US20040002448A1 - Macrocyclic peptides active against the hepatitis C virus - Google Patents
Macrocyclic peptides active against the hepatitis C virus Download PDFInfo
- Publication number
- US20040002448A1 US20040002448A1 US10/358,726 US35872603A US2004002448A1 US 20040002448 A1 US20040002448 A1 US 20040002448A1 US 35872603 A US35872603 A US 35872603A US 2004002448 A1 US2004002448 A1 US 2004002448A1
- Authority
- US
- United States
- Prior art keywords
- mmol
- compound
- mixture
- solution
- etoac
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- WXKFQWUHQWVRTG-UHFFFAOYSA-N [CH2-][OH+]C1CCCC1 Chemical compound [CH2-][OH+]C1CCCC1 WXKFQWUHQWVRTG-UHFFFAOYSA-N 0.000 description 30
- 0 *C1(NC(=O)[C@@H]2C[C@@H](OC3=CC([22*])=[W]C4=C3C=CC([21*])=C4)CN2C(=O)[C@@H]([3*])[2H]CC)CC1.[1*].[4*]C Chemical compound *C1(NC(=O)[C@@H]2C[C@@H](OC3=CC([22*])=[W]C4=C3C=CC([21*])=C4)CN2C(=O)[C@@H]([3*])[2H]CC)CC1.[1*].[4*]C 0.000 description 27
- IHHWHOQLONHZMR-UHFFFAOYSA-N [CH2-][C+]1=CSC(NC(C)=O)=N1 Chemical compound [CH2-][C+]1=CSC(NC(C)=O)=N1 IHHWHOQLONHZMR-UHFFFAOYSA-N 0.000 description 8
- CDEAGSIUQGMHSD-UHFFFAOYSA-N [CH2-][C+]1=CSC(NCC)=N1 Chemical compound [CH2-][C+]1=CSC(NCC)=N1 CDEAGSIUQGMHSD-UHFFFAOYSA-N 0.000 description 8
- OTMSDBZUPAUEDD-UHFFFAOYSA-N CC Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 7
- ARSLMHWBXROFQN-UHFFFAOYSA-N [CH2-][C+]1=NC=CS1 Chemical compound [CH2-][C+]1=NC=CS1 ARSLMHWBXROFQN-UHFFFAOYSA-N 0.000 description 6
- WRQJMEGRQMQMQW-UHFFFAOYSA-N [CH2-][OH+]C1CCC1 Chemical compound [CH2-][OH+]C1CCC1 WRQJMEGRQMQMQW-UHFFFAOYSA-N 0.000 description 6
- DJASYHUSBUPXPE-UHFFFAOYSA-N [CH2-][NH+]1C=CC=C1 Chemical compound [CH2-][NH+]1C=CC=C1 DJASYHUSBUPXPE-UHFFFAOYSA-N 0.000 description 4
- FEISPIJJLVWBQM-UHFFFAOYSA-N [CH2-][NH+]1C=CC=N1 Chemical compound [CH2-][NH+]1C=CC=N1 FEISPIJJLVWBQM-UHFFFAOYSA-N 0.000 description 4
- MFNQUFGJQAOLJU-UHFFFAOYSA-N [CH2-][C+]1=CSC(N)=N1 Chemical compound [CH2-][C+]1=CSC(N)=N1 MFNQUFGJQAOLJU-UHFFFAOYSA-N 0.000 description 3
- MKADJDUMYXWDEF-UHFFFAOYSA-N [CH2-][C+]1=CSC(NC(=O)OC)=N1 Chemical compound [CH2-][C+]1=CSC(NC(=O)OC)=N1 MKADJDUMYXWDEF-UHFFFAOYSA-N 0.000 description 3
- OBQLQBMDQUANNJ-UHFFFAOYSA-N [CH2-][C+]1=CSC(NC(C)C)=N1 Chemical compound [CH2-][C+]1=CSC(NC(C)C)=N1 OBQLQBMDQUANNJ-UHFFFAOYSA-N 0.000 description 3
- YFCKRKOXICSFSW-UHFFFAOYSA-N [CH2-][C+]1=CSC(NC)=N1 Chemical compound [CH2-][C+]1=CSC(NC)=N1 YFCKRKOXICSFSW-UHFFFAOYSA-N 0.000 description 3
- SDIIHTNFQYPPOD-UHFFFAOYSA-N [CH2-][C+]1=NC(C(C)C)=CS1 Chemical compound [CH2-][C+]1=NC(C(C)C)=CS1 SDIIHTNFQYPPOD-UHFFFAOYSA-N 0.000 description 3
- RVCYNVYEPADPQA-UHFFFAOYSA-N [CH2-][C+]1=NC(C)=CC=C1 Chemical compound [CH2-][C+]1=NC(C)=CC=C1 RVCYNVYEPADPQA-UHFFFAOYSA-N 0.000 description 3
- UQPWYRAFSLNDGU-UHFFFAOYSA-N C.C.C.C.C.C.C.C.[CH2-]/[C+]=C/COCCCC Chemical compound C.C.C.C.C.C.C.C.[CH2-]/[C+]=C/COCCCC UQPWYRAFSLNDGU-UHFFFAOYSA-N 0.000 description 2
- RSAIIBFKUJGUQI-UHFFFAOYSA-N [CH2-][C+]1=CC=CC=N1 Chemical compound [CH2-][C+]1=CC=CC=N1 RSAIIBFKUJGUQI-UHFFFAOYSA-N 0.000 description 2
- HEJCMISLNJKHEB-UHFFFAOYSA-N [CH2-][C+]1=CSC(C(C)C)=N1 Chemical compound [CH2-][C+]1=CSC(C(C)C)=N1 HEJCMISLNJKHEB-UHFFFAOYSA-N 0.000 description 2
- VRGDCCPVTSEGTA-UHFFFAOYSA-N [CH2-][C+]1=CSC(NC=O)=N1 Chemical compound [CH2-][C+]1=CSC(NC=O)=N1 VRGDCCPVTSEGTA-UHFFFAOYSA-N 0.000 description 2
- RJAYHGXITVTSJD-UHFFFAOYSA-N [CH2-][NH+]1C=CC(C)=N1 Chemical compound [CH2-][NH+]1C=CC(C)=N1 RJAYHGXITVTSJD-UHFFFAOYSA-N 0.000 description 2
- OLEZCQDWVNPPHJ-ZCFIWIBFSA-N [CH2-][NH2+][C@H](C)C(C)(C)C Chemical compound [CH2-][NH2+][C@H](C)C(C)(C)C OLEZCQDWVNPPHJ-ZCFIWIBFSA-N 0.000 description 2
- GYRLALFHYRTAGL-IRVZYSQHSA-M *.B.C.C.C=CCCCBr.C=CCCCSC(C)(C)[C@H](C)C(=O)O.C=CCCCSC(C)(C)[C@H](C)C(=O)OC.CC(C)(S)[C@H](N)C(=O)O.[Li]O Chemical compound *.B.C.C.C=CCCCBr.C=CCCCSC(C)(C)[C@H](C)C(=O)O.C=CCCCSC(C)(C)[C@H](C)C(=O)OC.CC(C)(S)[C@H](N)C(=O)O.[Li]O GYRLALFHYRTAGL-IRVZYSQHSA-M 0.000 description 1
- MQTKGLWAYYFIHB-QTFFFNEISA-N *.B.C.C.C=CCC[C@H](C)CC[C@H](C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N[C@]1(C(=O)OC)CC1C=C.CC1=CC2=NC(C)=CC(O)=C2C=C1.CC1=CC2=NC(C)=CC(O[C@@H]3C[C@H]4C(=O)N[C@]5(C(=O)O)CC5/C=C\CC[C@H](C)CC[C@H](NC(=O)OC5CCCC5)C(=O)N4C3)=C2C=C1.COC(=O)[C@@]12CC1/C=C\CC[C@H](C)CC[C@H](C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1/C=C\CC[C@H](C)CC[C@H](C)C(=O)N1C[C@H](OC3=C4C=CC(C)=CC4=NC(C)=C3)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1/C=C\CC[C@H](C)CC[C@H](N)C(=O)N1C[C@H](OC3=C4C=CC(C)=CC4=NC(C)=C3)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1/C=C\CC[C@H](C)CC[C@H](NC(=O)OC1CCCC1)C(=O)N1C[C@H](OC3=C4C=CC(C)=CC4=NC(C)=C3)C[C@H]1C(=O)N2.[2HH] Chemical compound *.B.C.C.C=CCC[C@H](C)CC[C@H](C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N[C@]1(C(=O)OC)CC1C=C.CC1=CC2=NC(C)=CC(O)=C2C=C1.CC1=CC2=NC(C)=CC(O[C@@H]3C[C@H]4C(=O)N[C@]5(C(=O)O)CC5/C=C\CC[C@H](C)CC[C@H](NC(=O)OC5CCCC5)C(=O)N4C3)=C2C=C1.COC(=O)[C@@]12CC1/C=C\CC[C@H](C)CC[C@H](C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1/C=C\CC[C@H](C)CC[C@H](C)C(=O)N1C[C@H](OC3=C4C=CC(C)=CC4=NC(C)=C3)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1/C=C\CC[C@H](C)CC[C@H](N)C(=O)N1C[C@H](OC3=C4C=CC(C)=CC4=NC(C)=C3)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1/C=C\CC[C@H](C)CC[C@H](NC(=O)OC1CCCC1)C(=O)N1C[C@H](OC3=C4C=CC(C)=CC4=NC(C)=C3)C[C@H]1C(=O)N2.[2HH] MQTKGLWAYYFIHB-QTFFFNEISA-N 0.000 description 1
- BDKNPFVEJKYANU-UHFFFAOYSA-N *.B.C.C.CC1=CC=CC(C(=O)Cl)=N1.CC1=CC=CC(C(=O)O)=N1.COC1=CC2=C(C=C1)C(O)=CC(C1=NC(C)=CC=C1)=N2.COC1=CC=C(C(C)=O)C(N)=C1.COC1=CC=C(C(C)=O)C(NC(=O)C2=NC(C)=CC=C2)=C1 Chemical compound *.B.C.C.CC1=CC=CC(C(=O)Cl)=N1.CC1=CC=CC(C(=O)O)=N1.COC1=CC2=C(C=C1)C(O)=CC(C1=NC(C)=CC=C1)=N2.COC1=CC=C(C(C)=O)C(N)=C1.COC1=CC=C(C(C)=O)C(NC(=O)C2=NC(C)=CC=C2)=C1 BDKNPFVEJKYANU-UHFFFAOYSA-N 0.000 description 1
- PJZLBECMTNDMLI-UHFFFAOYSA-N *.B.C.C.COC(=O)C1=NC=C(OC)C=C1.COC1=CC=C(C(C)=O)C(N)=C1.COC1=CC=C(C(C)=O)C(NC(=O)C2=NC=C(OC)C=C2)=C1.COC1=CN=C(C(=O)O)C=C1.COC1=CN=C(C2=NC3=C(C=CC(OC)=C3)C(O)=C2)C=C1 Chemical compound *.B.C.C.COC(=O)C1=NC=C(OC)C=C1.COC1=CC=C(C(C)=O)C(N)=C1.COC1=CC=C(C(C)=O)C(NC(=O)C2=NC=C(OC)C=C2)=C1.COC1=CN=C(C(=O)O)C=C1.COC1=CN=C(C2=NC3=C(C=CC(OC)=C3)C(O)=C2)C=C1 PJZLBECMTNDMLI-UHFFFAOYSA-N 0.000 description 1
- YKLICBUXGIFMNJ-MHPSQXBDSA-N *.B.C.C=CCCC1C[C@]1(NC(=O)[C@@H]1C[C@@H](OC2=CC(C3=CC=CC=C3)=NC3=CC(OC)=CC=C32)CN1C(=O)[C@@H](C)[C@@H](C)OCC=C)C(=O)OC.COC(=O)[C@@]12CC1CC/C=C/CO[C@H](C)[C@H](C)C(=O)N1C[C@H](OC3=CC(C4=CC=CC=C4)=NC4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.COC1=CC=C2C(=C1)N=C(C1=CC=CC=C1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3CC/C=C/CO[C@H](C)[C@H](C)C(=O)N2C1.COC1=CC=C2C(=C1)N=C(C1=CC=CC=C1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3CCCCCO[C@H](C)[C@H](C)C(=O)N2C1 Chemical compound *.B.C.C=CCCC1C[C@]1(NC(=O)[C@@H]1C[C@@H](OC2=CC(C3=CC=CC=C3)=NC3=CC(OC)=CC=C32)CN1C(=O)[C@@H](C)[C@@H](C)OCC=C)C(=O)OC.COC(=O)[C@@]12CC1CC/C=C/CO[C@H](C)[C@H](C)C(=O)N1C[C@H](OC3=CC(C4=CC=CC=C4)=NC4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.COC1=CC=C2C(=C1)N=C(C1=CC=CC=C1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3CC/C=C/CO[C@H](C)[C@H](C)C(=O)N2C1.COC1=CC=C2C(=C1)N=C(C1=CC=CC=C1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3CCCCCO[C@H](C)[C@H](C)C(=O)N2C1 YKLICBUXGIFMNJ-MHPSQXBDSA-N 0.000 description 1
- DCZPMWNUAYLXJM-DTGXLUCMSA-N *.B.C.C=CCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@H](OC2=CC(C3=CC=CC=C3)=NC3=CC(OC)=CC=C32)C[C@H]1C(=O)N[C@]1(C(=O)OCC)CC1C=C.CCOC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)NC(C)(C)C)C(=O)N1C[C@H](OC3=CC(C4=CC=CC=C4)=NC4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.CCOC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@H](OC3=CC(C4=CC=CC=C4)=NC4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.COC1=CC=C2C(=C1)N=C(C1=CC=CC=C1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)NC(C)(C)C)C(=O)N2C1 Chemical compound *.B.C.C=CCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@H](OC2=CC(C3=CC=CC=C3)=NC3=CC(OC)=CC=C32)C[C@H]1C(=O)N[C@]1(C(=O)OCC)CC1C=C.CCOC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)NC(C)(C)C)C(=O)N1C[C@H](OC3=CC(C4=CC=CC=C4)=NC4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.CCOC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@H](OC3=CC(C4=CC=CC=C4)=NC4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.COC1=CC=C2C(=C1)N=C(C1=CC=CC=C1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)NC(C)(C)C)C(=O)N2C1 DCZPMWNUAYLXJM-DTGXLUCMSA-N 0.000 description 1
- PZFJLONULANREV-DFFRKISFSA-N *.B.C.C=CCCC[C@H](C)C(=O)N1CC(Oc2ccnc3ccccc23)C[C@H]1C(=O)N[C@]1(C(=O)OC)C[C@H]1CCC=C.COC(=O)[C@@]12C[C@H]1CC/C=C/CCC[C@H](C)C(=O)N1CC(Oc3ccnc4ccccc34)C[C@H]1C(=O)N2.C[C@H]1CCC/C=C/CC[C@@H]2C[C@@]2(C(=O)O)NC(=O)[C@@H]2CC(Oc3ccnc4ccccc34)CN2C1=O.O=C1N[C@]2(C(=O)O)C[C@H]2CC/C=C/CCC[C@H](N=[Ac])C(=O)N2CC(Oc3ccnc4ccccc34)C[C@@H]12 Chemical compound *.B.C.C=CCCC[C@H](C)C(=O)N1CC(Oc2ccnc3ccccc23)C[C@H]1C(=O)N[C@]1(C(=O)OC)C[C@H]1CCC=C.COC(=O)[C@@]12C[C@H]1CC/C=C/CCC[C@H](C)C(=O)N1CC(Oc3ccnc4ccccc34)C[C@H]1C(=O)N2.C[C@H]1CCC/C=C/CC[C@@H]2C[C@@]2(C(=O)O)NC(=O)[C@@H]2CC(Oc3ccnc4ccccc34)CN2C1=O.O=C1N[C@]2(C(=O)O)C[C@H]2CC/C=C/CCC[C@H](N=[Ac])C(=O)N2CC(Oc3ccnc4ccccc34)C[C@@H]12 PZFJLONULANREV-DFFRKISFSA-N 0.000 description 1
- DWQOJDBPGWELKW-FRAVWECYSA-M *.B.C.C=CCS[C@@H](C)[C@H](C)C(=O)O.C=CCS[C@@H](C)[C@H](C)C(=O)OC.CC(=O)[S-].COC(=O)[C@@H](C)[C@@H](C)OS(=O)(=O)C1=CC=C(C)C=C1.COC(=O)[C@@H](C)[C@H](C)SC(C)=O.C[C@H](C(=O)O)[C@@H](C)O.[2HH].[K+] Chemical compound *.B.C.C=CCS[C@@H](C)[C@H](C)C(=O)O.C=CCS[C@@H](C)[C@H](C)C(=O)OC.CC(=O)[S-].COC(=O)[C@@H](C)[C@@H](C)OS(=O)(=O)C1=CC=C(C)C=C1.COC(=O)[C@@H](C)[C@H](C)SC(C)=O.C[C@H](C(=O)O)[C@@H](C)O.[2HH].[K+] DWQOJDBPGWELKW-FRAVWECYSA-M 0.000 description 1
- UBJUAOKVMDGGLH-SXOZJBJDSA-N *.B.C.COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](C)C(=O)N1C[C@H](O/C3=C/C(C(C)=O)=N\C4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](C)C(=O)N1C[C@H](O/C3=C/C(C(C)=O)=N\C4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.COC1=CC=C2C(O)=CC(C(C)=O)=NC2=C1.O=C(Cl)OC1CCCC1 Chemical compound *.B.C.COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](C)C(=O)N1C[C@H](O/C3=C/C(C(C)=O)=N\C4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](C)C(=O)N1C[C@H](O/C3=C/C(C(C)=O)=N\C4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.COC1=CC=C2C(O)=CC(C(C)=O)=NC2=C1.O=C(Cl)OC1CCCC1 UBJUAOKVMDGGLH-SXOZJBJDSA-N 0.000 description 1
- JKHGMYDYTMEQDV-UHFFFAOYSA-N *.B.C.COCCOCOC1=C2C=CC(OC)=CC2=NC(C(=O)OC)=C1.COCCOCOC1=C2C=CC(OC)=CC2=NC(C2=CN=CO2)=C1.COCCOCOC1=C2C=CC(OC)=CC2=NC(C2=CN=CO2)=C1.[H]C(=O)C1=CC(OCOCCOC)=C2C=CC(OC)=CC2=N1 Chemical compound *.B.C.COCCOCOC1=C2C=CC(OC)=CC2=NC(C(=O)OC)=C1.COCCOCOC1=C2C=CC(OC)=CC2=NC(C2=CN=CO2)=C1.COCCOCOC1=C2C=CC(OC)=CC2=NC(C2=CN=CO2)=C1.[H]C(=O)C1=CC(OCOCCOC)=C2C=CC(OC)=CC2=N1 JKHGMYDYTMEQDV-UHFFFAOYSA-N 0.000 description 1
- USTMLLOOIMFAIG-UHFFFAOYSA-N *.B.CC(C)(C)N=C=S.CC(C)(C)NC(=S)NC1CCCC1.NC(=S)NC1CCCC1.NC1CCCC1 Chemical compound *.B.CC(C)(C)N=C=S.CC(C)(C)NC(=S)NC1CCCC1.NC(=S)NC1CCCC1.NC1CCCC1 USTMLLOOIMFAIG-UHFFFAOYSA-N 0.000 description 1
- VYIFBPKQWNJACD-UHFFFAOYSA-M *.B.N#CS[K].NC(=S)NC1CC1.NC1CC1.O=C(Cl)C1=CC=CC=C1.O=C(NC(=S)NC1CC1)C1=CC=CC=C1 Chemical compound *.B.N#CS[K].NC(=S)NC1CC1.NC1CC1.O=C(Cl)C1=CC=CC=C1.O=C(NC(=S)NC1CC1)C1=CC=CC=C1 VYIFBPKQWNJACD-UHFFFAOYSA-M 0.000 description 1
- MWXYLJFOHBDVET-HDPWTFRZSA-N *.B[SH](C)C.C.C=CCCC1C[C@]1(NC(=O)[C@@H]1C[C@@H](OC2=CC(C3=CC=CC=C3)=NC3=CC(OC)=CC=C32)CN1)C(C)=O.C=CCCC1C[C@]1(NC(=O)[C@@H]1C[C@@H](OC2=CC(C3=CC=CC=C3)=NC3=CC(OC)=CC=C32)CN1C(=O)[C@@H](C)CCC)C(C)=O.CCC[C@H](C)C(=O)N1C[C@H](OC2=CC(C3=CC=CC=C3)=NC3=CC(OC)=CC=C32)C[C@H]1C(=O)N[C@]1(C(C)=O)CC1CCCCO.CCC[C@H](C)C(=O)O.CCl.C[C@@H](CCC(N)=O)C(=O)O.[2HH] Chemical compound *.B[SH](C)C.C.C=CCCC1C[C@]1(NC(=O)[C@@H]1C[C@@H](OC2=CC(C3=CC=CC=C3)=NC3=CC(OC)=CC=C32)CN1)C(C)=O.C=CCCC1C[C@]1(NC(=O)[C@@H]1C[C@@H](OC2=CC(C3=CC=CC=C3)=NC3=CC(OC)=CC=C32)CN1C(=O)[C@@H](C)CCC)C(C)=O.CCC[C@H](C)C(=O)N1C[C@H](OC2=CC(C3=CC=CC=C3)=NC3=CC(OC)=CC=C32)C[C@H]1C(=O)N[C@]1(C(C)=O)CC1CCCCO.CCC[C@H](C)C(=O)O.CCl.C[C@@H](CCC(N)=O)C(=O)O.[2HH] MWXYLJFOHBDVET-HDPWTFRZSA-N 0.000 description 1
- OOPGGVNLDSZDAX-UHFFFAOYSA-N *.CC1CCC(C)O1.COC1=CC2=C(C=C1)C(O)=CC(N)=N2.COC1=CC2=C(C=C1)C(O)=CC(N1C=CC=C1)=N2 Chemical compound *.CC1CCC(C)O1.COC1=CC2=C(C=C1)C(O)=CC(N)=N2.COC1=CC2=C(C=C1)C(O)=CC(N1C=CC=C1)=N2 OOPGGVNLDSZDAX-UHFFFAOYSA-N 0.000 description 1
- SPMBTBUISYKTIF-WKOCXQGCSA-N *.COC(=O)[C@@]12C[C@H]1CC/C=C/CCC[C@H](C)C(=O)N1CC(OC3=CC=NC4=C3C=CC=C4)C[C@H]1C(=O)N2.C[C@H]1CCCC(O)C(O)CC[C@@H]2C[C@@]2(C(=O)O)NC(=O)[C@@H]2CC(OC3=CC=NC4=C3C=CC=C4)CN2C1=O Chemical compound *.COC(=O)[C@@]12C[C@H]1CC/C=C/CCC[C@H](C)C(=O)N1CC(OC3=CC=NC4=C3C=CC=C4)C[C@H]1C(=O)N2.C[C@H]1CCCC(O)C(O)CC[C@@H]2C[C@@]2(C(=O)O)NC(=O)[C@@H]2CC(OC3=CC=NC4=C3C=CC=C4)CN2C1=O SPMBTBUISYKTIF-WKOCXQGCSA-N 0.000 description 1
- NZEIQPQWIDOXRN-GLQWWQRZSA-N B.C.C=CCO[C@H](C)[C@H](NC(=O)OC(C)(C)C)C(=O)O.C=CCO[C@H](C)[C@H](NC(=O)OC(C)(C)C)C(=O)OC.COC(=O)[C@@H](NC(=O)OC(C)(C)C)[C@@H](C)O.C[C@@H](O)[C@H](NC(=O)OC(C)(C)C)C(=O)O Chemical compound B.C.C=CCO[C@H](C)[C@H](NC(=O)OC(C)(C)C)C(=O)O.C=CCO[C@H](C)[C@H](NC(=O)OC(C)(C)C)C(=O)OC.COC(=O)[C@@H](NC(=O)OC(C)(C)C)[C@@H](C)O.C[C@@H](O)[C@H](NC(=O)OC(C)(C)C)C(=O)O NZEIQPQWIDOXRN-GLQWWQRZSA-N 0.000 description 1
- ARLNHOYDWXTJEZ-IGXRMXPISA-N BC(=O)N[C@H]1CCCCC/C=C\C2C[C@@]2(C(=O)O)NC(=O)[C@@H]2C[C@@H](OC3=CC(C4=NC(C)=NO4)=NC4=C3C=CC(OC)=C4)CN2C1=O Chemical compound BC(=O)N[C@H]1CCCCC/C=C\C2C[C@@]2(C(=O)O)NC(=O)[C@@H]2C[C@@H](OC3=CC(C4=NC(C)=NO4)=NC4=C3C=CC(OC)=C4)CN2C1=O ARLNHOYDWXTJEZ-IGXRMXPISA-N 0.000 description 1
- ARYJDONWCOOFAT-IGXRMXPISA-N BC(=O)N[C@H]1CCCCC/C=C\C2C[C@@]2(C(=O)O)NC(=O)[C@@H]2C[C@@H](OC3=CC(C4=NN=C(C)O4)=NC4=C3C=CC(OC)=C4)CN2C1=O Chemical compound BC(=O)N[C@H]1CCCCC/C=C\C2C[C@@]2(C(=O)O)NC(=O)[C@@H]2C[C@@H](OC3=CC(C4=NN=C(C)O4)=NC4=C3C=CC(OC)=C4)CN2C1=O ARYJDONWCOOFAT-IGXRMXPISA-N 0.000 description 1
- PVEKGLFCVQXNJY-UHFFFAOYSA-N Br.C.C.CC(C)NC(N)=S.CC(C)NC1=NC(C(=O)O)=CS1.CC(C)NC1=NC(C(=O)O)=CS1.COC1=CC(N)=C(C(C)=O)C=C1.COC1=CC(N)=C(C(C)=O)C=C1.COC1=CC(N)=CC=C1.COC1=CC(NC(=O)C2=CSC(NC(C)C)=N2)=C(C(C)=O)C=C1.COC1=CC=C2C(O)=CC(C3=CSC(NC(C)C)=N3)=NC2=C1.O=C(O)C(=O)CBr Chemical compound Br.C.C.CC(C)NC(N)=S.CC(C)NC1=NC(C(=O)O)=CS1.CC(C)NC1=NC(C(=O)O)=CS1.COC1=CC(N)=C(C(C)=O)C=C1.COC1=CC(N)=C(C(C)=O)C=C1.COC1=CC(N)=CC=C1.COC1=CC(NC(=O)C2=CSC(NC(C)C)=N2)=C(C(C)=O)C=C1.COC1=CC=C2C(O)=CC(C3=CSC(NC(C)C)=N3)=NC2=C1.O=C(O)C(=O)CBr PVEKGLFCVQXNJY-UHFFFAOYSA-N 0.000 description 1
- JUGZCWCXYZKIEO-UHFFFAOYSA-N C.C.C.C.C.C.C.C.C.C.[CH2-][CH+]CC=CCCCCCC Chemical compound C.C.C.C.C.C.C.C.C.C.[CH2-][CH+]CC=CCCCCCC JUGZCWCXYZKIEO-UHFFFAOYSA-N 0.000 description 1
- UCXXWYMMLPCRAI-UHFFFAOYSA-N C.C.C.C.C.C.C.C.C.[CH2-]/[C+]=C\CCCCCCC Chemical compound C.C.C.C.C.C.C.C.C.[CH2-]/[C+]=C\CCCCCCC UCXXWYMMLPCRAI-UHFFFAOYSA-N 0.000 description 1
- CNXKMWVAFOTSIS-UHFFFAOYSA-N C.C.C.C.C.C.C.C.C.[CH2-][C+]=CCCCC=CCC Chemical compound C.C.C.C.C.C.C.C.C.[CH2-][C+]=CCCCC=CCC CNXKMWVAFOTSIS-UHFFFAOYSA-N 0.000 description 1
- JRZFOTONCBVDSZ-UHFFFAOYSA-N C.C.C.C.C.C.C.C.[CH2-]/[C+]=C\CCCCCC Chemical compound C.C.C.C.C.C.C.C.[CH2-]/[C+]=C\CCCCCC JRZFOTONCBVDSZ-UHFFFAOYSA-N 0.000 description 1
- IWSKERJDVDMJID-AISPUDCNSA-N C.C.C.C.C.C.C.COC1=CC2=NC(C3=CC=CC=C3)=CC(O[C@@H]3C[C@H]4C(=O)N[C@]5(C(=O)O)CC5CCCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N4C3)=C2C=C1.[1*] Chemical compound C.C.C.C.C.C.C.COC1=CC2=NC(C3=CC=CC=C3)=CC(O[C@@H]3C[C@H]4C(=O)N[C@]5(C(=O)O)CC5CCCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N4C3)=C2C=C1.[1*] IWSKERJDVDMJID-AISPUDCNSA-N 0.000 description 1
- OBZIHBHPLAHIEE-UHFFFAOYSA-N C.C.C.C.C.C.[CH2-]/[C+]=C/CCCC Chemical compound C.C.C.C.C.C.[CH2-]/[C+]=C/CCCC OBZIHBHPLAHIEE-UHFFFAOYSA-N 0.000 description 1
- ZPELSZWAEGRYEZ-UHFFFAOYSA-N C.C.C.C.C.C.[CH2-][CH+]CCCCC Chemical compound C.C.C.C.C.C.[CH2-][CH+]CCCCC ZPELSZWAEGRYEZ-UHFFFAOYSA-N 0.000 description 1
- SYLRWIHWOAWVHJ-UHFFFAOYSA-N C.C.C.C.C.C1=CN=CC1.C1=CNC=C1.C1=CNC=N1.C1=CNC=N1.C1=CNN=C1.C1=COC=C1.C1=COC=N1.C1=COC=N1 Chemical compound C.C.C.C.C.C1=CN=CC1.C1=CNC=C1.C1=CNC=N1.C1=CNC=N1.C1=CNN=C1.C1=COC=C1.C1=COC=N1.C1=COC=N1 SYLRWIHWOAWVHJ-UHFFFAOYSA-N 0.000 description 1
- RKOKJJHRKXZOMU-CLHWYOSBSA-N C.C.C=CCCC(Br)CBr.C=CCCC1CC1(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C.C=CCCC1CC1(N)C(=O)OC(C)(C)C.C=CCCC1CC1(NC(=O)OCC[Si](C)(C)C)C(=O)OC(C)(C)C.C=CCCC1C[C@@]1(N)C(=O)OC(C)(C)C.C=CCC[C@@H]1C[C@]1(N)C(=O)OC(C)(C)C.CC(C)(C)OC(=O)CC(=O)OC(C)(C)C.[H]OC(=O)C1(C(=O)OC(C)(C)C)CC1CCC=C Chemical compound C.C.C=CCCC(Br)CBr.C=CCCC1CC1(C(=O)OC(C)(C)C)C(=O)OC(C)(C)C.C=CCCC1CC1(N)C(=O)OC(C)(C)C.C=CCCC1CC1(NC(=O)OCC[Si](C)(C)C)C(=O)OC(C)(C)C.C=CCCC1C[C@@]1(N)C(=O)OC(C)(C)C.C=CCC[C@@H]1C[C@]1(N)C(=O)OC(C)(C)C.CC(C)(C)OC(=O)CC(=O)OC(C)(C)C.[H]OC(=O)C1(C(=O)OC(C)(C)C)CC1CCC=C RKOKJJHRKXZOMU-CLHWYOSBSA-N 0.000 description 1
- CPRBDTFYEDVQHC-GPZOHOMCSA-N C.C.CCO/C(C)=N/C1=CC(OC)=CC=C1C(=O)OC.CCOC1=NC2=CC(OC)=CC=C2C(O)=C1.COC(=O)C1=CC=C(OC)C=C1N Chemical compound C.C.CCO/C(C)=N/C1=CC(OC)=CC=C1C(=O)OC.CCOC1=NC2=CC(OC)=CC=C2C(O)=C1.COC(=O)C1=CC=C(OC)C=C1N CPRBDTFYEDVQHC-GPZOHOMCSA-N 0.000 description 1
- IZZUQJSTJJUXAI-CMYCCFIZSA-N C.C=CCCC[C@H](C)C(=O)N1CC(Oc2ccnc3ccccc23)C[C@H]1C(=O)N[C@]1(C(=O)OC)C[C@H]1CCC=C.C=CCCC[C@H](C)C(=O)O.C=CCC[C@@H]1C[C@@]1(C)NC(=O)[C@@H]1CC(Oc2ccnc3ccccc23)CN1C.CC(C)(C)OC(=O)N1CC(Oc2ccnc3ccccc23)C[C@H]1C(=O)O.[H]N1CC(Oc2ccnc3ccccc23)C[C@H]1C(=O)N[C@]1(C(=O)OC)C[C@H]1CCC=C.[H][C@@]1(CCC=C)C[C@]1(N)C(=O)OC(C)(C)C Chemical compound C.C=CCCC[C@H](C)C(=O)N1CC(Oc2ccnc3ccccc23)C[C@H]1C(=O)N[C@]1(C(=O)OC)C[C@H]1CCC=C.C=CCCC[C@H](C)C(=O)O.C=CCC[C@@H]1C[C@@]1(C)NC(=O)[C@@H]1CC(Oc2ccnc3ccccc23)CN1C.CC(C)(C)OC(=O)N1CC(Oc2ccnc3ccccc23)C[C@H]1C(=O)O.[H]N1CC(Oc2ccnc3ccccc23)C[C@H]1C(=O)N[C@]1(C(=O)OC)C[C@H]1CCC=C.[H][C@@]1(CCC=C)C[C@]1(N)C(=O)OC(C)(C)C IZZUQJSTJJUXAI-CMYCCFIZSA-N 0.000 description 1
- ZEFZCPSZAOWOML-QSUHDREJSA-N C.CC(=O)O.CCC[C@H](C)C(=O)N1C[C@H](OC2=CC(C3=CC=CC=C3)=NC3=CC(OC)=CC=C32)C[C@H]1C(=O)N[C@]1(C(C)=O)CC1CCCC=O.COC1=CC=C2C(=C1)N=C(C1=CC=CC=C1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(C)=O)CC3CCCCN(C(C)=O)CC[C@H](C)C(=O)N2C1.COC1=CC=C2C(=C1)N=C(C1=CC=CC=C1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(C)=O)CC3CCCCNCC[C@H](C)C(=O)N2C1.F Chemical compound C.CC(=O)O.CCC[C@H](C)C(=O)N1C[C@H](OC2=CC(C3=CC=CC=C3)=NC3=CC(OC)=CC=C32)C[C@H]1C(=O)N[C@]1(C(C)=O)CC1CCCC=O.COC1=CC=C2C(=C1)N=C(C1=CC=CC=C1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(C)=O)CC3CCCCN(C(C)=O)CC[C@H](C)C(=O)N2C1.COC1=CC=C2C(=C1)N=C(C1=CC=CC=C1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(C)=O)CC3CCCCNCC[C@H](C)C(=O)N2C1.F ZEFZCPSZAOWOML-QSUHDREJSA-N 0.000 description 1
- OWXOTUJQKBSXCZ-UHFFFAOYSA-N C.CC(C)C(=O)CBr.CC(C)C1=CSC(C(=O)O)=N1.CCOC(=O)C(N)=O.CCOC(=O)C1=NC(C(C)C)=CS1.COC1=CC(N)=C(C(C)=O)C=C1.COC1=CC2=C(C=C1)C(O)=CC(C1=NC(C(C)C)=CS1)=N2.COC1=CC=C(C(C)=O)C(NC(=O)C2=NC(C(C)C)=CS2)=C1 Chemical compound C.CC(C)C(=O)CBr.CC(C)C1=CSC(C(=O)O)=N1.CCOC(=O)C(N)=O.CCOC(=O)C1=NC(C(C)C)=CS1.COC1=CC(N)=C(C(C)=O)C=C1.COC1=CC2=C(C=C1)C(O)=CC(C1=NC(C(C)C)=CS1)=N2.COC1=CC=C(C(C)=O)C(NC(=O)C2=NC(C(C)C)=CS2)=C1 OWXOTUJQKBSXCZ-UHFFFAOYSA-N 0.000 description 1
- KUQLKIUXGIZXHX-UHFFFAOYSA-N C.CN1C=CN=C1.CN1C=CN=C1C(=O)O.COC1=CC(N)=C(C(C)=O)C=C1.COC1=CC2=C(C=C1)C(O)=CC(C1=NC=CN1C)=N2.COC1=CC=C(C(C)=O)C(NC(=O)C2=NC=CN2C)=C1 Chemical compound C.CN1C=CN=C1.CN1C=CN=C1C(=O)O.COC1=CC(N)=C(C(C)=O)C=C1.COC1=CC2=C(C=C1)C(O)=CC(C1=NC=CN1C)=N2.COC1=CC=C(C(C)=O)C(NC(=O)C2=NC=CN2C)=C1 KUQLKIUXGIZXHX-UHFFFAOYSA-N 0.000 description 1
- BUDQDWGNQVEFAC-UHFFFAOYSA-N C1=COCCC1 Chemical compound C1=COCCC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 1
- GALLMPFNVWUCGD-UHFFFAOYSA-N C=CC1CC1(N)C(=O)O Chemical compound C=CC1CC1(N)C(=O)O GALLMPFNVWUCGD-UHFFFAOYSA-N 0.000 description 1
- AZLJLLRMKCMLEJ-NPPUIMKSSA-N C=CCCC(=O)CC[C@H](NC(=O)OC(C)(C)C)C(=O)N1CC(Oc2cc(-c3ccccc3)nc3cc(OC)ccc23)C[C@H]1C(=O)N[C@]1(C(=O)OC)CC1C=C.C=CCCC(=O)CC[C@H](NC(=O)OC(C)(C)C)C(=O)OC.COC(=O)[C@@]12C[C@H]1/C=C\CCC(=O)CC[C@H](NC(=O)OC(C)(C)C)C(=O)N1CC(Oc3cc(-c4ccccc4)nc4cc(OC)ccc34)C[C@H]1C(=O)N2.COc1ccc2c(OC3C[C@H]4C(=O)N[C@]5(C(=O)O)CC5/C=C\CCC(=O)CC[C@H](NC(=O)OC(C)(C)C)C(=O)N4C3)cc(-c3ccccc3)nc2c1.[H]N1CC(Oc2cc(-c3ccccc3)nc3cc(OC)ccc23)C[C@H]1C(=O)N[C@]1(C(=O)OC)CC1C=C Chemical compound C=CCCC(=O)CC[C@H](NC(=O)OC(C)(C)C)C(=O)N1CC(Oc2cc(-c3ccccc3)nc3cc(OC)ccc23)C[C@H]1C(=O)N[C@]1(C(=O)OC)CC1C=C.C=CCCC(=O)CC[C@H](NC(=O)OC(C)(C)C)C(=O)OC.COC(=O)[C@@]12C[C@H]1/C=C\CCC(=O)CC[C@H](NC(=O)OC(C)(C)C)C(=O)N1CC(Oc3cc(-c4ccccc4)nc4cc(OC)ccc34)C[C@H]1C(=O)N2.COc1ccc2c(OC3C[C@H]4C(=O)N[C@]5(C(=O)O)CC5/C=C\CCC(=O)CC[C@H](NC(=O)OC(C)(C)C)C(=O)N4C3)cc(-c3ccccc3)nc2c1.[H]N1CC(Oc2cc(-c3ccccc3)nc3cc(OC)ccc23)C[C@H]1C(=O)N[C@]1(C(=O)OC)CC1C=C AZLJLLRMKCMLEJ-NPPUIMKSSA-N 0.000 description 1
- UNWWGNITBPZXIQ-OEZHUUGISA-N C=CCCC(=O)CC[C@H](NC(=O)OC(C)(C)C)C(=O)OC.C=CCOC(=O)CC(=O)CC[C@H](NC(=O)OC(C)(C)C)C(=O)OC.C=CCOC(=O)CC=O.COC(=O)[C@H](CCC(=O)O)NC(=O)OC(C)(C)C.[MgH2] Chemical compound C=CCCC(=O)CC[C@H](NC(=O)OC(C)(C)C)C(=O)OC.C=CCOC(=O)CC(=O)CC[C@H](NC(=O)OC(C)(C)C)C(=O)OC.C=CCOC(=O)CC=O.COC(=O)[C@H](CCC(=O)O)NC(=O)OC(C)(C)C.[MgH2] UNWWGNITBPZXIQ-OEZHUUGISA-N 0.000 description 1
- SIKOJODOMOMFAX-UHFFFAOYSA-N C=CCCC1CC1(N)C(=O)O Chemical compound C=CCCC1CC1(N)C(=O)O SIKOJODOMOMFAX-UHFFFAOYSA-N 0.000 description 1
- JPMVHSAAQGMUNQ-GBOOVQHLSA-N C=CCCCC/C=C(\NC(C)=O)C(=O)OCC.C=CCCCCC(O)CO.C=CCCCCC=O.C=CCCCCC[C@@H](NC(C)=O)C(=O)OCC.C=CCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)O.CCOC(=O)C(NC(C)=O)C(=O)O.CCOC(=O)C(NC(C)=O)C(=O)OCC Chemical compound C=CCCCC/C=C(\NC(C)=O)C(=O)OCC.C=CCCCCC(O)CO.C=CCCCCC=O.C=CCCCCC[C@@H](NC(C)=O)C(=O)OCC.C=CCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)O.CCOC(=O)C(NC(C)=O)C(=O)O.CCOC(=O)C(NC(C)=O)C(=O)OCC JPMVHSAAQGMUNQ-GBOOVQHLSA-N 0.000 description 1
- HUQPPUWMFSBGSY-QUJMGQBTSA-N C=CCCCCCCBr.C=CCCCCCCC(=O)N1C(=O)OC[C@@H]1CC1=CC=CC=C1.C=CCCCCCCC(=O)O.C=CCCCCC[C@H](N=[N+]=[N-])C(=O)N1C(=O)OC[C@@H]1CC1=CC=CC=C1.C=CCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C(=O)OC[C@@H]1CC1=CC=CC=C1.C=CCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)O Chemical compound C=CCCCCCCBr.C=CCCCCCCC(=O)N1C(=O)OC[C@@H]1CC1=CC=CC=C1.C=CCCCCCCC(=O)O.C=CCCCCC[C@H](N=[N+]=[N-])C(=O)N1C(=O)OC[C@@H]1CC1=CC=CC=C1.C=CCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C(=O)OC[C@@H]1CC1=CC=CC=C1.C=CCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)O HUQPPUWMFSBGSY-QUJMGQBTSA-N 0.000 description 1
- LBPKFLBPTAVULV-KDGDKJEPSA-N C=CCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N[C@]1(C(=O)OC)CC1C=C.COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N2 Chemical compound C=CCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N[C@]1(C(=O)OC)CC1C=C.COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N2 LBPKFLBPTAVULV-KDGDKJEPSA-N 0.000 description 1
- OMJOVOTUGWQNKU-HNIFWMPASA-N C=CCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@H](OC2=C3C=CC(C)=CC3=NC(C(C)=O)=C2)C[C@H]1C(=O)N[C@]1(C(=O)OC)CC1C=C.COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@H](OC3=C4C=CC(C)=CC4=NC(C(C)=O)=C3)C[C@H]1C(=O)N2 Chemical compound C=CCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@H](OC2=C3C=CC(C)=CC3=NC(C(C)=O)=C2)C[C@H]1C(=O)N[C@]1(C(=O)OC)CC1C=C.COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@H](OC3=C4C=CC(C)=CC4=NC(C(C)=O)=C3)C[C@H]1C(=O)N2 OMJOVOTUGWQNKU-HNIFWMPASA-N 0.000 description 1
- WDZSHTRWVUDLLF-QJZGANMVSA-N C=CCC[C@@H](C)CC[C@H](NC(=O)OC(C)(C)C)C(=O)O.C=CCC[C@@H](C)CC[C@H](NC(C)=O)C(=O)OCC.CC(C)=CCC[C@@H](C)CC=O.CCOC(=O)[C@H](CC[C@H](C)CCC(O)C(C)(C)O)NC(C)=O.CCOC(=O)[C@H](CC[C@H](C)CCC=C(C)C)NC(C)=O.CCOC(=O)[C@H](CC[C@H](C)CCC=O)NC(C)=O Chemical compound C=CCC[C@@H](C)CC[C@H](NC(=O)OC(C)(C)C)C(=O)O.C=CCC[C@@H](C)CC[C@H](NC(C)=O)C(=O)OCC.CC(C)=CCC[C@@H](C)CC=O.CCOC(=O)[C@H](CC[C@H](C)CCC(O)C(C)(C)O)NC(C)=O.CCOC(=O)[C@H](CC[C@H](C)CCC=C(C)C)NC(C)=O.CCOC(=O)[C@H](CC[C@H](C)CCC=O)NC(C)=O WDZSHTRWVUDLLF-QJZGANMVSA-N 0.000 description 1
- RSJMHCSCZRPCML-FWVONAFASA-N CC(C)(C)OC(N[C@@](CCC/C=C/CC[C@H](C1)[C@]1(C(OC)=O)N1)(C=O)N(C[C@@H](C2)Oc3ccnc4c3cccc4)[C@@H]2C1=O)=O Chemical compound CC(C)(C)OC(N[C@@](CCC/C=C/CC[C@H](C1)[C@]1(C(OC)=O)N1)(C=O)N(C[C@@H](C2)Oc3ccnc4c3cccc4)[C@@H]2C1=O)=O RSJMHCSCZRPCML-FWVONAFASA-N 0.000 description 1
- HVVNCADYHRXEDR-KGMATJATSA-N CC(C)(C)OC(N[C@@](CCCC(C(CC[C@H](C1)[C@]1(C(O)=O)N1)O)O)(C=O)N(C[C@@H](C2)Oc3ccnc4c3cccc4)[C@@H]2C1=O)=O Chemical compound CC(C)(C)OC(N[C@@](CCCC(C(CC[C@H](C1)[C@]1(C(O)=O)N1)O)O)(C=O)N(C[C@@H](C2)Oc3ccnc4c3cccc4)[C@@H]2C1=O)=O HVVNCADYHRXEDR-KGMATJATSA-N 0.000 description 1
- HKZXDFQIKRFLMW-UHFFFAOYSA-N CC(C)C1C=CCCC1 Chemical compound CC(C)C1C=CCCC1 HKZXDFQIKRFLMW-UHFFFAOYSA-N 0.000 description 1
- VAAPKQNASNXGQY-UHFFFAOYSA-N CC(Nc1ncc(C)[s]1)=O Chemical compound CC(Nc1ncc(C)[s]1)=O VAAPKQNASNXGQY-UHFFFAOYSA-N 0.000 description 1
- YNYORQDPRBLCSF-UHFFFAOYSA-H CC1=CC(C)=C(N2C=CN(C3=C(C)C=C(C)C=C3C)C2C[Ru](Cl)(Cl)(=CC2=CC=CC=C2)[PH](C2CCCCC2)(C2CCCCC2)C2CCCCC2)C(C)=C1.Cl[Ru](Cl)(=CC1=CC=CC=C1)([PH](C1CCCCC1)(C1CCCCC1)C1CCCCC1)[PH](C1CCCCC1)(C1CCCCC1)C1CCCCC1.[H]C1=[Ru-](Cl)(Cl)([PH](C2CCCCC2)(C2CCCCC2)C2CCCCC2)[O+](C(C)C)C2=C1C=CC=C2 Chemical compound CC1=CC(C)=C(N2C=CN(C3=C(C)C=C(C)C=C3C)C2C[Ru](Cl)(Cl)(=CC2=CC=CC=C2)[PH](C2CCCCC2)(C2CCCCC2)C2CCCCC2)C(C)=C1.Cl[Ru](Cl)(=CC1=CC=CC=C1)([PH](C1CCCCC1)(C1CCCCC1)C1CCCCC1)[PH](C1CCCCC1)(C1CCCCC1)C1CCCCC1.[H]C1=[Ru-](Cl)(Cl)([PH](C2CCCCC2)(C2CCCCC2)C2CCCCC2)[O+](C(C)C)C2=C1C=CC=C2 YNYORQDPRBLCSF-UHFFFAOYSA-H 0.000 description 1
- OSGZXDNWWVOKNT-CIAQUZEISA-N CCNC1=NC(C2=NC3=C(C=CC(OC)=C3)C(O[C@@H]3CC4C(=O)N[C@]5(C(=O)O)CC5CCCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N4C3)=C2)=CS1 Chemical compound CCNC1=NC(C2=NC3=C(C=CC(OC)=C3)C(O[C@@H]3CC4C(=O)N[C@]5(C(=O)O)CC5CCCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N4C3)=C2)=CS1 OSGZXDNWWVOKNT-CIAQUZEISA-N 0.000 description 1
- LAEVGEXPHYGHLZ-LJNBLCGFSA-N CCNC1=NC(C2=NC3=C(C=CC(OC)=C3)C(O[C@@H]3C[C@H]4C(=O)N[C@]5(C(=O)O)CC5/C=C\CCCCC[C@H](NC(=O)OC5CCCC5)C(=O)N4C3)=C2)=CS1 Chemical compound CCNC1=NC(C2=NC3=C(C=CC(OC)=C3)C(O[C@@H]3C[C@H]4C(=O)N[C@]5(C(=O)O)CC5/C=C\CCCCC[C@H](NC(=O)OC5CCCC5)C(=O)N4C3)=C2)=CS1 LAEVGEXPHYGHLZ-LJNBLCGFSA-N 0.000 description 1
- HUYDIRYEFMWXOZ-FZPBBAPBSA-N CCNC1=NC(C2=NC3=C(C=CC(OC)=C3)C(O[C@@H]3C[C@H]4C(=O)N[C@]5(C(=O)O)CC5CCCCCCC[C@H](NC(=O)OC5CCCC5)C(=O)N4C3)=C2)=CS1 Chemical compound CCNC1=NC(C2=NC3=C(C=CC(OC)=C3)C(O[C@@H]3C[C@H]4C(=O)N[C@]5(C(=O)O)CC5CCCCCCC[C@H](NC(=O)OC5CCCC5)C(=O)N4C3)=C2)=CS1 HUYDIRYEFMWXOZ-FZPBBAPBSA-N 0.000 description 1
- HQRQLLDPYLHHRZ-NGMAJBNASA-N CNC1=NC(C2=NC3=C(C=CC(OC)=C3)C(O[C@@H]3C[C@H]4C(=O)N[C@]5(C(=O)O)CC5/C=C\CCCCC[C@H](NC(=O)OC5CCCC5)C(=O)N4C3)=C2)=CS1 Chemical compound CNC1=NC(C2=NC3=C(C=CC(OC)=C3)C(O[C@@H]3C[C@H]4C(=O)N[C@]5(C(=O)O)CC5/C=C\CCCCC[C@H](NC(=O)OC5CCCC5)C(=O)N4C3)=C2)=CS1 HQRQLLDPYLHHRZ-NGMAJBNASA-N 0.000 description 1
- CFEQUYXGFOZLGG-UHFFFAOYSA-N COC(=O)C1=NC2=CC(OC)=CC=C2C(O)=C1.COC1=CC=C2C(O)=CC(C3=NC(C)=NO3)=NC2=C1.COCCOCOC1=CC(C(=O)OC)=NC2=CC(OC)=CC=C21.COCCOCOC1=CC(C2=NC(C)=NO2)=NC2=CC(OC)=CC=C21 Chemical compound COC(=O)C1=NC2=CC(OC)=CC=C2C(O)=C1.COC1=CC=C2C(O)=CC(C3=NC(C)=NO3)=NC2=C1.COCCOCOC1=CC(C(=O)OC)=NC2=CC(OC)=CC=C21.COCCOCOC1=CC(C2=NC(C)=NO2)=NC2=CC(OC)=CC=C21 CFEQUYXGFOZLGG-UHFFFAOYSA-N 0.000 description 1
- ACPWDMBBRFUIJS-YXRLLDJJSA-N COC(=O)C1CCCCCCCCC[C@H](C)C(=O)N2C[C@@H](O)C[C@H]2C(=O)N1.COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1CCCCCCC[C@H](C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1CCCCCCC[C@H](C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N2.C[C@H]1CCCCCCCCCC(C(=O)O)NC(=O)[C@@H]2C[C@H](O)CN2C1=O Chemical compound COC(=O)C1CCCCCCCCC[C@H](C)C(=O)N2C[C@@H](O)C[C@H]2C(=O)N1.COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1CCCCCCC[C@H](C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1CCCCCCC[C@H](C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N2.C[C@H]1CCCCCCCCCC(C(=O)O)NC(=O)[C@@H]2C[C@H](O)CN2C1=O ACPWDMBBRFUIJS-YXRLLDJJSA-N 0.000 description 1
- JQGKHHKMIHGDSO-OVFNNLEZSA-N COC(=O)NC1=NC(C2=NC3=C(C=CC(OC)=C3)C(O[C@@H]3C[C@H]4C(=O)N[C@]5(C(=O)O)CC5/C=C\CCCCC[C@H](NC(=O)OC5CCCC5)C(=O)N4C3)=C2)=CS1 Chemical compound COC(=O)NC1=NC(C2=NC3=C(C=CC(OC)=C3)C(O[C@@H]3C[C@H]4C(=O)N[C@]5(C(=O)O)CC5/C=C\CCCCC[C@H](NC(=O)OC5CCCC5)C(=O)N4C3)=C2)=CS1 JQGKHHKMIHGDSO-OVFNNLEZSA-N 0.000 description 1
- HCIORPBMTDQZIE-ZPCCFCMBSA-N COC(=O)[C@@H]1C[C@@H](OC2=CC=NC3=C2C=CC(OC)=C3)CN1C(=O)OC(C)(C)C.COC(=O)[C@@H]1C[C@H](O)CN1C(=O)OC(C)(C)C.COC1=CC2=C(C=C1)C(O)=CC=N2 Chemical compound COC(=O)[C@@H]1C[C@@H](OC2=CC=NC3=C2C=CC(OC)=C3)CN1C(=O)OC(C)(C)C.COC(=O)[C@@H]1C[C@H](O)CN1C(=O)OC(C)(C)C.COC1=CC2=C(C=C1)C(O)=CC=N2 HCIORPBMTDQZIE-ZPCCFCMBSA-N 0.000 description 1
- FVICFSCFOQGWCN-DNAJAIDRSA-N COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N2.COC1=CC2=C(C=C1)/C(O[C@@H]1C[C@H]3C(=O)N[C@]4(C(=O)O)CC4/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N3C1)=C\C(C1=CSC(NC(C)C)=N1)=N/2.COC1=CC2=C(C=C1)C(O)=CC(C1=CSC(NC(C)C)=N1)=N2 Chemical compound COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N2.COC1=CC2=C(C=C1)/C(O[C@@H]1C[C@H]3C(=O)N[C@]4(C(=O)O)CC4/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N3C1)=C\C(C1=CSC(NC(C)C)=N1)=N/2.COC1=CC2=C(C=C1)C(O)=CC(C1=CSC(NC(C)C)=N1)=N2 FVICFSCFOQGWCN-DNAJAIDRSA-N 0.000 description 1
- VREOBIYSRXNGJR-ISWYMNBISA-M COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)OC1CCCC1)C(=O)N1C[C@H](O/C3=C/C(C(=O)C=[N+]=[N-])=N\C4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)OC1CCCC1)C(=O)N1C[C@H](O/C3=C/C(C(=O)CBr)=N\C4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)OC1CCCC1)C(=O)N1C[C@H](O/C3=C/C(C(=O)O[Na])=N\C4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)OC1CCCC1)C(=O)N1C[C@H](O/C3=C/C(C(C)=O)=N\C4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.F.[2HH] Chemical compound COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)OC1CCCC1)C(=O)N1C[C@H](O/C3=C/C(C(=O)C=[N+]=[N-])=N\C4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)OC1CCCC1)C(=O)N1C[C@H](O/C3=C/C(C(=O)CBr)=N\C4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)OC1CCCC1)C(=O)N1C[C@H](O/C3=C/C(C(=O)O[Na])=N\C4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)OC1CCCC1)C(=O)N1C[C@H](O/C3=C/C(C(C)=O)=N\C4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.F.[2HH] VREOBIYSRXNGJR-ISWYMNBISA-M 0.000 description 1
- HWJLGSZPLBETOC-QRLVFBIGSA-N COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)OC1CCCC1)C(=O)N1C[C@H](O/C3=C/C(C4=CSC(N)=N4)=N\C4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.COC1=CC=C2C(=C1)/N=C(C1=CSC(N)=N1)\C=C/2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC3CCCC3)C(=O)N2C1.[HH] Chemical compound COC(=O)[C@@]12CC1/C=C\CCCCC[C@H](NC(=O)OC1CCCC1)C(=O)N1C[C@H](O/C3=C/C(C4=CSC(N)=N4)=N\C4=CC(OC)=CC=C43)C[C@H]1C(=O)N2.COC1=CC=C2C(=C1)/N=C(C1=CSC(N)=N1)\C=C/2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC3CCCC3)C(=O)N2C1.[HH] HWJLGSZPLBETOC-QRLVFBIGSA-N 0.000 description 1
- PJAAEZAGZAIMHS-OSSOGDJPSA-N COC(=O)[C@@]12CC1CCCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N2.COC1=CC=C2C(=C1)/N=C(C1=CSC(N)=N1)\C=C/2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3CCCCCCC[C@H](NC(=O)OC3CCCC3)C(=O)N2C1 Chemical compound COC(=O)[C@@]12CC1CCCCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N1C[C@@H](O)C[C@H]1C(=O)N2.COC1=CC=C2C(=C1)/N=C(C1=CSC(N)=N1)\C=C/2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3CCCCCCC[C@H](NC(=O)OC3CCCC3)C(=O)N2C1 PJAAEZAGZAIMHS-OSSOGDJPSA-N 0.000 description 1
- PKCMPCWDLXVVJY-BVWRTZSASA-N COC(=O)[C@@]12C[C@@H]1/C=C\CCC(=O)CC[C@H](C)C(=O)N1CC(Oc3cc(-c4ccccc4)nc4cc(C)ccc34)C[C@H]1C(=O)N2.Cc1ccc2c(OC3C[C@H]4C(=O)N[C@]5(C(=O)O)C[C@@H]5/C=C\CCC(=O)CC[C@H](C)C(=O)N4C3)cc(-c3ccccc3)nc2c1 Chemical compound COC(=O)[C@@]12C[C@@H]1/C=C\CCC(=O)CC[C@H](C)C(=O)N1CC(Oc3cc(-c4ccccc4)nc4cc(C)ccc34)C[C@H]1C(=O)N2.Cc1ccc2c(OC3C[C@H]4C(=O)N[C@]5(C(=O)O)C[C@@H]5/C=C\CCC(=O)CC[C@H](C)C(=O)N4C3)cc(-c3ccccc3)nc2c1 PKCMPCWDLXVVJY-BVWRTZSASA-N 0.000 description 1
- KXKVLQRXCPHEJC-UHFFFAOYSA-N COC(C)=O Chemical compound COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 1
- JMZMROYUJRJAIO-UHFFFAOYSA-N COC1=CC2=C(C=C1)C(O)=CC(N)=N2.COC1=CC2=C(C=C1)C(OCC1=CC=CC=C1)=CC(Cl)=N2.COC1=CC2=C(C=C1)C(OCC1=CC=CC=C1)=CC(N)=N2.COC1=CC2=C(C=C1)C(OCC1=CC=CC=C1)=CC(O)=N2.COC1=CC=CC(N)=C1.COC1=CC=CC(N)=C1.Cl.[C-]#[N+]CC(=O)OCC Chemical compound COC1=CC2=C(C=C1)C(O)=CC(N)=N2.COC1=CC2=C(C=C1)C(OCC1=CC=CC=C1)=CC(Cl)=N2.COC1=CC2=C(C=C1)C(OCC1=CC=CC=C1)=CC(N)=N2.COC1=CC2=C(C=C1)C(OCC1=CC=CC=C1)=CC(O)=N2.COC1=CC=CC(N)=C1.COC1=CC=CC(N)=C1.Cl.[C-]#[N+]CC(=O)OCC JMZMROYUJRJAIO-UHFFFAOYSA-N 0.000 description 1
- PANQPUIFMXQGGS-ANCQHAHASA-N COC1=CC2=C(C=C1)C(O[C@@H]1C[C@H]3C(=O)N[C@]4(C(=O)O)CC4/C=C\CCCCC[C@H](NC(=O)OC4CCCC4)C(=O)N3C1)=CC(C1=CSC(NC(=O)OCC(C)C)=N1)=N2 Chemical compound COC1=CC2=C(C=C1)C(O[C@@H]1C[C@H]3C(=O)N[C@]4(C(=O)O)CC4/C=C\CCCCC[C@H](NC(=O)OC4CCCC4)C(=O)N3C1)=CC(C1=CSC(NC(=O)OCC(C)C)=N1)=N2 PANQPUIFMXQGGS-ANCQHAHASA-N 0.000 description 1
- TUOWTXFYRQVTSE-NTNXKPABSA-N COC1=CC2=C(C=C1)C(O[C@@H]1C[C@H]3C(=O)N[C@]4(C(=O)O)CC4/C=C\CCCCC[C@H](NC(=O)OC4CCCC4)C(=O)N3C1)=CC(C1=CSC(NC(C)=O)=N1)=N2 Chemical compound COC1=CC2=C(C=C1)C(O[C@@H]1C[C@H]3C(=O)N[C@]4(C(=O)O)CC4/C=C\CCCCC[C@H](NC(=O)OC4CCCC4)C(=O)N3C1)=CC(C1=CSC(NC(C)=O)=N1)=N2 TUOWTXFYRQVTSE-NTNXKPABSA-N 0.000 description 1
- PJZPDFUUXKKDNB-CJXSQDHESA-N COC1=CC2=C(C=C1)C(O[C@@H]1C[C@H]3C(=O)N[C@]4(C(=O)O)CC4/C=C\CCCCC[C@H](NC(=O)OC4CCCC4)C(=O)N3C1)=CC(C1=CSC(NC(C)C)=N1)=N2 Chemical compound COC1=CC2=C(C=C1)C(O[C@@H]1C[C@H]3C(=O)N[C@]4(C(=O)O)CC4/C=C\CCCCC[C@H](NC(=O)OC4CCCC4)C(=O)N3C1)=CC(C1=CSC(NC(C)C)=N1)=N2 PJZPDFUUXKKDNB-CJXSQDHESA-N 0.000 description 1
- WZIPTSLSJPNWIV-FZPBBAPBSA-N COC1=CC2=C(C=C1)C(O[C@@H]1C[C@H]3C(=O)N[C@]4(C(=O)O)CC4CCCCCCC[C@H](NC(=O)OC4CCCC4)C(=O)N3C1)=CC(C1=CSC(NC(C)=O)=N1)=N2 Chemical compound COC1=CC2=C(C=C1)C(O[C@@H]1C[C@H]3C(=O)N[C@]4(C(=O)O)CC4CCCCCCC[C@H](NC(=O)OC4CCCC4)C(=O)N3C1)=CC(C1=CSC(NC(C)=O)=N1)=N2 WZIPTSLSJPNWIV-FZPBBAPBSA-N 0.000 description 1
- WNADDQNMQXVFHV-GUMXKKIYSA-N COC1=CC2=C(C=C1)C(O[C@@H]1C[C@H]3C(=O)N[C@]4(C(=O)O)CC4CCCCCCC[C@H](NC(=O)OC4CCCC4)C(=O)N3C1)=CC(C1=CSC(NC(C)C)=N1)=N2 Chemical compound COC1=CC2=C(C=C1)C(O[C@@H]1C[C@H]3C(=O)N[C@]4(C(=O)O)CC4CCCCCCC[C@H](NC(=O)OC4CCCC4)C(=O)N3C1)=CC(C1=CSC(NC(C)C)=N1)=N2 WNADDQNMQXVFHV-GUMXKKIYSA-N 0.000 description 1
- KBWMMQHYYDCDRO-GUMXKKIYSA-N COC1=CC=C2C(=C1)/N=C(C1=NC(C(C)C)=CS1)\C=C/2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3CCCCCCC[C@H](NC(=O)OC3CCCC3)C(=O)N2C1 Chemical compound COC1=CC=C2C(=C1)/N=C(C1=NC(C(C)C)=CS1)\C=C/2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3CCCCCCC[C@H](NC(=O)OC3CCCC3)C(=O)N2C1 KBWMMQHYYDCDRO-GUMXKKIYSA-N 0.000 description 1
- GHHSNUOFOGXTGA-IEVMOELASA-N COC1=CC=C2C(=C1)/N=C(N1C=CC=C1)\C=C/2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3CCCCCCC[C@H](NC(=O)OC3CCCC3)C(=O)N2C1 Chemical compound COC1=CC=C2C(=C1)/N=C(N1C=CC=C1)\C=C/2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3CCCCCCC[C@H](NC(=O)OC3CCCC3)C(=O)N2C1 GHHSNUOFOGXTGA-IEVMOELASA-N 0.000 description 1
- LIWUUVCCZLNFDD-INCUHYFCSA-N COC1=CC=C2C(=C1)N=C(C1=CSC(NC3CC3)=N1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC3CCCC3)C(=O)N2C1 Chemical compound COC1=CC=C2C(=C1)N=C(C1=CSC(NC3CC3)=N1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC3CCCC3)C(=O)N2C1 LIWUUVCCZLNFDD-INCUHYFCSA-N 0.000 description 1
- IBBVFLYGFCGFOT-WPKUYOCDSA-N COC1=CC=C2C(=C1)N=C(C1=CSC(NC3CCC3)=N1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC3CCCC3)C(=O)N2C1 Chemical compound COC1=CC=C2C(=C1)N=C(C1=CSC(NC3CCC3)=N1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC3CCCC3)C(=O)N2C1 IBBVFLYGFCGFOT-WPKUYOCDSA-N 0.000 description 1
- QFUNXORWTHDTHQ-VXTCSDCJSA-N COC1=CC=C2C(=C1)N=C(C1=CSC(NC3CCCC3)=N1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC3CCCC3)C(=O)N2C1 Chemical compound COC1=CC=C2C(=C1)N=C(C1=CSC(NC3CCCC3)=N1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC3CCCC3)C(=O)N2C1 QFUNXORWTHDTHQ-VXTCSDCJSA-N 0.000 description 1
- ZMVGSGPPRXCIHS-WXDVHWTGSA-N COC1=CC=C2C(=C1)N=C(C1=CSC(NC3CCCCC3)=N1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC3CCCC3)C(=O)N2C1 Chemical compound COC1=CC=C2C(=C1)N=C(C1=CSC(NC3CCCCC3)=N1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC3CCCC3)C(=O)N2C1 ZMVGSGPPRXCIHS-WXDVHWTGSA-N 0.000 description 1
- CLEVEXNMFGSGDS-KLFQIYIKSA-N COC1=CC=C2C(=C1)N=C(C1=NC(C(C)C)=CS1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C1 Chemical compound COC1=CC=C2C(=C1)N=C(C1=NC(C(C)C)=CS1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C1 CLEVEXNMFGSGDS-KLFQIYIKSA-N 0.000 description 1
- YGLVVZWHCDZESB-WVLMTHSNSA-N COC1=CC=C2C(=C1)N=C(C1=NC=CN1C)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C1 Chemical compound COC1=CC=C2C(=C1)N=C(C1=NC=CN1C)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C1 YGLVVZWHCDZESB-WVLMTHSNSA-N 0.000 description 1
- PVBNAIRMXKTMNY-VMYNCKCISA-N COC1=CC=C2C(=C1)N=C(N1C=C(C)C=N1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C1 Chemical compound COC1=CC=C2C(=C1)N=C(N1C=C(C)C=N1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C1 PVBNAIRMXKTMNY-VMYNCKCISA-N 0.000 description 1
- QPUNNOCALCGZEN-VMYNCKCISA-N COC1=CC=C2C(=C1)N=C(N1C=CC(C)=N1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C1 Chemical compound COC1=CC=C2C(=C1)N=C(N1C=CC(C)=N1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C1 QPUNNOCALCGZEN-VMYNCKCISA-N 0.000 description 1
- NWVNIRNYZSSEAH-APZSFDFQSA-N COC1=CC=C2C(=C1)N=C(N1C=CC=N1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C1 Chemical compound COC1=CC=C2C(=C1)N=C(N1C=CC=N1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C1 NWVNIRNYZSSEAH-APZSFDFQSA-N 0.000 description 1
- PARUNSVMNWGMTP-SGMFRAHYSA-N COC1=CC=C2C(=C1)N=C(N1C=CN=C1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C1 Chemical compound COC1=CC=C2C(=C1)N=C(N1C=CN=C1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C1 PARUNSVMNWGMTP-SGMFRAHYSA-N 0.000 description 1
- MGRYRDPHJWDTIT-VMYNCKCISA-N COC1=CC=C2C(=C1)N=C(N1C=NC(C)=C1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C1 Chemical compound COC1=CC=C2C(=C1)N=C(N1C=NC(C)=C1)C=C2O[C@@H]1C[C@H]2C(=O)N[C@]3(C(=O)O)CC3/C=C\CCCCC[C@H](NC(=O)OC(C)(C)C)C(=O)N2C1 MGRYRDPHJWDTIT-VMYNCKCISA-N 0.000 description 1
- OUQMXTJYCAJLGO-UHFFFAOYSA-N Cc1c[s]c(N)n1 Chemical compound Cc1c[s]c(N)n1 OUQMXTJYCAJLGO-UHFFFAOYSA-N 0.000 description 1
- SMOKOTSWLUCDGG-UHFFFAOYSA-N Cc1c[s]c(NC(OC)=O)n1 Chemical compound Cc1c[s]c(NC(OC)=O)n1 SMOKOTSWLUCDGG-UHFFFAOYSA-N 0.000 description 1
- DGBNDUXTMUGNGM-UHFFFAOYSA-N Cc1c[s]c(NC)n1 Chemical compound Cc1c[s]c(NC)n1 DGBNDUXTMUGNGM-UHFFFAOYSA-N 0.000 description 1
- VZWOXDYRBDIHMA-UHFFFAOYSA-N Cc1ncc[s]1 Chemical compound Cc1ncc[s]1 VZWOXDYRBDIHMA-UHFFFAOYSA-N 0.000 description 1
- PAJPWUMXBYXFCZ-UHFFFAOYSA-N NC1(C(=O)O)CC1 Chemical compound NC1(C(=O)O)CC1 PAJPWUMXBYXFCZ-UHFFFAOYSA-N 0.000 description 1
- KYWMCFOWDYFYLV-UHFFFAOYSA-N OC(c1ncc[nH]1)=O Chemical compound OC(c1ncc[nH]1)=O KYWMCFOWDYFYLV-UHFFFAOYSA-N 0.000 description 1
- DYYSWVQVLFRJDK-UHFFFAOYSA-N [CH2-][C+]1=CC=C(OC)C=N1 Chemical compound [CH2-][C+]1=CC=C(OC)C=N1 DYYSWVQVLFRJDK-UHFFFAOYSA-N 0.000 description 1
- MIDZOXCQBYQKLW-UHFFFAOYSA-N [CH2-][C+]1=CN=C(NC(C)=O)S1 Chemical compound [CH2-][C+]1=CN=C(NC(C)=O)S1 MIDZOXCQBYQKLW-UHFFFAOYSA-N 0.000 description 1
- VGWZERJAJSXJJL-UHFFFAOYSA-N [CH2-][C+]1=CN=CO1 Chemical compound [CH2-][C+]1=CN=CO1 VGWZERJAJSXJJL-UHFFFAOYSA-N 0.000 description 1
- YDKAFPKKZHSHBK-UHFFFAOYSA-N [CH2-][C+]1=CSC(NC(=O)NC)=N1 Chemical compound [CH2-][C+]1=CSC(NC(=O)NC)=N1 YDKAFPKKZHSHBK-UHFFFAOYSA-N 0.000 description 1
- QRDARQNNIWHZSQ-UHFFFAOYSA-N [CH2-][C+]1=CSC(NC(=O)OCC(C)C)=N1 Chemical compound [CH2-][C+]1=CSC(NC(=O)OCC(C)C)=N1 QRDARQNNIWHZSQ-UHFFFAOYSA-N 0.000 description 1
- DNOHUGUAHSYZGW-UHFFFAOYSA-N [CH2-][C+]1=CSC(NC2CC2)=N1 Chemical compound [CH2-][C+]1=CSC(NC2CC2)=N1 DNOHUGUAHSYZGW-UHFFFAOYSA-N 0.000 description 1
- FRIXBYRYNYNZIU-UHFFFAOYSA-N [CH2-][C+]1=CSC(NC2CCC2)=N1 Chemical compound [CH2-][C+]1=CSC(NC2CCC2)=N1 FRIXBYRYNYNZIU-UHFFFAOYSA-N 0.000 description 1
- ASLSASSSWPFQGE-UHFFFAOYSA-N [CH2-][C+]1=CSC(NC2CCCC2)=N1 Chemical compound [CH2-][C+]1=CSC(NC2CCCC2)=N1 ASLSASSSWPFQGE-UHFFFAOYSA-N 0.000 description 1
- MOMVZMJZPSUBGA-UHFFFAOYSA-N [CH2-][C+]1=CSC(NC2CCCCC2)=N1 Chemical compound [CH2-][C+]1=CSC(NC2CCCCC2)=N1 MOMVZMJZPSUBGA-UHFFFAOYSA-N 0.000 description 1
- AWUWDRRVUXXJNX-UHFFFAOYSA-N [CH2-][C+]1=NC(C)=NO1 Chemical compound [CH2-][C+]1=NC(C)=NO1 AWUWDRRVUXXJNX-UHFFFAOYSA-N 0.000 description 1
- IPJSLVQFDLEOID-UHFFFAOYSA-N [CH2-][C+]1=NC=CN1C Chemical compound [CH2-][C+]1=NC=CN1C IPJSLVQFDLEOID-UHFFFAOYSA-N 0.000 description 1
- TVEKRMBZLSQBOI-UHFFFAOYSA-N [CH2-][C+]1=NN=C(C)O1 Chemical compound [CH2-][C+]1=NN=C(C)O1 TVEKRMBZLSQBOI-UHFFFAOYSA-N 0.000 description 1
- GUKKNVZJTXTEBF-UHFFFAOYSA-N [CH2-][C+]1C(C)(C)C1(C)C Chemical compound [CH2-][C+]1C(C)(C)C1(C)C GUKKNVZJTXTEBF-UHFFFAOYSA-N 0.000 description 1
- DUXBAJZGWJQWOD-UHFFFAOYSA-N [CH2-][C+]1CCC=CO1 Chemical compound [CH2-][C+]1CCC=CO1 DUXBAJZGWJQWOD-UHFFFAOYSA-N 0.000 description 1
- NFJPEKRRHIYYES-UHFFFAOYSA-N [CH2-][C+]1CCCC1 Chemical compound [CH2-][C+]1CCCC1 NFJPEKRRHIYYES-UHFFFAOYSA-N 0.000 description 1
- QMXMHZQRCZRBSD-UHFFFAOYSA-N [CH2-][NH+]1C=C(C)C=N1 Chemical compound [CH2-][NH+]1C=C(C)C=N1 QMXMHZQRCZRBSD-UHFFFAOYSA-N 0.000 description 1
- ZDPLYWSZGRDAGU-UHFFFAOYSA-N [CH2-][NH+]1C=CN=C1 Chemical compound [CH2-][NH+]1C=CN=C1 ZDPLYWSZGRDAGU-UHFFFAOYSA-N 0.000 description 1
- YDSDOWWNCRQHBX-UHFFFAOYSA-N [CH2-][NH+]1C=NC(C)=C1 Chemical compound [CH2-][NH+]1C=NC(C)=C1 YDSDOWWNCRQHBX-UHFFFAOYSA-N 0.000 description 1
- JGIHYKRGTGLLOK-UHFFFAOYSA-N [CH2-][OH+]C(C)C Chemical compound [CH2-][OH+]C(C)C JGIHYKRGTGLLOK-UHFFFAOYSA-N 0.000 description 1
- SOJSREOOEIUOHS-UHFFFAOYSA-N [CH2-][OH+]CC(C)C Chemical compound [CH2-][OH+]CC(C)C SOJSREOOEIUOHS-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06139—Dipeptides with the first amino acid being heterocyclic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Abstract
The present invention covers macrocyclic compounds of formula I active in-vitro and in cellular assays against the NS3 protease of the hepatitis C virus.

wherein W, R21, R22, R3, R4, D and A are as defined herein, or a pharmaceutically acceptable salt or ester thereof.
Description
- This application is a continuation of U.S. patent application Ser. No. 09/760,946, filed Jan. 16, 2001, which is a continuation-in-part of U.S. patent application Ser. No. 09/542,675 filed Apr. 3, 2000, now abandoned, which claims benefit of U.S. provisional application No. 60/128,011 filed Apr. 6, 1999, all of which are incorporated herein by reference in their entirety.
- The present invention relates to compounds, compositions, the preparation of such compounds and methods for the treatment of hepatitis C virus (HCV) infection. In particular, the present invention provides novel peptide analogues, pharmaceutical compositions containing such analogues and methods for using these analogues in the treatment of HCV infection.
- Hepatitis C virus (HCV) is the major etiological agent of post-transfusion and community-acquired non-A non-B hepatitis worldwide. It is estimated that over 170 million people worldwide are infected by the virus. A high percentage of carriers become chronically infected and many progress to chronic liver disease, so-called chronic hepatitis C. This group is in turn at high risk for serious liver disease such as liver cirrhosis, hepatocellular carcinoma and terminal liver disease leading to death. The mechanism by which HCV establishes viral persistence and causes a high rate of chronic liver disease has not been thoroughly elucidated. It is not known how HCV interacts with and evades the host immune system. In addition, the roles of cellular and humoral immune responses in protection against HCV infection and disease have yet to be established. Immunoglobulins have been reported for prophylaxis of transfusion-associated viral hepatitis, however, the Center for Disease Control does not presently recommend immunoglobulins treatment for this purpose. The lack of an effective protective immune response is hampering the development of a vaccine or adequate post-exposure prophylaxis measures, so in the near-term, hopes are firmly pinned on antiviral interventions.
- Various clinical studies have been conducted with the goal of identifying pharmaceutical agents capable of effectively treating HCV infection in patients afflicted with chronic hepatitis C. These studies have involved the use of interferon-alpha, alone and in combination with other antiviral agents. Such studies have shown that a substantial number of the participants do not respond to these therapies, and of those that do respond favorably, a large proportion were found to relapse after termination of treatment.
- Until a few years ago, interferon (IFN) was the only available therapy of proven benefit approved in the clinic for patients with chronic hepatitis C. However the sustained response rate is low, and interferon treatment also induces severe side-effects (i.e. retinopathy, thyroiditis, acute pancreatitis, depression) that diminish the quality of life of treated patients. Interferon in combination with ribavirin was originally approved for patients non-responsive to IFN alone. It has now been approved for naïve patients and presently constitutes the gold standard in HCV therapy. However, the side effects caused by IFN are not alleviated with this combination therapy.
- Therefore, a need exists for the development of effective antiviral agents for treatment of HCV infection that overcomes the limitations of existing pharmaceutical therapies.
- HCV is an enveloped positive strand RNA virus in the Flaviviridae family. The single strand HCV RNA genome is approximately 9500 nucleotides in length and has a single open reading frame (ORF) encoding a single large polyprotein of about 3000 amino acids. In infected cells, this polyprotein is cleaved at multiple sites by cellular and viral proteases to produce the structural and non-structural (NS) proteins. In the case of HCV, the generation of mature nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) is effected by two viral proteases. The first one, as yet poorly characterized, cleaves at the NS2-NS3 junction; the second one is a serine protease contained within the N-terminal region of NS3 (henceforth referred to as NS3 protease) and mediates all the subsequent cleavages downstream of NS3, both in cis, at the NS3-NS4A cleavage site, and in trans, for the remaining NS4A-NS4B, NS4B-NS5A, NS5A-NS5B sites. The NS4A protein appears to serve multiple functions, acting as a cofactor for the NS3 protease and possibly assisting in the membrane localization of NS3 and other viral replicase components. The complex formation of the NS3 protein with NS4A seems necessary to the processing events, enhancing the proteolytic efficiency at all of the sites. The NS3 protein also exhibits nucleoside triphosphatase and RNA helicase activities. NS5B is a RNA-dependent RNA polymerase that is involved in the replication of HCV.
- Patent application WO 97/06804 describes the (−) enantiomer of the nucleoside analogue cytosine-1,3-oxathiolane (also known as 3TC) as active against HCV. This compound, although reported as safe in previous clinical trials against HIV and HBV, has yet to be clinically proven active against HCV and its mechanism of action against the virus has yet to be reported.
- A general strategy for the development of antiviral agents is to inactivate virally encoded enzymes that are essential for the replication of the virus
- In this vein, intense efforts to discover compounds which inhibit the NS3 protease or RNA helicase of HCV have led to the following disclosures:
- U.S. Pat. No. 5,633,388 describes heterocyclic-substituted carboxamides and analogues as being active against HCV. These compounds are directed against the helicase activity of the NS3 protein of the virus but clinical tests have not yet been reported. A phenanthrenequinone has been reported by Chu et al., (Tet. Lett., (1996), 7229-7232) to have activity against the HCV NS3 protease in vitro. No further development on this compound has been reported.
- A paper presented at the Ninth International Conference on Antiviral Research, Urabandai, Fukyshima, Japan (1996) (Antiviral Research, (1996), 30, 1, A23 (abstract 19)) reports thiazolidine derivatives to be inhibitory to the HCV protease. Several studies have reported compounds inhibitory to other serine proteases, such as human leukocyte elastase. One family of these compounds is reported in WO 95/33764 (Hoechst Marion Roussel, 1995). The peptides disclosed in that application are morpholinylcarbonyl-benzoyl-peptide analogues that are structurally different from the peptides of the present invention.
- WO 98/17679 from Vertex Pharmaceuticals Inc. discloses inhibitors of serine protease, particularly, Hepatitis C virus NS3 protease
- Hoffman LaRoche (WO 98/22496; U.S. Pat. Nos. 5,866,684 & 6,018,020) has also reported hexapeptides that are proteinase inhibitors useful as antiviral agents for the treatment of HCV infection.
- Steinkühler et al. and Ingallinella et al. have published on NS4A-4B product inhibition (Biochemistry (1998), 37, 8899-8905 and 8906-8914).
- WO 97/43310 by Schering Corporation discloses 20 and 21 amino acid peptide sequences active against the HCV NS3 protease.
- WO 98/46597 by Emory University discloses peptides and peptidomimetics active in vitro against serine proteases.
- WO 98/46630 by Peptide Therapeutics Limited discloses depsipeptide substrate inhibiting the HCV NS3 protease.
- Finally, U.S. Pat. No. 5,869,253 discloses enzymatic RNA molecules that inhibit the HCV NS3 protease.
- None of the prior patent applications described above disclose suggest cyclic peptides active and selective against the Hepatitis C virus NS3 protease.
- WO 99/07733, WO 99/07734, WO 00/09543 and WO00/09558 disclose hexa to tetra-peptides and tripeptide analogs that inhibit the NS3 protease. However, these disclosures do not suggest or lead to the design of macrocyclic analogs of the present invention.
- WO 99/38888 published Aug. 5, 1999 by the Institute de Richerche di Biologia Moleculare (IRBM) discloses small peptides inhibitors of the HCV NS3 protease. Nothing in this disclosure suggest or indicates the cyclic nature of the peptides of the present invention. In addition, this PCT application was published after the priority date of the present application.
- WO 99/64442 by IRBM, also published after the priority date of this application, discloses oligopeptides with ketoacids at P1.
- WO 99/50230 from Vertex Pharmaceuticals (published on Oct. 7, 1999) was also published after the priority date of the present application. Even then, the publication does not remotely suggest any cyclic peptides of the present invention.
- One advantage of the present invention is that it provides macrocyclic peptides that are inhibitory to the NS3 protease of the hepatitis C virus.
- A further advantage of one aspect of the present invention resides in the fact that these peptides specifically inhibit the NS3 protease and do not show significant inhibitory activity against other serine proteases such as human leukocyte elastase (HLE), porcine pancreatic elastase (PPE), or bovine pancreatic chymotrypsin, or cysteine proteases such as human liver cathepsin B (Cat B).
- A further advantage of the present invention is that it provides small peptides of low molecular weight that are capable of penetrating cell membranes and inhibit the NS3 protease activity in cell culture.
- Still, a further advantage of the compounds of the present invention resides in the fact that they are active in both major genotypes found in clinical isolates (1a & 1b), strongly suggesting that these compound will be active against all presently known genotypes of HCV.
- Included in the scope of the invention are compounds of formula (I):

- wherein W is CH or N,
- R 21 is H, halo, C1-6 alkyl, C3-6 cycloalkyl, C1-6 haloalkyl, C1-6 alkoxy, C3-6 cycloalkoxy, hydroxy, or N(R23)2, wherein each R23 is independently H, C1-6 alkyl or C3-6 cycloalkyl; and
- R 22 is H, halo, C1-6 alkyl, C3-6 cycloalkyl, C1-6 haloalkyl, C1-6 thioalkyl, C1-6 alkoxy, C3-6 cycloalkoxy, C2-7 alkoxyalkyl, C3-6 cycloalkyl, C6 or 10 aryl or Het, wherein Het is a five-, six-, or seven-membered, saturated or unsaturated heterocycle, containing from one to four heteroatoms selected from nitrogen, oxygen and sulfur;
- said cycloalkyl, aryl or Het being substituted with R 24,
- wherein R 24 is H, halo, C1-6 alkyl, C3-6 cycloalkyl, C1-6 alkoxy, C3-6 cycloalkoxy, NO2, N(R25)2; NH—C(O)—R25, or NH—C(O)—NH—R25,
- wherein each R 25 is independently: H, C1-6 alkyl or C3-6 cycloalkyl;
- or R 24 is NH—C(O)—OR26 wherein R26 is C1-6 alkyl or C3-6 cycloalkyl;
- R 3 is hydroxy, NH2, or a group of formula —NH—R31, wherein R31 is C6 or 10 aryl, heteroaryl, —C(O)—R32, —C(O)—OR32, or —C(O)—NHR32,
- wherein R 32 is: C1-6 alkyl or C3-6 cycloalkyl;
- D is a 5 to 10-atom saturated or unsaturated alkylene chain optionally containing one to three heteroatoms independently selected from: O, S, or N—R 41, wherein
- R 41 is H, C1-6 alkyl, C3-6 cycloalkyl, or —C(O)—R42, wherein R42 is C1-6 alkyl, C3-6 cycloalkyl or C6 or 10 aryl; and wherein the atoms of the D chain that form part of the macrocyclic ring in structural formula (I) are numbered from left to right in structural formula (I) starting with position number 8.
- R 4 is H or from one to three substituents at any carbon atom of said chain D, said substituent independently selected from the group consisting of: C1-6 alkyl, C1-6 haloalkyl, C1-6 alkoxy, hydroxy, halo, amino, oxo, thio, or C1-6 thioalkyl and
- A is an amide of formula —C(O)—NH—R 5, wherein R5 is selected from the group consisting of: C1-8 alkyl, C3-6 cycloalkyl, C6 or 10 aryl or C7-16 aralkyl;
- or A is a carboxylic acid or a pharmaceutically acceptable salt or ester thereof.
- Included within the scope of this invention is a pharmaceutical composition comprising an anti-hepatitis C virally effective amount of a compound of formula I, or a therapeutically acceptable salt or ester thereof, in admixture with a pharmaceutically acceptable carrier medium or auxiliary agent.
- An important aspect of the invention involves a method of treating a hepatitis C viral infection in a mammal by administering to the mammal an anti-hepatitis C virally effective amount of the compound of formula I, or a therapeutically acceptable salt or ester thereof or a composition as described above.
- Another important aspect involves a method of inhibiting the replication of hepatitis C virus by exposing the virus to a hepatitis C NS3 protease-inhibiting amount of the compound of formula I, or a therapeutically acceptable salt or ester thereof or a composition as described above.
- Still another aspect involves a method of treating a hepatitis C viral infection in a mammal by administering thereto an anti-hepatitis C virally effective amount of a combination of the compound of formula I, or a therapeutically acceptable salt or ester thereof. According to one embodiment, the pharmaceutical compositions of this invention comprise an additional immunomodulatory agent. Examples of additional immunomodulatory agents include but are not limited to, α-, β-, and δ-interferons.
- According to an alternate embodiment, the pharmaceutical compositions of this invention may additionally comprise an antiviral agent. Examples of antiviral agents include, ribavirin and amantadine.
- According to another alternate embodiment, the pharmaceutical compositions of this invention may additionally comprise other inhibitors of HCV protease.
- According to yet another alternate embodiment, the pharmaceutical compositions of this invention may additionally comprise an inhibitor of other targets in the HCV life cycle, such as helicase, polymerase, metalloprotease or IRES.
- As used herein, the following definitions apply unless otherwise noted: The designation herein of a position within the D chain by position number(s), e.g. “position 10 of said D chain” or “D chain is substituted at position 8” or “double bond at position 13,14 of said D chain” or “D chain contains one double bond at position 11,12”, or similar language, means the position or positions within the D chain when the atoms of the D chain are numbered as set forth previously, i.e., the atoms of the D chain that form part of the macrocyclic ring in structural formula (I) are numbered from left to right in structural formula (I) starting with position number 8.
- With reference to the instances where (R) or (S) is used to designate the absolute configuration of a substituent, e.g. R 4 of the compound of formula I, the designation is done in the context of the whole compound and not in the context of the substituent alone.
- The designation “P1, P2, and P3” as used herein refer to the position of the amino acid residues starting from the C-terminus end of the peptide analogs and extending towards the N-terminus (i.e. P1 refers to position 1 from the C-terninus, P2: second position from the C-terminus, etc.) (see Berger A. & Schechter I., Transactions of the Royal Society London series B257, 249-264 (1970)).
- As used herein the term “1-aminocyclopropyl-carboxylic acid” (ACCA) refers to a compound of formula:

- As used herein the term “vinyl-ACCA” refers to a compound of formula:

- As used herein the term “homo-allyl-ACCA” refers to a compound of formula:

- The term “halo” as used herein means a halogen substituent selected from bromo, chloro, fluoro or iodo.
- The term “C 1-6 haloalkyl” as used herein means as used herein, either alone or in combination with another substituent, means acyclic, straight or branched chain alkyl substituents containing from 1 to six carbon atoms having one or more hydrogen substituted for a halogen selected from bromo, chloro, fluoro or iodo.
- The term “C 1-6 thioalkyl” as used herein means as used herein, either alone or in combination with another substituent, means acyclic, straight or branched chain alkyl substituents containing a thiol group such a thiopropyl.
- The term “C 1-6 alkyl” or “(lower)alkyl” as used herein, either alone or in combination with another substituent, means acyclic, straight or branched chain alkyl substituents containing from 1 to six carbon atoms and includes, for example, methyl, ethyl, propyl, butyl, hexyl, 1-methylethyl, 1-methylpropyl, 2-methylpropyl, 1,1-dimethylethyl.
- The term “C 3-6 cycloalkyl” as used herein, either alone or in combination with another substituent, means a cycloalkyl substituent containing from three to six carbon atoms and includes cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl. The term “unsaturated cycloalkyl” includes, for example, the substituent cyclohexenyl:

- The term “saturated or unsaturated alkylene” as used herein means a divalent alkyl substituent derived by the removal of one hydrogen atom from each end of a saturated or unsaturated straight or branched chain aliphatic hydrocarbon and includes, for example, —CH 2CH2C(CH3)2CH2CH2—, —CH2CH2CH═CHCH2CH2— or —CH2C≡CCH2CH2—. This alkyl chain may optionally contain a heteroatom such as oxygen (for example: CH3—CH2—O—CH2—).
- The term “C 1-6 alkoxy” as used herein, either alone or in combination with another substituent, means the substituent —O—C1-6 alkyl wherein alkyl is as defined above containing up to six carbon atoms. Alkoxy includes methoxy, ethoxy, propoxy, 1-methylethoxy, butoxy and 1,1-dimethylethoxy. The latter substituent is known commonly as tert-butoxy.
- The term “C 3-6 cycloalkoxy” as used herein, either alone or in combination with another substituent, means the substituent —O—C3-6 cycloalkyl containing from three to 6 carbon atoms.
- The term “C 1-6 alkoxyalkyl” as used herein, means the substituent C1-6 alkyl-O—C1-6 alkyl wherein alkyl is as defined above containing up to six carbon atoms. For example, methoxymethyl means —CH2—O—CH3.
- The term “C 2-7 acyl” as used herein, either alone or in combination with another substituent, means an C1-6 alkyl group linked through a carbonyl group such as —C(O)—C1-6 alkyl.
- The term “C 6 or C10 aryl” as used herein, either alone or in combination with another substituent, means either an aromatic monocyclic system containing 6 carbon atoms or an aromatic bicyclic system containing 10 carbon atoms. For example, aryl includes a phenyl or a naphthyl-ring system.
- The term “C 7-16 aralkyl” as used herein, either alone or in combination with another substituent, means an aryl as defined above linked through an alkyl group, wherein alkyl is as defined above containing from 1 to 6 carbon atoms. Aralkyl includes for example benzyl, and butylphenyl.
- The term “Het” as used herein, either alone or in combination with another substituent, means a monovalent substituent derived by removal of a hydrogen from a five-, six-, or seven-membered saturated or unsaturated (including aromatic) heterocycle containing from one to four heteroatoms selected from nitrogen, oxygen and sulfur. Examples of suitable heterocycles include: tetrahydrofuran, thiophene, diazepine, isoxazole, piperidine, dioxane, morpholine, pyrimidine or

- The term “Het” also includes a heterocycle as defined above fused to one or more other cycle be it a heterocycle or any other cycle. One such examples includes thiazolo[4,5-b]-pyridine.
- Although generally covered under the term “Het”, the term “heteroaryl” as used herein precisely defines an unsaturated heterocycle which is an aromatic system. Suitable example of heteroaromatic system include: quinoline, indole, pyridine,

- The term “pharmaceutically acceptable ester” as used herein, either alone or in combination with another substituent, means esters of the compound of formula I in which any of the carboxyl functions of the molecule, but preferably the carboxy terminus, is replaced by an alkoxycarbonyl function:

- in which the R moiety of the ester is selected from alkyl (e.g. methyl, ethyl, n-propyl, t-butyl, n-butyl); alkoxyalkyl (e.g. methoxymethyl); alkoxyacyl (e.g. acetoxymethyl); aralkyl (e.g. benzyl); aryloxyalkyl (e.g. phenoxymethyl); aryl (e.g. phenyl), optionally substituted with halogen, C 1-4 alkyl or C1-4 alkoxy. Other suitable prodrug esters are found in Design of prodrugs, Bundgaard, H. Ed. Elsevier (1985) incorporated herewith by reference. Such pharmaceutically acceptable esters are usually hydrolyzed in vivo when injected in a mammal and transformed into the acid form of the compound of formula I.
- With regard to the esters described above, unless otherwise specified, any alkyl moiety present advantageously contains 1 to 16 carbon atoms, particularly 1 to 6 carbon atoms. Any aryl moiety present in such esters advantageously comprises a phenyl group.
- In particular the esters may be a C 1-16 alkyl ester, an unsubstituted benzyl ester or a benzyl ester substituted with at least one halogen, C1-6 alkyl, C1-6 alkoxy, nitro or trifluoromethyl.
- The term “pharmaceutically acceptable salt” as used herein includes those derived from pharmaceutically acceptable bases. Examples of suitable bases include choline, ethanolamine and ethylenediamine. Na +, K+, and Ca++ salts are also contemplated to be within the scope of the invention (also see Pharmaceutical salts, Birge, S. M. et al., J. Pharm. Sci., (1977), 66, 1-19, incorporated herein by reference).
- R 1:
- Preferred embodiments of the present invention include compounds of formula I as described above, wherein the R 1 moiety is selected from the 2 different diastereoisomers where the 1-carbon center has the R configuration as represented by structures (i) and (ii):

- More preferably, the linker D is linked to R 1 in the configuration syn to A as represented by structure (ii).
- R 2:
- Preferred embodiments of the present invention include compounds of formula I as described above, wherein the R 2 moiety is

- wherein W is preferably N.
- Preferably, R 21 is H, C1-6 alkyl, C1-6 alkoxy, hydroxy, chloro, or N(R23)2 wherein R23 is preferably H or C1-6 alkyl. More preferably, R21 is H or C1-6 alkoxy. Most preferably, R21 is methoxy.
- Preferably R 22 is H, C1-6 thioalkyl, C1-6 alkoxy, phenyl or Het selected from the group consisting of:

- Preferably, R 24 is H, C1-6 alkyl, NH—R25, NH—C(O)—R25; or NH—C(O)—NH—R25 or NH—C(O)—OR26.
- More preferably R 22 is C1-4 alkoxy, phenyl or Het selected from the group consisting of:

- More preferably, R 24 is H, C1-6 alkyl, NH—R25, NH—C(O)—R25; or NH—C(O)—OR26.
- Most preferably R 22 is ethoxy, or Het selected from the group consisting of:

- Most preferably, R 24a is NH—R25, NH—C(O)—R25, or NH—C(O)—OR26. Most preferably, R24b is H or C1-6 alkyl.
- Preferably, each R 25 is independently: H, C1-6 alkyl, or C3-6 cycloalkyl. More preferably, R25 is C1-6 alkyl or C3-6 cycloalkyl. More preferably, R25 is C1-6 alkyl.
- Preferably, R 26 is C1-6 alkyl.
- R 3:
- Preferred embodiments of the present invention include compounds of formula I as described above, wherein the R 3 moiety is preferably an amide of formula NH—C(O)—R32, a urea of formula NH—C(O)—NH—R32, or a carbamate of formula NH—C(O)—OR32. More preferably, R3 is a carbamate or a urea. Most preferably, R3 is a carbamate.
- Preferably, R 32 is C1-6 alkyl, or C3-6 cycloalkyl. More preferably, R32 is C1-6 alkyl, or C4-6 cycloalkyl. Most preferably, R32 is tert-butyl, cyclobutyl or cyclopentyl.
- D:
- Preferred embodiments of the present invention include compounds of formula I, wherein linker D is a 6 to 8 atom saturated or unsaturated alkylene chain. More preferably, linker D is 7 atom chain.
- Preferably, the D chain contains one or two heteroatoms selected from: O, S, NH, N—C 1-6 alkyl or N—C2-7 acyl. More preferably, the D chain optionally contains one heteroatom selected from: NH, or N—C2-7 acyl, most preferably N(Ac), and is positioned at atom 10 of the chain. Most preferably, the chain containing a nitrogen atom is saturated.
- Alternatively, D contains one heteroatom selected from: O, or S. Preferably, when D is 7 atom in length, the O or S atom is at position 9 of the chain. Preferably, this chain is substituted with R 4, wherein R4 is H or C1-6 alkyl. More preferably, R4 is H or methyl. Most preferably, R4 is H or 8-(S)—Me. Even most preferably, D is saturated. Alternatively, D contains one double bond at position 11,12. Preferably, this double bond is trans.
- Alternatively, D is an all carbon saturated or unsaturated alkylene chain. In this case, D is preferably saturated and is 7 atom in length. More preferably, D is substituted with R 4, wherein R4 is H, oxo, thio, hydroxy, thioalkyl, alkoxy or alkyl. More preferably, R4 is H or C1-6 alkyl. Most preferably, R4 is H or methyl. Most preferably, R4 is H or 10-(S)—Me.
- Alternatively, D is an all carbon alkylene chain containing preferably one double bond and is 7 atom in length. More preferably, this double bond is at position 13,14 of the chain. Most preferably, this double bond is cis. Preferably, this D chain is substituted with R 4, wherein R4 is H, oxo, hydroxy, alkoxy or alkyl. More preferably, R4 is H or C1-6 alkyl. Even more preferably, R4 is H or methyl. Most preferably, R4 is H or 10-(S)—Me.
- A:
- Preferred embodiments of the present invention include compounds of formula I as described above, wherein A is a carboxylic acid.
- Specific Embodiments:
- Preferred embodiments of the present invention include compounds of formula I as described above, wherein R 2 is a quinoline substituent (i.e. W is N);
- R 3 is a group of formula —NH—C(O)—NHR32 or —NH—C(O)—OR32, wherein R32 is: C1-4 alkyl or C4-6 cycloalkyl;
- D is a 6 to 8 atom saturated or unsaturated alkylene chain linked to R 1 in configuration syn to A, optionally containing one or two heteroatoms independently selected from: O, S or N—R41, wherein R41 is C2-7 acyl;
- R 4 is H, or from one to three substituents independently selected from hydroxy or C1-6 alkyl; and
- A is a carboxylic acid, or a pharmaceutically acceptable salt or ester thereof.
- More preferably are compounds of formula I wherein R 1 is as defined above; R21 is H or methoxy;
- R 22 is C1-6 alkoxy, or Het selected from the group consisting of:

- wherein R 24a is H, C1-6 alkyl, NH—R25, NH—C(O)—R25, NH—C(O)—NH—R25,
- wherein R 25 is: H, C1-6 alkyl or C3-6 cycloalkyl;
- or R 24a is NH—C(O)—OR26, wherein R26 is C1-6 alkyl or C3-6 cycloalkyl;
- and R 24b is H or C1-6 alkyl;
- R 3 is a urea of the formula NH—C(O)—NHR32 or a carbamate of formula NH—C(O)—OR32, wherein R32 is C1-6 alkyl or C3-6 cycloalkyl;
- D is a C7-atom saturated or unsaturated alkylene chain optionally containing one double bond at position 11,12 or 13,14;
- said D chain optionally containing one heteroatom independently selected from: O, S, NH, N(Me), or N(Ac); and
- R 4 is H or C1-6 alkyl.
- Most preferably, are compounds of formula I wherein R 21 is methoxy, and R22 is ethoxy or:

- wherein R 24a is NH—(C1-4 alkyl), NH—C(O)—(C1-4 alkyl); or NH—C(O)—O—(C1-4 alkyl),; and
- D is saturated or contains one cis double bond at position 13,14.
- Finally, included within the scope of this invention are all compounds of formula I as presented in Tables 1 to 9.
- The pharmaceutical compositions of this invention may be administered orally, parenterally or via an implanted reservoir. Oral administration or administration by injection are preferred. The pharmaceutical compositions of this invention may contain any conventional non-toxic pharmaceutically-acceptable carriers or auxiliary agents such as adjuvants or vehicles. In some cases, the pH of the formulation may be adjusted with pharmaceutically acceptable acids, bases or buffers to enhance the stability of the formulated compound or its delivery form. The term parenteral as used herein includes subcutaneous, intracutaneous, intravenous, intramuscular, intraarticular, intrasynovial, intrasternal, intrathecal, and intralesional injection or infusion techniques.
- The pharmaceutical compositions may be in the form of a sterile injectable preparation, for example, as a sterile injectable aqueous or oleaginous suspension. This suspension may be formulated according to techniques known in the art using suitable dispersing or wetting agents (such as, for example. Tween 80) and suspending agents.
- The pharmaceutical compositions of this invention may be orally administered in any orally acceptable dosage form including, but not limited to, capsules, tablets, and aqueous suspensions and solutions. In the case of tablets for oral use, carriers which are commonly used include lactose and corn starch. Lubricating agents, such as magnesium stearate, are also typically added. For oral administration in a capsule form, useful diluents include lactose and dried corn starch. When aqueous suspensions are administered orally, the active ingredient is combined with emulsifying and suspending agents. If desired, certain sweetening and/or flavoring and/or coloring agents may be added.
- Other suitable vehicles or carriers for the above noted formulations and compositions can be found in standard pharmaceutical texts, e.g. in “Remington's Pharmaceutical Sciences”, 19 th ed., Mack Publishing Company, Easton, Pa., 1995.
- Dosage levels of between about 0.01 and about 100 mg/kg body weight per day, preferably between about 0.5 and about 75 mg/kg body weight per day of the protease inhibitor compounds described herein are useful in a monotherapy for the prevention and treatment of HCV mediated disease. Typically, the pharmaceutical compositions of this invention will be administered from about 1 to about 5 times per day or alternatively, as a continuous infusion. Such administration can be used as a chronic or acute therapy. The amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration. A typical preparation will contain from about 5% to about 95% active compound (w/w). Preferably, such preparations contain from about 20% to about 80% active compound.
- As the skilled artisan will appreciate, lower or higher doses than those recited above may be required. Specific dosage and treatment regimens for any particular patient will depend upon a variety of factors, including the activity of the specific compound employed, the age, body weight, general health status, sex, diet, time of administration, rate of excretion, drug combination, the severity and course of the infection, the patient's disposition to the infection and the judgment of the treating physician. Generally, treatment is initiated with small dosages substantially less than the optimum dose of the peptide. Thereafter, the dosage is increased by small increments until the optimum effect under the circumstances is reached. In general, the compound is most desirably administered at a concentration level that will generally afford antivirally effective results without causing any harmful or deleterious side effects.
- When the compositions of this invention comprise a combination of a compound of formula I and one or more additional therapeutic or prophylactic agent, both the compound and the additional agent should be present at dosage levels of between about 10 to 100%, and more preferably between about 10 and 80% of the dosage normally administered in a monotherapy regimen.
- When these compounds or their pharmaceutically acceptable salts are formulated together with a pharmaceutically acceptable carrier, the resulting composition may be administered in vivo to mammals, such as man, to inhibit HCV NS3 protease or to treat or prevent HCV virus infection. Such treatment may also be achieved using the compounds of this invention in combination with agents which include, but are not limited to: immunomodulatory agents, such as α-, β-, or γ-interferons; other antiviral agents such as ribavirin, amantadine; other inhibitors of HCV NS3 protease; inhibitors of other targets in the HCV life cycle such as helicase, polymerase, metalloprotease, or internal ribosome entry site (IRES); or combinations thereof. The additional agents may be combined with the compounds of this invention to create a single dosage form. Alternatively these additional agents may be separately administered to a mammal as part of a multiple dosage form.
- Accordingly, another embodiment of this invention provides methods of inhibiting HVC NS3 protease activity in mammals by administering a compound of the formula I, wherein the substituents are as defined above.
- In a preferred embodiment, these methods are useful in decreasing HCV NS3 protease activity in a mammal. If the pharmaceutical composition comprises only a compound of this invention as the active component, such methods may additionally comprise the step of administering to said mammal an agent selected from an immunomodulatory agent, an antiviral agent, a HCV protease inhibitor, or an inhibitor of other targets in the HCV life cycle such as helicase, polymerase, or metalloprotease. Such additional agent may be administered to the mammal prior to, concurrently with, or following the administration of the compositions of this invention.
- In an alternate preferred embodiment, these methods are useful for inhibiting viral replication in a mammal. Such methods are useful in treating or preventing HCV disease. If the pharmaceutical composition comprises only a compound of this invention as the active component, such methods may additionally comprise the step of administering to said mammal an agent selected from an immunomodulatory agent, an antiviral agent, a HCV protease inhibitor, or an inhibitor of other targets in the HCV life cycle. Such additional agent may be administered to the mammal prior to, concurrently with, or following the administration of the composition according to this invention.
- The compounds set forth herein may also be used as laboratory reagents. The Applicant provides for the first time compounds with a low molecular weight, that are highly active and specific against the HCV NS3 protease. Some of the present compounds may be instrumental in providing research tools for designing of viral replication assays, validation of animal assay systems and structural biology studies to further enhance knowledge of the HCV disease mechanisms.
- The compounds of this invention may also be used to treat or prevent viral contamination of materials and therefore reduce the risk of viral infection of laboratory or medical personnel or patients who come in contact with such materials (e.g. blood, tissue, surgical instruments and garments, laboratory instruments and garments, and blood collection or transfusion apparatuses and materials).
- Several ways of carrying the synthesis of acyclic intermediates of compounds of formula I are disclosed in WO 00/09543 and WO 00/09558 incorporated herein by reference.
- The compounds of the present invention are synthesized according to the general process illustrated in Schemes I, II and III (wherein PG is an appropriate protecting groups. [In all schemes presented below, D′ has the same definition as D but is 2 to 5 atom shorter].
- When the invention covers compounds of formula I wherein A is a N-substituted amide, vinyl-ACCA or homo-allyl ACCA (R 1) is coupled to an appropriate amine prior to the coupling to P2. Such coupling will be readily recognized by persons skilled in the art. As will be recognized by persons skilled in the art, such amide (A) is not protected but bears any relevant substituent R5 as defined above.
- The ring-closing reaction (macrocyclization) is carried out by either olefin metathesis (Scheme I) or when the linker contains a nitrogen atom, by reductive amination (Scheme II), or by peptide bond formation Scheme III.
- Details of these processes are presented below:
- A. Macrocyclisation via Olefin Metathesis

- Scheme I:
- There are several ways in which the coupling sequence can be carried out which can be easily recognized by persons skilled in the art. Starting with 4-(S)-hydroxyproline, the substituent at the 4-hydroxy can be incorporated via a Mitsunobu reaction (as described in Mitsunobu Synthesis, Jan. 1-28, 1981; Rano et al. Tet. Lett. 1994, 36, 3779-3792; Krchnak et al. Tet. Lett. 1995, 36, 6193-6196) before or after the macrocyclization. Alternatively the assembly can be done with the required 4-(R)-hydroxy-substituted proline as disclosed in the general processes of WO 00/09543 & WO 00/09558 (see below for specific examples of these fragments).
- Steps A, B, C: Briefly, the P1, P2, and P3 moieties can be linked by well known peptide coupling techniques and generally disclosed in WO 00/09543 & WO 00/09558.
- Step D: The formation of the macrocycle can be carried out via an olefin metathesis using a Ru-based catalyst such as the one reported by Miller, S. J.; Blackwell, H. E.; Grubbs, R. H. J. Am. Chem. Soc. 1996, 118, 9606-9614 (a); Kingsbury, J. S.; Harrity, J. P. A.; Bonitatebus, P. J.; Hoveyda, A. H. J. Am. Chem. Soc. 1999, 121, 791-799 (b) and Huang, J.; Stevens, E. D.; Nolan, S. P.; Petersen, J. L.; J. Am. Chem. Soc. 1999, 121, 2674-2678 (c). It will also be recognized that catalysts containing other transition metals such as Mo can be used for this reaction.

- Step E: Optionally, the double bond is reduced by standard hydrogenation methods well known in the art. When A′ is a protected carboxylic acid, it is also deprotected appropriately.
- B. Macrocyclization via Reductive Amination (for Linkers Containing N)
- When the linker contains a nitrogen atom, macrocyclization is achieved by reductive amination as shown in Scheme II to obtain inhibitors of general structure II.

- Step A: Hydroboration of the double bond following Brown's procedure (H. C. Brown and B. C. Subba Rao, J. Am. Che. Soc. 1959, 81, 6434-6437) followed by oxidation of the resulting alcohol (for example via Dess-Martin periodinate, J. Am. Chem. Soc. 1991, 113, 7277-7287) affords the corresponding aldehyde.
- Step B: Hydrogenation in the presence of acid leads to the removal of the amino protecting group followed by macrocyclization via reductive amination. The P3 unit used in this synthesis is easily obtained from a variety of amino acids, such as lysine, ornithine, glutamine (after a Hofmann reaction: Ber. 1881, 14, 2725) and others; these synthetic modifications are methods well known in the art.
- Step C: Optionally, the secondary amine in the linker D (formed after step D) is alkylated with alkyl halides or acetylated with alkyl or aryl acid chlorides using methodologies well known in the art to obtain inhibitors of general structure II. When A′ is a protected carboxylic acid, it is also deprotected appropriately.
- C. Macrocyclization via Lactam Formation
- Alternatively, it is understood that these macrocyclic compounds with general structure I and II can be synthesized in other ways. For example P1 and P3 can be first connected to the linker D, then coupled to P2 and the macrocyclization reaction can be a lactam formation in two possible ways as will be recognized by persons skilled in the art and as shown in Scheme III.

- Synthesis of P1
- The synthesis of inhibitors with general structure I and II requires the same P1 fragments:
- a) vinyl ACCA, the synthesis and resolution of which is described in WO 00/09543 & WO 00/09558 and co-pending applications Ser. No. 09/368,866 incorporated herein by reference in its entirety) or
- b) homoallyl ACCA (Example 1, compound 1f).
- Synthesis of P2
- Some of the P2 fragments used for the synthesis of compounds of formula I are described in WO 00/09543 & WO 00/09558 and co-pending applications Ser. No. 09/368,866 incorporated herein by reference in its entirety.
- Other P2 fragments are synthesized as follows:
- a. Synthesis of 2-“Het”-4-hydroxy-7-methoxyquinoline derivative
- (i) Approach from the corresponding “Het” carboxylic acid IVb

- The synthesis is carried out according to a modified procedure in Li et al. J. Med. Chem. 1994, 34, 3400-3407. Intermediate IVa where R21=OMe (Example 7, compound 7b) is prepared as described by Brown et al. J. Med. Chem. 1989, 32, 807-826.
- Step A: Intermediate IVa is coupled with heterocyclic carboxylic acids IVb under basic conditions with POCl 3 to activate the carboxylate group. A variety of carboxylic acids with general structure IVb are used for the preparation of inhibitors; these are either commercially available, synthesized as shown in scheme V, VI and VII, or synthesized individually using methods described in the specific examples.
- Step B: Ring-closure, followed by dehydration is achieved under basic conditions to obtain quinolines of general structure IVd.
- (i.a). Synthesis of “Het”-carboxylic acids of general formula IVb
- Synthesis of 2-(substituted)-amino-4-carboxy-aminothiazole derivatives (Vc)
- A modification of the procedure described by Berdikhina el al. Chem. Heterocycl. Compd. (Engl. Transl.) 1991, 4, 427-433 is used.

- A variety of 2-alkylaminothiazolyl-4-carboxylic acids, compounds of general structure Vc, are made using the general synthetic methodology outlined in Scheme V using thioureas (Va) with different alkyl substituents (R 25=alkyl group) and 3-bromopyruvic acid. This type of condensation reaction is well known in the art. Alternatively, the P2 fragment containing the 2-amino-substituted-thiazole derivatives are synthesized from the corresponding 2-carboxyl derivative as shown in scheme VI according to the procedure of: Unangst, P. C.; Connor, D. T. J. Heterocyc. Chem. 29, 5, 1992, 1097-1100.

- Examples of this process are disclosed in WO 00/09543 & WO 00/09558.
- Synthesis of 2-carboxy-4-substituted aminothiazole derivatives VIId
- A variety of 4-alkylthiazolyl-2-carboxylic acids, compounds of general structure VIId, is made using the general synthetic methodology outlined in Scheme VII.

- The procedure described by Janusz et al. J. Med. Chem. 1998, 41, 3515-3529 is used with modifications as described as follows: Ethyl thiooxamate (VIIa) is reacted with different □-bromoketones of general structure VIIb (R24=alkyl group) to form thiazolyl carboxylic acids of general structure VIId. This type of condensation reaction is well known in the art.
- Synthesis of 2-carboxy-(substituted)-imidazole derivative (VIIIb)
- A variety of alkylimidazolyl-2-carboxylic acids, compounds of general structure VIIIb, are made using the general synthetic methodology outlined in Scheme VIII.

- The procedure described by Baird et al. J. Amer. Chem. Soc. 1996, 118, 6141-6146. was used: an alkyl imidazole is deprotonated with a strong base (e.g. nBuLi) and then reacted with CO2 to form the carboxylic acid VIIIb. This type of condensation reaction is well known in the art.
- b. Synthesis of 4-hydroxy-7-methoxy-2-(imidazolyl or pyrazolyl)quinolines
- 4-Hydroxy-7-R 21 quinolines having an imidazolyl or pyrazolyl moiety at C2 are generally prepared using the methodology outlined in Scheme IX.

- The synthesis of the key intermediate, (wherein R 21=OMe) 4-benzyloxy-2-chloro-7-methoxyquinoline IXa is described in detail in Example 6 (compound 6e).
- Step A: At high temperatures, a variety of imidazoles, alkyl substituted imidazoles, pyrazoles or alkyl substituted pyrazoles can be used to displace the 2-chloro moiety of compound IXa giving compounds of general structure IXb.
- Step B: Upon removal of the benzyl protecting group from the 4-hydroxy moiety of the quinoline by standard hydrogenation methods, quinoline derivatives of general structure IXc are obtained.
- Synthesis of P3
- A variety of P3 fragments are synthesized containing the appropriate D linker extension for macrocyclization by olefin metathesis. In general P3 units containing a terminal olefin for metathesis are synthesized following the general schemes shown below (Schemes X, XI & XII).
- Synthesis of Linkers in Class A
- This general synthesis is used to make linkers that are all carbon based (no heteroatom) (Scheme X).

- The synthesis is performed according to the procedure of Evans el al. J. Am. Chem. Soc. 1990, 112, 4011-4030.
- The starting carboxylic acids (Xa) is commercially available or is prepared by know literature procedures familiar to those skilled in the art.
- Step A: The carboxylic acid Xa is activated with pivaloyl chloride and then reacted with the anion of Evans' chiral auxiliary 4(S)-4-(phenylmethyl)-2-oxazolidinone following well known chemistry (Review: D. J. Ager et al. Aldrichimica Acta 1997, 30, 3-11, and references therein) to obtain compounds of general structure Xb.
- Step B: Stereoselective □-azidation with trizylazide, of a chiral imide enolate such as those which would form from compounds with general structure Xb in the presence of a base like KHMDS, is also well known in the art (Review: D. J. Ager et al. Aldrichimica Acta 1997, 30, 3-11, and references therein).
- Step C: Reduction of the □-azide, catalyzed by SnCl 2, is followed by protection of the amine formed as its t-butyl carbamate gives intermediates of general structure Xc. These reactions are also well known in the art.
- Step D: Finally, the chiral auxiliary is hydrolyzed under basic conditions, such as a mixture of H 2O2 with LiOH, to produce the amino acid-type linkers of general structure Xe.
- Alternatively, P3 moieties having the same general structure Xe are synthesized following de procedure described by M. J. Burk et al. J. Am. Chem. Soc 1998, 120, 657-663 illustrated in Scheme XI. These compounds varied in the number of methylene units (—CH2—) along the linker (m=1 to 5) and the substitution of alkyl groups at R4, but did not contain a heteroatom.

- Step A: The monoacid compound XIb is prepared from commercially available diethyl 2-acetamidomalonate by standard ester hydrolysis under basic conditions.
- Step B: Knoevenagel-type condensation between an aldehyde of general structure XIc and compound XIb in the presence of a base, such as pyridine, and acetic anhydride leads to the formation of enamide intermediate XId having the Z stereochemistry around the newly formed double bond as shown.
- Step C: Regioselective and enantioselective catalytic hydrogenation of the enamide intermediate XId to the amino acid intermediate XIe is achieved using Burk's method.
- Step D: The nitrogen of the acetamido derivative XIe is then di-protected with the addition of a t-butyl carbamate substituent before the acetate group, as well as the ethyl ester, are hydrolyzed under standard basic condition to obtain P3 moieties of general structure XIf.
- Synthesis of Linkers in Class B
- General Structure of Linkers in Class B

- This general synthesis is used to make linkers containing oxygen or sulfur.

- Step A: Suitably protected amino acids, such Boc-(L)-serine methyl ester, Boc-(L)-threonine methyl ester or Boc-(L)-allothreonine methyl ester, are alkylated with allyl iodide in the presence of Ag 2O to give the methyl ester XIIb.
- Step B: Hydrolysis of the methyl ester under standard basic conditions yields the ether-type linkers of general structure XIIc (X═O).
- Step C: The sulfur analog is prepared from the same starting amino acid XIIa (appropriately protected as before) and its hydroxyl group is converted to a good leaving group (such as the tosylate intermediate XIId) using standard methodology well known in the art.
- Step D: The tosyl moiety is subsequently displaced with the anion of thioacetate leading to the formation of the thioester intermediate XIIe by inversion of the chiral center at the □-carbon.
- Step E: Hydrolysis of the thioester moiety under mild basic conditions yields the free thiol XIIf.
- Step F: Alkylation of the thiol moiety is easily achieved under basic conditions with allyl iodide.
- Step G: Finally, the sulfide analog XIIc (X═S) are obtained after hydrolysis of the methyl ester using standard procedures.
- Synthesis of R3 fragment:
- Examples of synthesis of fragments wherein R 3 is NH—R31 are disclosed in WO 00/09543.
- The present invention is illustrated in further detail by the following non-limiting examples. Other specific ways of synthesis or resolution can be found in WO 00/09543 & WO 00/09558 and in co-pending applications Ser. Nos. 09/368,670 and 09/368,866, all of which are hereby incorporated by reference.
- Temperatures are given in degrees Celsius. Solution percentages express a weight to volume relationship, and solution ratios express a volume to volume relationship, unless stated otherwise. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker 400 MHz spectrometer; the chemical shifts (δ) are reported in parts per million and are referenced to the internal deuterated solvent unless otherwise indicated. The NMR spectra of all final compounds (inhibitors) was recorded in DMSO-d 6 of their TFA salt unless otherwise indicated. Flash column chromatography was carried out on silica gel (SiO2) according to Still's flash chromatography technique (W. C. Still et al., J. Org. Chem., 1978, 43, 2923). Abbreviations used in the examples include Bn: benzyl; Boc: tert-butyloxycarbonyl [Me3COC(O)]; BSA: bovine serum albumin; Cbz: benzyloxycarbonyl; CHAPS: 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate; DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene; CH2Cl2=DCM: methylene chloride; DEAD: diethylazodicarboxylate; DIAD: diisopropylazodicarboxylate; DIPEA: diisopropylethylamine; DMAP: dimethylaminopyridine; DCC: 1,3-dicyclohexylcarbodiimide; DME: 1,2-dimethyoxyethane; DMF: dimethylformamide; DMSO: dimethylsulfoxide; DTT: dithiothreitol or threo-1,4-dimercapto-2,3-butanediol; DPPA: diphenylphosphoryl azide; EDTA: ethylenediaminetetraacetic acid; Et: ethyl; EtOH: ethanol; EtOAc: ethyl acetate; Et2O: diethyl ether; ESMS: electrospray mass spectrometry; HATU: O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; HPLC: high performance liquid chromatography; MS: mass spectrometry; MALDI-TOF: Matrix Assisted Laser Disorption Ionization-Time of Flight, FAB: Fast Atom Bombardment; LAH: lithium aluminum hydride; Me: methyl; MeOH: methanol; MES: (2-[N-morpholino]ethane-sulfonic acid); NaHMDS: sodium bis(trimethylsilyl)amide; NMM: N-methylmorpholine; NMMO: N-methylmorpholine oxide; NMP: N-methylpyrrolidine; Pr: propyl; Succ: 3-carboxypropanoyl; PNA: 4-nitrophenylamino or p-nitroanilide; TBAF: tetra-n-butylammonium fluoride; TBME: tert-butyl-methyl ether; tBuOK: potassium tert-butoxide; TBTU: 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate; TCEP: tris(2-carboxyethyl) phosphine hydrochloride; TFA: trifluoroacetic acid; THF: tetrahydrofuran; TIS: triisopropylsilane; TLC: thin layer chromatography; TMSE: trimethylsilylethyl; Tris/HCl: tris(hydroxymethyl)aminomethane hydrochloride.
- Synthesis of t-butyl-(1R,2R)/(1S,2S)-1-amino-2-homoallylcyclopropyl carboxylate (1f):

- A. To a suspension of benzyltriethylammonium chloride (5.08 g, 22.3 mmol.) in 50% aqueous NaOH (50 mL), 1,2-dibromo-5-hexene (1b, 8.10 g, 33.46 mmol) and di-t-butylmalonate (1a, 4.82 g, 22.30 mmol) were added in succession. The mixture was stirred vigorously at RT for 16 h, then diluted with H 2O and extracted with CH2Cl2 (3×50 mL). The organic layer was further washed with H2O (2×50 mL), brine/H2O (2/1, 2×50 mL, dried over MgSO4 and evaporated. The crude residue was purified by flash column chromatography on silica gel, using 3 to 5% EtOAc in hexane as the eluent, to obtain compound 1c in 38% yield (2.48 g).
- 1H NMR (CDCl3, 400 MHz): δ 1.19 (bd, J=7.9 Hz, 2H), 1.25-1.33 (m, 1H), 1.46 (s, 9H), 1.48 (s, 9H), 1.47-1.60 (m, 1H), 1.75-1.82 (m, 1H), 2.14-2.22 (m, 2H), 4.93-5.50 (m, 2H), 4.96 (dm, J=10.2 Hz, 1H), 5.18 (dm, J=17.2 Hz, 1H). ES(+)MS m/z 297 (M+H)+.
- B. To a suspension of potassium t-butoxide (5.75 g, 51.25 mmol) in anhydrous diethyl ether (150 mL) at 0°, H 2O was added (203 μL, 11.27 mmol) and the reaction mixture was stirred at 0° for 10 min. An ether solution of compound 1c (2.48 g in 10 mL diethyl ether, 10.25 mmol) was added and the mixture was stirred at RT for 5 h. The mixture was diluted with ice-cold H2O and extracted with diethyl ether (3×200 mL). The aqueous layer was acidified to pH 3.5-4 with ice-cold 10% aqueous citric acid and re-extracted with EtOAc (3×200 mL). The EtOAc layer was washed with H2O (2×100 mL), brine (100 mL), dried over MgSO4 and evaporated to give compound 1d in 85% yield based on the amount of recovered starting material.
- 1H NMR (CDCl3, 400 MHz): δ 1.51 (s, 9H), 1.64-1.68 (m, 1H), 1.68-1.75 (m, 1H), 1.77-1.88 (m, 1H), 1.96-2.01 (m, 1H), 2.03-2.22 (m, 3H), 5.01 (dm, J=6.4 Hz, 1H), 5.03 (dm, J=14.9 Hz, 1H), 5.72-5.83 (m, 1H). ES(+)MS: m/z 241 (M+H)+.
- C. To a solution of the acid 1d in anhydrous benzene (1.14 g in 25 mL benzene, 4.74 mmol), Et 3N (800 μL, 5.68 mmol) was added, followed by the addition of diphenylphosphoryl azide (1.13 mL, 5.21 mmol) and the mixture was heated to reflux for 3.5 h. Subsequently, trimethylsilylethanol (1.36 mL, 9.48 mmol.) was added and stirring at reflux was continued for an additional 4 h. The mixture was then cooled to RT, evaporated to half of its original volume, diluted with diethyl ether (30 mL) and washed with 5% aqueous NaHCO3 (2×30 mL), brine (50 mL), dried over MgSO4 and evaporated. The residual oil was chromatographed on silica gel using 10% EtOAc in hexane as the eluent to obtain pure compound 1e in 88% yield (1.49 g).
- 1H NMR (CDCl3, 400 MHz) δ 0.03 (s, 9H), 0.91-0.99 (m, 2H), 1.18-1.29 (m, 2H), 1.45 (bs, 11H), 1.56-1.72 (m, 2H), 2.02-2.18 (m, 2H), 4.12 (t, J=8.3 Hz, 2H), 4.93 (dm, J=10.2 Hz, 1H), 4.98 (dm, J=17.2 Hz, 1H), 5.07 (bs, 1H), 5.71-5.83 (m, 1H).
- D. To a solution of the cyclopropyl derivative 1e (1.19 g, 3.35 mmol, in 30 mL THF), t-Bu 4NF (6.7 mL of 1M in THF, 6.7 mmol.) was added and the mixture was first stirred at RT for 16 h and subsequently heated to reflux for 15 min. The solvent was carefully evaporated under low pressure (due to the high volatility of the free amine 1f, caution should be exercised during the evaporation of the solvent). The crude residue was re-dissolved in EtOAc (100 mL) and washed with H2O (2×50 mL), brine (50 mL), dried over MgSO4 and again the solvent was carefully evaporated. The crude product 1f (as a mixture of two enantiomers 1f′ and 1f″) was used for coupling with the P2 proline derivatives without further purification. Isolation of the P1P2 fragment having the desired stereochemistry at P1 was easily achieved at this stage using flash chromatography (example 21, fragment 21b).
- Synthesis of Boc-4(R)-[(7-methoxy-4-quinolinyl)oxy]proline (2c):

- 4-Hydroxy-7-methoxyquinoline (2b) was prepared according to the method described by Chun, M. W.; Olmstead, K. K.; Choi, Y. S.; Lee, C. O.; Lee, C.-K.; Kim, J. H.; Lee, J. Bioorg. Med. Chem. Lett. 1997, 7, 789. A solution of compound 2b (1.88 g, 10.73 mmol) and DEAD (3.4 mL, 21.46 mmol) in anhydrous THF were added to a stirring solution of protected cis-hydroxyproline 2a (2.63 g, 10.73 mmol) and triphenylphosphine (5.63 g, 21.46 mmol) in anhydrous THF (160 mL) at 0° under N2. The reaction mixture was allowed to warm-up to RT and stir for 14 h. The THF was then evaporated and the pure product 2c was isolated after flash column chromatography using 5% MeOH in EtOAc as the eluent, in 35% yield (1.5 g).
- 1H NMR (CDCl3, 400 MHz): δ 1.44 (s, 9H), 1.65 (bs, 1H), 2.34-2.43 (m, 1H), 2.63-2.76 (m, 1H), 3.78 (s, 3H), 3.75-3.85 & 3.89-3.99 (2m, 1H, 2 rotamers), 3.95 (s, 3H), 4.51 & 4.60 (2t, J=8 Hz, 1H, 2 rotamers), 5.15 (bs, 1H), 6.53-6.59 (m, 1H), 7.12-7.18 (dd, J=8.9 & 2.2 Hz, 1H), 7.36 (d, J=2.6 Hz, 1H), 8.03 (bd, J=9.2 Hz, 1H), 8.65 (bd, J=5.1 Hz, 1H).
- Synthesis of 2-ethoxy-4-hydroxy-7-methoxy quinoline (3c)

- The synthesis of Methyl-p-methoxyantranylate 3a was done as described in Katz et al. J. Org. Chem., 1953, 18, 1380-1400.
- The general synthesis for the quinoline derivative 3c is a modification of the procedure of Baccar et al. Indian Journal of Chemistry, 1995, Sat. B, 330-332.
- A. Methyl-p-methoxyantranylate 3a (3.069 g, 16.96 mmol) was dissolved in triethylorthoacetate (4.7 mL, 25.4 mmol), then a solution of anhydrous HCl (4 N/Dioxane, 50 μL, 0.6 mmol) was added. The resulting mixture was heated at reflux for 19 hours. The volatiles were then evaporated under vacuum to give product 3b (4.92 g, amber oil, quantitative yield) that was used as such for the next step.
- B. To a solution of the substrate 3b (assumed 16.96 mmol) in THF (34 mL) at −78° C. under nitrogen, was added LiHMDS (1 M/THF, 22 mL, 1.3 eq.). Shortly after the addition, the cold temperature bath was removed and the mixture was left to stir at ambient temperature for 1 hour, after which time, another portion of LiHMDS (16 mL) was added. The resulting mixture was stirred until complete disappearance of starting material (1 hour) by TLC (100% EtOAc, imidate R f=0.7, product Rf=0.2). HCl (4 N/dioxane, 10 mL) was then added and the mixture was concentrated under vacuum. The resulting paste was triturated from a mixture of EtOAc (10 mL) and aqueous NaH2PO4 (1 M, 10 mL) and sonicated. An abundant precipitate was formed, collected by filtration, washed with water and dried to afford the desired product 3c as a beige solid (3.117 g, 84% yield for 2 steps, >99% purity by HPLC).
- 1H NMR (400 MHz, DMSO-d) □ (ppm): 7.88 (d, J=8.9 Hz, 1H), 6.98 (br. s, 1H), 6.89 (br. d, J=8.6 Hz, 1H), 5.94 (br. s, 1H), 4.30 (br. s, 2H), 3.84 (s, 3H), 1.34 (t, J=7.0 Hz, 3H).
- Synthesis of 4-hydroxy-7-methoxy-2(3-methyl-1,2,4-oxadiazol-5-yl) quinoline (4d)

- A. To a solution of 2-carbomethoxy-4-hydroxy-7-methoxyquinoline 4a (the preparation of which is described in WO 00/09543 and WO 00/09558) (1 g, 4.29 mmol) in DMF (10 mL) under nitrogen was added NaH (60% in mineral oil, 190 mg, 4.98 mmol). The resulting mixture was stirred at ambient temperature for 1 hour, MEM chloride (455 μL, 4.98 mmol) was then added dropwise and the resulting mixture was stirred at ambient temperature for an extra 19.5 hours. The reaction mixture was diluted with EtOAc (100 mL), washed with H 2O (50 mL), brine (50 mL), dried with MgSO4, concentrated under vacuum to afford the crude reaction isolate (1.37 g). The latter was purified by flash column chromatography to afford product 4b (1.04 g, 75% yield) as a colorless oil.
- B. To a mixture of freshly activated 4 Å molecular sieve (500 mg) and acetamidoxime (248 mg, 3.35 mmol) was added THF (3 mL). The resulting mixture was stirred for 15 min. under nitrogen at ambient temperature, then NaH (60% in mineral oil, 124 mg, 3.24 mmol) was added by portions. The resulting suspension was stirred at ambient temperature for 1 hour, then ester 4b (500 mg, 1.56 mmol) was added in solution in THF (5 mL). The resulting mixture was heated at reflux for 1 hour then filtered over Celite, rinsing with EtOAc (3 portions of 20 mL) and concentrated under vacuum. The resulting crude mixture was purified by flash column chromatography (100% EtOAc) to afford product 4c (352 mg, 65% yield) as a white solid.
- C. To the MEM ether 4c (170 mg, 0.493 mmol) in THF (4 mL) was added aqueous HCl (1 N, 1 mL). The resulting mixture was stirred at ambient temperature for 1 hour then diluted with aqueous NaH 2PO4 (1 M, 50 mL). The solid formed was filtered, triturated with EtOAc, filtered and dried to afford the desired product (4d) (90 mg, 71% yield) as a white solid. MS (ES+) 258 (M+1), (ES−) 256 (M−1).
- 1H NMR (400 MHz, DMSO-d) □ (ppm): 8.03 (d, J=9.2 Hz, 1H), 7.38 (d, J=2.2 Hz, 1H), 7.06 (d, J=8.6 Hz, 1H), 6.85 (br. s, 1H), 3.88 (s, 3H), 2.64 (s, 3H).
- Synthesis of 4-hydroxy-7-methoxy-2(5-methyl-1,3,4-oxadiazol-2-yl) quinoline (5e)

- A. To substrate 4b (465 mg, 1.45 mmol) in ethanol (5 mL) was added anhydrous hydrazine (57 μL, 1.8 mmoL). The resulting solution was heated at reflux for 4 h, then concentrated under vacuum to afford product 5a (704 mg, quantitative crude yield) as a yellow solid which was used as such in the next step.
- B. Compound 5a (assumed 1.45 mmol) in triethylorthoacetate (5 mL) was heated at 100-110° C. under nitrogen. The resulting mixture was then diluted with EtOAc (100 mL), washed with aqueous saturated NaHCO 3 (50 mL), brine (50 mL), dried with MgSO4, concentrated under vacuum and purified by flash column chromatography (100% EtOAc). Compound 5b (359 mg, 61% yield for two steps) was obtained as a yellow oil. MS (ES+) 392 (m+1), (ES−) 390 (m−1).
- C. Compound 5b (333 mg, 0.852 mmol) was heated at 140° C. under high vacuum for 8.5 h and purified by flash column chromatography (100% EtOAc) to afford a mixture of 5b (116 mg, 35%, R f 0.5) and compound 5c (138 mg, 72% corrected yield, Rf 0.3). To a THF (4 mL) solution of compound 5c (138 mg, 0.4 mmol) was added aqueous HCl (1 N, 1 mL) and the resulting mixture was stirred until completion (30 min.). THF was evaporated under vacuum and aqueous NaH2PO4 (1 M, 2 mL) was added. The resulting suspension was sonicated, filtered and the solid was dried under high vacuum to afford the desired product 5d, (75 mg, 73% yield) as a beige solid. MS (ES+) 258 (m+1), (ES−) 256 (m−1). 1H NMR (400 MHz, DMSO-d): □ 8.03 (d, J=9.2 Hz, 1H), 7.39 (d, J=2.2 Hz, 1H), 7.06 (br. d, J=8.6 Hz, 1H), 6.85 (br. s, 1H), 3.88 (s, 3H), 2.64 (s, 3H).
- Synthesis of 4-benzyloxy-2-(chloro)-7-methoxyquinoline (6e)

- A. Commercially available Meta-anisidine (25 g, 0.20 mol) in dioxane (80 mL) was cooled down to 0° C. and anhydrous HCl (4 N/dioxane, 75 mL, 0.30 mol) was added. Then Et 2O (500 mL) was added and stirring was maintained for 1 hour. The beige solid was then filtered and dried under vacuum to afford salt 6a (31.88 g, 98% yield).
- B. To this salt was added ethylcyanoacetate (21.3 mL, 0.20 mol) and the mixture, in a flask equipped with a distillation head and a collecting flask, was heated to 280-300° C. Ethanol produced was collected to monitor the evolution of the reaction. At 9 mL of collected ethanol (theoretical amount 11.7 mL), heating was stopped, the reaction mixture cooled down to RT, diluted with water (200 mL)-EtOAc (200 mL) then stirred and aqueous NaH 2PO4 (300 mL) was added. After additional stirring for 1 h, filtration and drying, 6b was obtained (19.06 g, 84.5% purity, ˜50% yield) as a yellow solid and was used as such in the next reaction.
- C. Compound 6b (11.0 g, 57.8 mmol) in DMF (100 mL) at 0° C. was added to NaH (60% in mineral oil, 2.78 g, 115.6 mmol). The ice bath was then removed and the mixture was stirred at ambient temperature for 1 h, benzyl bromide (7.6 mL, 63.6 mmol) was then added and the reaction mixture was stirred for 16 hours. The solution was then diluted with EtOAc (220 mL)-hexane (220 mL) and the solid formed was filtered, triturated with aqueous saturated NaHCO 3 (110 mL), washed with water, hexane-EtOAc (1:1 ratio, 100 mL) and dried under high vacuum. Product 6c (5.6 g, 91% purity, 35% yield) was thus obtained as a yellow solid. To compound 6c (2.67 g, 9.52 mmol) in acetic acid (21 mL) was added iso-amyl nitrite (3.8 mL, 28.6 mmol) and the resulting mixture was stirred at ambient temperature and monitored by HPLC. More iso-amyl nitrite (1.3 mL, 9.52 mmol) was added after 2 hours and the mixture was left to stir over 90 hours (HPLC 81% product, 3% substrate). Water (100 mL) was added to the resulting suspension, which was then filtered. The brown solid collected was dried under high vacuum giving product 6d (2.35 g, 92% purity, 72% yield).
- D. To compound 6d (1.5 g, 4.39 mmol) was added phosphorous oxychloride (13 mL, 141 mmol) and the resulting mixture was heated at reflux for 1 hour then diluted with EtOAc (150 mL) and quenched at 0° C. slowly with aqueous NaOH (1 N, 150 mL) to pH 9. The two layers were separated and the organic layer was dried with MgSO 4 and concentrated under vacuum to afford a brown solid which was purified by flash column chromatography (15% EtOAc/hexane). Product 6e (819 mg, purity >99%, 62% yield) was obtained as a yellow solid.
- 1H NMR (400 MHz, CDCl3): □ 8.07 (d, J=9.2 Hz, 1H), 7.50-7.40 (m, 5H), 7.29 (d, J=2.5 Hz, 1H), 7.12 (dd, J=9.2, 2.5 Hz, 1H), 6.73 (s, 1H), 5.26 (s, 2H), 3.92 (s, 3H).
- Synthesis of 4-hydroxy-2-(1-imidazolyl)-7-methoxyquinoline (7b); 4-hydroxy-2-(4-methyl-1-imidazolyl)-7-methoxyquinoline (7d); 4-hydroxy-7-methoxy-2-(1-pyrazolyl)quinoline (7f); and 4-hydroxy-2-(3-methyl-1-pyrazolyl)-7-methoxyquinoline (7h).

- A. Compound 6e (423 mg, 1.41 mmol) and imidazole (400 mg, 5.88 mmol.) were heated at 110° C. for 20 h. The mixture was then diluted with EtOAc and washed with water and brine, dried with MgSO 4, concentrated under reduced pressure to afford compound 7a (422 mg, 96% purity, 90% yield) as a yellow solid. Compound 7a (319 mg, 0.963 mmol) with Pd (5%/C, 64 mg) in a mixture of ethanol (5 mL) and THF (5 mL) was purged and placed under one ATM. of hydrogen. After 7.5 h of stirring at ambient temperature, the reaction mixture was filtered, rinsed with a chloroform-methanol mixture, and concentrated to afford 7b (130 mg, 97.7% purity, 56% yield) as a yellow solid. MS (ES+) 242 (m+1), (ES−) 240 (m−1).
- 1H NMR (400 MHz, DMSO-d): □ 8.51 (s, 1H), 8.03 (d, J=8.9 Hz, 1H), 7.93 (s, 1H), 7.23 (d, J=1.9 Hz, 1H), 7.15 (s, 1H), 7.12 (dd, J=9.2, 2.2 Hz, 1H), 6.92 (br. s, 1H), 3.91 (s, 3H).
- B. Compound 6e (251 mg, 0.837 mmol) and 4-methylimidazole (344 mg, 4.19 mmol.) were heated at 110° C. for 20 h. The mixture was then diluted with EtOAc, washed with water and brine, dried with MgSO 4, and concentrated under reduced pressure to afford a crude containing a 10:1 mixture of 4-methyl and 5-methylimidazolyl isomer respectively. The major assumed desired isomer 11c, a white solid, (166 mg, 99% purity, 57% yield) was separated from a second more polar fraction (76 mg, 23% yield) containing a mixture of 4- and 5-methyl imidazolyl isomer by flash column chromatography (100% EtOAc). Compound 7c (163 mg, 0.472 mmol) with Pd (5%/C, 33 mg) in a mixture of ethanol (2.4 mL) and THF (5 mL) was purged and placed under one ATM. of hydrogen. After 18 h of stirring at ambient temperature, the reaction mixture was filtered, rinsed with a chloroform-methanol mixture, and concentrated to afford 7d (118 mg, 99% purity, 98% yield) as a white solid.
- 1H NMR (400 MHz, DMSO-d): □ 8.42 (br. s, 1H), 8.01 (d, J=9.2 Hz, 1H), 7.64 (br. s, 1H), 7.21 (br. s, 1H), 7.10 (d, J=8.9 Hz, 1H), 6.89 (br. s, 1H), 3.90 (s, 3H), 2.20 (s, 3H).
- C. Compound 6e (184 mg, 0.614 mmol) and pyrazole (209 mg, 3.07 mmol.) were heated at 110° C. for 17 h. The mixture was then diluted with EtOAc and washed with aqueous NaOH (1 N) and brine, dried with MgSO 4, concentrated under reduced pressure to afford a crude product which was purified by flash column chromatography (2:1 hexane-EtOAc) to afford 7e (103 mg, 50% yield) as a pale yellow solid. Compound 7e (103 mg, 0.311 mmol) with Pd (5%/C, 20 mg) in a mixture of ethanol (2 mL) and THF (2 mL) was purged and placed under one atm. of hydrogen. After 5.5 h of stirring at ambient temperature, the reaction mixture was filtered, rinsed with a chloroform-methanol mixture, and concentrated to afford 7f (77 mg, 99% purity, 99% yield) as a yellow solid. MS (ES+) 242 (m+1), (ES−) 240 (m−1).
- 1H NMR (400 MHz, DMSO-d): □ 8.72 (d, J=2.5 Hz, 1H), 8.31 (s, 1H), 8.00 (d, J=8.9 Hz, 1H), 7.83 (br. s, 1H), 7.43 (br. s, 1H), 7.24 (br. s, 1H), 7.10 (d, J=8.6 Hz, 1H), 6.59 (br. s, 1H), 3.90 (s, 3H).
- D. Compound 6e (217 mg, 0.724 mmol) and 4-methypyrazole (594 mg, 7.24 mmol.) were heated at 110° C. for 23 h. The mixture showing a 1:1 mixture of debenzylated compound 7h and benzylated product 7g was then diluted with EtOAc (2-3 mL) and filtered to afford the pure debenzylated product 7h (111 mg, 95% purity, 54% yield) as a white solid.
- 1H NMR (400 MHz, DMSO-d): □ 8.58 (d, J=2.6 Hz, 1H), 7.98 (d, J=9.2 Hz, 1H), 7.25 (br. s, 1H), 7.20 (s, 1H), 7.04 (br. d, J=9.2 Hz, 1H), 6.38 (s, 1H), 3.89 (s, 3H), 2.30 (s, 3H).
- Synthesis of 4-hydroxy-7-methoxy-2[4(2-isopropylaminothiazolyl)] quinoline (8f)
- Note: [A variety of 2-alkylaminothiazolyl substituents were made using the same synthetic scheme where compound 8b was replaced by other alkyl thioureas.]

- A. The protocol used for the conversion of m-anisidine to 8a was identical to that described in the literature: F. J. Brown et al. J. Med. Chem. 1989, 32, 807-826. However, the purification procedure was modified to avoid purification by chromatography. The EtOAc phase containing the desired product was treated with a mixture of MgSO4, charcoal and 5% w/w (based on expected mass) silica gel. After filtration on celite, the product was triturated with ether. Compound 8a was obtained as a pale brown solid in >99% purity (as confirmed by HPLC).
- B. A suspension of isopropyl thiourea (8b, 3.55 g, 30 mmol) and 3-bromopyruvic acid (8c, 5 g, 1 eq.) in dioxane (300 mL , 0.1 M) was heated to 80° C. Upon reaching 80° C. the solution became clear and soon after the product precipitated as a white solid. After 2 hours of heating, the solution was cooled to RT and the white precipitate was filtered to obtain compound 8d in high purity (>98% purity as confirmed by NMR) and 94% yield (7.51 g).
- C. A mixture of the carboxylic acid 8d (4.85 g, 18.2 mmol) and the aniline derivative 8a (3 g, 1 eq.) in pyridine (150 mL, 0.12 M) was cooled to −30° C. (upon cooling, the clear solution became partially a suspension). Phosphorus oxychloride (3.56 ml, 2.1 eq.) was then added slowly over a 5 min period. The reaction was stirred at −30° C. for 1 h, the bath was removed and the reaction mixture was allowed to warm-up to RT. After 1.5 h the reaction mixture was poured into ice, the pH was adjusted to 11 with aqueous 3N NaOH, extracted with CH 2Cl2, dried over anhydrous MgSO4, filtered and concentrated under vacuum. The beige solid was then purified by flash chromatography (45% EtOAc in hexane) to give compound 8e as a pale yellow solid in 73% yield (6.07 g).
- D. A solution of tBuOK (2.42 g, 21.6 mmol) in anhydrous tBuOH (40 ml, 0.14 M, distilled from Mg metal) was heated to reflux. Compound 8e (1.8 g, 5.4 mmol) was added portion-wise over 5 min and the dark red solution formed was stirred at reflux for an additional 20 min (completion of the reaction was monitored by HPLC). The mixture was cooled to RT and HCl was added (4 N in dioxane, 1.5 eq.). The mixture was then concentrated under vacuum, in order to assure that all of the HCl and dioxane were removed, the product was re-dissolved twice in CH 2Cl2 and dried under vacuum to finally obtain the HCl salt of compound 8f as a beige solid (1.62 g, 93% pure by HPLC). The product was then poured into a phosphate buffer (1N NaH2PO4, pH=˜4.5) and sonicated. The beige solid was filtered and dried under vacuum to give compound 8f (1.38 g, 81% yield) as a beige solid (91% pure by HPLC).
- 1H NMR (400 MHz, DMSO) δ 8.27 (s, 1H), 8.12 (d, 1H, J=9.2 Hz), 7.97 (br.s, 1H), 7.94 (s, 1H), 7.43 (s, 1H), 7.24 (dd, 1H, J=9.2, 2.2 Hz), 3.97 (m, 1H), 3.94 (s, 3H), 1.24 (d, 2H, J=6.4 Hz).
- Synthesis of 4-hydroxy-7-methoxy-2[2(4-isopropylthiazolyl)]quinoline (9f).
- Note: A variety of 2-(4-alkyl)-thiazolyl substituents were made using the same synthetic scheme where compound 9b was replaced by other α-bromoketones.

- A. To a solution of 3-methyl-butan-2-one (8 g, 93 mmol) in MEOH (100 mL) at −30° C., Br 2 (4.79 ml, 93 mmol, 1 eq.) was added dropwise over a period of 45 min. The resulting mixture was then stirred at RT for 90 min. Pentane was added and the solution washed with 5% aqueous NaHCO3, the organic layer was dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The resulting crude yellow oil, compound 9b, was used without further purification. A solution of ethyl thiooxamate (9a, 1.8 g, 13.5 mmol) and bromoketone derivative 9b (13.5 mmol.) in ethanol was stirred at 70° C. for 15 h. The mixture was then concentrated under vacuum and subsequently purified by flash column chromatography, using 15% EtOAc in hexane as the eluent, to obtain compound 9c (740 mg, 28% yield).
- B. A solution of compound 9c (700 mg, 3.5 mmol) in THF/MeOH/H 2O (3:1:1 ratio, 13 mL) was treated with LiOH H2O (148 mg, 3.5 mmol, 1 eq.) at RT for 5 h. The pH was then adjusted to 6 with 0.1N HCl and the mixture was concentrated to dryness under vacuum to obtain the acid 9d, which was used directly in the next step without further purification.
- C. A solution of 4-methoxy-2-amino-acetophenone (intermediate 8a, 570 mg, 3.45 mmol) and carboxylic acid derivative 9d (590 mg, 3.45 mmol, 1 eq.) in pyridine (30 mL) was cooled to −20 □C. POCl 3 (0.35 ml, 3.79 mmol, 1.1 eq.) was then added dropwise over a period of 5 min. The resulting solution was stirred at −10C. for 2 h. The reaction was quenched with the addition of H2O and the mixture was concentrated under vacuum. The residue was poured in a saturated aqueous solution of NaHCO3 and extracted with EtOAc. The organic layer was dried over anhydrous MgSO4, filtered and concentrated under vacuum. The crude product was purified by flash column chromatography, using 25% EtOAc in hexane as the eluent, to give compound 9e as a white solid (740 mg, 67% yield).

- D. tBuOK (518 mg, 2.1 eq.) was added to a suspension of compound 9e (700 mg, 2.2 mmol) in anhydrous tBuOH (11 mL). The resulting mixture was heated to 75 □C. for 7.5 h, the solution was then cooled to RT and acidified with the addition of HCl (4N HCl in dioxane, 2.5 mL). The mixture was concentrated under vacuum and the residue obtained was poured into a solution of 1N NaH 2PO4 and filtered. The solid material was then triturated with a small amount of EtOAc, filtered and dried under vacuum to obtain compound 9f as a pale beige solid (270 mg, 41% yield).
- 1H NMR (400 MHz, DMSO-d6) δ 8.00 (br. s, 1h), 7.60 (br. s, 1H), 7.51 (br. s, 1H), 7.43 (br. s, 1H), 7.29 (br. s, 1H), 7.14 (br. s, 1H), 6.95 (br. a, 1H), 3.90 (s, 3H), 3.15 (m, 1H), 1.33 (d, J=5.4 Hz, 6H).
- Synthesis of 4-hydroxy-2(1-methyl-2-imidazolyl)-7-methoxyquinoline (10d)

- A. A solution of N-methylimidazole 10a (5 g, 61 mmol) in 100 mL THF was cooled at −78C. n-BuLi (24.4 ml of a 2.5M/Et2O solution, 1 eq.) was added dropwise over 15 min. The resulting mixture was stirred 90 min. at −78C. then poured portionwise over excess solid CO2. The heterogeneous mixture was stirred 2 h and allowed to reach RT. 1N HCl was added to pH 5, the aqueous layer is separated and lyophilized. The residue thus obtained was extracted with EtOAc (to remove salts), dried (Na2SO4), filtered and concentrated under reduced pressure. 6.2 g (80% yield) of a white solid 10b was obtained.
- B. A solution of 4-methoxy-2-amino-acetophenone 8a (394 mg, 2.39 mmol) and the carboxylic acid derivative 10b (301 mg, 1 eq.) in pyridine (10 ml) was cooled to—20C. POCl3 (244 μl, 1.1 eq.) was then added dropwise over 5 min. The resulting solution was stirred at −10 □C. for 2.5 h. Water was then added and the mixture was concentrated under reduced pressure. The residue was poured in a saturated solution of NaHCO3 and extracted with EtOAc. The organic phase was dried (MgSO4), filtered and concentrated under reduced pressure. The product was purified by chromatography using silica gel (25% EtOAc/Hex) affording 530 mg (81% yield) of a pale yellow solid 10c.
- C. tBuOK (431 mg 2.1 eq.) was added to a suspension of the substrate 10c (500 mg, 1.8 mmol) in 8 ml of tBuOH. The resulting mixture was then heated to 75° C. for 7 h. The solution was allowed to reach room temperature overnight and 2.5 ml of HCl (4N/dioxane) was added. The mixture was concentrated under reduced pressure and the residue obtained was diluted with EtOAc. NaOH 1N was added until a pH of 7 was obtained. The organic phase was separated and dried (MgSO 4), filtered, and concentrated under reduce pressure to afford 145 mg of 10d (31% yield) as a pale beige solid. 1H NMR (400 MHz, DMSO-d): □ 7.99 (d, J=8.9 Hz, 1H), 7.49 (s, 1H), 7.37 (s, 1H), 7.18 (s, 1H), 6.92 (d, J=8.9 Hz, 1H), 6.31 (s, 1H), 3.87 (s, 3H), 3.84 (s, 3H).
- Synthesis of 4-hydroxy-2(1-pyrrolyl)-7-methoxyquinoline (11b)

- A. A solution of the substrate 11a (obtained from compound 6c after hydrogenolysis of the benzyl group with 5% Pd/C in ethanol-THF) (1 g, 5.25 mmol) and 2,5-dimethoxytetrahydro furan (0.68 ml, 1 eq.) in glacial acetic acid was refluxed for 4.5 h and allowed to reach RT. The mixture was then concentrated under reduced pressure. The residue was diluted with methanol and NaOH(aq.) 1N was added until pH 7 is reached. The product was purified by chromatography using silica gel (3% MeOH/CH 2Cl2, the residue was pre-adsorbed on silica gel). 140 mg (13% yield) of 11b as a white solid was obtained.
- 1H NMR (400 MHz, DMSO-d): □ 7.98 (d, J=9.2 Hz, 1H), 7.64 (s, 2H), 7.18 (d, J=2.5 Hz, 1H), 7.05 (br. d, J=7.9 Hz, 1H), 6.88 (br. s, 1H), 6.32 (s, 2H), 3.90 (s, 3H).
- Synthesis of 4-hydroxy-7-methoxy-2-(6-methyl-2-pyridyl)quinoline (12d)

- A. 6-Methylpicolinic acid 12a (411 mg, 3.0 mmol) and SOCl 2 (0.520 mL, 7.2 mmol, 2.4 eq.) were refluxed in benzene (5 mL) for 2 h. The solvent and excess SOCl2 were removed from the reaction mixture under vacuum and the residue was triturated with pentane. The solid material formed was filtered off and the filtrate concentrated to give the acid chloride 12b (500 mg, 2.6 mmol).
- B. To a solution of the crude acid chloride 12b in CH 2Cl2 (5 mL) at 0° C., a solution of the aniline 8a (344 mg, 2.08 mmol), DIPEA (1.45 mL, 8.35 mmol) and DMAP (61 mg, 0.5 mmol) in CH2Cl2 (10 mL) was added. The reaction mixture was stirred a RT for 16 h. The volatile components were removed under vacuum, the residue was dissolved in EtOAc and the solution was washed with 5% NaHCO3 (2×), H2O and brine. The organic layer was then dried over MgSO4 and concentrated under vacuum. The mixture was purified by flash column chromatography, using EtOAc/hexane (1:2) as the eluent, to obtain the amide 12c (490 mg, 82%).
- C. To a suspension of amide 12c (490 mg, 1.71 mmol) in t-BuOH (10 mL), tBuOK (410 mg, 3.43 mmol) was added and the mixture was stirred at 75° C. for 6 h and then at RT for 16 h. The mixture was then poured in phosphate buffer (175 mL, pH=7) and stirred for 30 min. The solid was triturated twice with ethyl acetate. The organic phase was washed with brine, dried over MgSO 4 and concentrated under vacuum. The solid obtained was triturated with EtOAc to give the quinoline derivative 12d (263 mg, 58%). 1H NMR: (CDCl3, 400 MHz): □ 2.68 (s, 3 H), 3,94 (s, 3 H), 6.85-6.88 (2d, J=8.68 & 9.5 Hz, 2 H), 6.94 (dd, J=8.9 & 2.2 Hz, 1 H), 7.27 (dd, J=6.7 & 1.9 Hz, 1 H), 7.73-7.79 (m, 2 H), 8.28 (d, J=8.9 Hz, 1 H), 10.3 (br s, 1 H).
- Synthesis of 4-hydroxy-7-methoxy-2-(5-methoxy-2-pyridyl)quinoline (13d)

- A. To a solution of compound 13a (623 mg, 3.73 mmol) in MeOH, NaOH (2M, 4.70 mL) was added and the reaction mixture was stirred at RT for 2 h. The solution was then acidified with HCl (6N, 2.2 mL) and concentrated to obtain compound 13b, which was used in the following step without purification.
- B. To a solution of the crude compound 13b (˜3.73 mmol) in pyridine (25 mL), the aniline 8a (500 mg, 3.03 mmol) was added and the solution was cooled to −25 ° C. before POCl 3 (0.35 mL, 3.73 mmol) was added. The reaction mixture was stirred at −10° C. for 1 h and then at 0° C. for 2 h. The mixture was then poured onto H2O and extracted with EtOAc (2-3×). The combined organic layers were washed with 5% NaHCO3 and brine, dried over MgSO4 and concentrated under vacuum. The crude material was purified by flash column chromatography, using EtOAc/hexane (1:2) as the eluent, to give the amide 13c (617 mg, 55%).
- C. To a suspension of the amide 13c (617 mg, 2.05 mmol) in anhydrous t-BuOH (10 mL), tBuOK (490 mg, 4.11 mmol) was added and the mixture was stirred at 75° C. for 6 h and then at RT for 16 h. The reaction mixture was poured in phosphate buffer (175 mL, pH=7) and stirred for 30 min. The solid material formed was filtered and triturated with EtOAc to give the quinoline derivative 13d (250 mg, 43%). 1H NMR: (DMSO, 400 MHz): □ 3.86 (s, 3 H), 3.94 (s, 3 H), 6.72 (bs, 1 H), 6.91 (dd, J=8.9 & 1.9 Hz, 1 H), 7.54 (d, J=1.9 Hz, 1 H), 7.60 (dd, J=8.9 & 2.9 Hz, 1 H), 7.97 (d, J=8.9 Hz, 1 H), 8.21 (d, J=8.6 Hz, 1 H), 8.48 (d, J=1.9 Hz, 1 H).
- Synthesis of 4-hydroxy-7-methoxy-2-(oxazol-5-yl) quinoline (14c):

- A. The protected quinoline derivative 4b from Example 4 (3.8 g, 11.8 mmol) was dissolved in CH 2Cl2 (60 mL) and cooled to −78° C. before diisobutylaluminum hydride (7.9 mL, 1 equiv., 1.5M in toluene) was added very slowly over 15 min. After stirring for 80 min, an additional amount of DIBAL was added (5.5 mL, 0.7 equiv., 1.5 M in toluene). After stirring at −78° C. a further 2 h, the reaction was carefully quenched with methanol (4 mL) at −78° C. and then poured into aqueous solution of Rochelle salt (1N K—Na tartrate). The thick paste was stirred with CH2Cl2 (300 mL) for 2 h until clear. The phases were separated and the organic phase dried (MgSO4), filtered and concentrated to give a white solid. Purification by flash chromatography (SiO2, 230-400 mesh) with 50% EtOAc/hexane gave aldehyde 14a as a white solid (2.5 g, 73%).
- B. To a stirred suspension of K 2CO3 (48 mg, 0.34 mmol) in MeOH (7 mL) was added toluenesulphonylmethylisocyanide (66 mg, 0.34 mmol). The reaction was heated to 45° C. and aldehyde 14a (0.10 g, 0.34 mmol) was added. The reaction mixture was heated to 80° C. for 16 h and then concentrated to dryness under vacuum. Purification was performed by flash chromatography (SiO2, 230-400 mesh) to afford the desired oxazole 14b (0.089 g, 80%). MS: 331.0 (M+H)+.
- C. The MEM protected hydroxyquinoline 14b was dissolved in THF (3 mL) and treated with aqueous HCl (1N, 1 mL). The reaction was stirred for 30 min at RT before being concentrated to dryness under vacuum. The residue was treated with phosphate buffer (3 mL, 1 N solution, pH 4.5) and stirred before the product was filtered out, washed with distilled water and dried overnight under high vacuum (60° C., 16 h). The desired hydroxy quinoline 14c was obtained as a tan colored solid (0.065 g, 100%). MS: 242.9 (M+H) +.
- 1H NMR (DMSO-d6): □ 8.65 (s, 1H), 8.02 (bs, 1H), 7.97 (d, J=8.9 Hz, 1H), 7.19 (s, 1H), 6.93 (d, J=7.9 Hz, 1H), 6.42 (bs, 1H), 3.87 (s, 3H). ES (+) MS: m/z 242.9 (M+H)+.
- Synthesis of (2S)-N-Boc-amino-non-8-enoic acid (15g)

- A. To a solution of commercially available diethyl 2-acetamidomalonate 15a (100 g, 0.46 mole) in dioxane (500 mL) was added aqueous sodium hydroxide (1 M, 1 eq., 460 mL) dropwise over 30 to 45 min. The resulting mixture was left to stir for 16.5 h, then dioxane was evaporated in vacuo, the aqueous solution was extracted with three portions of 300 mL of ethyl acetate and acidified to pH 1 with concentrated HCl. This solution was left to crystallize in an ice-water bath. After the appearance of a few crystals, the mixture was sonicated and an abundant precipitate appeared. Filtration and drying under vacuum afforded compound 15b, (62.52 g, 72% yield) as a white solid.
- B. To a magnetically stirred emulsion of commercially available 7-octene-1,2-diol 15c (25 g, 0.173 mole) and H 2O (100 mL), in a 1 L round bottom flask, an aqueous solution of sodium periodate (40.7 g, 0.190 moles, 1.1 eq., in 475 mL H2O) was added over a period of 20 min (slightly exothermic). The resulting mixture was stirred at room temperature for an additional 1 h (completion of reaction confirmed by TLC). The mixture was then decanted in a separatory funnel and the aqueous layer was separated from the organic layer. The aqueous solution was saturated with NaCl, decanted and separated from the organic fraction once more. The two organic fractions were combined, dried with sodium sulfate and filtered over a cotton plug (in a Pasteur pipette) to give compound 15d (15.135 g, colorless oil, 78% yield). The aqueous solution was extracted with CH2Cl2, dried with anhydrous MgSO4, and concentrated under vacuum (without heating, heptanal b.p.153° C.) to obtain an additional amount of compound 15d (1.957 g, colorless oil, 10% yield). Total yield 88%.
- C. To solid ethyl 2-acetamidomalonate 15b (7.57 g, 40 mmol.) was added 6-heptenal 15d (4.48 g, 40 mmol) in solution in pyridine (32 mL, 10 eq) over 1 min. The resulting solution was cooled in a 10° C. bath and acetic anhydride (12 mL, 3.2 eq.) was added over 4 min. The resulting orange solution was stirred for 3 h at RT and another portion of ethyl 2-acetamidomalonate 15b (2.27 g) was added. The resulting mixture was stirred at room temperature for an extra 11 h. Ice (60 mL) was then added and the solution was stirred for 1.5 h, then the mixture was diluted with 250 mL of water and extracted with two portions of ether. The etheral solution was washed with 1N HCl, sat. NaHCO 3, dried Na2SO4, concentrated and purified by flash chromatography (EtOAc 40%/hexane) to give compound 15e (4.8 g, 50% yield) as a pale yellow oil.
- D. To a degassed (argon bubbling for 30 min.) solution of Z-ethyl 2-acetamido-2,8-nonadienoate 15e (8.38 g, 35 mmol) in dry ethanol (70 mL) was added (S,S)-Et-DUPHOS Rh(COD)OTf (51 mg, S/C=496). The mixture was put under 30 psi of hydrogen (after 4 vacuum-H 2 cycles) and stirred on a Parr shaker for 2 h. The resulting mixture was evaporated to dryness to obtain the crude compound 15f, which was used in the subsequent step without purification.
- E. To a solution of crude (S)-ethyl 2-acetamido-8-nonenoate 15f (7.3 g, 30.3 mmol) in THF (100 mL), Boc 2O (13.2 g, 2 eq.) and DMAP (740 mg, 0.2 eq) were added, and the reaction mixture was heated at reflux for 2.5 h. Subsequently, most of the THF solvent was evaporated, the crude mixture was diluted with CH2Cl2 and washed with 1 N HCl in order to remove the DMAP. The organic layer was further extracted with saturated aqueous NaHCO3, dried with anhydrous Na2SO4 and concentrated under vacuum. The crude product was then diluted with THF (50 mL) and water (30 mL), LiOH.H2O (2.54 g, 2 eq.) was added and the resulting mixture was stirred at RT for 25 h (completion of the hydrolysis was confirmed by TLC). The reaction mixture was concentrated under vacuum to remove most of the THF solvent and diluted with CH2Cl2. The resulting solution was washed with 1 N HCl, dried with anhydrous Na2SO4 and concentrated under vacuum. In order to remove minor impurities and excess Boc2O, the crude product was purified by flash chromatography (using a solvent gradient from 100% hexane—100% EtOAc as the eluent). The titled compound 15g was obtained in high purity as a pale yellow oil (5.82 g, 71% yield). 1H NMR (DMSO, 400 MHz): □ 7.01 (d, J=8 Hz, 1H), 5.79 (tdd, Jt=6.7 Hz, Jd=17.0, 10.2 Hz, 1H), 5.00 (md, Jd=17.0 Hz, 1H), 4.93 (md, Jd=10.2 Hz, 1H), 3.83 (m, 1H), 2.00 (q, J=6.9 Hz, 2H), 1.65-1.5 (m, 2H), 1.38 (s, 9H), 1.35-1.21 (m, 6H).
- Alternative synthesis of (2S)-N-Boc-amino non-8-enoic acid (a5g):

- A. To a stirred suspension of finely cut Mg ribbons (0.55 g, 22.5 mmol) in dry THF (30 mL) containing dibromoethane (0.1 mL), 8-bromo-1-octene (15h, 2.52 mL, 15 mmol) was added dropwise over a period of 15 min, [the reaction is slightly exothermic]. After 30 min, the mixture was heated to 38° for 1 h and then cooled to −78° before it was added via a cannula onto an excess amount of solid CO 2. The mixture was diluted with diethyl ether (100 mL) and the solution was washed with brine (2×50 mL), dried over MgSO4 and evaporated. A crude oil was obtained which was purified by chromatography on silica gel using 15% EtOAc in hexanes as the eluent to give compound 15i in 62% yield(1.44 g).
- 1H NMR (CDCl3, 400 MHz) δ 1.31-1.42 (m, 6H), 1.60-1.69 (m, 2H), 2.02-2.09 (m, 2H), 2.35 (t, J=8.3 Hz, 2H), 4.99 (dm, J=10.0 Hz, 1H), 5.04 (dm, J=17.0 Hz, 1H), 5.75-5.86 (m, 1H).
- B. To a vigorously stirring solution of the carboxylic acid 15i (1.36 g, 8.7 mmol) in anhydrous THF (70 mL) at −78°, freshly distilled Et 3N (1.6 mL; 11.3 mmol) and pivaloyl chloride (1.18 mL, 9.58 mmol) were added via a syringe under anhydrous conditions. The mixture was stirred at −78° for 15 min and then at 0° for 45 min. The mixture was cooled again to −78° and then transferred via a cannula into an anhydrous solution of 4(S)-4-(phenylmethyl)-2-oxazolidinone lithium salt in THF at −78°; the lithium salt of the oxazolidinone reagent had been previously prepared by the slow addition of n-BuLi (2.00 M in hexanes, 7.85 mL, 15.7 mmol) into a THF (20 mL) solution of the oxazolidinone (2.78 g, 15.7 mmol) in THF at −78°.
- The reaction mixture was stirred at −78° for 15 min then at RT for 1.5 h. Finally, it was quenched with an aqueous solution of sodium bisulfate (100 mL of 1 M) and the THF evaporated to ¾ of its initial volume. The residue was extracted with EtOAc (2×150 mL) and the mixed organic layers were washed with 5% NaHCO 3 (3×50 mL), brined (2×50 mL), dried over MgSO4 and evaporated. The resulting crude oil was chromatographed on silica gel, using 15% EtOAc in Hexanes to obtain compound 15j in 68% yield (1.88 g).
- 1H NMR (CDCl3, 400 MHz) δ 1.35-1.47 (m, 6H), 1.67-1.74 (m, 2H), 2.02-2.09 (m, 2H), 2.65 (dd, J=13.4 & 9.9 Hz, 1H), 2.84-3.02 (m, 2H), 3.31 (dd, J=13.4 & 3.2 Hz, 1H), 4.13-4.22 (m, 2H), 4.62-4.71 (m, 1H), 4.93 (d, J=10.2 Hz, 1H), 5.00 (dd, J=17.2 & 1.6 Hz, 1H), 5.75-5.84 (m, 1H), 7.18-7.38 (m, 5H).
- C. To a stirred solution of KHMDS (0.8 M THF, 22 mL, 17.5 mmol) in dry THF (50 mL) at −78° was cannulated a solution of the acid derivative 15j (3.25 g, 10.30 mmol) in dry THF (40 mL) at −78°. The mixture was stirred at −78° for 45 min. To this mixture, a solution of trisylazide (3.67 g, 11.85 mmol) in dry THF (40 mL) at −78° was added. The mixture was stirred at −78° for 3 min then quenched with acetic acid (5 mL). Subsequently, it was stirred at RT for 1 h and 45 min and finally at 40° for 15 min. Most of the THF was evaporated. The residue was taken into EtOAc (100 mL) and the organic solution washed with H 2O (50 mL), 5% NaHCO3 (3×50 mL) and brine (50 mL), (MgSO4) and evaporated. The oil obtained was chromatographed on silica gel using Hexane/CH2Cl2 (1/1) as the eluent to give compound 15k (2.47 g, yield 67%).
- 1H NMR (CDCl3, 400 MHz) δ 1.32-1.45 (m, 6H), 1.45-1.6 (m, 1H), 1.75-1.88 (2, 2H, rotamers), 2.01-2.11 (m, 2H), 2.82-2.87 (m, 1H), 3.33 (dd, J=13.4 & 3.2 Hz, 1H), 4.10-4.28 (m, 2H), 4.62-4.72 (m, 1H), 4.90-5.05 (m, 3H), 5.73-5.88 (m, 1H), 7.17-7.38 (m, 5H).
- D. To a stirred solution of anhydrous SnCl 2 (2.61 g, 13.8 mmol) in anhydrous MeOH (80 mL), a solution of the azide 15k (2.45 g, 6.9 mmol) was cannulated at 0° in anhydrous MeOH (20 mL). The mixture was stirred at RT for 4 h. The MeOH was evaporated and the foamy material obtained was taken into dioxane/H2O (100 μL/20 μL) and treated with Boc2O (3.0 g, 13.8 mmol) and NaHCO3 (2.89 g, 34.5 mmol) (pH adjusted to 8 with more NaHCO3 if needed) and the mixture was stirred at RT for 16 h. Part of the dioxane was evaporated (˜50%) and the residue was extracted twice with EtOAc. The organic solution was washed with brine (2×50 mL), dried and evaporated. The residue obtained was chromatographed on silica gel using 20-25% EtOAc in hexane as eluent to give the compound 15l (1.75 g, yield 60%).
- 1H NMR (CDCl3, 400 MHz) δ 1.27-1.53 (m, 6H), 1.46 (s, 9H), 1.80 (m, 1H), 2.00-2.08 (m, 1H), 2.80 (t, J=12.1 Hz, 1H), 3.34 (d, 14.3 Hz, 1H), 4.17-4.23 (m, 2H), 4.60-4.66 (m, 1H), 4.93 (d, J=10.2 Hz, 1H), 5.05 (dd, J=17.2 & 1.9 Hz, 1H), 5.13 (bs, 1H), 5.38-5.43 (m, 1H), 5.74-5.84 (m, 1H), 7.22-7.36 (m, 5H).
- E. To a stirred solution at 90° of the N-Boc derivative 15l (1.74 g, 4.04 mmol) in THF/H 2O (75 mL/15 mL), H2O2 (30% v/w, 2.05 mL, 16.2 mmol) and LiOH.H2O (0.34 g, 8.1 mmol) were added and the solution was stirred at 0° for 1 h. The reaction was quenched with Na2SO3 (2.24 g in H2O, 15 mL, 17.8 mmol). The pH was adjusted to 4-5 with 10% aqueous citric acid and the mixture diluted with EtOAc. The aqueous fraction was extracted once more with EtOAc and the organic solution was washed twice with brine, dried and evaporated. The residue was chromatographed on silica gel using 20% hexane in EtOAc as the eluent to give the free carboxylic acid 15g (0.76 g, yield 70%). This compound was identical in all respects to the one obtained in example 15.
- Synthesis of (2S)-N-Boc-amino-5-oxo-non-8-enoic acid methyl ester (16d):

- This synthesis is based on methodology by T. Tsuda et al., J. Am. Chem. Soc., 1980, 102, 6381-6384.
- A. To a well stirred solution of the monoallyl ester of malonic acid (1.50 g, 10.4 mmol) in dry THF under N 2 (20 mL) at −78° n-Bu2Mg (0.9M/hexane, 5.8 mL, 5.2 mmol) was added dropwise over a period of 5 min. The heavy suspension was then stirred at RT for 1 h and evaporated to dryness (vacuum release under N2). The solid Mg salt 16b, was dried under vacuum for 1 h.
- Glutamic acid derivative 16a was first mixed with 1,1′-carbonylidiimidazole (1.65 g, 10.21 mmol) in anhydrous THF and the mixture was stirred at RT for 1 h in order to activate the free acid moiety. Subsequently, the activated glutamic acid derivative was cannulated into a solution of the Mg salt 16b and the reaction mixture obtained was stirred at RT for 16 h. It was then diluted with EtOAc and the organic solution was washed with 0.5 N ice-cold HCl, brined, dried and evaporated. The residue obtained was chromatographed on silica gel using 35-40% EtOAc in hexane as eluent to give compound 16c (1.85 g, yield 53%).
- 1H NMR (CDCl3, 400 MHz) δ 1.44 (s, 9H), 1.85-1.95 (m, 1H), 2.12-2.22 (m, 1H), 2.58-2.74 (m, 2H), 3.48 (s, 2H), 3.74 (s, 3H), 4.24-4.34 (m, 1H), 4.52 (dm, J=5.7 Hz, 2H), 5.09 (m, 1H), 5.25 (dm, J=10.2 Hz, 1H), 5.34 (dm, J=17.2 Hz, 1H), 5.91 (m, 1H).
- B. To a stirred solution of tetrakis (triphenylphosphine) Pd (0) (0.116 g, 5 mol %, 0.1 mmole) in dry DMF (7 mL) was added (via a siring and under a N 2 atmosphere) the diester 16c (0.687 g, 2 mmol) in dry DMF (3 mL). The mixture was stirred at RT for 3.5 h. The DMF was evaporated under reduced pressure and the residue diluted with EtOAc (20 mL). The EtOAc solution was washed with 0.5 N ice-cold HCl (5 mL), brine (10 mL), dried and evaporated. The residue was chromatographed on silica gel using 15-20% EtOAc in hexane as eluent to give compound 16d (0.253 g, yield 42%).
- 1H NMR (CDCl3, 400 MHz) δ 1.44 (s, 9H), 1.84-1.94 (m, 1H), 2.08-2.22 (m, 1H), 2.33 (dd, J=14.0 & 7.3 Hz, 2H), 2.45-2.55 (m, 4H), 3.74 (s, 3H), 4.28 (bm, 1H) 4.98 (dm, J=10.2 Hz, 1H), 5.03 (dm, J=17.2 Hz, 1H), 5.00-5.10 (m, 1H), 5.74-5.85 (m, 1H).
- Synthesis of (2S,5R)-N-Boc-2-amino-5-methyl-non-8-enoic acid (17f)

- A,B,C,D. Commercially available (R)-(+)-citronellal 17a was first converted to the amino acid derivative 17b following the same synthetic steps as those previously described in Example 15 for the conversion of aldehyde 15d to amino acid intermediate 15f.
- E. Compound 17b (0.675 g, 5.6 mmol) was dissolved in a mixture of tBuOH/acetone/H 2O (1:1:1, 18 mL) and placed in an ice bath (0° C.). NMMO (0.789 g, 6.74 mmol, 1.2 eq.) and OSO4 (2.5% w/w in tBuOH, 0.7 mL, 0.067 mmol., 0.012 eq) were added consecutively and the reaction mixture was stirred at RT for 4 h. Most of the acetone was removed by evaporation under vacuum and then the mixture was extracted with EtOAc. The organic layer was further washed with H2O and brine, dried over anhydrous MgSO4 and evaporated to dryness. The diol 17c was obtained in high purity after flash column chromatography using 1% EtOH in EtOAc as the eluent in 77% yield (0.575 g).
- F. To a solution of diol 17c (0.575 g, 1.73 mmol) in THF/H 2O (1:1, 20 mL) at 0° C., NaIO4 (0.48 g, 2.25 mmol, 1.3 eq.) was added and the reaction mixture was stirred at RT for 3.5 h. Most of the THF solvent was subsequently removed by evaporation under vacuum and the remaining mixture was extracted with EtOAc (2×100 mL). The combined organic layers were further washed with 5% aqueous citric acid solution (2×20 mL), 5% aqueous NaHCO3 (20 mL) and brine (2×50 mL), then the EtOAc solution was dried over anhydrous MgSO4 and evaporated to dryness under vacuum. The aldehyde intermediate 17d (0.47 g of crude product) was used in the next step without further purification.
- G. To a solution of Ph 3PCH3Br (925 mg, 2.6 mmol) in anhydrous toluene (15 mL), KHMDS (0.5M in toluene, 5.2 mL, 2.6 mmol) was added and the yellow suspension formed was stirred at RT for 30 min under N2. After that period, the suspension was first cooled to 0° C., a solution of the aldehyde 17d (0.47 g 1.73 mmol, dissolved in 15 mL of anhydrous THF) was added via a syringe and the mixture was allowed to warm-up to RT. After stirring at RT for 1 h, most of the THF was removed by evaporation under vacuum, EtOAc (100 mL) was added to the mixture and the organic layer was washed with H2O (30 mL), 5% aqueous NaHCO3 (30 mL) and brine (30 mL). The EtOAc solution was then dried over anhydrous MgSO4 and evaporated to dryness under vacuum. Pure compound 17e was isolated after purification by flash column chromatography on silica gel, using hexane:EtOAc (3:2) as the eluent, in 63% yield (0.29 g) for the two last steps.
- The hydrolysis of the ethyl ester and simultaneous exchange of the N-acetyl protecting group for a Boc in intermediate 17e to obtain compound 17f was carried out using the same procedure as that reported for the conversion of compound 15f to 15g (17f, 310 mg, quantitative). 1H NMR (CDCl3, 400 MHz): □ 0.88 (d, J=6.4 Hz, 3H), 1.18-1.28 (m, 2H), 1.35-1.48 (m, 3H), 1.45 (s, 9H), 1.64-1.74 (m, 1H), 1.81-1.89 (m, 1H), 1.94-2.12 (m, 2H), 4.28 (bd, J=˜3.2 Hz, 1H), 4.93 (dm, J=11.1 Hz, 1H), 5.00 (dm, J=16.8 Hz, 1H), 5.74-5.84 (m, 1H).
- Synthesis of N-Boc-O-allyl-(L)-threonine (18d)

- A. Boc-(L)-threonine 18a (500 mg, 2.28 mmol) was partially dissolved in CH 2Cl2/MeOH (8 mL/0.5 mL, respectively) at 0° C. A solution of diazomethane in diethyl ether was slowly added until the yellow color persisted, indicating the presence of excess diazomethane. Upon evaporation of the solvents, crude methyl ester 18b was obtained as a cloudy white oil (0.534 g).
- B. Intermediate 18b (311 mg, 1.33 mmol) was then dissolved in anhydrous diethyl ether (8 mL), Ag 20 was added (341 mg, 1.47 mmol) and freshly activated 4 Å molecular sieves (1 g). Finally, allyl iodide (134 μL, 1.47 mmol) was added to the reaction flask and the mixture was stirred at reflux. Two more portions of allyl iodide (45 □L, 0.50 mmol, each time) were added after a period of 20 h and 30 h, and stirring was continued for a total of 36 hours. Then the mixture was filtered through celite and purified by flash column chromatography on silica gel, using EtOAc/hexane (1:4) as the eluent, to give 73 mg (27% yield) of compound 18c as a clear oil.
- 1H NMR (CDCl3, 400MHz): δ 1.21 (d, J=6.0 Hz, 3H), 1.45 (s, 9H), 3.75 (s, 3H), 3.82-3.87 (m, 1H), 3.99-4.07 (m, 2H), 4.29 (dd, J=9.5 & 2.5 Hz, 1H), 5.14 (dm, J=10.5 Hz, 1H), 5.21 (dm, J=17.2 Hz, 1H), 5.75-5.84 (m, 1H).
- C. The ester compound 18c (99 mg, 0.362 mmol) was dissolved in a mixture of THF/MeOH/H 2O (2:1:1, 4 mL) and LiOH.H2O (61 mg, 1.45 mmol) was added. The solution was stirred at RT for 2 h, and was then acidified with 1N HCl to pH ˜3 before the solvents were removed under vacuum. The resulting oil, compound 18d was used as such for the synthesis of macrocyclic inhibitors.
- Synthesis of (2S, 3S)-N-Boc-2-amino-3(mercaptoallyl)butanoic acid (19e)

- A. Compound 19a (9.1 mmol) was dissolved in pyridine (5 mL) and the solution was cooled to 0° C. in an ice bath, tosyl chloride (2.3 g, 11.8 mmol, 1.3 eq.) was added in small portions and the reaction mixture was stirred at RT for 24 h. After that period, the reaction mixture was partitioned between diethyl ether (300 mL) and H 2O (100 mL). The ether layer was further washed with 0.2N HCl (6×100 mL) and brine (100 mL), dried over anhydrous MgSO4, filtered and concentrated to dryness under vacuum. Purification of the crude material by flash column chromatography, using hexane/EtOAc (gradient from 8:2 to 7:3 ratio) as the eluent, led to the isolation of tosyl derivative 19b in 85% yield (3.05 g).
- B. To solution of intermediate 19b (775 mg, 2 mmol) in anhydrous DMF (2.5 mL), potassium thioacetate (365 mg, 3.2 mmol, 1.6 eq.) was added and the reaction mixture was stirred at RT for 24 h. Most of the DMF was then evaporated under vacuum and the remaining mixture was partitioned between EtOAc and H 2O. The aqueous layer was re-extracted with EtOAc, the combined organic layers were washed with brine, dried over anhydrous MgSO4 and evaporated to dryness. Purification of the crude material by flash column chromatography using hexane/EtOAc (4:1 ratio) as the eluent, led to the isolation of compound 19c in 80% yield (465 mg).
- C. To a solution of thioester 19c (465 mg) in H 2O/EtOH (3:5 ratio, 8 mL), an aqueous solution of 0.2M NaOH (2.4 mL) was added and the mixture was stirred at RT for 1.5 h. Allyl iodide (0.292 mL, 3.2 mmol, 2 eq.) was then added and stirring was continued at RT for an additional 30 min. The reaction mixture was concentrated to half of its original volume and then extracted with EtOAc. The aqueous layer was acidified to pH=˜3 with cold aqueous 0.5N HCl and re-extracted with EtOAc. The combined organic layers were washed with brine, dried over anhydrous MgSO4 and evaporated to dryness under vacuum. The crude reaction mixture contained at least four products; all of the products were isolated after flash column chromatography on silica gel, using hexane/EtOAc (gradient from 9:1 to 3:1 ratio). The structure of the least polar compound (TLC Rf=0.68 in hex/EtOAc 4:1) corresponded to the desired product 19d (83 mg, 18% yield).
- 1H NMR (CDCl3, 400 MHz): □ 1.24 (d, J=7.0 Hz, 3H), 1.46 (s, 9H), 3.13-3.19 (m, 2H), 3.24-3.29 (m, 1H), 3.77 (s, 3H), 4.50 (dd, J=8.6 & 3.8 Hz, 1H), 5.12 (d, J=12.4 Hz, 1H), 5.15 (dd, J=18.4 & 1.3 Hz, 1H), 5.22 (bd, J=7.6 Hz, 1H), 5.75-5.85 (m, 1H).
- D. A solution of the methyl ester 19d (83 mg, 0.287 mmol) in MeOH/H 2O (3:1, 4 mL) was mixed with aqueous NaOH (0.2 N, 1.3 mL, 0.26 mmol) for 24 h at RT and for 1 h at 40° C. The reaction mixture was acidified with cold aqueous HCl (0.5 N HCl, at 0° C., pH=4-5), the MeOH was removed under vacuum and the remaining aqueous mixture was extracted with EtOAc. The organic solution was dried over MgSO4 and evaporated to dryness in order to obtain compound 19e. Compound 19e was used for the final synthesis of inhibitors without any further purification.
- Synthesis of (S)-N-Boc-2-amino-3-methyl-3(1-mercapto-4-butenyl)butanoic acid (20c)

- A. L-Penicillamine 20a (448 mg, 3 mmol) was dissolved in DMF/DMSO (5:1 ratio, 6 mL), 4-bromopentene (0.46 mL, 4.5 mmol, 1.5 eq.) and CsOH.H 20 (1.0 g, 6 mL, 2 eq.) were added and the reaction mixture was stirred at RT. After 24 h, Boc2O (820 mg, 3.75 mmol, 1.25 eq.) was added to the mixture and stirring was continued for an additional 12 h. The DMF was subsequently removed under vacuum, the remaining mixture was diluted with cold aqueous 0.5N HCl adjusting the pH=˜4-5 and then extracted with EtOAc (2×50 mL). The organic layer was washed with brine (2×), dried over anhydrous MgSO4 and evaporated to dryness to give the crude carboxylic acid 20b.
- B. Purification of 20b turned out to be difficult, thus the crude product was first treated with diazomethane to form the corresponding methyl ester 20c, and then purified by flash column chromatography, using hexane/EtOAc (9:1) as the eluent, to obtain 190 mg (20% yield) of the pure methyl ester 20c.
- 1H NMR (400 MHz, CDCl3): □ 1.35 (s, 3H), 1.37 (s, 3H), 1.44 (s, 9H), 1.59-1.67 (m, 2H), 2.11-2.17 (m, 2H), 2.51-2.60 (m, 2H), 3.74 (s, 3H), 4.29 (d, J=8.6 Hz, 1H), 4.98 (dm, J=10.5 Hz, 1H), 5.03 (dm, J=19 Hz, 1H), 5.35 (bd, J=7 Hz, 1H), 5.72-5.83 (m, 1H).
- C. The ester was subsequently dissolved in THF/MeOH/H 2O (2:2:1, 5mL), LiOH.H2O (50 mg, 2.0 mmol, 2 eq.) was added and the reaction mixture stirred at 40° C. for 4 h to hydrolyze the ester 20c back to the acid 20b. The reaction mixture was acidified with 0.5N HCl to pH=4-5, the THF and MeOH were evaporated to dryness and the remaining aqueous solution was extracted with EtOAc. The EtOAc layer was dried over anhydrous MgSO4, and evaporated to dryness to give compound 20b, which was used in the subsequent synthesis of macrocyclic inhibitors without further purification.
- The general procedure for coupling reactions done in solution and specific examples thereof are described in WO 00/09543 and WO 00/09558.
- These procedures have been used for the synthesis of the intermediate dipeptides 26c, 30a and tripeptides 23a, 24a, 31a, 32a, and 33a.
- Synthesis of Acyclic Tripeptide 21e

- A. To a solution of the proline derivative 21a (prepared from commercially available Boc-4(R)-hydroxyproline and 4-chloro-quinoline as described in WO 00/05543 and WO 00/09558) (1.32 g, 3.68 mmol) and the crude homoallyl ACCA 1f (˜3.35 mmol) in CH 2Cl2 (10 mL), NMM (1.21 mL, 10.05 mmol) and HATU (1.53, 4.02 mmol) were added in succession and the suspension was stirred at RT for 18 h. After that period, the solvent was evaporated and the crude reaction mixture was redissolved in EtOAc (30 mL). The solution was washed with 5% aqueous NaHCO3 (2×10 mL), brine (10 mL), dried over MgSO4 and evaporated. The crude product was purified my chromatography on silica gel using 8% diethyl ether in EtOAc as the eluent to obtain the desired diastereomer of compound 21b in 20% yield (the absolute stereochemistry was not determined).
- 1H NMR (CDCl3, 400 MHz): δ 0.93 & 1.01 (t, J=8.3 Hz, 1H, rotamers in ratio of 3:7), 1.14-1.35 (m, 2H), 1.44 (s, 9H), 1.45 (s, 9H), 1.50-1.82 (m, 4H), 2.08-2.24 (m, 2H), 2.32 (bs, 0.7H), 2.63 (bs, 0.75H), 2.93 (bs, 0.75H), 3.16 (m, 0.25H), 3.77 (bs, 1.5H), 3.88 (bs, 0.5H), 4.4-4.55 (m, 1H), 4.98 (d, J=10.2 Hz, 1H), 5.03 (dd, J=17.2 & 1.6 Hz, 1H), 5.24 (bs, 1H), 5.75-5.88 (m, 1H), 6.57 & 6.78 (2bs, 1H, 2 rotamers), 7.42-7.58 (m, 3H), 7.63-7.73 (m, 2H), 8.04 (d, J=8.3 Hz, 1H), 8.11 (d, J=8.3 Hz, 1H), 8.74 (d, J=5.1 Hz, 1H).
- B. To a solution of the dipeptide 21b (137 mg, 0.248 mmol) in dry CH 2Cl2, a solution of HCl in dioxane (4M, 4 mL) was added and the mixture was stirred at RT for 1.5 h. The solvent was then evaporated and the residue dried under high vacuum to give the free amino acid. The mixture was dissolved in diethyl ether/MeOH (3 μL/2 μL) and treated with a slight excess of diazomethane dissolved in diethyl ether. After 30 min, the excess diazomethane was destroyed with the addition of HCl (4M in dioxane) and the mixture was evaporated to dryness to obtain the HCl salt of compound 21c which was used in the next step without any purification.
- C. To a stirred suspension of the crude dipeptide 21c (0.23 g, 0.48 mmole) in CH 2Cl2 (25 mL) was added in succession the (2S)-N-Boc-amino-hept-6-enoic acid 21d (0.151 g, 0.62 mmol), NMM (210 μL, 1.91 mmol) and HATU (0.236 g, 0.62 mmole) and the mixture was stirred at RT for 16 h (the pH was adjusted to ˜8 with NNM after 1 h if needed). The CH2Cl2 was evaporated, the residue taken into EtOAc (50 mL) and the organic solution washed with 5% NaHCO3 (2×20 mL), brine (2×20 mL), dried and evaporated. The crude compound obtained was chromatographed on silica gel (50 mL, 2% EtOH/EtOAc) to give compound 21e (0.139 g, yield 46%).
- 1H NMR (CDCl3, 400 MHz, rotamers in 6:1 ratio) chemical shifts of major rotamer δ 1.21-1.27 (m, 1H), 1.36 (s, 9H), 1.45-1.81 (4m, 7H), 2.20-2.22 (m, 4H), 2.28-2.37 (m, 1H), 2.90-2.99 (m, 1H), 3.66 (s, 3H), 3.94-3.98 (m, 1H), 4.29 (bd, J=9.9 Hz, 1H), 4.46-4.50 (m, 1H), 4.81 (dd, J=8.3 & 5.4 Hz, 1H), 4.92-5.06 (m, 4H), 5.16 (d, J=8.3 Hz 1H), 5.37 (m, 1H), 5.70-5.84 (m, 2H), 6.82 (d, J=5.1 Hz, 1H), 7.47-7.55 (m, 2H), 7.71 (dt, J=7.0 & 1.3 Hz, 1H), 8.03 (d, J=8.6 Hz, 1H), 8.17 (d, J=8.0 Hz, 1H), 8.78 (d, J=5.1 Hz, 1H).
- General Procedure for Macrocyclization Via Olefin Metathesis
- In all cases, the tri-peptide diene was dissolved in CH 2Cl2 at a concentration of 0.01M and the solution was deoxygenated by the bubbling of argon (˜1 h for a volume of 500 mL). A solution of catalyst (5-30 mol %, dissolved in a small amount of degassed CH2Cl2) is added and the reaction mixture is refluxed until all starting material was converted to product(s) as indicated by TLC and HPLC. The crude reaction mixtures were subsequently concentrated to near dryness and filtered through a short pad of silica gel, eluting first with CH2Cl2 to remove most of the catalyst and then with EtOAc in order to elute all of the macrocyclic produc(s) (most of the time as a single diastereomer). The crude product(s) from each reaction is analyzed by chiral HPLC on a CHIRALCEL OJ-R column (purchased from Chiral Technologies Inc, 0.46 □×15 cm), using an isocratic solvent mixture of 70% H2O+0.06% TFA−30% CH3CN+0.06% TFA at 205 nm. The major macrocyclic product(s) was fully characterized by: 1H, COSY, TOCSY, and ROESY NMR data in order to confirm its structure and stereochemistry.
- Synthesis of Macrocyclic Intermediate (23b)

- A solution of diene 23a (4.0 g, 7.88 mmol) in dry CH 2Cl2 (800 mL, Aldrich-anhydrous) was deoxygenated by bubbling Ar for 2 h. Hoveyda's catalyst (262 mg, 0.434 mmol, 5.5 mol %) was then added as a solid and the reaction was refluxed under an Ar balloon. After 28 h, the red-orange solution was evaporated to an amorphous solid and then purified by flash column chromatography over silica gel. The initial solvent system was 10% EtOAc in CH2Cl2. Once the catalyst was eluted from the column, the solvent was changed to pure EtOAc. Elution of the catalyst from the column was evident from its color. The macrocyclic product 23b was isolated as a colorless foam which was re-dissolved in CH2Cl2/hexane (˜1:2). Evaporation of the solvent afforded a white powder (3.362 g, 89% yield).
- !H NMR (CDCl3, 400 MHz): δ 1.20-1.50 (m, 6H), 1.43 (s, 9H), 1.53 (dd, J=9.5 & 5.4, 1H), 1.61-1.70 (m, 1H), 1.76-1.90 (m, 2H), 2.05-2.26 (m, 4H), 2.45 (d, J=14.3, 1H), 3.67 (s, 3H), 3.71 (d, J=11.1, 1H), 3.90 (dd, J=11.1 & 4.3, 1H), 4.43-4.53 (m, 2H), 4.76 (d, J=8.6, 1H), 4.86 (bd, J=9.8, 1H), 5.20-5.23 (m, 2H), 5.57 (dt, J=7.0 & 9.8, 1H), 7.32 (bs, 1H).
- Synthesis of Macrocyclic Intermediate (24b):

- A solution of diene 24a (2.76 g, 3.82 mmol) in anhydrous CH 2Cl2 (600 mL, anhydrous) was deoxygenated by bubbling Ar for 1.5 h. A solution of Hoveyda's catalyst (117 mg, 0.19 mmol, 0.05 eq) in anhydrous and degassed CH2Cl2 (8 mL) was added via cannula and the reaction was stirred at reflux under an Ar balloon. After 20 h, the reaction mixture was approximately 50% completed, at which point a second portion of catalyst was added (117 mg) and the stirring was continued for an additional 16 h. The solution was then concentrated to ˜100 mL, applied to the top of a pad of silica gel (6×10 cm) and the catalyst was first recovered by eluting with CH2Cl2. Compound 24b was washed off the pad of silica with 3% MeOH in EtOAc and re-purified by flash column chromatography using EtOAc/hexane (2:1) to obtain 70% yield of a slightly olive-tinted white solid (1.85 g, 94% pure by HPLC).
- 1H NMR (400 MHz, DMSO-d6) □ 8.69 (s, 1H), 8.13 (d, J=9.2 Hz, 1H), 7.50-7.44 (m, 2H), 7.17 (dd, J=9.2, 2.2 Hz, 1H), 7.04 (d, J=6.4 Hz, 1H), 5.60-5.56 (m, 1H), 5.52 (dd, J=9.2 Hz, 1H), 5.25 (dd, J=9.2 Hz, 1H), 4.59 (d, J=11 Hz, 1H), 4.44 (dd, J=9.2 Hz, 1H), 4.05-3.98 (m, 1H), 3.94 (s, 3H), 3.92 (s, 3H), 3.89-3.82 (m, 1H), 3.55 (s, 3H), 2.64-2.53 (m, 1H), 2.46 (d, J=7.3 Hz, 1H), 2.40-2.31 (m, 1H), 2.21 (dd, J=8.9 Hz, 1H), 1.78-1.65 (m, 2H), 1.55 (dd, J=4.8 Hz, 1H), 1.485 (dd, J=4.8 Hz, 1H), 1.41-1.30 (m, 7H), 1.16 (s, 9H). MS; es+: 795.4 (M+H)+.
- Synthesis of Compound 202 & 203 (Table 2)

- A. The diene compound 21e (0.130 g, 0.205 mmol) was cyclized using catalytic amounts of bis-(tricyclohexylphosphine) benzylidene ruthenium IV dichloride (Grubb's catalyst, supra) (52 mg, 0.064 mmol) in CH 2Cl2 (60 mL) under reflux for 2 h to give after chromatography on silica gel (50 mL, 3% EtOH/EtOAc) compound 25a (60.1 mg, yield 48%).
- 1H NMR (CDCl3, 400 MHz) δ 1.22-1.30 (m, 2H), 1.35 (s, 9H), 1.44-2.35 (m, 13H), 3.07-3.14 & 3.16-3.24 (2m, 1H, rotamers in 1:3 ratio), 3.69 (s, 3H), 3.96-4.04 (m, 1H0, 4.42-4.50 (m, 1H), 4.95-5.04 (m, 1H), 5.05-5.15 (m, 1H), 5.20-5.30 (m, 1H), 5.55-5.65 (m, 1H), 6.75-6.79 (2d, J=5.4 Hz, 1H, rotamers in 1:3 ratio), 7.36 (s, 1H), 7.46-7.50 (m, 1H), 8.03 (d, J=8.3 Hz, 1H), 8.13 & 8.17 (2d, J=8.0 Hz, 1H, rotamers in 1:3 ratio), 8.77 (d, J=5.1 Hz, 1H).
- B. The ester moiety of the macrocyclic compound 25a (0.0156 g, 0.026 mmol) was hydrolyzed with LiOH.H 2O (8.7 mg, 0.206 mmol) in THF/MeOH/H2O (4 mL /2 mL/2 mL). The crude product was purified by C18 reversed phase HPLC on a Whatman (Partisil 10,0DS3) 50/2.4 cm column using a solvent gradient from 5% aqueous CH3CN to 100% CH3CN to obtain pure compound 202 as an amorphous white solid (11.8 mg).
- 1H NMR (DMSO, 400 MHz): δ 1.12 (s, 9H), 1.20-1.24 (m, 2H), 1.32-1.40 (m, 3H), 1.58-1.62 (m, 2H), 1.68-1.78 (m, 3H), 1.95-2.02 (m, 1H), 2.08-2.18 (m, 2H), 2.42-2.59 (m, 2H), 3.97-4.00 (bd, J=9.8 Hz, 2H), 4.47 (t, J=8.6 Hz, 1H), 4.58 (d, J=11.8 Hz, 1H), 5.22-5.29 (m, 1H), 5.46-5.54 (m, 1H), 5.66 (s, 1H), 7.12 (d, J=6.0 Hz, 1H), 7.49 (d, J=3.5 Hz, 1H), 7.68 (dd, J=7.3 Hz, 1H), 7.98 (dd, J=7.0 Hz, 1H), 8.08 (d, J=8.3 Hz, 1H), 8.21 (s, 1H), 8.35 (d, J=8.3 Hz, 1H), 9.08 (d, J=5 Hz, 1H).
- C. The macrocyclic compound 25a (20 mg, 0.033 mmol) in dry CH 2Cl2 (1 mL) was stirred in presence of 4M HCl/dioxane (5 mL) for 1 h. The mixture was evaporated and dried carefully. The residue was re-dissolved in CH2Cl2/DMF (3 mL/1 mL) and treated with NMM (14.5 μL, 0.132 mmol) and acetic anhydride (7.0 μL, 0.073 mmol) and stirred at RT for 14 h. The mixture was evaporated and dried under high vacuum. The residue was then dissolved in a mixture of THF/MeOH/H2O (4 mL/2 mL/2 mL) and stirred overnight with LiOH.2H2O (11 mg, 0.264 mmol). The residue isolated after acidification to pH=3 with 1N ice-cold HCl was purified by C18 reversed phase HPLC using a solvent gradient from 0-40% aqueous CH3CN (0.06% TFA) in order to isolated pure compound 203 as an amorphous white solid (12 mg).
- 1H NMR (50 mM Na2PO4 buffer, pH=6.0, 600 MHz): δ 1.22-1.27 (m, 2H), 1.38-1.43 (m, 2H), 1.58-1.64 (m, 2H), 1.67-1.76 (m, 2H), 1.77-1.84 (m, 1H), 1.92-1.99 (m, 1H), 2.22-2.08 (m, 1H), 2.12-2.27 (m, 1H), 2.22-2.27 (m, 1H), 2.60-2.67 (m, 1H, Pro-β′), 2.83-2.89 (m, 1H, Pro-β), 4.32 (dd, J=12.1 & 3.5 Hz, 1H, Pro-δ′), 4.41 (dd, J=12.1 & 7.3 Hz, 1H), 4.56 (bd, J=8.0 Hz, 1H, Pro-δ), 4.62 (dd, J=8.9 Hz, 1H, Pro-α), 5.40-5.46 (m, 1H), 5.55-5.61 (m, 1H), 5.73 (bs, 1H, Pro-γ), 7.41 (d, J=6.3 Hz, 1H), 7.64 (bs, 1H, Acca-NH), 7.80 (dd, J=7.9 Hz, 1H), 8.03 (dd, J=8.0 Hz, 1H), 8.07 (d, J=9.5 Hz, 1H), 8.16 (d, J=7 Hz, 1H, AcNH), 8.36 (d, J=8.3 Hz, 1H), 8.90 (d, J=6.0 Hz, 1H).
- Synthesis of Compound 508 (Table 5)


- A. A solution of Boc-protected L-glutamine 26a (4.93 g, 20 mmol) and iodobenzene diacetate (7.73 g, 24 mmol, 1.2 eq.) in EtOAc/CH 3CN/H2O (2:2:1, 60 mL), was stirred at 16° C. for 1 h and at 20° C. for 3 h. The reaction mixture was then diluted with H2O (20 mL), the EtOAc and CH3CN solvents were removed under vacuum and the remaining aqueous mixture was extracted with diethyl ether (3×50 mL) and EtOAc (50 mL) in order to remove most of the impurities. The aqueous layer (containing the amine intermediate) was then concentrated to dryness, the remaining material was re-dissolved in 10% Na2CO3 (30 mL), cooled to 0° C. in an ice bath and a solution of benzyl chloroformate (3.3 mL, 20.4 mmol, 1.02 eq.) dioxane (40 mL) was slowly added (˜10 min). The reaction mixture was stirred at 0° C. for 1 h and at RT for 2 h. The mixture was then diluted with H2O (50 mL), extracted with cold (˜5° C.) diethyl ether (3×50 mL), acidified with 4M HCl to pH=3-4 and extracted with EtOAc (3×50 mL). The combined organic layers were dried over anhydrous MgSO4 and evaporated to dryness under vacuum. The crude material was purified by flash column chromatography, using EtOAc/Hexane/AcOH (7:2.9:0.1) to obtain compound 26b in 43% overall yield (3.04 g).
- B. Dipeptide intermediate 26c (250 mg, 0.41 mmol), compound 26b (171 mg, 0.49 mmol, 1.2 eq.) and HATU (185 mg, 0.49 mmol, 1.2 eq.) were dissolved in CH 2Cl2 (6 mL) and DIPEA (0.29 mL, 1.62 mmol, 4 eq.) was added. The reaction mixture was stirred at RT for 14 h, then the CH2Cl2 was evaporated under vacuum and the crude material re-dissolved in EtOAc. The EtOAc solution was washed with aqueous 5% NaHCO3 and brine, dried over anhydrous MgSO4 and evaporated to dryness. Compound 26d was obtained after purification of the crude material by flash column chromatography, using EtOAc/hexane (4:1) as the eluent, in 98% yield (338 mg).
- C. A solution of compound 26d (335 mg, 0.394 mmol) in THF (5 mL) was cooled to 0° C. and a solution of BH 3 in dimethyl sulfide (0.12 mL of 10M solution, 1.2 mmol, 3 eq.) was added. The reaction mixture was allowed to warm-up to RT and stir for 1 h. Then it was cooled again to 0° C. before an aqueous solution of NaOH (0.8 mL of 2.5 M solution, 1.97 mmol, 5 eq) was added slowly over a period of 15 min, followed by the slow addition (˜15 min) of an aqueous solution of H2O2 (0.8 mL of an 8.8 M solution, 6.9 mmol, 17.5 eq.). The reaction mixture was allowed to warm-up to RT and stir for 1 h. After that period, the reaction mixture was acidified to pH ˜4 in order to quench the excess BH3, then aqueous NaHCO3 was added to adjust the pH=˜9-10, the THF was removed under vacuum and the crude material was partitioned between H2O and EtOAc. The aqueous layer was re-extracted with EtOAc, the combined organic layers were washed with brine, dried over anhydrous MgSO4 and evaporated to dryness under vacuum. The crude material was purified by flash column chromatography, using EtOAc/hexane/NH4OH (8:2:0.5) as the eluent, to obtain pure compound 26e in 57% yield (192 mg).
- D. To a solution of compound 26e in CH 2Cl2 (8 mL), Dess-Martin periodinate (195 mg, 97%, 0.33 mmol, 1.5 eq) was added and the reaction mixture was stirred at RT for 1.5 h. The reaction was quenched with the addition of aqueous Na2S2O3 (3 mL of 5% solution), then saturated aqueous NaHCO3 (5 mL) was added and the mixture was stirred at RT for 15 min. Finally, the reaction crude was extracted with EtOAc, the organic layer was washed with aqueous 5% NaHCO3 and brine, dried over anhydrous MgSO4 and evaporated under vacuum to give 188 mg of aldehyde 28f which was used in the next step without further purification.
- E. A solution of compound 26f (188 mg, 0.22 mmol), CH 3CO2H (38 μL) and Pd(OH)2 (25 mg) in ethanol (5 mL) was stirred to RT under H2 at atmospheric pressure for 16 h. After that period, more H2 gas, Pd(OH)2 (180 mg) and CH3CO2H (154 μL) and were added to the flask and stirring was continued for an additional 24 h. The mixture was then filtered and the solvent evaporated to dryness, the crude macrocyclic product was purified by flash column chromatography, using CHCl3/MeOH/AcOH (10:2:1), to obtain compound 26g in ˜30% yield (48 mg).
- F. A mixture of compound 26g (22 mg, 0.031 mmol), DIPEA (27 μL, 0.155 mmol, 5 eq.) and acetic anhydride (8.7 μL, 0.093 mmol, 3 eq.) in CH 2Cl2 (5 mL) was stirred at RT for 16 h. The CH2Cl2 was then removed under vacuum, a mixture of THF/MeOH/H2O (2:2:1, 5 mL) and LiOH.2H2O (13 mg, 0.31 mmol, 10 eq.) were added and the hydrolysis reaction was allowed to proceed for 68 h at RT and 2 h at 50° C. The reaction mixture was then acidified (pH=˜4) and purified by reversed phase HPLC to obtain the final compound 508 (˜6 mg, ˜26% yield for the last 2 steps).
- 1H NMR (DMSO, 400 MHz) of 508 (mixture of rotamers confirmed by COSY, TOCSY and ROESY NMR data): δ 1.18 (s, 9H), 1.09-1.85 (overlapping m, 11H), 1.95 (s, 3H), 2.30 (m, 1H), 2.63 (m, 1H), 3.18-4.14 (overlapping m, 6H), 3.96 (s, 3H), 4.44 (m, 1H), 4.62 & 4.69 (2d, J=11.8 Hz, 1H, rotamers), 5.82 (bs, 1H), 7.20 (m, 2H), 7.53 (bs, 1H), 7.67 (bs, 4H), 8.19 (bs, 3H), 8.61 (s, 1H).
- Synthesis of the Saturated Macrocyclic Intermediate (27a)

- A. The unsaturated macrocyclic intermediate 23b (3.50 g, 7.30 mmol) was dissolved in EtOAc (30 mL) and 700 mg (20% w/w) of 5% Rh on alumina was added. The mixture was stirred under H 2 gas at atmospheric pressure and at RT for 1.5 h. After that period, HPLC analysis confirmed the complete conversion of starting material to two products, the desired product 27a and a minor product (8% of the total mass) which was later identified to be compound 27b, formed from opening of the cyclopropane ring. The reaction mixture was filtered and concentrated to give a light green color solid (3.47 g). The solid was co-evaporated twice with EtOH to remove all of the EtOAc (the presence of EtOAc interferes in the next step). Separation of compound 27a from 27b by chromatography proved to be very difficult, thus an alternative method was devised based on the relative rates of hydrolysis of their respective methyl ester moieties.
- B. The crude mixture of compounds 27a and 27b (3.47 g) was dissolved in THF:MeOH (1:1, 20 mL), an aqueous solution of LiOH.H 2O (24 mg in 5 mL H2O, 8% eq) was added and the reaction mixture was stirred at RT for 16 h (complete hydrolysis of the side product 27b to its corresponding acid 27c was confirmed by HPLC). The reaction mixture was concentrated under vacuum in order to remove most of the THF and MeOH and partitioned between H2O (100 mL) and EtOAc (300 mL). The organic layer was washed with 0.5 N NaOH (3×100 m), brine (100 mL), 10% aqueous citric acid (2×100 mL), brine (100 mL), dried over anhydrous MgSO4, filtered and concentrated to dryness. The desired product 27a was obtained in high purity (>90% by HPLC) as a light green foam and in 93% overall yield (3.28 g) for the two steps.
- 1H NMR: (400 MHz, CDCl3): □ 1.1-1.38 (m, 13 H), 1.42 (s, 9 H), 1.51-1.57 (m, 1H), 1.63-1.67 (dd, J=8.0 & 5.1 Hz, 1 H), 1.81-1.87 (m, 1 H), 1.92-1.99 (m, 1 H), 2.02-2.08 (m, 1 H), 2.62 (d, J=14 Hz, 1 H), 3.4 (d, J=8.3, 1H), 3.65 (s, 3H), 4.01 (dd, J=10.8 & 4.1 Hz, 1 H), 4.42-4.48 (m, 1 H), 4.51-4.55 (m, 1 H), 4.87 (d, J=8.6 Hz, 1 H), 5.14 (d, J=8.6 Hz, 1 H), 7.97 (br s, 1 H).
- Synthesis of Compound #741 (Table 7)

- Quinoline derivative 8f was attached to the pre-formed macrocyclic compound 23b via a Mitsunobu reaction. The quinoline derivative 8f (30 mg, 0.095 mmol) was dissolved in THF, then macrocycle 23b (45.6 mg, 1 eq.) and PPh 3 (49.8 mg, 2 eq.) are then added. The resulting mixture is cooled to 0° C. DIAD (37.4 μl, 2 eq.) is then added dropwise. The solution is stirred 1 hour at 0 C then stirred overnight at room temperature. The mixture was then diluted with EtOAc (15 ml), washed with a saturated solution of NaHCO3 (15 ml), followed by brine. The solution was dried with MgSO4, filtered and concentrated in vacuo. 202 mg of a yellow oil was obtained. The product was purified by flash chromatography on silica gel (100% EtOAc). The product still contained DIAD byproducts after the purification. The resulting product obtained contained 55% w/w of the desired product, so the yield was declared to be 62%.
- The ester intermediate (46 mg, 0.06 mmol) was dissolved in a mixture of THF/MeOH/H 2O (2:1:1 ratio, 2 mL), LiOH.H2O (20 mg, 0.48 mmol) was added and the solution was stirred at RT. After a period of 16 h, analysis of the reaction mixture by HPLC indicated that the hydrolysis was complete. The organic solvents were removed under vacuum and the remaining crude material dissolved in DMSO was purified by C18 reversed phase HPLC to give pure inhibitor 741.
- 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.67 (s, 1H), 8.29-8.14 (m, 2H), 8.08-7.97 (m, 1H), 7.91-7.78 (m, 1H), 7.74 (s, 1H), 7.31-7.20 (m, 1H), 7.10 (d, J=5.7 Hz, 1H), 5.82-5.71 (m, 1H), 5.58-5.47 (m, 1H), 5.32-5.23 (m, 1H), 4.74-4.64 (m, 1H), 4.55-4.47 (m, 1H), 4.23-4.06 (m, 1H), 4.04-3.94 (m, 1H), 3.97 (s, 3H), 3.92-3.85 (m, 1H), 2.70-2.55 (m, 2H), 2.53-2.36 (m, 2H), 2.20-2.09 (m, 1H), 1.80-1.62 (m, 2H), 1.56-1.43 (m, 2H), 1.42-1.29 (m, 6H), 1.27 (d, J=3.2 Hz, 3H), 1.25 (d, J=2.9 Hz, 3H), 1.12 (s, 9H). MS: 763.1 (M+1), 761.1 (M−1).
- Synthesis of Compound 205 (Table 2)

- To a solution of the macrocyclic compound 25a (21 mg, 0.035 mmol) in t-butanol/H 2O) (1.5 mL/1.5 mL) at 0°, a solution of OsO4 in t-butanol (0.36 mL of a 35% w/v, 0.035 mmol) was added and the mixture was stirred at RT for 1 h. The mixture was diluted with EtOAc (20 mL) and the organic solution washed with 5% NaHCO3 (2×10 mL), brine (2×10 mL), dried and evaporated to dryness. The crude compound was taken into THF/MeOH/H2O (3 mL/1.5 mL/1.5 mL) and stirred in presence of LiOH.H2O (13 mg, 0.28 mmol) for 16 h. The mixture was acidified to pH 4 with 0.5 N ice-cold HCl, evaporated and purified by C18 reversed phase HPLC using a solvent gradient from H2O (0.06% TFA) to 40% aqueous CH3CN (0.06% TFA). The syn diol 205 was isolated in high purity as amorphous white solid. compound #205: 1H NMR (DMSO, 400 MHz): δ 1.01 (s, 9H), 1.06-1.30 (m, 9H), 1.48-1.68 (m, 3H), 1.78-1.88 (m, 1H), ≈2.2-2.5 (2m, 2H), 3.78-3.82 (m, 1H), 3.86-3.90 (m, 1H), 4.39 (t, J=8.9 Hz, 1H), 4.61 (d, J=11.4 Hz, 1H), 5.60 (bs, 1H, Pro-γ), 7.03 (d, J=6.0 Hz, 1H), 7.40 (bs, 1H), 7.58-7.62 (m, 1H), 7.87-7.91 (m, 1H), 8.00 (d, J=8.3 Hz, 1H), 8.24 (d, J=8.6 Hz, 1H), 8.60 (s, 1H), 8.99 (bs, 1H). EMS (negative ionization mode): m/z 625 (M−H)−.
- Synthesis of Compound 214 & 218 (Table 2)


- A. A solution of the keto-nonenoate ester 16d (0.180 g, 0.6 mmol) in MeOH/H 2O (5 mL /2 mL) was stirred at RT in presence of LiOH.H2O (50 mg, 1.2 mmol) for 1 h. The solution was acidified to pH 6 with 0.5 N ice-cold HCl and most of the MeOH was evaporated. The residue was then dissolved in EtOAc (30 mL) and the solution was washed with 0.5 N ice-cold HCl (10 mL), brined (10 mL), dried and evaporated. The crude residue was then re-dissolved in CH2Cl2 (10 mL) and reacted with the P1-P2 fragment 30a (0.337 g, 0.6 mmol) in the presence of HATU (233 mg, 0.612 mmol) and DIPEA (420 μL, 2.4 mmol) over a period of 16 h at RT. The reaction mixture was chromatographed on silica gel using EtOAc/hexane (1/1) as the eluent, to isolate the pure compound 30b (0.370 g, yield 83%, Purity >95% by HPLC).
- 1H NMR (CDCl3, 400 MHz) δ 1.41 (s, 9H), 1.45-1.54 (m, 1H), 1.58-1.62 (m, 1H), 1.73-1.77 (m, 1H), 1.86-1.91 (m, 1H), 2.16 (dd, J=17.8 & 8.6 Hz, 1H), 2.26-2.43 (2m, 2H), 2.46-2.58 (m, 2H), 2.64-2.81 (m, 1H), 2.85-2.92 & 2.95-3.03 (2m, 1H, rotamers in 1:3 ratio), 3.67 (s, 3H), 3.95 (s, 3H), 4.10-4.18 (m, 1H), 4.20-4.30 (m, 1H), 4.40-4.55 (m, 1H), 4.80-4.88 (m, 1H), 4.92-5.10 (m, 2H), 5.14 (dd, J=10.2 & 1.6 Hz, 1H), 5.24-5.38 (m, 4H), 5.42-5.54 (m, 1H), 5.68-5.86 (m, 2H), 7.04-7.14 (m, 2H), 7.42-7.64 (m, 5H), 7.92-8.12 (m, 3H).
- B. The diene 30b (0.370 g, 0.49 mmol) was cyclized in the presence of the bis-(tricyclohexylphosphine) benzylidene ruthenium IV dichloride catalyst (0.125 mg, 0.15 mmol) in CH 2Cl2 (distilled from CaH2 and degassed with argon for 30 min) over a period of 2 h at reflux. The compound was obtained as a mixture of stereoisomers (30c and 30d 1:1 ratio) after flash column chromatography on silica gel using EtOAc/Hexane (3/1) in 35% yield (0.124 g).
- 1H NMR of mixture 30c & 30d (CDCl3, 400 MHz) δ 1.44 (s, 4H) & 1.37 (s, 4H), 1.60 (m, 2H), 1.83 (m, 0.5H), 2.01 (m, 1H), 2.09 (m, 1H), 2.42 (m, 5H), 2.73 (m, 2H), 3.26 (m, 0.5H), 3.69 (s, 1.5H), 3.76 (s, 1.5H), 3.96 (s, 3H), 4.10 (m, 1H), 4.24 (m, 0.5H), 4.10 (m, 0.5H), 4.58 (m, 1H), 4.73 (m, 1H), 4.89 (m, 0.5H), 4.97 (m, 0.5H), 5.30 (m, 0.5H), 5.44 (m, 2H), 5.64 (m, 1H), 7.1-7.0 (m, 3H), 7.47 (m, 4H), 8.08-7.98 (m, 3H).
- C,D. Hydrolysis of the methyl esters 30c and 30d (24 mg, 0.033 mmol) was carried out in THF/MeOH/H 2O (1 mL/0.5 mL/0.5 mL) with LiO.H2O (11 mg, 0.246 mmol) over a period of 16 h at RT. After that period the reaction mixture was acidified to pH 4-5 and chromatographed on a C18 reversed phase HPLC column using a solvent gradient from H2O (0.06% TFA) to 50% aqueous CH3CN (0.06% TFA). The desired compounds 214 and 218 were isolated from the mixture of the two compounds in high purity (94% pure by HPLC) in 15% yield (3 mg). compound 214: 1H NMR (DMSO, 400 MHz) δ 1.15 (s, 9H), 1.48-1.54 (m, 2H), 1.65-1.74 (m, 1H), 1.77-1.85 (m, 1H), 2.12-2.25 (m, 4H), 2.27-2.34 (m, 1H), 2.61-2.68 (m, 1H), 2.87 (bt, J=11.5 Hz, 1H), 3.92 (dd, J=9.2 & 1.5 Hz, 1H, Pro-δ), 3.97 (s, 3H, —OCH3), 4.14-4.20 (m, 1H), 4.52 (t, J=7.8 Hz, 1H, Pro-α), 4.66 (d, J=11.8 Hz, 1H, Pro-δ), 5.45 (t, J=9.9 Hz, 1H), 5.51-5.58 (m, 1H), 5.82 (bs, 1H, Pro-γ), 7.09 (d, J=6.0 Hz, 1H, BocNH), 7.26 (bs, 1H), 7.53 (s, 1H), 7.67 (bs, 3H), 8.16 (d, J=2 Hz, 1H), 8.18 (s, 1H), 8.83 (s, 1H, ACCA-NH).
- compound 218: 1H NMR(DMSO, 400 MHz): δ 1.06-1.10 (m, 1H), 1.18 (s, 9H), 1.52-1.55 (m, 1H), 1.62-1.80 (m, 1H), 2.10-2.68 (overlapping, 9H), 3.90 (bd, J=8.3 Hz, 1H), 3.96 (s, 3H, OCH3), 4.20-4.27 (m, 1H), 4.58-4.63 (m, 1H, Pro-δ), 4.66 (dd, J=8.3 Hz, 1H, Pro-α) 4.88 (dd, J=10.2 Hz, 1H), 5.18-5.26 (m, 1H), 5.73-5.79 (m, 1H, Pro-γ), 7.01 (d, J=6.4 Hz, 1H), 7.23 (bs, 1H), 7.50 (bs, 1H), 7.66 (bs, 3H), 8.20 (bs, 2H), 8.53 (s, 1H).
- Synthesis of Compound 209 (Table 2)

- A. The diene 31a (249 mg, 0.330 mmol) was dissolved in 30 mL of anhydrous CH 2Cl2 and the solution was degassed with argon for 15 min. The catalyst bis-(tricyclohexylphosphine) benzylidene ruthenium IV dichloride (82 mg, 0.100 mmol) was dissolved in 3 mL of anhydrous and degassed CH2Cl2 and added to the diene solution. The reaction mixture was refluxed for 2 h under N2. The solution was concentrated and purified by flash column chromatography to obtain compound 31b as a brown solid in 71% yield (171 mg).
- 1H NMR (CDCl3, 400 MHz): δ 1.22-1.44 (m, 10H), 1.42 (s, 9H), 1.66-1.74 (m, 1H), 1.87-1.97 (m, 2H), 2.13-2.28 (m, 3H), 2.32-2.39 (m, 1H), 3.08-3.16 (m, 1H), 3.41 (s, 3H), 4.07-4.22 (m, 3H), 4.28-4.34 (m, 1H), 4.58-4.64 (m, 1H), 4.95-4.99 (m, 1H), 5.22-5.29 (m, 2H), 5.38-5.43 (m, 1H), 5.48-5.56 (m, 1H), 7.00-7.12 (m, 3H), 7.43-7.55 (m, 4H), 7.97-8.11 (m, 3H). ES(+)MS: m/z 727.4 (M+H)+.
- B. Compound 31b (0.117 mmol) was stirred in a solution of HCl (1 mL of 4N in dioxane) for 30 min and concentrated to dryness. The solid was taken up in CH 2Cl2 (2 mL), Et3N (82 μL, 0.585 mmol) and t-butylisocyanate (35 mg, 0.351 mmol) were successively added. After stirring at RT for 20 h, the mixture was concentrated to dryness and the crude compound 31c was used in the final hydrolysis step without further purification.
- C. Compound 31c (85 mg, 0.117 mmol) was dissolved in THF/MeOH/H 2O (2 mL/1 mL/1 mL), LiOH.H2O (39 mg, 0.936 mmol) was added and the solution was stirred for 20 h at RT. After that period, acetic acid (1 mL) was added and the solution was concentrated to remove the MeOH and THF. The pure compound 209 was isolated after purification of the crude by C18 reverse phase HPLC (25 mg, ˜31% yield).
- 1H NMR (DMSO, 400 MHz): δ 1.04 (s, 9H), 1.15-1.24 (m, 2H), 1.30-1.40 (m, 5H), 1.44-1.51 (m, 2H), 1.54-1.68 (m, 1H), 1.75-1.88 (m, 1H), 2.18 (dd, J=17.2 & 8.5 Hz, 1H), 2.32-2.45 (m, 1H, Pro-β), 2.54-2.62 (m, 1H), 2.65-2.68 (m, 1H, Pro-β), 3.91 (dd, J=11.1 & 3.5 Hz, 1H, Pro-δ), 3.96 (s, 3H, —OCH3), 4.17-4.23 (m, 1H), 4.47 (dd, J=8.6, 1H, Pro-α), 4.67 (bd, J=7.9 Hz, 1H, Pro-δ), 5.30 (dd, J=9.5 Hz, 1H), 5.52 (bdd, J=19 & 8.3, 1H), 5.68 (s, 1H), 5.78 (bs, 1H, Pro-γ), 5.94 (bs, 1H), 7.21 (bs, 1H), 7.51 (bs, 1H), 7.66 (bs, 4H), 8.19 (s, 2H), 8.40 (d, J=7 Hz, 1H), 8.61 (s, 1H, ACCA—NH). ES(+)MS: m/z 698.3 (M+H)+.
- Synthesis of Compounds #404 and #407 (Table 4)

- A. The diene 32a (84 mg, 0.11 mmol) was dissolved in anhydrous CH 2Cl2 (11 mL) and the solution was degassed over a period of 15 min. with a flow of argon. The bis-(tricyclohexylphosphine) benzylidene ruthenium IV dichloride catalyst (19 mg, 0.023 mmol) was first dissolved in 1 mL of degassed CH2Cl2 and then it was transferred to the reaction flask via cannula. The reaction mixture was stirred for 2 h at reflux. The solvent was then removed under vacuum and the reaction mixture was purified by flash column chromatography on silica gel, using EtOAc/hexane (1:1) as the eluent, to give the macrocyclic compound 32b as a yellow oil (33 mg, 41% yield).
- B. The ester intermediate 32b (33 mg, 0.045 mmol) was dissolved in a mixture of THF/MeOH/H 2O (2:1:1 ratio, 2 mL), LiOH.H2O (8 mg, 0.18 mmol) was added and the solution was stirred at RT. After a period of 16 h, analysis of the reaction mixture by HPLC indicated that the hydrolysis was incomplete. Thus an additional amount of LiOH.H2O (4 mg, 0.09 mmol) was added and the solution was stirred at RT for a total of 36 h. Finally, the solution was acidified with a small aliquot of acetic acid, the organic solvents were removed under vacuum and the remaining crude material was purified by C18 reversed phase HPLC to give pure inhibitor 404.
- 1H NMR (DMSO, 400 MHz): δ 1.21 (d, J=6.0 Hz, 3H, Me), 1.36 (s, 9H, Boc), 1.1-1.4 (3m, 3H), 1.66 (m, 1H), 1.80 (m, 1H), 2.10 (m, 2H), 2.57 (m, 2H), 3.90 (m, 4H), 4.47 (bd, J=12.7 Hz, 1H), 4.58 (bd, J=7.3, 1H), 4.66 (dd, J=8.0 Hz, 1H), 5.57 (m, 1H), 5.66 (m, 1H), 5.83 (bs, 1H), 6.18 (bd, J=6.9 Hz, 1H), 7.25 (bd, J=7.3 Hz, 1H), 7.56 (bs, 1H), 7.70 (m, 4H), 8.22 (bd, J=2.9 Hz, 2H), 8.29 (bs, J=9.2 Hz, 1H).
- C. Inhibitor 404 (15 mg, 0.021 mmol) was dissolved in ethanol (2 mL) and Pd 10%/C (2 mg) was added. The mixture was stirred under hydrogen at RT for 16 h. After filtration, the mixture was purified by C18 reversed phase HPLC to give inhibitor 407 as a white solid (10 mg, 66% yield)
- 1H NMR (DMSO, 400 MHz): □ 1.04 (m, 1H), 1.17 (d, J=6.0 Hz, 3 H), 1.35 (s, 9H), 1.25-1.75 (m, 12 H), 2.32-2.45 (m, 1 H), 3.40-3.50 (m, 2 H), 3.74-3.83 (m, 1H), 3.85-3.93 (m, 1H), 3.97 (s, 3H), 4.27-4.36 (dd, J=21.1 & 8.6 Hz, 1H), 4.54 (dd, J=7.95 & 7.95 Hz, 1H), 5.64 (d, J=8.3 Hz, 1H), 5.82 (br s, 1H), 7.27-7.33 (m, 1H), 7.53-7.57 (bs, 1 H), 7.60-7.74 (m, 4 H), 8.13-8.27 (m, 3 H), 8.30-8.35 (br s, 1H).
- Synthesis of Compound #824 (Table 8)

- A. Compound 33a (˜0.55 mmol) was dissolved in CH 2Cl2 (100 mL) and the solution was degassed carefully before a sample of Hoveyda's catalyst (17 mg, 0.028 mmol, 0.05 eq.) was added. The solution was then stirred under reflux for 5 h. The reaction mixture was concentrated and purified by flash column chromatography, using a solvent gradient of CH2Cl2/EtOAc (from 3:2 to 2:3 ratio), to give compound 33b in 72% yield (194 mg).
- B. To a solution of compound 33b (70 mg, 0.142 mmol), 2-ethoxy-4-hydroxy-7-methoxyquinoline 3c (63 mg, 0.284 mmol, 2 eq.) and Ph 3P (186 mg, 0.71 mmol, 5 eq.) in anhydrous THF (15 mL) at 0° C., DIAD (140 □L, 0.71 mmol, 5 eq.) was added slowly over a period of 20 min. The reaction mixture was allowed to warm-up to RT and to stir at RT for 2.5 h. Subsequently, the THF was evaporated under vacuum and the crude product was purified by flash column chromatography, using a solvent gradient of hexane/EtOAc (from 7:3 to 1:1 ratio). Pure compound 33c was isolated in 73% yield (72 mg).
- C. Compound 33c (72 mg, 0.104 mmol) was mixed with CH 2Cl2 (5 mL) and 4M HCl in dioxane (5 mL) and the mixture was allowed to stir at RT for 1.5 h in order to cleave the Boc protecting group and obtain the HCl salt of intermediate 33d. The reaction crude reaction mixture was evaporated to dryness under vacuum, dried under vacuum to assure the removal of all HCl and used in the next step without purification.
- D. To a solution of cyclopentanol (29 □L, 0.32 mmol) in THF (10 mL), a solution of phosgene in toluene (1.93 M, 274 □L, 0.528 mmol) was added dropwise and the mixture was stirred at R.T. for 2 h to form the cyclopentyl chloroformate reagent. After that period, approximately half of the solvent was removed by evaporation under vacuum, the remaining light yellow solution was diluted by the addition of CH 2Cl2 (5 mL) and reconcentrated to half of its original volume, in order to assure the removal of all excess phosgene. The above solution of the cyclopentyl chloroformate reagent was further diluted with THF (10 mL), cooled to 0° C. and added to the solid compound 33d (0.104 mmol) at 0° C. Et3N (75 μL, 0.534 mmol, 5.2 eq.) was added to the reaction mixture and stirring was continued at 0° C. for 1.5 h. The solvents were removed under vacuum and the crude material purified by flash column chromatography, using EtOAc/hexane (1:1) as the eluent, to obtain compound 33e in almost quantitative yield (75 mg).
- E. Hydrolysis of the methyl ester was achieved by reacting compound 33e (75 mg, 0.11 mmol) with LiOH.H 2O (35 mg, 0.84 mmol, 8 eq.) in a solvent mixture of THF/MeOH/H2O (2:2:1 ratio, 7.5 mL) at 50° C. for 2.5 h. Upon completion of the hydrolysis, the mixture was acidified to pH=4.5 and the solvents were evaporated to dryness under vacuum. The crude product was purified by C18 reversed phase preparative HPLC, using a solvent gradient of H2O to 58% aqueous CH3CN (with 0.06% TFA), to obtain inhibitor #824 as a white amorphous solid (45 mg, 65% yield).
- 1H NMR of the Na+ salt of #824 (DMSO, 400 MHz): □ 0.88 (d, J=6.7 Hz, 3H), 0.95-1.70 (overlapping resonances, 17H), 1.37 (t, J=7 Hz, 3H), 2.00-2.10 (m, 1H), 2.10-2.33 (m, 3H), 2.38-2.44 (m, 1H), 3.80-3.85 (m, 1H), 3.85 (s, 3H), 4.02-4.08 (m, 1H), 4.42 (q, J=7 Hz, 2H), 4.35-4.44 (m, 1H), 4.50 (d, J=10.8 Hz, 1H), 4.63 (bs, 1H), 5.28 (dd, J=9.5 Hz, 1H), 5.38 (bs, 1H), 5.42-5.49 (m, 1H), 6.37 (s, 1H), 6.87 (dd, J=8.9 & 2.2 Hz, 1H), 7.07 (d, J=2.2 Hz, 1H), 7.28 (d, J=7.0 Hz, 1H), 7.90 (d, J=8.9 Hz, 1H), 8.57 (s, 1H).
- Synthesis of Compound #812 (Table 8)



- A. To a solution of the macrocyclic intermediate 23b (13.05 g, 27.2 mmol, 1.0 eq.), Ph 3P (14.28 g, 54.4 mmol, 2.0 eq) and 2-carboxymethoxy-4-hydroxy-7-methoxyquinoline (WO 00/09543 & WO 00/09558) (6.67 g, 28.6 mmol, 1.05 eq) in THF (450 mL) at 0° C., DIAD (10.75 mL, 54.6 mmol, 2.0 eq) was added dropwise over a period of 15 min. The ice bath was then removed and the reaction mixture was stirred at RT for 3 h. After the complete conversion of starting material to products, the solvent was evaporated under vacuum, the remaining mixture diluted with EtOAc, washed with saturated NaHCO3 (2×) and brine (1×), the organic layer was dried over anhydrous MgSO4, filtered and evaporated to dryness. Pure compound 34a was obtained after flash column chromatography; the column was eluted first with hexane/EtOAc (50:50), followed by CHCl3/EtOAc (95:5) to remove Ph3PO and DIAD byproducts and elution of the impurities was monitored by TLC. Finally, the desired product 34a was eluted from the column with CHCl3/EtOAc (70:30). Usually, the chromatography step had to be repeated 2-3 times before compound 34a could be isolated in high purity as a white solid with an overall yield of 68% (12.8 g, 99.5% pure by HPLC).
- B. To a solution of the Boc-protected intermediate 34a (1.567 g) in CH 2Cl2 (15 mL), 4N HCl in dioxane (12 mL) was added and the reaction mixture was stirred at RT for 1 h. [In the event that a thick gel would form half way through the reaction period, an additional 10 mL CH2Cl2 was added.] Upon completion of the deprotection the solvents were evaporate to dryness to obtain a yellow solid and a paste like material. The mixture was redissolved in approximately 5% MeOH in CH2Cl2 and re-evaporated to dryness under vacuum to obtain compound 34b as a yellow solid, which was used in the next step without any purification.
- C. To a solution of cyclopentanol (614 □L, 6.76 mmoL) in THF (15 mL), a solution of phosgene in toluene (1.93 M, 5.96 mL, 11.502 mmol) was added dropwise and the mixture was stirred at R.T. for 2 h to form the cyclopentyl chloroformate reagent (z). After that period, approximately half of the solvent was removed by evaporation under vacuum, the remaining light yellow solution was diluted by the addition of CH 2Cl2 (5 mL) and concentrated to half of its original volume, in order to assure the removal of all excess phosgene. The above solution of the cyclopentyl chloroformate reagent was further diluted with THF (15 mL) and added to the amine-2HCl salt 34b. The mixture was cooled to 0° C. in an ice bath, the pH was adjusted to ˜8.5-9 with the addition of Et3N (added dropwise) and the reaction mixture was stirred at 0° C. for 1 h. After that period, the mixture was diluted with EtOAc, washed with water (1×), saturated NaHCO3 (2×), H2O (2×) and brine (1×). The organic layer was dried over anhydrous MgSO4, filtered and evaporated under vacuum to obtain a yellow-amber foam. Compound 34c was obtained as a white foam after purification by flash column chromatography (using a solvent gradient from 30% hexane to 20% hexane in EtOAc as the eluent) in 80% yield (1.27 g) and >93% purity.
- D. The dimethyl ester 34c (1.17 g) was dissolved in a mixture of THF/MeOH/H 2O (20 mL, 2:1:1 ratio), and an aqueous solution of NaOH (1.8 mL, 1N, 1 eq.) was added. The reaction mixture was stirred at RT for 1 h before it was evaporated to dryness to obtain the sodium salt 34d as a white solid (˜1.66 mmol). Compound 34d was used in the next step without purification.
- E. The crude sodium salt 34d (1.66 mmoL) was dissolved in THF (17 mL), Et 3N was added and the mixture was cooled to 0° C. in an ice bath. Isobutylchloroformate (322 □l, 2.5 mmol) was added dropwise and the mixture was stirred at 0° C. for 75 min. After that period, diazomethane (15 mL) was added and stirring was continued at 0° C. for 30 min and then at RT for an additional 1 h. Most of the solvent was evaporated to dryness under vacuum, the remaining mixture was diluted with EtOAc, washed with saturated NaHCO3 (2×), H2O (2×) and brine (1×), dried over anhydrous MgSO4, filtered and evaporated to dryness to obtain compound 34e as a light yellow foam (1.2 g, ˜1.66 mmol). The diazoketone intermediate 34e was used in the next step without purification.
- F. The diazoketone 34e (1.2 g, 1.66 mmoL) dissolved in THF (17 mL) was cooled to 0° C. in an ice bath. A solution of aqueous HBr (48%, 1.24 mL) was added dropwise and the reaction mixture was stirred at 0° C. for 1 h. The mixture was then diluted with EtOAc, wash with saturated NaHCO 3 (2×), H2O (2×) and brine (1×), the organic layer was dried over anhydrous MgSO4, filtered and evaporated to dryness to obtain the □-bromoketone intermediate 34f as a light yellow foam (˜1.657 mmol).
- G. To a solution of the bromoketone 34f (600 mg, 0.779 mmol) in isopropanol (5 mL), thiourea (118 mg, 1.55 mmol) was added and the reaction mixture was placed in a pre-heated oil bath at 75° C. where it was allowed to stir for 1 hr. The isopropanol was then removed under vacuum and the product dissolved in EtOAc (100 mL). The solution was washed with saturated NaHCO 3 and brine, the organic layer was dried over anhydrous Na2SO4, filtered and evaporated to afford the crude product 34g (522 mg) as a red-brown solid. This material was used in the final step without any further purification.
- H. The crude methyl ester 34g (122 mg, 0.163 mmol) was dissolved in a solution of THF/MeOH/H 2O (2:1:1 ratio, 4 mL) and saponified using LiOH.H2O (89 mg, 2.14 mmol). The hydrolysis reaction was carried out over a 12-15 h period at RT. The solvents were then removed under vacuum and the crude product purified by C18 reversed phase HPLC, using a solvent gradient from 10% CH3CN in H2O to 100% CH3CN, to afford the HCV protease inhibitor #812 as a yellow solid (24 mg, 20% overall yield for the conversion of intermediate 34f to inhibitor #812).
- 1H NMR (400 MHz, DMSO-d6) □ 8.63 (s, 1H), 8.26-8.15 (m, 2H), 7.79 (bs, 1H), 7.72 (bs, 1H), 7.50 (bs, 2H), 7.33-7.25 (m, 2H), 5.77 (bs, 1H), 5.52 (dd, J=8.3 Hz, 1H), 5.27 (dd, J=9.2 Hz, 1H), 4.64 (d, J=10.8 Hz, 1H), 4.50 (dd, J=8.3 Hz, 1H), 4.39-4.31 (m, 1H), 4.08-3.99 (m, 2H), 3.94 (s, 3H), 3.87 (d, J=9.5 Hz, 2H) 2.65-2.53 (m, 2H), 2.46-2.36 (m, 2H), 2.20-2.12 (dd, J=8.6 Hz, 1H), 1.80-1.64 (m, 2H) 1.63-1.06 (m, 14H). MS; es+: 733.2 (M+H)+, es−: 731.2 (M−H)−.
- Using the same procedure as described in example 34 but reacting bromoketone 34f with commercially available N-methylthiourea gave # 811 (Table 8)

- 1H NMR (400 MHz, DMSO-d6): □ 8.63 (s, 1H), 8.20 (s, 1H), 8.18 (s, 1H), 8.12-7.93 (m, 1H), 7.88-7.69 (m , 2H), 7.32-7.24 (m, 2H), 5.82-5.75 (m, 1H), 5.52 (ddd, J=8.1 Hz, 1H), 5.28 (dd, J=9.9 Hz, 1H), 4.67-4.61 (m, 1H), 4.51 (dd, J=8.8 Hz, 1H), 4.44-4.37 (m, 1H), 4.08-4.00 (m, 1H), 3.96 (s, 3H), 3.89 (m, 1H), 3.04 (d, J=4.1 Hz, 3H), 2.65-2.37 (m, 3H), 2.16 (m, 1H), 1.77-1.65 (m, 2H), 1.63-1.11 (m, 17H). MS; es+: 747.2 (M+H)+, es−: 745.3 (M−H)−.
- Using the same procedure as described in example 34 but reacting bromoketone 34f with commercially available N-ethylthiourea gave # 810 (Table 8)

- 1H NMR (400 MHz, DMSO-d6): □ 8.63 (s, 1H), 8.27 (bs, 1H), 8.20 (d, J=9.0 Hz, 1H), 8.13-8.07 (m, 1H), 7.86 (bs, 1H), 7.78 (s, 1H), 7.33-7.25 (m, 2H), 5.81 (bs, 1H), 5.54 (dd, J=8.8 Hz, 1H), 5.28 (dd, J=9.7 Hz, 1H), 4.65 (d, J=12.4 Hz, 1H), 4.51 (dd, J=8.8 Hz, 1H), 4.38 (bs, 1H), 4.03 (m, 1H), 3.97 (s, 3H), 3.92-3.87 (m, 1H), 3.54-3.46 (m, 2H)), 2.68-2.65 (m, 2H), 2.47-2.38 (m, 1H), 2.15 (dd, J=8.6 Hz, 1H), 1.78-1.65 (m, 2H), 1.60-1.12 (m, 17H), 1.25 (t, J=7.3Hz, 3H). MS; es+: 783.2 (M+Na)+, es−: 761.2 (M+H)+.
- Using the same procedure as described in example 34 but reacting bromoketone 34f with commercially available N-iso-propylthiourea gave # 822

- 1H NMR (400 MHz, DMSO-d6) δ 8.63 (s, 1H), 8.33-8.23 (bs, 1H), 8.21 (d, J=9.2 Hz, 1H), 8.04 (d, J=8.3 Hz, 1H), 7.86 (bs, 1H), 7.77 (s, 1H), 7.35-7.23 (m, 2H), 5.81 (bs, 1H), 5.52 (dd, J=8.5 Hz, 1H), 5.27 (dd, J=9.2 Hz, 1H), 4.65 (d, J=11.8 Hz, 1H), 4.51 (dd, J=7.6 Hz, 1H), 4.37 (bs, 1H), 4.15 (bs, 1H), 4.07-3.98 (m, 2H), 3.97 (s, 3H), 3.88 (d, J=8.9 Hz, 1H), 2.60-2.53 (m, 2H), 2.47-2.37 (m, 2H), 2.19-2.10 (dd, J=9.2 Hz, 1H), 1.80-1.64 (m, 2H), 1.63-1.29 (m, 13H), 1.27 and 1.25 (2×d, J=6.5 Hz, 6H), 1.23-1.09 (m, 2H). MS; es+: 775.0 (M+H)+, es−: 772.9 (M−H)−.
- Using the same procedure as described in example 34 but reacting bromoketone 34f with commercially available N-acetylthiourea gave # 809

- 1H NMR (400 MHz, DMSO-d6): □ 8.62 (s, 1H), 8.30 (bs, 1H), 8.17 (d, J=8.9 Hz, 1H), 7.62 (bs, 1H), 7.52 (bs, 1H), 7.28 (d, J=6.4 Hz, 1H), 7.21 (bs, 1H), 5.63 (bs, 1H), 5.54 (dd, J=8.1 Hz, 1H), 5.28 (dd, J=9.5Hz, 1H), 4.62 (d, J=12.1 Hz, 1H), 4.56-4.46 (m, 2H), 4.11-4.04 (m, 1H), 3.95 (s, 3H), 3.93-3.88 (m, 1H), 2.62-2.54 (m, 1H), 2.45-2.36 (m, 1H), 2.22 (s, 3H), 2.21-2.13 (m, 1H), 1.79-1.69 (m, 2H), 1.65-1.30 (m, 16H), 1.26-1.12 (m, 2H). MS; es+: 775.3 (M+H)+, es−: 773.3 (M−H)−.
- To a stirred solution of the 2-amino-4-thiazolyl intermediate 34g (0.24 g, 0.32 mmol) in CH 2Cl2 (5 mL) at RT was added DIPEA (0.55 mL, 3.18 mmol, 10 eq) and methyl chloroformate (0.13 mL, 1.6 mmol, 5 eq). The reaction mixture was stirred for 6.5 h before being concentrated under vacuum. The crude isolated material was then hydrolyzed to the desired carboxylic acid as described in Example 34 to yield compound # 818

- 1H NMR (400 MHz, DMSO-d6): □ 8.61 (s, 1H), 8.21-8.07 (m, 2H), 7.61-7.38 (m, 2H), 7.26 (d, J=6.4 Hz, 1H), 7.19-7.10 (m, 1H), 5.60-5.47 (m, 2H), 5.27 (dd, J=9.2 Hz, 1H), 4.63-4.53 (m, 1H), 4.47 (d, J=7.9 Hz, 1H), 4.13-4.04 (m, 1H), 3.93 (s, 3H), 3.92-3.87 (m, 2H), 3.79 (s, 3H), 2.42-2.30 (m, 2H), 2.17 (dd, J=9.2 Hz, 1H), 1.81-1.68 (m, 2H), 1.63-1.29 (m, 16H), 1.23-1.10 (m, 2H). MS; es+: 791.1 (M+H)+, es−: 789.1 (M−H)−.
- Following the conditions described above in example 34E, but using isobutyl chloroformate, gave the analogous substituted carbamate intermediate. The crude isolated material was then hydrolyzed to the desired compound # 819

- 1H NMR (400 MHz, DMSO-d6): □ 8.62 (s, 1H), 8.47-8.27 (bs, 1H), 8.18 (d, J=8.6 Hz, 1H), 7.69-7.60 (m, 1H), 7.60-7.51 (m, 1H), 7.28 (d, J=6.7 Hz, 1H), 7.28-7.19 (m, 1H), 5.70-5.60 (m, 1H), 5.52 (dd, J=8.3 Hz, 1H), 5.27 (dd, J=9.8 Hz, 1H), 4.63 (d, J=11.8 Hz, 1H), 4.53-4.44 (m, 2H), 4.10-3.99 (m, 1H), 4.04 (d, J=6.7 Hz, 2H), 3.95 (s, 3H), 3.94-3.87 (m, 1H), 2.65-2.53 (m, 1H), 2.46-2.34 (m, 1H), 2.16 (dd, J=8.1 Hz, 1H), 2.03-1.91 (m, 1H), 1.79-1.09 (m, 20H), 0.95 (d, J=6.7 Hz, 6H). MS; es+: 833.2 (M+H)+, es−: 831.2 (M−H)−.
- Synthesis of Compound #908
- Starting with derivative 27a and using the same chemistry as described in example 34, the following saturated macrocycle, compound # 908 (Table 9) was obtained.

- 1H NMR (400 MHz, DMSO-d6): □ 8.47 (s, 1H), 8.16 (d, J=10 Hz, 1H), 8.15-8.07 (m, 1H), 7.82-7.63 (m, 2H), 7.53-7.43 (m, 2H), 7.33-7.22 (m, 1H), 7.13 (d, J=7 Hz, 1H), 5.77-5.65 (m, 1H), 4.62-4.52 (m, 2H), 4.50-4.4 (m, 1H), 4.20-4.10 (m, 1H), 3.94 (s, 3H), 3.89-3.83 (m, 1H), 2.59-2.53 (m, 1H), 2.48-2.40 (m, 1H), 1.79-1.0 (m, 25H);). MS; es+: 735.2 (M+H)+, es−: 733.2 (M−H)−.
- Synthesis of Compound #909
- Using the same procedure as described in example 35 but using available N-acetylthiourea gave compound # 909 (Table 9).

- 1H NMR (400 MHz, DMSO-d6): □ 8.53-8.41 (m, 2H), 8.20 (d, J=9.2 Hz, 1H), 7.68 (bs, 1H), 7.68 (bs, 1H), 7.27 (dd, J=9.2 Hz, 1H), 7.15 (d, J=6.4 Hz, 1H), 5.67 (bs, 1H), 4.65-4.50 (m, 3H), 4.44-4.37 (m, 1H), 4.21-4.13 (m, 1H), 3.96 (s, 3H), 3.99-3.86 (m, 1H), 2.62-2.39 (m, 2H), 2.24 (s, 3H), 1.78-1.67 (m, 3H), 1.67-1.01 (m, 22H). MS; es+: 798.0 (M+Na)+, es−: 777.0 (M+H)+.
- Synthesis of Compound #910
- Using the same procedure as described in example 35 but using available N-ethylthiourea gave compound # 910 (Table 9).

- 1H NMR (400 MHz, DMSO-d6): □ 8.47 (s, 1H), 8.29 (bs, 1H), 8.20 (d, J=9.2 Hz, 1H), 8.09 (bs, 1H), 7.87 (s, 1H), 7.77 (s, 1H), 7.32 (dd, J=9.2 Hz, 1H), 7.14 (dd, J=6.7 Hz, 1H), 5.78 (bs, 1H), 4.58 (dd, J=8.1 Hz, 2H), 4.43 (bs, 1H), 4.18-4.12 (m, 1H), 3.97 (s, 3H), 3.87 (d, J=8.9 Hz, 1H), 3.55-3.46 (m, 2H), 2.63-2.53 (m, 1H), 2.47-2.41 (m, 1H), 1.78-1.00 (m, 25H), 1.25 (t, J=7.3 Hz, 3H).). MS; es+: 763.1 (M+H)+, es−: 761.1 (M−H)−.
- Synthesis of Compound #911
- Using the same procedure as described in example 35 but using available N-iso-propylthiourea gave compound # 911 (Table 9).

- 1H NMR (400 MHz, DMSO-d6): □ 8.47 (s, 1H), 8.29-8.19 (m, 1H), 8.19 (d, J=9.2 Hz, 1H), 8.09-8.0 (m, 1H), 7.83 (bs, 1H), 7.74 (bs, 1H), 7.31 (d, J=8 Hz, 1H), 7.14 (d, J=6.4 Hz, 1H), 5.76 (bs, 1H), 4.64-4.53 (m, 2H), 4.44 (bs, 1H), 4.22-4.09 (m, 3H), 3.97 (s, 3H), 3.87 (d, J=8.6 Hz, 1H), 2.63-2.58 (m, 1H), 2.46-2.41 (m, 1H), 1.79-1.10 (m, 24H), 1.27 and 1.26 (2×d, J=6.5 Hz, 6H). MS; es+: 777.0 (M+H)+, es−: 775.0 (M−H)−.
- Synthesis of Compound #716

- 1H NMR (400 MHz, DMSO-d6): δ (ppm): 8.62 (s, 1H), 8.13 (d, J=9.2 Hz, 1H), 7.64-7.54 (m, 2H), 7.47 (d, J=2.6 Hz, 1H), 7.16 (dd, J=9.2, 2.2 Hz, 1H), 7.03 (d, J=6.0 Hz, 1H), 5.63 (s, 1H), 5.52 (q, J=9.9 Hz, 1H), 5.26 (t, J=8.9 Hz, 1H),4.62 (d, J=11.45, 1H), 4.45 (dd, J=9.2, 8.27 Hz, 1H),4.02 (m, 1H), 3.93 (s, 3H), 3.7 (dd, J=7.6, 1.0 Hz, 1H), 2.66 (s, 3H), 2.55-2.65 (m, 1H), 2.35-2.45 (m, 1H), 2.17 (q, J=8.6 Hz, 1H), 1.65-1.75 (m, 2H), 1.5-1.35 (m, 7H), 1.15 (s, 9H). MS: 705. (M+1), 703 (M−1)
- Synthesis of Compound #717

- 1H NMR (400 MHz, DMSO-d6): δ (ppm): 8.62 (s, 1H), 8.15 (d, J=8.9 Hz, 1H), 7.62 (s, 1H), 7.49 (s, 1H), 7.19 (dd, J=9.2, 2.2 Hz, 1H), 7.02 (d, J=5.4 Hz, 1H), 5.64 (s, 1H), 5.52 (q, J=9.9 Hz, 1H), 5.26 (t, J=9.2 Hz, 1H),4.63 (d, J=11.44, 1H), 4.45 (t, J=9.2 Hz, 1H), 3.94 (s, 3H), 3.9-3.8 (m, 1H),2.7-2.55 (m, 1H), 2.4-2.3 (m, 1H), 2.18 (q, J=8.9 Hz, 1H), 1.75-1.65 (m, 2H), 1.5-1.2 (m, 7H), 1.14 (s, 9H). MS: 705. (M+1), 703 (M−1).
- Synthesis of Compound #718

- 1H NMR (400 MHz, DMSO-d6): δ (ppm): 9.55 (s, 1H), 8.63 (s, 1H), 8.43 (s, 1H), 8.13 (d, J=9.2 Hz, 1H), 7.66 (s, 1H), 7.46 (s, 1H), 7.32 (d, J=2.6 Hz, 1H), 7.10-7.07 (m, 2H), 5.64-5.54 (m, 1H), 5.59-5.48 (m, 1H), 5.33-5.23 (m, 1H), 4.73-4.61 (m, 1H), 4.45 (dd, J=7.5, 9.1 Hz, 1H), 4.09-4.00 (m, 1H), 3.92 (s, 3H), 3.93-3.83 (m, 1H), 2.67-2.55 (m, 2H), 2.53-2.43 (m, 1H), 2.42-2.31 (m, 1H), 2.23-2.12 (m,1H), 1.81-1.66 (m, 2H), 1.52-1.42 (m, 2H), 1.42-1.25 (m, 6H), 1.21 (s, 9H). MS: 689.3 (M+1), 687.3 (M−1)
- Synthesis of Compound #722

- 1H NMR (400 MHz, DMSO-d6): □ (ppm): 9.70 (s, 1H), 8.64 (s, 1H), 8.26 (s, 1H), 8.14 (d, J=9.2 Hz, 1H), 7.45 (s, 1H), 7.30 (d, J=2.5 Hz, 1H), 7.14-7.06 (m, 2H), 5.60-5.54 (m, 1H), 5.58-5.48 (m, 1H), 5.31-5.23 (m, 1H), 4.71-4.62 (m, 1H), 4.49-4.40 (m, 1H), 4.08-3.99 (m, 1H), 3.92 (s, 3H), 3.92-3.84 (m, 1H), 2.69-2.54 (m, 2H), 2.53-2.46 (m, 1H), 2.42-2.31 (m, 1H), 2.37 (s, 3H), 2.22-2.13 (m, 1H), 1.81-1.64 (m, 2H), 1.54-1.42 (m, 2H), 1.42-1.27 (m, 6H), 1.22 (s, 9H). MS: 703.3 (M+1), 701.3 (M−1)
- Synthesis of Compound #733

- 1H NMR (400 MHz, DMSO-d6): □ (ppm): 8.75 (m, 1H), 8.62 (s, 1H), 8.06 (d, J=9.2 Hz, 1H), 7.88-7.87 (m, 1H), 7.48 (s, 1H), 7.28 (d, J=2.6 Hz, 1H), 7.05-7.00 (m, 2H), 6.64-6.63 (m, 1H), 5.62-5.58 (m, 1H), 5.55-5.49 (m, 1H), 5.28-5.24 (m, 1H), 4.64-4.61 (m, 1H), 4.48-4.44 (m, 1H), 4.07-4.03 (m, 1H), 3.91 (s, 3H), 3.92-3.85 (m, 1H), 2.67-2.54 (m, 2H),2.53-2.45 (m, 1H), 2.41-2.34 (m, 1H), 2.20-2.14 (m, 1H), 1.75-1.69 (m, 2H), 1.50-1.43 (m, 2H), 1.41-1.32 (m, 6H), 1.17 (s, 9H). MS: 689.3 (M+1), 687.2 (M−1)
- Synthesis of Compound # 703

- 1H NMR (400 MHz, DMSO-d6): □ 8.50 (s, 1H), 8.19 (s, 1H), 8.17 (s, 1H), 8.11-8.00 (m, 1H), 7.88-7.77 (m, 1H), 7.73 (s, 1H), 7.25 (d, J=8.6 Hz, 1H), 6.93 (d, J=6 Hz, 1H), 5.89-5.68 (m, 1H), 4.62 (d, J=11 Hz, 1H), 4.53 (dd, J=8.3 Hz, 1H), 4.16-4.07 (m, 1H), 3.96 (s, 3H), 3.88 (bd, J=9.5 Hz, 1H), 3.53-3.43 (m, 2H), 2.63-2.51 (m, 1H), 2.46-2.36 (m, 1H), 1.81-1.62 (m, 2H), 1.60-1.01 (m, 15H), 1.24 (t, J=7.4 Hz, 3H), 1.17 (s, 9H). MS; es+: 751.1(M+H)+, es−: 749.1-(M−H)−.
- Synthesis of Compound #734

- 1H NMR (400 MHz, DMSO-d6): δ (ppm): 8.62 (s, 1H), 8.54 (s, 1H), 8.04 (d, J=9.2 Hz, 1H), 7.70 (s, 1H), 7.43 (s, 1H), 7.24 (d, J=2.6 Hz, 1H), 7.05-6.98 (m, 2H), 5.57-5.54 (m, 1H), 5.55-5.48 (m, 1H), 5.28-5.24 (m, 1H), 4.63-4.59 (m, 1H), 4.47-4.43 (m, 1H), 4.13-3.99 (m, 1H), 3.90 (s, 3H), 3.92-3.83 (m, 1H), 2.67-2.55 (m, 2H), 2.53-2.46 (m, 1H), 2.43-2.31 (m, 1H), 2.22-2.15 (m, 1H), 2.15 (3H), 1.75-1.70 (m, 2H), 1.51-1.42 (m, 2H), 1.41-1.28 (m, 6H), 1.17 (s, 9H). MS: 703.2 (M+1), 701.3 (M−1)
- Synthesis of Compound #738

- 1H NMR (400 MHz, DMSO-d6): δ (ppm): 8.64 (d, J=2.5 Hz, 1H), 8.62 (s, 1H), 8.04 (d, J=9.2 Hz, 1H), 7.39 (s, 1H), 7.24 (d, J=2.5 Hz, 1H), 7.04 (d, J=6.0 Hz, 1H), 6.99 (dd, J=2.2, 9.2 Hz, 1H), 6.43 (d, J=2.2 Hz, 1H), 5.62-5.57 (m, 1H), 5.56-5.47 (m, 1H), 5.31-5.22 (m, 1H), 4.65-4.56 (m, 1H), 4.45 (dd, J=7.6,8.9 Hz, 1H), 4.07-4.00 (m, 1H), 3.90 (s, 3H), 3.88-3.84 (m, 1H), 2.68-2.56 (m, 2H), 2.54-2.43 (m, 1H), 2.42-2.31 (m, 1H), 2.34 (s, 3H), 2.24-2.14 (m, 1H), 1.80-1.64 (m, 2H), 1.52-1.43 (m, 2H), 1.43-1.27 (m, 6H), 1.18 (s, 9H). MS: 703.2 (M+1), 701.2 (M−1)
- Synthesis of Compound #725

- 1H NMR (400 MHz, DMSO-d6): □ (ppm): 8.62 (s, 1H), 8.10 (d, J=9.2 Hz, 1H), 7.57 (s, 1H), 7.49 (s, 1H), 7.35 (d, J=2.2 Hz, 1H), 7.09-7.03 (m, 2H), 5.65-5.61 (m, 1H), 5.55-5.49 (m, 1H), 5.28-5.24 (m, 1H), 4.62-4.57 (m, 1H), 4.49-4.45 (m, 1H), 4.08-4.01 (m, 1H), 3.93 (s, 3H), 3.92-3.86 (m, 1H), 3.20-3.14 (m, 1H), 2.65-2.56 (s, 1H), 2.53-2.47 (m, 1H) 2.42-2.35 (m, 1H), 2.22-2.15 (m, 1H), 1.79-1.68 (m, 2H), 1.50-1.43 (m, 2H), 1.41-1.28 (m, 12H), 1.18 (s, 9H). MS:748.2 (M+1), 746.2 (M−1)
- Synthesis of Compound #726

- 1H NMR (400 MHz, DMSO-d6): □ (ppm): 8.64 (s, 1H), 8.10 (d, J=9.5 Hz, 1H), 7.83-7.76 (m, 2H), 7.60 (s, 1H), 7.44-7.42 (m, 1H), 7.18-7.01 (m, 2H), 5.56-5.49 (m, 2H), 5.29-5.24 (m, 1H), 4.66-4.63 (m, 1H), 4.47-4.42 (m, 1H), 4.28 (s, 3H), 4.06-4.02 (m, 2H), 3.94 (s, 3H), 3.93-3.86 (m, 1H), 2.66-2.55 (m, 2H), 2.42-2.31 (m, 2H), 2.22-2.14 (m, 1H), 1.79-1.65 (m, 2H), 1.52-1.27 (m, 7H), 1.22 (s, 9H). MS: 703.2 (M+1), 701.3 (M−1)
- Synthesis of Compound #906

- 1H NMR (400 MHz, DMSO-d6): □ (ppm): 8.46 (s, 1H), 8.06 (d, J=9.2 Hz, 1H), 7.57 (s, 1H), 7.49 (s, 1H), 7.34 (m, 1H), 7.14-7.05 (m, 2H), 5.63-5.58 (m, 1H), 4.66-4.61 (m, 1H), 4.54-4.44 (m, 2H),4.23-4.18 (m, 1H) 3.93 (s, 3H), 3.92-3.88 (m, 1H), 3.21-3.14 (m, 1H), 2.44-2.33 (m, 1H), 1.35 (d, J=7 Hz, 6H), 1.73-1.01 (m, 26H) MS: 762.0 (M+1), 759.9 (M−1)
- Synthesis of Compound #907

- 1H NMR (400 MHz, DMSO-d6): □ (ppm): 8.46 (s, 1H), 7.98 (d, J=8.9 Hz, 1H), 7.91-7.89 (m, 2H), 7.23-7.21 (m, 2H), 7.07-7.00 (m, 2H), 6.35-6.32 (m, 2H), 5.64-5.58 (m, 1H), 4.65-4.61 (m, 1H), 4.53-4.47 (m, 2H),4.24-4.19 (m, 1H) 3.90 (s, 3H), 3.86-3.84 (m, 1H), 2.40-2.33 (m, 1H), 1.73-1.01 (m, 26H). MS: 702.0 (M+1), 699.9 (M−1)
- Compound #825
- Using the same procedure as described in Example 34 but, in step G, using N-cyclopropylthiourea gave compound #825.

- 1H NMR (400 MHz,DMSO-d6): δ 8.55 (bs, 1H), 8.38 (bs, 1H), 8.02 (d, J=8.9 Hz, 1H), 7.53 (s, 1H), 7.42 (s, 1H), 7.28 (d, J=1.6 Hz, 11H), 7.12 (bs, 1H), 7.04 (d, J=8.3 Hz, 1H), 5.52-5.37 (m, 2H), 5.34-5.13 (m, 1H), 4.75-4.64 (m, 1H), 4.41 (d, J=15.9, 8.9 Hz, 1H), 4.33-4.18 (m, 1H), 4.09-3.98 (m, 1H), 3.98-3.83 (m, 1H), 3.91 (s, 3H), 2.57-2.43 (m, 1H), 2.06 (s, 3H), 1.78-1.18 (m, 19H), 1.16-1.12 (m, 1H), 0.78-0.72 (m, 2H), 0.61-0.54 (m, 2H). MS; es+: 773.4(M+H)+, es−: 771.5 (M−H)−.
- Synthesis of N-cyclopropylurea

- A: The KSCN was first pumped overnight under high vacuum prior to use. Then, to a solution of the KSCN (4.60 g; 47.33 mmoL) in acetone (35 mL), at 0 C., was added dropwise the benzoylchloride (5.0 mL; 43.03 mmoL). The milky solution was stirred in an ice bath for 1.5 h, then, the cyclopropylamine (3.2 mL; 46.04 mmoL) was added dropwise to the light yellow opaque mixture. The reaction mixture was stirred for 1.5 h at 0 C, then, another 500 μL cyclopropylamine (7.22 mmoL) was added and the reaction mixture stirred at RT for 30 min. at which time the reaction was determined to be complete by HPLC. The reaction mixture was poured into ice/H 2O (300 mL), stirred for 5 min. and the light yellow solid was filtered, washed several times with H2O and dried under vacuum to provide the intermediate (6.62 g).
- B: The intermediate (6.62 g) was suspended in 2N NaOH (50 mL) and heated to reflux for 15 min. HPLC indicated the complete conversion of the intermediate to the product. The solution was cooled to RT, saturated with solid NaCl and extracted into EtOAc (3×). The combined EtOAc extracts were washed with H 2O (2×) and brine (1×), dried (MgSO4), filtered and evaporated to obtain the crude product as an off-white solid. The crude product was triturated in hexane/EtOAc 5/5 to provide the N-cyclopropyl thiourea as a white crystalline-like solid (2.5 g; 50% yield over 2 steps).
- 1H NMR (400 MHz,DMSO-d6): 7.92 (bs, 1H), 7.61 (bs, 1H), 7.13 (bs, 1H), 2.39 (bs, 1H), 0.67-0.63 (m, 2H), 0.51-0.44 (m, 2H). MS; es+116.9 (M+H)+, es−: 114.8 (M−H)−.
- Synthesis of Compound #827

- Using the same procedure as described in Example 34 up to and including step H, but in step G, using N-cyclopentylthiourea gave compound #827.
- Synthesis of N-cyclopentylurea

- A: To a solution of t-butyl isothiocyanate (5.0 mL; 39.41 mmoL) in CH 2Cl2 (200 mL) was added cyclopentylamine (4.67 mL; 47.29 mmoL) followed by DIEA and the reaction mixture was stirred at RT for 2 h. The mixture was diluted with EtOAc, washed with 10% citric acid (2×), saturated NaHCO3 (2×), H2O (2×) and brine (1×). The organic layer was dried over anhydrous MgSO4, filtered and evaporated to dryness to obtain the t-butyl-cyclopentylthiourea as a white solid (3.70 g; 47% yield).
- B: The t-butyl-cyclopentylthiourea (3.70 g) was dissolved in concentrated HCl (46 mL). The dark yellow solution was set to a gentle reflux. After 40 min, the reaction mixture was allowed to cool. The volume was concentrated to approx. half under reduced pressure, cooled in ice and basified to pH 9.5 with solid and saturated NaHCO 3. The product was extracted into EtOAc (3×), the combined EtOAc extracts were washed with H2O (2×) and brine (1×). The organic layer was dried over anhydrous MgSO4, filtered and evaporated to dryness to obtain the crude N-cyclopentylthiourea as a beige solid (2.46 g crude). Trituration of the crude material in hexane/EtOAc 95/5 provided, after filtration, the N-cyclopentylthiourea as a white solid (2.38; 90% yield).
- 1H NMR (400 MHz, DMSO-d6): 7.58 (bs, 1H), 7.19 (bs, 1H), 6.76 (bs, 1H), 4.34 (bs, 1H), 1.92-1.79 (m,2H), 1.66-1.55 (m, 2H), 1.55-1.30 (m,4H). MS; es+ 144.9(M+H)+, es−: 142.8 (M−H)−. (NA SALT) 1H NMR (400 MHZ,DMSO-D6): δ 8.02 (D, J=9.2 Hz, 1H), 7.90 (D, J=6.4 Hz, 1H), 7.76 (S, 1H), 7.44 (BS, 2H), 7.27 (D, J=1.9 HZ, 1H), 7.11 (D, J=7.0 Hz, 1H), 7.03 (D, J=9.2 Hz, 1H), 5.48 (DD, J=18.4,9.9 Hz, 1H), 5.43 (BS, 1H), 5.15 (DT, J=17.8, 7.63 Hz, 1H), 4.70 (BS, 1H), 4.49-4.34 (M, 2H), 4.34-4.25 (M, 1H), 4.13-4.03 (M, 1H), 3.99-3.86 (M, 1H), 3.90 (S, 3H), 2.58-2.44 (M, 1H), 2.42-2.32 (M, 1H), 2.15-1.93 (M, 4H), 1.83-1.14 (M, 24H), 1.14-1.12 (M, 1H). MS; ES+: 801.4(M+H)+, ES−: 799.3 (M−H)−.
- Compound # 826

- 1H NMR (400 MHz,DMSO-d6):8.22 (d, J=6.0 Hz, 1H), 8.02 (d, J=9.2 Hz, 1H), 7.75 (s, 1H), 7.46 (s, 1H), 7.44 (s, 1H), 7.27 (d, J=1.9 Hz, 1H), 7.11 (d, J=6.7Hz, 1H), 7.02 (dd, J=9.5, 1.9 Hz, 1H), 5.54-5.41 (m, 1H), 5.44 (s, 1H), 5.14 (dd, J=15.9, 9.9 Hz, 1H), 4.75-4.66 (m, 1H), 4.48-4.34 (m, 2H), 4.34-4.26 (m, 1H), 4.12-4.02 (m, 2H), 3.90 (s, 3H), 2.57-2.46 (m, 1H), 2.42-2.31 (m, 3H), 2.12-1.95 (m, 4H), 1.82-1.20 (m, 20H), 1.13-1.02 (m, 1H). MS; es+: 787.4(M+H)+, es−: 785.4 (M−H)−.
- Compound #828

- 1H NMR (400 MHz,DMSO-d6):8.02 (d, J=9.2 Hz, 1H), 7.86 (bs, 1H), 7.78 (d, J=7.3 Hz, 1H), 7.43 (s, 2H), 7.27 (d, J=2.2 Hz, 1H), 7.12 (d, J=6.9 Hz, 1H), 7.03 (dd, J=9.2, 1.9 Hz, 1H), 5.57-5.40 (m, 1H), 5.40 (s, 1H), 5.26-5.17 (m, 1H), 4.70 (bs, 1H), 4.52-4.35 (m, 2H), 4.29-4.23 (m, 1H), 4.18-4.00 (m, 1H), 3.90 (s, 3H), 3.87-3.65 (m, 1H), 2.42-2.32 (m, 1H), 2.19-2.10 (m, 1H), 2.07-1.96 (m, 3H), 1.82-0.95 (m, 28H). MS; es+: 815.4(M+H)+, es−: 813.4 (M−H)−.
- Full-length NS3-NS4A heterodimer protein fluorogenic assay
- The NS2-NS5B-3′non coding region was cloned by RT-PCR into the pCR®3 vector (Invitrogen) using RNA extracted from the serum of an HCV genotype 1b infected individual (provided by Dr. Bernard Willems, Hôpital St-Luc, Montréal, Québec, Canada). The NS3-NS4A region (NS3-NS4AFL) was then subdloned by PCR into the pFastBac™ HTa baculovirus expression vector (Gibco/BRL). The vector sequence includes a region encoding a 28-residue N-terminal sequence which contains a hexahistidine tag. The Bac-to-Bac™ baculovirus expression system (Gibco/BRL) was used to produce the recombinant baculovirus. His-NS3-NS4AFL was expressed by infecting 10 6 Sf21 cells/mL with the recombinant baculovirus at a multiplicity of infection of 0.1-0.2 at 27°. Authentic auto-proteolysis occurs during expression to produce a non covalent and stable NS3-NS4A protein complex (referred to as full-length “FL”). The infected culture was harvested 48 to 64 h later by centrifugation at 4°. The cell pellet was homogenized in 50 mM NaPO4, pH 7.5, 40% glycerol (w/v), 2 mM β-mercaptoethanol, in presence of a cocktail of protease inhibitors. His-NS3-NS4AFL was then extracted from the cell lysate with 1.5% NP-40, 0.5% Triton X-100, 0.5M NaCl, and a DNase treatment. After ultracentrifugation, the soluble extract was diluted 4-fold and bound on a Pharmacia Hi-Trap Ni-chelating column. The His-NS3-NS4AFL was eluted in a >90% pure form (as judged by SDS-PAGE), using a 50 to 400 mM imidazole gradient. The His-NS3-NS4AFL was stored at −80° in 50 mM sodium phosphate, pH 7.5, 10% (w/v) glycerol, 0.5 M NaCl, 0.25 M imidazole, 0.1% NP-40. It was thawed on ice and diluted just prior to use.
- The protease activity of His-NS3-NS4AFL was assayed in 50 mM Tris-HCl, pH 8.0, 0.25 M sodium citrate, 0.01% (w/v) n-dodecyl-β-D-maltoside, 1 mM TCEP. Five (5) μM of the internally quenched substrate anthranilyl-DDIVPAbu[C(O)—O]-AMY(3-NO 2)TW-OH (SEQ. ID NO.: 1) in presence of various concentrations of inhibitor were incubated with 1.5 nM of His-NS3-NS4AFL for 45 min at 23°. The final DMSO concentration did not exceed 5.25%. The reaction was terminated with the addition of 1M MES, pH 5.8. Fluorescence of the N-terminal product was monitored on a Perkin-Elmer LS-50B fluorometer equipped with a 96-well plate reader (excitation wavelength: 325 nm; emission wavelength: 423 nm). The % inhibition was calculated with the following equation:
- 100−[(fluoinh−fluoblank)/(fluoctl−fluoblank)×100]
- A non-linear curve fit with the Hill model was applied to the inhibition-concentration data, and the 50% effective concentration (IC 50) was calculated by the use of SAS software (Statistical Software System; SAS Institute, Inc. Cary, N.C.).
- Recombinant HCV NS3 Protease Radiometric Assay
- The substrate used for the HCV NS3 protease radiometric assay, DDIVPC-SMSYTW (SEQ. ID NO.: 2), is cleaved between the cysteine and the serine residues by the enzyme. The sequence DDIVPC-SMSYTW (SEQ. ID NO.: 2) corresponds to the NS5A/NS5B natural cleavage site in which the cysteine residue in P2 has been substituted for a proline. The peptide substrate DDIVPC-SMSYTW (SEQ. ID NO.: 2) and the tracer biotin-DDIVPC-SMS[ 125I-Y]TW (SEQ. ID NO.: 3) were incubated with the recombinant NS3 protease in the absence or in the presence of inhibitors. The separation of substrate from products was performed by adding avidin-coated agarose beads to the assay mixture followed by filtration. The amount of SMS[125I-Y]TW (SEQ. ID NO.: 4) product found in the filtrate (with or without inhibitor) allowed for the calculation of the percentage of substrate conversion and of the percentage of inhibition.
- A. Reagents
- Tris and Tris-HCl (UltraPure) were obtained from Life Technologies. Glycerol (UltraPure), MES and BSA were purchased from Sigma®. TCEP was obtained from Pierce, DMSO from Aldrich® and NaOH from Anachemia®.
- Assay buffer: 50 mM Tris-HCl, pH 7.5, 30% (w/v) glycerol, 2% (w/v) CHAPS, 1 mg/mL BSA, 1 mM TCEP (TCEP added just prior to use from a 1 M stock solution in water).
- Substrate: DDIVPC-SMSYTW (SEQ. ID NO.: 2), 25 μM final concentration (from a 2 mM stock solution in DMSO stored at −20° C. to avoid oxidation).
- Tracer: reduced mono-iodinated substrate(biotin-DDIVPC-SMS[ 125I-Y]TW (SEQ. ID NO.: 3)) (≈1 nM final concentration).
- HCV NS3 protease type 1b, 25 nM final concentration (from a stock solution in 50 mM sodium phosphate, pH 7.5, 10% glycerol, 300 mM NaCl, 5 mM DTT, 0.01% NP-40).
- B. Protocol
- The assay was performed in a 96-well polypropylene plate. Each well contained: 20 μL substrate/tracer in assay buffer; 10 μL±inhibitor in 20% DMSO/assay buffer; 10 μL NS3 protease 1b.
- Blank (no inhibitor and no enzyme) and control (no inhibitor) were also prepared on the same assay plate.
- The enzymatic reaction was initiated by the addition of the enzyme solution and the assay mixture was incubated for 60 min at 23° C. under gentle agitation. Twenty (20) μL of 0.025 N NaOH were added to quench the enzymatic reaction. Twenty (20) μL of avidin-coated agarose beads (purchased from Pierce®) were added in a Millipore® MADP N65 filtration plate. The quenched assay mixture was transferred to the filtration plate, and incubated for 60 min at 23° C. under gentle agitation.
- The plates were filtered using a Millipore® MultiScreen Vacuum Manifold Filtration apparatus, and 40 μL of the filtrate was transferred to an opaque 96-well plate containing 60 μL of scintillation fluid per well.
- The filtrates were counted on a Packard® TopCount instrument using a 125I-liquid protocol for 1 minute.
- The % inhibition was calculated with the following equation:
- 100−[(countsinh−countsblank)/(countsctl−countsblank)×100]
- A non-linear curve fit with the Hill model was applied to the inhibition-concentration data, and the 50% effective concentration (IC 50) was calculated by the use of SAS software (Statistical Software System; SAS Institute, Inc., Cary, N.C.).
- Specificity Assays
- The specificity of the compounds was determined against a variety of serine proteases: human leukocyte elastase, porcine pancreatic elastase and bovine pancreatic α-chymotrypsin and one cysteine protease: human liver cathepsin B. In all cases a 96-well plate format protocol using a chromogenic substrate specific for each enzyme was used. Each assay included a 1 h enzyme-inhibitor pre-incubation at RT followed by addition of substrate and hydrolysis to ≈30% conversion as measured on a UV Thermomax® microplate reader or a fluorescence Perkin-Elmer® LS50B plate reader. Substrate concentrations were kept as low as possible compared to K M to reduce substrate competition. Compound concentrations varied from 300 to 0.06 μM depending on their potency.
- The final conditions for each assay were as follows:
- 50 mM Tris-HCl pH 8, 0.5 M Na 2SO4, 50 mM NaCl, 0.1 mM EDTA, 3% DMSO, 0.01% Tween-20 with;
- [100 μM Succ-AAPF-pNA (SEQ. ID NO.: 5) and 250 pM α-chymotrypsin], [133 μM Succ-AAA-pNA and 8 nM porcine elastase], [133 μM Succ-AAV-pNA and 8 nM leukocyte elastase]; or
- [100 mM NaHPO 4 pH 6, 1 mM EDTA, 3% DMSO, 1 mM TCEP, 0.01% Tween-20, 4 μM Z-FR-AMC (7-amino-4-methylcoumarin) and 0.5 nM cathepsin B (the stock enzyme was activated in buffer containing 20 mM TCEP before use)].
- A representative example is summarized below for porcine pancreatic elastase:
- In a polystyrene flat-bottom 96-well plate (Cellwells, Corning) were added using a Biomek liquid handler (Beckman):
- 40 μL of assay buffer (50 mM Tris-HCl pH 8, 1 M Na 2SO4, 50 mM NaCl, 0.1 mM EDTA);
- 20 μL of enzyme solution (50 mM Tris-HCl pH 8, 50 mM NaCl, 0.1 mM EDTA, 0.02% Tween-20, 40 nM porcine pancreatic elastase); and
- 20 μL of inhibitor solution (50 mM Tris-HCl, pH 8, 50 mM NaCl, 0.1 mM EDTA, 0.02% Tween-20, 1.5 mM-0.3 μM inhibitor, 15% v/v DMSO).
- After 60 min pre-incubation at RT, 20 μL of substrate solution (50 mM Tris-HCl, pH 8, 0.5 M Na 2SO4, 50 mM NaCl, 0.1 mM EDTA, 665 μM Succ-AAA-pNA) were added to each well and the reaction was further incubated at RT for 60 min after which time the absorbance was read on the UV Thermomax® plate reader. Rows of wells were allocated for controls (no inhibitor) and for blanks (no inhibitor and no enzyme).
- The sequential 2-fold dilutions of the inhibitor solution were performed on a separate plate by the liquid handler using 50 mM Tris-HCl pH 8, 50 mM NaCl, 0.1 mM EDTA, 0.02% Tween-20, 15% (v/v) DMSO. All other specificity assays were performed in a similar fashion.
- The percentage of inhibition was calculated using the formula:
- [1−((UVinh−UVblank)/(UVctl−UVblank))]×100
- A non-linear curve fit with the Hill model was applied to the inhibition-concentration data, and the 50% effective concentration (IC 50) was calculated by the use of SAS software (Statistical Software System; SAS Institute, Inc., Cary, N.C.).
- NS3 Protease Cell-Based Assay
- This assay is done with Huh-7 cells, a human cell line derived from a hepatoma, co-transfected with 2 DNA constructs:
- one (called NS3) expressing part of the HCV non-structural polyprotein fused to the tTA protein through an NS5A-NS5B cleavage site in the following order: NS3-NS4A-NS4B-NS5A-(NS5B)tTA where (NS5B) represents the 6 first amino acids of NS5B. This polyprotein is expressed under the control of the CMV promoter,
- the other (called SEAP) expressing the reporter protein, secreted alkaline phosphatase (SEAP), under the regulation of a tTA-responsive promoter.
- The first construct leads to the expression of a polyprotein from which the different mature proteins are released through cleavage by the NS3 protease. It is believed that the mature viral proteins forms a complex at the membrane of the endoplasmic reticulum. tTA is a fusion protein, described by Gossen and Bujard (Proc. Natl. Acad. Sci. USA 89 (1992): 5547-5551), which contains a DNA-binding domain and a transcriptional activator. Release of the tTA protein requires an NS3-dependent cleavage at the NS5A-NS5B cleavage site between NS5A and itself. This last cleavage allows tTA to migrate to the nucleus and transactivate the SEAP gene. Therefore, reduction of NS3 proteolytic activity leads to confinement of tTA to the cytoplasm and concomitant decrease in SEAP activity.
- To control for cellular activities other than inhibition of NS3 protease which are due to the compound, a parallel co-transfection is done with a construct expressing tTA alone and the same reporter construct such that SEAP activity is independent of the NS3 protease.
- Protocol of the assay: Huh-7 cells, grown in CHO-SFMII (Life Technologies)+10% FCS (fetal calf serum) were co-transfected with the two DNA constructs in the following proportions:
- 7 μg NS3+500 ng SEAP+800 μl FuGENE (Boehringer Mannheim) per 4×10 6 Huh-7 cells. After 5 hours at 37° C., the cells were washed, trypsinized and plated (at 80 000 cells/well) in 96-well plates containing a range of concentrations of the compounds to be tested. After a 24-hour incubation period, the SEAP activity in the medium was measured with the Phospha-Light kit (Tropix).
- Analysis of the percent inhibition of SEAP activity with respect to compound concentration was performed with the SAS software to obtain the EC 50.
- The following tables list compounds representative of the invention.
- All compounds listed in Tables 1 to 9 were found to be active in the enzymatic assay presented in Example 48. A number accompanied by an asterisk (*) represents enzymatic activity obtained with the radiometric assay presented in Example 49 with IC 50's under 50 μM. In these enzymatic assays, the following grading was used: A≧1 μM; 1 μM>B>0.1 μM; and C≦0.1 μM.
- Several compounds were tested in the specificity assays presented in Example 50 and were found to be specific for the NS3 protease. In general, the results from the different specificity assays are the following: HLE>300 μM; PPE>300 μM; α-Chym.>300 μM; Cat. B>300 μM; indicating that these compounds are highly specific toward the HCV NS3 protease and are not expected to exhibit serious side-effects.
- In addition, certain of these compounds were tested in the cell-based assay described in Example 51 and were found to have activity with an EC 50 below 10 μM, strongly indicating that these compounds can cross the cell membrane. Particularly, compounds of Tables 7, 8 and 9 have been assessed in the cellular assay and the result indicated in the last column. In this cellular assay, the following coding was used: A>1 μM; B≦1 μM.
- The following abbreviations are used within the following tables: MS: Electrospray mass spectral data; m/z MH + except when otherwise indicated by an asterisk*=m/z MH−; Ac: acetyl; Bn: benzyl; Boc: tert-butyloxycarbonyl; Ph: phenyl; Pr: propyl.
TABLE 1 
single stereoisomer at R1 Double bond position 13- between 12 R1 bond enzyme Cpd. # and 13 stereochem R22 MS activity. 101 12,13-trans 1R, position phenyl 685.8 A* 13 syn to amide 102 none 1R, position phenyl 687.2 C 13 syn to acid 103 none 1R position phenyl 687.2 A* 13 syn to amide -
TABLE 2 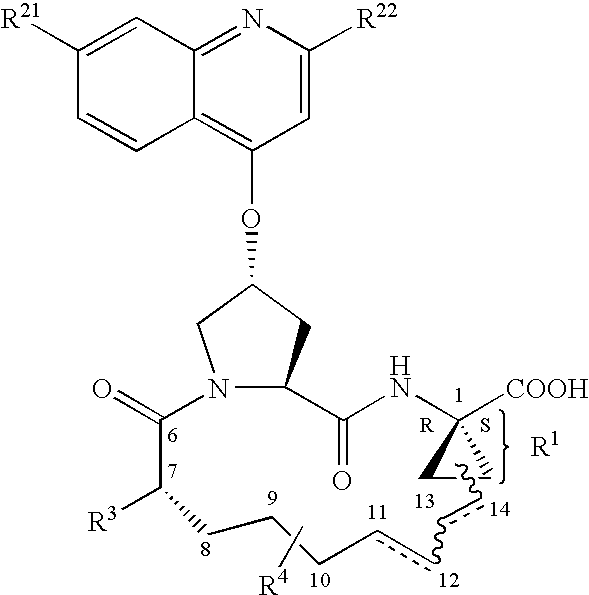
single stereoisomer at R1 position 14- Cpd double R1 bond enz. # R3 R4 bond stereochem R21 R22 MS act. 202 NH— H 11,12-trans 1R or 1S, H H 593.7 B Boc position 14 syn to acid 203 NH- H 11,12-trans 1R or 1S, H H 535.6 A acetyl position 14 syn to acid 205 NH— 11-OH none 1R or 1S, H H 627.7 B Boc 12-OH position 14 cis syn to acid 206 NH— H 13,14-cis 1R, position 14 H H 593.7 C Boc syn to acid 207 NH— H 13,14-cis 1R, OMe H 623.7 C Boc position 14 syn to acid 208 NH— H 13,14-cis 1R, OMe phenyl 699.8 C Boc position 14 syn to acid 209 NH— H 13,14-cis 1R, OMe phenyl 698.8 C C(O)— position 14 NH-tBu syn to acid 210 NH— H 13,14-cis 1S, position OMe phenyl 699.8 A* Boc 14 syn to acid 211 NH2 H 13,14-cis 1R, OMe phenyl 599.7 C position 14 syn to acid 213 OH H 13,14-cis 1R, OMe H 524.6 B (one position 14 isomer) syn to acid 214 NH— 10-oxo 13,14-cis 1R, OMe phenyl 713.8 C Boc position 14 syn to acid 215 NH— H none 1R, OMe phenyl 701.8 C Boc position 14 syn to acid 217 NH— 10-OH 13,14-cis 1R, OMe phenyl 715.8 C Boc (mixt position 14 dia syn to acid stereo) 218 NH— 10-oxo 13,14-cis 1R, position OMe phenyl 713.8 C Boc 14 syn to amide 219 NH—Ac H none 1R, OMe phenyl 643.2 C position 14 syn to acid 220 NH—Boc H 13,14-cis 1R, position 14 syn to amide OMe 
706.2 C -
TABLE 3 
single stereoisomer at R1 D—R1 Cpd. bond enz # R3 —D— stereochem R21 R22 MS act. 301 NH—Boc 
1R or 1S, D syn to acid H H 621.7 B 302 NH—Boc 
1R, D syn to amide OMe Ph 671 A 303 NH—Boc 
1R, D syn to amide OMe Ph 701.3 B 304 NH—Boc 
1R, D syn to acid OMe Ph 711.1 C 305 HO 
1R, D syn to acid OMe Ph 602.2 B 306 NH—Boc 
1R, D syn to amide OMe Ph 673.2 A* 307 NH—Boc 
1R, D syn to acid OMe 
777 C 308 NH—Ac 
1R, D syn to acid OMe OEt 609.2 C -
TABLE 4 
position 14-R1 bond syn to the acid 11, 12 Cpd double enzyme # R4 9-X bond MS activity. 401 H CH2 trans 699.3 C 402 H CH2 cis 699.4 B 403 H O trans 701.3 C 404 
O trans 715.3 B 405 
O trans 715.2 C 406 H O none 703.3 C 407 
O none 717.3 B 408 
O none 717.3 C 409 
O cis 715.2 B 410 
S trans 731.3 C 411 
S cis 731.3 A* 412 8-(Me)2 9-S cis 745.3 A -
TABLE 5 
position 14-R1 bond syn to the acid Cpd enzyme # 10-X 11-X 12-X MS activity. 501 CH2 O CH2 703.2 C 502 CH2 CH2 CH2 701 C 503 CH2 CH2 NH 702.3 A 504 CH2 CH2 N(Me) 716.3 A* 505 CH2 CH2 N(CO)Me 744.3 B 506 CH2 CH2 N(CO)Ph 806.3 B 507 NH CH2 CH2 702.3 C 508 N(CO)Me CH2 CH2 744.3 C -
TABLE 6 
position 14-R1 bond syn to the acid enzyme Cpd # R21 R22 MS activity. 601 N(Me)2 
776.2 C 602 OH (CF3) 675.2* C 603 OMe 
690.1 C -
TABLE 7 
position 14-R1 bond syn to the acid X9; X10; 13, 14 double Cpd # R4 and X11 bond R22 MS cell. act. 701 H X9 = CH2 Cis phenyl 701.3 A X10 = CH2 X11 = O 702 H CH2(All) Cis 
763.1 B 703 H CH2(All) None 
751.4 B 704 H CH2(All) Cis 
700.3 B 705 H CH2(All) Cis 
706.2 B 707 H CH2(All) Cis 
748.2 B 708 H CH2(All) Cis 
749.2 B 709 H CH2(All) None 
765.2 B 710 H CH2(All) None 
750.1 B 711 H CH2(All) None 
702.2 B 712 H CH2(All) Cis —OEt 667.3 B 713 H CH2(All) None 
708.1 B 714 H CH2(All) None —OEt 669.3 B 715 H CH2(All) Cis 
688.3 B 716 H CH2(All) Cis 
705.3 A 717 H CH2(All) Cis 
705.2 B 718 H CH2(All) Cis 
689.3 B 719 H CH2(All) Cis 
714.2 B 720 H CH2(All) None 
751.2 B 721 H CH2(All) None 
716.3 B 722 H CH2(All) Cis 
703.3 B 723 H CH2(All) None 
690.3 B 724 H CH2(All) None 
781.1 B 725 H CH2(All) Cis 
748.2 B 726 H CH2(All) Cis 
703.2 B 727 H CH2(All) Cis —CH2—OMe 667.3 A 728 H CH2(All) Cis Me 637.3 A 729 H CH2(All) Cis 
735.2 B 730 H CH2(All) None 
780.1 B 731 H CH2(All) Cis 
721.1 B 732 H CH2(All) Cis 
705.3 A 733 H CH2(All) Cis 
689.3 B 734 H CH2(All) Cis 
703.2 B 735 H CH2(All) Cis 
749.2 B 736 H CH2(All) Cis 
779.2 B 737 H CH2(All) Cis 
730.2 B 738 H CH2(All) Cis 
703.2 B 739 10-(R) CH2(All) None Ph 715.2 B Me 740 10-(S) CH2(All) none Ph 715.3 B Me 741 H CH2(All) Cis 
763.1 B -
TABLE 8 
position 14-R1 bond syn to the acid, double bond 13, 14: cis Cpd # R32 R4 R22 MS cell. act. 801 
H 
761.2 B 803 n-Pr H OEt 637.3 A 804 
H 
790.3 B 805 
H 
700.1 B 806 
H OEt 663.2 A 807 
H OEt 679.3 B 808 
H OEt 653.2 A 809 
H 
775.1 B 810 
H 
761.2 B 811 
H 
747.2 B 812 
H 
733.2 B 813 
H OEt 691.3 B 814 
H 
718.3 B 815 
H 
726.3 B 816 
H 
776.3 B 817 
H 
760.2 B 818 
H 
791.1 B 819 
H 
833.2 B 820 
H 
747.2 B 821 
H 
700.9 B 822 
H 
775.4 B 823 
H 
715.2 B 824 
10-(R) Me OEt 693.0 B 825 
H 
773.4 B 826 
H 
787.4 B 827 
H 
801.4 B 828 
H 
815.4 B -
TABLE 9 
position 14-R1 bond syn to the acid Cpd # R32 R4 R22 MS cell. act. 901 
H OEt 681.3 B 902 
H 
719.9 B 903 
H 
705.9 B 904 
H 
703.0 B 905 
H 
689.0 B 906 
H 
762.0 B 907 
H 
702.0 B 908 
H 
735.2 B 909 
H 
777.0 B 910 
H 
763.1 B 911 
H 
777.0 B 912 
H 
748.9 B 913 
H 
762.9 B 914 
H 
749.0 B 915 
H 
751.1 B 916 
10 (R) Me OEt 695.2 B -
-
1 5 1 11 PRT Artificial Sequence Substrate for NS3-NS4A heterodimer protein fluorogenic assay 1 Asp Asp Ile Val Pro Xaa Ala Met Tyr Thr Trp 1 5 10 2 12 PRT Artificial Sequence Substrate for recombinant HCV NS3 protease radiometric assay 2 Asp Asp Ile Val Pro Cys Ser Met Ser Tyr Thr Trp 1 5 10 3 12 PRT Artificial Sequence Tracer for NS3 protease assay 3 Asp Asp Ile Val Pro Cys Ser Met Ser Tyr Thr Trp 1 5 10 4 6 PRT Artificial Sequence NS3 protease C-cleavage product 4 Ser Met Ser Tyr Thr Trp 1 5 5 4 PRT Artificial Sequence Artifical substrate for chymotrypsin assay 5 Ala Ala Pro Phe 1
Claims (1)
1. A compound of formula (I):

wherein W is CH or N,
R21 is H, halo, C1-6 alkyl, C3-6 cycloalkyl, C1-6 haloalkyl, C1-6 alkoxy, C3-6 cycloalkoxy, hydroxy, or N(R23)2, wherein each R23 is independently H, C1-6 alkyl or C3-6 cycloalkyl;
R22 is H, halo, C1-6 alkyl, C3-6 cycloalkyl, C1-6 haloalkyl, C1-6 thioalkyl, C1-6 alkoxy, C3-6 cycloalkoxy, C2-7 alkoxyalkyl, C3-6 cycloalkyl, C6 or 10 aryl or Het, wherein Het is a five-, six-, or seven-membered saturated or unsaturated heterocycle containing from one to four heteroatoms selected from nitrogen, oxygen and sulfur;
said cycloalkyl, aryl or Het being substituted with R24,
wherein R24 is H, halo, C1-6 alkyl, C3-6 cycloalkyl, C1-6 alkoxy, C3-6 cycloalkoxy, NO2, N(R25)2, NH—C(O)—R25 or NH—C(O)—NH—R25, wherein each R25 is independently: H, C1-6 alkyl or C3-6 cycloalkyl;
or R24 is NH—C(O)—OR26 wherein R26 is C1-6 alkyl or C3-6 cycloalkyl;
R3 is hydroxy, NH2, or a group of formula —NH—R31, wherein R31 is C6 or 10 aryl, heteroaryl, —C(O)—R32, —C(O)—NHR32 or —C(O)—OR32,
wherein R32 is C1-6 alkyl or C3-6 cycloalkyl;
D is a 5 to 10-atom saturated or unsaturated alkylene chain optionally containing one to three heteroatoms independently selected from: O, S, or N—R41, wherein
R41 is H, C1-6 alkyl, C3-6 cycloalkyl or —C(O)—R42, wherein R42 is C1-6 alkyl, C3-6 cycloalkyl or C6 or 10 aryl;
R4 is H or from one to three substituents at any carbon atom of said chain D, said substituent independently selected from the group consisting of: C1-6 alkyl, C1-6 haloalkyl, C1-6 alkoxy, hydroxy, halo, amino, oxo, thio and C1-6 thioalkyl, and
A is an amide of formula —C(O)—NH—R5, wherein R5 is selected from the group consisting of: C1-8 alkyl, C3-6 cycloalkyl, C6 or 10 aryl and C7-16 aralkyl;
or A is a carboxylic acid or a pharmaceutically acceptable salt or ester thereof.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/358,726 US20040002448A1 (en) | 1999-04-06 | 2003-02-05 | Macrocyclic peptides active against the hepatitis C virus |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12801199P | 1999-04-06 | 1999-04-06 | |
| US54267500A | 2000-04-03 | 2000-04-03 | |
| US09/760,946 US6608027B1 (en) | 1999-04-06 | 2001-01-16 | Macrocyclic peptides active against the hepatitis C virus |
| US10/358,726 US20040002448A1 (en) | 1999-04-06 | 2003-02-05 | Macrocyclic peptides active against the hepatitis C virus |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/760,946 Continuation US6608027B1 (en) | 1999-04-06 | 2001-01-16 | Macrocyclic peptides active against the hepatitis C virus |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20040002448A1 true US20040002448A1 (en) | 2004-01-01 |
Family
ID=27736913
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/760,946 Expired - Lifetime US6608027B1 (en) | 1999-04-06 | 2001-01-16 | Macrocyclic peptides active against the hepatitis C virus |
| US10/358,726 Abandoned US20040002448A1 (en) | 1999-04-06 | 2003-02-05 | Macrocyclic peptides active against the hepatitis C virus |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/760,946 Expired - Lifetime US6608027B1 (en) | 1999-04-06 | 2001-01-16 | Macrocyclic peptides active against the hepatitis C virus |
Country Status (1)
| Country | Link |
|---|---|
| US (2) | US6608027B1 (en) |
Cited By (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20030224977A1 (en) * | 2002-01-30 | 2003-12-04 | Boehringer Ingelheim (Canada) Ltd. | Macrocyclic peptides active against the hepatitis C virus |
| US20040229777A1 (en) * | 2003-03-27 | 2004-11-18 | Boehringer Ingelheim International Gmbh | Crystalline phases of a potent HCV inhibitor |
| US20050054562A1 (en) * | 2003-06-18 | 2005-03-10 | Tranzyme Pharma Inc. | Macrocyclic antagonists of the motilin receptor |
| US20050075279A1 (en) * | 2002-10-25 | 2005-04-07 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis C virus |
| US20050080005A1 (en) * | 2003-09-22 | 2005-04-14 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis C virus |
| US20050192212A1 (en) * | 2004-01-21 | 2005-09-01 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis C virus |
| US20050201303A1 (en) * | 2004-03-09 | 2005-09-15 | Siemens Information And Communication Networks, Inc. | Distributed voice conferencing |
| US20050267018A1 (en) * | 2003-10-14 | 2005-12-01 | Blatt Lawrence M | Macrocyclic compounds as inhibitors of viral replication |
| US20070060510A1 (en) * | 2003-04-18 | 2007-03-15 | Enanta Pharmaceuticals, Inc. | Quinoxalinyl macrocyclic hepatitis C serine protease inhibitors |
| WO2008008502A1 (en) * | 2006-07-13 | 2008-01-17 | Achillion Pharmaceuticals, Inc. | 4-amino-4-oxobutanoyl peptides as inhibitors of viral replication |
| US20080019942A1 (en) * | 2006-07-05 | 2008-01-24 | Intermune, Inc | Novel inhibitors of hepatitis c virus replication |
| US20090104001A1 (en) * | 2007-10-19 | 2009-04-23 | Kochheiser Michael A | Non-dimpling fastener |
| WO2009070692A1 (en) * | 2007-11-29 | 2009-06-04 | Enanta Pharmaceuticals, Inc. | C5-substituted, proline-derived, macrocyclic hepatitis c serine protease inhibitors |
| US20090148407A1 (en) * | 2005-07-25 | 2009-06-11 | Intermune, Inc. | Novel Macrocyclic Inhibitors of Hepatitis C Virus Replication |
| US20090155209A1 (en) * | 2007-05-03 | 2009-06-18 | Blatt Lawrence M | Novel macrocyclic inhibitors of hepatitis c virus replication |
| US20090202480A1 (en) * | 2008-02-04 | 2009-08-13 | Idenix Pharmaceuticals, Inc. | Macrocyclic serine protease inhibitors |
| US20090269305A1 (en) * | 2008-04-15 | 2009-10-29 | Intermune, Inc. | Novel macrocyclic inhibitors of hepatitis c virus replication |
| US20090297476A1 (en) * | 2007-05-10 | 2009-12-03 | Intermune, Inc. | Novel peptide inhibitors of hepatitis c virus replication |
| US20100144608A1 (en) * | 2008-09-11 | 2010-06-10 | Yiyin Ku | Macrocyclic hepatitis C serine protease inhibitors |
| US20100152103A1 (en) * | 2008-12-10 | 2010-06-17 | Achillion Pharmaceuticals, Inc. | 4-amino-4-oxobutanoyl peptide cyclic analogues, inhibitors of viral replication |
| US20100168384A1 (en) * | 2009-06-30 | 2010-07-01 | Abbott Laboratories | Anti-viral compounds |
| US20100216725A1 (en) * | 2008-12-10 | 2010-08-26 | Achillion Pharmaceuticals, Inc. | 4-amino-4-oxobutanoyl peptides as inhibitors of viral replication |
| US20100221217A1 (en) * | 2009-02-27 | 2010-09-02 | Intermune, Inc. | Therapeutic composition |
| US20100292219A1 (en) * | 2007-02-26 | 2010-11-18 | Achillion Pharmaceuticals, Inc. | Tertiary amine substituted peptides useful as inhibitors of hcv replication |
| US20110081315A1 (en) * | 2009-09-28 | 2011-04-07 | Intermune, Inc. | Novel macrocyclic inhibitors of hepatitis c virus replication |
| US8119592B2 (en) | 2005-10-11 | 2012-02-21 | Intermune, Inc. | Compounds and methods for inhibiting hepatitis C viral replication |
| US8377962B2 (en) | 2009-04-08 | 2013-02-19 | Idenix Pharmaceuticals, Inc. | Macrocyclic serine protease inhibitors |
| WO2014058794A1 (en) | 2012-10-08 | 2014-04-17 | Abbvie Inc. | Compounds useful for making hcv protease inhibitors |
| US8937041B2 (en) | 2010-12-30 | 2015-01-20 | Abbvie, Inc. | Macrocyclic hepatitis C serine protease inhibitors |
| US8951964B2 (en) | 2010-12-30 | 2015-02-10 | Abbvie Inc. | Phenanthridine macrocyclic hepatitis C serine protease inhibitors |
| US9006423B2 (en) | 2013-03-15 | 2015-04-14 | Achillion Pharmaceuticals Inc. | Process for making a 4-amino-4-oxobutanoyl peptide cyclic analogue, an inhibitor of viral replication, and intermediates thereof |
| US9085607B2 (en) | 2013-03-15 | 2015-07-21 | Achillion Pharmaceuticals, Inc. | ACH-0142684 sodium salt polymorph, composition including the same, and method of manufacture thereof |
| US9115175B2 (en) | 2013-03-14 | 2015-08-25 | Achillion Pharmaceuticals, Inc. | Processes for Producing sovaprevir |
| US9227952B2 (en) | 2013-03-15 | 2016-01-05 | Achillion Pharmaceuticals, Inc. | Sovaprevir polymorphs and methods of manufacture thereof |
| US9284307B2 (en) | 2009-08-05 | 2016-03-15 | Idenix Pharmaceuticals Llc | Macrocyclic serine protease inhibitors |
| US9333204B2 (en) | 2014-01-03 | 2016-05-10 | Abbvie Inc. | Solid antiviral dosage forms |
| US9353100B2 (en) | 2011-02-10 | 2016-05-31 | Idenix Pharmaceuticals Llc | Macrocyclic serine protease inhibitors, pharmaceutical compositions thereof, and their use for treating HCV infections |
| US10201584B1 (en) | 2011-05-17 | 2019-02-12 | Abbvie Inc. | Compositions and methods for treating HCV |
Families Citing this family (170)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BR9712544B1 (en) * | 1996-10-18 | 2013-10-22 | SERINE PROTEASE INHIBITORS, PHARMACEUTICAL COMPOSITION UNDERSTANDING THE SAME AND ITS USES | |
| US6323180B1 (en) | 1998-08-10 | 2001-11-27 | Boehringer Ingelheim (Canada) Ltd | Hepatitis C inhibitor tri-peptides |
| US6608027B1 (en) * | 1999-04-06 | 2003-08-19 | Boehringer Ingelheim (Canada) Ltd | Macrocyclic peptides active against the hepatitis C virus |
| SK18862001A3 (en) * | 1999-06-25 | 2002-11-06 | Basf Aktiengesellschaft | Corynebacterium glutamicum genes encoding metabolic pathway proteins |
| SV2003000617A (en) | 2000-08-31 | 2003-01-13 | Lilly Co Eli | INHIBITORS OF PROTEASA PEPTIDOMIMETICA REF. X-14912M |
| CA2369711A1 (en) * | 2002-01-30 | 2003-07-30 | Boehringer Ingelheim (Canada) Ltd. | Macrocyclic peptides active against the hepatitis c virus |
| US20040138109A1 (en) * | 2002-09-30 | 2004-07-15 | Boehringer Ingelheim Pharmaceuticals, Inc. | Potent inhibitor of HCV serine protease |
| EP1558730B1 (en) * | 2002-10-29 | 2008-01-30 | Boehringer Ingelheim International GmbH | Inhibitor-resistant hcv ns3 protease |
| US20050159345A1 (en) * | 2002-10-29 | 2005-07-21 | Boehringer Ingelheim International Gmbh | Composition for the treatment of infection by Flaviviridae viruses |
| US7601709B2 (en) * | 2003-02-07 | 2009-10-13 | Enanta Pharmaceuticals, Inc. | Macrocyclic hepatitis C serine protease inhibitors |
| RS20050741A (en) * | 2003-04-02 | 2007-06-04 | Boehringer Ingelheim International Gmbh., | Pharmaceutical compositions for hepatitis c viral protease inhibitors |
| WO2004089974A1 (en) * | 2003-04-10 | 2004-10-21 | Boehringer Ingelheim International Gmbh | Process for the preparation of macrocyclic compounds by ruthenium complex catalysed metathesis reaction |
| US7148347B2 (en) * | 2003-04-10 | 2006-12-12 | Boehringer Ingelheim International Gmbh | Process for preparing macrocyclic compounds |
| WO2004094452A2 (en) * | 2003-04-16 | 2004-11-04 | Bristol-Myers Squibb Company | Macrocyclic isoquinoline peptide inhibitors of hepatitis c virus |
| WO2004096285A2 (en) * | 2003-04-25 | 2004-11-11 | Gilead Sciences, Inc. | Anti-infective phosphonate conjugates |
| US20050261237A1 (en) * | 2003-04-25 | 2005-11-24 | Boojamra Constantine G | Nucleoside phosphonate analogs |
| US7470724B2 (en) * | 2003-04-25 | 2008-12-30 | Gilead Sciences, Inc. | Phosphonate compounds having immuno-modulatory activity |
| WO2005002626A2 (en) * | 2003-04-25 | 2005-01-13 | Gilead Sciences, Inc. | Therapeutic phosphonate compounds |
| US7452901B2 (en) * | 2003-04-25 | 2008-11-18 | Gilead Sciences, Inc. | Anti-cancer phosphonate analogs |
| NZ542342A (en) * | 2003-04-25 | 2009-05-31 | Gilead Sciences Inc | Antiviral phosphonate analogs |
| US7407965B2 (en) * | 2003-04-25 | 2008-08-05 | Gilead Sciences, Inc. | Phosphonate analogs for treating metabolic diseases |
| EP1620109A2 (en) * | 2003-04-25 | 2006-02-01 | Gilead Sciences, Inc. | Kinase inhibitor phosphonate conjugates |
| US20090247488A1 (en) * | 2003-04-25 | 2009-10-01 | Carina Cannizzaro | Anti-inflammatory phosphonate compounds |
| CN101410120A (en) * | 2003-04-25 | 2009-04-15 | 吉里德科学公司 | Anti-inflammatory phosphonate compounds |
| US7427636B2 (en) * | 2003-04-25 | 2008-09-23 | Gilead Sciences, Inc. | Inosine monophosphate dehydrogenase inhibitory phosphonate compounds |
| US7432261B2 (en) * | 2003-04-25 | 2008-10-07 | Gilead Sciences, Inc. | Anti-inflammatory phosphonate compounds |
| CN1791599A (en) * | 2003-05-21 | 2006-06-21 | 贝林格尔.英格海姆国际有限公司 | Hepatitis c inhibitor compounds |
| US7273851B2 (en) * | 2003-06-05 | 2007-09-25 | Enanta Pharmaceuticals, Inc. | Tri-peptide hepatitis C serine protease inhibitors |
| US7125845B2 (en) * | 2003-07-03 | 2006-10-24 | Enanta Pharmaceuticals, Inc. | Aza-peptide macrocyclic hepatitis C serine protease inhibitors |
| MY148123A (en) | 2003-09-05 | 2013-02-28 | Vertex Pharma | Inhibitors of serine proteases, particularly hcv ns3-ns4a protease |
| CN1889970B (en) * | 2003-10-14 | 2012-06-13 | F·霍夫曼-罗须公司 | Macrocyclic carboxylic acids and acylsulfonamides as inhibitors of HCV replication |
| RS20060259A (en) | 2003-10-14 | 2008-08-07 | Intermune Inc., | Macrocyclic carboxylic acids and acylsulfonamides as inhibitors of hcv replication |
| WO2005044279A1 (en) * | 2003-10-24 | 2005-05-19 | Gilead Sciences, Inc. | Purine nucleoside phosphonate conjugates |
| WO2005044308A1 (en) * | 2003-10-24 | 2005-05-19 | Gilead Sciences, Inc. | Phosphonate analogs of antimetabolites |
| US7494660B2 (en) * | 2003-10-27 | 2009-02-24 | Vertex Pharmaceuticals Incorporated | HCV NS3-NS4A protease resistance mutants |
| MXPA06005683A (en) * | 2003-11-20 | 2006-12-14 | Schering Corp | Depeptidized inhibitors of hepatitis c virus ns3 protease. |
| EP1699558B1 (en) * | 2003-12-08 | 2009-03-11 | Boehringer Ingelheim International GmbH | Removal of ruthenium by-product by supercritical fluid processing |
| US20050153990A1 (en) * | 2003-12-22 | 2005-07-14 | Watkins William J. | Phosphonate substituted kinase inhibitors |
| BRPI0418031A (en) * | 2003-12-22 | 2007-04-17 | Gilead Sciences Inc | phosphonate-substituted kinase inhibitors |
| WO2005063751A1 (en) * | 2003-12-22 | 2005-07-14 | Gilead Sciences, Inc. | 4’-substituted carbovir-and abacavir-derivatives as well as related compounds with hiv and hcv antiviral activity |
| DE602005011402D1 (en) * | 2004-01-28 | 2009-01-15 | Boehringer Ingelheim Pharma | TRANSITION METAL END PRODUCTS CONTAINING REACTION SOLUTIONS |
| NZ549223A (en) * | 2004-02-27 | 2010-10-29 | Schering Corp | Sulfur compounds as inhibitors of hepatitis C virus NS3 serine protease |
| BRPI0508095A (en) * | 2004-02-27 | 2007-07-17 | Schering Corp | compounds as inhibitors of hepatitis c virus ns3 serine protease |
| KR20060124725A (en) * | 2004-02-27 | 2006-12-05 | 쉐링 코포레이션 | Inhibitors of hepatitis c virus ns3 protease |
| AU2005222056A1 (en) * | 2004-02-27 | 2005-09-22 | Schering Corporation | Novel compounds as inhibitors of hepatitis C virus NS3 serine protease |
| US7816326B2 (en) * | 2004-02-27 | 2010-10-19 | Schering Corporation | Sulfur compounds as inhibitors of hepatitis C virus NS3 serine protease |
| WO2005087730A1 (en) * | 2004-02-27 | 2005-09-22 | Schering Corporation | 3,4-(cyclopentyl)-fused proline compounds as inhibitors of hepatitis c virus ns3 serine protease |
| JP4654239B2 (en) | 2004-03-15 | 2011-03-16 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Method for preparing macrocyclic dipeptide suitable for treatment of hepatitis C virus infection |
| EA012389B1 (en) * | 2004-03-30 | 2009-10-30 | Интермун, Инк. | Macrocyclic compounds as inhibitors of viral replication |
| US7399749B2 (en) * | 2004-05-20 | 2008-07-15 | Schering Corporation | Substituted prolines as inhibitors of hepatitis C virus NS3 serine protease |
| EP1753775B1 (en) * | 2004-05-25 | 2012-12-26 | Boehringer Ingelheim International GmbH | Process for preparing acyclic hcv protease inhibitors |
| BRPI0513426A (en) | 2004-07-16 | 2007-11-27 | Gilead Sciences Inc | Amino Acid Preparation Process Useful in the Preparation of Peptide Receptor Modulators |
| CA2573346C (en) * | 2004-07-20 | 2011-09-20 | Boehringer Ingelheim International Gmbh | Hepatitis c inhibitor peptide analogs |
| UY29016A1 (en) * | 2004-07-20 | 2006-02-24 | Boehringer Ingelheim Int | ANALOGS OF INHIBITING DIPEPTIDES OF HEPATITIS C |
| AU2005330489B2 (en) | 2004-07-27 | 2011-08-25 | Gilead Sciences, Inc. | Nucleoside phosphonate conjugates as anti HIV agents |
| US7597884B2 (en) | 2004-08-09 | 2009-10-06 | Alios Biopharma, Inc. | Hyperglycosylated polypeptide variants and methods of use |
| WO2006026352A1 (en) * | 2004-08-27 | 2006-03-09 | Schering Corporation | Acylsulfonamide compounds as inhibitors of hepatitis c virus ns3 serine protease |
| EP1794179A1 (en) * | 2004-09-17 | 2007-06-13 | Boehringer Ingelheim International Gmbh | Ring-closing metathesis process in supercritical fluid |
| CA2578356C (en) | 2004-09-24 | 2013-05-28 | Boehringer Ingelheim Pharmaceuticals, Inc. | A new class of surfactant-like materials |
| WO2007001406A2 (en) * | 2004-10-05 | 2007-01-04 | Chiron Corporation | Aryl-containing macrocyclic compounds |
| WO2006052391A2 (en) * | 2004-10-14 | 2006-05-18 | Rigel Pharmaceuticals, Inc. | Heterocyclic inhibitors of ires-mediated translation and methods of use thereof |
| DE102005002336A1 (en) * | 2005-01-17 | 2006-07-20 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Process for conducting continuous olefin-ring closure metathesis in compressed carbon dioxide |
| WO2006096652A2 (en) | 2005-03-08 | 2006-09-14 | Boehringer Ingelheim International Gmbh | Process for preparing macrocyclic compounds |
| AU2006242475B2 (en) * | 2005-05-02 | 2011-07-07 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| US20070237818A1 (en) * | 2005-06-02 | 2007-10-11 | Malcolm Bruce A | Controlled-release formulation of HCV protease inhibitor and methods using the same |
| AU2006252519B2 (en) | 2005-06-02 | 2012-08-30 | Merck Sharp & Dohme Corp. | HCV protease inhibitors in combination with food |
| AU2006252553B2 (en) * | 2005-06-02 | 2012-03-29 | Merck Sharp & Dohme Corp. | Combination of HCV protease inhibitors with a surfactant |
| US7608592B2 (en) * | 2005-06-30 | 2009-10-27 | Virobay, Inc. | HCV inhibitors |
| US7470664B2 (en) * | 2005-07-20 | 2008-12-30 | Merck & Co., Inc. | HCV NS3 protease inhibitors |
| TWI382025B (en) * | 2005-07-29 | 2013-01-11 | Tibotec Pharm Ltd | Macrocylic inhibitors of hepatitis c virus |
| PE20070211A1 (en) * | 2005-07-29 | 2007-05-12 | Medivir Ab | MACROCYCLIC COMPOUNDS AS INHIBITORS OF HEPATITIS C VIRUS |
| TWI383980B (en) * | 2005-07-29 | 2013-02-01 | Tibotec Pharm Ltd | Macrocylic inhibitors of hepatitis c virus |
| AU2006275605B2 (en) * | 2005-08-01 | 2011-01-06 | Merck Sharp & Dohme Corp. | Macrocyclic peptides as HCV NS3 protease inhibitors |
| NZ565540A (en) | 2005-08-02 | 2011-06-30 | Vertex Pharma | Inhibitors of serine proteases |
| EP2364970A1 (en) | 2005-08-19 | 2011-09-14 | Vertex Pharmaceuticals Incorporated | Processes and intermediates |
| US8399615B2 (en) | 2005-08-19 | 2013-03-19 | Vertex Pharmaceuticals Incorporated | Processes and intermediates |
| US7964624B1 (en) | 2005-08-26 | 2011-06-21 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases |
| AR055395A1 (en) | 2005-08-26 | 2007-08-22 | Vertex Pharma | INHIBITING COMPOUNDS OF THE ACTIVITY OF SERINA PROTEASA NS3-NS4A OF HEPATITIS C VIRUS |
| ES2364426T3 (en) * | 2005-09-09 | 2011-09-02 | Boehringer Ingelheim International Gmbh | METAL PROCESS WITH CLOSURE OF THE RING FOR THE PREPARATION OF MACROCYCLIC PEPTIDES. |
| US7705138B2 (en) | 2005-11-11 | 2010-04-27 | Vertex Pharmaceuticals Incorporated | Hepatitis C virus variants |
| EP1951748B1 (en) | 2005-11-11 | 2013-07-24 | Vertex Pharmaceuticals, Inc. | Hepatitis c virus variants |
| KR101576159B1 (en) | 2005-11-16 | 2015-12-09 | 에스*바이오 피티이 리미티드 | Heteroalkyl linked pyrimidine derivatives |
| GB0602663D0 (en) * | 2006-02-09 | 2006-03-22 | Univ Cardiff | Removal of catalyst species |
| JP5436864B2 (en) | 2006-02-27 | 2014-03-05 | バーテックス ファーマシューティカルズ インコーポレイテッド | Co-crystal containing VX-950 and pharmaceutical composition containing the same |
| EP1993994A2 (en) | 2006-03-16 | 2008-11-26 | Vertex Pharmceuticals Incorporated | Deuterated hepatitis c protease inhibitors |
| GB0609492D0 (en) * | 2006-05-15 | 2006-06-21 | Angeletti P Ist Richerche Bio | Therapeutic agents |
| US8268776B2 (en) | 2006-06-06 | 2012-09-18 | Enanta Pharmaceuticals, Inc. | Macrocylic oximyl hepatitis C protease inhibitors |
| US9526769B2 (en) * | 2006-06-06 | 2016-12-27 | Enanta Pharmaceuticals, Inc. | Macrocylic oximyl hepatitis C protease inhibitors |
| GB0612423D0 (en) * | 2006-06-23 | 2006-08-02 | Angeletti P Ist Richerche Bio | Therapeutic agents |
| UY30437A1 (en) * | 2006-06-26 | 2008-01-31 | Enanta Pharm Inc | QUINOXALINIL MACROCECLIC INHIBITORS OF SERINE PROTEASE VIRUS OF HEPATITIS C |
| US20090035271A1 (en) * | 2007-08-01 | 2009-02-05 | Ying Sun | Tetrazolyl macrocyclic hepatitis c serine protease inhibitors |
| US7718612B2 (en) * | 2007-08-02 | 2010-05-18 | Enanta Pharmaceuticals, Inc. | Pyridazinonyl macrocyclic hepatitis C serine protease inhibitors |
| CN101674844A (en) * | 2006-08-04 | 2010-03-17 | 英安塔制药有限公司 | tetrazolyl macrocyclic hepatitis c serine protease inhibitors |
| US7662779B2 (en) * | 2006-08-11 | 2010-02-16 | Enanta Pharmaceuticals, Inc. | Triazolyl macrocyclic hepatitis C serine protease inhibitors |
| US7687459B2 (en) * | 2006-08-11 | 2010-03-30 | Enanta Pharmaceuticals, Inc. | Arylalkoxyl hepatitis C virus protease inhibitors |
| US20090098085A1 (en) * | 2006-08-11 | 2009-04-16 | Ying Sun | Tetrazolyl acyclic hepatitis c serine protease inhibitors |
| CA2666814A1 (en) * | 2006-08-21 | 2008-05-29 | United Therapeutics Corporation | Combination therapy for treatment of viral infections |
| JP2010507656A (en) * | 2006-10-24 | 2010-03-11 | メルク エンド カムパニー インコーポレーテッド | HCV NS3 protease inhibitor |
| US8309540B2 (en) * | 2006-10-24 | 2012-11-13 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| US8138164B2 (en) * | 2006-10-24 | 2012-03-20 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| ES2444575T3 (en) * | 2006-10-27 | 2014-02-25 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| KR101615500B1 (en) | 2006-10-27 | 2016-04-27 | 머크 샤프 앤드 돔 코포레이션 | HCV NS3 protease inhibitors |
| US20100081672A1 (en) * | 2006-12-07 | 2010-04-01 | Schering Corporation | Ph sensitive matrix formulation |
| WO2008075103A1 (en) * | 2006-12-20 | 2008-06-26 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Antiviral indoles |
| GB0625345D0 (en) * | 2006-12-20 | 2007-01-31 | Angeletti P Ist Richerche Bio | Therapeutic compounds |
| GB0625349D0 (en) * | 2006-12-20 | 2007-01-31 | Angeletti P Ist Richerche Bio | Therapeutic compounds |
| AU2008205116A1 (en) * | 2007-01-08 | 2008-07-17 | Phenomix Corporation | Macrocyclic hepatitis C protease inhibitors |
| NZ579295A (en) | 2007-02-27 | 2012-03-30 | Vertex Pharma | Inhibitors of serine proteases |
| SI2114924T1 (en) | 2007-02-27 | 2012-06-29 | Vertex Pharma | Co-crystals and pharmaceutical compositions comprising the same |
| US7906513B2 (en) * | 2007-04-26 | 2011-03-15 | Enanta Pharmaceuticals, Inc. | Hydrazide-containing hepatitis C serine protease inhibitors |
| WO2008134397A1 (en) * | 2007-04-26 | 2008-11-06 | Enanta Pharmaceuticals, Inc. | Aza-tripeptide hepatitis c serine protease inhibitors |
| US20080317712A1 (en) * | 2007-04-26 | 2008-12-25 | Deqiang Niu | Arylpiperidinyl and arylpyrrolidinyl tripeptide hepatitis c serine protease inhibitors |
| WO2008134395A1 (en) * | 2007-04-26 | 2008-11-06 | Enanta Pharmaceuticals, Inc. | Aza-peptide macrocyclic hepatitis c serine protease inhibitors |
| US20080279821A1 (en) * | 2007-04-26 | 2008-11-13 | Deqiang Niu | Arylpiperidinyl and arylpyrrolidinyl macrocyclic hepatitis c serine protease inhibitors |
| US20080292587A1 (en) * | 2007-04-26 | 2008-11-27 | Ying Sun | Oximyl dipeptide hepatitis c protease inhibitors |
| US20080274082A1 (en) * | 2007-04-26 | 2008-11-06 | Yonghua Gai | Oximyl hydroxyamic analogs as hepatitis c virus protease inhibitor |
| US20080286233A1 (en) * | 2007-04-26 | 2008-11-20 | Ying Sun | Piperizinyl macrocyclic hepatitis c serine protease inhibitors |
| US20090123423A1 (en) * | 2007-04-26 | 2009-05-14 | Yonghua Gai | Hydroxyamic analogs as hepatitis c virus serine protease inhibitor |
| US7910587B2 (en) * | 2007-04-26 | 2011-03-22 | Enanta Pharmaceuticals, Inc. | Quinoxalinyl dipeptide hepatitis C virus inhibitors |
| ES2381410T3 (en) | 2007-05-04 | 2012-05-28 | Vertex Pharmceuticals Incorporated | Combination therapy for the treatment of HCV infections |
| US20090005387A1 (en) * | 2007-06-26 | 2009-01-01 | Deqiang Niu | Quinoxalinyl macrocyclic hepatitis c virus serine protease inhibitors |
| CA2692145C (en) * | 2007-06-29 | 2015-03-03 | Gilead Sciences, Inc. | Antiviral compounds |
| AP2874A (en) | 2007-06-29 | 2014-03-31 | Gilead Sciences Inc | Antiviral compounds |
| CA2693533A1 (en) * | 2007-07-17 | 2009-01-22 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Macrocyclic indole derivatives for the treatment of hepatitis c infections |
| WO2009010804A1 (en) * | 2007-07-19 | 2009-01-22 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | Macrocyclic compounds as antiviral agents |
| CN101835774B (en) * | 2007-08-30 | 2014-09-17 | 弗特克斯药品有限公司 | Co-crystals and pharmaceutical compositions comprising the same |
| US8383583B2 (en) | 2007-10-26 | 2013-02-26 | Enanta Pharmaceuticals, Inc. | Macrocyclic, pyridazinone-containing hepatitis C serine protease inhibitors |
| WO2009064955A1 (en) | 2007-11-14 | 2009-05-22 | Enanta Pharmaceuticals, Inc. | Macrocyclic tetrazolyl hepatitis c serine protease inhibitors |
| WO2009070689A1 (en) * | 2007-11-29 | 2009-06-04 | Enanta Pharmaceuticals, Inc. | Bicyclic, c5-substituted proline derivatives as inhibitors of the hepatitis c virus ns3 protease |
| MX2010006209A (en) | 2007-12-05 | 2010-08-10 | Enanta Pharm Inc | Quinoxalinyl derivatives. |
| WO2009079353A1 (en) * | 2007-12-14 | 2009-06-25 | Enanta Pharmaceuticals, Inc. | Triazole-containing macrocyclic hcv serine protease inhibitors |
| WO2009100225A1 (en) | 2008-02-07 | 2009-08-13 | Virobay, Inc. | Inhibitors of cathepsin b |
| CA2719008A1 (en) * | 2008-03-20 | 2009-09-24 | Enanta Pharmaceuticals, Inc. | Fluorinated macrocyclic compounds as hepatitis c virus inhibitors |
| AU2009241445A1 (en) * | 2008-04-28 | 2009-11-05 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| WO2009148923A1 (en) | 2008-05-29 | 2009-12-10 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2009152051A1 (en) * | 2008-06-12 | 2009-12-17 | Phenomix Corporation | Synthesis of a macrocyclic hcv protease inhibitor |
| WO2010005986A1 (en) | 2008-07-08 | 2010-01-14 | Gilead Sciences, Inc. | Salts of hiv inhibitor compounds |
| RS53420B (en) * | 2008-07-22 | 2014-12-31 | Msd Italia S.R.L. | Combinations of a macrocyclic quinoxaline compound which is an hcv ns3 protease inhibitor with other hcv agents |
| US8338565B2 (en) * | 2008-08-20 | 2012-12-25 | Ensemble Therapeutics Corporation | Macrocyclic compounds for inhibition of tumor necrosis factor alpha |
| WO2010033466A1 (en) * | 2008-09-16 | 2010-03-25 | Phenomix Corporation | Macrocyclic inhibitors of hepatitis c protease |
| ME01831B (en) | 2008-09-16 | 2014-12-20 | Boehringer Ingelheim Int | Crystalline forms of a 2-thiazolyl-4-quinolinyl-oxy derivative, a potent hcv inhibitor |
| KR20110054003A (en) * | 2008-09-17 | 2011-05-24 | 베링거 인겔하임 인터내셔날 게엠베하 | Combination of hcv ns3 protease inhibitor with interferon and ribavirin |
| EP2341924A4 (en) | 2008-10-02 | 2013-01-23 | David Gladstone Inst | Methods of treating hepatitis c virus infection |
| US20100086519A1 (en) * | 2008-10-03 | 2010-04-08 | The Charlotte-Mecklenburg Hospital Authority D/B/A Carolinas Medical Center | Treatment of Hepatitis C Infection With Metalloporphyrins |
| US20100272674A1 (en) * | 2008-12-04 | 2010-10-28 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| CN102300871A (en) * | 2008-12-19 | 2011-12-28 | 吉里德科学公司 | Hcv ns3 protease inhibitors |
| US8102720B2 (en) * | 2009-02-02 | 2012-01-24 | Qualcomm Incorporated | System and method of pulse generation |
| US8975247B2 (en) | 2009-03-18 | 2015-03-10 | The Board Of Trustees Of The Leland Stanford Junion University | Methods and compositions of treating a flaviviridae family viral infection |
| US8936781B2 (en) | 2009-05-13 | 2015-01-20 | Enanta Pharmaceuticals, Inc. | Macrocyclic compounds as hepatitis C virus inhibitors |
| WO2011014487A1 (en) | 2009-07-30 | 2011-02-03 | Merck Sharp & Dohme Corp. | Hepatitis c virus ns3 protease inhibitors |
| WO2011049908A2 (en) * | 2009-10-19 | 2011-04-28 | Enanta Pharmaceuticals, Inc. | Bismacrokyclic compounds as hepatitis c virus inhibitors |
| KR20120106942A (en) | 2009-10-30 | 2012-09-27 | 베링거 인겔하임 인터내셔날 게엠베하 | Dosage regimens for hcv combination therapy comprising bi201335, interferon alpha and ribavirin |
| SG181797A1 (en) | 2009-12-18 | 2012-07-30 | Idenix Pharmaceuticals Inc | 5,5-fused arylene or heteroarylene hepatitis c virus inhibitors |
| WO2011150190A2 (en) * | 2010-05-26 | 2011-12-01 | Anacor Pharmaceuticals, Inc. | Hcv inhibitor compounds and methods of use thereof |
| AU2011330970B2 (en) | 2010-11-18 | 2016-10-20 | Metalysis Limited | Electrolysis apparatus |
| CN103415530A (en) | 2011-03-09 | 2013-11-27 | Jitsubo株式会社 | Novel cross-linked peptides containing non-peptide cross-linked structure, method for synthesizing cross-linked peptides, and novel organic compound used in method |
| US20120252721A1 (en) | 2011-03-31 | 2012-10-04 | Idenix Pharmaceuticals, Inc. | Methods for treating drug-resistant hepatitis c virus infection with a 5,5-fused arylene or heteroarylene hepatitis c virus inhibitor |
| US8957203B2 (en) * | 2011-05-05 | 2015-02-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8691757B2 (en) * | 2011-06-15 | 2014-04-08 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| BR112014013649A2 (en) | 2011-12-06 | 2020-10-27 | The Board Of Trustees Of The Leland Stanford Junior University | methods and agents for the treatment of viral diseases and uses of said agents |
| CN107964006A (en) | 2012-01-11 | 2018-04-27 | 艾伯维公司 | It is used to prepare the method for HCV protease inhibitor |
| US9598433B2 (en) * | 2012-11-02 | 2017-03-21 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2014138374A1 (en) | 2013-03-08 | 2014-09-12 | Boehringer Ingelheim International Gmbh | Oral combination therapy for treating hcv infection in specific patient sub-population |
| US9365615B2 (en) | 2013-09-09 | 2016-06-14 | Jitsubo Co., Ltd. | Cross-linked peptides containing non-peptide cross-linked structure, method for synthesizing cross-linked peptides, and novel organic compound used in method |
| US20160229866A1 (en) | 2013-09-20 | 2016-08-11 | Idenix Pharmaceuticals Inc. | Hepatitis c virus inhibitors |
| US10449210B2 (en) | 2014-02-13 | 2019-10-22 | Ligand Pharmaceuticals Inc. | Prodrug compounds and their uses |
| EP3114122A1 (en) | 2014-03-05 | 2017-01-11 | Idenix Pharmaceuticals LLC | Solid forms of a flaviviridae virus inhibitor compound and salts thereof |
| WO2015134561A1 (en) | 2014-03-05 | 2015-09-11 | Idenix Pharmaceuticals, Inc. | Pharmaceutical compositions comprising a 5,5-fused heteroarylene flaviviridae inhibitor and their use for treating or preventing flaviviridae infection |
| JP2017520545A (en) | 2014-07-02 | 2017-07-27 | リガンド・ファーマシューティカルズ・インコーポレイテッド | Prodrug compounds and their use |
| SI3661937T1 (en) | 2017-08-01 | 2021-11-30 | Gilead Sciences, Inc. | Crystalline forms of ethyl ((s)-((((2r,5r)-5-(6-amino-9h-purin-9-yl)-4-fluoro-2,5-dihydrofuran-2-yl)oxy)methyl)(phenoxy)phosphoryl)-l-alaninate (gs-9131) for treating viral infections |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6608027B1 (en) * | 1999-04-06 | 2003-08-19 | Boehringer Ingelheim (Canada) Ltd | Macrocyclic peptides active against the hepatitis C virus |
Family Cites Families (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3139421A (en) | 1960-03-14 | 1964-06-30 | Parke Davis & Co | Azo compounds and methods for producing same |
| CA2032559C (en) | 1989-12-28 | 2001-11-06 | Kiyofumi Ishikawa | Endothelin antagonistic cyclic pentapeptides |
| US5721210A (en) | 1990-07-09 | 1998-02-24 | Tanabe Seiyaku Co., Ltd. | Cyclic cell adhesion modulation compounds |
| US5192746A (en) | 1990-07-09 | 1993-03-09 | Tanabe Seiyaku Co., Ltd. | Cyclic cell adhesion modulation compounds |
| US5610054A (en) | 1992-05-14 | 1997-03-11 | Ribozyme Pharmaceuticals, Inc. | Enzymatic RNA molecule targeted against Hepatitis C virus |
| WO1994015958A2 (en) | 1993-01-08 | 1994-07-21 | Tanabe Seiyaku Co., Ltd. | Peptide inhibitors of cell adhesion |
| US5830888A (en) | 1993-04-16 | 1998-11-03 | Monsanto Company | Macrocyclic retroviral protease inhibitors |
| MY147327A (en) | 1995-06-29 | 2012-11-30 | Novartis Ag | Somatostatin peptides |
| GB9517022D0 (en) | 1995-08-19 | 1995-10-25 | Glaxo Group Ltd | Medicaments |
| US5633388A (en) | 1996-03-29 | 1997-05-27 | Viropharma Incorporated | Compounds, compositions and methods for treatment of hepatitis C |
| CA2254122A1 (en) | 1996-05-10 | 1997-11-20 | Schering Corporation | Synthetic inhibitors of hepatitis c virus ns3 protease |
| BR9712544B1 (en) | 1996-10-18 | 2013-10-22 | SERINE PROTEASE INHIBITORS, PHARMACEUTICAL COMPOSITION UNDERSTANDING THE SAME AND ITS USES | |
| GB9623908D0 (en) | 1996-11-18 | 1997-01-08 | Hoffmann La Roche | Amino acid derivatives |
| AU7127298A (en) | 1997-04-14 | 1998-11-11 | Emory University | Serine protease inhibitors |
| GB9707659D0 (en) | 1997-04-16 | 1997-06-04 | Peptide Therapeutics Ltd | Hepatitis C NS3 Protease inhibitors |
| JP2002512625A (en) | 1997-05-29 | 2002-04-23 | メルク エンド カンパニー インコーポレーテッド | Heterocyclic amide compounds as cell adhesion inhibitors |
| EP1012180B1 (en) | 1997-08-11 | 2004-12-01 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c inhibitor peptide analogues |
| AU757783B2 (en) | 1997-08-11 | 2003-03-06 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis C inhibitor peptides |
| CA2260499A1 (en) | 1998-01-29 | 1999-07-29 | Sumitomo Pharmaceuticals Company Limited | Pharmaceutical compositions for the treatment of ischemic brain damage |
| IT1299134B1 (en) | 1998-02-02 | 2000-02-29 | Angeletti P Ist Richerche Bio | PROCEDURE FOR THE PRODUCTION OF PEPTIDES WITH PROTEAS INHIBITING THE NS3 PROTEASIS OF THE HCV VIRUS, PEPTIDES SO OBTAINABLE AND PEPTIDES |
| WO1999050230A1 (en) | 1998-03-31 | 1999-10-07 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases, particularly hepatitis c virus ns3 protease |
| GB9812523D0 (en) | 1998-06-10 | 1998-08-05 | Angeletti P Ist Richerche Bio | Peptide inhibitors of hepatitis c virus ns3 protease |
| AR022061A1 (en) | 1998-08-10 | 2002-09-04 | Boehringer Ingelheim Ca Ltd | INHIBITING PEPTIDES OF HEPATITIS C, A PHARMACEUTICAL COMPOSITION CONTAINING THEM, THE USE OF THE SAME TO PREPARE A PHARMACEUTICAL COMPOSITION, THE USE OF AN INTERMEDIATE PRODUCT FOR THE PREPARATION OF THESE PEPTIDES AND A PROCEDURE FOR THE PREPARATION OF ANOGRAPH . |
| US6323180B1 (en) | 1998-08-10 | 2001-11-27 | Boehringer Ingelheim (Canada) Ltd | Hepatitis C inhibitor tri-peptides |
| EP1268525B1 (en) | 2000-04-05 | 2008-12-31 | Schering Corporation | Macrocyclic ns3-serine protease inhibitors of hepatitis c virus comprising n-cyclic p2 moieties |
| AR030558A1 (en) | 2000-04-19 | 2003-08-27 | Schering Corp | MACROCICLIC COMPOUNDS, WHICH INCLUDE ENANTIOMERS, STEROISOMERS, ROTAMERS AND TAUTOMEROS, SALTS AND PHARMACEUTICALLY ACCEPTABLE SOLVATOS, PHARMACEUTICAL COMPOSITION, A METHOD FOR THE PREPARATION AND THE USE OF SUCH COMPUTERS FOR THE INHIBITION, THE SERIES |
| AR029851A1 (en) | 2000-07-21 | 2003-07-16 | Dendreon Corp | NEW PEPTIDES AS INHIBITORS OF NS3-SERINA PROTEASA DEL VIRUS DE HEPATITIS C |
| AU2001276988B2 (en) | 2000-07-21 | 2007-01-25 | Dendreon Corporation | Peptides as NS3-serine protease inhibitors of hepatitis C virus |
| SV2003000617A (en) | 2000-08-31 | 2003-01-13 | Lilly Co Eli | INHIBITORS OF PROTEASA PEPTIDOMIMETICA REF. X-14912M |
-
2001
- 2001-01-16 US US09/760,946 patent/US6608027B1/en not_active Expired - Lifetime
-
2003
- 2003-02-05 US US10/358,726 patent/US20040002448A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6608027B1 (en) * | 1999-04-06 | 2003-08-19 | Boehringer Ingelheim (Canada) Ltd | Macrocyclic peptides active against the hepatitis C virus |
Cited By (90)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20030224977A1 (en) * | 2002-01-30 | 2003-12-04 | Boehringer Ingelheim (Canada) Ltd. | Macrocyclic peptides active against the hepatitis C virus |
| US20060089300A1 (en) * | 2002-10-25 | 2006-04-27 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis C virus |
| US7504378B2 (en) | 2002-10-25 | 2009-03-17 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis C virus |
| US20050075279A1 (en) * | 2002-10-25 | 2005-04-07 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis C virus |
| US20040229777A1 (en) * | 2003-03-27 | 2004-11-18 | Boehringer Ingelheim International Gmbh | Crystalline phases of a potent HCV inhibitor |
| US7368452B2 (en) | 2003-04-18 | 2008-05-06 | Enanta Pharmaceuticals, Inc. | Quinoxalinyl macrocyclic hepatitis C serine protease inhibitors |
| US20070060510A1 (en) * | 2003-04-18 | 2007-03-15 | Enanta Pharmaceuticals, Inc. | Quinoxalinyl macrocyclic hepatitis C serine protease inhibitors |
| US20050054562A1 (en) * | 2003-06-18 | 2005-03-10 | Tranzyme Pharma Inc. | Macrocyclic antagonists of the motilin receptor |
| US8497242B2 (en) | 2003-06-18 | 2013-07-30 | Tranzyme Pharma Inc. | Processes for intermediates for macrocyclic compounds |
| US8129561B2 (en) | 2003-06-18 | 2012-03-06 | Tranzyme Pharma Inc. | Processes for intermediates for macrocyclic compounds |
| US9181298B2 (en) | 2003-06-18 | 2015-11-10 | Ocera Therapeutics, Inc. | Intermediates for macrocyclic compounds |
| US10040751B2 (en) | 2003-06-18 | 2018-08-07 | Ocera Therapeutics, Inc. | Intermediates for macrocyclic compounds |
| US20050080005A1 (en) * | 2003-09-22 | 2005-04-14 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis C virus |
| US7642235B2 (en) | 2003-09-22 | 2010-01-05 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis C virus |
| US20050267018A1 (en) * | 2003-10-14 | 2005-12-01 | Blatt Lawrence M | Macrocyclic compounds as inhibitors of viral replication |
| US20090111969A1 (en) * | 2003-10-14 | 2009-04-30 | Blatt Lawrence M | Macrocyclic compounds as inhibitors of viral replication |
| US7491794B2 (en) | 2003-10-14 | 2009-02-17 | Intermune, Inc. | Macrocyclic compounds as inhibitors of viral replication |
| US20090105471A1 (en) * | 2003-10-14 | 2009-04-23 | Blatt Lawrence M | Macrocyclic compounds as inhibitors of viral replication |
| US20090111982A1 (en) * | 2003-10-14 | 2009-04-30 | Blatt Lawrence M | Macrocyclic compounds as inhibitors of viral replication |
| US20050192212A1 (en) * | 2004-01-21 | 2005-09-01 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis C virus |
| US7749961B2 (en) | 2004-01-21 | 2010-07-06 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis C virus |
| US8036358B2 (en) * | 2004-03-09 | 2011-10-11 | Siemens Enterprise Communications, Inc. | Distributed voice conferencing |
| US20050201303A1 (en) * | 2004-03-09 | 2005-09-15 | Siemens Information And Communication Networks, Inc. | Distributed voice conferencing |
| US8299021B2 (en) | 2005-07-25 | 2012-10-30 | Intermune, Inc. | Macrocyclic inhibitors of hepatitis C virus replication |
| US7829665B2 (en) | 2005-07-25 | 2010-11-09 | Intermune, Inc. | Macrocyclic inhibitors of hepatitis C virus replication |
| EP2305696A3 (en) * | 2005-07-25 | 2011-10-12 | Intermune, Inc. | Macrocyclic inhibitors of Hepatitis C virus replication |
| US20090148407A1 (en) * | 2005-07-25 | 2009-06-11 | Intermune, Inc. | Novel Macrocyclic Inhibitors of Hepatitis C Virus Replication |
| US8119592B2 (en) | 2005-10-11 | 2012-02-21 | Intermune, Inc. | Compounds and methods for inhibiting hepatitis C viral replication |
| US20100209391A1 (en) * | 2006-07-05 | 2010-08-19 | Intermune, Inc. | Novel inhibitors of hepatitis c virus replication |
| US20080019942A1 (en) * | 2006-07-05 | 2008-01-24 | Intermune, Inc | Novel inhibitors of hepatitis c virus replication |
| US7781474B2 (en) | 2006-07-05 | 2010-08-24 | Intermune, Inc. | Inhibitors of hepatitis C virus replication |
| US20090048297A1 (en) * | 2006-07-13 | 2009-02-19 | Achillion Pharmaceuticals | 4-amino-4-oxobutanoyl peptides as inhibitors of viral replication |
| US9610317B2 (en) | 2006-07-13 | 2017-04-04 | Achillion Pharmaceuticals, Inc. | 4-amino-4-oxobutanoyl peptides as inhibitors of viral replication |
| EA017448B1 (en) * | 2006-07-13 | 2012-12-28 | Ачиллион Фармасьютикалз, Инк. | 4-amino-4-oxobutanoyl peptides as inhibitors of viral replication |
| US9233136B2 (en) | 2006-07-13 | 2016-01-12 | Achillion Pharmaceuticals, Inc. | 4-amino-4-oxobutanoyl peptides as inhibitors of viral replication |
| EP2374812A3 (en) * | 2006-07-13 | 2012-05-02 | Achillion Pharmaceuticals, Inc. | 4-amino-4-oxobutanoyl Peptides as Inhibitors of Viral Replication |
| US7906619B2 (en) | 2006-07-13 | 2011-03-15 | Achillion Pharmaceuticals, Inc. | 4-amino-4-oxobutanoyl peptides as inhibitors of viral replication |
| AU2007272839B2 (en) * | 2006-07-13 | 2012-05-24 | Achillion Pharmaceuticals, Inc. | 4-amino-4-oxobutanoyl peptides as inhibitors of viral replication |
| WO2008008502A1 (en) * | 2006-07-13 | 2008-01-17 | Achillion Pharmaceuticals, Inc. | 4-amino-4-oxobutanoyl peptides as inhibitors of viral replication |
| US8785378B2 (en) | 2006-07-13 | 2014-07-22 | Achillion Pharmaceuticals, Inc. | Macrocyclic peptides as inhibitors of viral replication |
| US8435984B2 (en) | 2007-02-26 | 2013-05-07 | Achillion Pharmaceuticals, Inc. | Tertiary amine substituted peptides useful as inhibitors of HCV replication |
| US20100292219A1 (en) * | 2007-02-26 | 2010-11-18 | Achillion Pharmaceuticals, Inc. | Tertiary amine substituted peptides useful as inhibitors of hcv replication |
| US20090155209A1 (en) * | 2007-05-03 | 2009-06-18 | Blatt Lawrence M | Novel macrocyclic inhibitors of hepatitis c virus replication |
| US7932277B2 (en) | 2007-05-10 | 2011-04-26 | Intermune, Inc. | Peptide inhibitors of hepatitis C virus replication |
| US20090297476A1 (en) * | 2007-05-10 | 2009-12-03 | Intermune, Inc. | Novel peptide inhibitors of hepatitis c virus replication |
| US20090104001A1 (en) * | 2007-10-19 | 2009-04-23 | Kochheiser Michael A | Non-dimpling fastener |
| WO2009070692A1 (en) * | 2007-11-29 | 2009-06-04 | Enanta Pharmaceuticals, Inc. | C5-substituted, proline-derived, macrocyclic hepatitis c serine protease inhibitors |
| US8263549B2 (en) | 2007-11-29 | 2012-09-11 | Enanta Pharmaceuticals, Inc. | C5-substituted, proline-derived, macrocyclic hepatitis C serine protease inhibitors |
| US20090175822A1 (en) * | 2007-11-29 | 2009-07-09 | Moore Joel D | C5-substituted, proline-derived, macrocyclic hepatitis c serine protease inhibitors |
| US20090202480A1 (en) * | 2008-02-04 | 2009-08-13 | Idenix Pharmaceuticals, Inc. | Macrocyclic serine protease inhibitors |
| US8003659B2 (en) | 2008-02-04 | 2011-08-23 | Indenix Pharmaceuticals, Inc. | Macrocyclic serine protease inhibitors |
| US8093379B2 (en) | 2008-02-04 | 2012-01-10 | Idenix Pharmaceuticals, Inc. | Macrocyclic serine protease inhibitors |
| US20100016578A1 (en) * | 2008-02-04 | 2010-01-21 | Idenix Pharmaceuticals, Inc. | Macrocyclic serine protease inhibitors |
| US8048862B2 (en) | 2008-04-15 | 2011-11-01 | Intermune, Inc. | Macrocyclic inhibitors of hepatitis C virus replication |
| US20090269305A1 (en) * | 2008-04-15 | 2009-10-29 | Intermune, Inc. | Novel macrocyclic inhibitors of hepatitis c virus replication |
| US8642538B2 (en) | 2008-09-11 | 2014-02-04 | Abbvie, Inc. | Macrocyclic hepatitis C serine protease inhibitors |
| US20100144608A1 (en) * | 2008-09-11 | 2010-06-10 | Yiyin Ku | Macrocyclic hepatitis C serine protease inhibitors |
| US9309279B2 (en) | 2008-09-11 | 2016-04-12 | Abbvie Inc. | Macrocyclic hepatitis C serine protease inhibitors |
| US8420596B2 (en) | 2008-09-11 | 2013-04-16 | Abbott Laboratories | Macrocyclic hepatitis C serine protease inhibitors |
| EP2364309A2 (en) * | 2008-12-10 | 2011-09-14 | Achillion Pharmaceuticals, Inc. | New 4-amino-4-oxobutanoyl peptides as inhibitors of viral replication |
| US9133115B2 (en) | 2008-12-10 | 2015-09-15 | Achillion Pharmaceuticals, Inc. | 4-amino-4-oxobutanoyl peptides as inhibitors of viral replication |
| EP2364309A4 (en) * | 2008-12-10 | 2012-08-29 | Achillion Pharmaceuticals Inc | New 4-amino-4-oxobutanoyl peptides as inhibitors of viral replication |
| US20100152103A1 (en) * | 2008-12-10 | 2010-06-17 | Achillion Pharmaceuticals, Inc. | 4-amino-4-oxobutanoyl peptide cyclic analogues, inhibitors of viral replication |
| US8614180B2 (en) | 2008-12-10 | 2013-12-24 | Achillion Pharmaceuticals, Inc. | 4-amino-4-oxobutanoyl peptides as inhibitors of viral replication |
| EP2364310A4 (en) * | 2008-12-10 | 2012-05-16 | Achillion Pharmaceuticals Inc | 4-amino-4-oxobutanoyl peptide cyclic analogues, inhibitors of viral replication |
| US20100216725A1 (en) * | 2008-12-10 | 2010-08-26 | Achillion Pharmaceuticals, Inc. | 4-amino-4-oxobutanoyl peptides as inhibitors of viral replication |
| EP2364310A2 (en) * | 2008-12-10 | 2011-09-14 | Achillion Pharmaceuticals, Inc. | 4-amino-4-oxobutanoyl peptide cyclic analogues, inhibitors of viral replication |
| US8735345B2 (en) | 2009-02-27 | 2014-05-27 | Hoffmann La Roche Inc. | Therapeutic composition |
| US20100221217A1 (en) * | 2009-02-27 | 2010-09-02 | Intermune, Inc. | Therapeutic composition |
| US8377962B2 (en) | 2009-04-08 | 2013-02-19 | Idenix Pharmaceuticals, Inc. | Macrocyclic serine protease inhibitors |
| US8993595B2 (en) | 2009-04-08 | 2015-03-31 | Idenix Pharmaceuticals, Inc. | Macrocyclic serine protease inhibitors |
| US8232246B2 (en) | 2009-06-30 | 2012-07-31 | Abbott Laboratories | Anti-viral compounds |
| US20100168384A1 (en) * | 2009-06-30 | 2010-07-01 | Abbott Laboratories | Anti-viral compounds |
| US9284307B2 (en) | 2009-08-05 | 2016-03-15 | Idenix Pharmaceuticals Llc | Macrocyclic serine protease inhibitors |
| US20110081315A1 (en) * | 2009-09-28 | 2011-04-07 | Intermune, Inc. | Novel macrocyclic inhibitors of hepatitis c virus replication |
| US8951964B2 (en) | 2010-12-30 | 2015-02-10 | Abbvie Inc. | Phenanthridine macrocyclic hepatitis C serine protease inhibitors |
| US8937041B2 (en) | 2010-12-30 | 2015-01-20 | Abbvie, Inc. | Macrocyclic hepatitis C serine protease inhibitors |
| US9353100B2 (en) | 2011-02-10 | 2016-05-31 | Idenix Pharmaceuticals Llc | Macrocyclic serine protease inhibitors, pharmaceutical compositions thereof, and their use for treating HCV infections |
| US10201584B1 (en) | 2011-05-17 | 2019-02-12 | Abbvie Inc. | Compositions and methods for treating HCV |
| US10201541B1 (en) | 2011-05-17 | 2019-02-12 | Abbvie Inc. | Compositions and methods for treating HCV |
| WO2014058794A1 (en) | 2012-10-08 | 2014-04-17 | Abbvie Inc. | Compounds useful for making hcv protease inhibitors |
| US9481708B2 (en) | 2013-03-14 | 2016-11-01 | Achillion Pharmaceuticals, Inc. | Process for producing Sovaprevir |
| US9115175B2 (en) | 2013-03-14 | 2015-08-25 | Achillion Pharmaceuticals, Inc. | Processes for Producing sovaprevir |
| US9540346B2 (en) | 2013-03-15 | 2017-01-10 | Achillion Pharmaceuticals, Inc. | Sovaprevir polymorphs and methods of manufacture thereof |
| US9227952B2 (en) | 2013-03-15 | 2016-01-05 | Achillion Pharmaceuticals, Inc. | Sovaprevir polymorphs and methods of manufacture thereof |
| US9085607B2 (en) | 2013-03-15 | 2015-07-21 | Achillion Pharmaceuticals, Inc. | ACH-0142684 sodium salt polymorph, composition including the same, and method of manufacture thereof |
| US9006423B2 (en) | 2013-03-15 | 2015-04-14 | Achillion Pharmaceuticals Inc. | Process for making a 4-amino-4-oxobutanoyl peptide cyclic analogue, an inhibitor of viral replication, and intermediates thereof |
| US9333204B2 (en) | 2014-01-03 | 2016-05-10 | Abbvie Inc. | Solid antiviral dosage forms |
| US9744170B2 (en) | 2014-01-03 | 2017-08-29 | Abbvie Inc. | Solid antiviral dosage forms |
| US10105365B2 (en) | 2014-01-03 | 2018-10-23 | Abbvie Inc. | Solid antiviral dosage forms |
Also Published As
| Publication number | Publication date |
|---|---|
| US6608027B1 (en) | 2003-08-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6608027B1 (en) | Macrocyclic peptides active against the hepatitis C virus | |
| EP1169339B1 (en) | Macrocyclic peptides inhibiting the hepatitis c virus ns3 protease | |
| CA2369711A1 (en) | Macrocyclic peptides active against the hepatitis c virus | |
| EP1673385B1 (en) | Macrocyclic peptides active against the hepatitis c virus | |
| US7504378B2 (en) | Macrocyclic peptides active against the hepatitis C virus | |
| US6767991B1 (en) | Hepatitis C inhibitor peptides | |
| JP5433573B2 (en) | Macrocyclic compounds as antiviral agents | |
| JP5377290B2 (en) | Macrocyclic compounds as antiviral agents | |
| EP2477980A1 (en) | Hcv protease inhibitors | |
| JP6342922B2 (en) | Hepatitis C virus inhibitor | |
| SA00210014B1 (en) | Macrocyclic peptides actvet the hepatitis c virus | |
| CN102020698B (en) | Hepatitis c virus protease inhibitor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| STCB | Information on status: application discontinuation |
Free format text: ABANDONED -- FAILURE TO RESPOND TO AN OFFICE ACTION |