in the specification set forth below.
-
-
Figure 1 shows a synthetic scheme for a representative amino acid building block of the invention. -
Figure 2 shows a synthetic scheme for another representative amino acid building block of the invention. -
Figure 3 shows a synthetic scheme for a representative building block of the invention. -
Figure 4 shows a synthetic scheme for another representative building block of the invention. -
Figure 5 shows a synthetic scheme for a representative tether building block of the invention. -
Figure 6 shows a synthetic scheme for another representative tether building block of the invention -
Figure 7 shows a general scheme for the solid phase synthesis of macrocyclic compounds of the present invention. -
Figure 8 shows a general scheme for the solution phase synthesis of macrocyclic compounds of the present invention of the invention. -
Figure 9 shows a synthetic scheme for a representative compound of the present invention. -
Figure 10 shows a synthetic scheme for another representative compound of the present invention. -
Figure 11 shows a synthetic scheme for another representative amino acid building block of the invention. -
Figure 12 shows a synthetic scheme for representative compounds of the present invention. -
Figure 13 presents graphs depicting pharmacokinetic parameters for exemplary compounds of the present invention, specifically after intravenous administration of 2.0 mg/kg compound 502 with cyclodextrin (panel A), after subcutaneous injection of 2.0 mg/kg compound 502 without cyclodextrin (panel B), after intravenous administration of 1.9 mg/kg compound 552 with cyclodextrin (panel C), after subcutaneous administration of 2.0 mg/kg compound 552 without cyclodextrin (panel D), after oral administration of 8.0 mg/kg compound 552 with cyclodextrin (panel E), after intravenous administration of 2.0 mg/kg compound 563 with cyclodextrin (panel F), and after oral administration of 7.7 mg/kg compound 563 with cyclodextrin (panel G). -
Figure 14 shows a synthetic scheme for a representative tether building block of the invention. -
Figure 15 shows a synthetic scheme for another representative tether building block of the invention. -
Figure 16 shows a synthetic scheme for another representative tether building block of the invention. -
Figure 17 shows a synthetic scheme for a representative compound of the present invention. -
Figure 18 presents graphs depicting the effect of a representative compound of the present invention on motilin-induced contractions in shrew colon (panel A) and [Nle13]-motilin-induced contractions in shrew colon (panel B). -
Figure 19 presents a graph depicting the effect of a representative compound of the present invention in a dog model of chemotherapy-induced diarrhea. - The foregoing and other aspects of the present invention will now be described in more detail with respect to other embodiments described herein. It should be appreciated that the invention can be embodied in different forms and should not be construed as limited to the embodiments set forth herein. Rather, these embodiments are provided so that this disclosure will be thorough and complete, and will fully convey the scope of the invention to those skilled in the art.
- The terminology used in the description of the invention herein is for the purpose of describing particular embodiments only and is not intended to be limiting of the invention. As used in the description of the invention and the appended claims, the singular forms "a", "an" and "the" are intended to include the plural forms as well, unless the context clearly indicates otherwise. Additionally, as used herein, the term "and/or" includes any and all combinations of one or more of the associated listed items and may be abbreviated as "/".
- Unless otherwise defined, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs.
- All publications, U.S. patent applications, U.S. patents and other references cited herein are incorporated by reference in their entireties.
- The term "alkyl" refers to straight or branched chain saturated or partially unsaturated hydrocarbon groups having from 1 to 20 carbon atoms, in some
instances 1 to 8 carbon atoms. The term "lower alkyl" refers to alkyl groups containing 1 to 6 carbon atoms. Examples of alkyl groups include, but are not limited to, methyl, ethyl, isopropyl, tert-butyl, 3-hexenyl, and 2-butynyl. By "unsaturated" is meant the presence of 1, 2 or 3 double or triple bonds, or a combination of the two. Such alkyl groups may also be optionally substituted as described below. - When a subscript is used with reference to an alkyl or other hydrocarbon group defined herein, the subscript refers to the number of carbon atoms that the group may contain. For example, C2-C4 alkyl indicates an alkyl group with 2, 3 or 4 carbon atoms.
- The term "cycloalkyl" refers to saturated or partially unsaturated cyclic hydrocarbon groups having from 3 to 15 carbon atoms in the ring, in some
instances 3 to 7, and to alkyl groups containing said cyclic hydrocarbon groups. Examples of cycloalkyl groups include, but are not limited to, cyclopropyl, cyclopropylmethyl, cyclopentyl, 2-(cyclohexyl)ethyl, cycloheptyl, and cyclohexenyl. Cycloalkyl as defined herein also includes groups with multiple carbon rings, each of which may be saturated or partially unsaturated, for example decalinyl, [2.2.1]-bicycloheptanyl or adamantanyl. All such cycloalkyl groups may also be optionally substituted as described below. - The term "aromatic" refers to an unsaturated cyclic hydrocarbon group having a conjugated pi electron system that contains 4n+2 electrons where n is an integer greater than or equal to 1. Aromatic molecules are typically stable and are depicted as a planar ring of atoms with resonance structures that consist of alternating double and single bonds, for example benzene or naphthalene.
- The term "aryl" refers to an aromatic group in a single or fused carbocyclic ring system having from 6 to 15 ring atoms, in some
instances 6 to 10, and to alkyl groups containing said aromatic groups. Examples of aryl groups include, but are not limited to, phenyl, 1-naphthyl, 2-naphthyl and benzyl. Aryl as defined herein also includes groups with multiple aryl rings which may be fused, as in naphthyl and anthracenyl, or unfused, as in biphenyl and terphenyl. Aryl also refers to bicyclic or tricyclic carbon rings, where one of the rings is aromatic and the others of which may be saturated, partially unsaturated or aromatic, for example, indanyl or tetrahydronaphthyl (tetralinyl). All such aryl groups may also be optionally substituted as described below. - The term "heterocycle" or "heterocyclic" refers to saturated or partially unsaturated monocyclic, bicyclic or tricyclic groups having from 3 to 15 atoms, in some
instances 3 to 7, with at least one heteroatom in at least one of the rings, said heteroatom being selected from O, S or N. Each ring of the heterocyclic group can contain one or two O atoms, one or two S atoms, one to four N atoms, provided that the total number of heteroatoms in each ring is four or less and each ring contains at least one carbon atom. The fused rings completing the bicyclic or tricyclic heterocyclic groups may contain only carbon atoms and may be saturated or partially unsaturated. The N and S atoms may optionally be oxidized and the N atoms may optionally be quatemized. Heterocyclic also refers to alkyl groups containing said monocyclic, bicyclic or tricyclic heterocyclic groups. Examples of heterocyclic rings include, but are not limited to, 2- or 3-piperidinyl, 2- or 3-piperazinyl, 2- or 3-morpholinyl. All such heterocyclic groups may also be optionally substituted as described below - The term "heteroaryl" refers to an aromatic group in a single or fused ring system having from 5 to 15 ring atoms, in some
instances 5 to 10, which have at least one heteroatom in at least one of the rings, said heteroatom being selected from O, S or N. Each ring of the heteroaryl group can contain one or two O atoms, one or two S atoms, one to four N atoms, provided that the total number of heteroatoms in each ring is four or less and each ring contains at least one carbon atom. The fused rings completing the bicyclic or tricyclic groups may contain only carbon atoms and may be saturated, partially unsaturated or aromatic. In structures where the lone pair of electrons of a nitrogen atom is not involved in completing the aromatic pi electron system, the N atoms may optionally be quaternized or oxidized to the N-oxide. Heteroaryl also refers to alkyl groups containing said cyclic groups. Examples of monocyclic heteroaryl groups include, but are not limited to pyrrolyl, pyrazolyl, pyrazolinyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, thiadiazolyl, isothiazolyl, furanyl, thienyl, oxadiazolyl, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, and triazinyl. Examples of bicyclic heteroaryl groups include, but are not limited to indolyl, benzothiazolyl, benzoxazolyl, benzothienyl, quinolinyl, tetrahydroisoquinolinyl, isoquinolinyl, benzimidazolyl, benzopyranyl, indolizinyl, benzofuranyl, isobenzofuranyl, chromonyl, coumarinyl, benzopyranyl, cinnolinyl, quinoxalinyl, indazolyl, purinyl, pyrrolopyridinyl, furopyridinyl, thienopyridinyl, dihydroisoindolyl, and tetrahydroquinolinyl. Examples of tricyclic heteroaryl groups include, but are not limited to carbazolyl, benzindolyl, phenanthrollinyl, acridinyl, phenanthridinyl, and xanthenyl. All such heteroaryl groups may also be optionally substituted as described below. - The term "hydroxy" refers to the group -OH.
- The term "alkoxy" refers to the group -ORa, wherein Ra is alkyl, cycloalkyl or heterocyclic. Examples include, but are not limited to methoxy, ethoxy, tert-butoxy, cyclohexyloxy and tetrahydropyranyloxy.
- The term "aryloxy" refers to the group -ORb wherein Rb is aryl or heteroaryl. Examples include, but are not limited to phenoxy, benzyloxy and 2-naphthyloxy.
- The term "acyl" refers to the group -C(=O)-Rc wherein Rc is alkyl, cycloalkyl, heterocyclic, aryl or heteroaryl. Examples include, but are not limited to, acetyl, benzoyl and furoyl.
- The term "amino acyl" indicates an acyl group that is derived from an amino acid.
- The term "amino" refers to an -NRdRe group wherein Rd and Re are independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, heterocyclic, aryl and heteroaryl. Alternatively, Rd and Re together form a heterocyclic ring of 3 to 8 members, optionally substituted with unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl, unsubstituted heteroaryl, hydroxy, alkoxy, aryloxy, acyl, amino, amido, carboxy, carboxyalkyl, carboxyaryl, mercapto, sulfinyl, sulfonyl, sulfonamido, amidino, carbamoyl, guanidino or ureido, and optionally containing one to three additional heteroatoms selected from O, S or N.
- The term "amido" refers to the group -C(=O)-NRfRg wherein Rf and Rg are independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, heterocyclic, aryl and heteroaryl. Alternatively, Rf and Rg together form a heterocyclic ring of 3 to 8 members, optionally substituted with unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl, unsubstituted heteroaryl, hydroxy, alkoxy, aryloxy, acyl, amino, amido, carboxy, carboxyalkyl, carboxyaryl, mercapto, sulfinyl, sulfonyl, sulfonamido, amidino, carbamoyl, guanidino or ureido, and optionally containing one to three additional heteroatoms selected from O, S or N.
- The term "amidino" refers to the group -C(=NRh)NRiRj wherein Rh is selected from the group consisting of hydrogen, alkyl, cycloalkyl, heterocyclic, aryl and heteroaryl; and Ri and Rj are independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, heterocyclic, aryl and heteroaryl. Alternatively, Ri and Rj together form a heterocyclic ring of 3 to 8 members, optionally substituted with unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl, unsubstituted heteroaryl, hydroxy, alkoxy, aryloxy, acyl, amino, amido, carboxy, carboxyalkyl, carboxyaryl, mercapto, sulfinyl, sulfonyl, sulfonamido, amidino, carbamoyl, guanidino or ureido, and optionally containing one to three additional heteroatoms selected from O, S or N.
- The term "carboxy" refers to the group -CO2H.
- The term "carboxyalkyl" refers to the group -CO2Rk, wherein Rk is alkyl, cycloalkyl or heterocyclic.
- The term "carboxyaryl" refers to the group -CO2Rm, wherein Rm is aryl or heteroaryl.
- The term "cyano" refers to the group -CN.
- The term "formyl" refers to the group -C(=O)H, also denoted -CHO.
- The term "halo," "halogen" or "halide" refers to fluoro, fluorine or fluoride, chloro, chlorine or chloride, bromo, bromine or bromide, and iodo, iodine or iodide, respectively.
- The term "hydroxymethyl" refers to the group -CH2OH.
- The term "mercapto" refers to the group -SRn wherein Rn is hydrogen, alkyl, cycloalkyl, heterocyclic, aryl or heteroaryl.
- The term "nitro" refers to the group -NO2.
- The term "oxo" refers to the bivalent group =O, which is substituted in place of two hydrogen atoms on the same carbon to form a carbonyl group.
- The term "sulfinyl" refers to the group -S(=O)Rp wherein Rp is alkyl, cycloalkyl, heterocyclic, aryl or heteroaryl.
- The term "sulfonyl" refers to the group -S(=O)2-Rq1 wherein Rq1 is alkyl, cycloalkyl, heterocyclic, aryl or heteroaryl.
- The term "trifluoromethyl" refers to the group -CF3.
- The term "aminosulfonyl" refers to the group -NRq2-S(=O)2-Rq3 wherein Rq2 is hydrogen, alkyl, cycloalkyl, heterocyclic, aryl or heteroaryl; and Rq3 is alkyl, cycloalkyl, heterocyclic, aryl or heteroaryl.
- The term "sulfonamido" refers to the group -S(=O)2-NRrRS wherein Rr and Rs are independently selected from the group consisting of hydrogen, alkyl, cycloalkyl, heterocyclic, aryl or heteroaryl. Alternatively, Rr and Rs together form a heterocyclic ring of 3 to 8 members, optionally substituted with unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl, unsubstituted heteroaryl, hydroxy, alkoxy, aryloxy, acyl, amino, amido, carboxy, carboxyalkyl, carboxyaryl, mercapto, sulfinyl, sulfonyl, sulfonamido, amidino, carbamoyl, guanidino or ureido, and optionally containing one to three additional heteroatoms selected from O, S or N.
- The term "carbamoyl" refers to a group of the formula -N(Rt)-C(=O)-ORu wherein Rt is selected from hydrogen, alkyl, cycloalkyl, heterocyclic, aryl or heteroaryl; and Ru is selected from alkyl, cycloalkyl, heterocylic, aryl or heteroaryl.
- The term "guanidino" refers to a group of the formula -N(Rv)-C(=NRw)-NRxRy wherein Rv, Rw, Rx and Ry are independently selected from hydrogen, alkyl, cycloalkyl, heterocyclic, aryl or heteroaryl. Alternatively, Rx and Ry together form a heterocyclic ring or 3 to 8 members, optionally substituted with unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl, unsubstituted heteroaryl, hydroxy, alkoxy, aryloxy, acyl, amino, amido, carboxy, carboxyalkyl, carboxyaryl, mercapto, sulfinyl, sulfonyl, sulfonamido, amidino, carbamoyl, guanidino or ureido, and optionally containing one to three additional heteroatoms selected from O, S or N.
- The term "ureido" refers to a group of the formula -N(Rz)-C(=O)-NRaaRbb wherein Rz, Raa and Rbb are independently selected from hydrogen, alkyl, cycloalkyl, heterocyclic, aryl or heteroaryl. Alternatively, Raa and Rbb together form a heterocyclic ring of 3 to 8 members, optionally substituted with unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl, unsubstituted heteroaryl, hydroxy, alkoxy, aryloxy, acyl, amino, amido, carboxy, carboxyalkyl, carboxyaryl, mercapto, sulfinyl, sulfonyl, sulfonamido, amidino, carbamoyl, guanidino or ureido, and optionally containing one to three additional heteroatoms selected from O, S or N.
- The term "optionally substituted" is intended to expressly indicate that the specified group is unsubstituted or substituted by one or more suitable substituents, unless the optional substituents are expressly specified, in which case the term indicates that the group is unsubstituted or substituted with the specified substituents. As defined above, various groups may be unsubstituted or substituted (i.e., they are optionally substituted) unless indicated otherwise herein (e.g., by indicating that the specified group is unsubstituted).
- The term "substituted" when used with the terms alkyl, cycloalkyl, heterocyclic, aryl and heteroaryl refers to an alkyl, cycloalkyl, heterocyclic, aryl or heteroaryl group having one or more of the hydrogen atoms of the group replaced by substituents independently selected from unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl, unsubstituted heteroaryl, hydroxy, alkoxy, aryloxy, acyl, amino, amido, carboxy, carboxyalkyl, carboxyaryl, halo, oxo, mercapto, sulfinyl, sulfonyl, sulfonamido, amidino, carbamoyl, guanidino, ureido and groups of the formulas -NRccC(=O)Rdd, -NReeC(=NRff)Rgg, -OC(=O)NRhhRii. -OC(=O)Rjj, -OC(=O)ORkk, -NRmmSO2Rnn, or -NRppSO2NRqqPrr wherein Rcc, Rdd, Ree, Rff, Rgg, Rhh, Rii, Rjj , Rmm, Rpp, Rqq and Rrr are independently selected from hydrogen, unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl or unsubstituted heteroaryl; and wherein Rkk and Rnn are independently selected from unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl or unsubstituted heteroaryl. Alternatively, Rgg and Rhh, Rjj and Rkk or Rpp and Rqq together form a heterocyclic ring of 3 to 8 members, optionally substituted with unsubstituted alkyl, unsubstituted cycloalkyl, unsubstituted heterocyclic, unsubstituted aryl, unsubstituted heteroaryl, hydroxy, alkoxy, aryloxy, acyl, amino, amido, carboxy, carboxyalkyl, carboxyaryl, mercapto, sulfinyl, sulfonyl, sulfonamido, amidino, carbamoyl, guanidino or ureido, and optionally containing one to three additional heteroatoms selected from O, S or N. In addition, the term "substituted" for aryl and heteroaryl groups includes as an option having one of the hydrogen atoms of the group replaced by cyano, nitro or trifluoromethyl.
- A substitution is made provided that any atom's normal valency is not exceeded and that the substitution results in a stable compound. Generally, when a substituted form of a group is present, such substituted group is preferably not further substituted or, if substituted, the substituent comprises only a limited number of substituted groups, in some
instances - When any variable occurs more than one time in any constituent or in any formula herein, its definition on each occurrence is independent of its definition at every other occurrence. Also, combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- A "stable compound" or "stable structure" refers to a compound that is sufficiently robust to survive isolation to a useful degree of purity and formulation into an efficacious therapeutic agent.
- The term "amino acid" refers to the common natural (genetically encoded) or synthetic amino acids and common derivatives thereof, known to those skilled in the art. When applied to amino acids, "standard" or "proteinogenic" refers to the genetically encoded 20 amino acids in their natural configuration. Similarly, when applied to amino acids, "unnatural" or "unusual" refers to the wide selection of non-natural, rare or synthetic amino acids such as those described by Hunt, S. in Chemistry and Biochemistry of the Amino Acids, Barrett, G.C., Ed., Chapman and Hall: New York, 1985.
- The term "residue" with reference to an amino acid or amino acid derivative refers to a group of the formula:

- The term "fragment" with respect to a dipeptide, tripeptide or higher order peptide derivative indicates a group that contains two, three or more, respectively, amino acid residues.
- The term "amino acid side chain" refers to any side chain from a standard or unnatural amino acid, and is denoted RAA. For example, the side chain of alanine is methyl, the side chain of valine is isopropyl and the side chain of tryptophan is 3-indolylmethyl.
- The term "agonist" refers to a compound that duplicates at least some of the effect of the endogenous ligand of a protein, receptor, enzyme or the like.
- The term "antagonist" refers to a compound that inhibits at least some of the effect of the endogenous ligand of a protein, receptor, enzyme or the like.
- The term "variant" when applied to a receptor is meant to include dimers, trimers, tetramers, pentamers and other biological complexes containing multiple components. These components can be the same or different.
- The term "peptide" refers to a chemical compound comprised of two or more amino acids covalently bonded together.
- The term "peptidomimetic" refers to a chemical compound designed to mimic a peptide, but which contains structural differences through the addition or replacement of one of more functional groups of the peptide in order to modulate its activity or other properties, such as solubility, metabolic stability, oral bioavailability, lipophilicity, permeability, etc. This can include replacement of the peptide bond, side chain modifications, truncations, additions of functional groups, etc. When the chemical structure is not derived from the peptide, but mimics its activity, it is often referred to as a "non-peptide peptidomimetic."
- The term "peptide bond" refers to the amide [-C(=O)-NH-] functionality with which individual amino acids are typically covalently bonded to each other in a peptide.
- The term "protecting group" refers to any chemical compound that may be used to prevent a potentially reactive functional group, such as an amine, a hydroxyl or a carboxyl, on a molecule from undergoing a chemical reaction while chemical change occurs elsewhere in the molecule. A number of such protecting groups are known to those skilled in the art and examples can be found in "Protective Groups in Organic Synthesis," Theodora W. Greene and Peter G. Wuts, editors, John Wiley & Sons, New York, 3rd edition, 1999 [ISBN 0471160199]. Examples of amino protecting groups include, but are not limited to, phthalimido, trichloroacetyl, benzyloxycarbonyl, tert-butoxycarbonyl, and adamantyloxycarbonyl. In some embodiments, amino protecting groups are carbamate amino protecting groups, which are defined as an amino protecting group that when bound to an amino group forms a carbamate. In other embodiments, amino carbamate protecting groups are allyloxycarbonyl (Alloc), benzyloxycarbonyl (Cbz), 9-fluorenylmethoxycarbonyl (Fmoc), tert-butoxycarbonyl (Boc) and α,α-dimethyl-3,5-dimethoxybenzyloxycarbonyl (Ddz). For a recent discussion of newer nitrogen protecting groups: Theodoridis, . Examples of hydroxyl protecting groups include, but are not limited to, acetyl, tert-butyldimethylsilyl (TBDMS), trityl (Trt), tert-butyl, and tetrahydropyranyl (THP). Examples of carboxyl protecting groups include, but are not limited to methyl ester, tert-butyl ester, benzyl ester, trimethylsilylethyl ester, and 2,2,2-trichloroethyl ester.
- The term "solid phase chemistry" refers to the conduct of chemical reactions where one component of the reaction is covalently bonded to a polymeric material (solid support as defined below). Reaction methods for performing chemistry on solid phase have become more widely known and established outside the traditional fields of peptide and oligonucleotide chemistry.
- The term "solid support," "solid phase" or "resin" refers to a mechanically and chemically stable polymeric matrix utilized to conduct solid phase chemistry. This is denoted by "Resin," "P-" or the following symbol:

- Examples of appropriate polymer materials include, but are not limited to, polystyrene, polyethylene, polyethylene glycol, polyethylene glycol grafted or covalently bonded to polystyrene (also termed PEG-polystyrene, TentaGel™, Rapp, W.; Zhang, L.; Bayer, E. In Innovations and Perspectives in Solid Phase Synthesis. Peptides, Polypeptides and Oligonucleotides; Epton, R., Ed.; SPCC Ltd.: Birmingham, UK; p 205); polyacrylate (CLEAR™), polyacrylamide, polyurethane, PEGA [polyethyleneglycol poly(N,N-dimethylacrylamide) co-polymer, Meldal, M. Tetrahedron Lett. 1992, 33, 3077-3080], cellulose, etc. These materials can optionally contain additional chemical agents to form cross-linked bonds to mechanically stabilize the structure, for example polystyrene cross-linked with divinylbenzene (DVB, usually 0.1-5%, preferably 0.5-2%). This solid support can include as non-limiting examples aminomethyl polystyrene, hydroxymethyl polystyrene, benzhydrylamine polystyrene (BHA), methylbenzhydrylamine (MBHA) polystyrene, and other polymeric backbones containing free chemical functional groups, most typically, -NH2 or -OH, for further derivatization or reaction. The term is also meant to include "Ultraresins" with a high proportion ("loading") of these functional groups such as those prepared from polyethyleneimines and cross-linking molecules (Barth, M.; Rademann, J. J. Comb. Chem. 2004, 6, 340-349). At the conclusion of the synthesis, resins are typically discarded, although they have been shown to be able to be reused such as in Frechet, J.M.J.; Haque, K.E. Tetrahedron Lett. 1975, 16, 3055.
- In general, the materials used as resins are insoluble polymers, but certain polymers have differential solubility depending on solvent and can also be employed for solid phase chemistry. For example, polyethylene glycol can be utilized in this manner since it is soluble in many organic solvents in which chemical reactions can be conducted, but it is insoluble in others, such as diethyl ether. Hence, reactions can be conducted homogeneously in solution, then the product on the polymer precipitated through the addition of diethyl ether and processed as a solid. This has been termed "liquid-phase" chemistry.
- The term "linker" when used in reference to solid phase chemistry refers to a chemical group that is bonded covalently to a solid support and is attached between the support and the substrate typically in order to permit the release (cleavage) of the substrate from the solid support. However, it can also be used to impart stability to the bond to the solid support or merely as a spacer element. Many solid supports are available commercially with linkers already attached.
- Abbreviations used for amino acids and designation of peptides follow the rules of the IUPAC-IUB Commission of Biochemical Nomenclature in J. Biol. Chem. 1972, 247, 977-983. This document has been updated: Biochem. J., 1984, 219, 345-373; Eur. J. Biochem., 1984, 138, 9-37; 1985, 152, 1 ; Internat. J. Pept. Prot. Res., 1984, 24, following p 84; J. Biol. Chem., 1985, 260, 14-42; Pure Appl. Chem., 1984, 56, 595-624; Amino Acids and Peptides, 1985, 16, 387-410; and in Biochemical Nomenclature and Related Documents, 2nd edition, Portland Press, 1992, pp 39-67. Extensions to the rules were published in the JCBN/NC-IUB Newsletter 1985, 1986, 1989; see Biochemical Nomenclature and Related Documents, 2nd edition, Portland Press, 1992, pp 68-69.
- The term "effective amount" or "effective" is intended to designate a dose that causes a relief of symptoms of a disease or disorder as noted through clinical testing and evaluation, patient observation, and/or the like, and/or a dose that causes a detectable change in biological or chemical activity. The detectable changes may be detected and/or further quantified by one skilled in the art for the relevant mechanism or process. As is generally understood in the art, the dosage will vary depending on the administration routes, symptoms and body weight of the patient but also depending upon the compound being administered.
- Administration of two or more compounds "in combination" means that the two compounds are administered closely enough in time that the presence of one alters the biological effects of the other. The two compounds can be administered simultaneously (concurrently) or sequentially. Simultaneous administration can be carried out by mixing the compounds prior to administration, or by administering the compounds at the same point in time but at different anatomic sites or using different routes of administration. The phrases "concurrent administration", "administration in combination", "simultaneous administration" or "administered simultaneously" as used herein, means that the compounds are administered at the same point in time or immediately following one another. In the latter case, the two compounds are administered at times sufficiently close that the results observed are indistinguishable from those achieved when the compounds are administered at the same point in time.
- The term "pharmaceutically active metabolite" is intended to mean a pharmacologically active product produced through metabolism in the body of a specified compound.
- The term "solvate" is intended to mean a pharmaceutically acceptable solvate form of a specified compound that retains the biological effectiveness of such compound. Examples of solvates, without limitation, include compounds of the invention in combination with water, isopropanol, ethanol, methanol, DMSO, ethyl acetate, acetic acid, or ethanolamine.
- The term "simultaneously" or "concurrently" is intended to mean, when applied to a disease or disorder, that the diseases or disorders occur at the same point in time for at least some of the time the subject is suffering from the diseases or disorders, but not necessarily the entire time.
- The term "sequentially" is intended to mean, when applied to a disease or disorder, that the diseases or disorders do not occur at the same point in time, but occur immediately after each other.
- Novel macrocyclic compounds of the present invention include macrocyclic compounds comprising a building block structure including a tether component that undergoes cyclization to form the macrocyclic compound. The building block structure can comprise amino acids (standard and unnatural) and a tether component as described herein. The tether component can be selected from compounds that result in the following structures:





- Macrocyclic compounds of the present invention further include those of formula I:


Ar is selected from the group consisting of:
R2 is selected from the group consisting of lower alkyl, substituted lower alkyl, cycloalkyl and substituted cycloalkyl;
R3, R4, R5, and R6 are independently selected from the group consisting of hydrogen, lower alkyl and substituted lower alkyl;
R7 is selected from the group consisting of hydrogen, lower alkyl, hydroxy and amino;
R10a and R10b are independently selected from the group consisting of hydrogen, lower alkyl and substituted lower alkyl;
X1, X2, X6, X7, X8 and X9 are independently selected from the group consisting of hydrogen, halogen, trifluoromethyl and lower alkyl;
X3, X4 and X5 are independently selected from the group consisting of hydrogen, hydroxyl, alkoxy, halogen, trifluoromethyl and lower alkyl;
L1, L2, L3 and L4 are independently selected from the group consisting of CH and N; with the proviso that the total number of nitrogens in the ring must be 0, 1, 2 or 3;
L5 and L6 are independently selected from the group consisting of O, CR8aR8b and NR9; wherein R8a and R8b are independently selected from the group consisting of hydrogen and lower alkyl; and R9 is selected from the group consisting of hydrogen, lower alkyl, formyl, acyl and sulfonyl. - The present invention includes isolated compounds. An isolated compound refers to a compound that, in some embodiments, comprises at least 10%, at least 25%, at least 50% or at least 70% of the compounds of a mixture. In some embodiments, the compound, pharmaceutically acceptable salt thereof or pharmaceutical composition containing the compound exhibits a statistically significant binding and/or antagonist activity when tested in biological assays at the human motilin receptor.
- In the case of compounds, salts, or solvates that are solids, it is understood by those skilled in the art that the inventive compounds, salts, and solvates may exist in different crystal or polymorphic forms, all of which are intended to be within the scope of the present invention and specified formulas.
- The compounds of formula I disclosed herein have asymmetric centers. The inventive compounds may exist as single stereoisomers, racemates, and/or mixtures of enantiomers and/or diastereomers. All such single stereoisomers, racemates, and mixtures thereof are intended to be within the scope of the present invention. In particular embodiments, however, the inventive compounds are used in optically pure form. The terms "S" and "R" configuration as used herein are as defined by the IUPAC 1974 Recommendations for Section E, Fundamentals of Stereochemistry (Pure Appl. Chem. 1976, 45, 13-30.)
- Unless otherwise depicted to be a specific orientation, the present invention accounts for all stereoisomeric forms. The compounds may be prepared as a single stereoisomer or a mixture of stereoisomers. The non-racemic forms may be obtained by either synthesis or resolution. The compounds may, for example, be resolved into the component enantiomers by standard techniques, for example formation of diastereomeric pairs via salt formation. The compounds also may be resolved by covalently bonding to a chiral moiety. The diastereomers can then be resolved by chromatographic separation and/or crystallographic separation. In the case of a chiral auxiliary moiety, it can then be removed. As an alternative, the compounds can be resolved through the use of chiral chromatography. Enzymatic methods of resolution could also be used in certain cases.
- As generally understood by those skilled in the art, an "optically pure" compound is one that contains only a single enantiomer. As used herein, the term "optically active" is intended to mean a compound comprising at least a sufficient excess of one enantiomer over the other such that the mixture rotates plane polarized light. Optically active compounds have the ability to rotate the plane of polarized light. The excess of one enantiomer over another is typically expressed as enantiomeric excess (e.e.). In describing an optically active compound, the prefixes D and L or R and S are used to denote the absolute configuration of the molecule about its chiral center(s). The prefixes "d" and "I" or (+) and (-) are used to denote the optical rotation of the compound (i.e., the direction in which a plane of polarized light is rotated by the optically active compound). The "I" or (-) prefix indicates that the compound is levorotatory (i.e., rotates the plane of polarized light to the left or counterclockwise) while the "d" or (+) prefix means that the compound is dextrarotatory (i.e., rotates the plane of polarized light to the right or clockwise). The sign of optical rotation, (-) and (+), is not related to the absolute configuration of the molecule, R and S.
- A compound of the invention having the desired pharmacological properties will be optically active and, can be comprised of at least 90% (80% e.e.), at least 95% (90% e.e.), at least 97.5% (95% e.e.) or at least 99% (98% e.e.) of a single isomer.
- Likewise, many geometric isomers of double bonds and the like can also be present in the compounds disclosed herein, and all such stable isomers are included within the present invention unless otherwise specified. Also included in the invention are tautomers and rotamers of formula I, II and/or III.
- The use of the following symbols refers to substitution of one or more hydrogen atoms of the indicated ring with the defined substituent R.

- The use of the following symbol indicates a single bond or an optional double bond:

- The compounds of formula I can be synthesized using traditional solution synthesis techniques or solid phase chemistry methods. Synthetic methods for this general type of macrocyclic structure are described in Intl. Pat. Appls.
WO 01/25257 WO 2004/111077 ,WO 2005/012331 ,WO 2005/012332 ,WO 2006/009645 andWO 2006/009674 , including purification procedures described inWO 2004/111077 andWO 2005/012331 . Representative procedures for compounds of the invention are presented in the Examples. - Reagents and solvents were of reagent quality or better and were used as obtained from various commercial suppliers unless otherwise noted. DMF, DCM (CH2Cl2), DME, CH3CN and THF used are of DriSolv® (EMD Chemicals, Inc., part of Merck KGaA, Darmstadt, Germany) or synthesis grade quality except for (i) deprotection, (ii) resin capping reactions and (iii) washing. NMP used for the amino acid (AA) coupling reactions is of analytical grade. DMF was adequately degassed by placing under vacuum for a minimum of 30 min prior to use. Homogeneous catalysts were obtained from Strem Chemicals, Inc. (Newbury Port, MA, USA). Cbz-, Boc- and Fmoc-protected amino acids and side chain protected derivatives, including those of N-methyl and unnatural amino acids, were obtained from commercial suppliers or synthesized through standard methodologies known to those in the art. Ddz-amino acids were either synthesized by standard methods, or obtained commercially from Orpegen (Heidelberg, Germany) or Advanced ChemTech (Louisville, KY, USA). Bts-amino acids were synthesized by established procedures. Analytical TLC was performed on pre-coated plates of silica gel 60F254 (0.25 mm thickness) containing a fluorescent indicator and were visualized using the method(s) and reagent(s) indicated, for example using ultraviolet light (UV) and/or ceric-molybdic acid (CMA) solution (prepared by mixing 100 mL of sulfuric acid, 10 g ceric ammonium sulfate and 25 g ammonium molybdate).
- The term "concentrated/evaporated/removed under reduced pressure" indicates removal of solvent and volatile components utilizing a rotary evaporator under either water aspirator pressure (typically 10-30 torr) or the stronger vacuum provided by a mechanical oil vacuum pump ("high vacuum," typically ≤1 torr) as appropriate for the solvent being removed. Drying of a compound "in vacuo" or under "high vacuum" refers to drying using an oil vacuum pump at low pressure (≤1 torr). "Flash chromatography" was performed using silica gel 60 (230-400 mesh, EMD Chemicals, Still, W. C.; Kahn, M.; Mitra, A. J. Org. Chem. 1978, 43, 2923-2925) and is a procedure well-known to those in the art. "Dry pack" indicates chromatography on silica gel that has not been pre-treated with solvent, generally applied on larger scales for purifications where a large difference in Rf exists between the desired product and any impurities. For solid phase chemistry processes, "dried in the standard manner" is that the resin is dried first in air (1 h), and subsequently under vacuum (oil pump usually) until full dryness is attained (~30 min to O/N). Glassware used in air and water sensitive reactions were dried in an oven at least O/N and cooled in a desiccator prior to use.
- Amino acids, Boc- and Fmoc-protected amino acids and side chain protected derivatives, including those of N-methyl and unnatural amino acids, were obtained from commercial suppliers [for example: Advanced ChemTech (Louisville, KY, USA), Bachem (Bubendorf, Switzerland), ChemImpex (Wood Dale, IL, USA), Novabiochem (subsidiary of Merck KGaA, Darmstadt, Germany), PepTech (Burlington, MA, USA), Synthetech (Albany, OR, USA), AstaTech (Bristol, PA, USA)] or synthesized through standard methodologies known to those in the art. Ddz-amino acids were either obtained commercially from Orpegen (Heidelberg, Germany) or Advanced ChemTech (Louisville, KY, USA) or synthesized using standard methods utilizing Ddz-OPh or Ddz-N3. (Birr, C.; Lochinger, W.; Stahnke, G.; Lang, P. Justus Liebigs Ann. Chem. 1972, 763, 162-172.) Bts-amino acids were synthesized by known methods. (Vedejs, E.; Lin, S.; Klapara, A.; Wang, J. J. Am. Chem. Soc. 1996, 118, 9796-9797; Vedejs, E.; Kongkittingam, C. J.Org. Chem. 2000, 65, 2309-2318; also Intl. Pat. Appl. Nos.
WO 01/25257 WO 2004/111077 ) N-Alkyl amino acids, in particular N-methyl amino acids, are commercially available from multiple vendors (Bachem, Novabiochem, Advanced ChemTech, ChemImpex, Peptech). In addition, N-alkyl amino acid derivatives were accessed via literature methods. (Hansen, D. W., Jr.; Pilipauskas, D. J. Org. Chem. 1985, 50, 945-950.) Aziridine-2-carboxylic acid was constructed using reported methods from the literature (Baldwin, J. E. et al. Tetrahedron 1993, 49, 6309-6330; Nakajima, K. et al. Bull. Chem. Soc. Jpn. 1978, 51, 1577-1578.) 3-Chlorotyrosine was synthesized using the literature method. (Yu, G.; Mason, H.J.; Galdi, K.; et al. Synthesis 2003, 403-407.) - Other amino acids have been constructed specifically for use in these macrocycles, such as those of formula II:




- Methods for the construction of representative amino acids of this type are provided in the Examples. Some of these amino acids can be considered as mimics of arginine and/or its guanidine moiety (Peterlin-Masic, L.; Kikelj, D. Tetrahedron 2001, 57, 7073 and references cited therein).
- Tethers were obtained from the methods previously described in Intl. Pat. Appl. Nos.
WO 01/25257 WO 2004/111077 ,WO 2005/012331 ,WO 2006/009645 andWO 2006/009674 . Procedures for the synthesis of representative tethers as described herein are also presented in the Examples below. Tethers useful for the compounds of the present invention can include suitable tethers as described in the references cited above as well as known and/or novel tethers described herein. Exemplary tethers include, but are not limited to, the following:





- Specific procedures applicable for the synthesis of exemplary compounds of the invention using these tethers are provided in the Examples.
- Specific solid phase techniques for the synthesis of the macrocyclic compounds of the invention have been described in Intl. Pat. Appl. Publ. Nos.
WO 01/25257 WO 2004/111077 ,WO 2005/012331 andWO 2005/012332 . Solution phase synthesis routes, including methods amenable to larger scale manufacture, were described in Intl. Pat. Appl. Publ. Nos.WO 2006/009645 andWO 2006/009674 . These methods for representative compounds of the present invention are further described in the Examples. Table 1 presents a summary of the synthesis of 90 representative compounds of the present invention. The reaction methodology employed for the construction of the macrocyclic molecule is indicated inColumn 2 and relates to the particular scheme of the synthetic strategy, for example, use of the solid phase strategy as shown inFigure 7 or the solution phase approach as shown inFigure 8 and described in Example 9. Columns 3-6 indicate the individual amino acid and tether building blocks employed for each compound, utilizing either standard nomenclature or referring to the building block designations presented elsewhere in this application. AA1 refers to the building block that forms the -NR10a-CH(CHR7Ar)-C(=O)- portion of the compounds of formula I; AA2 refers to the building block that forms the -NH-CHR1-C(=O)- portion of the compounds of formula I; AA1 refers to the building block that forms the -NH-CHR2-C(=O)- portion of the compounds of formula I; Tether refers to the building block that forms the remaining portion of the compound of formula I that between NR10a and NR10b, typically including NR10a It should be noted that for 1-substituted tethers, for example T38, T90, T91, T92, T93, T94, T95, T96, T97, T98 and T99, the reaction chemistry for its introduction inverts the stereochemistry at that center. Hence, (R)-substituted tether building blocks result in compounds of formula I with an (S)-stereocenter. Likewise, (S)-substituted tether building blocks result in compounds of formula I with an (R)-stereocenter. Tethers T90 and T91 are introduced along with AA1 as fragments F2 and F1 respectively as described in Examples 6 and 5. The relevant deprotection and coupling protocols required for the assembly of the cyclization precursors are performed utilizing standard procedures and those described inWO 2004/111077 ,WO 2006/009645 andWO 2006/009674 as appropriate for the nature of the building block. The final macrocycles are obtained after application of the appropriate deprotection sequences. If any reaction(s) was(were) required to be carried out post-cyclization, they are listed inColumn 7. All of the macrocycles presented in Table 1 were purified and met internal acceptance criteria.Table 1. Synthesis of Representative Compounds of the Invention Compound No.1 Strategy AA1 AA2 AA3 Tether Post-cyclization Reaction(s) 501 Solution phase Bts-(D)Phe(3Cl)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 17, then Example 28 502 Solution phase Bts-(D)Phe(3CF3)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 17, then Example 28 503 Solution phase Bts-(D)Phe(3CF3)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 17, then Example 13 504 Solution phase Bts-(D)Phe(3CF3)-OBn Boc-(D)Val Boc-AA5 Boc-T40(R) Example 17, then Example 13 505 Solution phase Bts-(D)Phe(3CF3)-OBn Boc-(D)Val Boc-AA5 Boc-T40(S) Example 17, then Example 13 506 Solid phase Bts-(D)Phe(3Cl) Boc-(D)Val Boc-Abu(CN) Boc-T9 Example 33 507 Solid phase Bts-(D)Phe(3C1) Ddz-(D)Val Ddz-Dab(Boc) Ddz-T9 Example 17, then Example 13 508 Solution phase Bts-(D)Phe(3Cl)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 17, then Example 28 509 Solid phase Bts-(D)Phe(3Cl) Boc-(D)Val Boc-AA1 Boc-T9 Example 29 510 Solid phase Bts-(D)Phe(3Cl) Boc-(D)Val Boc-AA2 Boc-T9 none 511 Solid phase Bts-(D)Phe(3Cl) Boc-(D)Val Boc-AA3 Boc-T9 none 512 Solution phase Bts-(D)Phe(3Cl)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 17, then Example 25 513 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val H-Nva-OBn Boc-T38(S) none 514 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val Boc-AA5 Boc-T40(R) Example 17, then Example 28 515 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val Boc-AA5 Boc-T40(S) Example 17, then Example 28 516 Solution phase Bts-(D)Phe(3Cl)-OBn Boc-(D)Val Boc-AA5 Boc-T38(R) Example 17, then Example 13 517 Solution phase Bts-(D)Phe(3Cl)-OBn Boc-(D)Val Boc-AA5 Boc-T38(S) Example 17, then Example 13 518 Solid phase Bts-(D)Phe(3Cl) Boc-(D)Val Boc-Ala(CN) Boc-T9 Example 33 519 Solution phase Bts-(D)Phe(3Cl)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 17, then Example 12 520 Solution phase Bts-(D)Phe(3Cl)-OBn Boc-(D)Val H-Cpa-OBn Boc-T39 none 521 Solution phase Bts-(D)Phe(3Cl)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 17, then Example 12 522 Solution phase Bts-(D)Phe(3Cl)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 17, then Example 14 523 Solution phase Bts-(D)Phe(3CF3)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 24 524 Solution phase Bts-(D)Phe(3CF3)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 17, then Example 26 525 Solution phase Bts-(D)Phe(3CF3)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 17, then Example 18 526 Solution phase Bts-(D)Phe(3CF3)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 21 527 Solution phase Bts-(D)Phe(3CF3)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 23 528 Solution phase Bts-(D)Phe(3CF3)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 22 529 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val Boc-AA5 Boc-T38(R) Example 17, then Example 28 530 Solution phase F1 (with Tether) Boc-(D)Val H-Nva-OBn F1 (with AA1) none 531 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val H-Cpa-OBn Boc-T40 none 532 Solution phase Bts-(D)Phe(3CF3)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 17, then Example 19, then Example 20 533 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val Boc-AA5 Boc-T38(R) Example 17, then Example 19, then Example 21 534 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val Boc-AA5 Boc-T38(R) Example 17, then Example 18 535 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val Boc-AA5 Boc-T38(R) Example 17, then Example 26 536 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val Boc-AA5 Boc-T38(R) Example 21 537 Solution phase Bts-(D)Phe(3CF3)-OBn Boc-(D)Val Boc-AA5 Boc-T9 none 538 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val Boc-AA5 Boc-T38(R) none 539 Solution phase Bts-(D)Phe(3CF3)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 17, then Example 19 540 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val Boc-AA5 Boc-T38(R) Example 17, then Example 19 541 Solution phase F2 (with Tether) Boc-(D)Val H-Nva-OBn F2 (with AA.) none 542 Solid phase Bts-(D)Phe(3CF3) Ddz-(D)Val Ddz-Dap(Boc) Ddz-T9 none 543 Solid phase Bts-(D)Phe(3F) Ddz-(D)Val Ddz-Dap(Boc) Ddz-T38(R) none 544 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val Boc-AA5 Boc-T81(1R,8S) Example 17 545 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val Boc-AA5 Boc-T81(1R,8S) none 546 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val ' Boc-AA5 Boc-T81(1R,8S) Example 17, then Example 25 547 Solution phase Bts-(D)Phe(3CF3)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 17, then Example 25 548 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val Boc-AA5 Boc-T38(R) Example 17, then Example 25 549 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val Boc-AA5 Boc-T38(R) Example 17, then Example 28, then methylation under standard conditions 550 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val Boc-AA5 Boc-T81(lR,8S) Example 17, then Example 26 551 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val H-Nva-OBn Boc-T38(R) none 552 Solution phase Bts-(D)Tyr(3C1)(Bts)-OMe Boc-(D)Val H-Nva-OBut Boc-T38(R) none 553 Solution phase Bts-(D)Tyr(3tBu)-OBn Boc-(D)Val H-Nva-OBn Boc-T9 none 554 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val H-Cpa-OBn Boc-T38(R) none 555 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Cpg H-Cpa-OBn Boc-T38(R) none 556 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val H-Leu-OBn Boc-T38(R) none 557 Solution phase BLs-(D)Phe(3F)-OBn Boc-(D)Ala H-Nva-OBn Boc-T38(R) none 558 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Abu H-Nva-OBn Boc-T38(R) none 559 Solution phase Bts-(D)Phe(4F)-OBn Boc-(D)Val H-Cpa-OBn Boc-T38(R) none 560 Solution phase Bts-(D)Phe(4F)-OBn Boc-(D)Val H-Nva-OBn Boc-T38(R) none 561 Solution phase Bts-(D)Phe(3,4diF)-OBn Boc-(D)Val H-Nva-OBn Boc-T38(R) none 562 Solution phase Bts-(D)Phe(30Me)-OBn Boc-(D)Val H-Nva-OBn Boc-T38(R) none 563 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val H-Nva-OBn Boc-T92(R) none 564 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val H-Nva-OBn Boc-T93(R) none 565 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val H-Nva-OBn Boc-T95(R) none 566 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Cpg H-Cpa-OBn Boc-T40(S) none 567 Solution phase Bts-(D)Phe(4F)-OBn Boc-(D)Val H-Cpa-OBn Boc-T40(S) none 568 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val H-Ser(But)-OBn Boc-T38(R) none 569 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val H-Ser(OMe)-OBn Boc-T38(R) none 570 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val H-Nva-OBn Boc-T81(1R,8S) none 571 Solution phase Bts-(D)2Thi-OBn Boc-(D)Val H-Nva.OBn Boc-T38(R) none 572 Solution phase Bts-(D)3Thi-OBn Boc-(D)Val H-Nva-OBn Boc-T38(R) none 573 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val H-Nva-OBn Boc-T94(R) none 574 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val H-Nva-OBn Boc-T96(R) none 575 Solution phase Bts-(D)Phe(4CF3)-OBn Boc-(D)Val H-Nva-OBn Boc-T9 none 576 Solution phase Bts-(D)Phe(4Cl)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 17, then Example 30 577 Solution phase Bts-(D)Phe(4Cl)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 17, then Example 31 578 Solution phase Bts-(D)Tyr(OBut)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 17, then Example 33 579 Solution phase Bts-(D)Tyr(OBut)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 17, then Example 32 580 Solution phase Bts-(D)Phe(4C1)-OBn Boc-(D)Val Boc-AA5 Boc-T9 Example 17, then Example 28 581 Solution phase Bts-(D)Phe(4C1)-OBn Boc-(D)Val Boc-A.AS Boc-T9 Example 17, then Example 32 582 Solid phase Bts-(D)Tyr(3,5diI)-OBn Boc-(D)Val Boc-Nva Boc-T9 none 583 Solution phase Bts-(D)Tyr(OMe)-OBn Boc-(D)Val H-Hse-OBn Boc-T9 none 584 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D) Val H-Cpa-OBn Boc-T9 none 585 Solution phase Bts-(D)Phe(3Cl)-OBn Boc-(D)Val H-Cpa-OBn Boc-T40(S) none 586 Solution phase Bts-(D)Phe(3Cl)-OBn Boc-(D)Val H-Cpa-OBn Boc-T40(R) none 587 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val H-Cpa-OBn Boc-T40(R) none 588 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val H-Cpa-OBn Boc-T40(S) none 589 Solution phase Bts-(D)Phe(3F)-OBn Boc-(D)Val H-Cpa-OBn Boc-T38(S) none 590 Solution phase Bts-(D)Phe(3Cl)-OBn Boc-(D)Val H-Cpa-OBn Boc-T58 none 1 In instances where diastereomeric products were isolated after purification, the individual isomers are denoted with letters (a,b,c) after the compound identifier. - The table directly below presents analytical data obtained for compounds 501-589 (Table 2), as determined by LC-MS analysis of the purified products. These compounds were further examined for their ability to interact at the human motilin receptor utilizing the biological test methods described herein.
Table 2: Analytical Data for Representative Compounds of the Invention Compound No. Molecular Formula MW Calc (g/mol) MS [(M+H)+] Found 501 C30H42N5O4Cl 572.1 572 502 C31H42N5O4F3 605.7 606 503 C30H40N7O4F3 619.7 620 504 C31H42N7O4F3 633.7 634 505 C31H42N7O4F3 633.7 634 506 C30H41N6O4Cl 585.1 585 507 C30H42N7O4Cl 600.2 600 508 C31H44N5O4Cl 586.2 586 509 C31H39N6O4SCl 627.2 627 510 C31H39N6O4Cl 595.1 595 511 C31H39N6O4Cl 595.1 595 512 C29H40N5O4Cl 558.1 558 513 C31H43N4O4F 554.7 555 514 C31H44N5O4F 569.7 570 515 C31H44N5O4F 569.7 570 516 C30H42N7O4Cl 600.2 600 517 C30H42N7O4Cl 600.2 600 518 C29H39N6O4Cl 571.1 571 519 C31H42N7O4Cl 612.2 612 520 C31H43N4O4Cl 571.2 571 521 C32H44N7O4Cl 626.2 626 522 C31H41N8O4Cl 625.2 611 523 C32H41N8O4F3 658.7 659 524 C33H40N7O4F3 655.7 656 525 C33H44N5O5F3 647.7 648 526 C32H40N7O4F3 643.7 644 527 C31H39N8O4F3 644.7 645 528 C33H42N7O4F3 657.7 658 529 C31H44N5O4F 569.7 570 530 C33H47N4O4F 582.7 583 531 C31H43N4O4F 554.7 555 532 C33H44N5O4F3 631.7 632 533 C33H46N5O4F 595.7 596 534 C33H46N5O5F 611.7 612 535 C33H42N7O4F 619.7 620 536 C32H42N7O4F 607.7 608 537 C29H36N7O4F3 603.6 604 538 C29H38N7O4F 567.7 568 539 C33H40N5O4F3 627.7 628 540 C33H42N5O4F 591.7 592 541 C31H43N4O5F 570.7 571 542 C29H38N5O4F3 577.6 578 543 C29H40N5O4F 541.7 542 544 C30H42N5O4 555.7 556 545 C30H40N7O4F 581.7 582 546 C31H44N5O4F 569.7 570 547 C30H40N5O4F3 591.7 592 548 C30H42N5O4F 555.7 556 549 C32H46N5O4F 583.7 584 550 C34H44N7O4F 633.8 634 551 C31H43N4O4F 554.7 555 552 C31H43N4O5Cl 587.1 587 553 C34H50N4O5 594.8 595 554 C32H43N4O4F 566.7 567 555 C32H41N4O4F 564.7 565 556 C32H45N4O4F 568.7 569 557 C29H39N4O4F 526.6 527 558 C30H41N4O4F 540.7 541 559 C32H43N4O4F 566.7 567 560 C31H43N4O4F 554.7 555 561 C31H42N4O4F2 572.7 573 562 C32H46N4O5 566.7 567 563 C31H42N4O4F2 572.7 573 564 C31H42N4O4F2 572.7 573 565 C30H42N5O4F 555.7 556 566 C32H41N4O4F 564.7 565 567 C32H43N4O4F 566.7 567 568 C29H39N4O5F 542.6 543 569 C30H41N4O5F 556.7 557 570 C32H45N4O4F 568.7 569 571 C29H42N4O4S 542.7 543 572 C29H42N4O4S 542.7 543 573 C30H42N5O4F 555.7 556 574 C30H42N5O4F 555.7 556 575 C31H41N4O4F3 590.7 591 576 C29H40N5O6SCl 622.2 622 577 C29H39N6O5Cl 587.1 587 578 C29H41N7O5 567.7 568 579 C31H45N5O5 567.7 568 580 C30H42N5O4Cl 572.1 572 581 C31H44N5O4Cl 586.2 586 582 C30H40N4O5I2 790.5 791 583 C30H42N4O6 554.7 555 584 C31H41N4O4F 552.7 553 585 C32H43N4O4Cl 583.2 583 586 C32H43N4O4Cl 583.2 583 587 C32H43N4O4F 566.7 567 588 C32H43N4O4F 566.7 567 589 C32H43N4O4F 566.7 567 590 C33H45N4O4Cl 597.2 597 Notes 1. Molecular formulas and molecular weights are calculated automatically from the structure via ActivityBase software (ID Business Solutions, Ltd., Guildford, Surrey, UK).
2. [M+H]+ obtained from LC-MS analysis using standard methods.
3. All analyses conducted on material after preparative purification. - 1H and 13C NMR spectra were recorded on a
Varian Mercury 300 MHz spectrometer (Varian, Inc., Palo Alto, CA) and are referenced internally with respect to the residual proton signals of the solvent unless otherwise noted. 1H NMR data are presented, using the standard abbreviations, as follows: chemical shift (δ) in ppm (multiplicity, integration, coupling constant(s)). The following abbreviations are used for denoting signal multiplicity: s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, b or br = broad, and m = multiplet. Information about the conformation of the molecules in solution can be determined utilizing appropriate two-dimensional NMR techniques known to those skilled in the art. (Martin, G.E.; Zektzer, A.S. Two-Dimensional NMR Methods for Establishing Molecular Connectivity: A Chemist's Guide to Experiment Selection, Performance, and Interpretation, John Wiley & Sons: New York, 1988, ISBN 0471187070.) - HPLC analyses were performed on a Waters Alliance® system 2695 running at 1 mL/min using an Xterra® MS C18 column (or comparable) 4.6 x 50 mm (3.5 µm) and the indicated gradient method. A Waters 996 PDA provided UV data for purity assessment (Waters Corporation, Milford, MA). An LCPackings (Dionex Corporation, Sunnyvale, CA) splitter (50:40:10) allowed the flow to be separated in three parts. The first part (50%) was diverted to a mass spectrometer (Micromass® Platform II MS equipped with an APCI probe) for identity confirmation. The second part (40%) went to an evaporative light scattering detector (ELSD, Polymer Laboratories, now part of Varian, Inc., Palo Alto, CA, PL-ELS-1000™) for purity assessment and the last portion (10%) went to a chemiluminescence nitrogen detector (CLND, Antek® Model 8060, Antek Instruments, Houston, TX, part of Roper Industries, Inc., Duluth, GA) for quantitation and purity assessment. Data was captured and processed utilizing the most recent version of the Waters Millennium® software package.
- Enantiomeric and diastereomeric purity were assessed using appropriate chiral HPLC columns using a Waters Breeze system (or comparable). Although other packing materials can be utilized, particularly useful columns for these analyses are: Chiralpak AS-RH and Chiralcel OD-RH (Chiral Technologies, West Chester, PA, USA).
- Preparative HPLC purifications were performed on final deprotected macrocycles using the Waters FractionLynx® system, on an XTerra® MS C18 column (or comparable) 19 x 100 mm (5 µm). The injections were done using an At-Column-Dilution configuration with a Waters 2767 injector/collector and a Waters 515 pump running at 2 mL/min. The mass spectrometer, HPLC, and mass-directed fraction collection are controlled via MassLynx® software version 3.5 with FractionLynx®. Fractions (13 x 125 mm tubes) shown by MS analysis to contain the product were evaporated under reduced pressure, most typically on a centrifugal evaporator system (Genevac® HT-4 (Genevac Inc, Valley Cottage, NY), ThermoSavant Discovery®, SpeedVac® or comparable (Thermo Electron Corporation, Waltham, MA) or, alternatively, lyophilized. Compounds were then thoroughly analyzed by LC-MS-UV-ELSD-CLND analysis for identity confirmation, purity and quantity assessment.
- Automated medium pressure chromatographic purifications were performed on an Isco CombiFlash® 16x system with disposable silica or C18 cartridges that permitted up to sixteen (16) samples to be run simultaneously (Teledyne Isco, Inc., Lincoln, NE). MS spectra were recorded on a Waters Micromass® Platform II or ZQ™ system. HRMS spectra were recorded with a VG Micromass ZAB-ZF spectrometer. Chemical and biological information were stored and analyzed utilizing the ActivityBase® database software (ID Business Solutions Ltd., Guildford, Surrey, UK).
- General methods for the HPLC determination of stereoisomeric purity were employed according to techniques known to those skilled in the art and further optimized for the compounds of the present invention.
-
- 1. Isocratic plateau of 40 min at 35% ACN, 65% of a 50 mM solution of CH3COONH4 in H2O.
- 2. 5 min gradient to 70% ACN, 30% of a 50 mM solution of CH3COONH4 in H2O.
- 3. Isocratic plateau of 10 min at 70% ACN, 30% of a 50 mM solution of CH3COONH4 in H2O.
- 4. 5 min gradient to 35% ACN, 65% of a 50 mM solution of CH3COONH4 in H2O.
- 5. Isocratic plateau of 10 min at 35% ACN, 65% of a 50 mM solution of CH3COONH4 in H2O.
- 6. Flow: 0.5 mL/min
- 7. Column temperature: room temperature
- 8. Sample temperature: room temperature
-
- 1. Isocratic plateau of 40 min at 40% ACN, 60% of a
solution 50 mM of CH3COONH4 in H2O. - 2. 5 min gradient to 70% ACN, 30% of a
solution 50 mM of CH3COONH4 in H2O. - 3. Isocratic plateau of 10 min at 70% ACN, 30% of a
solution 50 mM of CH3COONH4 in H2O. - 4. 5 min gradient to 40% ACN, 60% of a
solution 50 mM of CH3COONH4 in H2O. - 5. Isocratic plateau of 10 min at 40% ACN, 60% of a
solution 50 mM of CH3COONH4 in H2O. - 6. Flow: 0.5 mL/min
- 7. Column temperature: room temperature
- 8. Sample temperature: room temperature
-
- 1. 40 min isocratic 55%/45% of ACN/ 50 mM CH3COONH4 in H2O
- 2. 5 min gradient to 70%/30% of ACN/ 50 mM CH3COONH4 in H2O
- 3. 10 min isocratic 70%/30% of ACN/ 50 mM CH3COONH4 in H2O
- 4. 5 min gradient to 55%/44% of ACN/ 50 mM CH3COONH4 in H2O
- 5. 10 min isocratic 55%/45% of ACN/ 50 mM CH3COONH4 in H2O
- 6. Flow: 0.5 mL/min
- 7. Column temperature: room temperature
- 8. Sample temperature: room temperature
-
- 1. 40 min isocratic 27%/73% of ACN/ 50 mM CH3COONH4 in H2O
- 2. 5 min gradient to 70%/30% of ACN/ 50 mM CH3COONH4 in H2O
- 3. 10 min isocratic 70%/30% of ACN/ 50 mM CH3COONH4 in H2O
- 4. 5 min gradient to 27%/73% of ACN/ 50 mM CH3COONH4 in H2O
- 5. 10 min isocratic 27%/73% of ACN/ 50 mM CH3COONH4 in H2O
- 6. Flow: 0.5 mL/min
- 7. Column temperature: room temperature
- 8. Sample temperature: room temperature
- The compounds of the present invention were evaluated for their ability to interact at the human motilin receptor utilizing a competitive radioligand binding assay, fluorescence assay or Aequorin functional assay as described below. Such methods can be conducted in a high throughput manner to permit the simultaneous evaluation of many compounds.
- Specific assay methods for the motilin receptor and their use in generally identifying agonists and antagonists thereof are known. (Peeters, T.L.; Depoortere, I.; Macielag, M.J.; Marvin, M.S.; Florance, J.R.; Galdes, A. Biochem. Biophys. Res. Comm. 1994, 198, 411-416; Farrugia, G.; Macielag, M.J.; Peeters, T.L.; Sarr, M.G.; ; Galdes, A.; Szurszewski, J.H. Am. J. Physiol. 1997, 273, G404-G412; Depoortere, I.; Macielag, M.J.; Galdes, A.; Peeters, T.L. Eur. J. Pharmacol. 1995, 286, 241-247; Poitras, P.; Miller, P.; Gagnon, D.; St-Pierre, S. Biochem. Biophys. Res. Comm. 1994, 205, 449-454; Haramura, M.; Okamachi, A.; Tsuzuki, K.; Yogo, K.; Ikuta, M.; Kozono, T.; Takanashi, H.; Murayama, E. J. Med. Chem. 2002, 45, 670-675; Beavers, M.P.; Gunnet, J.W.; Hageman, W.; Miller, W.; Moore, J.B.; Zhou, L.; Chen, R.H.K.; Xiang, A.; Urbanski, M.; Combs, D.W.; Mayo, K.H.; Demarest, K.T. Drug Des. Disc. 2001, 17, 243-251.)
- Appropriate methods for determining the functional activity, pharmacological profile and in vivo activity of compounds of the present invention that interact at the human motilin receptor are also described below.
- The competitive binding assay at the human motilin receptor was carried out analogously to assays described in the literature.
-
- Membranes were prepared from CHO cells stably transfected with the human motilin receptor and utilized at a quantity of 1.5 µg/assay point. [PerkinElmer™ SignalScreen Product #6110544]
- [125I]-Motilin (PerkinElmer, #NEX-378); final concentration: 0.04-0.06 nM
- Motilin (Bachem™, #H-4385); final concentration: 1 µM
- Multiscreen Harvest plates-GFB (Millipore™, #MAHFB1H60)
- Deep-well polypropylene titer plate (Beckman Coulter™, #267006)
- TopSeal-A (PerkinElmer, #6005185)
- Bottom seal (Millipore, #MATAH0P00)
- MicroScint-0 (PerkinElmer, #6013611)
- Binding Buffer: 50 mM Tris-HCl (pH 7.4), 10 mM MgCl2, 1 mM EDTA, 0.1% BSA
-
- 150 µL of membranes diluted in binding buffer
- 10 µL of compound diluted in binding buffer
- 10 µL of radioligand ([125I]-Motilin) diluted in binding buffer
- 10, 5.0, 2.0, 1.0, 0.50, 0.20, 0.10, 0.050, 0.020, 0.010, 0.0050 µM.
- Compounds were provided frozen on dry ice at a stock concentration of 10 mM diluted in 100% DMSO and stored at -20°C until the day of testing. On the test day, compounds were allowed to thaw at room temperature and than diluted in assay buffer according to the desired test concentrations. Under these conditions, the maximum final DMSO concentration in the assay was 0.5%.
- In deep-well plates, diluted cell membranes (1.5 µg/mL) are combined with 10 µL of either binding buffer (total binding, N=5), 1 µM motilin (non-specific binding, N=3) or the appropriate concentration of test compound. The reaction is initiated by addition of 10 µL of [125I]-motilin (final conc. 0.04 - 0.06 nM) to each well. Plates are sealed with TopSeal-A, vortexed gently and incubated at room temperature for 2 hours. The reaction is arrested by filtering samples through pre-soaked (0.3% polyethyleneimine, 2 h) Multiscreen Harvest plates using a Tomtec Harvester, washed 9 times with 500 µL of cold 50 mM Tris-HCl (pH 7.4), and than plates are air-dried in a fumehood for 30 minutes. A bottom seal is applied to the plates prior to the addition of 25 µL of MicroScint-0 to each well. Plates are than sealed with TopSeal-A and counted for 30 sec per well on a TopCount Microplate Scintillation and Luminescence Counter (PerkinElmer) where results are expressed as counts per minute (cpm).
- Data are analyzed by GraphPad™ Prism® (GraphPad Software, San Diego, CA) using a variable slope non-linear regression analysis. Ki values were calculated using a Kd value of 0.16 nM for [125I]-motilin (previously determined during membrane characterization).

- Binding activity at the motilin receptor for representative compounds of the present invention is shown in Table 3. Compound structures for Table 3 are presented with the various groups as defined for the general structure of formula I.
Table 3: Binding Activity at the Human Motilin Receptor for Representative Compounds of the Invention Compound No. Ki (nM) Compound No. Ki (nM) 501a B 543 B 501b B 544 A 502a A 545 A 502b B 546 A 503a A 547 A 503b B 548 A 503c A 549 C 504 B 550 A 505 A 551 A 506a B 552 A 506b B 553 A 507 B 554a A 508 B 554b C 509 B 554c B 510 B 555 A 511a B 556 A 511b C 557 A 512a B 558 A 512b C 559 A 513 B 560 A 514 C 561 A 515a A 562 A 515b B 563 B 516a A 564 A 516b B 565 A 516c C 566 A 517a C 567 A 517b C 568 B 518 B 569 A 519a B 570 A 519b B 571 A 520 B 572 A 521 B 573 B 522 B 574 B 523 B 575 B 524 B 576 C 525 B 577 C 526 B 578a C 527 B 578b D 528 B 579 C 529 A 580a B 530 B 580b B 531 B 581 B 532 A 582a C 533 B 582b D 534 B 583 C 535 B 584 B 536 B 585 A 537 A 586 B 538 A 587 B 539 A 588 A 540 A 589a C 541 A 589b D 542 B 590 B a Binding activity determined using standard method, Ki values are expressed as follows: A ≤10 nM, B ≤ 100 nM, C ≤1000 nM, D > 1000 nM - The evaluation of compounds of the invention for functional activity can be conducted according to literature methods or as described below. (Carreras, C.W.; Siani, M.A.; Santi, D.V.; Dillon, S.B. Anal. Biochem. 2002, 300, 146-151.)
-
- Membranes were prepared using AequoScreen™ (EUROSCREEN, Belgium) cell lines expressing the human motilin receptor (cell line ES-380-A; receptor accession #AF034632). This cell line is constructed by transfection of the human motilin receptor into CHO-K1 cells co-expressing Gα16 and the mitochondrially targeted Aequorin (Ref. #ES-WT-A5).
- Motilin (Bachem, #H-4385)
- Assay buffer: DMEM-F12 (Dulbeccoe's Modified Eagles Medium) with 15 mM HEPES and 0.1% BSA (pH 7.0)
- Coelenterazine (Molecular Probes™, Leiden, The Netherlands)
- 10, 3.16, 1.0, 0.316, 0.10 µM.
- Compounds were provided as dry films at a quantity of approximately 1.2 µmol in pre-formatted 96-well plates. Compounds were dissolved in 100% DMSO at a concentration of 10 mM and stored at -20°C until further use. Daughter plates were prepared at a concentration of 500 µM in 30% DMSO with 0.1% BSA and stored at -20°C until testing. On the test day, compounds were allowed to thaw at room temperature and than diluted in assay buffer according to the desired test concentrations. Under these conditions, the maximum final DMSO concentration in the assay was 0.6%.
- Cells are collected from culture plates with Ca2+ and Mg2+-free phosphate buffered saline (PBS) supplemented with 5 mM EDTA, pelleted for 2 minutes at 1000 x g, resuspended in assay buffer (see above) at a density of 5 x 106 cells/mL and incubated overnight in the presence of 5 µM coelenterazine. After loading, cells were diluted with assay buffer to a concentration of 5 x 105 cells/mL.
- For agonist testing, 50 µL of the cell suspension was mixed with 50 µL of the appropriate concentration of test compound or motilin (reference agonist) in 96-well plates (duplicate samples). The emission of light resulting from receptor activation was recorded using the Functional Drug Screening System 6000 'FDSS 6000' (Hamamatsu Photonics K.K., Japan).
- For antagonist testing, an approximate EC80 concentration of motilin (i.e. 0.5 nM; 100 µL) was injected onto the cell suspension containing the test compounds (duplicate samples) 15-30 minutes after the end of agonist testing and the consequent emission of light resulting from receptor activation was measured as described in the paragraph above.
- Results are expressed as Relative Light Units (RLU). Concentration response curves were analyzed using GraphPad™ Prism® (GraphPad Software, San Diego, CA) by non-linear regression analysis (sigmoidal dose-response) based on the equation E=Emax/(1+EC50/C)n where E is the measured RLU value at a given agonist concentration (C), Emax is the maximal response, EC50 is the concentration producing 50% stimulation and n is the slope index. For agonist testing, results for each concentration of test compound were expressed as percent activation relative to the signal induced by motilin at a concentration equal to the EC80 (i.e. 0.5 nM). For antagonist testing, results for each concentration of test compound were expressed as percent inhibition relative to the signal induced by motilin at a concentration equal to the EC80 (i.e. 0.5 nM).
- A wider evaluation of receptor selectivity can be obtained through the use of "ExpresSProfile", a broad target screen offered commercially by CEREP (Poitiers, France). In this screen, a single-point (10 µM) binding assay is performed across 50 individual receptors from four distinct target classes: non-peptide G-Protein Coupled Receptors (GPCRs), peptide GPCRs, ion channels and amine transporters.
- Binding activity at the motilin receptor for representative compounds of the present invention is shown in Table 4. Compound structures for Table 4 are presented in Table 3 with the various groups as defined for the general structure of formula I.
Table 4: Functional Activity at the Motilin Receptor for Representative Compounds of the Invention Compound No. IC50 (nM)a Compound No. IC50(nM)a 502 B 543 C 503 C 544 A 504 C 545 A 505 B 546 A 511 C 547 B 512 C 548 B 513 C 550 A 515 B 552 A 516 C 555 A 529 B 557 B 530 C 563 B 532 C 564 A 533 B 565 B 534 B 568 B 535 B 569 B 536 C 570 A 537 A 571 B 538 A 572 A 539 A 573 B 540 A 574 D 541 B 585 B 542 B 586 C a Functional activity determined using standard method, IC50 values are expressed as follows: A ≤1 nM, B ≤10 nM, C ≤100 nM, D ≤1000 nM -
- Membranes were prepared from CHO cells stably transfected with the human motilin receptor and utilized at a quantity of 1.5 µg/assay point.
-
- GTPγS (Sigma, #G-8634)
- [35S]-GTPγS (PerkinElmer, #NEX-030H)
- Motilin (Bachem, #H-4385)
- 96-well FlashPlate microplates (PerkinElmer, #SMP200)
- Deep-well polypropylene titer plate (Beckman Coulter, #267006)
- TopSeal-A (PerkinElmer, #6005185)
- Assay Buffer: 50 mM Tris (pH 7.4), 100 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 1 µM GDP, 0.1% BSA
-
- 25 µL of compound diluted in assay buffer
- 25 µL of assay buffer (agonist assay) or 0.6 µM motilin (0.1 µM final concentration) diluted in assay buffer (antagonist assay)
- 100 µL of [35S]-GTPγS diluted in assay buffer
- 50, 20, 10, 5.0, 2.0, 1.0, 0.50, 0.20, 0.10, 0.050, 0.020, 0.010 µM.
- Compounds were provided frozen on dry ice at a stock concentration of 10 mM diluted in 100% DMSO and stored at -20°C until the day of testing. On the test day, compounds were allowed to thaw at room temperature and than diluted in assay buffer according to the desired test concentrations. Under these conditions, the maximum final DMSO concentration in the assay was 0.5%.
- CHO membranes were immobilized into 96-well FlashPlate microplates. Test compound, GTPγS, motilin and [35S]-GTPγS were combined in each well according to the Assay Volumes described above.
- For the assay to measure agonist activity, an additional 25 µL of buffer was added to each well in addition to 25 µL of either buffer (basal value, N=4), 1.0 µM (final conc.) motilin (Emax value, N=3), 25 µM (final conc.) GTPγS (non-specific value, N=4), or the appropriate concentration of test compound (N=3).
- For the assay to measure antagonist activity, an additional 25 µL of either buffer (unstimulated control) or motilin (0.10 µM final conc.) is added to each well, in addition to either 25 µL of buffer (basal value, N=3), 1.0 µM (final conc.) motilin (Emax value, N=3), 25 µM (final conc.) GTPγS (non-specific value, N=4), or the appropriate concentration of test compound (N=3).
- The reaction is initiated by addition of 100 mL of [35S]-GTPγS to each well. Each plate is sealed (TopSeal-A) and incubated in the dark at room temperature for 150 min. Then, plates are counted for 30 seconds per well on the TopCount NXT.
- Data were analyzed by GraphPad™ Prism® 3.0 (GraphPad Software, San Diego, CA) using non-linear regression analysis (sigmoidal dose-response) for the calculation of IC50/EC50 values.

- Where Top and Bottom correspond to the top and bottom values of the dose-response curve calculated by GraphPad Prism.
- Evaluation of compounds of the invention for ex vivo activity was conducted on strips of rabbit duodenum according to literature methods. (Van Assche, G.; Depoortere, I.; Thijs, T.; Janssens, J.J.; Peeters, T.L. Eur. J. Pharmacol. 1997, 337, 267-274; Matthijs, G.; Peeters, T. L.; Vantrappen, G. Naunyn-Schmiedeberg's Arch. Pharmacol. 1989, 339, 332-339.) Related methods can also be employed for this type of study. (Tomomasa, T.; Yagi, H.; Kimura, S.; Snape, W.J., Jr.; Hyman, P.E. Pediatric Res. 1989, 26, 458-461; Takanishi, H.; Yogo, K.; Ozaki, M.; Akima, M.; Koga, H.; Nabata, H. J. Pharm. Exp. Ther. 1995, 273, 624-628.)
- Duodenal segments were vertically suspended in organ chambers of 10 mL filled with Krebs buffer and connected to an isotonic force transducer, with a preload of 1 g. After a stabilization period, the muscle strips were challenged with 10-4 M acetylcholine and washed. This was repeated until a stable maximal contraction was obtained (2-3 times), with an interval of at least 20 minutes.
- After a stable base line was reached, test compounds were added to the bath. After a 15 minute incubation, a dose response to motilin was recorded by adding logarithmically increasing concentrations of motilin to the bath (
final concentration 10-9 to 10-6 M). A blank experiment (no test compound present) was also performed. At the end of the dose response curve, a supramaximal dose of acetylcholine (10-4 M) was given and this response was used as a reference (100% contraction). - The results of experiments at different concentrations of test compound were combined and analyzed to derive the pA2 value from the Schild plot.
- The in vivo efficacy of the compounds of the present invention can be evaluated utilizing appropriate animal models. Rat models have been utilized for evaluation of compounds in chemotherapy-induced diarrhea (CID). (Takasuna, K.; Kasai, Y.; Kitano, Y.; et al. Jpn. J. Cance Res. 1995, 86, 978-984; Tavakkolizadeh, A.; Shen, R.; Abraham, P.; et al. J. Surg. Res. 2000, 91, 77-82; Horikawa, M.; Kato, Y.; Sugiyama. Pharm. Res. 2002, 19, 1345-1353.) Similarly, mouse models have been used to test agents for amelioration of gastrointestinal toxicity of chemotherapeutic agents. (Boushey RP, Yusta B, Drucker DJ. Cancer Res 2001, 61, 687-93; Zhao, J.; Huang, L.; Belmar, N.; Buelow, R.; Fong, T. Clin. Canc. Res. 2004, 10, 2851-2859.) Additionally, dogs are known to suffer from diarrhea during treatment with common chemotherapeutic agents. (Kawato, Y.; Sekiguchi, M.; Akahane, K.; Tsutomi, Y.; Hirota, Y.; Kuga, H.; Suzuki, W.; Hakusui, H.; Sato, K. J. Pharm. Pharmacol. 1993, 45, 444-448 ; Kato, T.; Shimamoto, Y.; Uchida, J.; Ohshimo, H.; Abe, M.; Shirasaka, T.; Fukushima, M. Anticancer Res. 2001, 21, 1705-12.) However, rats and mice do not possess a functional motilin receptor and would not be appropriate animal models for a motilin antagonist, but could be used to differentiate effects at the motilin receptor from non-specific effects. The dog is an appropriate model species as it possesses a functional motilin receptor and has been used previously for studies involving motilin modulation. Other species appropriate for such an evaluation include hamster, pig, rabbit, shrew, guinea pig and opossum. In addition, methods analogous to those described for rats and mice could be adapted to canine or other models to test the compounds of the invention. Hence, the effect of treatment with compounds of the invention on the frequency and severity of incidences of diarrhea in a dog or other animal model can be used as a measure of the effectiveness of the compounds of the invention.
- The objective of this study was to determine the efficacy of representative macrocyclic compounds of the present invention in chemotherapy-induced diarrhea (CID) following intravenous administration to the male beagle dog (Canis familiaris) for up to 32 days. Comparison with existing agents utilized for this condition, octreotide and loperamide, was an additional objective.
- Twenty-one (21) male naïve Beagle dogs were randomly assigned to 7 groups and treated as follows:
Table 5. Treatment Groups Group Numbers Group Designation Dose Level (mg/kg/day) Dose Concentration (mg/mL) Routes Animal Numbers (male) TA1° TA2/ TA1 TA2/ TA1 TA2/ Comparator Comparator Comparator 1 Positive Control 4/8/6 0 0.5/4/ 0 IF/IV/ NA 3 (Irinotecan) 2 IV 2 Compound 5526 5.0b 2 0.33 IV IF 3 Low Dose 3 Compound 5526 15.0b 2 1.00 IV IF 3 High Dose 4 Compound 5526 5.0c 2 0.33 IV IF 3 Pre-Dose 5 Vehicle Control d0 0 0 0 IV IF 3 6 Octreotide 6 0.02 l e2 0.1 IV SC 3 7 Loperamide 6 0.18f 2 0.1 IV PO 3 Abbreviations: IF, intravenous infusion; IV, intravenous bolus injection; NA, not applicable; TA1, Irinotecan (ready-to-use solution for Group I and irinotecan hydrochloride for Groups 2 to 4, 6 and 7); TA2,Compound 552.Notes: a TA1 (irinotecan solution or irinotecan hydrochloride) was used to induce diarrhea in the animals in Groups 1 to 4, 6 and 7. For the animals inGroup 1, treatment with TA1 consisted of three (3) cycles:Cycle 1, irinotecan solution was administered at a dose level of 4 mg/kg/day by one hour infusion via implanted catheter for 5 consecutive days followed by a 9-day rest period.Cycle 2, irinotecan solution was administered by one hour infusion via implanted catheter for four consecutive days at a dose level of 4 mg/kg/day and by bolus injection (over approximately one minute) at a dose level of 8 mg/kg/day via implanted catheter for the fifth day followed by a 5-day rest period; forCycle 3, one spare animal was added toGroup 1. Irinotecan formulation was prepared with irinotecan hydrochloride powder and administered to the four animals by bolus injection via implanted catheter at a dose level of 4 mg/kg/day for five consecutive days. These four animals were observed for 9 days and subjected to necropsy following an overnight period without food.For the animals in Groups 2 to 4, 6 and 7, irinotecan formulation was prepared with irinotecan hydrochloride powder and administered by bolus injection via implanted catheter up to two treatment cycles for a 28-day study period. In each treatment cycle, animals were treated with a single daily dose at 6 mg/kg for 5 consecutive days followed by a 9-day rest period without treatment. Before commencing treatment with irinotecan (Group 4) or upon onset of irinotecan-induced diarrhea (Group Group 2 to 4), octreotide (Group 6) or loperamide (Group 7) as detailed below. Upon completion of the treatment/observation periods and following an overnight period without food, all surviving animals were subjected to necropsy.b TA2 (compound 552) was administered by infusion to all the animals in the group via an implanted catheter over 45 minutes. The treatment with compound 552 started on the day of onset of diarrhea, if diarrhea was observed in any of the three animals from the group in the morning or on the following day if diarrhea was observed in the afternoon. Treatment withcompound 552 then continued throughout the remainder of the 28-day study period. The daily dosage (i.e. 5.0 and 15.0 mg/kg/day forGroups Groups compound 552.c TA2 (compound 552) was administered to all three animals in the group following the same procedures for Groups compound 552.d The animals in the Vehicle Control group (5% dextrose for injection, USP) were treated with the vehicles for irinotecan and compound 552. The vehicle for irinotecan was administered by a slow bolus injection (approximately over one minute) via infusion line for up to two treatment cycles. In each treatment cycle, animals were treated with single daily doses for 5 consecutive days followed by a 9-day rest period. The vehicle forcompound 552 was administered by following the dosing procedures forcompound 552 administration or 23 days (fromDay 6 to Day 28.e All the animals in the group were dosed with octreotide by subcutaneous injection on the day of onset of irinotecan-induced diarrhea, if diarrhea was observed in any of the three animals from the group in the morning or on the following day if diarrhea was observed in the afternoon. The treatment was maintained throughout the remainder of the 28-day study period. The daily dosage (0.021 mg/kg/day) was divided into three equal doses (0.007 mg/kg/occasion) and administered by subcutaneous injection early in the morning, noon and late in the afternoon, respectively. Each administration was separated by at least 4 hours (±30 minutes). f All the animals in the group were dosed with loperamide by oral gavage on the day of onset of irinotecan-induced diarrhea, if diarrhea was observed in any of the three animals from the group in the morning or on the following day if diarrhea was observed in the afternoon. The treatment was maintained throughout the remainder of the 28-day study period. The daily dosage (0.18 mg/kg/day) was divided into three equal doses (0.06 mg/kg/occasion) and administered by oral gavage approximately early in the morning, noon and late in the afternoon, respectively. Each administration was separated by at least 4 hours (±30 minutes). - Dose levels for irinotecan (Seminars in Oncology 1996, 23, 11-20) as well as loperamide and octreotide (The ) were selected based on the indicated literature data.
- The test compounds, controls and comparators were prepared at appropriate intervals. All formulations intended for administration to the test animals were stored refrigerated at 2-8°C. Irinotecan was obtained as a sterile solution for intravenous injection containing irinotecan hydrochloride trihydrate at a concentration of 20 mg/mL, ready to use. The irinotecan formulation for dosing the animals (0.5 mg/mL) in Group 1 (
Cycles - For the remaining treatment with irinotecan (i.e.
Cycle 3 forGroup 1;Groups 2 to 4;Groups 6 and 7), a stock irinotecan formulation was prepared from irinotecan solid (irinotecan hydrochloride) and used for preparation of a dose formulation as described below: - The following protocol was employed. The procedure is for preparation of 10 mL. This procedure may be scaled as necessary:
- 1. Accurately weigh/measure 450 mg sorbitol and 9 mg of lactic acid and place in an appropriate size container.
- 2. Gradually add sterile water for injection to the container to approximately 90% of the final volume. Mix well.
- 3. Measure pH. Adjust pH to 3.0 -3.8 with sodium hydroxide and/or hydrochloric acid, if necessary.
- 4. Quality sufficient to the final volume. Mix well.
- The following protocol was employed:
- 1. An appropriate amount of Irinotecan hydrochloride will be weighed accurately and placed in an appropriate size sterile container.
- 2. An appropriate volume of vehicle solution (approximately 90% of final volume) will be added to the test article powder. Mix on a stir plate until Irinotecan hydrochloride is completely dissolved. Water bath (i.e. 45-55 °C) or short term sonication may be used to facilitate dissolution.
- 3. Measure pH. Adjust pH to 3.0 -3.8 with sodium hydroxide and/or hydrochloric acid, if necessary.
- 4. Quality sufficient to the final volume. Mix well.
- 5. The above solution will be sterile filtered with a 0.22 µm filter into a sterile container. Duplicate samples (1.0 mL each) will be obtained and stored frozen (approximately -80 C) pending shipment. The stock solution will be stored refrigerated (2-8 °C), if not used immediately after preparation, and used within one week of preparation.
- 6. The formulation for dosing the animals is achieved by dilution of the stock formulation with 5% dextrose for injection, USP. The dosing formulation is stored refrigerated (2 to 8°C), protected from light and used within 48 hours.
- Octreotide was provided as a sterile lyophilisate and reconstituted with sterile water for injection to yield a stock solution (10 mg/mL). The stock solution was stored refrigerated (2 to 8°C). On each day of dosing, the stock solution is diluted with sterile water for injection to a final concentration of 0.1 mg/mL. Loperamide hydrochloride was dissolved in saline for injection to yield a final concentration of loperamide base of 0.1 mg/mL (Note: 1 g of loperamide hydrochloride = 0.929 g of loperamide base. A correction factor of 1.076 was used for formulation preparation). Loperamide hydrochloride solution is prepared weekly.
Compound 552 was formulated as a solution with 10% hydroxypropyl-13-cyclodextrin (97%) in water. In order to verify the concentration of the test articles in the formulations, representative samples (1.0 mL in duplicate, except for octreotide, where only 0.1 mL each will be collected) were collected at appropriate intervals during the study. - A total of 21 males and 2 spare males were obtained from a commercial supplier (for example Marshall BioResources, North Rose, New York) with
ages 5 to 7 months old at onset of treatment and weights 6-9 kg at onset of treatment. The animals were acclimated between receipt of the animals and the start of treatment to accustom the dogs to the laboratory environment prior to the start of the study approximately 3 weeks forGroup Group 2 to 4, and 3 weeks forGroup 5 to 7. Each dog was housed in a stainless steel cage equipped with an automatic watering system supplemented by water bottles as appropriate. Each cage was clearly labeled with a color-coded cage card indicating the study, group and animal numbers, sex and dose level. Each animal was uniquely identified by the supplier by means of a tattoo on the ventral aspect of one pinna. The animal room environment was controlled (targeted ranges: temperature 21 ± 3°C,relative humidity 50 ± 20%, 12 hours light, 12 hours dark, 10-15 air changes per hour) except during designated procedures. Temperature and relative humidity were monitored continuously and recorded 4 times daily. - A standard certified commercial dog chow (for example 400 g of Teklad Certified 25% Lab Dog Diet #8727C) was available to each dog once daily during a minimum two-hour feeding period but up to a maximum of 4 hours. During acclimation, the animals were slowly acclimated to the two-hour feeding regimen. Concentrations of the constituents of the diet and contaminants (e.g., heavy metals, aflatoxin, organophosphates, pesticides and chlorinated hydrocarbons) were controlled and routinely measured by the manufacturers. Municipal tap water (which has been purified by reverse osmosis, ultraviolet light and further filtered with a 0.2µm filter) was provided to the animals ad libitum except during designated procedures.
- During the pretreatment period, all the dogs were prepared for surgery approximately 2-3 weeks before starting treatment. These animals were food deprived overnight prior to the surgical procedure and water was removed on the morning of surgery to insert an intravenous catheter. The dogs were injected intramuscularly with a pre-anesthetic cocktail (e.g. butorphanol, acepromazine and glycopyrrolate) for preparation of the surgical sites. Before and during surgery, animals were anesthetized by isoflurane inhalation. The intravenous catheter consisted of a length of medical-grade tubing inserted into the femoral vein and advanced into the vena cava. The catheter was exteriorized at the nape of the neck using a trocar which created a subcutaneous tunnel from the inguinal area to the dorso-cervical area. During the recovery from anesthesia, each animal was dressed in a clean jacket. The catheter was attached to a swivel device, via a jacket and tether system, and connected to a syringe by another section of medical-grade tubing. The pumps and reservoirs were contained in a specially designed box positioned on the exterior of the cage.
- Each dog received an intramuscular injection of antibiotic (for example benzanthine penicillin G and procaine penicillin G) prophylactically one day prior to surgery and 2 days post-surgery. A topical antiseptic was applied to the surgical site once daily, as needed. Throughout the study period, the catheter exit site, localized in the nape of the neck, was inspected daily. When necessary, the site was shaved, cleansed free of debris and painted with a topical antiseptic/antibiotic. The jacket was changed monthly or more frequently, if deemed necessary. Following recovery from surgery, animals received 0.9% Sodium Chloride for Injection, USP at a rate of 2.5 mL/h until the start of infusion of
compound 552. The syringes/bags containing the saline solution were changed with appropriate frequency during the pretreatment period. - For the animals in
Group 1, treatment with test article No. 1 (irinotecan) consisted of three (3) cycles:Cycle 1, irinotecan was administered at a dose level of 4 mg/kg/day by one hour infusion via implanted catheter for 5 consecutive days followed by a 9-day rest period.Cycle 2, irinotecan was administered by one hour infusion via implanted catheter for four consecutive days at a dose level of 4 mg/kg/day and by bolus injection (approximately over one minute) at a dose level of 8 mg/kg/day via implanted catheter for the fifth day followed by a 5-day rest period;Cycle 3, one spare animal was added toGroup 1. Irinotecan formulation was prepared with irinotecan hydrochloride powder and administered to the four animals by bolus injection via implanted catheter at a dose level of 4 mg/kg/day for five consecutive days. For the animals inGroups 2 to 4, 6 and 7, irinotecan formulation that was prepared with irinotecan hydrochloride powder was administered by bolus injection via implanted catheter up to two treatment cycles for a 28-day study period. In each treatment cycle, animals were treated with a single daily dose at 6 mg/kg for 5 consecutive days followed by a 9-day rest period without treatment. The control/vehicle article for irinotecan was administered to all dogs inGroup 5 by following the same dosing procedures for irinotecan administration. The dose volume was 8 mL/kg forCycle 1 and the first 4 days ofCycle 2 forGroup Cycle 2 forGroup Groups 2 to 4, 6 and 7. The actual volume infused to each dog was calculated and adjusted based on the most recent practical body weight of each animal. - The test article No. 2 (compound 552) was intravenously infused via an implanted catheter over 45 minutes to all the animals in
Group 4 four days before the start of TA1 (irinotecan) treatment and throughout the 28-day study period. By following the same dosing procedure, TA2 (compound 552) was administered to all the animals inGroups Groups 2 to 4 respectively) was administered by two equal doses (i.e. 2.5, 7.5 and 2.5 mg/kg/occasion forGroups 2 to 4 respectively) which were separate by at least 6 h. Each dose was infused at a dose volume of 10 mL/kg/h. The vehicle forcompound 552 was administered to all dogs inGroup 5 by following the same dosing procedures and regimen forcompound 552 administration for 23 days (fromDay 6 to Day 28). The actual volume infused was calculated and adjusted based on the most recent practical body weight of each animal. The syringes/bags were changed at appropriate intervals as necessary and the weights recorded prior to the start and at the end of the infusion. - Octreotide was administered by subcutaneous injection to all animals in
Group 6 starting on the same day of diarrhea onset, if diarrhea was observed in any one of the three animals from the group in the morning, or on the following day of diarrhea onset, if diarrhea was observed in any one of the three animals in the goup in the afternoon. The treatment was maintained throughout the remainder of the 28-day study period. The daily dosage (0.021 mg/kg/day) was divided into three equal doses (0.007 mg/kg/occasion) and administered by subcutaneous injection approximately early in the morning, noon and late in the afternoon, respectively. Each administration was separated by at least 4 hours (±30 minutes). The dose volume for each administration was 0.07 mL/kg. The actual dose volume was calculated and adjusted based on the most recent practical body weight of each animal. - Loperamide was administered by oral gavage to all animals in
Group 7 starting on the same day of diarrhea onset, if diarrhea was observed in any one of the three animals of the group in the morning, or on the following day of diarrhea onset, if diarrhea was observed in anyone of the three animals in the group in the afternoon. The treatment was maintained throughout the remainder of the 28-day study period. The daily dosage (0.18 mg/kg/day) was divided into three equal doses (0.06 mg/kg/occasion) and administered by oral gavage approximately early in the morning, noon and late in the afternoon, respectively. Each administration was separated at least 4 h (±30 min). The dose volume for each administration was 0.6 mL/kg. The actual dose volume was calculated and adjusted based on the most recent practical body weight of each animal. - During the treatment periods of the study, the infusion line in each animal was maintained by infusion of 0.9% Sodium Chloride for Injection USP at a rate of 2.5 mL/h between each treatment of irinotecan,
compound 552 or vehicles. - Cage-side clinical signs (ill health, behavioral changes, stool changes etc.) were recorded once daily during the acclimation period except on detailed clinical examination days. Cage-side clinical signs were recorded twice a day (am and pm) during the treatment/observation period except on the days of a detailed clinical examination where they were recorded once (PM). A detailed clinical examination of each dog was performed once pretreatment, weekly during the treatment/observation periods and before necropsy. Particular attention was paid to the surgical sites.
- Observations of stool consistency was recorded twice daily one week prior to first treatment of irinotecan and throughout the 28-day study period and graded as follows:
Table 6. Observations of Stool Consistency. Stool Observation Grade Normal 1 loose 2 Liquid 3 Liquid with mucous or blood etc 4 - Whenever possible, number of vomits and discharges per day, severity of diarrhea was recorded as well as stool consistency. Symptoms of diarrhea-induced dehydration are treated by infusion of Ringer solution at the discretion of a veterinarian.
- Body weights were recorded for all animals once prior to group assignment, and approximately one week prior to initiation of treatment. Body weights were recorded for all animals up to 1 day prior to dosing and twice weekly thereafter during the treatment/observation periods, as well as terminally prior to necropsy (fasted). Food consumption was measured daily during the week prior to treatment and throughout the treatment/observation periods. A series of blood samples (approximately 2.0 mL each) were removed from each dog in
Groups compound 552 and after the second (pm) infusion on the last day of treatment withcompound 552. (Note: the second (pm) infusion on the first day of treatment withcompound 552 was performed after completion of the last blood sampling). For this purpose, each dog was bled by venipuncture and the samples collected into tubes containing the anticoagulant, K2 EDTA. Tubes were placed on wet ice pending processing. On each occasion, samples were collected prior to the start of infusion, 5, 15, 30, 45, 50, 65, 95, 135 minutes, 3 and 6 hours after the start of infusion ofcompound 552. Following collection, the samples were centrifuged at approximately 1500xg, approximately 4°C, for at least 10 minutes and the resulting plasma recovered and dispensed into two aliquots and stored frozen (at approximately -80°C) pending shipment. - Numerical data obtained during the conduct of the study was subjected to calculation of group means and standard deviations and will be reported along with all individual numerical and non numerical results. The data was analyzed for homogeneity of variance using Levene Median and for normality using Kolmogorov-Smimov tests. Homogeneous data was analyzed using the Analysis of Variance and the significance of intergroup differences was analyzed using Dunnett's test. Heterogeneous data was analyzed using Kruskal-Wallis test and the significance of intergroup differences between the control and treated groups assessed using Dunn's test. A significance level of p ≤0.05 was reported.
- As shown in
Figure 19 ,compound 552, a potent and selective motilin antagonist, demonstrated superior efficacy in the treatment of irinotecan-induced CID in dogs versus the current standard of care. The compound proved to be more effective, with a quicker onset and longer duration of action as well. - Plasma concentrations of
compound 552 in male beagle dogs receiving low and high intravenous doses ofcompound 552 as part of the above experiment were determined. Plasma samples were prepared for analysis by solid phase extraction and analyzed by LC-MS/MS. The average Cmax observed at 30 min after the start of the 45-min infusion was about 1.5 and 1.8 µg/mL on the first and last day of treatment withcompound 552, respectively, for the animals receiving 2.5 mg/kg. The average Cmax observed for the high dose group administered with 7.5 mg/kg occurred between 15 and 45 min after the start of the 45-min infusion and was about 3.4 and 5.0 µg/mL on the first and last day of treatment withcompound 552, respectively. The average AUC0-t was similar on the first (78,233 ng.min/mL) and last day (83,047 ng.min/mL) of dosing for the low dose group (2.5 mg/kg/occasion) and was proportionally higher on the first day (246,073 ng.min/mL) of dosing for the high dose group (7.5 mg/kg/occasion). The average AUC0-t was higher (380,758 ng.min/mL) on the last day of dosing for the high dose group, however the higher average value-resulted from one animal exhibiting particularly high AUC0-t. The plasma concentration data demonstrated dose-related exposure forcompound 552 in beagle dogs during the CID efficacy study. - Alternative treatment regimens can also be employed to induce diarrhea in these model.
- Cytochrome P450 enzymes are implicated in the phase I metabolism of drugs. The majority of drug-drug interactions are metabolism-based and, moreover, these interactions typically involve inhibition of cytochrome P450s. Six CYP450 enzymes (CYPIA2, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4) appear to be commonly responsible for the metabolism of most drugs and the associated drug-drug interactions. Assays to determine the binding of compounds of the invention to the various metabolically important isoforms of cytochrome P450 metabolizing enzymes are commercially available, for example NoAb BioDiscoveries (Mississaugua, ON, Canada) and Absorption Systems (Exton, PA, USA). As well, a number of appropriate methods have been described or reviewed in the literature. (White, R.E. Ann. Rev. Pharmacol. Toxicol. 2000, 40, 133-157; Li, A.P. Drug. Disc. ; Turpeinen, M.; Korhonen, L.E. Tolonen, A.; et al. Eur. J. Pharm. Sci. 2006, 29, 130-138.)
- The key aspects of the experimental method were as follows:
- Assay was performed on microsomes (Supersomes®, BD Gentest, Becton-Dickinson) prepared from insect cells expressing individual human CYP-450 subtypes, specifically:
- CYP subtypes: 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4
- Two substrates are typically tested for CYP-3A4 as this enzyme exhibits complex inhibition kinetics
- Assays monitored, via fluorescence detection, the formation of a fluorescent metabolite following incubation of the microsomes with a specific CYP substrate.
- Compounds of the present invention were tested in duplicate samples at eight test concentrations using 3-fold serial dilutions (concentration range of 0.0457 to 100 µM).
- For each CYP-450 enzyme, a specific inhibitor was tested in duplicate at eight concentrations as a positive control.
- The concentration of the inhibitor or test compound that inhibited metabolite formation by 50% (IC50) was calculated by non-linear regression analysis of the % inhibition vs. log concentration (M) curve.
- Results for representative compounds of the invention are summarized in Table 5.
Table 7. Rabbit Duodenum Contractile Assay Results for Representative Compounds of the Invention Compound Contractile Potency (pA2) Binding Ki (nM) 502 7.92 A 551 8.29 A 553 8.71 A a Binding activity is expressed as follows: A ≤10 nM, B ≤100 nM, C
≤1000nM, D > 1000 nM - The pharmacokinetic and pharmacodynamic properties of drugs are largely a function of the reversible binding of drugs to plasma or serum proteins such as albumin and α1-acid glycoprotein. In general, only unbound drug is available for diffusion or transport across cell membranes, and for interaction at the pharmacological target. On the other hand, drugs with low plasma protein binding generally have large volumes of distribution and rapid clearance since only unbound drug is available for glomerular filtration and, in some cases, hepatic clearance. Thus, the extent of plasma protein binding can influence efficacy, distribution and elimination. The ideal range for plasma protein binding is in the range of 87-98% for most drug products.
- Protein binding studies were performed using human plasma. Briefly, 96-well microplates were used to incubate various concentrations of the test article for 60 min at 37°C. Bound and unbound fractions are separated by equilibrium dialysis, where the concentration remaining in the unbound fraction is quantified by LC-MS or LC-MS-MS analysis. Drugs with known plasma protein binding values such as quinine (~35%), warfarin (~98%) and naproxen (~99.7%) were used as reference controls.
- Results for representative compounds of the invention are summarized in Table 6.
Table 8: Inhibition of CYP P450 Isozymes by Representative Compounds of the Invention Compound IC50 (µM)a 3A4 3A4 1A2 2A6 2B6 2C8 2C9 2C19 2D6 2E1 (BQ) (BFC) 551 10.4 2.37 n/a n/a n/a n/a 26.9 37.0 30.1 >100 553 3.48 0.0192 n/a n/a n/a >100 44.5 n/a 80.8 >100 a n/a indicates IC50 value much above 100 µM - The Caco-2 cell line, derived from a human colorectal carcinoma, has become an established in vitro model for the prediction of drug absorption across the human intestine. (Sun, D.; Yu, L.X.; Hussain, M.A.; Wall, D.A.; Smith, R.L.; Amidon, G.L. Curr. Opin. Drug Discov. Devel. 2004, 7, 75-85; Bergstrom, C.A. Basic Clin. Pharmacol. Toxicol. 2005, 96, 156-61; Balimane, P.V.; Han, Y.H.; Chong, S. AAPS J. 2006, 8, E1-13; Shah, P.; Jogani, V.; Bagchi, T.; Misra, A. Biotechnol. Prog. 2006, 22, 186-198) When cultured on semi-permeable membranes, Caco-2 cells differentiate into a highly functionalized epithelial barrier with remarkable morphological and biochemical similarity to the small intestinal columnar epithelium. Fully differentiated cell monolayers can be used to assess the membrane transport properties of novel compounds. In addition, the apparent permeability coefficients (Papp) obtained from Caco-2 cell transport studies have been shown to reasonably correlate with human intestinal absorption.
- Assays to determine the permeability of compounds of the invention utilizing Caco-2 cells are commercially available, for example NoAb BioDiscoveries (Mississaugua, ON, Canada) and Absorption Systems (Exton, PA, USA).
- Alternatively, parallel artificial membrane permeability assays (PAMPA) can be utilized to assess intestinal permeability. (Avdeef, A. Expert Opin. Drug. Metab. Toxicol. 2005,1, 325-42.)
- Permeability across the Caco-2 cell layer was determined by growing the cells on a membrane placed between two (donor and acceptor) chambers. Drug candidates are typically added to the apical (A) side of the cell layer and their appearance in the basolateral (B) side is measured over incubation time. Permeability in this direction represents intestinal absorption. Permeability may also be determined from the basolateral to the apical side of the Caco-2 cell. A higher apical to basolateral Papp, compared to the basolateral to apical Papp, is indicative of carrier-mediated transport. P-gp mediated transport is suggested when a higher basolateral to apical Papp is observed relative to the apical to basolateral Papp.
- Permeability (10 µM) for compounds of the invention in the apical to basolateral and basolateral to apical direction were tested in duplicate. Samples will be collected from the donor and acceptor chambers at the beginning (0 min) and following 60 min of incubation at 37°C and stored frozen at -70°C until bioanalysis. Samples for each test compound generated from the Caco-2 permeability assay were further analyzed by LC-MS-MS. The permeability of [3H]-mannitol and [3H]-propranolol were determined in parallel as controls.
- The permeability coefficient (Papp) of each compound and radiolabeled standard was determined using the following equation:

- Results for representative compounds of the invention are summarized in Table 7.
Table 9: Caco-2 Permeability Assays for Representative Compounds of the Invention Compound Papp A to B cm/sec x10-6 Papp B to A cm/sec x10-6 BtoA/AtoB 502 5 11 2 508 1 5 5 511 15 17 1 512 0.3 11 37 515 3 9 3 531 19 18 1 554 15 18 1 580 5 7 1 590 2 11 6 - The pharmacokinetic behavior of compound of the invention can be ascertained by methods well known to those skilled in the art. (Wilkinson, G. R. "Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination" in Goodman & Gilman's The Pharmacological Basis of Therapeutics, Tenth Edition, Hardman, J.G.; Limbird, L.E., Eds., McGraw Hill, Columbus, OH, 2001, .) The following method was used to investigate the pharmacokinetic parameters (elimination half-life, total plasma clearance, etc.) for intravenous, subcutaneous and oral administration of compounds of the present invention.
- Rats: male, Sprague-Dawley (~250 g)
- Rats/Treatment Group: 6 (2 subsets of 3 rats each, alternate bleeds)
- Each sample of test compound was sent in solution in a formulation (such as with cyclodextrin) appropriate for dosing. It will be appreciated by one skilled in the art that appropriate modifications to this protocol can be made as required to adequately test the properties of the compound under analysis.
-
- 1. Intravenous (i.v.): 2 mg/kg
- 2. Subcutaneous(s.c): 2 mg/kg
- 3. Oral (p.o.): 8 mg/kg
-
- 1. Same protocol for all dosing groups
- 2. For each group, 2 subsets (A and B) of 3 rats/subset
- At the time intervals indicated above, 0.7 mL of blood were collected from each animal. It is expected that this volume of blood will yield a sample of at least 0.3 mL of plasma. EDTA was used as an anti-coagulant for whole blood collection. Whole blood samples were chilled and immediately processed by centrifugation to obtain plasma.
- Plasma samples were stored frozen (-70°C) until analysis. Analytical detection of parent compound in plasma samples performed by LC-MS after an appropriate preparation protocol: extraction using solid phase extraction (SPE) cartridges (Oasis MCX, Oasis HLB) or liquid-liquid extraction.
- Column: Atlantis dC18 from Waters 2.1 x 30 mm
- Mobile phases:
- A: 95% MeOH, 5% water, 0.1% TFA
- B: 95% water, 5% MeOH, 0.1% TFA
- Flow: 0.5 mL/min
- Gradient (linear):
Time min A B 0 30% 70% 0.5 30% 70% 2.8 100% 0% 3.8 100% 0% 4.0 30% 70% 5.0 30% 70% - The analyte was quantitated based upon a standard curve and the method validated with internal standards.
Table 12. Pharmacokinetic Parameters for Representative Compounds of the Invention Compound Mode of Elimination Clearance Bioavailability Administrationa (t1/2, min) (mL/min/kg) (oral)b 502 i.v. 102 57 na 502 s.c. 146 79 na 551 i.v. 139 33 na 551 p.o.d 70-125 6% 552 i.v. 24 45 na 552 s.c. 103 39 na 552 p.o.c 307 10% 563 i.v. 55 27 na 563 p.o.c 117 21% 568 s.c. 132 42 na 568 i.v. 25 65 na a i.v. = intravenous (10 time points over 150 min); s.c. = subcutaneous (10 time points over 360 min), p.o. = oral (10 time points over 240 min)
b na = not applicable
c Done in the presence of 10% beta-cyclodextrin
d Done without cyclodextrin - Results of the time courses for
compounds 502, 552 and 563 are provided inFigures 13A-13G . - Previous studies have demonstrated that motilin stimulates the contractile activity in smooth muscle strips isolated from the rabbit gastric antrum (Van Assche, G.; Depoortere, I.; Thijs, T.; Janssens, J.J.; Peeters, T.L. Concentration-dependent stimulation of cholinergic motor nerves or smooth muscle by [Nle13]-motilin in the isolated rabbit gastric antrum. Eur. J. Pharmacol. 1997, 337, 267-274.) or from the circular muscle of-the duodenum as described in Method 3-D above. Motilin (threshold concentration of 10-8 M) enhances contractions induced by electrical field stimulation (4 Hz) by a post-ganglionic interaction with the cholinergic neurotransmission. At higher concentrations, motilin causes a tonic contraction interacting directly with the antral smooth muscle. Analogously, the effects of motilin and [Nle13]-motilin (Calbiochem), as selective agonists of the motilin receptor, on smooth muscle strips isolated from the gastric antrum, the circular muscle of the duodenum and the colon of the shrew were investigated. Immunohistochemical studies have shown that endocrine cells in the GI tract of the musk shrew express motilin (Kanamori, Y.; Nakazawa, S.; Kitoh, J.; Hoshino, M. The distribution of endocrine cells in the mucosa of the gastrointestinal tract of the house musk shrew, Suncus murinus (Insectivora). Cell. Tissue Res. 1989, 258, 365-71; Kitamura, N.; Yamada, J.; Watanabe, T.; Yamashita, T. An immunohistochemical study on the distribution of endocrine cells in the gastrointestinal tract of the musk shrew, Suncus murinus. Histol Histopathol. 1990, 5, 83-88.) Shrews were fasted for 8-10 h, euthanized with CO2 inhalation, and the stomach, the duodenum and colon isolated immediately and placed in Krebs buffer aerated with 95% O2 and 5% CO2. Specifically, mucosa-free circular muscle strips were isolated from the antral region, the duodenum and the colon using a dissecting microscope. Isometric contractions were recorded in organ baths following equilibration at optimal tension using the PowerLab data acquisition system. Electrical field stimulation (EFS: 0.5 ms pulse duration, 1-32 Hz pulse frequency) was used to induce neurally mediated contractions. In particular, four colonic segments (10-12 mm) were prepared from each animal. The segments were suspended at 1 g initial tension in 10-mL organ baths and equilibrated for 90 min. Experiments were performed to investigate the effect of motilin and [Nle13]-motilin (10-9-10-7 M): a) on the basal tone and the development of spontaneous phasic contractions; b) on neurally mediated contractile responses induced by EFS. The contractile effects induced by motilin or [Nle13]-motilin applied at a bath concentration of 0.3 µM reached a maximum within the first 4 min of treatment and diminished within 8-10 min. The results confirm that motilin receptors are involved in the regulation of GI muscle contractility in the shrew.
- To define the effect of test compounds on different regions of the GI tract of the shrew (gastric antrum, ileum and colon) and to evaluate the antagonist affinity of test compounds in this model system, the ability of varying concentrations of test compound to antagonize the motilin or [Nle13]-motilin-induced contractions was measured in organ baths as described above.
Compound 552 dose-dependently inhibited the contractions induced in isolated colonic segments from the shrew by activation of the motilin receptor by motilin and [Nle13] motilin as depicted inFigure 18 . Compound 552 (0.01 µM-10 µM) caused a dose-dependent inhibition of the contractile response to 0.3 µM motilin or to 0.3 µM [Nle13]-motilin. For each experiment, data are mean ± SEM and the number of animals was as indicated for each treatment.Compound 552 did not cause a significant change in basal contractile activity (data not shown). - Animal models can be used to investigate the effect of treatment with representative compounds of the present invention on amelioration of the symptoms or prevention of the development of diarrhea induced in different manners, in particular by stress or by PGE2. Since rats and mice do not express motilin receptors, the musk shrew (Suncus murinus) is one appropriate animal model expressing the motilin receptor and suitable for this work. In addition, opossum, rabbit or the pig are also suitable animal models for this type of evaluation.
- Laboratory musk shrews (Suncus murinus) were used in the study. Musk shrews do not live in social groups and have aggressive behavior when placed together, thus the animals were housed singly in Plexiglas cages with pine shavings and shredded paper for bedding (for appropriate handling procedures see Temple, J.L. The musk shrew (Suncus murinus): a model species for studies of nutritional regulation of reproduction. ILAR J. 2004, 45, 25-34). The temperature was kept at 70-72°F and a 14:10 h light:dark cycle maintained throughout the study. Since the shrews are most active at dawn and dusk, the experiments were started within the first hour of the light-on phase. Food and water was provided ad libitum in accordance with the species requirements: Food: 3 part Purina Cat Chow and 1 part Mink Chow (for example Wisconsin "Mink Complete Pellets-Grow-Fur": crude protein < 34%, crude fat >20%, crude fiber >4%). The food was placed in a small plastic dish within the cage. Water: Slightly acidic (pH 5.5) distilled water, to reduce contamination of tap water supply. The cages and water bottles were changed weekly. All animals were acclimated to the animal facility for at least 1 week prior to initiating the study.
- First, information regarding normal food intake and fecal pellet output in the shrew was obtained. Animals were acclimated to handling and daily food intake and fecal pellet output measured in a group of 12 shrews. The animals were brought to the laboratory:in their home cages and left there for 3 h. During this period, each animal was taken out of the cage and covered with a folded towel. The skin on the top of the back was gently pressed and lifted to mimic the procedure used to administer a subcutaneous injection. Then the animal was returned in the home cage and the fecal pellet output followed for 2 h. The data served as a reference to naïve animals when establishing the model of PGE2-induced defecation. When acclimated to handling, the shrews were randomly assigned in two groups (n=6 per group) and subjected to different procedures to induce fecal pellet output reminiscent of diarrhea. In the first group, the shrews were placed in individual restraint cages for 1 h at room temperature. Fecal output (number of pellets) was measured during 1 h of immobilization and within 1 h after the restraint stress. The consistency was evaluated using a 3 level scoring system: 0, normal; 1, soft; 2 unformed. (Saito, T.; Mizutani, F.; Iwanaga, Y.; Morikawa, K.; Kato, H. Laxative and anti-diarrheal activity of polycarbophil in mice and rats. Jpn. J. Pharmacol. 2002, 89, 133-141.) In the second group, an increase in fecal output was induced by administration of PGE2 (0.3 mg/kg, i.p.) and changes in stool consistency monitored for 2 h. Stool consistency was evaluated using the 3 level scoring system. Based on the literature results obtained by Saito in mice, soft or unformed stool should be produced within 15-30 min. after PGE2 dosing. The time measured from PGE2 administration to the induction of diarrhea (first appearance of soft watery stool) was also recorded. In addition, wet weight/dry weight ratio of the fecal pellets was measured prior to and after administration of PGE2 or restraint.
- To investigate the effect of a test compound or its vehicle on accelerated defecation induced by restraint stress, as a model of stress-induced diarrhea, the animals (8 per dose level) were treated with test compound (0.1, 1 and 10 mg/kg s.c. or other appropriate doses and mode of administration) or vehicle. Animals were placed in individual restraint cages for 1 h at room temperature and fecal output was measured as described above. The animals had free access to food and water prior to the experiment.
- To investigate the effect of a test compound or its vehicle on PGE2-induced diarrhea, the animals were randomly assigned to groups of 8 and each group treated with test compound (0.1, 1 and 10 mg/kg s.c. or other appropriate doses and mode of administration) or vehicle. Diarrhea was induced by administration of PGE2 (0.3 mg/kg, i.p.) and changes in stool consistency were evaluated as described above. The animals were acclimated with free access to food and water.
- All values are presented means ± SE from 8 successful experiments for each treatment group. The statistical significance of values was determined by one-way ANOVA and Dunnett's multiple-comparison test (differences from the vehicle control). Probabilities of <5% (p < 0.05) are considered significant.
- A number of animal models of inflammation and, in particular, inflammatory diseases and disorders of the GI tract, are well-established in the art and can be used to evaluate the efficacy of representative compounds of the present invention on treating gastrointestinal inflammation. (Wirtz, S.; Neurath, M.F. Int. J. Colorectal. Dis. 2000, 15, 144-160; Powrie, F.; Ulbig, H. Novartis Found. Symp. 2004, 263, 164-178; Jurjus, A.R.; Khoury, N.N.; Reimund, J.-M. J. Pharmacol. Toxicol. ; Eckmann, L. Ann. NY Acad Sci. 2006, 1072, 28-38; Byrne, F.R.; Viney, J.L. Curr. Opin. Drug Disc. Develop. 2006, 9, 207-217.)
- The macrocyclic compounds of the present invention or pharmacologically acceptable salts thereof according to the invention may be formulated into pharmaceutical compositions of various dosage forms. To prepare the pharmaceutical compositions of the invention, one or more compounds, including optical isomers, enantiomers, diastereomers, racemates or stereochemical mixtures thereof, or pharmaceutically acceptable salts thereof as the active ingredient is intimately mixed with appropriate carriers and additives according to techniques known to those skilled in the art of pharmaceutical formulations.
- A pharmaceutically acceptable salt refers to a salt form of the compounds of the present invention in order to permit their use or formulation as pharmaceuticals and which retains the biological effectiveness of the free acids and bases of the specified compound and that is not biologically or otherwise undesirable. Examples of such salts are described in Handbook of Pharmaceutical Salts: Properties, Selection, and Use, Wermuth, C.G. and Stahl, P.H. (eds.), Wiley-Verlag Helvetica Acta, Zurich, 2002 [ISBN 3-906390-26-8]. Examples of such salts include alkali metal salts and addition salts of free acids and bases: Examples of pharmaceutically acceptable salts, without limitation, include sulfates, pyrosulfates, bisulfates, sulfites, bisulfites, phosphates, monohydrogenphosphates, dihydrogenphosphates, metaphosphates, pyrophosphates, chlorides, bromides, iodides, acetates, propionates, decanoates, caprylates, acrylates, formates, isobutyrates, caproates; heptanoates, propiolates, oxalates, malonates, succinates, suberates, sebacates, fumarates, maleates, butyne-1,4-dioates, hexyne-1,6-dioates, benzoates, chlorobenzoates, methylbenzoates, dinitrobenzoates, hydroxybenzoates, methoxybenzoates, phthalates, xylenesulfonates, phenylacetates, phenylpropionates, phenylbutyrates, citrates, lactates, γ-hydroxybutyrates, glycollates, tartrates, methanesulfonates, ethane sulfonates, propanesulfonates, toluenesulfonates, naphthalene-1-sulfonates, naphthalene-2-sulfonates, and mandelates.
- If an inventive compound is a base, a desired salt may be prepared by any suitable method known to those skilled in the art, including treatment of the free base with an inorganic acid, such as, without limitation, hydrochloric acid, hydrobromic acid, hydroiodic, carbonic acid, sulfuric acid, nitric acid, phosphoric acid, and the like, or with an organic acid, including, without limitation, formic acid, acetic acid, propionic acid, maleic acid, succinic acid, mandelic acid, fumaric acid, malonic acid, pyruvic acid, oxalic acid, stearic acid, ascorbic acid, glycolic acid, salicylic acid, pyranosidyl acid, such as glucuronic acid or galacturonic acid, alpha-hydroxy acid, such as citric acid or tartaric acid, amino acid, such as aspartic acid or glutamic acid, aromatic acid, such as benzoic acid or cinnamic acid, sulfonic acid, such as p-toluenesulfonic acid, methanesulfonic acid, ethanesulfonic acid, 2-hydroxyethanesulfonic acid, benzenesulfonic acid, cyclohexyl-aminosulfonic acid or the like.
- If an inventive compound is an acid, a desired salt may be prepared by any suitable method known to the art, including treatment of the free acid with an inorganic or organic base, such as an amine (primary, secondary, or tertiary); an alkali metal or alkaline earth metal hydroxide; or the like. Illustrative examples of suitable salts include organic salts derived from amino acids such as glycine, lysine and arginine; ammonia; primary, secondary, and tertiary amines such as ethylenediamine, N,N'-dibenzylethylenediamine, diethanolamine, choline, and procaine, and cyclic amines, such as piperidine, morpholine, and piperazine; as well as inorganic salts derived from sodium, calcium, potassium, magnesium, manganese, iron, copper, zinc, aluminum, and lithium.
- The carriers and additives used for such pharmaceutical compositions can take a variety of forms depending on the anticipated mode of administration. Thus, compositions for oral administration may be, for example, solid preparations such as tablets, sugar-coated tablets, hard capsules, soft capsules, granules, powders and the like, with suitable carriers and additives being starches, sugars, binders, diluents, granulating agents, lubricants, disintegrating agents and the like. Because of their ease of use and higher patient compliance, tablets and capsules represent the most advantageous oral dosage forms for many medical conditions.
- Similarly, compositions for liquid preparations include solutions, emulsions, dispersions, suspensions, syrups, elixirs, and the like with suitable carriers and additives being water, alcohols, oils, glycols, preservatives, flavoring agents, coloring agents, suspending agents, and the like. Typical preparations for parenteral administration comprise the active ingredient with a carrier such as sterile water or parenterally acceptable oil including polyethylene glycol, polyvinyl pyrrolidone, lecithin, arachis oil or sesame oil, with other additives for aiding solubility or preservation may also be included. In the case of a solution, it can be lyophilized to a powder and then reconstituted immediately prior to use. For dispersions and suspensions, appropriate carriers and additives include aqueous gums, celluloses, silicates or oils.
- The pharmaceutical compositions according to embodiments of the present invention include those suitable for oral, rectal, topical, inhalation (e.g., via an aerosol) buccal (e.g., sub-lingual), vaginal, topical (i.e., both skin and mucosal surfaces, including airway surfaces), transdermal administration and parenteral (e.g., subcutaneous, intramuscular, intradermal, intraarticular, intrapleural, intraperitoneal, intrathecal, intracerebral, intracranially, intraarterial, or intravenous), although the most suitable route in any given case will depend on the nature and severity of the condition being treated and on the nature of the particular active agent which is being used.
- Compositions for injection will include the active ingredient together with suitable carriers including propylene glycol-alcohol-water, isotonic water, sterile water for injection (USP), emulPhor™-alcohol-water, cremophor-EL™ or other suitable carriers known to those skilled in the art. These carriers may be used alone or in combination with other conventional solubilizing agents such as ethanol, propylene glycol, or other agents known to those skilled in the art.
- Where the macrocyclic compounds of the present invention are to be applied in the form of solutions or injections, the compounds may be used by dissolving or suspending in any conventional diluent. The diluents may include, for example, physiological saline, Ringer's solution, an aqueous glucose solution, an aqueous dextrose solution, an alcohol, a fatty acid ester, glycerol, a glycol, an oil derived from plant or animal sources, a paraffin and the like. These preparations may be prepared according to any conventional method known to those skilled in the art.
- Compositions for nasal administration may be formulated as aerosols, drops, powders and gels. Aerosol formulations typically comprise a solution or fine suspension of the active ingredient in a physiologically acceptable aqueous or non-aqueous solvent. Such formulations are typically presented in single or multidose quantities in a sterile form in a sealed container. The sealed container can be a cartridge or refill for use with an atomizing device. Alternatively, the sealed container may be a unitary dispensing device such as a single use nasal inhaler, pump atomizer or an aerosol dispenser fitted with a metering valve set to deliver a therapeutically effective amount, which is intended for disposal once the contents have been completely used. When the dosage form comprises an aerosol dispenser, it will contain a propellant such as a compressed gas, air as an example, or an organic propellant including a fluorochlorohydrocarbon or fluorohydrocarbon.
- Compositions suitable for buccal or sublingual administration include tablets, lozenges and pastilles, wherein the active ingredient is formulated with a carrier such as sugar and acacia, tragacanth or gelatin and glycerin.
- Compositions for rectal administration include suppositories containing a conventional suppository base such as cocoa butter.
- Compositions suitable for transdermal administration include ointments, gels and patches.
- Other compositions known to those skilled in the art can also be applied for percutaneous or subcutaneous administration, such as plasters.
- Further, in preparing such pharmaceutical compositions comprising the active ingredient or ingredients in admixture with components necessary for the formulation of the compositions, other conventional pharmacologically acceptable additives may be incorporated, for example, excipients, stabilizers, antiseptics, wetting agents, emulsifying agents, lubricants, sweetening agents, coloring agents, flavoring agents, isotonicity agents, buffering agents, antioxidants and the like. As the additives, there may be mentioned, for example, starch, sucrose, fructose, dextrose, lactose, glucose, mannitol, sorbitol, precipitated calcium carbonate, crystalline cellulose, carboxymethylcellulose, dextrin, gelatin, acacia, EDTA, magnesium stearate, talc, hydroxypropylmethylcellulose, sodium metabisulfite, and the like.
- In some embodiments, the composition is provided in a unit dosage form such as a tablet or capsule.
- In further embodiments, the present invention provides kits including one or more containers comprising pharmaceutical dosage units comprising an effective amount of one or more compounds of the present invention.
- The present invention further provides prodrugs comprising the compounds described herein. The term "prodrug" is intended to mean a compound that is converted under physiological conditions or by solvolysis or metabolically to a specified compound that is pharmaceutically active. The "prodrug" can be a compound of the present invention that has been chemically derivatized such that, (i) it retains some, all or none of the bioactivity of its parent drug compound, and (ii) it is metabolized in a subject to yield the parent drug compound. The prodrug of the present invention may also be a "partial prodrug" in that the compound has been chemically derivatized such that, (i) it retains some, all or none of the bioactivity of its parent drug compound, and (ii) it is metabolized in a subject to yield a biologically active derivative of the compound. Known techniques for derivatizing compounds to provide prodrugs can be employed. Such methods may utilize formation of a hydrolyzable coupling to the compound.
- The present invention further provides that the compounds of the present invention may be administered in combination with a therapeutic agent used to prevent and/or treat metabolic and/or endocrine disorders, gastrointestinal disorders, cardiovascular disorders, obesity and obesity-associated disorders, central nervous system disorders, genetic disorders, hyperproliferative disorders and inflammatory disorders. Exemplary agents include analgesics (including opioid analgesics), anesthetics, antifungals, antibiotics, antiinflammatories (including nonsteroidal anti-inflammatory agents), anthelmintics, antiemetics, antihistamines, antihypertensives, antipsychotics, antiarthritics, antitussives, antivirals, cardioactive drugs, cathartics, chemotherapeutic agents (such as DNA-interactive agents, antimetabolites, tubulin-interactive agents, hormonal agents, and agents such as asparaginase or hydroxyurea), corticoids (steroids), antidepressants, depressants, diuretics, hypnotics, minerals, nutritional supplements, parasympathomimetics, hormones (such as corticotrophin releasing hormone, adrenocorticotropin, growth hormone releasing hormone, growth hormone, thyrptropin-releasing hormone and thyroid stimulating hormone), sedatives, sulfonamides, stimulants, sympathomimetics, tranquilizers, vasoconstrictors, vasodilators, vitamins and xanthine derivatives.
- Subjects suitable to be treated according to the present invention include, but are not limited to, avian and mammalian subjects, and are preferably mammalian. Mammals of the present invention include, but are not limited to, canines, felines, bovines, caprines, equines, ovines, porcines, rodents (e.g. rats and mice), lagomorphs, primates, humans, and the like, and mammals in utero. Any mammalian subject in need of being treated according to the present invention is suitable. Human subjects are preferred. Human subjects of both genders and at any stage of development (i.e., neonate, infant, juvenile, adolescent, adult) can be treated according to the present invention.
- Illustrative avians according to the present invention include chickens, ducks, turkeys, geese, quail, pheasant, ratites (e.g., ostrich) and domesticated birds (e.g., parrots and canaries), and birds in ovo.
- The present invention is primarily concerned with the treatment of human subjects, but the invention can also be carried out on animal subjects, particularly mammalian subjects such as mice, rats, dogs, cats, livestock and horses for veterinary purposes. The invention can be further carried out for drug screening and drug development purposes.
- In therapeutic use for treatment of conditions in mammals (i.e. humans or animals) for which an antagonist of the motilin receptor is effective, the compounds of the present invention or an appropriate pharmaceutical composition thereof may be administered in an effective amount. Since the activity of the compounds and the degree of the therapeutic effect vary, the actual dosage administered will be determined based upon generally recognized factors such as age, condition of the subject, route of delivery and body weight of the subject. The dosage can be from about 0.1 to about 100 mg/kg, administered orally 1-4 times per day. In addition, compounds can be administered by injection at approximately 0.01 - 20 mg/kg per dose, with administration 1-4 times per day. Treatment could continue for weeks, months or longer. Determination of optimal dosages for a particular situation is within the capabilities of those skilled in the art.
- The compounds of formula I of the present invention can be used for the prevention and treatment of a range of gastrointestinal disorders characterized by hypermotility or hypermotilinemia.
- In particular embodiments, the macrocyclic compounds of the present invention can be used to treat diarrhea, cancer treatment-related diarrhea, chemotherapy-induced diarrhea, radiation enteritis, radiation-induced diarrhea, stress-induced diarrhea, chronic diarrhea, AIDS-related diarrhea, C. difficile associated diarrhea, traveller's diarrhea, diarrhea induced by graph versus host disease and other types of diarrhea.
- In other embodiments, the present invention is directed to a method of treating irritable bowel syndrome, inflammatory bowel disease, dyspepsia, functional gastrointestinal disorders, chemotherapy-induced nausea and vomiting (emesis), post-operative nausea and vomiting, Crohn's disease, gastroesophogeal reflux disorders, ulcerative colitis, pancreatitis, infantile hypertrophic pyloric stenosis, carcinoid syndrome, malabsorption syndrome, diabetes mellitus, obesity, postgastroenterectomy syndrome, atrophic colitis or gastritis, gastric stasis, gastrointestinal dumping syndrome, celiac disease, short bowel syndrome, cachexia and eating disorders leading to obesity in humans and other mammals comprising administering a therapeutically effective amount of a compound of formula I.
- As used herein, "treatment" is not necessarily meant to imply cure or complete abolition of the disorder or symptoms associated therewith.
- The compounds of the present invention can further be utilized for the preparation of a medicament for the treatment of a range of medical conditions involving gastrointestinal motility disorders.
- Further embodiments of the present invention will now be described with reference to the following examples. It should be appreciated that these examples are for the purposes of illustrating embodiments of the present invention, and do not limit the scope of the invention.
- Standard Procedure for the Synthesis of Boc-Dap(thiazol-2-yl) (Boc-AA1,
Figure 1 ) Step 1-1. [2-Hydroxy-1-(methoxy-methyl-carbamoyl)-ethyl]-carbamic acid tert-butyl ester (AA1-1). To a solution of Boc-Ser-OH (AA1-0, 3.0 g, 0.015 mol) in DMF (40 mL) was added DIPEA (2.6 mL, 15.0 mmol) and HBTU (5.53 g, 15.0 mmol), then the mixture stirred at room temperature until a homogeneous solution was obtained. N,O-Dimethylhydroxylamine hydrochloride (1.60 g, 16.5 mmol) and DIPEA (2.85 mL, 16.0 mmol) were then added. The solution was stirred at room temperature O/N. The mixture was quenched addition of a saturated aqueous solution of NaHCO3 at 0°C, then extracted with ethyl acetate. The organic phase was dried over Na2SO4 and concentrated under reduced pressure to dryness. Flash chromatography using ethyl acetate as eluent furnished AA1-1 in 85% yield.
TLC (100% ethyl acetate): Rf= 0.40 (CMA). - Step 1-2. Thiazol-2-yl-carbamic acid benzyl ester (AA1-2). To a stirred solution of 2-aminothiazole (AA1-A, 3.0 g, 30.0 mmol) and triethylamine (6.30 mL, 45.0 mmol) at 0°C. was added benzyl chloroformate (5 mL, 36 mmol). The reaction was stirred at RT O/N. The reaction mixture was washed with a saturated aqueous solution of NaHCO3, then water and concentrated to dryness under reduced pressure. The resulting yellow solid was crystallized from ethanol to afford AA1-2 in 70% yield.
- Step 1-3. Boc-Dap(Z-thiazol-2-yl) (Boc-AA1) This procedure is based on that presented for other substrates in Gautam Panda et al. . A mixture of Boc-Ser-Weinreb amide AA1-1 (3.0 g, 12.0 mmol) and triphenylphosphine (4.75 g, 18 mmol) in anhydrous THF (100 mL) was cooled in an ice bath. A solution of DIAD (3.63 g, 18 mmol) in THF (20 mL) was added to this mixture dropwise. After mixing of the solution for 5 min, AA1-2 (5.65 g, 24 mmol) in THF (50 mL) was slowly added. The mixture was allowed to warm to room temperature and stirred O/N. The solvent was removed under reduced pressure. The crude residue was purified by flash chromatography (petroleum ether:ethyl acetate, 8:2) to provide AA1-3 in 40% yield.
TLC (petroleum ether /ethyl acetate 8:2): Rf = 0.30 (CMA) - The next reaction sequence was based on the procedure described in the literature (Evans, D. A. et al. Tetrahedron Lett. 1987, 28, 6141.) To a solution of AA1-3 (1.0 g, 2.15 mmol) in THF (50 mL) and H2O (10 mL) was added LiOH (150 mg, 3.22 mmol) and the reaction stirred at 0°C for 12 h. The reaction mixture was quenched with saturated aqueous NH4Cl and extracted with ethyl acetate. Flash chromatography (ethyl acetate:methanol, 8:2) furnished an 80% yield of Boc-AA1.
TLC (ethyl acetate:methanol, 8:2): Rf = 0.40 (CMA);
1H NMR (CD3OD): δ 1.28 (s, 9H), 4.48 (m, 2H), 4.63 (m, 1H), 5.30 (s, 2H), 7.09 (m, 1H), 7.34-7.39 (m, 4H), 7.47 (m, 1H);
LC-MS (Grad_A4): tR = 7.67 minutes; mass calculated for C19H23N3O6S: 421.4674, found: 421 -

- Step 2-1. Boc-Serine-β-lactone (AA2-1). This procedure is based on that found in the literature (Vederas, J.C.; et al. J. Am. Chem. Soc. 1987, 109, 4649-4659). Into a dry 250 mL 3-neck flask equipped with a mechanical stirrer under a nitrogen atmosphere was added triphenylphosphine (4.5 g, 17.1 mmol, 1.1 eq), followed by 100 mL of an anhydrous THF:CH3CN (1:9) mixture. The mixture was stirred until a solution was obtained, then cooled to -55°C (bath temperature) and dimethylazodicarboxylate (DMAD, 1.9 mL, 17.1 mmol, 1.1 eq) added dropwise over 10 min. After the addition, the mixture was stirred for 20 min and a solution of Boc-Ser-OH (3.18 g, 15.5 mmol, 1.0 eq.) in 50 mL of anhydrous THF:CH3CN (1:9) was added dropwise over 30 min. The mixture was stirred at -55°C for 1.5 h, then the bath was removed and the solution allowed to warm slowly to room temperature. Once the mixture reached room temperature, the solvent was evaporated under reduced pressure. The resulting yellow oil was purified by flash chromatography [gradient, hexanes:EtOAc, (80:20) to (60:40)] to give 2.10 g of AA2-1 as a white solid in 72% yield. Purification of the crude material is best performed on the same day as the reaction to avoid decomposition. DCM can be added to help dissolve the crude residue.
TLC (Hex/EtOAc, 60/40): Rf= 0.55 (CMA) - Step 2-2. 1-Trimethylsilylimidazole (AA2-2). HMDS (11.46 mL, 0.055 mol, 1.5 eq) was added dropwise over 20 min to a solution of imidazole (5.0 g, 0.074 mol, 2 eq) in 150 ml of toluene under argon, then the reaction mixture heated to reflux for 3 h. The reaction mixture was then concentrated to dryness under reduced pressure. Distillation of the residue gave AA2-2 as a colorless oil in 85%. For best results, this product should be stored at 0°C under argon.
- Step 2-3. Boc-imidazol-1-yl-Ala (AA2). Similar to the method described in Vederas, J.C.; et al. J. Am. Chem. Soc. 1985, 107, 7105-7109, trimethylsilylimidazole AA2-2 (0.488 g, 3.50 mmol, 1.3 eq) in dry CH3CN (10 mL) was treated dropwise over 5 min with AA2-1 (0.5 g, 2.67 mmol, 1 eq) in dry CH3CN (10 mL) under argon, then the mixture stirred for 28 h. The reaction was then cooled in an ice-water bath and 10 ml of a cold aqueous solution of ammonium chloride (0.1 M) added. The mixture was brought to room temperature and stirred for 5 min. The aqueous phase was extracted with DCM (3 x 50 mL) and submitted to chromatography on C18 cartridges (10 g) [gradient, water:methanol, (100:00) to (90:10)] to give 0.48 g of AA2 as a white solid in 55% yield.
TLC (ethyl acetate:methanol:water, 8:2:1): Rf= 0.30 (CMA);
1H NMR (D2O): δ 1.20 (s, 9H), 4.20 (m, 2H), 4.50 (M, 1H), 7.30 (s, 1H), 7.32 (s, 1H), 8.50 (s, 1H);
LC-MS (Grad_A4): tR = 3.02 min; mass calculated for C11H17N3O4: 255.2704; found: 255. -

- The procedure is based on that described in the literature (Vederas, J.C.; et al. J.Am. Chem. Soc. 1985, 107, 7105-7109). Pyrazole (0.80 g, 11.5 mmol, 1.5 eq) in dry CH3CN (10 mL) was treated dropwise over 5 min with lactone AA3-1 (synthesized as described previously, 0.50 g, 2.67 mmol, 1.0 eq) in dry CH3CN (10 mL) under argon and the resulting mixture stirred for 12 h at 50°C. The reaction was then concentrated to dryness under reduced pressure and the crude residue purified by flash chromatography (ethyl acetate:methanol, 8:2) to give 1.0 g (60% yield) of AA3 as a white solid.
TLC (ethyl acetate:methanol, 8:2): Rf= 0.30 (CMA);
LC-MS (Grad_A4): tR = 5.14 min; mass calculated for C11H17N3O4: 255.2704, found: 255. - Standard Procedure for the Synthesis of Ddz-Dap(Boc-Me)-OH (Ddz-AA4,
Figure 2 ) Step 4-1. Z-Serine-β-lactone (AA4-2). This intermediate was prepared in an analogous fashion to that described previously for the Boc derivative AA3-1. In a dry 250 mL 3-neck flask equipped with a mechanical stirrer under nitrogen atmosphere was added triphenylphosphine (4.5 g, 17.1 mmol, 1.1 eq.), followed by 100 mL of an anhydrous THF:CH3CN (1:9) solvent mixture. The mixture was stirred until a solution was obtained and then cooled to -55°C (bath temperature) and dimethylazodicarboxylate (DMAD, 1.9 mL, 17.1 mmol, 1.1 eq) was added dropwise over 10 min. After completion of the addition, the mixture was stirred for 20 min, then a solution of Z-Ser-OH (AA4-1, 3.7 g, 15.5 mmol, 1.0 eq) in 50 mL of anhydrous THF:CH3CN (1:9) was added dropwise over 30 min. The reaction mixture was stirred at -55°C for 1.5 h, then the cooling bath removed and the solution allowed to warm slowly to room temperature. Once the mixture reached room temperature, the solvent was evaporated under reduced pressure. The resulting yellow oil was purified by flash chromatography [gradient, hexanes/EtOAc, (80:20) to (60:40)] to give 2.5 g of AA4-2 as a white solid in 72% yield. Purification of the crude oil is preferentially performed the same day to avoid decomposition. DCM can be added to help solubilize the residue.
TLC (hexanes/EtOAc (60/40): Rf= 0.55 (UV, CMA) - Step 4-2. Z-Dap(Me)-OH hydrochloride salt (AA4-3). A dry 500 mL round bottom flask under nitrogen atmosphere was charged with 60 ml of anhydrous CH3CN followed by N-methyl trimethylsilylamine (AA4-0, synthesized as described in Step 4-6, 1.9 mL, 13.3 mmol, 1.5 eq.). A solution of AA4-2 (2.0 g, 8.9 mmol, 1.0 eq) in 40 mL of anhydrous MeCN was added to the flask and the mixture was stirred under nitrogen at room temperature until TLC indicates no trace of starting material [hexanes:EtOAc (60:40), UV, CMA]. The mixture was then cooled to 0°C and 180 mL of cold 0.1 M HCI added. The mixture was allowed to warm to room temperature and stirred for 30 min. The solvent was evaporated under reduced pressure, the resulting residue azeotroped twice with toluene then dried under vacuum to give 3.1 g of crude AA4-3 as a light yellow foam. The crude material was used without any purification.
- Step 4-3. Z-Dap(Boc-Me)-OH (AA4-4). To a flask containing the crude AA4-3 (8.9 mmol, 1.0 eq based on theoretical yield) was added 90 mL of anhydrous DCM. The mixture was cooled to 0°C and diisopropylethylamine (DIPEA, 7.8 mL, 44.5 mmol, 5.0 eq) added dropwise, which helps to solubilize the crude substrate. Di-tert-butyl dicarbonate (2.1 g, 9.8 mmol, 1.1 eq) was added in one portion, then the mixture warmed to room temperature and stirred O/N. Solvent was evaporated under reduced pressure and the resulting residue taken up in 50 mL of an Et2O: NaOH (1 M, 1:1) mixture. The pH was confirmed to be between 9 and 10 and adjusted to this level if it was not. The separated aqueous phase was washed with Et2O (2 x 25 mL), acidified to
pH 2 with 1 M HCl and extracted with EtOAc (3 x 25 mL). The combined organic phases were washed with brine (2 x 25 mL), dried over MgSO4, filtered, evaporated under reduced pressure and dried under vacuum to give 2.5 g (80% yield) of AA4-4 as a white foam. - Step 4-4. H-Dap(Boc-Me)-OH (AA4-5). A solution of AA4-4 (2.5 g, 7.1 mmol, 1.0 eq.) in 75 mL of MeOH was added to 10% palladium on activated carbon (250 mg, 10% w/w) under a nitrogen atmosphere. Hydrogen was then bubbled into the mixture until completion of the reaction. [TLC BuOH:AcOH:H2O (4:1:1), UV, ninhydrin]. The mixture was then purged with nitrogen, filtered on a Celite® (World Minerals Inc., Santa Barbara, CA) pad and rinsed well with MeOH (3 x 25 mL). The filtrate was evaporated under reduced pressure and dried under vacuum to give 1.5 g (93%) of AA5-5 as a pale yellow solid.
- Step 4-5. Ddz-Dap(Boc-Me)-OH (Ddz-AA4). To a solution of AA4-5 (1.9 g, 8.7 mmol, 1.0 eq) in 20 mL of MeOH was added Triton B (4.3 mL, 9.5 mmol, 1.1 eq). The mixture was stirred 30 min at room temperature, then concentrated under reduced pressure. The residue was azeotroped with toluene (2x) to remove excess H2O and MeOH. The resulting slurry was dissolved in 20 mL of anhydrous DMF and Ddz-OPh (3.1 g, 9.5 mmol, 1.1 eq) added in one portion. The mixture was stirred 36-48 h at 50°C under nitrogen. DMF was then removed under high vacuum and the residue diluted in H2O (50 mL). The resulting aqueous phase (checked to ensure pH was 9 and adjusted if necessary) was washed with Et2O until TLC indicates no phenol in the aqueous phase. The basic aqueous phase was cooled to 0°C, Et2O (50 mL) added, and the aqueous phase acidified with citrate buffer (1.0 M, pH 3.5). The separated aqueous phase was extracted with Et2O (2 x 50 mL). The combined organic phases were then washed sequentially with citrate buffer (2 x 50 mL), H2O (2 x 50 mL) and brine (1 x 50 mL), dried with MgSO4, filtered and concentrated under reduced pressure to afford 2.5 g (65%) of Ddz-AA4 as a pale brown solid.
1H NMR (DMSO-d6): δ 12.65 (s, 1H), 7.45-7.35 (dd, 1H), 6.50 (s, 2H), 6.35 (2, 1H), 4.10 (q, 1H), 3.70 (s, 6H), 3.65-3.55 (dd, 1H), 3.25-3.15 (dd, 1H), 2.75 (d, 3H), 1.60 (d, 6H), 1.35 (s, 9H). - Step 4-6. Synthesis of N-methyl trimethylsilylamine (AA4-0). Methylamine (25.0 mL, 0.56 mol, 2.5 eq) was condensed at -20°C, then added to 250 mL of anhydrous Et2O at - 20°C under a nitrogen atmosphere. Freshly distilled (from LAH) chlorotrimethylsilane (28.1 mL, 0.22 mol, 1.0 eq) was added dropwise to the solution at -20°C. A white precipitate was formed. After completion of the addition, the reaction was warmed to room temperature and stirred for 4 h. The mixture was then cooled to 0°C and filtered cold. The filtered salts are washed with anhydrous Et2O (2 x 50 mL), then the filtrate distilled through a 45 cm Vigreux column to remove Et2O. The residue is then redistilled on a 15 cm Vigreux column to isolate 9.7 g (44%) of N-methyl trimethylsilylamine (bp 68-72°C) as a colorless liquid.
- Step 5-1. 3-(3-Fluorophenyl)-2-hydroxy-propionic acid benzyl ester (F1-2). Sodium nitrite (3.77 g, 54.6 mmol) was added in portions to a solution of L-3-fluorophenylalanine (F1-1, 5.0 g, 27.3 mmol) in acetic acid (82 mL) which is maintained at room temperature by immersion in an ice-water bath bearing a thermocouple. The solution is stirred at room temperature for 1 hr, then concentrated under reduced pressure. Water was then added to the residue and the product extracted with Et2O, dried over MgSO4, filtrated and concentrated under reduced pressure to yield 2.57g (42%) of the corresponding acid. The crude product was dissolved in toluene (120 mL), then p-toluenesulfonic acid (pTSA, 216 mg, 1.14 mmol) and BnOH (5.62 mL, 56.9 mmol) added and the solution refluxed with a Dean-Stark apparatus O/N. The resulting solution was cooled to room temperature, concentrated under reduced pressure and purified by flash chromatography (gradient, 100% hexanes to hexanes/ethyl acetate, 4/1) to yield 1.63 g (52%) of benzyl ester F1-2 along with an impure fraction (3.14 g).
TLC: Rf= 0.5 (4/1 hexanes/ethyl acetate);
1H NMR (CDCl3): δ 2.77 (br s, 1H, OH); 2.95 (dd, J= 4.39 and 13.67 Hz, 1H, CH 2CHOH); 3.11 (dd, J= 6.45 and 13.78 Hz, 1H, CH 2CHOH); 4.48 (m, 1H, CH2CHOH); 5.18 (s, 2H, PhCH 2O); 6.86 to 6.93 (m, 3H); 7.17 to 7.20 (m, 1H); 7.31 to 7.39 (m, 5H);
LC-MS (Grad_A4): tR = 7.16 main; (M+Na)+ 297. - Step 5-2. {3-[2-(2-Amino-3-methyl-butoxy)-phenyl]-propyl}-carbamic acid tert-butyl ester (F1-5). Diisopropyl azodicarboxylate (DIAD, 3.13 mL, 15.9 mmol) is added dropwise to a well-stirred solution of triphenylphosphine (4.17 g, 15.9 mmol) in THF (150 mL) at 0°C under nitrogen. The mixture is stirred at 0°C for 30 min, then warmed to room temperature. The PPh3-DIAD adduct should precipitate as a white solid. Cbz-valinol (F1-4,3.43 g, 14.5 mmol) in THF (150 mL) was added to this solution followed by iodophenol (F1-3, 3.18 g, 14.5 mmol) and the resulting solution was stirred O/N under nitrogen. The solution was then concentrated under reduced pressure, and the crude product purified by flash chromatography (gradient, 9/1 to 8/2 hexanes/ethyl acetate) to yield the corresponding adduct (2.98 g, 47%). This was dissolved in CH3CN (70 mL) and Boc-propargylamine (1.31 g, 8.48 mmol) added to the solution. Argon was bubbled into this solution for 15 min, then Et3N (23 mL) added. Argon was bubbled into the resulting solution for 5 min, then CuI (45 mg) and PdCl2(PPh3)2 (145 mg) added and the resulting solution was stirred under argon O/N. The solution was concentrated under reduced pressure, dissolved with Et2O, washed sequentially with aqueous citrate buffer (2x), saturated aqueous NaHCO3 (2x), and brine (1x), dried over MgSO4, filtered and concentrated under reduced pressure. The resulting oil was purified by flash chromatography (8/2 hexanes/ethyl acetate) to yield the corresponding alkyne (2.39 g, 76%). The crude product can also be used without further purification in the next reaction.
1H NMR (CDCl3): δ 1.01 (d, J= 6.7 Hz, 3H, CH 3CH); 1.02 (d, J= 6.7 Hz, 3H, CH 3CH); 1.22 to 1.28 (m, 1H); 1.43 (s, 9H, (CH3)3C); 2.01 to 2.16 (m, 1H, CHCH(CH 3)2); 3.79 to 3.84 (m, 1H); 3.99 to 4.03 (m, 1H); 4.08 to 4.15 (m, 2H); 5.04 (br. S, 1H); 5.12 (s, 2H, PhCH 2O); 5.28 (d, J= 9.4 Hz, 1H); 6.83 (d, J= 8.2 Hz, 1H); 6.90 (t, J= 7.6 Hz, 1H); 7.21 to 7.37 (m, 7H);
LC-MS (Grad_A4): tR = 12.45 min, (M+H-Boc)+ 367. - The alkyne (2.39 g, 5.13 mmol) was dissolved in 95% EtOH (50 mL). PtO2 (116 mg) was added and the solution purged with H2 (2x), then pressurized under H2 (120 psi) and stirred O/N. The resulting solution was depressurized, filtered over Celite (World Minerals Inc., Santa Barbara, CA), washed with ethyl acetate and concentrated under reduced pressure. The residue was dissolved in ethyl acetate (50 mL). 10% Pd/C (500 mg) was added and the solution purged with H2 (2x), then pressurized under H2 (120 psi) and stirred O/N. The resulting mixture was depressurized, filtered over Celite® (World Minerals Inc., Santa Barbara, CA), washed with ethyl acetate, concentrated under reduced pressure and the residue purified by flash chromatography (gradient, 100% ethyl acetate to 10% MeOH/90% ethyl acetate) to yield the corresponding amine F1-5 (1.15 g, 67%).
1H NMR (CDCl3): δ 0.99 (d, J= 6.39 Hz, 6H, (CH 3)2CH); 1.44 (s, 9H, (CH 3)3C); 1.79 (quint., J= 6.45 Hz, (CH3)2CH); 2.66 (t, J= 7.62 Hz, 2H); 2.98 to 3.00 (m, 1H); 3.09 to 3.11 (m, 2H); 3.80 (t, J= 8.21 Hz); 3.99 (dd); 5.04 to 5.06 (m); 6.82 to 6.91 (m, 2H); 7.11 to 7.18 (m, 2H). - Step 5-3. 2-{1-[2-(3-tert-Butoxycarbonylaminopropyl)-phenoxymethyl]-2-methyl-propyl-amino}-3-(3-fluorophenyl)-propionic acid benzyl ester F1. Freshly distilled (CF3SO2)2O (29.5 µL, 175 µmol) was added to a solution of the alcohol F1-2 (45 mg, 164 µmol) in CH2Cl2 at 0°C over 5 min, followed by 2,6-lutidine (22 µL, 189 µmol). The resulting solution was stirred for 10 min at 0°C, then DIPEA (32 µL, 181 µmol) added immediately followed by a solution of the amine F1-5 (55 mg, 164 µmol) in CH2Cl2 (1 mL) over 15 min. The resulting solution was stirred at room temperature under nitrogen O/N. Then CH2Cl2 was added and the organic solution washed sequentially with water, saturated aqueous NaHCO3, and brine, dried over MgSO4, filtered and concentrated under reduced pressure. The resulting oil was purified by flash chromatography to yield the benzyl ester F1 (40 mg, 41 %).
- The related fragment F2 was made following the same synthetic route as described for fragment F1 as illustrated in
Figure 4 . - Standard Procedure for the Synthesis of Protected Tether Ddz-T38(S) (
Figure 5 ) Step 7-1. Iodo alcohol T38-2(S). To a solution of 2-iodophenol (T38-1, 19.0 g, 86.2 mmol, 1.0 eq) in acetone (150 mL), potassium carbonate (13.1 g, 94.8 mmol, 1.1 eq) was added, followed by (S)-(-)-propylene oxide (T38-A, 30.1 mL, 0.431 mol, 5.0 eq). The mixture was stirred at 75°C in a sealed high pressure flask overnight. The mixture was cooled down to room temperature, filtered and the filter rinsed twice with acetone. The combined filtrate and rinses were concentrated under reduced pressure and the resulting residue dissolved in Et2O (150 mL). The organic layer was washed with 1 N NaOH (3 x 100 mL) and brine (2 x 100 mL), dried over magnesium sulfate, filtered and evaporated under reduced pressure to give 23.9 g of T38-2(S) as a yellow oil in quantitative yield. Use of (R)-(+)-propylene oxide in this step results in formation of the enantiomeric protected tether [Ddz-T38(R)] via the same procedure. - Step 7-2. Ddz-amino-alkyne [T38-4(S)]. Into a solution of T38-2(S) (23.9 g, 86.0 mmol, 1.0 eq) and Ddz-propargylamine (T38-3, 29.8 g, 107.5 mmol, 1.25 eq) in CH3CN (130 mL) was bubbled argon for 10 min. Distilled (after being stirred O/N with CaH2) triethylamine (45 mL) was then added to the solution and the mixture purged by bubbling with argon for 5 min. Recrystallized copper (I) iodide (572 mg, 3.0 mmol, 0.035 eq, Organometallic in Synthesis, A Manual, Manfred Schlosser, 2nd edition, 2002, p.669.) and trans-dichlorobis(triphenylphosphine) palladium (II) (2.1 g, 3.0 mmol, 0.035 eq) were added and the reaction mixture stirred O/N under argon at room temperature. The reaction was monitored by TLC [(EtOAc/Hex, 60/40), Rf =0.62, UV, CMA]. The mixture was filtered through a silica gel pad and rinsed with EtOAc. The combined filtrate and rinses were concentrated under reduced pressure until dryness and the residual oil diluted with a mixture of DCM:Et2O (15:85, 300 mL). The organic phase was washed sequentially with 1.0 M citrate buffer (3 x 150 mL), saturated NaHCO3 (2 x 150 mL, CAUTION, pressure is generated), brine (1 x 150 mL), and then dried with magnesium sulfate, filtered and evaporated under reduced pressure. The crude product was purified by flash chromatography [gradient, EtOAc:Hex:Et3N (30:70:0.5) to EtOAc:Hex:Et3N (60:40:0.5)] to give 35.0 g of T38-4(S) as a brown syrup in quantitative yield.
- Step 7-3. Ddz-Amino-alcohol [Ddz-T38(S)]. To a solution of T38-4(S) (35.0 g, 81.9 mmol, 1.0 eq) in 95% EtOH (400 mL) under nitrogen was added platinum oxide (1.86 g, 8.2 mmol, 0.1 eq). Hydrogen was bubbled into the mixture O/N while stirring. Reaction was monitored by 1H NMR until completion. When the reaction was complete, nitrogen was bubbled for 10 min to remove the excess of hydrogen. The mixture was then filtered through a Celite® (World Minerals Inc., Santa Barbara, CA) pad and washed with 95% EtOH until no material was eluting [TLC (EtOAc/Hex, 60/40)]. The solvent was removed under reduced pressure. The product was diluted in DCM (300 mL) and MP-TMT scavenger resin (5 eq, based on amount of catalyst) was added. The mixture was stirred O/N, filtered and the resin rinsed twice with DCM. The combined filtrate and rinses were evaporated under reduced pressure to afford 34.1 g (89%) of Ddz-T38(S) as a yellow to orange oil.
1H NMR (CDCl3): δ 7.20-7.10, (m, 2H), 6.95-6.80 (m, 2H), 6.55 (bs, 2H), 6.35 (s, 1H), 5.18 (bt, 1H), 4.12 (m, 1H), 3.95 (m, 2H), 3.80 (s, 6H), 3.15 (bq, 2H), 2.65 (t, 2H), 1.98 (bs, 2H), 1.65 (bs, 6H), 1.25 (m, 3H).
13C NMR (CDCl3): δ 160.8, 156.6, 155.8, 149.6, 130.4, 127.5, 121.3, 111.7, 103.2, 98.4, 80.7, 73.5, 66.6, 55.5, 40.2, 30.5, 29.3, 29.1, 27.3, 19.5.
LC-MS (Grad_A4): tR = 8.46 min - Note that T38(S) is used to make compounds of formula I with an (R)-stereocenter. Similarly, T38(R) is used to synthesize compounds of formula I with an (S)-stereocenter as described further in Examples 10 and 11.
- Additionally, protected derivatives of tethers T92 and T93 can be obtained using analogous methods to that of Example 7 starting from 3-fluoro-2-iodophenol and 6-fluoro-2-iodophenol, respectively, or the corresponding 2-bromophenols.
- Standard Procedure for the Synthesis of Protected Tether Boc-T81(1R,8S) (
Figure 6 ) Step 8-1. 3-(2-Methoxy-phenyl)-2-methyl-acrylic acid ethyl ester (T81-2). To a suspension of sodium hydride (65% in oil, 26.4 g, 661 mmol, washed thoroughly with hexanes to remove oil) in THF (746 mL) at 0°C was added (EtO)2P(O)CH(Me)CO2Et (144 mL, 661 mmol). The mixture was stirred at room temperature for 30 min, then the solution cooled to 0°C and aldehyde T81-1 (60.0 g, 441 mmol) slowly added. The reaction was stirred during O/N with monitoring by TLC [ (ethyl acetate:hexanes, 2:7), Rf = 0.49 (UV, CMA)]. A saturated solution of ammonium chloride was added and the aqueous phase extracted with Et2O (3x), dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (ethyl acetate/hexanes, 2/8) to give T81-2 as a yellow oil (97-100% yield). - Step 8-2. 3-(2-Methoxy-phenyl)-2-methyl-propionic acid ethyl ester (T81-3). To a solution of T81-2 (7.3 g, 23 mmol) in 95% ethanol (100 ml) was carefully added 10% Pd/C (730 mg). Hydrogen was bubbled through the solution, then the solution stirred under hydrogen atmosphere for O/N. The suspension was filtered through a pad of silica gel and washed with ethyl acetate. The combined filtrate and washes were concentrated under reduced pressure to give T81-3 which was suitable for use in the next step without any further purification (yield: 86-90%).
- Step 8-3. 3-(2-Methoxy-phenyl)-2-methyl-propan-1-ol (T81-4). To a solution of T81-3 (55.1 g, 248 mmol) in THF (1.2 L) at 0°C was added LiAlH4 (18.8 g, 496 mmol) in portions. After the addition was completed, the solution was stirred 15 min at 0°C and monitored by TLC [(30% EtOAc, 70% hexanes), Rf = 0.30 (UV, CMA)] until complete. An addition of water (18.8 mL) was effected very slowly (under a nitrogen flow) followed by a solution of 15% sodium hydroxide (18.8 mL), then water (56 mL). The residual salt was filtered and the filtrate concentrated under reduced pressure to give alcohol T81-4 (99%) which was used without any further purification in the next step.
- Step 8-4. Acetic acid 3-(2-methoxy-phenyl)-2-methyl-propyl ester [T81-5(S)] and 3-(2-Methoxy-phenyl)-2-methyl-propan-1-ol [T81-5(R)]. Enzyme Amano PS (1.7 g) and racemic alcohol T81-4 (30.7, 0.67 mol) were mixed with TBME (292 mL) in a reaction flask containing a magnetic stirring bar with no cap. In another flask, TBME was placed in a dessicator and used to restore the volume of reaction after 24 h (due to loss by evaporation). The mixture was stirred for 24 h in a dessicator in the presence of saturated MgCl2 (5-10 mL of a saturated solution of MgCl2 in water was prepared and stored in the dessicator without any cap). After 24 h, the pre-equilibrated solvent was added to the reaction flask to compensate for loss by evaporation. Vinyl acetate (56 mL, 3.7 equiv) was added to the reaction flask, which was immediately sealed with a septum. The mixture was stirred at room temperature until a conversion of 42% was obtained as determined by LC-MS (aliquot starting after 2 h up to 5.5 h at which time the reaction was typically complete). The reaction was filtered through a medium fritted glass-sintered funnel. The residue was washed with hexanes and the combined filtrate and washes were concentrated under reduced pressure. The residue was purified by flash chromatography (gradient, ethyl acetate/hexanes, 10/90 to 50/50) to give the acetate T81-5(S) (18.3 g) and the alcohol T81-5(R). To determine the enantiomeric purity, a small amount of acetate T81-5(S) was hydrolyzed by stirring with K2CO3 (3 eq) in MeOH for 1 h. (For workup, water was added and MeOH evaporated under reduced pressure, then the aqueous phase extracted with Et2O. The combined organic phases were dried over MgSO4 and concentrated to give the alcohol in quantitative yield.) The crude alcohol was analyzed using LC-MS with a chiral HPLC column to determine the %ee.
- The alcohol T81-5(R) was retreated with the Amano PS with the same procedure used for the racemic alcohol except that the reaction was permitted to proceed until a conversion of 20% as determined by LC-MS (aliquots starting at 2 h, usually reached this level after 8.5 h). Then the mixture was treated as described for T81-5(S) to give the alcohol T81-6(R) (14.1 g) and the acetate T81-5(S). (J. Chem. Soc., Perkin Trans. 1 2000, 367.) This alcohol can be substituted for the T81-6(S) isomer in Step 8-6 below and transformed into T81 containing an 8R-stereocenter.
- Step 8-5. 3-(2-Methoxy-phenyl)-2-methyl-propan-1-ol [T81-6(S)]. To a solution of the acetate T81-5(S) (3.0 g, 13.4 mmol, 1.0 eq) in DCM (65 mL) at -30°C was added a solution of 1 M BBr3 in DCM (33.8 mL, 34 mmol, 2.5 eq). The mixture was stirred at 0°C for 3 h, with monitoring by TLC [(30%AcOEt, 70% hexanes), Rf = 0.30 (UV, CMA)]. Methanol (135 mL, 4x the volume of BBr3) was added slowly at 0°C, followed by water. The mixture was stirred O/N. The aqueous phase was extracted with DCM (3 x 150 mL). The organic phase was dried over MgSO4, filtrated and concentrated under reduced pressure. The residue was purified by flash chromatography (ethyl acetate/hexanes, 8/2, Rf = 0.3) to give 2.25 g of T81-6(S) (98%).
- Step 8-6. 3-[2-(2-Hydroxy-propoxy)-phenyl]-2-methyl-propan-1-ol [T81-7(1R,8S)]. To the phenol T81-6(S) (1.4 g, 8.42 mmol, 1.0 eq) in DMF (16 mL) was added potassium carbonate (1.6 g, 8.42 mmol, 1 eq), and (R)-methylcarbonate (T81-A, 0.73 mL, 8.42 mmol, 1.0 eq). The resulting mixture was stirred at 85°C for 72 h with monitoring of the reaction by TLC [(ethyl acetate/hexanes: 2/8); Rf = 0.30, UV, CMA]. The mixture was cooled to room temperature and brine was added. The aqueous phase was extracted with ethyl acetate and the combined organic phases were extracted with brine. The organic phase was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by flash chromatography (ethyl acetate/hexanes, 2/8, Rf =0.30) to give T81-7(1R,8S) (1.45 g, 76 %). Use of (S)-methylcarbonate in this reaction led to T81(1S,8S) containing a (1S)-stereocenter through the same transformations as described below for the corresponding (1R)-isomer.
- Step 8-7. Toluene-4-sulfonic acid 3-[2-(2-hydroxy-propoxy)-phenyl]-2-methyl-propyl ester [T81-8(1R,8S)]. To a solution of the alcohol T81-7(1R,8S) (2.0 g, 8.9 mmol, 1.0 eq) in pyridine:dichloromethane (1:5, 25 mL) at 0°C was added Ts-Cl (1.90 g, 9.8 mmol, 1.1 eq) in 10 mL dichloromethane dropwise, and the resulting mixture stirred at 0°C degree O/N with monitoring of the reaction by TLC [(ethyl acetate:hexanes, 3:7) Rf = 0.50 (UV)]. Water was added and the aqueous phase was extracted with CH2Cl2. The combined organic phases were washed 3 times with a solution of 0.1 N HCl, then dried over MgSO4 and concentrated under reduced pressure. The residue was purified by flash chromatography (ethyl acetate/hexanes, 3/7, Rf= 0.5) to give T81-8(1R,8S) (2.29 g, 70%). A small amount of di-tosylated compound was also isolated in ~10% yield.
- Step 8-8. 1-[2-(3-Azido-2-methyl-propyl)-phenoxy]-propan-2-ol [T81-9(1R,8S)]. To a solution of the tosylate T81-8(1R,8S) (2.2 g, 5.85 mmol, 1.0 eq) in DMF (25 mL) was added NaN3 (1.85 g, 0.03 mol, 5 eq) and the resulting mixture was stirred at 50°C for 24 h or until complete. The reaction was monitored by TLC [(ethyl acetate:hexanes, 3:7), Rf= 0.35 (UV, CMA)]. Water was added and the aqueous phase was extracted with Et2O. The combined organic phases were washed with brine, then dried over MgSO4 and concentrated under reduced pressure. The residue was purified by flash chromatography (ethyl acetate/hexanes, 3/7) to give T81-9((1R,8S) (1.2 g, 80%).
- Step 8-9. 1-[2-(3-Amino-2-methyl-propylpphenoxy]-propan-2-ol [T81-10 (1R,8S)]. To a solution of T81-9((1R,8S) (1.20 g, 4.8 mmol, 1.0 eq) in ethyl acetate (25 mL) was carefully added 10% Pd/C (0.12 g). Hydrogen was bubbled continuously through the solution and the resulting mixture stirred at room temperature O/N. The reaction was monitored by TLC [(ethyl acetate/ hexanes; 3/7), Rf = baseline, UV, CMA]. The solution was filtered through a pad of Celite® (World Minerals Inc., Santa Barbara, CA) and rinsed with ethyl acetate. The combined filtrate and rinses were evaporated under reduced pressure to give the crude amine T81-10((1R,8S) (1.12 g, 98%).
- Step 8-10. 13-[2-(2-Hydroxy-propoxy)-phenyl]-2-methyl-propyl}-carbamic acid tert-butyl ester [Boc-T81 (1R,8S)]. To a solution of T81-10(1R,8S) (1.12 g, 5.01 mmol, 1.0 eq) in THF:H2O (1:1, 20 mL) was added K2CO3 (1.43 g, 10.0 mmol, 2.0 eq) and Boc2O (3.30 g, 15.0 mmol, 3 eq). The mixture was stirred at room temperature O/N. The reaction was monitored by TLC [(ethyl acetate/hexanes, 4/6). Rf= 0.50 (UV, ninhydrin)]. Aqueous citrate buffer was added and the mixture extracted with Et2O (3x). The combined organic phases were washed sequentially with citrate buffer, water and brine. The organic phase was dried over MgSO4 and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (ethyl acetate/hexanes, 4/6) to give Boc-T81(1R,8S) (1.0 g, 75%).
1H NMR (CDCl3): δ 0.9 (d, 3H), 1.25 (d, 3H), 1.42 (s, 9H), 1.90 (m, 1H), 2.30 (m, 1H), 2.80-3.05 (m, 4H), 3.82 (m, 1H), 4.01 (m, 1H), 4.25 (m, 1H), 6.85 (m, 2H), 7.15 (m, 2H);
LC-MS (Grad_A4): tR = 8.52 min; Mass found: 323;
13C NMR (CDCl3): δ 19.01 29.10, 35.10, 36.20,38.10, 46.20, 62.20, 70.00, 80.10, 115.20, 120.15, 128.10, 130.00, 132.10, 158.50.
Note that T81(1R;8S) and T81(1R,8R) are used to make compounds of formula I with a (1S)-stereocenter. Similarly, T81(1S,8S) and T81(1S,8R) are used to synthesize compounds of formula I with a (1R)-stereocenter. - Step 9-1. In a round bottom flask equipped with a Dean-Stark apparatus, was suspended amino acid G-1 (0.2 mol, 1.0 eq) in toluene (1 L per 0.2 mol, solvent/wash amounts in the procedure are scaled for smaller or larger scales). p-Toluenesulfonic acid (pTSA, 1.2 eq) and benzyl alcohol (5.0 eq) were added and the mixture was stirred at reflux 16-18 h. The mixture was cooled down to room temperature and a white precipitate was formed. The precipitate was diluted with MTBE (500 mL), filtered and triturated with MTBE (3 x 500 mL). The solid was dried under high vacuum for 16-18 h to give the intermediate tosylate salt.
- The tosylate salt was taken up in an aqueous solution of 1 M Na2CO3 (200 mL). The basic aqueous phase was extracted with EtOAc (4 x 150 mL) and the combined organic phases were washed with brine (1 x 100 mL), dried over MgSO4, filtered, concentrated under reduced pressure and dried in vacuo to give the free amino-ester G2.
- As will be appreciated by those skilled in the art, alternative esters to the benzyl ester can also be employed for the procedure with modification of the deprotection protocol.
- Step 9-2. To a solution of freshly free based G-2 (1.0 eq.) in DCM (90-100 mL) was added triethylamine (TEA, 1.1 eq), pyridine (3.4 eq) and Bts-Cl (2.0 eq). The reaction was stirred O/N at room temperature. The reaction mixture was diluted with DCM (100 mL), washed sequentially with aqueous citrate buffer (2x), brine (1x), dried over MgSO4, filtered and concentrated under reduced pressure. The resulting residue was purified by flash chromatography to give Bts protected amino-ester G-3.
- Step 9-3. Protected tether G-4 (1.5 eq.), triphenylphosphine (8.5g, 32.4 mmol, 1.5 eq) and G-3 (1.0 eq) were dissolved in anhydrous THF (100 mL). The solution was cooled to 0°C in an ice-water bath and diisopropylazodicarboxylate (DIAD, 1.45 eq) was added dropwise. The preformed DIAD:PPh3 adduct (Example 38) could be used for this step as well. After addition, the mixture was stirred 1 h at 0°C, then warmed up slowly to room temperature and stirred for 16-18 h under nitrogen. THF was removed under reduced pressure and the crude residue was purified by flash chromatography to give alkylated amino acid ester G-5.
- Note that when tether G-4 contains a hydroxy group bonded to a chiral carbon atom, inversion occurs at that center. Hence, an (S)-stereocenter in the tether leads to compounds of formula I with an (R)-stereocenter at that chiral carbon atom. Likewise, tethers with an (R)-stereocenter lead to compounds of formula I with an (S)-stereocenter at that chiral carbon atom.
- Step 9-4. To a solution of G-5 (1.0 eq) in DMF (75 mL), was added solid sodium carbonate (5.0 eq), followed by 2-mercaptoethanol (5.0 eq). The mixture was stirred for 16-18 h at room temperature. Reaction was monitored by HPLC (Grad_A4). Water (150 mL) was added and the aqueous phase extracted with EtOAc (3 x 75 mL). The combined organic phases were washed with brine (1 x 100mL), dried over MgSO4, filtered and concentrated under reduced pressure. The crude residue was purified by flash chromatography to give debetsylated amino acid ester G-6. It is important to make sure no benzothiazole:mercaptoethanol adduct is left as determined by LC-MS after the reaction as this side product has been shown to inhibit the subsequent hydrogenolysis reaction.
- Step 9-5. This step is described for approximately 15 mmol scale. A round bottom flask under nitrogen atmosphere, was charged with 10% Pd/C (50% wet, 10% by weight) and 95% EtOH (70 mL), followed by a solution of G-6 (15 mmol, 1.0 eq) in
EtOH 95% (70 mL). The mixture was stirred under hydrogen bubbling using a gas dispersion tube for 4 hours. A precipitate formed in the mixture after 2 h. EtOAc or THF (100 mL) was added to obtain a stirring suspension. The mixture was purged with nitrogen for 10 min to remove the excess hydrogen, filtered on a Celite® (World Minerals Inc., Santa Barbara, CA) pad and washed with EtOAc and MeOH until TLC indicated there was no more material eluting. The solvent was evaporated under reduced pressure and the resulting solid was dried under high vacuum to give amino acid G-7. - Step 9-6. For a salt of the amino acid benzyl ester (G-8, 1 eq, described for 15 mmol scale), the solid was taken up in an aqueous solution of 1 M Na2CO3 (50 mL). The basic aqueous phase was extracted with EtOAc (4 x 50 mL) and the combined organic phases were washed with brine (1 x 600 mL), dried over MgSO4, filtered, concentrated under reduced pressure and dried under high vacuum 4-6 h to give the free amino-ester G-9. Step 9-7. G-9 (commercially available or synthesized as described in Step 9-1 or 9-6, 15.5 mmol, 1.0 eq) and the protected amino acid (G-10, 1.05 eq) were dissolved in anhydrous THF/CH2Cl2 (1:1) (80 mL). 6-Cl-HOBt (1.0 eq) and DIPEA (5.0 eq) were then added. The mixture was cooled to 0°C in an ice-water bath and EDCI (1.1 eq) was added. The mixture was stirred 1 h at 0°C, allowed to warm to room temperature and stirred 16-18 h. Solvent was evaporated under reduced pressure and the residue was dissolved in EtOAc (100 mL). The organic phase was washed sequentially with an aqueous solution of citrate buffer (1 M, pH 3.5) (2 x 50 mL), H2O (1 x 50 mL), an aqueous solution of saturated NaHCO3 (2 x 50 mL) and brine (1 x 50 mL). The organic phase was dried over MgSO4, filtered, concentrated under reduced pressure and dried under high vacuum 16-18 h to give crude dipeptide.
- The crude dipeptide (1.0 eq) was dissolved in a 3.3 M HCl solution in EtOAc (prepared by bubbling gaseous HCl into EtOAc at 0°C, 10 eq) and the mixture was stirred for 1 h at room temperature and a white precipitated formed. The precipitate was filtered and washed sequentially with EtOAc (1 x 50 mL) and MTBE (1 x 50 mL), then dried under high vacuum 16-18 h to give hydrochloride salt G-11.
- Step 9-8. To a solution of acid fragment G-7 (1.0 eq) and dipeptide hydrochloride salt G-11 (1.05 eq) in anhydrous THF/DCM (1:1) (10 mL, described for 1.50 mmol) at 0°C was added DIPEA (5.0 eq), followed by HATU (1.05 eq). The mixture was stirred 1 h at 0°C, allowed to warm to room temperature and stirred 16-18 h under nitrogen. Solvent was removed under reduced pressure and the residue dissolved in EtOAc (50 mL). The organic phase was washed sequentially with an aqueous solution of citrate buffer (1 M, pH 3.5) (2 x 25 mL), an aqueous solution of saturated NaHCO3 (2 x 25 mL) and brine (1 x 25 mL). The organic phase was dried over MgSO4, filtered and concentrated under reduced pressure. The crude residue was purified by flash chromatography to give the desired protected alkylated tripeptide G12.
- Step 9-9. A round bottom flask under a nitrogen atmosphere was charged with 10% Pd/C (50% wet, 10% weight) and 95
% EtOH 95% (5 mL, described for 1.30 mmol), followed by a solution of G-12 (1.0 eq) in 95% EtOH (8 mL). The mixture was stirred under hydrogen bubbling for 4 h. The reaction mixture turned very thick after 2-3 h. THF was added to dissolve, at least partially, the suspension. The mixture was purged with nitrogen for 5 min to remove the excess of hydrogen, filtered on a Celite® (World Minerals Inc., Santa Barbara, CA) pad and washed with THF until there was no further material eluting. The solvent was evaporated under reduced pressure and the resulting solid dried under high vacuum to give acid G-13. - Step 9-10. Boc protected G-13 (1.0 eq, described for 1.30 mmol) was dissolved in TFA/TES/DCM (33/3/64) (9.8 eq, approximately 3 mL). The mixture was stirred 30 min at room temperature and evaporated under reduced pressure. The residue was co-evaporated twice with THF, then dried under high vacuum for 16-18 h to give macrocyclic precursor G-14.
- Step 9-11. To a solution of G-14 (1.0 eq, described for 1.30 mmol) in anhydrous THF (sufficient to form a 25 mM solution, although solutions of 10 mM concentration are also acceptable) were added DIPEA (5.0 eq) and DEPBT (1.1 eq). The mixture was stirred at room temperature 16-18 h. THF was evaporated under reduced pressure and the residue was taken up in 1 M Na2CO3 (aq):EtOAc (1:1) (50 mL). The separated basic aqueous phase was washed with brine (2 x 25 mL), dried over MgSO4, filtered and evaporated under reduced pressure. The crude residue was purified by flash chromatography or reverse phase HPLC to give the macrocycle G-15. Table 13 below lists the yields of this final macrocyclization step for other representative compounds of the invention that have been synthesized according to this standard procedure.
Table 13. Representative Yields for Step 9-11 Compound Macrocyclization Yielda Concentration 513 73% 25 mM 554 75% 25 mM 555 78% 25 mM 557 76% 25 mM 558 72% 25 mM 559 83% 10 mM 560 73% 10 mM 561 55% 10 mM 562 87% 10 mM a Yield determined after silica gel purification, based on linear precursor prior Boc deprotection. - Step 10-1. Chlorination of H-(D)Tyr-OMe. To a solution of H-(D)Tyr-OMe (free base, 10-1,0.11 mol, 21.7 g) in glacial acetic acid (550 mL) at 5°C was added SO2Cl2 (12.1 mL, 0.15 mmol, 1.35 eq) dropwise. After 1 h stirring, Et2O was added and the product precipitated. It was filtered and washed with Et2O, to give 20.8 g (85.4%) of 10-2.
LC-MS (UV, CLND, ELSD): 98.2/98.4/99.4; tR = 3.39 min, (M+H)+ 229. - Step 10-2. Double Bts protection. To a solution of 10-2, HCl salt in DCM (50 mL) was added pyridine (5.8 mL, 68 mmol, 6.8 eq) and Et3N (3.0 mL, 22 mmol, 2.2 eq), followed by solid Bts-Cl (9.3 g, 40 mmol, 4 eq). The mixture was then stirred for 16 h at rt. It was diluted with DCM (100 mL), washed sequentially with citrate buffer (2 x 50 mL), saturated aqueous sodium bicarbonate (2 x 50 mL), brine once then dried on MgSO4 and concentrated. The product was purified by flash chromatography (gradient, EtOAc:hexanes 30:70 to 50:50) to give 10-3 as an off-white solid (5.3 g, 85%). Alternatively, the product can be isolated in quantitative yield by trituration with 1.0 M HCl/EtOAc.
1H NMR (CDCl3): consistent with structure
LC-MS (UV, CLND, ELSD): 94.2/95.2/100; tR = 8.88 min, (M+H)+ 624 - Step 10-3. Fukuyama-Mitsunobu. To a solution of 10-3 (3.2 g, 5.1 mmol), Boc-T38(R) (2.2 g, 7.2 mmol, 1.4 eq), PPh3 (1.9 g, 7.2 mmol, 1.4 eq) in THF (25 mL) at 0°C under N2, was added DIAD (1.5 mL, 7.2 mmol, 1.4 eq) dropwise. After the addition, the mixture was stirred at rt for 16 h then concentrated under reduced pressure. The residue was purified by flash chromatography (gradient, EtOAc:hexanes, 30:70 to 45:55) to give 10-4 as a yellow oil that solidified on standing (5.7 g, > 100%). This material can be used directly in the next step. Alternatively, the PPh3 and DIAD can be reduced to 1.1 eq initially, then an additional 0.3-0.4 eq of each added later if required to drive the reaction to completion.
LC-MS (UV, CLND, ELSD): 91/80/100; tR =14.53 min, (M+H)+ 229 - Step 10-4. Debetsylation. To a solution of 10-4 (5.2 g) in DMF (50 mL) was added sodium carbonate (3.3 g, 31 mmol, 5 eq) and 2-mercaptoethanol (2.2 mL, 31 mmol, 5 eq). The mixture was then stirred for 16 h under N2, then DMF was evaporated and the mixture was diluted with water (200 mL) and pH was adjusted to 8. The aqueous layer was extracted with EtOAc (3 x 100 mL). The combined organic layers were washed with brine, then dried with MgSO4 and concentrated under reduced pressure. The product was purified by flash chromatography (EtOAc:hexanes, gradient, 40:60 to 70:30) to give 10-5 as a yellow oil, 4.98 g. LC/MS indicated that this material contained a significant quantity of an adduct of benzothiazole:mercaptoethanol (84% by UV). For higher reaction concentrations, the reaction can also be performed in THF/H2O (1:2) as solvent with LiOH as base.
- Step 10-5. Ester hydrolysis. To a solution of the crude 10-5 (4.98 g, contaminated with benzothiazolemercaptoethanol adduct) in a 1:1 mixture of THF:water (40 mL total), was added 2.6 g of LiOH-H2O (62 mmol). The mixture was stirred for 16 h at rt, then THF was removed under reduced pressure. To the mixture was added 200 mL water and the pH adjusted to 6. A white precipitate formed, which was filtered off and washed with cold water (2 x 40 mL), then cold MTBE (2 x 40 mL) and triturated with iPrOH:heptane (1:2). An off-white solid, 10-6, was obtained (1.94 g, 75% for the last three steps). The product can also be recrystallized in 95% EtOH using heptane to induce solid formation.
LC-MS (UV, CLND, ELSD): 65/98/100 (16% benzothiazole adduct remains, tR =
5.05 min): tR = 6.48 min, (M+H)+ 507. - Step 10-6. Fragment coupling. To a mixture of 10-6 (1.94 g, 3.8 mmol, 1.0 eq) and 10-7 (synthesized as described in Step 10-9, 1.24 g, 4.6 mmol, 1.2 eq) in DMF (20 mL) were added HATU (1.75 g, 4.6 mmol, 1.2 eq) and DIPEA (4.0 ml, 23 mmol, 5 eq.). The mixture was stirred for 16 h at rt under N2, then the solvent evaporated under reduced pressure. The residue was taken up in water (20 mL), the pH adjusted to 8-9 and the product extracted with EtOAc (2 x 20 mL) and DCM (2 x 20 mL). THF was added to the combined organic layers to dissolve the suspended solids, and the mixture was dried on MgSO4 and concentrated under reduced pressure. The product was purified by flash chromatography (gradient, EtOAc:hexanes, 40:60 to 60:40) to give a 10-8 as a yellowish oil (1.5 g, 52%). Repeated reactions provided a crude yield of as high as 73.6%.
LC-MS (UV, CLND, ELSD): 90.7/100/98.7; tR = 11.33 min; (M+H)+ 761. - Step 10-7. Deprotections. To 10-8 (1.5 g, 2.0 mmol) was added a mixture of TFA (70 mL), DCM (30 mL) and Et3SiH (3 mL). The mixture was stirred at rt for 2 h, then concentrated under reduced pressure. It was evaporated twice with DCM, then dried in vacuo. The crude residue, 10-9, was azeotroped with THF (2x) prior to macrocyclization in Step X-8.
- Step 10-8. Macrocyclization. To the above crude 10-9 in THF (100 mL) was added DEPBT (1.2 g, 4.0 mmol, 2 eq) and DIPEA (4.2 mL, 24 mmol, 6 eq) and the mixture stirred at rt under N2 for 16-86 h. THF was removed and the product purified by flash chromatography (100% EtOAc) to give
compound 552 as a white foam (0.80 g, 68.3% for two steps). Similar yields (70.9%) and purity (89.3/91.1/100, UV, CLND, ELSD) were obtained by modifying the above procedure to replace the flash chromatography with passage through a silica gel pad.
LC-MS (UV, CLND, ELSD): 92.9/100/98.6; tR = 5.84 min; (M+H)+ 586. - Step 10-9. Hydrochloride salt formation.
Compound 552 was dissolved in acetonitrile (60 mg.mL) and 1.5 eq. 0.5 M HCl (aq) added to immediately form a solid precipitate. The solid was collected by filtration and washed with acetonitrile. The resulting HCl salt ofcompound 552 can be recrystallized one or more times from 95% EtOH using heptane to induce solid formation. - Step 10-10. Dipeptide hydrogenolysis

- A mixture of dipeptide 10-10 (formed via standard methods from Cbz-Val-OH and H-Nva-OtBu, 2.4 g, 6.0 mmol) and 10% Pd/C (50% wet, 0.5 g) in 95% EtOH (150 mL) was shaken under 30 psi hydrogen in a Parr apparatus. After 6 h, TLC indicated the reaction is over. The mixture was filtered on a well packed Celite® (World Minerals Inc., Santa Barbara, CA) plug and washed with ethanol, then the combined filtrate and washings concentrated under reduced pressure to provide 10-7 sufficiently pure for us in Step 10-6 above..
1H NMR: consistent with structure
LC-MS: tR = 5.86 min; (M+H)+ 272. - Step 11-1. In a round bottom flask equipped with a Dean-Stark apparatus, was suspended H-(D)Phe(3F)-OH (11-1, 35.6 g, 194 mmol, 1.0 eq) in toluene (1 L). paraToluenesulfonic acid (44.3 g, 233 mmol, 1.2 eq), benzyl alcohol (101 mL, 970 mmol, 5.0 eq) were added and the mixture was stirred at reflux 16-18 hours. The mixture was cooled down to room temperature and a white precipitate formed. The precipitate was diluted with MTBE (500 mL), filtered and triturated with MTBE (3 x 500 mL). The solid was dried under high vacuum for 16-18 h to give the tosylate salt as a white solid (85.1 g, 98.6%).
- The tosylate salt (85.1 g) was taken up in an aqueous solution of 1 M Na2CO3 (210 mL). The basic aqueous phase was extracted with EtOAc (4 x 150 mL), then the combined organic phases were washed with brine (1 x 100 mL), dried over MgSO4, filtered, concentrated under reduced pressure and dried under high vacuum to give the free amino-ester 11-2 as a clear oil (49.5 g, 95%).
- Step 11-2. To a solution of freshly free based 11-2 (49.5 g, 181 mmol, 1.0 eq) in DCM (90 mL) was added triethylamine (27.8 mL, 616 mmol, 1.1 eq), pyridine (49.8 mL, 616 mmol, 3.4 eq) and Bts-Cl (84.7 g, 363 mmol, 2.0 eq). The reaction was stirred O/N at room temperature during which time the solution turned orange. The reaction mixture was diluted with DCM (100mL), washed sequentially with citrate buffer (2x) and brine (1x), dried over MgSO4, filtered and concentrated under reduced pressure. The residual oil was purified by flash chromatography [gradient, hexanes:EtOAc (8:2) to hexanes:EtOAc (7:3)] to give Bts protected amino-ester 11-3 as a yellowish solid (63.4 g, 74.4%).
TLC [hexanes:EtOAc (1:1)]: Rf= 0.30 (UV, CMA). - Step 11-3. Boc-T38(R) tether (11-4, 10.0 g, 32.4 mmol, 1.5 eq), triphenylphosphine (8.5 g, 32.4 mmol, 1.5 eq) and Bts-(D)Phe(3F)-OBn (11-3, 10.2 g, 21.6 mmol, 1.0 eq) were dissolved in anhydrous THF (108 mL). The solution was cooled to 0°C in an ice-water bath and diisopropylazodicarboxylate (DIAD, 6.2 mL, 31.3 mmol, 1.45 eq) was added dropwise. After addition, the mixture was stirred 1 h at 0°C, then warmed up slowly to room temperature and stirred for 16-18 h under nitrogen. THF was removed under reduced pressure and the crude residue was purified by flash chromatography [gradient, hexanes/EtOAc (9:1) to hexanes/EtOAc (75:25)] to give alkylated amino acid ester 11-5 as a pale yellow solid (15.1 g, 91%).
TLC [hexanes:EtOAc (1:1)]: Rf= 0.67 (UV, CMA). - Step 11-4. To a solution of alkylated amino acid 11-5 (14.0 g, 18.4 mmol, 1.0 eq) in DMF (74 mL), was added sodium carbonate (9.8 g, 92.0 mmol, 5.0 eq), followed by 2-mercaptoethanol (6.5 mL, 92.0 mmol, 5.0 eq). The mixture was stirred for 16-18 h at room temperature. Reaction was monitored by HPLC [(Grad_A4): 9.69 min (product), 15.37 min (starting material)]. Water (150 mL) was added and the aqueous phase extracted with EtOAc (3 x 75mL). The combined organic phases were washed with brine (1 x 100 mL), dried over MgSO4, filtered and concentrated under reduced pressure. The crude residue was purified by dry pack silica gel chromatography [hexanes:EtOAc (8:2)] to give debetsylated amino acid ester 11-6 as a yellow oil (7.95 g, 76%). It is very important to make sure no benzothiazole:mercaptoethanol adduct is left by LC-MS after the reaction. This side product has been known to inhibit the subsequent hydrogenolysis reaction.
- Step 11-5. A round bottom flask under nitrogen atmosphere was charged with 10% Pd/C (50% wet, 785 mg, 10% by weight) and 95% EtOH (70 mL), followed by a solution of amino acid ester 11-6 (7.85 g, 13.9 mmol, 1.0 eq) in 95% EtOH (70 mL). The mixture was stirred under hydrogen bubbling using a gas dispersion tube for 4 h. A precipitate formed after 2 h. EtOAc (100 mL) was added to obtain a stirring suspension. The reaction mixture was purged with nitrogen for 10 min to remove the excess of hydrogen, filtered on a Celite® (World Minerals Inc., Santa Barbara, CA) pad and washed with EtOAc and MeOH until there was no further material eluting. The solvent was evaporated under reduced pressure and the resulting solid dried under high vacuum to give amino acid 11-7 as a white solid (6.2 g, 94%).
TLC [hexanes/EtOAc (1:1)]: Rf= baseline (UV, CMA);
LC-MS (Grad_A4): 7.04 min. - Step 11-6. Commercially available H-Leu-OBn tosylate salt (11-8, 6.1 g) was taken up in an aqueous solution of 1 M Na2CO3 (50 mL). The basic aqueous phase was extracted with EtOAc (4 x 50 mL) and the combined organic phases were washed with brine (1 x 580 mL), dried over MgSO4, filtered, concentrated under reduced pressure and dried under vacuum 4-6 h to give the free amino-ester 11-9 as a pale yellow oil (3.44 g, 100%).
- Step 11-7. Freshly free based H-Leu-OBn (11-9, 3.44 g, 15.5 mmol, 1.0 eq) and Boc-(D)Val-OH (11-10, 3.54 g, 16.3 mmol, 1.05 eq) were dissolved in anhydrous THF:CH2Cl2 (1:1, 81 mL). 6-Cl-HOBt (2.63 g, 15.5 mmol, 1.0 eq) and DIPEA (14.2 mL, 81.5 mmol, 5.0 eq) were added. The mixture was cooled to 0°C in an ice-water bath and EDC (3.28 g, 17.1 mmol, 1.1 eq) was added. The mixture was stirred 1 hour at 0°C, allowed to warm to room temperature and stirred 16-18 h. Solvent was evaporated under vacuum and the residue was dissolved in EtOAc (100 mL). The organic phase was washed sequentially with an aqueous solution of citrate buffer (1 M, pH 3.5) (2 x 50 mL), H2O (1 x 50 mL), an aqueous solution of saturated NaHCO3 (2 x 50 mL) and brine (1 x 50 mL). The organic phase was dried over MgSO4, filtered, concentrated under reduced pressure and dried under high vacuum 16-18 h to give crude dipeptide as a pale yellow solid (6.6 g, > 100% crude).
TLC [hexanes/EtOAc (1:1)]: Rf= 0.40 (UV, CMA). - The crude dipeptide (6.6 g, 15.7 mmol, 1.0 eq) was dissolved in a 3.3M HCl solution in EtOAc (prepared by bubbling gaseous HCl in EtOAc at 0°C, 43.6 mL, 157 mmol, 10 eq). The mixture was stirred for 1 h at room temperature and a white precipitate formed. The precipitate was filtered and washed with EtOAc (1 x 50 mL) and MTBE (1 x 50 mL), then dried under vacuum 16-18 h to give hydrochloride salt 11-11 as a white solid (4.38 g, 79% for 2 steps).
TLC [hexanes:EtOAc (1:1)]: Rf= baseline (UV, ninhydrin);
LC-MS (Grad_A4): 6.59 min. - Step 11-8. To a solution of 11-7 (750 mg, 1.58 mmol, 1.0 eq) and 11-11 (591 g, 1.66 mmol, 1.05 eq) in anhydrous THF:DCM (1:1, 10.5 mL) at 0°C was added DIPEA (1.4 mL, 7.9 mmol, 5.0 eq), followed by HATU (631 mg, 1.66 mmol, 1.05 eq). The mixture was stirred 1 h at 0°C, allowed to warm to room temperature and stirred 16-18 h under nitrogen. Solvent was removed under reduced pressure and the residue was dissolved in EtOAc (50 mL). The organic phase was washed with an aqueous solution of citrate buffer (1 M, pH 3.5, 2 x 25 mL), an aqueous solution of saturated NaHCO3 (2 x 25 mL) and brine (1 x 25 mL). The organic phase was dried over MgSO4, filtered, then concentrated under reduced pressure. The crude residue was purified by flash chromatography [gradient, hexanes:EtOAc (75:25) to hexanes:EtOAc (6:4)] to give the desired protected alkylated tripeptide 11-12 as a pale yellow gummy solid (988 mg, 81%).
TLC [hexanes/EtOAc (1:1)]: Rf= 0.43 (UV, CMA). - Step 11-9. A round bottom flask under nitrogen atmosphere, was charged with Pd/
C 10% (50% wet, 208 mg, 10% by weight) and 95% EtOH (5 mL), followed by a solution of benzyl ester X-12 (1.04 g, 1.34 mmol, 1.0 eq) in 95% EtOH (8.4 mL). The mixture was stirred under hydrogen bubbling for 4 h. The reaction mixture turned very thick after 2-3 h and THF was added to dissolve the suspension. The mixture was purged with nitrogen for 5 min to remove the excess of hydrogen, filtered on a Celite® (World Minerals Inc., Santa Barbara, CA) pad and washed with THF until there was no further material eluting. The solvent was evaporated under reduced pressure and the resulting solid was dried under high vacuum to give acid 11-13 as a white solid (911 mg, 100%).
TLC: [hexanes/EtOAc (1: 1)]: Rf = baseline (UV, CMA). - Step 11-10. Boc protected alkylated tripeptide 11-13 (911 mg, 1.33 mmol, 1.0 eq) was dissolve in a TFA/TES/DCM (33/3/64, 3.0 mL, 12.9 mmol, 9.8 eq). The mixture was stirred 30 min at room temperature and evaporated under reduced pressure. The oily residue was co-evaporated twice with THF, then dried under high vacuum for 16-18 h to give macrocyclic precursor 11-14 as a pale yellow gummy solid (1.27 g, > 100%).
TLC: [EtOAc:MeOH (9:1)]: Rf = baseline (UV, ninhydrin). - Step 11-11. To a solution of macrocyclic precursor 11-14 [1.27 g, 1.33 mmol (based on quantity obtained after hydrogenolysis), 1.0 eq) in anhydrous THF (53.6 mL, for a 25 mM solution) were added DIPEA (1.17 mL, 6.79 mmol, 5.0 eq) and DEPBT (440 mg, 1.47 mmol, 1.1 eq). The mixture was stirred at room temperature 16-18 h. THF was evaporated under reduced pressure and the residue was taken up in 1 M Na2CO3:EtOAc (1:1, 50 mL). The separated basic aqueous phase was washed with brine (2 x 25 mL), dried over MgSO4, filtered and evaporated under reduced pressure. The crude residue was purified by flash chromatography [gradient; 100% EtOAc to EtOAc:MeOH (98:2)] to give
macrocycle 556 as a pale yellow solid (617 mg, 81%).
TLC [EtOAc/MeOH (9:1)]: Rf = 0.45 (UV, CMA);
LC/MS (Grad_B4): 10.88 min. -

- The synthesis is based upon that in the literature for introduction of this functionality (St Laurent, D. R.; et al. Bioorg. Med. Chem. 1995, 3, 1145). To a solution of primary amine macrocycle 12-1 (0.030 mmol) in 15 mL anhydrous THF, was added the thioether reagent 12-A or 12-B (0.30 mmol, 10 eq, 65 mg) followed by Et3N (0.60 mmol, 20 eq, 0.083 mL). The mixture was stirred at reflux under nitrogen for 16 h, then evaporated to dryness under reduced pressure. LC-MS analysis indicated in both cases a mixture of Boc-protected (12-2) and Boc-deprotected (12-3) macrocyclic products. The Boc-deprotected product was isolated from the mixture by MS-triggered reverse phase HPLC. Alternatively, the mixture could be treated with HCl in EtOAc to cleave the Boc group prior to the purification. The general procedure was applied for the representative macrocycles 519 (12-3, m =1) and 521 (12-3, m = 2).
-

- To a solution of the fully deprotected macrocycle 13-1 (7.0 mg, 12.9 µmol, 1.0 eq) in anhydrous THF (1.5 mL) was added Et3N (18 µL, 129 µmol, 10 eq) followed by guanidylation reagent 13-A (10.1 mg, 25.8 µmol, 2.0 eq). (Fechtinger, K.; Zapf, C.; Sings, H. L.; Goodman, M. J. Org. Chem. 1998, 63, 3804.) The mixture was stirred O/N at room temperature under nitrogen atmosphere. The solvent was evaporated under reduced pressure and the crude residue purified by flash chromatography [gradient, MeOH/DCM (1:99) to (5:95)] to provide 13-2. After evaporation of the desired fractions, a solution of TFA/Et3SiH/DCM (50/3/47, 2 mL) was added to the doubly Boc protected macrocycle product 13-2 in a 20 mL vial. The mixture was stirred for 2 h at room temperature, then concentrated under reduced pressure to give the desired guanidine analog 13-3 as its TFA salt. The crude product was purified by reverse phase HPLC.
TLC [MeOH:DCM (1:9)]: Rf= 0.57 (UV, CMA). -

- The general procedure is exemplified for the representative macrocycle 522. To a solution of macrocycle 14-1 (0.01 mmol) in EtOH (3 mL) was added triethylamine (0.1 mmol, 10 eq) and the reagent 14-A. The mixture was stirred at 55°C for 16 h, then concentrated under reduced pressure. The product was purified using the ISCO CombiFlash® (Teledyne Isco, Inc., Lincoln, NE, USA), then directly subjected to the next step. The intermediate thioether was dissolved in 7 N NH3 in MeOH, then stirred at 55°C for 16 h in a pressure bottle. The solvent was evaporated and the residue subjected to preparative HPLC to provide the cyanoguanidine macrocycle 522.
- Step 15-1. (Panda, G.; Rao, N. V. Synlett 2004, 714.) To a stirred suspension of Boc-L-Ser-OH (15-1, 5.0 g, 24.5 mmol, 1.0 eq) in 160 mL of anhydrous DMF at room temperature was added N,N-diisopropylethylamine (DIPEA, 4.25 mL, 24.5 mmol, 1.0 eq). To the mixture was added HBTU (9.30 g, 24.5 mmol, 1.0 eq) and the reaction stirred vigorously at room temperature for 5-10 min, then cooled in an ice-water bath and N,O-dimethyl hydroxylamine HCl salt (2.65 g, 27 mmol, 1.1 eq) added followed by N,N-diisopropylethylamine (4.70 mL, 27 mmol, 1.1 eq). The mixture was stirred for 1 h, then the ice bath removed and the reaction stirred O/N at room temperature. The reaction mixture was cooled to 0°C and saturated aqueous NaHCO3 solution (200 mL) was added slowly. The reaction mixture was extracted with ethyl acetate (4 x 200 mL). The combined organic extracts were washed with brine, dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude residue was purified by flash chromatography (100% EtOAc) to afford 15-2 as a colorless solid (5.50 g, 90%).
TLC (100% EtOAc): Rf = 0.40 (ninhydrin). - Step 15-2. To a mixture of the alcohol 15-2 (5.0 g, 20 mmol, 1 eq) and triethylamine (5.60 mL, 40.3 mmol, 2 eq) in 100 mL of anhydrous dichloromethane under N2, was added freshly distilled methanesulfonyl chloride (2.35 mL, 30 mmol, 1.5 eq) dropwise over 5 min. The reaction was allowed to warm to room temperature and stirred for 3 h. The mixture was washed sequentially with cold water, 1 M citrate buffer, saturated aqueous sodium bicarbonate, and brine. The organic layer was dried over magnesium sulfate and concentrated under reduced pressure to provide 15-3 (98% combined yield), sufficient for use in the following step. This compound should be prepared and used in the same day. Step 15-3. To a solution of 15-3 (5.0 g, 15.3 mmol, 1 eq) in 60 ml of anhydrous DMF was added sodium azide (3.0 g, 46 mmol, 3 eq) under an argon atmosphere and the mixture stirred at 50°C for 12 h. ( CAUTION! Such quantities of NaN3 should be handled carefully.). The reaction mixture was cooled to 0°C and water (100 mL) added very slowly. The reaction mixture was extracted with ethyl acetate (3 x 100 mL). The combined organic extracts were washed with brine (2 × 200 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The oily residue was purified by flash chromatography (hexanes:EtOAc, 1:1), to afford Boc-AA5 as a colorless solid (3.35 g, 80%).
TLC [hexanes:EtOAc (1:1)]: Rf= 0.5 (ninhydrin);
1H NMR (CDCl3,): δ 1.42 (s, 9H), 3.21 (s, 3H), 3.45-3.55 (m, 2H), 3.75 (s, 3H), 4.80 (m, 1H), 5.50 (br s, 1H);
LC-MS: tR = 5.66 min; [M+H]+: 275. -

- To a solution of 537 (1 g, 1.66 mmol, 1 eq) in THF:Water (1:1, 10 mL) at room temperature was added Boc2O (1.08g, 4.97 mmol, 1 eq) and K2CO3 (0.352 g, 3.33 mmol, 2 eq). The mixture was stirred at room temperature for 72 h. The solution was extracted with ethyl acetate, then the organic layers washed with brine, dried over magnesium sulfate and concentrated to dryness under reduced pressure. The resulting residue was purified by flash column chromatography (ethyl acetate:hexanes, 1:1) to give the protected macrocycle Boc-537 (75% yield).
-

- To a solution of macrocycle (537, synthesized via the General Procedure of Example 9, 1.0 g, 1.65 mmol, 1 eq) in THF (10 mL) at room temperature was added triphenylphosphine (0.440 g, 1.65 mmol, 1 eq), and 1 ml of water. The mixture was stirred at 40°C for overnight reaction. The reaction mixture was concentrated to dryness under reduced pressure and the aqueous phase was extracted with ethyl acetate (3 times), dried over MgSO4, filtered and concentrated. The residue was purified by flash column chromatography on silica gels (ethyl acetate:methanol, 9:1) to give 17-2 (90% yield). Similarly, this procedure applied to compound 538 (also synthesized via the General Procedure of Example 9, 0.940 g, 1.65 mmol, 1 eq) gave 17-13 (90% yield), and applied to compound 545 (also synthesized via the General Procedure of Example 9, 0.960 g, 1.65 mmol, 1 eq) gave 17-20 (90% yield).
- Such amino macrocycles can also be synthesized by assembly with appropriately protected Dap derivatives using Example 9 or the solid phase procedure illustrated in
Figure 7 . -

- The procedure is exemplified for the representative macrocycle 525. To a solution of macrocycle 17-2 (50 mg, 86.5 µmol, 1 eq) in acetonitrile/trimethylorthoformate (TMOF, 2 mL/0.5 mL) at room temperature was added glutaraldehde (35 µl, 86.5 µmol, 1 eq), NaBH3CN (8.10 mg, 0.13 mmol, 1 eq) and 1 drop of acetic acid. The mixture was stirred at room temperature O/N. The solution was cooled to 0°C and 1 mL of a solution of NaOH (25%) was slowed added. The reaction was stirred during 5 min. The aqueous phase was extracted with ethyl acetate (3x), then the combined organic phases washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (100% EtOAc to give 525 (50% yield). Compound 534 was synthesized from 17-13 using the standard procedure of Example 18 in 50% yield.
-

- To a solution of macrocycle 17-2 (50 mg, 86.5 µmol, 1 eq) in acetonitrile/TMOF (2 mL/0.5 mL) at room temperature was added
butane 1,4-dialdehyde (7.5 mg, 86.5 µmol, 1 eq), and 1 drop of acetic acid. The mixture was stirred at room temperature O/N. The solution was cooled to 0°C and 1 mL of a solution of 25% NaOH was slowed added. The reaction was stirred for 5 min, then the aqueous phase extracted with ethyl acetate (3x). The combined organic phases were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (100% EtOAc) to give macrocycle 19-1 (50% yield). Compound 540 was synthesized from 17-13 using the standard procedure of Example 19 in 50% overall yield for the three step sequence. -

- To a solution of macrocycle 19-1 (50 mg, 86.5 µmol, 1 eq) in 95% ethanol (2 mL) at room temperature was added PtO2 (10 mg, 20% weight), and 1 drop of acetic acid. The mixture was stirred at room temperature under a hydrogen atmosphere using a balloon O/N. The PtO2 was filtered through Celite® (World Minerals Inc., Santa Barbara, CA) and washed with ethanol. All solvents were removed under reduced pressure. The resulting residue was purified by HPLC to give 20-1 (98% yield).
-

- The procedure is exemplified for the representative macrocycle 526 and is based upon the literature procedure for this functional group (Rostovstev, V.; Green, L. K.; Fokin, V. V.; Sharpless, K. B. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596 and references cited). To a solution of macrocycle 537 (100 mg, 0.165 mmol, 1 eq), CuI (3.4 mg, 0.0165 mmol, 0.1 eq) and DIPEA (30 µL, 0.165 mmol, 1 eq) in acetonitrile/methanol (2 mL/0.5 mL) at room temperature. The reaction was cooled to -78°C and an excess of propylene gas was bubbled (over 10 eq) into the solution and the flask sealed. The mixture was stirred at room temperature O/N. The reaction was cooled to 0°C, propylene was released slowly, and Cu was filtered through Celite® (World Minerals Inc., Santa Barbara, CA) and washed with acetonitrile. All solvents were removed under reduced pressure. The resulting residue was purified by purified by flash chromatography (ethyl acetate:methanol, 95:05) to give 526 (76% yield).
-

- The procedure is exemplified for the representative macrocycle 528. To a solution of 537 (100 mg, 0.165 mmol, 1 eq) in toluene (1 mL) was added 2-butyne (3 mL, excess) at room temperature and the flask sealed. The mixture was stirred at 140°C for 48 h. The reaction was cooled to 0°C, 2-butyne was released slowly and all solvents removed under reduced pressure. The residue was purified by purified by flash chromatography (100% ethyl acetate) to give 528 (65% yield).
-

- The procedure is exemplified for the representative macrocycle 527. To a solution of 537 (100 mg, 0.165 mmol, 1 eq) in DMSO (1 mL) was addedZnBr2 (37 mg, 0.165 mmol, 1eq), acetonitrile (5 mL, excess) at room temperature. The mixture was stirred at 140°C for 48 h. The reaction was cooled to room temperature, then solvents were removed under reduced pressure. The resulting residue was purified by purified by flash chromatography on silica gels (100% ethyl acetate) to give 527 (35%yield).
-

- The procedure is exemplified for the representative macrocycles 523. To a solution of 537 (100 mg, 0.165 mmol, 1 eq) were added Boc-propargylamine (28 mg, 0.184 mmol, 1.1 eq), DIPEA (30 µL, 0.165 mmol, 1 eq), and CuI (3.4 mg, 0.0165 mmol, 0.1 eq) in acetonitrile/methanol (2 mL/0.5 mL) was stirred at room temperature O/N. The solvents were removed under reduced pressure and the resulting residue purified by purified by flash chromatography (ethyl acetate:methanol, 95:05) to give 24-1 (80% yield). 24-1 was treated with 2 mL of HCl in dioxane (4 N, 0.79 mmol, 20 eq) and the mixture stirred at room temperature for 1 h. The reaction mixture was concentrated to dryness under reduced pressure to give 523 (98% yield).
-

- Step 25-1. To a solution of 17-2 (1.0 g, 1.73 mmol, 1 eq) in DCM (10 mL) at room temperature was added Bts-Cl (0.41 g, 1.73 mmol, 1 eq), and triethylamine (0.175g, 1.73 mmol, 1 eq). The mixture was stirred at room temperature O/N. The reaction mixture was washed with 0.1 N HCl, dried over magnesium sulfate and concentrated to dryness under reduced pressure. The resulting residue was purified by flash chromatography (EtOAc:hexanes, 1:1) to give 25-1 (95% yield).
- Step 25-2. To a solution of 25-1 (50 mg, 64.5 µmol, 1 eq) in THF (10 mL) at room temperature was added methanol (26 µL, 0.645 mmol, 10 eq), the pre-formed triphenylphosphine-DIAD adduct (synthesized as described in Example 38, 32 mg, 0.071 mmol, 1.1 eq). The mixture was stirred at room temperature O/N. The reaction mixture was concentrated to dryness under reduced pressure. The residue was purified by flash chromatography (ethyl acetate:hexanes, 1:1) to give 25-2 (98% yield).
- Step 25-3. 25-2 (50 mg) was dissolved in ethanol:THF (1:1,2 mL) at room temperature, treated with thiophenol resin (1 g) and agitated on an orbital shaker for 2 h. The resin was filtered off and washed several times with ethanol:THF. The filtrate and washings were removed under reduced pressure and the crude residue subjected to HPLC purification to give 25-3 (50% yield).
- Compound 546 was synthesized from 17-20 using the standard procedure of Example 25 in ~44% overall yield for the three step sequence.
- Compound 548 was synthesized from 17-13 using the standard procedure of Example 25 in 40% overall yield for the three step sequence.
- Monomethylamino-containing macrocycles are also synthesized using building block Boc-AA4 in solid phase methods as illustrated in
Figure 7 or in solution phase procedures as described in Example 9. -

- The procedure is exemplified for the representative macrocycle 524. To a solution of 17-2 (0.10 g, 0.173 mmol, 1 eq) in DMF (1.7 mL) at room temperature was added 2-bromopyrimidine (0.136 g, 0.865 mmol, 5 eq), and K2CO3 (60 mg, 0.432 mmol, 2.5 eq). The mixture was stirred at 70°C O/N, then cooled to RT. The reaction mixture was diluted with 10 mL of ethyl acetate, washed with 0.1 N HCl and brine, then dried over magnesium sulfate and concentrated to dryness under reduced pressure. The resulting residue was purified by flash chromatography (ethyl acetate:hexanes, 1:1) to give 524 (96% yield). Compound 535 was synthesized from 17-13 using the standard procedure of Example 26 in 96% yield.
- Compound 550 was synthesized from X-13 using the standard procedure of Example 26 in 80% yield.
- Step 27-1. To a solution of 27-1 (synthesized from the corresponding macrocycle containing a free secondary amine and a protected primary amine by treatment with Bts-Cl similarly to as described in Step 11-2 followed by deprotection of the Boc under standard conditions, 3.3 g, 3.63 mmol, 1 eq) in TFA (50 mL, 0.653 mol, 180 eq) at room temperature was added polystyrene (PS) sulfonamide resin (Argonaut Technologies, now part of Biotage AB, Uppsala, Sweden, 1.1 mmol/g,, 16.8 g, 18.2 mmol, 5 eq). The mixture was stirred at 70°C for 2 h. The resin was filtered off and washed with TFA and DCM several times. The filtrate and washings were removed under reduced pressure. The resulting residue was purified by flash chromatography (100% ethyl acetate) to give 27-2 (70% yield).
- Step 27-2. To a solution of 27-2 (1.2 g, 1.55 mmol, 1 eq) in acetonitrile/TMOF (6 mL/2 mL) at room temperature was added formaldehyde (0.426 ml, 15.5 mmol, 10 eq), NaBH3CN (97 mg, 15.5 mmol, 10 eq) and 1 drop of acetic acid. The reaction was stirred at room temperature for 2 h. The solution was cooled to 0°C, then 1 mL of a solution of 25% NaOH slowly added and the reaction stirred for 5 min. The aqueous phase was extracted with ethyl acetate (3x), washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (ethyl acetate:methanol, 95:5) to give 27-3 (75% yield).
- Step 27-3. To a solution of 27-3 (1.33 g, 1.66 mmol, 1 eq) in DMF (10 mL) at room temperature was added mercaptopropanoic acid (0.9 g, 8.30 mmol, 5 eq), and K2CO3 (1.15 g, 8.30 mmol, 5 eq). The mixture was stirred at room temperature for 12 h. The reaction mixture was diluted with ethyl acetate and water, then extracted with ethyl acetate. The combined organic layers were washed with brine, dried over magnesium sulfate and concentrated to dryness under reduced pressure. The resulting residue was purified by flash chromatography (ethyl acetate:methanol, 9:1) to give 27-4 (80% yield).
- Step 28-1. To a solution of 538 (0.200 g, 0.352 mmol, 1 eq) in DCM (2 mL) at room temperature were added trifluoroacetic anhydride (TFAA, 122 µL, 0.882 mmol, 2.5 eq) and triethylamine (TEA, 250 µL, 1.76 mmol, 5 eq). The reaction was stirred at 0°C for 1 h. The mixture was then diluted with 10 mL of dichloromethane and washed with 0.1 N HCl and brine, dried over magnesium sulfate and concentrated to dryness under reduced pressure. The residue was purified by flash chromatography (ethyl acetate:hexanes, 1:1) to give 28-1 (35% yield)
- Step 28-2. To a solution of 28-1 (66 mg, 0.0994 mmol, 1 eq) in ethyl acetate (5 mL) at room temperature was added Pd/C (15 mg, 20% by weight), and 1 drop of acetic acid. The mixture was stirred at room temperature under a hydrogen atmosphere using a balloon O/N. The Pd/C was filtered through Celite® (World Minerals Inc., Santa Barbara, CA) and washed with ethyl acetate. The filtrate and washings were removed under reduced pressure to provide 28-2.
- Step 28-3. The crude 28-2 was dissolved in acetonitrile/TMOF (6 mL/2 mL) at room temperature and formaldehyde (22 µL, 0.0995 mmol, 3 eq), NaBH3CN (6.15 mg, 0.0298 mmol, 2 eq) and 1 drop of acetic acid were added. The mixture was stirred at room temperature for 2 h. The solution was cooled to 0°C and 1 mL of a solution of 25% NaOH was slowly added and the mixture stirred for 5 min. The aqueous phase was extracted with ethyl acetate (3x), washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was directly dissolved in methanol (5 mL) at room temperature, K2CO3 (15 mg) added and the mixture stirred at room temperature for 1 h. The solution was cooled to 0°C and 1 mL of a solution of 0.1 N HCl was slowly added and the mixture stirred for 5 min. The aqueous phase was extracted with ethyl acetate (3x), washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude residue was subjected to HPLC purification to give 549 (< 5 mg).
-

- To a solution of 29-1 (36 mg, 1 eq) in TFA (10 mL, 180 eq) at room temperature was added polystyrene (PS) sulfonamide resin (Argonaut Technologies, now part of Biotage AB, Uppsala, Sweden, 1.1 mmol/g, 0.5 g, 5 eq). The mixture was stirred at 70°C for 2 h. The resin was filtered off and washed with TFA and DCM several times. The filtrate and washings were removed under reduced pressure. The resulting residue was purified by flash chromatography (100% ethyl acetate) to give 509 (50%).
-

- The procedure is exemplified for the synthesis of compound 576. To a solution of the Bts protected macrocycle 30-1 (50 mg, 67.6 µmol, 1.0 eq) in anhydrous DCM (4 mL) was added Et3N (188 µL, 1.35 mmol, 20 eq) followed by DMAP (1.6 mg, 13.5. µmol, 0.2 eq). The mixture was cooled to 0°C in an ice-water bath and MsCl (105 µL, 1.35 mmol, 20 eq.) added. The mixture was stirred for 1 h at 0°C, warmed to room temperature and stirred O/N under nitrogen atmosphere. The solvent was evaporated under reduced pressure and the Bts protecting group was removed from 30-2 using polymer bound thiophenol (1 g of freshly prepared resin) in a solution of THF:95% EtOH (1:1) for 2 h. Filtration and evaporation of the solvents under reduced pressure gave crude 576 which was purified by reverse phase HPLC.
TLC [MeOH:DCM (5:95)]: Rf = 0.24 (UV, CMA). -

- The procedure is exemplified for the synthesis of compound 577. To a solution of the Bts protected macrocycle 31-1 (synthesized from the corresponding macrocycle containing a free secondary amine and a protected primary amine by treatment with Bts-Cl similarly to as described in Step 11-2 followed by deprotection of the Boc under standard conditions, 40 mg, 54.0 µmol, 1.0 eq) in anhydrous DCM (3.5 mL) was added Et3N (150 µL, 1.08 mmol, 20 eq) followed by DMAP (1.3 mg, 10.8 µmol, 0.2 eq). The mixture cooled to 0°C in an ice-water bath and TMS isocyanate (143 µL, 1.08 mmol, 20 eq) then added. The mixture was stirred for 1 h at 0°C, warmed to room temperature and stirred O/N under nitrogen atmosphere. The reaction was monitored by HPLC-MS. The solvent was evaporated under reduced pressure and the crude residue purified by flash chromatography [gradient, DCM to MeOH:DCM (1:9)]. After evaporation of the combined product-containing fractions, the Bts protecting group was removed from 31-2 using polymer bound thiophenol (1 g of freshly prepared resin) in a solution of THF:95%EtOH (1:1) for 2 h. Filtration and evaporation of the solvents under reduced pressure gave crude 577 which was purified by reverse phase HPLC.
-

- The procedure is exemplified by the synthesis of the representative compound 581. To a solution of 32-1 (60 mg, 81.1 µmol, 1.0 eq) in TMOF:MeCN (1:1, 5 mL) was added acetone (148 µL, 2.02 mmol, 25 eq) followed by NaBH3CN (126 mg, 2.02 mmol, 25 eq) and glacial acetic acid (11.6 µL, 202.8 umol, 2.5 eq). The mixture was stirred at room temperature O/N under a nitrogen atmosphere. A solution of sodium hydroxide (1 M) was then added until pH 10-12 and the resulting aqueous phase extracted with DCM (3x). The combined organic phases were washed once with brine, dried over MgSO4, filtered and evaporated under reduced pressure to give crude 32-2 which was purified by reverse phase HPLC. Removal of the Bts group was achieved as described for Example 31.
- Use of other aliphatic aldehydes and ketones in place of acetone in this procedure leads to other analogous alkylated amine products.
-

- The procedure is exemplified by the synthesis of the representative compound 506 and is also applicable for representative compound 518 starting from 33-2. Nitrile 33-1 (obtained from solid phase synthesis with appropriate amino acid building blocks according to the procedure outlined in
Figure 7 , 218 mg crude) was dissolved in a solution of 1.25 M HCl in EtOH (25 mL) at 0°C and gaseous HCl was bubbled into the solution for 30 min. The mixture was then stirred at room temperature for 2 h. The solution was evaporated under reduced pressure, then dried under high vacuum. The crude residue was dissolved in a solution of 2 M ammonia in ethanol (25 mL) and the mixture stirred at room temperature O/N. The solution was evaporated under reduced pressure and purified by reverse phase HPLC to give 33-3 (17.4 mg). Removal of the Bts group was achieved as described for Example 31. - Step 34-1. 3-(2-(R) Hydroxyethyl-1-oxy)-2-bromopyridine [T94-2(R)]. 2-Bromo-3-pyridinol T94-1, 2.0 g, 12 mmol, 1 eq) was dissolved in DMF (anhydrous, 50 mL) at room temperature under an atmosphere of nitrogen. Potassium carbonate (1.6 g, 12 mmol, 1 eq) and (R)-methyl carbonate (1 mL, 12 mmol, 1 eq) was added and the mixture stirred vigorously for 15 min. The reaction was then heated to 85°C using an oil-bath for 72 h. To the mixture was added 100 mL of water and the aqueous layer extracted with ethyl acetate (5 x 100 mL). The combined organic layers were washed with brine (200 mL) and dried over MgSO4. The organic layer was concentrated under reduced pressure. The resulting residue was subjected to flash chromatography (100% ethyl acetate) to obtain T94-2(R) as a pale-yellow oil (2.12 g, 76%).
TLC (100% ethyl acetate): Rf: 0.55 (UV, CMA) - Step 34-2. 3-(2-(R) Hydroxyethyl-1-oxy)-2-(1-tert-butoxycarbonylamino-prop-2-yn-3-yl)pyridine [T94-3(R)]. T94-2(R) (1 g, 4.3 mmol, 1 eq) and Boc-propargylamine derivative (1.1 g, 6.46 mmol, 1.5 eq) were dissolved in Et3N (distilled over CaH2):DMF (1:3, 15 mL) and the reaction mixture was degassed by bubbling argon through the solution. trans-Dichloro-bis(triphenylphosphine)palladium(II) (91 mg, 3 mol%, 0.13 mmol) and freshly recrystallized copper(I) iodide (28.5 mg, 0.15 mmol, 3.5 mol%) were added and the mixture was warmed to 50°C and stirred O/N. The solvent was removed under reduced pressure (oil pump vacuum) and the resulting residue purified by flash chromatography (100% ethyl acetate) to obtain T94-3(R) as a pale brown solid (1.12 g, 85%).
TLC (100% ethyl acetate): Rf: 0.33 (UV, CMA) - Step 34-3. 3-(2-(R)Hydroxyethyl-1-oxy)-2-(1-tert-butoxycarbonylaminoprop-3-yl)pyridine [Boc-T94(R)]. The acetylenic compound T94-3(R) (5.0 g, 16 mmol) was dissolved in EtOH (80 mL), then PtO2 (4 mol%, 150 mg) was added and the mixture stirred under a hydrogen atmosphere (a balloon full of hydrogen gas was used) O/N. After this period of time, the reaction mixture was filtered through a pad of Celite® (World Minerals Inc., Santa Barbara, CA), washed with THF, then the combined filtrate and washings were concentrated under reduced pressure to afford a relatively pure (by 1H NMR), but still colored, sample of Boc-T94(R) in a quantitative yield. Further purification can be achieved by subjecting this material to flash chromatography (100% ethyl acetate). The product Boc-T94(R) has the same Rf as the starting material and hence, 1H NMR is the best way to distinguish them.
TLC (100% ethyl acetate): Rf: 0.33 (UV, CMA)
1H NMR (CDCl3): δ 1.30 (d, 3H), 1.4 (s, 9H), 1.90 (m, 2H), 2.80 (m, 2H), 3.15 (m, 2H), 3.80-3.90 (m, 2H), 4.150 (m, 1H), 5,01 (m, 1H), 7.10 (m, 2H), 8.01 (sb, 1H).
13C NMR (CDCl3): δ 19.69, 28.62, 29.77, 40.25, 66.07, 73,62, 76.90, 77.32, 79.18, 118.21, 122.19, 128.80, 132.49, 140.74, 151.63, 153.08, 156.41.
LC-MS(Condition Grad_A4): tR = 4.71 min; [M+H]+ 411.
Note that T94(R) is used to make compounds of formula I with an (S)-stereocenter. Similarly, T94(S) is used to synthesize compounds of formula I with an (R)-stereocenter. - The sequence was based upon reaction methodology reported in the literature. (Lin, N. Bioorg. Med. ; Swindell, C. Heterocycles 1986, 24, 3373-3377; Shimano, Masanao, Tetrahedron Lett. 1998, 39, 4363-4366; Snieckus, V. J. Org. Chem. 1985, 50, 5436-5438.)
- Step 35-1. A solution of 3-hydroxypyridine (T95-1, 14.0 g, 0.147 mmol) in THF/DMF (315 mL/315 mL) was cooled to 0°C, then potassium tert-butoxide (24.7 g, 221 mmol) added in small portions. The mixture was stirred for 5 min, then MOM-Cl (13.0 mL, 162 mmol) added dropwise over 10 min. A saturated aqueous solution of ammonium chloride was added and the THF removed under reduced pressure. The aqueous phase was then extracted with Et2O (3x). The combined organic phases were dried over MgSO4, filtered and concentrated under reduced pressure. The crude T95-2 (orange oil) was used as obtained in the next step.
- Step 35-2. A solution of T95-2 (6.0 g, 43 mmol) and TMEDA (7.8 mL, 52 mmol) in THF (175 mL) was cooled to -78°C and agitated with a mechanical stirrer. nBuLi (1.56 M in hexane, 31.4 mL, 49 mmol) was added dropwise. A sticky orange thick syrup was formed and dissolved over time. The solution was stirred for 1 h then I2 (14.2 g, 56 mmol) was added and the solution stirred another 1 h at -78°C then warmed up to rt O/N. Water was added to the reaction and the THF removed under reduced pressure. The aqueous phase was extracted with Et2O (3x). The organic phases were combined, dried over MgSO4, filtered and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (gradient, 8/2 EtOAc/Hex to 6/4 EtOAc/Hex) to give 7.0 g (68%) of T95-3 as a yellowish solid.
- Step 35-3. To a solution of T95-3 (6.5 g, 24.6 mmol) in DCM (49 mL) at rt was added TFA (49 mL). The mixture was stirred for 2 h at rt. The solvent was removed under reduced pressure and re-treated with the same reagents under the same conditions for an additional hour. The reaction was monitored by 1H NMR spectroscopy on a worked-up reaction aliquot. A saturated aqueous solution of sodium bicarbonate was added and the aqueous phase extracted with EtOAc (3x). The combined organic phases were dried over MgSO4, filtered and concentrated under reduced pressure to give 5.1 g (94%) of T95-4 as a yellowish solid, which was used as such for the following step.
- Step 35-4. To a solution of 3-hydroxy-4-iodopyridine (5.09 g, 23.0 mmol) in DMF (95 mL) were added (R)-propylene carbonate (T95-A, 2.35 g, 23.0 mmol)) and potassium carbonate (3.18 g, 23.0 mmol). The solution was stirred at 85°C for 72 h. Water was added and the aqueous phase extracted with EtOAc (3x). The combined organic phases were washed with brine (1x) and water (1x), dried over MgSO4, filtered and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (100% EtOAc) to give 1.4 g (24%) of T95-4(R).
- Step 35-5. A solution of iodide T95-4(R) (1.27 g, 4.56 mmol) and Boc-propargylamine (1.06 g, 6.83 mmol) in CH3CN/TEA (13.2 mL/5.4 mL) was degassed for 5 min with argon, then Pd(Cl)2(PPh3)2 (95 mg) and Cul (25 mg) were added. The solution was heated at 50°C O/N. The solvent was removed under reduced pressure and the resulting residue was purified by flash chromatography (100% EtOAc) to give 1.23 g (89%) of T95-5(R).
- Step 35-6. To a solution of T95-5(R) (935 mg, 3.05 mmol) in 95% EtOH (20 mL) was added PtO2 (28 mg, 0.122 mmol). The solution was saturated with hydrogen (H2 gas bubbled into the reaction mixture for 2 h), then stirred O/N under a hydrogen atmosphere. The mixture was filtered through Celite® (World Minerals Inc., Santa Barbara, CA), washed several times with EtOAc, then MeOH. The filtrate was concentrated under reduced pressure and the residue thus obtained was suitable for use in the construction of macrocyclic compounds of the present invention without any further purification (see Example 37).
LC-MS (Grad B4): tR = 4.83 min - Note that T95(R) is used to make compounds of formula I with an (S)-stereocenter as described further in Example 37. Similarly, T95(S) is used to synthesize compounds of formula I with an (R)-stereocenter.
- The sequence was based upon reaction methodology reported in the literature. (Meana, A.Synlett 2003, 1678-1682; Canibano, V. Synthesis 2001, 2175-2179.)
- Step 36-1. To a solution of 4-hydroxypyridine (T96-1, 7.0 g, 74 mmol) in CCl4 (360 mL) at rt was added NBS (26.2 g, 0.147 mol). The solution was stirred for 24 h in the dark (covered with aluminum foil). The mixture was concentrated under reduced pressure and the resulting residue triturated with MeOH, then with acetone to give 18.9 g (100%) of T96-2.
- Step 36-2. To a solution of T96-2 (8.0 g, 31.3 mmol) in THF (307 mL) at -90°C was added slowly a solution of n-BuLi (1.15 M in hexanes, 71 mL). The mixture was stirred for 40 min, then water (10 eq) was added and the mixture warmed to room temperature. The solvent was removed under reduced pressure and the resulting residue purified by flash chromatography (acetone:MeOH, 10:1) to give 3.22 g (60%) of T 96-3.
- Step 36-3. T 96-3 (3.22 g, 18.4 mmol), PPh3 (4.82 g, 18.4 mmol) and the mono-PMB ether of (R)-glycerol (T96-A, synthesized from the corresponding diol using standard methods, 1.8 g, 9.2 mmol) were dissolved in THF (14 mL). The mixture was cooled to 0°C, then DIAD (3.5 mL, 17.9 mmol) added dropwise maintaining the temperature at 0°C. The solution was stirred at 0°C for 1 h, then at rt O/N. The mixture was concentrated under reduced pressure and the residue purified by flash chromatography (50/50 EtOAc/Hex) to yield T96-4(R) (42%).
- Step 36-4. To a solution of T96-4(R) (1.25 g, 3.56 mmol) in DIPEA (20 mL) was added Boc-propargylamine and the mixture degassed with argon for 10 min. Triphenylphosphine (121 mg), CuI (29 mg) and Pd(Cl)2PPH3 (160 mg) were then added. The solution was stirred at 70°C O/N. The reaction mixture was concentrated under reduced pressure and the resulting residue purified by flash chromatography (gradient, 40% to 90% EtOAc/Hexane) to give 1.3 g (80%) of T96-5(R).
- Step 36-5. Pd/C (10%, 50% wet, 628 mg) was added to a solution of T96-5(R) (900 mg) and AcOH (1.5 eq) in 95% EtOH (20 mL). The solution was saturated with H2 at 1300 psi. The mixture was filtered through Celite® (World Minerals Inc., Santa Barbara, CA) and washed several times with EtOAc, then with MeOH. The combined filtrate and washings were concentrated under reduced pressure and the crude Boc-T96(R) obtained was used in the synthesis of macrocyclic compounds of the invention without any further purification.
1H NMR (CDCl3): δ 1.4 (d, CH3), 1.5 (s, Boc), 1.8 (t, CH2), 2.6 (t, CH2), 3.15 (t, CH2), 4.0 (m, 2xCH), 4.3 (m, 1H), 4.98 (1H, NH), 6.8 (d, 1H aromatic), 8.3(d, 1H), 8.4 (s, 1H aromatic). - Note that T96(R) is used to make compounds of formula I with an (S)-stereocenter. Similarly, T96(S) is used to synthesize compounds of formula I with an (R)-stereocenter.
- Step 37-1. Boc-T95(R) (901 mg, 2.90 mmol), triphenylphosphine (762 mg, 2.90 mmol) and Bts-(D)Phe(3F)-OBn (976 mg, 2.07 mmol) were dissolved in anhydrous THF (10 mL). The solution was cooled to 0°C in an ice-water bath and diisopropylazodicarboxylate (DIAD, 551 µL) was added dropwise. Once the addition was completed, the mixture was stirred for 1 h at 0°C, then warmed up slowly to room temperature and stirred for an additional 16-18 h under nitrogen. The THF was removed under reduced pressure and the crude residue obtained purified by flash chromatography (gradient, 50/50 EtOAc/Hexane to 80/20 EtOAc/Hex) to give 858 mg of 37-1. Yield for this step is typically 80-90%.
- Step 37-2. To a solution of alkylated amino acid 37-1 (778 mg, 1.02 mmol) in DMF (6 mL) was added sodium carbonate (519 mg, 5 eq), followed by 2-mercaptoethanol (346 µL, 5 eq). The mixture was stirred for 16-18 hours at room temperature. Water was added and the aqueous phase was extracted with EtOAc (3x). The combined organic phases were washed with brine (1x), dried over MgSO4, filtered and concentrated under reduced pressure. The crude residue was purified by flash chromatography (
gradient 6/3 EtOAc/Hexane to 100% EtOAc) to give 552 mg of 37-2 (96%). - Step 37-3. Pd/C (10%, 50% wet, 628 mg) was carefully added to a solution of 37-2 (502 mg, 0.888 mmol) and AcOH (2 drops) in 95% EtOH (6 mL). The solution was saturated with hydrogen (H2 gas bubbled into the reaction mixture for 2 h), then stirred O/N under H2 atmosphere. The mixture was filtered through Celite® (World Minerals Inc., Santa Barbara, CA) and washed several times with EtOAc, then with MeOH. The filtrate was concentrated under reduced pressure and the crude 37-3 (94%) thus obtained was used in the next step without any further purification.
- Step 37-4. To a solution of the amino acid zwitterion 37-3 (293 mg, 0.617 mmol) and H-D-Val-Nva-OBn hydrochloride salt (422 mg, 1.23 mmol) in anhydrous THF (3.0 mL) at 0°C was added DIPEA (1.1 mL, 6.17 mmol), followed by HATU (469 mg, 1.23 mmol). The mixture was stirred for 16-18 h under nitrogen. The solvent was removed under reduced pressure and the resulting residue dissolved in EtOAc. The organic phase was washed sequentially with an aqueous solution of citrate buffer (1 M, pH 3.5, 2x), a saturated aqueous of sodium bicarbonate (2x) and brine (2x). The organic phase was dried over MgSO4, filtered and concentrated under reduced pressure. The residue thus obtained was purified by flash chromatography (80/20 EtOAc /Hex) to give 290 mg of 37-4 (62%).
- Step 37-5. Pd/C (10%, 50% wet, 28 mg) was added to a solution of 37-4 (284 mg, 0.373 mmol) in 95% EtOH (4 mL). The solution was saturated with hydrogen (H2 gas bubbled into the reaction mixture for 2 h), then stirred O/N under a H2 atmosphere. The mixture was filtered through Celite® (World Minerals Inc., Santa Barbara, CA) and washed several times with EtOAc, then with MeOH. The filtrate was concentrated under reduced pressure and the crude 37-5 obtained, 256 mg (100%), was used in the next step without any further purification.
- Step 37-6. Boc protected alkylated tripeptide 37-5 (256 mg) was dissolved in a TFA/TES/DCM (33/3/64, 17 mL) mixture. The reaction was stirred I h at room temperature, then concentrated under reduced pressure. The resulting oily residue was co-evaporated with DCM (3x), then THF (3x) and dried under reduced pressure to give macrocyclic precursor 37-6.
- Step 37-7. To a solution of 37-6 (0.380 mmol) in anhydrous THF (33 mL) were added DIPEA (330 µL, 5 eq) and DEPBT (136 mg, 1.2 eq). The mixture was stirred at room temperature 16-18 h. THF was evaporated under reduced pressure, the residue taken up in EtOAc and washed with brine (2x). The organic solution was dried over MgSO4, filtered and concentrated under reduced pressure. The crude residue obtained was purified by flash chromatography (95/5 DCM/MeOH) to give the macrocycle 565 (65%).
LC/MS (Grad_B4): tR = 5.26 min. -

- Diisopropyl azodicarboxylate (DIAD, 1 eq) is added dropwise to a well-stirred solution of triphenylphosphine (1 eq) in THF (0.4 M) at 0°C under nitrogen. After completion of the addition, the mixture is stirred at 0°C for an additional 30 min. The white solid obtained is collected by filtration (use medium sized fritted filters) and washed with cold anhydrous THF until the washes are colorless. Finally, the white precipitate is washed once with anhydrous Et2O. The adduct is then dried well in vacuo, and stored under nitrogen. (It is important to store this reagent under anhydrous conditions!) The adduct can be used in place of the separately added reagents in Mitsunobu-type reactions.
- Formulations designed to enhance the solubility of drugs often result in both increasing oral bioavailability and clinical efficacy. The most popular approaches incorporate the active lipophilic component into inert lipid vehicles (Christopher J. H. Porter & al. Lipid and lipid-based formulation; optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug. Disc. 2007, 6, 231-246; Aungst, B.J. Novel formulation strategies for improving oral bioavailability of drugs with poor membrane permeation or presystemic metabolism. J. Pharm. Sci. 1993, 81, 979-987), such as oils (Burcham, D.L.; Maurin, M.B.; Hausner, E.A.; Huang, S.M. Improved oral bioavailability of the ), surfactant dispersions (Serajuddin, A.T.M.; Sheen, P.-C.; Mufson, D.; Bernstein, D.F.; Augustine, M.A. Effect of vehicle amphiphilicity on the dissolution and bioavailability of a poorly water-soluble drug from solid dispersion. J. Pharm. Sci. 1988, 77, 414-417), self-emulsifying formulations, (Charman, S.A.; Charman, W.N.; Rogge, M.C.; Wilson, T.D.; Dutko, F.J.; Pouton, C.W. Self-emulsifying drug delivery systems: formulation and biopharmaceutical evaluation of an investigational lipophilic compound. Pharm. Res. 1992, 9, 87-93; Craig, D.Q.M.; Lievens, H.S.R.; Pitt, K.G.; Storey, D.E. An investigation into the physicochemical properties of self-emulsifying systems using low frequency dielectric spectroscopy, surface tension measurements and particle size analysis. Int. J. Pharm. 1993, 96 147-155; Shah, N.H.; Carvajal, M.T.; Patel, C.L; Infeld, M.H.; Malick, A.W. Self-emulsifyinj drug delivery systems (SEDDS) with polyglycolysed glycerides for improving in vitro dissolution and oral absorption of lipophilic drugs. Int. J. Pharm. 1994, 106, 15-23) and emulsions (Palin K.J.; Phillips, A.J.; Ning, A. The oral absorption of cefoxitin from oil and emulsion vehicles in rats. Int. J. Pharm. 1986, 33, 99-104; Kararli, T.T.; Needham, T.E.; Grifan, M.; Schoenhard, G. Ferro, L.J.; Alcorn, L. Oral delivery of a renin inhibitor compound using emulsion formulations Pharm. Res. 1992, 9, 888-893).
- The pharmacokinetic parameters of representative formulations are determined through established methods known to those skilled-in the art as previously referenced in Method 3-I. Results with representative formulations for a representative compound of the present invention are shown below.
Table 14. Oral Bioavailability in Rat of Representative Formulations of Compound 552Vehicle 5% HP-β-CD in water 5% HP-β-CD in water Cremophor EL/ Lysorbate 80/ Soybean oilOleic Acid/ Labrafil Dosing Route IV oral oral oral n (rats) 3 3 3 3 Dose (mg/kg) 2 8 8 8 Dose volume (mL/kg) 4 16 1 1 Dosing solution concentration (mg/mL) 0.5 0.5 8 8 t1/2 (min) 45 ± 13 - - - Cmax (ng/mL) 1719 ± 299 369 ± 97 229 ± 162 143 ± 77 Tmax (min) 5 5/15 15/30 30/60 Clearance (mL/min/kg) 55 ±10 - - - AUCinf (ng.min/mL) 36977 ± 6980 15291 ± 6811 23806 ± 2141 21412 ± 10039 F (%) (individual results) - 6,15,9 18, 15, 16 7, 21, 15 F (%) (average) - 10 ± 5 16 ± 1 14 ± 7 - The invention may be further understood with reference to the following non-limiting clauses:
- 1. A compound of formula I:

- Y is


- R1 is selected from the group consisting of lower alkyl and cycloalkyl;
- R2 is selected from the group consisting of lower alkyl, substituted lower alkyl, cycloalkyl and substituted cycloalkyl;
- R3, R4, R5, and F6 are independently selected from the group consisting of hydrogen, lower alkyl and substituted lower alkyl;
- R7 is selected from the group consisting of hydrogen, lower alkyl, hydroxy and amino;
- R10a and R10b are independently selected from the group consisting of hydrogen, lower alkyl and substituted lower alkyl;
- X1, X2, X6, X7, X8 and X9 are independently selected from the group consisting of hydrogen, halogen, trifluoromethyl and lower alkyl;
- X3, X4 and X5 are independently selected from the group consisting of hydrogen, hydroxyl, alkoxy, halogen, trifluoromethyl and lower alkyl;
- L1, L2, L3 and L4 are independently selected from the group consisting of CH and N; with the proviso that the total number of nitrogens in the ring must be 0, 1, 2 or 3; and
- L5 and L6 are independently selected from the group consisting of O, CR8aR8b and NR9; wherein R8a and R8b are independently selected from the group consisting of hydrogen and lower alkyl; and R9 is selected from the group consisting of hydrogen, lower alkyl, formyl, acyl and sulfonyl.
- Y is
- 2. The compound of
clause 1, wherein Ar is selected from the group consisting of:


- 3. The compound of
clause 1, wherein R1 is selected from the group consisting of methyl, ethyl, isopropyl and cyclopropyl. - 4. The compound of
clause 1, wherein R2 is selected from the group consisting of (CH2)mCH3, (CH2)n1R11, CH2N3, (CH2)n2R12R13, (CH2)n3C(=NR14)NR14R16, (CH2)n4NR17C(=NR18)NR19R20, and (CH2)n5NHC(=O)NR21R22 wherein m is 0, 1, 2, or 3; n1, n2, n3, n4 and n5 are independently 1, 2, 3 or 4; R11, R13, R16, R17, R20, R21 and R22 are independently selected from the group consisting of hydrogen and lower alkyl; R12, R15 and R19 are independently selected from the group consisting of hydrogen, lower alkyl, carboxyalkyl, carboxyaryl and sulfonyl; R14 and R18 are independently selected from the group consisting of hydrogen, lower alkyl, carboxyalkyl, carboxyaryl, sulfonyl and cyano. - 5. The compound of
clause 1, wherein R2 is selected from the group consisting of:


- 6. The compound of
clause 1, wherein L1, L2, L3 and L4 are each CH, L5 is O and L6 is CH2. - 7. The compound of
clause 1, wherein R3, R4, R5 and R6 are each hydrogen;
or R3 is methyl and R4, R5 and R6 are each hydrogen; or R3 is hydroxymethyl and R4, R5 and R6 are each hydrogen; or R3, R4 and R6 are each hydrogen and R5 is methyl; or R3 and R5 are each methyl and R4 and R6 are each hydrogen. - 8. The compound of
clause 1, wherein Y is:
- 9. The compound of
clause 1 having the following structure:















































- 10. A compound having the following structure:

wherein R25 is selected from the group consisting of hydrogen, alkyl, aryl, cycloalkyl, heterocyclic and heteroaryl; R26 is selected from the group consisting of hydrogen, alkyl, aryl, acyl, carboxyalkyl, carboxyaryl, sulfonyl and a standard protecting group used for amino acids; R27 is selected from the group consisting of hydrogen and alkyl; and R28 is selected from the group consisting of


- 11. A compound having the following structure:

wherein X10 and X11 are independently selected from the group consisting of hydrogen, halogen, trifluoromethyl and lower alkyl;
R40, R41, R42, and R43 are independently selected from the group consisting of hydrogen, lower alkyl and substituted lower alkyl;
R44 is selected from the group consisting of hydrogen, alkyl, acyl, carboxyalkyl, carboxyaryl, sulfonyl and a standard protecting group for an amine functional group;
R45 is selected from the group consisting of hydrogen and alkyl;
R46 is selected from the group consisting of hydrogen, alkyl, acyl, sulfonyl and a standard protecting group for a hydroxyl functional group;
L11, L12, L13 and L14 are independently selected from the group consisting of CH and N; with the proviso that the total number of nitrogens in the ring must be 0, 1, 2 or 3; and
L15 and L16 are independently selected from the group consisting of O, CR47R48 and NR49; wherein R47 and R48 are independently selected from the group consisting of hydrogen and lower alkyl; and R49 is selected from the group consisting of hydrogen, lower alkyl, formyl, acyl and sulfonyl. - 12. The compound of
clause 11 having the following structure:



wherein PG1 is selected from the group consisting of hydrogen and a protecting group for an amine functional group, and PG2 is selected from the group consisting of hydrogen and a protecting group for a hydroxy functional group. - 13. A macrocyclic compound, comprising:
- (a) a building block structure; and
- (b) a compound of formula I of
clause 1.
- 14. A method of using a compound of
clause 11 to synthesize a compound of formula I. - 15. A pharmaceutical composition comprising:
- (a) a compound of formula (I) of
clause 1; and - (b) a pharmaceutically acceptable carrier, excipient or diluent.
- (a) a compound of formula (I) of
- 16. A pharmaceutical composition comprising:
- (a) a compound of
clause 9; and - (b) a pharmaceutically acceptable carrier, excipient or diluent.
- (a) a compound of
- 17. A pharmaceutical composition comprising:
- (a) a compound of
clause 13; and - (b) a pharmaceutically acceptable carrier, excipient or diluent.
- (a) a compound of
- 18. A method of suppressing gastrointestinal motility comprising administering to a subject suffering from one or more disorders caused by gastrointestinal hypermotility or hypermotilinemia an effective amount of a compound of formula I:

- Y is

- Ar is selected from the group consisting of:

- R2 is selected from the group consisting of lower alkyl, substituted lower alkyl, cycloalkyl and substituted cycloalkyl;
- R3, R4, R5, and R6 are independently selected from the group consisting of hydrogen, lower alkyl and substituted lower alkyl;
- R7 is selected from the group consisting of hydrogen, lower alkyl, hydroxy and amino;
- R10a and R10b are independently selected from the group consisting of hydrogen, lower alkyl and substituted lower alkyl;
- X1, X2, X6, X7, X8 and X9 are independently selected from the group consisting of hydrogen, halogen, trifluoromethyl and lower alkyl;
- X3, X4 and X5 are independently selected from the group consisting of hydrogen, hydroxyl, alkoxy, halogen, trifluoromethyl and lower alkyl;
- L1, L2, L3 and L4 are independently selected from the group consisting of CH and N; with the proviso that the total number of nitrogens in the ring must be 0, 1, 2 or 3; and
- L5 and L6 are independently selected from the group consisting of O, CR8aR8b and NR9; wherein R8a and R8b are independently selected from the group consisting of hydrogen and lower alkyl; and R9 is selected from the group consisting of hydrogen, lower alkyl, formyl, acyl and sulfonyl.
- Y is
- 19. The method of
clause 14, wherein the compound has any of the structures ofclause 9, or an optical isomer, enantiomer, diastereomer, racemate or stereochemical mixture thereof. - 20. The method of
clause 18, wherein the compound is administered orally. - 21. The method of
clause 18, wherein the compound is administered parenterally. - 22. The method of
clause 18, wherein the subject is a mammal. - 23. The method of
clause 18, wherein the subject is a human. - 24. The method of
clause 18, wherein the compound is co-administered with an additional agent useful for suppressing gastrointestinal motility. - 25. The method of
clause 18, wherein the gastrointestinal disorder is selected from diarrhea, cancer treatment-related diarrhea, cancer-induced diarrhea, chemotherapy-induced diarrhea, radiation enteritis, radiation-induced diarrhea, stress-induced diarrhea, chronic diarrhea, AIDS-related diarrhea, C. difficile associated diarrhea, traveller's diarrhea, diarrhea induced by graph versus host disease, dyspepsia, irritable bowel syndrome, chemotherapy-induced nausea and vomiting (emesis) and post-operative nausea and vomiting and functional gastrointestinal disorders. - 26. A method of treating a disorder associated with abnormal stomach or intestinal absorption in a subject comprising administering a therapeutically effective amount of a compound of formula I:

- Y is

- Ar is selected from the group consisting of:

- R1 is selected from the group consisting of lower alkyl and cycloalkyl;
- R2 is selected from the group consisting of lower alkyl, substituted lower alkyl, cycloalkyl and substituted cycloalkyl;
- R3, R4, R5, and R6 are independently selected from the group consisting of hydrogen, lower alkyl and substituted lower alkyl;
- R7 is selected from the group consisting of hydrogen, lower alkyl, hydroxy and amino;
- R10a and R10b are independently selected from the group consisting of hydrogen, lower alkyl and substituted lower alkyl;
- X1, X2, X6, X7, X8 and X9 are independently selected from the group consisting of hydrogen, halogen, trifluoromethyl and lower alkyl;
- X3, X4 and X5 are independently selected from the group consisting of hydrogen, hydroxyl, alkoxy, halogen, trifluoromethyl and lower alkyl;
- L1, L2, L3 and L4 are independently selected from the group consisting of CH and N; with the proviso that the total number of nitrogens in the ring must be 0, 1, 2 or 3; and
- L5 and L6 are independently selected from the group consisting of O, CR8aR8b and NR9; wherein R8a and R8b are independently selected from the group consisting of hydrogen and lower alkyl; and R9 is selected from the group consisting of hydrogen, lower alkyl, formyl, acyl and sulfonyl.
- Y is
- 27. The method of clause 26, wherein the compound has any of the structures of
clause 9, or an optical isomer, enantiomer, diastereomer, racemate or stereochemical mixture thereof. - 28. The method of clause 26, wherein the disorder is short bowel syndrome or celiac disease.
- 29. The method of clause 26, wherein the disorder is cachexia.
- 30. The method of clause 29, wherein the disorder is cancer-related cachexia, AIDS-related cachexia or renal disease-related cachexia.
- 31. The method of clause 26, wherein the subject is a mammal.
- 32. The method of clause 26, wherein the subject is a human.
- 33. The method of clause 26, wherein the subject is treated with an additional compound that modulates stomach or intestinal absorption.
- 34. A method of treating a disorder associated with inflammation of the gastrointestinal tract in a subject comprising administering a therapeutically effective amount of a compound of formula 1:

- Y is

- Ar is selected from the group consisting of:

- R1 is selected from the group consisting of lower alkyl and cycloalkyl;
- R2 is selected from the group consisting of lower alkyl, substituted lower alkyl, cycloalkyl and substituted cycloalkyl;
- R3, R4, R5, and R6 are independently selected from the group consisting of hydrogen, lower alkyl and substituted lower alkyl;
- R7 is selected from the group consisting of hydrogen, lower alkyl, hydroxy and amino;
- R10a and R10b are independently selected from the group consisting of hydrogen, lower alkyl and substituted lower alkyl;
- X1, X2, X6, X7, X8 and X9 are independently selected from the group consisting of hydrogen, halogen, trifluoromethyl and lower alkyl;
- X3, X4 and X5 are independently selected from the group consisting of hydrogen, hydroxyl, alkoxy, halogen, trifluoromethyl and lower alkyl;
- L1, L2, L3 and L4 are independently selected from the group consisting of CH and N; with the proviso that the total number of nitrogens in the ring must be 0, 1, 2 or 3; and
- L5 and L6 are independently selected from the group consisting of O, CR8aR8b and NR9; wherein R8a and R8b are independently selected from the group consisting of hydrogen and lower alkyl; and R9 is selected from the group consisting of hydrogen, lower alkyl, formyl, acyl and sulfonyl.
- Y is
- 35. The method of clause 34, wherein the compound has any of the structures of
clause 9, or an optical isomer, enantiomer, diastereomer, racemate or stereochemical mixture thereof. - 36. The method of clause 34, wherein the disorder is located in the stomach or intestine.
- 37. The method of clause 34, wherein the disorder is inflammatory bowel disease, ulcerative colitis, Crohn's disease or pancreatitis.
- 38. The method of clause 34, wherein the subject is a mammal.
- 39. The method of clause 34, wherein the subject is a human.
- 40. The method of clause 34, wherein the subject is treated with an additional compound that modulates inflammation.
- The foregoing is illustrative of the present invention, and is not to be construed as limiting thereof. The invention is defined by the following claims, with equivalents of the claims to be included therein.
| Time (min.) relative to Dose Administration | |||||||||||
|
| Pre-dose | 1 | 5 | 20 | 60 | 90 | 120 | 180 | 240 | 300 | |
| Subset A | √ | √ | √ | √ | √ | ||||||
| Subset B | √ | √ | √ | √ | √ | √ | |||||
| Time (min.) relative to Dose Administration | ||||||||||
| Subset ID | Pre-dose | 5 | 15 | 30 | 60 | 90 | 120 | 180 | 270 | 360 |
| Subset A | √ | √ | √ | √ | √ | |||||
| Subset B | √ | √ | √ | √ | √ | |||||
Claims (16)
- A compound having one of the following structures:










for use in a method of suppressing gastrointestinal motility in a subject, or in treating a disorder associated with abnormal stomach or intestinal absorption in a subject, or in a method of treating a disorder associated with inflammation of the gastrointestinal tract in a subject. - The compound of claim 1, wherein:the suppression of gastrointestinal motility is in a subject suffering from a gastrointestinal disorder selected from diarrhoea, dyspepsia, irritable bowel syndrome, chemotherapy-induced nausea and vomiting (emesis), post-operative nausea and vomiting and functional gastrointestinal disorders;the disorder associated with abnormal stomach or intestinal absorption is short bowel syndrome, celiac disease or cachexia; andthe disorder associated with inflammation of the gastrointestinal tract is located in the stomach or intestine, or is inflammatory bowel disease, ulcerative colitis, Crohn's disease or pancreatitis.
- The compound of claim 2, wherein the gastrointestinal disorder is selected from diarrhea, dyspepsia, irritable bowel syndrome, chemotherapy-induced nausea and vomiting (emesis) and post-operative nausea and vomiting.
- The compound of claim 3, wherein the diarrhea is cancer treatment-related diarrhea, cancer-induced diarrhea, chemotherapy-induced diarrhea, radiation enteritis, radiation-induced diarrhea, stress-induced diarrhea, chronic diarrhea, AIDS-related diarrhea, C. difficile associated diarrhea, traveller's diarrhea or diarrhea induced by graft versus host disease.
- The compound of claim 3, wherein the gastrointestinal disorder is selected from dyspepsia and irritable bowel syndrome.
- The compound of claim 2, wherein the disorder associated with abnormal stomach or intestinal absorption is celiac disease
- The compound of claim 2, wherein the cachexia is cancer-related cachexia, AIDS-related cachexia or renal disease-related cachexia.
- The compound of claim 2, wherein the disorder associated with inflammation of the gastrointestinal tract is inflammatory bowel disease, ulcerative colitis, Crohn's disease or pancreatitis.
- The compound of any one of claims 1 to 8, wherein the compound is of number 505, 511, 515, 516, 523, 525, 530, 531, 540, 550, 553, 561, 562, 566, 573 or 578.
- Use of a compound of number 505, 511, 515, 516, 523, 525, 530, 531, 540, 550, 553, 561, 562, 566, 573, 578, 504, 507, 508, 512, 581, 585, 586, 590, 535, 580, 501 or 559, as defined in claim 1, or an optical isomer, enantiomer, diastereomer, racemate or stereochemical mixture thereof, or a pharmaceutically acceptable salt, hydrate or solvate thereof, in the manufacture of a medicament for use in a method of suppressing gastrointestinal motility in a subject, in treating a disorder associated with abnormal stomach or intestinal absorption in a subject, or in a method of treating a disorder associated with inflammation of the gastrointestinal tract in a subject.
- The use of claim 10, wherein the method is as defined in any one of claims 2 to 9.
- The compound of any one of claims 1 to 9, or the use of claim 10 or claim 11, wherein the subject is a mammal and/or a human.
- A compound of number 505, 511, 515, 516, 523, 525, 530, 531, 540, 550, 553, 561, 562, 566, 573, 578, 504, 507, 508, 512, 581, 585, 586, 590, 535, 580, 501 or 559, as defined in claim 1, or an optical isomer, enantiomer, diastereomer, racemate or stereochemical mixture thereof, or a pharmaceutically acceptable salt, hydrate or solvate thereof.
- The compound of claim 13, which is a compound of number 505, 511, 515, 516, 523, 525, 530, 531, 540, 550, 553, 561, 562, 566, 573 or 578.
- A pharmaceutical composition comprising:(a) a compound of number 505, 511, 515, 516, 523, 525, 530, 531, 540, 550, 553, 561, 562, 566, 573, 578, 504, 507, 508, 512, 581, 585, 586, 590, 535, 580, 501 or 559, as defined in claim 1, or an optical isomer, enantiomer, diastereomer, racemate or stereochemical mixture thereof, or a pharmaceutically acceptable salt, hydrate or solvate thereof; and(b) a pharmaceutically acceptable carrier, excipient or diluent.
- The composition of claim 15, wherein the compound is of number 505, 511, 515, 516, 523, 525, 530, 531, 540, 550, 553, 561, 562, 566, 573 or 578.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US82523706P | 2006-09-11 | 2006-09-11 | |
| EP20070838009 EP2054429B1 (en) | 2006-09-11 | 2007-09-11 | Macrocyclic antagonists of the motilin receptor for treatment of gastrointestinal dysmotility disorders |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP07838009.4 Division | 2007-09-11 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP2431380A2 true EP2431380A2 (en) | 2012-03-21 |
| EP2431380A3 EP2431380A3 (en) | 2013-07-03 |
Family
ID=39030996
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP20070838009 Not-in-force EP2054429B1 (en) | 2006-09-11 | 2007-09-11 | Macrocyclic antagonists of the motilin receptor for treatment of gastrointestinal dysmotility disorders |
| EP20110075225 Withdrawn EP2431380A3 (en) | 2006-09-11 | 2007-09-11 | Macrocyclic antagonist of the motilin receptor for treatment of gastrointestinal dysmotility disorders |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP20070838009 Not-in-force EP2054429B1 (en) | 2006-09-11 | 2007-09-11 | Macrocyclic antagonists of the motilin receptor for treatment of gastrointestinal dysmotility disorders |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US9133235B2 (en) |
| EP (2) | EP2054429B1 (en) |
| JP (1) | JP5684474B2 (en) |
| CN (1) | CN101528765B (en) |
| CA (1) | CA2662897C (en) |
| WO (1) | WO2008033328A2 (en) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4928261B2 (en) | 2003-06-18 | 2012-05-09 | トランザイム・ファーマ・インコーポレイテッド | Macrocyclic antagonist of motilin receptor |
| US7491695B2 (en) * | 2003-06-18 | 2009-02-17 | Tranzyme Pharma Inc. | Methods of using macrocyclic modulators of the ghrelin receptor |
| US20080287371A1 (en) * | 2007-05-17 | 2008-11-20 | Tranzyme Pharma Inc. | Macrocyclic antagonists of the motilin receptor for modulation of the migrating motor complex |
| JP5545215B2 (en) * | 2008-08-29 | 2014-07-09 | Jsr株式会社 | Method for producing N-carboxyamino acid anhydride and amino acid carbamate compound |
| JP2010078398A (en) * | 2008-09-25 | 2010-04-08 | Saitama Univ | Method for screening digestive tract mobility substance |
| EP2491004A4 (en) * | 2009-10-23 | 2013-07-03 | Tranzyme Pharma Inc | Macrocyclic inhibitors of serine protease enzymes |
| WO2011053821A1 (en) | 2009-10-30 | 2011-05-05 | Tranzyme Pharma, Inc. | Macrocyclic ghrelin receptor antagonists and inverse agonists and methods of using the same |
| US8795979B2 (en) | 2011-01-18 | 2014-08-05 | General Atomics | Hydrolase enzyme substrates and uses thereof |
| WO2012121248A1 (en) * | 2011-03-10 | 2012-09-13 | 国立大学法人名古屋大学 | Iodoarene derivative, method for producing optically active spirolactone compound using same, and method for producing optically active cyclization adduct |
| CN102557989B (en) * | 2011-12-19 | 2013-12-25 | 深圳翰宇药业股份有限公司 | Ulimorelin intermediate and Ulimorelin preparation method |
| BR112017014750A2 (en) * | 2015-01-09 | 2018-06-19 | Jaguar Animal Health | methods for treating diarrhea in pets |
| CN111747979B (en) * | 2020-07-07 | 2021-10-08 | 新乡海滨药业有限公司 | Preparation method of meropenem key intermediate N-trimethylsilylimidazole |
Citations (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4677097A (en) | 1984-10-05 | 1987-06-30 | Kitasato Kenkyusho | Compositions for inducing contractile motility of the gastrointestinal tract |
| US4920102A (en) | 1988-04-18 | 1990-04-24 | Eli Lilly And Company | Method for treating gastrointestinal disorders |
| US5008249A (en) | 1985-08-31 | 1991-04-16 | Kitasato Kenkyusho | Therapeutic method of stimulating digestive tract contractile motion in mammals |
| US5175150A (en) | 1985-08-31 | 1992-12-29 | Kitasato, Kenkyusho | Erythromycin derivative |
| US5418224A (en) | 1992-01-07 | 1995-05-23 | Kali-Chemie Pharma Gmbh | 4,13-dioxabicyclo[8.2.1]tridecenone compounds and pharmaceutical compositions containing them |
| JPH07138284A (en) | 1993-11-19 | 1995-05-30 | Chugai Pharmaceut Co Ltd | Motilin antagonist |
| US5422341A (en) | 1993-08-06 | 1995-06-06 | Ohmeda Pharmaceutical Products Division Inc. | Motilin-like polypeptides with gastrointestinal motor stimulating activity |
| US5432261A (en) | 1989-01-06 | 1995-07-11 | Sanwa Kagaku Kenkyusho Co. Ltd. | Motlin-like polypeptide and use thereof |
| US5459049A (en) | 1989-01-06 | 1995-10-17 | Sanwa Kagaku Kenkyushko Co., Ltd. | Motilin-like polypeptide and use thereof |
| US5470830A (en) | 1993-08-06 | 1995-11-28 | Ohmeda Pharmaceutical Products Division Inc. | Motilin-like polypeptides that inhibit gastrointestinal motor activity |
| US5470961A (en) | 1992-03-19 | 1995-11-28 | Takeda Chemical Ind., Ltd. | 14 and/or 15-hydroxy 6,9 hemiacetal erythromycin derivatives |
| US5523401A (en) | 1991-04-09 | 1996-06-04 | Abbott Laboratories | Macrocyclic lactam prokinetic agents |
| US5538961A (en) | 1991-04-09 | 1996-07-23 | Abbott Laboratories | Macrocyclic lactam prokinetic agents |
| US5578579A (en) | 1992-01-21 | 1996-11-26 | Abbott Laboratories | 4"-deoxyerythromycin derivatives |
| US5658888A (en) | 1992-05-26 | 1997-08-19 | Chugai Seiyaku Kabushiki Kaisha | Erythromycin derivatives |
| JPH09249620A (en) | 1996-03-13 | 1997-09-22 | Taisho Pharmaceut Co Ltd | Arylalkane derivative |
| US5695952A (en) | 1986-09-12 | 1997-12-09 | Kyowa Hakko Kogyo Co., Ltd. | Method for producing Leu13 !motilin |
| US5712253A (en) | 1996-06-18 | 1998-01-27 | Abbott Laboratories | Macrocyclic 13-membered ring derivatives of erythromycins A and B |
| US5721353A (en) | 1986-09-12 | 1998-02-24 | Kyowa Hakko Kogyo Co., Ltd. | DNAs coding for LCU13 ! motilin |
| US5734012A (en) | 1996-05-16 | 1998-03-31 | Ohmeda Pharmaceutical Products Division Inc. | Cyclic motilin-like polypeptides with gastrointestinal motor stimulating activity |
| WO1998042840A1 (en) | 1997-03-24 | 1998-10-01 | Zymogenetics, Inc. | Motilin homologs |
| WO1999009053A1 (en) | 1997-08-15 | 1999-02-25 | Chugai Seiyaku Kabushiki Kaisha | Phenethylamine derivatives |
| WO1999021846A1 (en) | 1997-10-28 | 1999-05-06 | Ortho-Mcneil Pharmaceutical Inc. | Cyclopentene derivatives useful as antagonists of the motilin receptor |
| US5912235A (en) | 1996-10-24 | 1999-06-15 | Solvay Pharmaceuticals Gmbh | 10, 13, 15-trioxatricyclo 9.2.1.1.9.6 !-pentadecan one derivatives, method for their production and pharmaceutical compositions containing them |
| US5922849A (en) | 1996-11-22 | 1999-07-13 | Abbott Laboratories | Process for preparation of N-demethyl-4"-deoxy-erthromycins A and B |
| WO1999064436A1 (en) | 1998-06-12 | 1999-12-16 | Merck & Co., Inc. | Cloning and identification of the motilin receptor |
| WO2000017231A1 (en) | 1998-09-24 | 2000-03-30 | Chugai Seiyaku Kabushiki Kaisha | Ethylamine derivatives |
| US6077943A (en) | 1996-03-01 | 2000-06-20 | Takeda Chemical Industries, Ltd. | Method of producing erythromycin derivative |
| US6084079A (en) | 1998-05-15 | 2000-07-04 | Keyes; Robert F. | Process for preparing N-demethyl-N-alkyl erythromycin derivatives |
| WO2000044770A1 (en) | 1999-01-28 | 2000-08-03 | Chugai Seiyaku Kabushiki Kaisha | Substituted phenethylamine derivatives |
| US6100239A (en) | 1996-11-26 | 2000-08-08 | Chugai Seiyaku Kabushiki Kaisha | 13-membered ring macrolide compound, medicine containing the same, and process for producing the same |
| US6165985A (en) | 1998-02-13 | 2000-12-26 | Solvay Pharmaceuticals Gmbh | 11-acetyl-12,13-dioxabicyclo[8.2.1]-tridecenone derivatives, processes for their preparation and pharmaceutical compositions comprising them |
| WO2001000830A1 (en) | 1999-06-30 | 2001-01-04 | Zymogenetics, Inc. | Sgip peptides |
| WO2001025257A2 (en) | 1999-10-04 | 2001-04-12 | Neokimia, Inc. | Combinatorial synthesis of libraries of macrocyclic compounds useful in drug discovery |
| WO2001060833A2 (en) | 2000-02-18 | 2001-08-23 | Kosan Biosciences, Inc. | Motilide compounds |
| WO2001068620A1 (en) | 2000-03-13 | 2001-09-20 | Ortho-Mcneil Pharmaceutical, Inc. | Novel cyclopentene derivatives useful as antagonists of the motilin receptor |
| WO2001068621A1 (en) | 2000-03-13 | 2001-09-20 | Ortho-Mcneil Pharmaceutical, Inc. | Novel cyclohexene derivatives useful as antagonists of the motilin receptor |
| WO2001068622A1 (en) | 2000-03-13 | 2001-09-20 | Ortho-Mcneil Pharmaceutical, Inc. | Novel cyclobutene derivatives useful as antagonists of the motilin receptor |
| US20010041791A1 (en) | 1997-03-24 | 2001-11-15 | Zymogenetics, Inc. | Motilin homologs |
| WO2001085694A2 (en) | 2000-05-05 | 2001-11-15 | Ortho-Mcneil Pharmaceutical, Inc. | Substituted diamide derivatives useful as motilin antagonists |
| WO2002013727A1 (en) | 2000-08-16 | 2002-02-21 | Edwards Lifesciences Corporation | Stent for implantation into the carotid artery formed by process using intraluminal mapping |
| WO2002016404A1 (en) | 2000-08-24 | 2002-02-28 | Chugai Seiyaku Kabushiki Kaisha | Cyclic peptide derivative |
| US6380158B1 (en) | 1997-03-24 | 2002-04-30 | Zymogenetics, Inc. | Motilin homologs |
| US6403775B1 (en) | 1998-10-28 | 2002-06-11 | Kosan Biosciences, Inc. | Erythronolide compounds |
| WO2002051855A2 (en) | 2000-12-01 | 2002-07-04 | Kosan Biosciences, Inc. | Motilide compounds |
| US6420521B1 (en) | 1999-06-30 | 2002-07-16 | Zymogenetics, Inc. | Short gastrointestinal peptides |
| US20020094962A1 (en) | 2000-12-01 | 2002-07-18 | Gary Ashley | Motilide compounds |
| WO2002059141A1 (en) | 2001-01-25 | 2002-08-01 | Chugai Seiyaku Kabushiki Kaisha | Peptide derivatives |
| WO2002064092A2 (en) | 2001-02-15 | 2002-08-22 | Kosan Biosciences, Inc. | Method for evaluating therapeutic efficacy |
| WO2002064623A1 (en) | 2001-02-12 | 2002-08-22 | Chugai Seiyaku Kabushiki Kaisha | Process for preparation of peptide derivatives |
| WO2002092592A1 (en) | 2001-05-10 | 2002-11-21 | Solvay Pharmaceuticals Gmbh | Novel 1-amidomethylcarbonyl-piperidine derivatives, method and intermediate products for the production thereof and medicament containing said compounds |
| WO2004019879A2 (en) | 2002-08-29 | 2004-03-11 | Kosan Biosciences, Inc. | Motilide compounds |
| WO2004111077A1 (en) | 2003-06-18 | 2004-12-23 | Tranzyme Pharma Inc. | Macrocyclic antagonists of the motilin receptor |
| WO2005012331A1 (en) | 2003-07-31 | 2005-02-10 | Tranzyme Pharma | Spatially-defined macrocyclic compounds useful for drug discovery |
| WO2005012332A1 (en) | 2003-07-31 | 2005-02-10 | Tranzyme Pharma | Spatially-defined macrocycles incorporating peptide bond surrogates |
| WO2005018576A2 (en) | 2003-08-26 | 2005-03-03 | Kosan Biosciences, Inc. | N-desmethyl-n-substituted-11-deoxyerythromycin compounds |
| US20050065156A1 (en) | 2003-09-17 | 2005-03-24 | Li James J. | Compounds useful as motilin agonists and method |
| WO2005027637A1 (en) | 2003-09-17 | 2005-03-31 | Bristol-Myers Squibb Company | Compounds useful as motilin agonists and method |
| US6939861B2 (en) | 1999-04-16 | 2005-09-06 | Kosan Biosciences, Inc. | Amido macrolides |
| WO2006009674A1 (en) | 2004-06-18 | 2006-01-26 | Tranzyme Pharma, Inc. | Macrocyclic modulators of the ghrelin receptor |
| WO2006009645A1 (en) | 2004-06-18 | 2006-01-26 | Tranzyme Pharma, Inc. | Methods of using macrocyclic modulators of the ghrelin receptor |
| WO2006070937A1 (en) | 2004-12-28 | 2006-07-06 | Chugai Seiyaku Kabushiki Kaisha | Drugs for the treatment and/or prophylaxis of gastroparesis symptom |
| US20060270616A1 (en) | 2005-05-24 | 2006-11-30 | Yaoquan Liu | Motilide compounds |
| US20060287243A1 (en) | 2005-06-17 | 2006-12-21 | Navneet Puri | Stable pharmaceutical compositions including motilin-like peptides |
| WO2006138023A1 (en) | 2005-06-17 | 2006-12-28 | Baxter International Inc. | Stable buffered, pharmaceutical compositions including motilin-like peptides |
| US7211568B2 (en) | 2003-12-18 | 2007-05-01 | Kosan Biosciences Incorporated | 9-Desoxoerythromycin compounds as prokinetic agents |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006046977A1 (en) | 2004-10-26 | 2006-05-04 | Tranzyme Pharma, Inc. | Macrocyclic ghrelin receptor antagonists and methods of using the same |
| JP5122446B2 (en) | 2005-06-13 | 2013-01-16 | トランザイム・ファーマ,インコーポレイテッド | Macrocyclic ghrelin receptor antagonists and inverse agonists and methods of use thereof |
-
2007
- 2007-09-11 CA CA2662897A patent/CA2662897C/en not_active Expired - Fee Related
- 2007-09-11 CN CN200780033494.1A patent/CN101528765B/en not_active Expired - Fee Related
- 2007-09-11 WO PCT/US2007/019705 patent/WO2008033328A2/en active Application Filing
- 2007-09-11 EP EP20070838009 patent/EP2054429B1/en not_active Not-in-force
- 2007-09-11 EP EP20110075225 patent/EP2431380A3/en not_active Withdrawn
- 2007-09-11 US US12/440,802 patent/US9133235B2/en active Active
- 2007-09-11 JP JP2009527455A patent/JP5684474B2/en not_active Expired - Fee Related
Patent Citations (115)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4677097A (en) | 1984-10-05 | 1987-06-30 | Kitasato Kenkyusho | Compositions for inducing contractile motility of the gastrointestinal tract |
| US5008249A (en) | 1985-08-31 | 1991-04-16 | Kitasato Kenkyusho | Therapeutic method of stimulating digestive tract contractile motion in mammals |
| US5175150A (en) | 1985-08-31 | 1992-12-29 | Kitasato, Kenkyusho | Erythromycin derivative |
| US5695952A (en) | 1986-09-12 | 1997-12-09 | Kyowa Hakko Kogyo Co., Ltd. | Method for producing Leu13 !motilin |
| US5721353A (en) | 1986-09-12 | 1998-02-24 | Kyowa Hakko Kogyo Co., Ltd. | DNAs coding for LCU13 ! motilin |
| US6018037A (en) | 1986-09-12 | 2000-01-25 | Kyowa Hakko Kogyo Co., Ltd | DNA coding for (Leu13) motilin |
| US4920102A (en) | 1988-04-18 | 1990-04-24 | Eli Lilly And Company | Method for treating gastrointestinal disorders |
| US5432261A (en) | 1989-01-06 | 1995-07-11 | Sanwa Kagaku Kenkyusho Co. Ltd. | Motlin-like polypeptide and use thereof |
| US5459049A (en) | 1989-01-06 | 1995-10-17 | Sanwa Kagaku Kenkyushko Co., Ltd. | Motilin-like polypeptide and use thereof |
| US5523418A (en) | 1991-04-09 | 1996-06-04 | Abbott Laboratories | Macrocyclic lactam prokinetic agents |
| US5523401A (en) | 1991-04-09 | 1996-06-04 | Abbott Laboratories | Macrocyclic lactam prokinetic agents |
| US5538961A (en) | 1991-04-09 | 1996-07-23 | Abbott Laboratories | Macrocyclic lactam prokinetic agents |
| US5554605A (en) | 1991-04-09 | 1996-09-10 | Abbott Laboratories | Macrocyclic lactam prokinetic agents |
| US5418224A (en) | 1992-01-07 | 1995-05-23 | Kali-Chemie Pharma Gmbh | 4,13-dioxabicyclo[8.2.1]tridecenone compounds and pharmaceutical compositions containing them |
| US5578579A (en) | 1992-01-21 | 1996-11-26 | Abbott Laboratories | 4"-deoxyerythromycin derivatives |
| US5470961A (en) | 1992-03-19 | 1995-11-28 | Takeda Chemical Ind., Ltd. | 14 and/or 15-hydroxy 6,9 hemiacetal erythromycin derivatives |
| US5854407A (en) | 1992-03-19 | 1998-12-29 | Takeda Chemical Corporation | 15-hydroxy 6,9-hemiacetal erythromycin compounds |
| US5658888A (en) | 1992-05-26 | 1997-08-19 | Chugai Seiyaku Kabushiki Kaisha | Erythromycin derivatives |
| US5470830A (en) | 1993-08-06 | 1995-11-28 | Ohmeda Pharmaceutical Products Division Inc. | Motilin-like polypeptides that inhibit gastrointestinal motor activity |
| US5422341A (en) | 1993-08-06 | 1995-06-06 | Ohmeda Pharmaceutical Products Division Inc. | Motilin-like polypeptides with gastrointestinal motor stimulating activity |
| JPH07138284A (en) | 1993-11-19 | 1995-05-30 | Chugai Pharmaceut Co Ltd | Motilin antagonist |
| US6077943A (en) | 1996-03-01 | 2000-06-20 | Takeda Chemical Industries, Ltd. | Method of producing erythromycin derivative |
| JPH09249620A (en) | 1996-03-13 | 1997-09-22 | Taisho Pharmaceut Co Ltd | Arylalkane derivative |
| US5734012A (en) | 1996-05-16 | 1998-03-31 | Ohmeda Pharmaceutical Products Division Inc. | Cyclic motilin-like polypeptides with gastrointestinal motor stimulating activity |
| US5712253A (en) | 1996-06-18 | 1998-01-27 | Abbott Laboratories | Macrocyclic 13-membered ring derivatives of erythromycins A and B |
| US5912235A (en) | 1996-10-24 | 1999-06-15 | Solvay Pharmaceuticals Gmbh | 10, 13, 15-trioxatricyclo 9.2.1.1.9.6 !-pentadecan one derivatives, method for their production and pharmaceutical compositions containing them |
| US5922849A (en) | 1996-11-22 | 1999-07-13 | Abbott Laboratories | Process for preparation of N-demethyl-4"-deoxy-erthromycins A and B |
| US6100239A (en) | 1996-11-26 | 2000-08-08 | Chugai Seiyaku Kabushiki Kaisha | 13-membered ring macrolide compound, medicine containing the same, and process for producing the same |
| US20040254345A1 (en) | 1997-03-24 | 2004-12-16 | Zymogenetics, Inc. | Motilin homologs |
| US6838438B2 (en) | 1997-03-24 | 2005-01-04 | Zymogenetics, Inc. | Motilin Homologs |
| US20050106146A1 (en) | 1997-03-24 | 2005-05-19 | Sheppard Paul O. | Antibodies to motilin homologs |
| US6380158B1 (en) | 1997-03-24 | 2002-04-30 | Zymogenetics, Inc. | Motilin homologs |
| US20010041791A1 (en) | 1997-03-24 | 2001-11-15 | Zymogenetics, Inc. | Motilin homologs |
| WO1998042840A1 (en) | 1997-03-24 | 1998-10-01 | Zymogenetics, Inc. | Motilin homologs |
| WO1999009053A1 (en) | 1997-08-15 | 1999-02-25 | Chugai Seiyaku Kabushiki Kaisha | Phenethylamine derivatives |
| US6255285B1 (en) | 1997-08-15 | 2001-07-03 | Chugai Seiyaku Kabushiki Kaisha | Phenethylamine derivatives |
| US5972939A (en) | 1997-10-28 | 1999-10-26 | Ortho-Mcneil Pharmaceutical, Inc. | Cyclopentene derivatives useful as antagonists of the motilin receptor |
| WO1999021846A1 (en) | 1997-10-28 | 1999-05-06 | Ortho-Mcneil Pharmaceutical Inc. | Cyclopentene derivatives useful as antagonists of the motilin receptor |
| US6165985A (en) | 1998-02-13 | 2000-12-26 | Solvay Pharmaceuticals Gmbh | 11-acetyl-12,13-dioxabicyclo[8.2.1]-tridecenone derivatives, processes for their preparation and pharmaceutical compositions comprising them |
| US6084079A (en) | 1998-05-15 | 2000-07-04 | Keyes; Robert F. | Process for preparing N-demethyl-N-alkyl erythromycin derivatives |
| WO1999064436A1 (en) | 1998-06-12 | 1999-12-16 | Merck & Co., Inc. | Cloning and identification of the motilin receptor |
| US6720433B2 (en) | 1998-09-24 | 2004-04-13 | Chugai Seiyaku Kabushiki Kaisha | Ethylamine derivatives |
| US6586630B1 (en) | 1998-09-24 | 2003-07-01 | Chugai Seiyaku Kabushiki Kaisha | Ethylamine derivatives |
| WO2000017231A1 (en) | 1998-09-24 | 2000-03-30 | Chugai Seiyaku Kabushiki Kaisha | Ethylamine derivatives |
| US6403775B1 (en) | 1998-10-28 | 2002-06-11 | Kosan Biosciences, Inc. | Erythronolide compounds |
| WO2000044770A1 (en) | 1999-01-28 | 2000-08-03 | Chugai Seiyaku Kabushiki Kaisha | Substituted phenethylamine derivatives |
| US6939861B2 (en) | 1999-04-16 | 2005-09-06 | Kosan Biosciences, Inc. | Amido macrolides |
| US20030176640A1 (en) | 1999-06-30 | 2003-09-18 | Zymogenetics, Inc. | SGIP peptides |
| WO2001000830A1 (en) | 1999-06-30 | 2001-01-04 | Zymogenetics, Inc. | Sgip peptides |
| US6420521B1 (en) | 1999-06-30 | 2002-07-16 | Zymogenetics, Inc. | Short gastrointestinal peptides |
| US20050208626A1 (en) | 1999-06-30 | 2005-09-22 | Zymogenetics, Inc. | SGIP peptides |
| WO2001025257A2 (en) | 1999-10-04 | 2001-04-12 | Neokimia, Inc. | Combinatorial synthesis of libraries of macrocyclic compounds useful in drug discovery |
| US6750205B2 (en) | 2000-02-18 | 2004-06-15 | Kosan Biosciences, Inc. | Motilide compounds |
| US20030220271A1 (en) | 2000-02-18 | 2003-11-27 | Gary Ashley | Motilide compounds |
| US20020025936A1 (en) | 2000-02-18 | 2002-02-28 | Gary Ashley | Motilide compounds |
| WO2001060833A2 (en) | 2000-02-18 | 2001-08-23 | Kosan Biosciences, Inc. | Motilide compounds |
| US6562795B2 (en) | 2000-02-18 | 2003-05-13 | Kosan Biosciences, Inc. | Motilide compounds |
| US6667309B2 (en) | 2000-03-13 | 2003-12-23 | Ortho-Mcneil Pharmaceutical, Inc. | Cyclobutene derivatives useful as antagonists of the motilin receptor |
| US6384031B2 (en) | 2000-03-13 | 2002-05-07 | Ortho-Mcneil Pharmaceutical, Inc. | Cyclobutene derivatives useful as antagonists of the motilin receptor |
| US20010056106A1 (en) | 2000-03-13 | 2001-12-27 | Chen Robert H. | Novel cyclobutene derivatives useful as antagonists of the motilin receptor |
| US6423714B2 (en) | 2000-03-13 | 2002-07-23 | Ortho Mcneil-Pharmaceutical, Inc.. | Cyclohexene derivatives useful as antagonists of the motilin receptor |
| US20020103238A1 (en) | 2000-03-13 | 2002-08-01 | Chen Robert H. | Novel cyclobutene derivatives useful as antagonists of the motilin receptor |
| US6624165B2 (en) | 2000-03-13 | 2003-09-23 | Ortho-Mcneil Pharmaceutical, Inc. | Cyclopentene derivatives useful as antagonists of the motilin receptor |
| US20020111484A1 (en) | 2000-03-13 | 2002-08-15 | Chen Robert H. | Novel cyclopentene derivatives useful as antagonists of the motilin receptor |
| WO2001068620A1 (en) | 2000-03-13 | 2001-09-20 | Ortho-Mcneil Pharmaceutical, Inc. | Novel cyclopentene derivatives useful as antagonists of the motilin receptor |
| WO2001068621A1 (en) | 2000-03-13 | 2001-09-20 | Ortho-Mcneil Pharmaceutical, Inc. | Novel cyclohexene derivatives useful as antagonists of the motilin receptor |
| US20010041701A1 (en) | 2000-03-13 | 2001-11-15 | Chen Robert H. | Novel cyclopentene derivatives useful as antagonists of the motilin receptor |
| WO2001068622A1 (en) | 2000-03-13 | 2001-09-20 | Ortho-Mcneil Pharmaceutical, Inc. | Novel cyclobutene derivatives useful as antagonists of the motilin receptor |
| US20020002192A1 (en) | 2000-03-13 | 2002-01-03 | Chen Robert H. | Novel cyclohexene derivatives useful as antagonists of the motilin receptor |
| US6392040B2 (en) | 2000-03-13 | 2002-05-21 | Ortho-Mcneil Pharmaceutical, Inc. | Cyclopentene compounds useful as antagonists of the motilin receptor |
| US6511980B2 (en) | 2000-05-05 | 2003-01-28 | Ortho Mcneil Pharmaceutical, Inc. | Substituted diamine derivatives useful as motilin antagonists |
| US20050148584A1 (en) | 2000-05-05 | 2005-07-07 | Johnson Sigmond G. | Novel substituted diamine derivatives useful as motilin antagonists |
| US6967199B2 (en) | 2000-05-05 | 2005-11-22 | Ortho-Mcneil Pharmaceutical, Inc. | Substituted diamine derivatives useful as motilin antagonists |
| WO2001085694A2 (en) | 2000-05-05 | 2001-11-15 | Ortho-Mcneil Pharmaceutical, Inc. | Substituted diamide derivatives useful as motilin antagonists |
| US20030203906A1 (en) | 2000-05-05 | 2003-10-30 | Johnson Sigmong G. | Novel substituted diamine derivatives useful as motlin antagonists |
| US20020013352A1 (en) | 2000-05-05 | 2002-01-31 | Johnson Sigmond G. | Novel substituted diamine derivatives useful as motilin antagonists |
| US20070054888A1 (en) | 2000-05-05 | 2007-03-08 | Johnson Sigmond G | Novel substituted diamine derivatives useful as motilin agonists |
| WO2002013727A1 (en) | 2000-08-16 | 2002-02-21 | Edwards Lifesciences Corporation | Stent for implantation into the carotid artery formed by process using intraluminal mapping |
| WO2002016404A1 (en) | 2000-08-24 | 2002-02-28 | Chugai Seiyaku Kabushiki Kaisha | Cyclic peptide derivative |
| US7018981B2 (en) | 2000-08-24 | 2006-03-28 | Chugai Seiyaku Kabushiki Kaisha | Cyclic motilin receptor antagonists |
| US20030191053A1 (en) | 2000-08-24 | 2003-10-09 | Hiroharu Matsuoka | Cyclic peptide derivative |
| US20020094962A1 (en) | 2000-12-01 | 2002-07-18 | Gary Ashley | Motilide compounds |
| US20040147461A1 (en) | 2000-12-01 | 2004-07-29 | Gary Ashley | Motilide compounds |
| WO2002051855A2 (en) | 2000-12-01 | 2002-07-04 | Kosan Biosciences, Inc. | Motilide compounds |
| WO2002059141A1 (en) | 2001-01-25 | 2002-08-01 | Chugai Seiyaku Kabushiki Kaisha | Peptide derivatives |
| WO2002064623A1 (en) | 2001-02-12 | 2002-08-22 | Chugai Seiyaku Kabushiki Kaisha | Process for preparation of peptide derivatives |
| US6875576B2 (en) | 2001-02-15 | 2005-04-05 | Kosan Biosciences, Inc. | Method for evaluating therapeutic efficacy |
| WO2002064092A2 (en) | 2001-02-15 | 2002-08-22 | Kosan Biosciences, Inc. | Method for evaluating therapeutic efficacy |
| US20020192709A1 (en) | 2001-02-15 | 2002-12-19 | Christopher Carreras | Method for evaluating therapeutic efficacy |
| US20040152732A1 (en) | 2001-05-10 | 2004-08-05 | Solvay Pharmaceuticals Gmbh | 1-amidomethylcarbonyl-piperidine compounds, methods and intermediate products for the production thereof and pharmaceutical formulations containing said compounds |
| WO2002092592A1 (en) | 2001-05-10 | 2002-11-21 | Solvay Pharmaceuticals Gmbh | Novel 1-amidomethylcarbonyl-piperidine derivatives, method and intermediate products for the production thereof and medicament containing said compounds |
| WO2004019879A2 (en) | 2002-08-29 | 2004-03-11 | Kosan Biosciences, Inc. | Motilide compounds |
| US20040138150A1 (en) | 2002-08-29 | 2004-07-15 | Santi Daniel V. | Motilide compounds |
| US6946482B2 (en) | 2002-08-29 | 2005-09-20 | Kosan Biosciences, Inc. | Motilide compounds |
| US20050054562A1 (en) | 2003-06-18 | 2005-03-10 | Tranzyme Pharma Inc. | Macrocyclic antagonists of the motilin receptor |
| WO2004111077A1 (en) | 2003-06-18 | 2004-12-23 | Tranzyme Pharma Inc. | Macrocyclic antagonists of the motilin receptor |
| WO2005012332A1 (en) | 2003-07-31 | 2005-02-10 | Tranzyme Pharma | Spatially-defined macrocycles incorporating peptide bond surrogates |
| WO2005012331A1 (en) | 2003-07-31 | 2005-02-10 | Tranzyme Pharma | Spatially-defined macrocyclic compounds useful for drug discovery |
| WO2005018576A2 (en) | 2003-08-26 | 2005-03-03 | Kosan Biosciences, Inc. | N-desmethyl-n-substituted-11-deoxyerythromycin compounds |
| US20050119195A1 (en) | 2003-08-26 | 2005-06-02 | Christopher Carreras | N-desmethyl-N-substituted-11-deoxyerythromycin compounds |
| US20050065156A1 (en) | 2003-09-17 | 2005-03-24 | Li James J. | Compounds useful as motilin agonists and method |
| US20050080116A1 (en) | 2003-09-17 | 2005-04-14 | Li James J. | Compounds useful as motilin agonists and method |
| WO2005027908A1 (en) | 2003-09-17 | 2005-03-31 | Bristol-Myers Squibb Company | Compounds useful as motilin agonists and method |
| WO2005027637A1 (en) | 2003-09-17 | 2005-03-31 | Bristol-Myers Squibb Company | Compounds useful as motilin agonists and method |
| US7262195B2 (en) | 2003-09-17 | 2007-08-28 | Bristol-Myers Squibb Company | Compounds useful as motilin agonists and method |
| US7211568B2 (en) | 2003-12-18 | 2007-05-01 | Kosan Biosciences Incorporated | 9-Desoxoerythromycin compounds as prokinetic agents |
| WO2006009674A1 (en) | 2004-06-18 | 2006-01-26 | Tranzyme Pharma, Inc. | Macrocyclic modulators of the ghrelin receptor |
| WO2006009645A1 (en) | 2004-06-18 | 2006-01-26 | Tranzyme Pharma, Inc. | Methods of using macrocyclic modulators of the ghrelin receptor |
| WO2006070937A1 (en) | 2004-12-28 | 2006-07-06 | Chugai Seiyaku Kabushiki Kaisha | Drugs for the treatment and/or prophylaxis of gastroparesis symptom |
| WO2006127252A1 (en) | 2005-05-24 | 2006-11-30 | Pfizer, Inc. | Motilide compounds |
| US20060270616A1 (en) | 2005-05-24 | 2006-11-30 | Yaoquan Liu | Motilide compounds |
| WO2006138023A1 (en) | 2005-06-17 | 2006-12-28 | Baxter International Inc. | Stable buffered, pharmaceutical compositions including motilin-like peptides |
| US20060293243A1 (en) | 2005-06-17 | 2006-12-28 | Navneet Puri | Stable, buffered, pharmaceutical compositions including motilin-like peptides |
| WO2006138026A2 (en) | 2005-06-17 | 2006-12-28 | Baxter International Inc. | Stable pharmaceutical compositions including motilin-like peptides |
| US20060287243A1 (en) | 2005-06-17 | 2006-12-21 | Navneet Puri | Stable pharmaceutical compositions including motilin-like peptides |
Non-Patent Citations (275)
| Title |
|---|
| "Biochemical Nomenclature and Related Documents", 1992, PORT1AND· PRESS, pages: 39 - 67 |
| "Biochemical Nomenclature and Related Documents, 2nd edition,", 1992, PORTLAND PRESS, pages: 68 - 69 |
| AKASHI, Y.J., SPRINGER, J., ANKER, S.D., CURR. HEART FAIL. REP., vol. 2, 2005, pages 198 - 203 |
| AL-ABRI, S.S., BEECHING, N.J., NYE, F.J., LANCET INFECT. DIS., vol. 5, 2005, pages 349 - 360 |
| ALAEDINI, A., GREEN, P.H.R., ANN. INTERN. MED., vol. 142, 2005, pages 289 - 298 |
| AMINO ACIDS AND PEPTIDES, vol. 16, 1985, pages 387 - 410 |
| ANDERSEN, V., CAMILLERI, M., DRUGS, vol. 66, 2006, pages 1073 - 1088 |
| ANKER, S.D., STEINBORN, W., STRASSBURG, S. ANN. MED., vol. 36, 2004, pages 518 - 529 |
| ARBUCKLE, R.B., HUBER, S.L., ZACKER, C., THE ONCOLOGIST, vol. 5, 2000, pages 250 - 259 |
| ASAKAWA, A., INUI, A., MOMOSE, K.M. ET AL., PEPTIDES, vol. 19, 1998, pages 987 - 990 |
| AUNGST, B.J.: "Novel formulation strategies for improving oral bioavailability of drugs with poor membrane permeation or presystemic metabolism", J. PHARM. SCI., vol. 82, 1993, pages 979 - 987 |
| AVDEEF, A., EXPERT OPIN. DRUG. METAB. TOXICOL., vol. 1, 2005, pages 325 - 42 |
| BALDWIN, J. E. ET AL., TETRAHEDRON, vol. 49, 1993, pages 6309 - 6330 |
| BALIMANE, P.V., HAN, Y.H., CHONG, S., AAPS J., vol. 8, 2006, pages EL-13 |
| BARTH, M., RADEMANN, J., J. COMB. CHEM., vol. 6, 2004, pages 340 - 349 |
| BARTLETT, J.G., N. ENGL. J. MED., vol. 346, 2002, pages 334 - 339 |
| BEAVERS, M.P., GUNNET, J.W., HAGEMAN, W., MILLER, W., MOORE, J.B., ZHOU, L., CHEN, R.H.K., XIANG, A., URBANSKI, M., COMBS, D.W., DRUG DES. DISC., vol. 17, 2001, pages 243 - 251 |
| BEAVERS, M.P., GUNNET, J.W., HAGEMAN, W., MILLER, W., MOORE, J.B., ZHOU, L., CHEN, R.H.K., XIANG, A., URBANSKI, M., COMBS, D.W., DRUG DESIGN DISC., vol. 17, 2001, pages 243 - 251 |
| BENSON, A.B., III; AJANI, J.A., CATALANO, R.B. ET AL., J. CLIN. ONCOL., vol. 22, 2004, pages 2918 - 2926 |
| BERGSTROM, C.A., BASIC CLIN. PHARMACOL. TOXICOL., vol. 96, 2005, pages 156 - 61 |
| BESTERMAN, H.S., ADRIAN, T.E., BLOOM, S.R. ET AL., DIGESTION, vol. 24, 1982, pages 195 - 208 |
| BESTERMAN, H.S., ADRIAN, T.E.., MALLINSON, C.N., GUT, vol. 23, 1982, pages 854 - 861 |
| BESTERMAN, H.S., CHRISTOFIDES, N.D., WELSBY, P.D. ET AL., GUT, vol. 24, 1983, pages 665 - 671 |
| BESTERMAN, H.S., COOK, G.C., SARSON, D.L. ET AL., BR. MED J., vol. 17, 1979, pages 1252 - 1255 |
| BESTERMAN, H.S., J. CLIN. PATHOL., vol. 8, 1978, pages 76 - 84 |
| BESTERMAN, H.S., MALLINSON, C.N. ET AL., SCAND. J. GASTROENTEROL., vol. 18, 1983, pages 845 - 852 |
| BIOCHEM. J., vol. 219, 1984, pages 345 - 373 |
| BIRR, C., LOCHINGER, W., STAHNKE, G., LANG, P., JUSTUS LIEBIGS ANN. CHEM., vol. 763, 1972, pages 162 - 172 |
| BOTHA, C., LIBBY, G., BR. J. HOSP. MED. (LOND., vol. 67, 2006, pages 344 - 349 |
| BOUSHEY RP, YUSTA B, DRUCKER DJ., CANCER RES, vol. 61, 2001, pages 687 - 93 |
| BUCHMAN, A.L., GASTROENTEROLOGY, vol. 130, no. 1, 2006, pages S5 - S15 |
| BUCHMAN, A.L., SCOLAPIO, J., FRYER, J., GASTROENTEROLOGY, vol. 124, 2003, pages 1111 - 1134 |
| BURCHAM, D.L., MAURIN, M.B., HAUSNER, E.A., HUANG, S.M., IMPROVED ORAL BIOAVAILABILITY OF THE HYPOCHOLESTEROLEMIC DMP 565 IN DOGS, BIOPHARM. DRUG DISPOS., vol. 18, 1997, pages 737 - 742 |
| BYRNE, F.R., VINEY, J.L., CURR. OPIN. DRUG DISC. DEVELOP., vol. 9, 2006, pages 207 - 217 |
| CANIBANO, V., SYNTHESIS, 2001, pages 2175 - 2179 |
| CARRERAS, C.W., BURLINGAME, M., CARNEY, J. ET AL., CAN. J. GASTROENTEROL., vol. 19, 2005, pages 15 |
| CARRERAS, C.W., LIU, Y., CHEN, Y. ET AL., GASTROENTEROLOGY, vol. 128, 2005, pages A464 |
| CARRERAS, C.W., SIANI, M.A., SANTI, D.V., DILLON, S.B., ANAL. BIOCHEM., vol. 300, 2002, pages 146 - 151 |
| CHAND, N., MIHAS, A.A., J CLIN. GASTROENTEROL., vol. 40, 2006, pages 3 - 14 |
| CHARMAN, S.A., CHARMAN, W.N., ROGGE, M.C., WILSON, T.D., DUTKO, F.J., POUTON, C.W.: "Self-emulsifying drug delivery systems: formulation and biopharmaceutical evaluation of an investigational lipophilic compound", PHARM. RES., vol. 9, 1992, pages 87 - 93 |
| CHEN, C.L., ORR, W.C., VERLINDEN, M.H. ET AL., ALIMENT. PHARMACOL. THER., vol. 16, 2002, pages 749 - 757 |
| CHEN, H., CHEN, L., WANG, J.J., WEI, H.J., YUNG, W.H., NEUROREPORT, vol. 18, 2007, pages 1345 - 1349 |
| CHOI, M.G., CAMILLERI, M., BURTON, D.D., JOHNSON, S., EDMONDS, A., J. PHARMACOL. EXP. THER., vol. 285, 1998, pages 37 - 40 |
| CHOUNG, R.S., LOCKE, G.R. III, SCHLECK, C.D., ZINSMETISTER, A.R., TALLEY, N.J., AM. J. GASTROENTEROL., vol. 102, 2007, pages 1983 - 1989 |
| CHRISTOPHER J. H. PORTER: "Lipid and lipid-based formulation; optimizing the oral delivery of lipophilic drugs", NAT. REV. DRUG. DISC., vol. 6, 2007, pages 231 - 246 |
| CHUA, A.S., WORLD J. GASTROENTEROL., vol. 12, 2006, pages 2656 - 2659 |
| COHEN, J., WEST, A.B., BINI, E.J., GASTROENTEROL. CLIN. NORTH AM., vol. 30, 2001, pages 637 - 664 |
| CRAIG, D.Q.M., LIEVENS, H.S.R., PITT, K.G., STOREY, D.E.: "An investigation into the physicochemical properties of self-emulsifying systems using low frequency dielectric spectroscopy, surface tension measurements and particle size analysis", INT. J. PHARM., vol. 96, 1993, pages 147 - 155 |
| CREMONINI, F., DELGADO-AROS, S., TALLEY, N.J., BEST PRACT. RES. CLIN. GASTROENTEROL., vol. 18, 2004, pages 717 - 733 |
| CREMONINI, F., TALLEY, N.J., NAT. CLIN. PRACT. GASTROENTEROL. HEPATOL., vol. 2, 2005, pages 82 - 88 |
| DEPOORTERE, I., MACIELAG, M.J., ; GALDES, A., PEETERS, T.L., EUR. J. PHARMACOL., vol. 286, 1995, pages 241 - 247 |
| DEPOORTERE, I., MACIELAG, M.J., GALDES, A., PEETERS, T.L., EUR. J. PHARMACOL., vol. 286, 1995, pages 241 - 247 |
| DEPOORTERE, I., PEETERS, T.L., AM. J. PHYSIOL., vol. 272, 1997, pages G994 - G999 |
| DEPOORTERE, I., VAN ASSCHE, G., PEETERS, T.L., NEUROGASTROENTEROL. MOTIL., vol. 13, 2001, pages 55 - 63 |
| DIBAISE, J.K., YOUNG, R.J., VANDERHOOF, J.A., AM. J. GASTROENTEROL., vol. 99, 2004, pages 1823 - 1832 |
| DUPONT, H.L., GASTROENTEROL. CLIN. NORTH AM., vol. 35, 2006, pages 337 - 353 |
| ECLQNANN, L., ANN. NY ACAD SCI., vol. 1072, 2006, pages 28 - 38 |
| EUR. J. BIOCHEM., vol. 138, 152, 1984, pages 9 - 37,1 |
| EVANS, D. A. ET AL., TETRAHEDRON LETT., vol. 28, 1987, pages 6141 |
| FAGHIH, R., NELLANS, H.N., PLATTNER,: J.J., DRUGS OF THE FUTURE, vol. 23, 1998, pages 861 - 872 |
| FARRUGIA, G., MACIELAG, M.J., PEETERS, T.L., SARR, M.G., GALDES, A., SZURSZEWSKI, J.H., AM. J. PHYSIOL., vol. 273, 1997, pages G404 - G412 |
| FARTHING, M.J.G., DIG. DIS., vol. 24, 2006, pages 47 - 58 |
| FARTHING, M.J.G., EXP. OPIN. INVEST. DRUGS, vol. 13, 2004, pages 777 - 785 |
| FEIGHNER, S.D., TAN, C.P., MCKEE, K.K. ET AL., SCIENCE, vol. 284, 1999, pages 2184 - 2188 |
| FLOWERS, M.E., KANSU, E., SULLIVAN, K.M., HEMATOL ONCOL CLIN NORTH AM., vol. 13, 1999, pages 1091 - 1112 |
| FRECHET, J.M.J., HAQUE, K.E., TETRAHEDRON LETT., vol. 16, 1975, pages 3055 |
| FROST. F, CRAUN, G.F., CALDERON, R.L., EMERG. INFECT. DIS., vol. 4, 1998, pages 619 - 625 |
| GAGNON, B., BRUERA, E., DRUGS, vol. 55, 1998, pages 675 - 688 |
| GAN, T.J., J. AM. MED. ASSOC., vol. 287, 2002, pages 1233 - 1236 |
| GAUTAM PANDA ET AL., SYNLETT, vol. 4, 2004, pages 714 - 716 |
| GIBSON, R.J., KEEFE, D.M.K., SUPPORT. CARE CANCER, vol. 14, 2006, pages 890 - 900 |
| GILKIN, R.J., JR. CLIN. THER., vol. 27, 2005, pages 1696 - 1709 |
| GINSBURG, P.M., THULUVATH, P.J., LIVER TRANSPL., vol. 11, 2005, pages 881 - 890 |
| GOLDBERG-ARNOLD, R.J., GABRAIL, N., RAUT, M., KIM, R., SUNG, J.C.Y., ZHOU, Y. J., SUPPORT. ONCOL., vol. 3, 2005, pages 227 - 232 |
| GOLEMBIEWSKI, J., CHEMIN, E., CHOPRA, T., AM. J. HEALTH-SYST. PHARM., vol. 62, 2005, pages 1247 - 1260 |
| GREEN, P.H.R., JABRI, B., ANN. REV. MED., vol. 57, 2006, pages 207 - 221 |
| GREENBERG, G.R., BUCHAN, A.M., MCLEOD, R.S., PRESTON, P., COHEN, Z., GUT, vol. 30, 1989, pages 1721 - 1730 |
| GRUNBERG, S.M., J. SUPPORT. ONCOL., vol. 2, 2004, pages 1 - 12 |
| GWEDE, C.K., SEM. NURSING ONCOL., vol. 19, 2003, pages 6 - 10 |
| HABIB, A.S., GAN, T.J., CAN. J. ANESTH., vol. 51, 2004, pages 326 - 341 |
| HANSEN, D. W., JR., PILIPAUSKAS, D., J. ORG. CHEM., vol. 50, 1985, pages 945 - 950 |
| HARAMURA, M., OKAMACHI, A., TSUZUKI, K., YOGO, K., IKUTA, M., KOZONO, T., TAKANASHI, H., MURAYAMA, E., CHEM. PHARM. BULL., vol. 49, 2001, pages 40 - 43 |
| HARAMURA, M., OKAMACHI, A., TSUZUKI, K., YOGO, K., IKUTA, M., KOZONO, T., TAKANASHI, H., MURAYAMA, E., J. MED. CHEM., vol. 45, 2002, pages 670 - 675 |
| HARAMURA, M., TSUZUKI, K., OKAMACHI, A. ET AL., BIOORG. MED. CHEM., vol. 10, 2002, pages 1805 - 1811 |
| HARAMURA, M., TSUZUKI, K., OKAMACHI, A. ET AL., CHEM. PHARM. BULL., vol. 47, 1999, pages 1555 - 1559 |
| HARDMAN, J.G.; LIMBIRD, L.E.: "Goodman & Gilman's The Pharmacological Basis of Therapeutics, Tenth Edition", 2001, MCGRAW HILL, article WILKINSON, G. R.: "Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination" |
| HASLER, W.L., HELDSINGER, A., CHUNGAL, O.Y., AM. J. PHYSIOL., vol. 262, 1992, pages G50 - G55 |
| HERRSTEDT, J., DOMBERNOWSKY, P., BASIC CLIN. PHARMACOL. TOXICOL., vol. 101, 2007, pages 143 - 150 |
| HILL, I.D., CURR. TREAT. OPTIONS GASTROENTEROL., vol. 9, 2006, pages 399 - 408 |
| HORIKAWA, M., KATO, Y., SUGIYAMA, PHARM. RES., vol. 19, 2002, pages 1345 - 1353 |
| HULL, M.W., BECK, P.L., CAN. FAM. PHYS., vol. 50, 2004, pages 1536 - 1540 |
| ILAR J., vol. 45, 2004, pages 25 - 34 |
| INATOMI, N., SATO, F., ITOH, Z., OMURA, S.: "Mode of action of macrolides with motilin agonistic activity - motilides. Macrolide Antibiotics, 2nd edition,", 2002, ACADEMIC PRESS, pages: 501 - 531 |
| INTERNAT. J. PEPT. PROT. RES., vol. 24, 1984, pages 84 |
| INUI, A., CA CANCER J. CLIN., vol. 52, 2002, pages 72 - 91 |
| ITOH, Z., AIZAWA, I., SEKIGUCHI, T., CLIN. GASTROENTEROL., vol. 11, 1982, pages 497 - 521 |
| ITOH, Z., PEPTIDES, vol. 18, 1997, pages 593 - 608 |
| ITOH, Z., SEKIGUCHI, T., SCAND. J. GASTROENTEROL., vol. 82, 1983, pages 121 - 134 |
| ITOH, Z.,: "Motilin", 1990, ACADEMIC PRESS |
| J. BIOL. CHEM., vol. 247, 1972, pages 977 - 983 |
| J. BIOL. CHEM., vol. 260, 1985, pages 14 - 42 |
| J. CHEM. SOC., PERKIN TRANS., vol. 1, 2000, pages 367 |
| JACKSON, C., BUCHMAN, A.L., CURR. GASTROENTEROL. REP., vol. 7, 2005, pages 373 - 378 |
| JCBN/NC-IUB NEWSLETTER, 1985 |
| JOHNSON, S.G., GUNNET, J.W., MOORE, J.B. ET AL., BIOORG. MED. CHEM. LETT., vol. 16, 2006, pages 3362 - 3366 |
| JONES, R.B., ROBINS, G.G., HOWDLE, P.D., CURR. OPIN. GASTROENTEROL., vol. 22, 2006, pages 117 - 123 |
| JORDAN, K., KASPER, C., SCHMOLL, H.-J., EUR. J. CANC., vol. 41, 2005, pages 199 - 205 |
| JURJUS, A.R., KHOURY, N.N., REIMUND, J.-M. J., PHARMACOL. TOXICOL. METHODS, vol. 50, 2004, pages 81 - 92 |
| KACHEL, G.W., FRASE, L.L., DOMSCHKE, W., CHEY, W.Y., KREJS, G.J., GASTROENTEROLOGY, vol. 87, 1984, pages 550 - 556 |
| KAMERLING, I.M., VAN HAARST, A.D., BURGGRAAF, J., SCHOEMAKER, R.C., BIEMOND, I., HEINZERLING, H., JONES, R., COHEN, A.F., MASCLEE,, AM. J. PHYSIOF GASTROINTEST. LIVER PHYSIOL., vol. 284, 2003, pages G776 - G781 |
| KAMERLING, I.M.C., VAN HAARST, A.D., BURGGRAAF, J. ET AL., BR. J. CLIN. PHARMACOL., vol. 57, 2003, pages 393 - 401 |
| KAMM, M.A., EUR. J: SURG., vol. 583, 1998, pages 37 - 40 |
| KANAMORI, Y., NAKAZAWA, S., KITOH, J., HOSHINO, M.: "The distribution of endocrine cells in the mucosa of the gastrointestinal tract of the house musk shrew, Suncus murinus (Insectivora", CELL. TISSUE RES., vol. 258, 1989, pages 365 - 71 |
| KARARLI, T.T., NEEDHAM, T.E., GRIFÆN, M., SCHOENHARD, G., ERRO, L.J., ALCORN, L.: "Oral delivery of a renin inhibitor compound using emulsion formulations", PHARM. RES., vol. 9, 1992, pages 888 - 893 |
| KATO, T., SHIMAMOTO, Y., UCHIDA, J., OHSHIMO, H., ABE, M., SHIRASAKA, T., FUKUSHIMA, M., ANTICANCER RES., vol. 21, 2001, pages 1705 - 12 |
| KAWATO, Y., SEKIGUCHI, M., AKAHANE, K., TSUTOMI, Y., HIROTA, Y., KUGA, H., SUZUKI, W., HAKUSUI, H., SATO, K. J., PHARM. PHARMACOL., vol. 45, 1993, pages 444 - 448 |
| KELLY, C.P., POTHOULAKIS, C., LAMONT, J.T., N. ENGL. J. MED., vol. 330, 1994, pages 257 - 262 |
| KEM, K.A., NORTON, J.A., JPEN, vol. 12, 1980, pages 286 - 298 |
| KENNEDY, H.J., SARSON, D.L., BLOOM, S.R. ET AL., DIGESTION, vol. 24, 1982, pages 133 - 136 |
| KITAMURA, N., YAMADA, J., WATANABE, T., YAMASHITA, T.: "An immunohistochemical study on the distribution of endocrine cells in the gastrointestinal tract of the musk shrew, Suncus murinus", HISTOL HISTOPATHOL., vol. 5, 1990, pages 83 - 88 |
| KLEIBEUKER, J.H., THIJS, J.C., CURR.. OPIN. GASTROENTEROL., vol. 20, 2004, pages 546 - 550 |
| KOGA, H., TAKANASHI, H., ITOH, Z., OMURA, S., DRUGS OF THE FUTURE, vol. 27, 2002, pages 255 - 272 |
| KONING, F., GASTROENTEROLOGY, vol. 129, 2005, pages 1294 - 1301 |
| KONTUREK, S.J., DEMBINSKI, A., KROL, R., WUNSCH, E., SCAND. J. GASTROENTEROL., vol. 11, 1976, pages 57 - 61 |
| KOVAC, A.L., DRUG SAFETY, vol. 26, 2003, pages 227 - 259 |
| KOVAC, A.L., DRUGS, vol. 59, 2000, pages 213 - 243 |
| KUSANO, M., SEKIGUCHI, T., KAWAMURA, O., KIKUCHI, K., MIYAZAKI, M., TSUNODA, T., HORIKOSHI, T., MORI, M., AM. J. GASTROENTEROL., vol. 92, 1997, pages 481 - 484 |
| KYNE, L., FARRELL, R.J., KELLY, C.P., GASTROENTEROL. CLIN. N. AM., vol. 30, 2001, pages 753 - 777 |
| LACY, B.E., DE LEE, R. J, CLIN. GASTROENTEROL., vol. 39, 2005, pages S230 - S242 |
| LAMIAN, V., RICH, A., MA, Z., LI, J., SEETHALA, R., GORDON, D., DUBAQUIE, Y., MOL. PHARMACOL., vol. 69, 2006, pages 109 - 118 |
| LEE, K.L., SHIRATORI, K., CHEN, Y.F., CHANG, T.-M., CHEY, W.Y., AM. J. PHYSIOL., vol. 14, 1986, pages G759 - 764 |
| LI, A.P., DRUG. DISC. TODAY, vol. 6, 2001, pages 357 - 366 |
| LI, J.J., CHAO, H.G., WANG, H. ET AL., J. MED. CHEM., vol. 47, 2004, pages 1704 - 1708 |
| LIMA, A.A.M., CURR. OPIN. INFECT. DIS., vol. 14, 2001, pages 547 - 552 |
| LIN, N., BIOORG, MED. CHEM. LETT, vol. 8, 1998, pages 249 - 254 |
| LINDLEY, C.M., HIRSCH, J.D., O'NEILL, C.V., TRANSAU, M.D, GILBERT, C.S., OSTERHAUS, J.T., QUAL. LIFE RES., vol. 1, 1992, pages 331 - 340 |
| LIPINSKI, C.A., LOMBARDO, F., DOMINY, B.W., FEENEY, P.J., ADV. DRUG DELIVERY REV., vol. 23, 1997, pages 3 - 25 |
| LIU, M., DONG, L., DUAN, Z., ZHU, W.-Y., CUI, Y., LEI, L., J. MED. COLLEGES PLA, vol. 20, 2005, pages 321 - 326 |
| LUIKING, Y.C., PEETERS, T.L., STOLK, M.F., NIEUWENHUIJS, V.B., PORTINCASA, P., DEPOORTERE, I., VAN BERGE HENEGOUWEN, G.P.; AKKERMA, GUT, vol. 42, 1998, pages 830 - 835 |
| M.D. ANDERSON, SYMPTOM INVENTORY, CANCER, vol. 89, no. 7, 2000, pages 1634 - 1646 |
| MACNAUGHTON, W.K., ALIMENT. PHARMACOL. THER., vol. 14, 2000, pages 523 - 528 |
| MAGEE, D.F., NARUSE, S. J., PHYSIOL., vol. 355, 1984, pages 441 - 447 |
| MAHADEVA, S., GOH, K.L., WORLD J. GASTROENTEROL., vol. 12, 2006, pages 2661 - 2666 |
| MALNICK, S.D.H., ZIMHONY, O., ANN. PHARMACOTHER., vol. 36, 2002, pages 1767 - 1775 |
| MARSAULT, E., BENAKLI, K., BEAUBEIN, S., SAINT-LOUIS, C., DEZIEL, R., FRASER, G., BIOORG MED. CHEM. LETT., vol. 17, 2007, pages 4187 - 4190 |
| MARSAULT, E., HOVEYDA, H.R., PETERSON, M.L., SAINT-LOUIS, C., LANDRY, A., VÉZINA, M., OUELLET, L., WANG, Z., RAMASESHAN, M., BEAU, J. MED. CHEM., vol. 49, 2006, pages 7190 - 7197 |
| MARTIN, G.E., ZEKTZER, A.S.: "Two-Dimensional NMR Methods for Establishing Molecular Connectivity: A Chemist's Guide to Experiment Selection, Performance, and Interpretation", 1988, JOHN WILEY & SONS |
| MARTIN, M., ONCOLOGY, vol. 53, 1996, pages 26 - 31 |
| MASSAD, T., JARVET, J., TANER, R. ET AL., J. BIOMOL. NMR, vol. 38, 2007, pages 107 - 123 |
| MATSUURA, B., DONG, M., MILLER, L.J., J. BIOL. CHEM., vol. 277, 2002, pages 9834 - 9839 |
| MATSUURA, B., DONG, M., NAIK, S., MILLER, L.J., ONJI, M., J. BIOL. CHEM., vol. 281, 2006, pages 12390 - 12396 |
| MATTHIJS, G., PEETERS, T. L., VANTRAPPEN, G., NAUNYN-SCHMIEDEBERG'S ARCH. PHARMACOL., vol. 339, 1989, pages 332 - 339 |
| MAXION-BERGEMANN, S., THIELECKE, F., ABEL, F., BERGEMANN, R., PHARMACOECONOMICS, vol. 24, 2006, pages 21 - 37 |
| MCCALLUM, R.W., CYNSHI, O., ALIMENT. PHARMACOL. THER., vol. 26, 2007, pages 107 - 116 |
| MCNALLY, M.A., TALLEY, N.J., CURR. TREATMENT OPT. GASTROENTEROL., vol. 10, 2007, pages 157 - 168 |
| MEANA, A., SYNLETT, 2003, pages 1678 - 1682 |
| MELDAL, M., TETRAHEDRON LETT., vol. 33, 1992, pages 3077 - 3080 |
| MILLER, P., TRUDEL, L., ST-PIERRE, S., TAKANASHI, H., POITRAS, P., PEPTIDES, vol. 21, 2000, pages 283 - 287 |
| MISIAKOS, E.P., MACHERAS, A., KAPETANAKIS, T., LIAKAKOS, T., J. CLIN. GASTROENTEROL., vol. 41, 2007, pages 5 - 18 |
| MITSELOS, A., DEPOORTERE, I., PEETERS, T.L., BIOCHEM. PHARMACOL., vol. 73, 2007, pages 115 - 124 |
| MIZUTA, Y., SHIKUWA, S., ISOMOTO, H., MISHIMA, R., AKAZAWA, Y., MASUDA, J., OMAGARI, K., TAKESHIMA, F., KOHNO, S. J., GASTROENTEROL, vol. 41, 2006, pages 1025 - 1040 |
| MONKEMULLER K, MALFERTHEINER P., WORLD J. GASTROENTEROL., vol. 12, 2006, pages 2694 - 2700 |
| MORLEY, J.E., J. GERONTOLOGY, SER. A: BIOL. SCI. MED. SCI., vol. 58A, 2003, pages 131 - 137 |
| MUEGGE, I., MED. RES. REV., vol. 23, 2003, pages 302 - 321 |
| MULVIHILL, S.J.; ET AL.: "Basic and Clinical Endocrinology, 4th edition,", 1994, APPLETON & LANGE, pages: 551 - 570 |
| NAKAJIMA, K. ET AL., BULL. CHEM. SOC. JPN., vol. 51, 1978, pages 1577 - 1578 |
| NARITA, T., OKABE, N., HANE, M. ET AL., J. VET. PHARMACOL. . THER., vol. 29, 2006, pages 569 - 577 |
| NELSON, D.K., DIG. DIS. SCI., vol. 41, 1996, pages 2006 - 2015 |
| NETZER, P., SCHMITT, B., INAUEN, W., ALIMENT. PHARMACOL. THER., vol. 16, 2002, pages 1481 - 1490 |
| NGUYEN, N.P., ANTOINE, J.E., DUTTA, S., KARLSSON, U., SALLAH, S., CANCER, vol. 95, 2002, pages 1151 - 1163 |
| NORTH, C.S., ALPERS, D.H., THOMPSON, S.J., SPITZNAGEL, E.L., DIG. DIS. SCI., vol. 41, 1996, pages 633 - 640 |
| O'BRIEN, B.E, KAKLAMANI, V.G., BENSON, A.B., CLIN. COLORECTAL CANC., vol. 4, 2005, pages 375 - 381 |
| O'DONOHUE, T.L. ET AL., PEPTIDES, vol. 2, 1981, pages 467 - 477 |
| OHMAN, L., SIMREN, M., DIG. LIVER DIS., vol. 39, 2007, pages 201 - 215 |
| OLDFIELD, E.C., REV. GASTROENTEROL. DISORD., vol. 2, 2002, pages 176 - 88 |
| OLFIELD, E.C., REV. GASTROENTEROL. DISORD., vol. 6, 2006, pages 79 - 96 |
| OSOBA, D., ZEE, B., WARR, D. ET AL., SUPPORT. CARE CANCER, vol. 5, 1997, pages 303 - 313 |
| OZAKI, K., SUDO, H., MURAMATSU, H., YOGO, K., KAMEI, K., KOGA, H., ITOH, Z., OMURA, S., TAKANASHI, H., INFLAMMOPHARMACOLOGY, vol. 15, 2007, pages 36 - 42 |
| OZAKI, K.I., YOGO, K., SUDO, H., ONOMA, M., KAMEI, K., AKIMA, M., KOGA, H., ITOH, Z., OMURA, S., TAKANASHI, H., PHARMACOLOGY, vol. 79, 2007, pages 223 - 235 |
| PALIN, K.J., PHILLIPS, A.J., NING, A.: "The oral absorption of cefoxitin from oil and emulsion vehicles in rats", INT. J. PHARM., vol. 33, 1986, pages 99 - 104 |
| PANDA, G., RAO, N. V., SYNLETT, 2004, pages 714 |
| PARK, M.I., FERBER, I., CAMILLERI, M. ET AL., NEUROGASTROENTEROL. MOTIL, vol. 18, 2006, pages 28 - 36 |
| PEETERS, T.L., BORMANS, V., VANTRAPPEN, G., REGUL. PEPT., vol. 23, 1988, pages 171 - 182 |
| PEETERS, T.L., CURR. OPIN. INVESTIG. DRUGS., vol. 2, 2001, pages 555 - 557 |
| PEETERS, T.L., DEPOORTERE, I., MACIELAG, M.J., MARVIN, M.S., FLORANCE, J.R., GALDES, A., BIOCHEM. BIOPHYS. RES. COMM., vol. 198, 1994, pages 411 - 416 |
| PEETERS, T.L., GASTROENTEROLOGY, vol. 105, 1993, pages 1886 - 1899 |
| PEETERS, T.L., MACIELAG, M.J., DEPOORTERE, I. ET AL., PEPTIDES, vol. 13, 1992, pages 1103 - 1107 |
| PEETERS, T.L., NEUROGASTROENTEROL. MOTIL., vol. 18, 2006, pages 1 - 5 |
| PEETERS, T.L., TANG, M., PEPTIDES, vol. 28, 2007, pages 625 - 631 |
| PEETERS, T.L., VANTRAPPEN, G., JANSSENS, J., GASTROENTEROLOGY, vol. 79, 1980, pages 716 - 719 |
| PETERLIN-MASIC, L., KIKELJ, D., TETRAHEDRON, vol. 57, 2001, pages 7073 |
| POITRAS, P., MILLER, P., DICKNER, M., MAO, Y.K., DANIEL, E.E., ST-PIERRE, S., TRUDEL, L., PEPTIDES, vol. 17, 1996, pages 701 - 707 |
| POITRAS, P., MILLER, P., GAGNON, D., ST-PIERRE, S., BIOCHEM. BIOPHYS. RES. COMM., vol. 205, 1994, pages 449 - 454 |
| POWRIE, F., ULBIG, H., NOVARTIS FOUND. SYMP., vol. 263, 2004, pages 164 - 178 |
| PURE APPL. CHEM., vol. 45, 1976, pages 13 - 30 |
| PURE APPL. CHEM., vol. 56, 1984, pages 595 - 624 |
| ROSENFELD, D.J., GARTHWAITE, T.L., PHYSIOL. BEHAV., vol. 39, 1987, pages 753 - 756 |
| ROSS, W.A., COURIEL, D., CURR. OPIN. GASTROENTEROL., vol. 21, 2005, pages 64 - 69 |
| ROSTOVSTEV, V., GREEN, L. K., FOKIN, V. V., SHARPLESS, K. B., ANGEW. CHEM. INT. ED. ENGL., vol. 41, 2002, pages 2596 |
| ROTHENBERG, M.L., MEROPOL, N.J., POPLIN, E.A., VANCUTSEM, E., WADLER, S. J., CLIN. ONCOL., vol. 19, 2001, pages 3801 - 3807 |
| SAAD, R.J., CHEY, W.D., ALIMENT. PHARMACOL. THER., vol. 24, 2006, pages 475 - 492 |
| SAITO, T., MIZUTANI, F., IWANAGA, Y., MORIKAWA, K., KATO, H.: "Laxative and anti-diarrheal activity of polycarbophil in mice and rats", JPN. J. PHARMACOL., vol. 89, 2002, pages 133 - 141 |
| SAITO, Y.A., SCHOENFELD, P., LOCKE, G.R., AM. J. GASTROENTEROL., vol. 97, 2002, pages 1910 - 1915 |
| SALAT, P., PARIKH, V., IND. J. PHARMACOL., vol. 31, 1999, pages 333 - 339 |
| SALTZ, L.B., J. SUPPORT. ONCOL., vol. 1, 2003, pages 35 - 46 |
| SANDHAM, D.A., PLANNKUCHE, H.-J., ANN. REP. MED. CHEM., vol. 41, 2006, pages 211 - 219 |
| SATOH, M., SAKAI, T., SANO, I. ET AL., J. PHARMACOL. EXP. THER., vol. 271, 1994, pages 574 - 579 |
| SAVINO, R., GRASSINO, E.C., FISSORE, M.F. ET AL., CLIN. ENDOCRINOL., vol. 65, 2006, pages 158 - 162 |
| SCHILLER, L.R., CURR. TREAT. OPTIONS GASTROENTEROL., vol. 8, 2005, pages 259 - 266 |
| SCHOENFELD, P., GASTROENTEROL. CLIN. NORTH AM., vol. 34, 2005, pages 319 - 335 |
| SCHROEDER, M.S., AM. FAM. PHYS., vol. 71, 2005, pages 921 - 928 |
| SCOLAPIO, J.S., CURR. OPIN. GASTROENTEROL., vol. 20, 2004, pages 143 - 145 |
| SEMINARS IN ONCOLOGY, vol. 23, 1996, pages 11 - 20 |
| SERAJUDDIN, A.T.M., SHEEN, P.-C., MUFSON, D., BERNSTEIN, D.F., AUGUSTINE, M.A.: "Effect of vehicle amphiphilicity on the dissolution and bioavailability of a poorly water-soluble drug from solid dispersion", J. PHARM. SCI., vol. 77, 1988, pages 414 - 417 |
| SESTAK, K., CURR. HIV RES., vol. 3, 2005, pages 199 - 205 |
| SHAH, N.H., CARVAJAL, M.T., PATEL, C.I., INFELD, M.H., MALICK, A.W.: "Self-emulsifying drug delivery systems (SEDDS) with polyglycolysed glycerides for improving in vitro dissolution and oral absorption of lipophilic drugs", INT. J. PHARM., vol. 106, 1994, pages 15 - 23 |
| SHAH, P., JOGANI, V., BAGCHI, T., MISRA, A., BIOTECHNOL. PROG., vol. 22, 2006, pages 186 - 198 |
| SHARMA, R., TOBIN, P., CLARKE, S.J., LANCET ONCOL., vol. 6, 2005, pages 93 - 102 |
| SHIMANO MASANAO, TETRAHEDRON LETT., vol. 39, 1998, pages 4363 - 4366 |
| SIMREN, M., BJORNSSON, E.S., ABRAHAMSSON, H., NEUROGASTROENTEROL. MOTIL., vol. 17, 2005, pages 51 - 57 |
| SMITH, M.L., DIG. LIVER DIS., vol. 37, 2005, pages 547 - 558 |
| SNIECKUS, V., J. ORG. CHEM., vol. 50, 1985, pages 5436 - 5438 |
| SPILLER, R., NEUROGASTROENTEROL. MOTIL., vol. 18, 2006, pages 1045 - 1055 |
| SPRINGER ,J., FILIPPATOS, G., AKASHI, Y.J., ANKER, S.D., CURR. OPIN. CARDIOL., vol. 21, 2006, pages 229 - 233 |
| ST LAURENT, D. R. ET AL., BIOORG. MED. CHEM., vol. 3, 1995, pages 1145 |
| STANGHELLINI, V., DE PONTI, F., DE GIORGIO, R. ET AL., DRUGS, vol. 63, 2003, pages 869 - 892 |
| STEM, J., IPPOLITI, C., SEM. ONCOL. NURS., vol. 19, 2003, pages 11 - 16 |
| STILL, W. C., KAHN, M., MITRA, A., J. ORG. CHEM., vol. 43, 1978, pages 2923 - 2925 |
| SUKHOTNIK, I., CORAN, A.G. ET AL., PEDIATR. SURG. INT., vol. 21, 2005, pages 947 - 953 |
| SUN, D., YU, L.X., HUSSAIN, M.A., WALL, D.A., SMITH, R.L., AMIDON, G.L., CURR. OPIN. DRUG DISCOV. DEVEL., vol. 7, 2004, pages 75 - 85 |
| SUZUKI, H., NISHIZAWA, T., HIBI, T., J. GASTROENTEROL., vol. 41, 2006, pages 513 - 523 |
| SWINDELL, C., HETEROCYCLES, vol. 24, 1986, pages 3373 - 3377 |
| TACK, J., BASIC PRACT. RES. CLIN. GASTROENTEROL., vol. 21, 2007, pages 633 - 644 |
| TACK, J., BISSCHOPS, R., SAMELLI, G., GASTROENTEROLOGY, vol. 127, 2004, pages 1239 - 1255 |
| TAKANASHI, H., YOGO, K., OZAKI, K., KOGA, H., ITOH, Z., OMURA, S., PHARMACOLOGY, vol. 79, 2007, pages 137 - 148 |
| TAKANASHI, H., YOGO, K., OZAKI, M., AKIMA, M., KOGA, H., NABATA, H., J. PHARM. EXP. THER., vol. 273, 1995, pages 624 - 628 |
| TAKANISHI, H., YOGO, K., OZAKI, M., AKIMA, M., KOGA, H., NABATA, H. J, PHARM. EXP. THER., vol. 273, 1995, pages 624 - 628 |
| TAKASUNA, K., KASAI, Y., KITANO, Y. ET AL., JPN. J. CANCE RES., vol. 86, 1995, pages 978 - 984 |
| TAKESHITA E, MATSUURA B, DONG M, MILLER LJ, MATSUI H, ONJI M. J., GASTROENTEROL., vol. 41, 2006, pages 223 - 230 |
| TALLEY, N.J., GABRIEL, S.E., HARMSEN, W.S. ET AL., GASTROENTEROLOGY, vol. 109, 1995, pages 1736 - 1741 |
| TALLEY, N.J., INTERN. MED. J., vol. 36, 2006, pages 724 - 728 |
| TALLEY, N.J., VAKIL, N. ET AL., AM. J. GASTROENTEROL., vol. 100, 2005, pages 2324 - 2337 |
| TALLEY, N.J., VAKIL, N., MOAYYEDI, P., GASTROENTEROLOGY, vol. 129, 2005, pages 1756 - 1780 |
| TALLEY, N.J., VERLINDEN, M., GEENAN, D.J. ET AL., GUT, vol. 49, 2001, pages 395 - 401 |
| TALLEY, N.J., VERLINDEN, M., SNAPE, W. ET AL., ALIMENT. PHARMACOL. THER., vol. 14, 2000, pages 1653 - 1661 |
| TAVAKKOLIZADEH, A., SHEN, R., ABRAHAM, P. ET AL., 1. SURG. RES., vol. 91, 2000, pages 77 - 82 |
| THE ONCOLOGIST, vol. 3, 1998, pages 50 - 53 |
| THEODORA W. GREENE AND PETER G. WUTS: "Protective Groups in Organic Synthesis, 3rd edition", 1999, JOHN WILEY & SONS |
| THEODORIDIS, G., TETRAHEDRON, vol. 56, 2000, pages 2339 - 2358 |
| THIELEMANS, L., DEPOORTERE, I., PERRET, J. ET AL., J. PHARMACOL. EXP. THER., vol. 313, 2005, pages 1397 - 1405 |
| THIELEMANS, L., DEPOORTERE, I., VAN ASSCHE, G., BENDER, E., PEETERS, T.L., BRAIN RES., vol. 895, 2001, pages 119 - 128 |
| THOM, K., FORREST, G., CURR. OPIN. GASTROENTEROL., vol. 22, 2006, pages 18 - 23 |
| TISDALE, M.J. J., NATL. CANCER INST., vol. 89, 1997, pages 1763 - 1773 |
| TOMOMASA, T., YAGI, H., KIMURA, S., SNAPE, W.J., JR., HYMAN, P.E., PEDIATRIC RES., vol. 26, 1989, pages 458 - 461 |
| TRAMÈR, M.R., BEST PRACT. RES. CLIN. ANAESTHESIOL., vol. 18, 2004, pages 693 - 701 |
| TURPEINEN, M., KORHONEN, L.E., TOLONEN, A. ET AL., EUR. J. PHARM. SCI., vol. 29, 2006, pages 130 - 138 |
| VAN ASSCHE, G., DEPOORTERE, I., THIJS, T, JANSSENS, J.J., PEETERS, T.L., EUR. J. PHARMACOL., vol. 337, 1997, pages 267 - 274 |
| VAN ASSCHE, G., DEPOORTERE, I., THIJS, T., JANSSENS, J.J., PEETERS, T.L.: "Concentration-dependent stimulation of cholinergic motor nerves or smooth muscle by [Nle13]-motilin in the isolated rabbit gastric antrum", EUR. J. PHARMACOL., vol. 337, 1997, pages 267 - 274 |
| VANDERHOOF, J.A., J. PED. GASTROENTEROL. NUTRI., vol. 39, 2004, pages 5768 - 5771 |
| VANDERHOOF, J.A., YOUNG, R.J., THOMPSON, J.S., PEDIATRIC DRUGS, vol. 5, 2003, pages 625 - 631 |
| VEBER, D.F., JOHNSON, S.R., CHENG, H.-Y., SMITH, B.R., WARD, K.W., KOPPLE, K.D., J. MED. CHEM., vol. 45, 2002, pages 2615 - 2623 |
| VEDEJS, E., KONGKITTINGAM, C., J.ORG. CHEM., vol. 65, 2000, pages 2309 - 2318 |
| VEDEJS, E., LIN, S., KLAPARA, A., WANG, J., J. AM. CHEM. SOC., vol. 118, 1996, pages 9796 - 9797 |
| VEDERAS, J.C. ET AL., J. AM. CHEM. SOC., vol. 107, 1985, pages 7105 - 7109 |
| VEDERAS, J.C. ET AL., J. AM. CHEM. SOC., vol. 109, 1987, pages 4649 - 4659 |
| VOTH, D.E., BALLARD, J.D., CLIN. MICROBIOL. REV., vol. 18, 2005, pages 247 - 263 |
| WEBER, F.H., JR., RICHARDS; R.D., MCCALLUM, R.W., AM. J. GASTROENTEROL., vol. 88, 1993, pages 485 - 490 |
| WERMUTH, C.G. AND STAHL, P.H.: "Handbook of Pharmaceutical Salts: Properties, Selection, and Use", 2002, WILEY-VERLAG HELVETICA ACTA |
| WESTERBERG, D.P., GILL, J.M., DAVE, B. ET AL., J. AM. OSTEOPATH. ASSN., vol. 106, 2006, pages 145 - 151 |
| WESTERGAARD, H., SEM. GASTROINTEST. DIS., vol. 13, 2002, pages 210 - 220 |
| WHITE, R.E., ANN. REV. PHARMACOL. TOXICOL., vol. 40, 2000, pages 133 - 157 |
| WIRTZ, S., NEURATH, M.F., INT. J. COLORECTAL. DIS., vol. L5, 2000, pages 144 - 160 |
| WU, Y.J., CURR. PHARM. DES., vol. 6, 2000, pages 181 - 223 |
| YU, G., MASON, H.J., GALDI, K. ET AL., SYNTHESIS, 2003, pages 403 - 407 |
| ZHAO, J., HUANG, L., BELMAR, N., BUELOW, R., FONG, T., CLIN. CANC. RES., vol. 10, 2004, pages 2851 - 2859 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2431380A3 (en) | 2013-07-03 |
| US9133235B2 (en) | 2015-09-15 |
| JP2010503620A (en) | 2010-02-04 |
| US20100093720A1 (en) | 2010-04-15 |
| CN101528765A (en) | 2009-09-09 |
| EP2054429A2 (en) | 2009-05-06 |
| CN101528765B (en) | 2015-04-22 |
| WO2008033328A3 (en) | 2008-07-24 |
| WO2008033328A2 (en) | 2008-03-20 |
| EP2054429B1 (en) | 2013-11-06 |
| JP5684474B2 (en) | 2015-03-11 |
| CA2662897A1 (en) | 2008-03-20 |
| CA2662897C (en) | 2017-11-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2054429B1 (en) | Macrocyclic antagonists of the motilin receptor for treatment of gastrointestinal dysmotility disorders | |
| US10258602B2 (en) | Macrocyclic ghrelin receptor modulators and methods of using the same | |
| EP2210612B1 (en) | Macrocyclic antagonists of the motilin receptor | |
| US8334256B2 (en) | Pharmaceutical salts of macrocyclic modulators of the ghrelin receptor | |
| US7491695B2 (en) | Methods of using macrocyclic modulators of the ghrelin receptor | |
| USRE42013E1 (en) | Macrocyclic modulators of the ghrelin receptor | |
| USRE42624E1 (en) | Methods of using macrocyclic modulators of the ghrelin receptor | |
| US20090198050A1 (en) | Macrocyclic Modulators of the Ghrelin Receptor | |
| US20080287371A1 (en) | Macrocyclic antagonists of the motilin receptor for modulation of the migrating motor complex | |
| US20180110824A1 (en) | Macrocyclic Modulators of the Ghrelin Receptor | |
| US8921521B2 (en) | Macrocyclic modulators of the Ghrelin receptor | |
| WO2011146845A1 (en) | Modified macrocyclic ghrelin receptor modulators and methods of using the same | |
| JP5739766B2 (en) | Use of macrocyclic modulators of ghrelin receptor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AC | Divisional application: reference to earlier application |
Ref document number: 2054429 Country of ref document: EP Kind code of ref document: P |
|
| AK | Designated contracting states |
Kind code of ref document: A2 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LI LT LU LV MC MT NL PL PT RO SE SI SK TR |
|
| RAP1 | Party data changed (applicant data changed or rights of an application transferred) |
Owner name: TRANZYME PHARMA INC. |
|
| RAP1 | Party data changed (applicant data changed or rights of an application transferred) |
Owner name: TRANZYME PHARMA, INC. |
|
| PUAL | Search report despatched |
Free format text: ORIGINAL CODE: 0009013 |
|
| AK | Designated contracting states |
Kind code of ref document: A3 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LI LT LU LV MC MT NL PL PT RO SE SI SK TR |
|
| RIC1 | Information provided on ipc code assigned before grant |
Ipc: C07K 5/087 20060101AFI20130524BHEP Ipc: A61P 1/00 20060101ALI20130524BHEP Ipc: C07D 213/65 20060101ALI20130524BHEP Ipc: C07C 271/16 20060101ALI20130524BHEP Ipc: C07D 213/64 20060101ALI20130524BHEP |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: THE APPLICATION IS DEEMED TO BE WITHDRAWN |
|
| 18D | Application deemed to be withdrawn |
Effective date: 20140104 |