WO1992017215A1 - Contrast media - Google Patents
Contrast media Download PDFInfo
- Publication number
- WO1992017215A1 WO1992017215A1 PCT/EP1992/000698 EP9200698W WO9217215A1 WO 1992017215 A1 WO1992017215 A1 WO 1992017215A1 EP 9200698 W EP9200698 W EP 9200698W WO 9217215 A1 WO9217215 A1 WO 9217215A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- medium
- multinuclear
- cooh
- complexes
- chem
- Prior art date
Links
- 239000002872 contrast media Substances 0.000 title claims abstract description 43
- 229940039231 contrast media Drugs 0.000 title description 10
- 229910052751 metal Inorganic materials 0.000 claims abstract description 52
- 239000002184 metal Substances 0.000 claims abstract description 52
- 238000000034 method Methods 0.000 claims abstract description 49
- 229910052721 tungsten Inorganic materials 0.000 claims abstract description 43
- 229910052750 molybdenum Inorganic materials 0.000 claims abstract description 36
- 230000002708 enhancing effect Effects 0.000 claims abstract description 25
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 claims abstract description 20
- 239000010937 tungsten Substances 0.000 claims abstract description 20
- 125000004429 atom Chemical group 0.000 claims description 70
- 239000003446 ligand Substances 0.000 claims description 47
- 150000001875 compounds Chemical class 0.000 claims description 37
- 229910052760 oxygen Inorganic materials 0.000 claims description 30
- -1 dicarboxymethyl Chemical group 0.000 claims description 27
- 229910052757 nitrogen Inorganic materials 0.000 claims description 27
- 239000013522 chelant Substances 0.000 claims description 26
- RAEOEMDZDMCHJA-UHFFFAOYSA-N 2-[2-[bis(carboxymethyl)amino]ethyl-[2-[2-[bis(carboxymethyl)amino]ethyl-(carboxymethyl)amino]ethyl]amino]acetic acid Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(=O)O)CCN(CCN(CC(O)=O)CC(O)=O)CC(O)=O RAEOEMDZDMCHJA-UHFFFAOYSA-N 0.000 claims description 24
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 23
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 claims description 23
- 150000003839 salts Chemical class 0.000 claims description 20
- 229910052717 sulfur Inorganic materials 0.000 claims description 20
- 239000002253 acid Substances 0.000 claims description 17
- 229910052714 tellurium Inorganic materials 0.000 claims description 17
- 229910052711 selenium Inorganic materials 0.000 claims description 16
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 15
- 229910052702 rhenium Inorganic materials 0.000 claims description 14
- 229960001124 trientine Drugs 0.000 claims description 13
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 12
- 150000001412 amines Chemical class 0.000 claims description 11
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 11
- NBZBKCUXIYYUSX-UHFFFAOYSA-N iminodiacetic acid Chemical compound OC(=O)CNCC(O)=O NBZBKCUXIYYUSX-UHFFFAOYSA-N 0.000 claims description 11
- 229910052740 iodine Inorganic materials 0.000 claims description 11
- 229910052747 lanthanoid Inorganic materials 0.000 claims description 11
- 150000002602 lanthanoids Chemical class 0.000 claims description 11
- 239000001301 oxygen Substances 0.000 claims description 11
- 229920000768 polyamine Polymers 0.000 claims description 11
- 229910052720 vanadium Inorganic materials 0.000 claims description 11
- 229910052739 hydrogen Inorganic materials 0.000 claims description 10
- 239000001257 hydrogen Substances 0.000 claims description 10
- 229910052742 iron Inorganic materials 0.000 claims description 10
- 229910052758 niobium Inorganic materials 0.000 claims description 10
- VILCJCGEZXAXTO-UHFFFAOYSA-N 2,2,2-tetramine Chemical compound NCCNCCNCCN VILCJCGEZXAXTO-UHFFFAOYSA-N 0.000 claims description 9
- 229910052688 Gadolinium Inorganic materials 0.000 claims description 9
- QPCDCPDFJACHGM-UHFFFAOYSA-N N,N-bis{2-[bis(carboxymethyl)amino]ethyl}glycine Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(=O)O)CCN(CC(O)=O)CC(O)=O QPCDCPDFJACHGM-UHFFFAOYSA-N 0.000 claims description 9
- 150000005846 sugar alcohols Polymers 0.000 claims description 9
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 8
- 125000000022 2-aminoethyl group Chemical group [H]C([*])([H])C([H])([H])N([H])[H] 0.000 claims description 8
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 8
- 229910052794 bromium Inorganic materials 0.000 claims description 8
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 claims description 8
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 claims description 8
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 8
- 229910052703 rhodium Inorganic materials 0.000 claims description 8
- 239000005864 Sulphur Substances 0.000 claims description 7
- 238000002059 diagnostic imaging Methods 0.000 claims description 7
- 229910052698 phosphorus Inorganic materials 0.000 claims description 7
- XNCSCQSQSGDGES-UHFFFAOYSA-N 2-[2-[bis(carboxymethyl)amino]propyl-(carboxymethyl)amino]acetic acid Chemical compound OC(=O)CN(CC(O)=O)C(C)CN(CC(O)=O)CC(O)=O XNCSCQSQSGDGES-UHFFFAOYSA-N 0.000 claims description 6
- RPNUMPOLZDHAAY-UHFFFAOYSA-N Diethylenetriamine Chemical compound NCCNCCN RPNUMPOLZDHAAY-UHFFFAOYSA-N 0.000 claims description 6
- 125000003118 aryl group Chemical group 0.000 claims description 6
- 229910052713 technetium Inorganic materials 0.000 claims description 6
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 claims description 5
- 150000001720 carbohydrates Chemical class 0.000 claims description 5
- 125000004122 cyclic group Chemical group 0.000 claims description 5
- 229910052755 nonmetal Inorganic materials 0.000 claims description 5
- 125000004437 phosphorous atom Chemical group 0.000 claims description 5
- 239000007983 Tris buffer Substances 0.000 claims description 4
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 claims description 4
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 4
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 4
- 230000001225 therapeutic effect Effects 0.000 claims description 4
- 239000003937 drug carrier Substances 0.000 claims description 3
- 238000004519 manufacturing process Methods 0.000 claims description 3
- 125000000962 organic group Chemical group 0.000 claims description 3
- 229920000570 polyether Polymers 0.000 claims description 3
- ITWBWJFEJCHKSN-UHFFFAOYSA-N 1,4,7-triazonane Chemical compound C1CNCCNCCN1 ITWBWJFEJCHKSN-UHFFFAOYSA-N 0.000 claims description 2
- 125000002853 C1-C4 hydroxyalkyl group Chemical group 0.000 claims description 2
- 241001465754 Metazoa Species 0.000 claims description 2
- 125000003277 amino group Chemical group 0.000 claims description 2
- 125000004103 aminoalkyl group Chemical group 0.000 claims description 2
- ZRAOLHHYMQLCAW-UHFFFAOYSA-N tris(1h-pyrazol-5-yl) borate Chemical compound C1=CNN=C1OB(OC1=NNC=C1)OC=1C=CNN=1 ZRAOLHHYMQLCAW-UHFFFAOYSA-N 0.000 claims description 2
- NOIXNOMHHWGUTG-UHFFFAOYSA-N 2-[[4-[4-pyridin-4-yl-1-(2,2,2-trifluoroethyl)pyrazol-3-yl]phenoxy]methyl]quinoline Chemical compound C=1C=C(OCC=2N=C3C=CC=CC3=CC=2)C=CC=1C1=NN(CC(F)(F)F)C=C1C1=CC=NC=C1 NOIXNOMHHWGUTG-UHFFFAOYSA-N 0.000 claims 1
- 239000004721 Polyphenylene oxide Substances 0.000 claims 1
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 abstract description 12
- 239000011733 molybdenum Substances 0.000 abstract description 12
- 238000003384 imaging method Methods 0.000 abstract description 11
- 230000005298 paramagnetic effect Effects 0.000 abstract description 11
- 229910001385 heavy metal Inorganic materials 0.000 abstract description 9
- 238000002595 magnetic resonance imaging Methods 0.000 abstract 1
- 238000012285 ultrasound imaging Methods 0.000 abstract 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 126
- 239000000243 solution Substances 0.000 description 113
- 229910001868 water Inorganic materials 0.000 description 102
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 52
- 239000007787 solid Substances 0.000 description 46
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 45
- 238000002360 preparation method Methods 0.000 description 38
- 239000000047 product Substances 0.000 description 33
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 32
- 238000006243 chemical reaction Methods 0.000 description 30
- 238000001914 filtration Methods 0.000 description 28
- 239000000543 intermediate Substances 0.000 description 27
- 239000003921 oil Substances 0.000 description 27
- 235000019198 oils Nutrition 0.000 description 27
- 239000000203 mixture Substances 0.000 description 26
- 239000000725 suspension Substances 0.000 description 26
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 25
- QXNVGIXVLWOKEQ-UHFFFAOYSA-N Disodium Chemical compound [Na][Na] QXNVGIXVLWOKEQ-UHFFFAOYSA-N 0.000 description 25
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 24
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 23
- 239000008367 deionised water Substances 0.000 description 21
- 229910021641 deionized water Inorganic materials 0.000 description 21
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 20
- 239000010949 copper Substances 0.000 description 20
- 239000011669 selenium Substances 0.000 description 20
- 239000011734 sodium Substances 0.000 description 20
- 239000002244 precipitate Substances 0.000 description 19
- 125000000217 alkyl group Chemical group 0.000 description 16
- XEEYBQQBJWHFJM-UHFFFAOYSA-N iron Substances [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 16
- 150000002739 metals Chemical class 0.000 description 16
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- 125000004014 thioethyl group Chemical group [H]SC([H])([H])C([H])([H])* 0.000 description 15
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 14
- XLYOFNOQVPJJNP-ZSJDYOACSA-N heavy water Substances [2H]O[2H] XLYOFNOQVPJJNP-ZSJDYOACSA-N 0.000 description 14
- 239000002904 solvent Substances 0.000 description 14
- 238000003756 stirring Methods 0.000 description 14
- 150000002500 ions Chemical class 0.000 description 13
- 229910020628 SiW12O40 Inorganic materials 0.000 description 12
- 238000007792 addition Methods 0.000 description 12
- 239000000706 filtrate Substances 0.000 description 12
- 230000002829 reductive effect Effects 0.000 description 12
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical class CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 11
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 11
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 11
- 230000015572 biosynthetic process Effects 0.000 description 11
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 10
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 10
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 10
- 238000001816 cooling Methods 0.000 description 10
- 230000009467 reduction Effects 0.000 description 10
- 150000001793 charged compounds Chemical class 0.000 description 9
- 229910021645 metal ion Inorganic materials 0.000 description 9
- 230000003595 spectral effect Effects 0.000 description 9
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 8
- 238000005341 cation exchange Methods 0.000 description 8
- 239000003795 chemical substances by application Substances 0.000 description 8
- 239000011572 manganese Substances 0.000 description 8
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 8
- 238000010992 reflux Methods 0.000 description 8
- 239000000126 substance Substances 0.000 description 8
- 229910052796 boron Inorganic materials 0.000 description 7
- 229910052802 copper Inorganic materials 0.000 description 7
- 239000013078 crystal Substances 0.000 description 7
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 7
- 238000005160 1H NMR spectroscopy Methods 0.000 description 6
- 229920005654 Sephadex Polymers 0.000 description 6
- 239000012507 Sephadex™ Substances 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- 239000011575 calcium Substances 0.000 description 6
- 150000001768 cations Chemical class 0.000 description 6
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 6
- 238000010511 deprotection reaction Methods 0.000 description 6
- 239000000839 emulsion Substances 0.000 description 6
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 6
- 230000001965 increasing effect Effects 0.000 description 6
- 229920002521 macromolecule Polymers 0.000 description 6
- 229910052748 manganese Inorganic materials 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- 229910052708 sodium Inorganic materials 0.000 description 6
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 6
- 238000006467 substitution reaction Methods 0.000 description 6
- 229910052715 tantalum Inorganic materials 0.000 description 6
- 229910052723 transition metal Inorganic materials 0.000 description 6
- 150000003624 transition metals Chemical class 0.000 description 6
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 5
- 229910052692 Dysprosium Inorganic materials 0.000 description 5
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 5
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 5
- 238000005481 NMR spectroscopy Methods 0.000 description 5
- 229910008940 W(CO)6 Inorganic materials 0.000 description 5
- 229910052782 aluminium Inorganic materials 0.000 description 5
- 150000001408 amides Chemical class 0.000 description 5
- 229910052804 chromium Inorganic materials 0.000 description 5
- 239000011651 chromium Substances 0.000 description 5
- 230000000536 complexating effect Effects 0.000 description 5
- 238000001212 derivatisation Methods 0.000 description 5
- 238000001493 electron microscopy Methods 0.000 description 5
- 239000003995 emulsifying agent Substances 0.000 description 5
- 229910052732 germanium Inorganic materials 0.000 description 5
- 230000007062 hydrolysis Effects 0.000 description 5
- 238000006460 hydrolysis reaction Methods 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 229910052759 nickel Inorganic materials 0.000 description 5
- 230000003647 oxidation Effects 0.000 description 5
- 238000007254 oxidation reaction Methods 0.000 description 5
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 5
- 229910052707 ruthenium Inorganic materials 0.000 description 5
- 229910052710 silicon Inorganic materials 0.000 description 5
- 229910052718 tin Inorganic materials 0.000 description 5
- 231100000419 toxicity Toxicity 0.000 description 5
- 230000001988 toxicity Effects 0.000 description 5
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 4
- 0 CCC(N(*)*)=O Chemical compound CCC(N(*)*)=O 0.000 description 4
- 229910014813 CaC2 Inorganic materials 0.000 description 4
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 4
- 229910052689 Holmium Inorganic materials 0.000 description 4
- 101000935117 Homo sapiens Voltage-dependent P/Q-type calcium channel subunit alpha-1A Proteins 0.000 description 4
- 208000036758 Postinfectious cerebellitis Diseases 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- 102100025330 Voltage-dependent P/Q-type calcium channel subunit alpha-1A Human genes 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 239000012298 atmosphere Substances 0.000 description 4
- 229910052788 barium Inorganic materials 0.000 description 4
- TZCXTZWJZNENPQ-UHFFFAOYSA-L barium sulfate Chemical compound [Ba+2].[O-]S([O-])(=O)=O TZCXTZWJZNENPQ-UHFFFAOYSA-L 0.000 description 4
- 235000014633 carbohydrates Nutrition 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 238000010828 elution Methods 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 238000004992 fast atom bombardment mass spectroscopy Methods 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 229910052736 halogen Inorganic materials 0.000 description 4
- 150000002367 halogens Chemical group 0.000 description 4
- 125000001072 heteroaryl group Chemical group 0.000 description 4
- 125000005842 heteroatom Chemical group 0.000 description 4
- 230000007935 neutral effect Effects 0.000 description 4
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 4
- 229910000027 potassium carbonate Inorganic materials 0.000 description 4
- KXXXUIKPSVVSAW-UHFFFAOYSA-K pyranine Chemical compound [Na+].[Na+].[Na+].C1=C2C(O)=CC(S([O-])(=O)=O)=C(C=C3)C2=C2C3=C(S([O-])(=O)=O)C=C(S([O-])(=O)=O)C2=C1 KXXXUIKPSVVSAW-UHFFFAOYSA-K 0.000 description 4
- 125000001453 quaternary ammonium group Chemical group 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 238000002604 ultrasonography Methods 0.000 description 4
- 239000011701 zinc Substances 0.000 description 4
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 3
- IZQAUUVBKYXMET-UHFFFAOYSA-N 2-bromoethanamine Chemical compound NCCBr IZQAUUVBKYXMET-UHFFFAOYSA-N 0.000 description 3
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 3
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 3
- JPVYNHNXODAKFH-UHFFFAOYSA-N Cu2+ Chemical class [Cu+2] JPVYNHNXODAKFH-UHFFFAOYSA-N 0.000 description 3
- 229920000858 Cyclodextrin Polymers 0.000 description 3
- 229910052691 Erbium Inorganic materials 0.000 description 3
- 229910052693 Europium Inorganic materials 0.000 description 3
- VTLYFUHAOXGGBS-UHFFFAOYSA-N Fe3+ Chemical class [Fe+3] VTLYFUHAOXGGBS-UHFFFAOYSA-N 0.000 description 3
- OYHQOLUKZRVURQ-HZJYTTRNSA-N Linoleic acid Chemical compound CCCCC\C=C/C\C=C/CCCCCCCC(O)=O OYHQOLUKZRVURQ-HZJYTTRNSA-N 0.000 description 3
- 239000005642 Oleic acid Substances 0.000 description 3
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- 235000021314 Palmitic acid Nutrition 0.000 description 3
- 229910052777 Praseodymium Inorganic materials 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 3
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 3
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 3
- 235000021355 Stearic acid Nutrition 0.000 description 3
- YOUIDGQAIILFBW-UHFFFAOYSA-J Tungsten(IV) chloride Inorganic materials Cl[W](Cl)(Cl)Cl YOUIDGQAIILFBW-UHFFFAOYSA-J 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 229910052783 alkali metal Inorganic materials 0.000 description 3
- 150000001340 alkali metals Chemical class 0.000 description 3
- 230000029936 alkylation Effects 0.000 description 3
- 238000005804 alkylation reaction Methods 0.000 description 3
- 229910052787 antimony Inorganic materials 0.000 description 3
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 description 3
- 159000000009 barium salts Chemical class 0.000 description 3
- 229910052791 calcium Inorganic materials 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 235000012000 cholesterol Nutrition 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 229910052733 gallium Inorganic materials 0.000 description 3
- 229910052738 indium Inorganic materials 0.000 description 3
- 238000004255 ion exchange chromatography Methods 0.000 description 3
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 3
- 229910052746 lanthanum Inorganic materials 0.000 description 3
- OYHQOLUKZRVURQ-IXWMQOLASA-N linoleic acid Natural products CCCCC\C=C/C\C=C\CCCCCCCC(O)=O OYHQOLUKZRVURQ-IXWMQOLASA-N 0.000 description 3
- 235000020778 linoleic acid Nutrition 0.000 description 3
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 3
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 3
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 3
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 3
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- 235000021317 phosphate Nutrition 0.000 description 3
- 150000003003 phosphines Chemical class 0.000 description 3
- 229920001467 poly(styrenesulfonates) Polymers 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 125000003226 pyrazolyl group Chemical group 0.000 description 3
- 230000002285 radioactive effect Effects 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 238000002390 rotary evaporation Methods 0.000 description 3
- 239000002002 slurry Substances 0.000 description 3
- 239000001632 sodium acetate Substances 0.000 description 3
- 235000017281 sodium acetate Nutrition 0.000 description 3
- 239000012279 sodium borohydride Substances 0.000 description 3
- 229910000033 sodium borohydride Inorganic materials 0.000 description 3
- 229910052938 sodium sulfate Inorganic materials 0.000 description 3
- 239000011343 solid material Substances 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 239000003381 stabilizer Substances 0.000 description 3
- 239000008117 stearic acid Substances 0.000 description 3
- PORWMNRCUJJQNO-UHFFFAOYSA-N tellurium atom Chemical compound [Te] PORWMNRCUJJQNO-UHFFFAOYSA-N 0.000 description 3
- 229910052716 thallium Inorganic materials 0.000 description 3
- 229910052719 titanium Inorganic materials 0.000 description 3
- PBYZMCDFOULPGH-UHFFFAOYSA-N tungstate Chemical compound [O-][W]([O-])(=O)=O PBYZMCDFOULPGH-UHFFFAOYSA-N 0.000 description 3
- 229910052725 zinc Inorganic materials 0.000 description 3
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 2
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 2
- QBPPRVHXOZRESW-UHFFFAOYSA-N 1,4,7,10-tetraazacyclododecane Chemical compound C1CNCCNCCNCCN1 QBPPRVHXOZRESW-UHFFFAOYSA-N 0.000 description 2
- GKQHIYSTBXDYNQ-UHFFFAOYSA-M 1-dodecylpyridin-1-ium;chloride Chemical compound [Cl-].CCCCCCCCCCCC[N+]1=CC=CC=C1 GKQHIYSTBXDYNQ-UHFFFAOYSA-M 0.000 description 2
- WLZVJRRVASQFRV-UHFFFAOYSA-N 1-trityl-1,4,7-triazonane Chemical compound C1CNCCNCCN1C(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 WLZVJRRVASQFRV-UHFFFAOYSA-N 0.000 description 2
- RZESKRXOCXWCFX-UHFFFAOYSA-N 2-[bis[2-[carboxymethyl-[2-(methylamino)-2-oxoethyl]amino]ethyl]amino]acetic acid Chemical compound CNC(=O)CN(CC(O)=O)CCN(CC(O)=O)CCN(CC(O)=O)CC(=O)NC RZESKRXOCXWCFX-UHFFFAOYSA-N 0.000 description 2
- LDLCZOVUSADOIV-UHFFFAOYSA-N 2-bromoethanol Chemical compound OCCBr LDLCZOVUSADOIV-UHFFFAOYSA-N 0.000 description 2
- FXFXEPOUQLENRY-UHFFFAOYSA-N 2-tert-butyl-4-imino-3,3-dimethylbutanoic acid Chemical compound CC(C)(C)C(C(=O)O)C(C)(C)C=N FXFXEPOUQLENRY-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- 229910052695 Americium Inorganic materials 0.000 description 2
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical class NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 2
- 229910052684 Cerium Inorganic materials 0.000 description 2
- LZZYPRNAOMGNLH-UHFFFAOYSA-M Cetrimonium bromide Chemical compound [Br-].CCCCCCCCCCCCCCCC[N+](C)(C)C LZZYPRNAOMGNLH-UHFFFAOYSA-M 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 2
- FCKYPQBAHLOOJQ-UHFFFAOYSA-N Cyclohexane-1,2-diaminetetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)C1CCCCC1N(CC(O)=O)CC(O)=O FCKYPQBAHLOOJQ-UHFFFAOYSA-N 0.000 description 2
- UNXHWFMMPAWVPI-QWWZWVQMSA-N D-threitol Chemical compound OC[C@@H](O)[C@H](O)CO UNXHWFMMPAWVPI-QWWZWVQMSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- 239000002616 MRI contrast agent Substances 0.000 description 2
- 239000007832 Na2SO4 Substances 0.000 description 2
- 229910052779 Neodymium Inorganic materials 0.000 description 2
- 229910052781 Neptunium Inorganic materials 0.000 description 2
- 229910018828 PO3H2 Inorganic materials 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical group [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- 229910052772 Samarium Inorganic materials 0.000 description 2
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- STSCVKRWJPWALQ-UHFFFAOYSA-N TRIFLUOROACETIC ACID ETHYL ESTER Chemical compound CCOC(=O)C(F)(F)F STSCVKRWJPWALQ-UHFFFAOYSA-N 0.000 description 2
- 229910052776 Thorium Inorganic materials 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- 229910052770 Uranium Inorganic materials 0.000 description 2
- 229910052769 Ytterbium Inorganic materials 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 125000002947 alkylene group Chemical group 0.000 description 2
- 238000005576 amination reaction Methods 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 125000000129 anionic group Chemical group 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 150000004982 aromatic amines Chemical class 0.000 description 2
- 229910052785 arsenic Inorganic materials 0.000 description 2
- PWHCIQQGOQTFAE-UHFFFAOYSA-L barium chloride dihydrate Chemical compound O.O.[Cl-].[Cl-].[Ba+2] PWHCIQQGOQTFAE-UHFFFAOYSA-L 0.000 description 2
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 2
- 230000001588 bifunctional effect Effects 0.000 description 2
- LLSDKQJKOVVTOJ-UHFFFAOYSA-L calcium chloride dihydrate Chemical compound O.O.[Cl-].[Cl-].[Ca+2] LLSDKQJKOVVTOJ-UHFFFAOYSA-L 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- YMKDRGPMQRFJGP-UHFFFAOYSA-M cetylpyridinium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCC[N+]1=CC=CC=C1 YMKDRGPMQRFJGP-UHFFFAOYSA-M 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- 239000000539 dimer Substances 0.000 description 2
- WJJMNDUMQPNECX-UHFFFAOYSA-N dipicolinic acid Chemical compound OC(=O)C1=CC=CC(C(O)=O)=N1 WJJMNDUMQPNECX-UHFFFAOYSA-N 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 239000012153 distilled water Substances 0.000 description 2
- XJWSAJYUBXQQDR-UHFFFAOYSA-M dodecyltrimethylammonium bromide Chemical compound [Br-].CCCCCCCCCCCC[N+](C)(C)C XJWSAJYUBXQQDR-UHFFFAOYSA-M 0.000 description 2
- 238000000921 elemental analysis Methods 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 230000037406 food intake Effects 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- UIWYJDYFSGRHKR-UHFFFAOYSA-N gadolinium atom Chemical compound [Gd] UIWYJDYFSGRHKR-UHFFFAOYSA-N 0.000 description 2
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 2
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 2
- 230000005283 ground state Effects 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 239000003456 ion exchange resin Substances 0.000 description 2
- 229920003303 ion-exchange polymer Polymers 0.000 description 2
- 229910052745 lead Inorganic materials 0.000 description 2
- 125000005647 linker group Chemical group 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 150000002678 macrocyclic compounds Chemical class 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 2
- 150000002829 nitrogen Chemical class 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 150000002892 organic cations Chemical class 0.000 description 2
- 239000007800 oxidant agent Substances 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 2
- 239000011574 phosphorus Chemical group 0.000 description 2
- 229910052697 platinum Inorganic materials 0.000 description 2
- 229920000962 poly(amidoamine) Polymers 0.000 description 2
- 239000013460 polyoxometalate Substances 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 2
- 239000011347 resin Substances 0.000 description 2
- 229920005989 resin Polymers 0.000 description 2
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 235000009518 sodium iodide Nutrition 0.000 description 2
- 159000000000 sodium salts Chemical class 0.000 description 2
- GRVFOGOEDUUMBP-UHFFFAOYSA-N sodium sulfide (anhydrous) Chemical compound [Na+].[Na+].[S-2] GRVFOGOEDUUMBP-UHFFFAOYSA-N 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- NSCLYPFRBKULHE-UHFFFAOYSA-N tert-butyl 2-[2-[2-[bis(2-hydroxyethyl)amino]ethyl-[2-[2-[bis(2-hydroxyethyl)amino]ethyl-[2-[bis[2-[(2-methylpropan-2-yl)oxy]-2-oxoethyl]amino]ethyl]amino]ethyl]amino]ethyl-[2-[(2-methylpropan-2-yl)oxy]-2-oxoethyl]amino]acetate Chemical compound CC(C)(C)OC(=O)CN(CC(=O)OC(C)(C)C)CCN(CCN(CCO)CCO)CCN(CCN(CCO)CCO)CCN(CC(=O)OC(C)(C)C)CC(=O)OC(C)(C)C NSCLYPFRBKULHE-UHFFFAOYSA-N 0.000 description 2
- ZLUHJGIGYVPYJN-UHFFFAOYSA-N tert-butyl 2-[2-[2-aminoethyl-[2-[2-aminoethyl-[2-[bis[2-[(2-methylpropan-2-yl)oxy]-2-oxoethyl]amino]ethyl]amino]ethyl]amino]ethyl-[2-[(2-methylpropan-2-yl)oxy]-2-oxoethyl]amino]acetate Chemical compound CC(C)(C)OC(=O)CN(CC(=O)OC(C)(C)C)CCN(CCN)CCN(CCN)CCN(CC(=O)OC(C)(C)C)CC(=O)OC(C)(C)C ZLUHJGIGYVPYJN-UHFFFAOYSA-N 0.000 description 2
- BNWCETAHAJSBFG-UHFFFAOYSA-N tert-butyl 2-bromoacetate Chemical compound CC(C)(C)OC(=O)CBr BNWCETAHAJSBFG-UHFFFAOYSA-N 0.000 description 2
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical compound [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 description 2
- AGGKEGLBGGJEBZ-UHFFFAOYSA-N tetramethylenedisulfotetramine Chemical compound C1N(S2(=O)=O)CN3S(=O)(=O)N1CN2C3 AGGKEGLBGGJEBZ-UHFFFAOYSA-N 0.000 description 2
- 125000003396 thiol group Chemical group [H]S* 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- TUQOTMZNTHZOKS-UHFFFAOYSA-N tributylphosphine Chemical compound CCCCP(CCCC)CCCC TUQOTMZNTHZOKS-UHFFFAOYSA-N 0.000 description 2
- RXJKFRMDXUJTEX-UHFFFAOYSA-N triethylphosphine Chemical compound CCP(CC)CC RXJKFRMDXUJTEX-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- KPGXUAIFQMJJFB-UHFFFAOYSA-H tungsten hexachloride Chemical compound Cl[W](Cl)(Cl)(Cl)(Cl)Cl KPGXUAIFQMJJFB-UHFFFAOYSA-H 0.000 description 2
- 238000000870 ultraviolet spectroscopy Methods 0.000 description 2
- 229910052727 yttrium Inorganic materials 0.000 description 2
- 229910052726 zirconium Inorganic materials 0.000 description 2
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 description 1
- SKOUCJIEPRPEEZ-UHFFFAOYSA-N 1,1-bis(1-methylimidazol-2-yl)ethanol Chemical compound CN1C=CN=C1C(C)(O)C1=NC=CN1C SKOUCJIEPRPEEZ-UHFFFAOYSA-N 0.000 description 1
- ZKWQSBFSGZJNFP-UHFFFAOYSA-N 1,2-bis(dimethylphosphino)ethane Chemical compound CP(C)CCP(C)C ZKWQSBFSGZJNFP-UHFFFAOYSA-N 0.000 description 1
- VYMPLPIFKRHAAC-UHFFFAOYSA-N 1,2-ethanedithiol Chemical compound SCCS VYMPLPIFKRHAAC-UHFFFAOYSA-N 0.000 description 1
- WPWHSFAFEBZWBB-UHFFFAOYSA-N 1-butyl radical Chemical compound [CH2]CCC WPWHSFAFEBZWBB-UHFFFAOYSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- URDCARMUOSMFFI-UHFFFAOYSA-N 2-[2-[bis(carboxymethyl)amino]ethyl-(2-hydroxyethyl)amino]acetic acid Chemical compound OCCN(CC(O)=O)CCN(CC(O)=O)CC(O)=O URDCARMUOSMFFI-UHFFFAOYSA-N 0.000 description 1
- FCKYPQBAHLOOJQ-UWVGGRQHSA-N 2-[[(1s,2s)-2-[bis(carboxymethyl)amino]cyclohexyl]-(carboxymethyl)amino]acetic acid Chemical compound OC(=O)CN(CC(O)=O)[C@H]1CCCC[C@@H]1N(CC(O)=O)CC(O)=O FCKYPQBAHLOOJQ-UWVGGRQHSA-N 0.000 description 1
- DUKPLGYBRQILLM-UHFFFAOYSA-K 2-[bis[2-[carboxylatomethyl-(2-morpholin-4-yl-2-oxoethyl)amino]ethyl]amino]acetate;gadolinium(3+) Chemical compound [Gd+3].C1COCCN1C(=O)CN(CC([O-])=O)CCN(CC(=O)[O-])CCN(CC([O-])=O)CC(=O)N1CCOCC1 DUKPLGYBRQILLM-UHFFFAOYSA-K 0.000 description 1
- VAOPNARVTSNDLQ-UHFFFAOYSA-N 2-[bis[2-[carboxymethyl-(2-morpholin-4-yl-2-oxoethyl)amino]ethyl]amino]acetic acid Chemical compound C1COCCN1C(=O)CN(CC(O)=O)CCN(CC(=O)O)CCN(CC(O)=O)CC(=O)N1CCOCC1 VAOPNARVTSNDLQ-UHFFFAOYSA-N 0.000 description 1
- QFGOEJMTJKHKAF-UHFFFAOYSA-N 2-[carboxymethyl(2-chloroethyl)amino]acetic acid Chemical compound OC(=O)CN(CCCl)CC(O)=O QFGOEJMTJKHKAF-UHFFFAOYSA-N 0.000 description 1
- JRJGALCVMHPBJW-UHFFFAOYSA-N 2-[carboxymethyl(2-chloroethyl)amino]acetic acid;hydrochloride Chemical compound Cl.OC(=O)CN(CCCl)CC(O)=O JRJGALCVMHPBJW-UHFFFAOYSA-N 0.000 description 1
- PUAQLLVFLMYYJJ-UHFFFAOYSA-N 2-aminopropiophenone Chemical class CC(N)C(=O)C1=CC=CC=C1 PUAQLLVFLMYYJJ-UHFFFAOYSA-N 0.000 description 1
- MJLVLHNXEOQASX-UHFFFAOYSA-M 2-bromo-3,3-dimethylbutanoate Chemical compound CC(C)(C)C(Br)C([O-])=O MJLVLHNXEOQASX-UHFFFAOYSA-M 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- BAQCROVBDNBEEB-KSDLJXBFSA-N 3-acetamido-5-[acetyl(methyl)amino]-2,4,6-triiodo-n-[(2s,3r,4r,5s,6r)-2,4,5-trihydroxy-6-(hydroxymethyl)oxan-3-yl]benzamide Chemical compound CC(=O)N(C)C1=C(I)C(NC(C)=O)=C(I)C(C(=O)N[C@@H]2[C@H]([C@H](O)[C@@H](CO)O[C@@H]2O)O)=C1I BAQCROVBDNBEEB-KSDLJXBFSA-N 0.000 description 1
- QWMFKVNJIYNWII-UHFFFAOYSA-N 5-bromo-2-(2,5-dimethylpyrrol-1-yl)pyridine Chemical compound CC1=CC=C(C)N1C1=CC=C(Br)C=N1 QWMFKVNJIYNWII-UHFFFAOYSA-N 0.000 description 1
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 1
- 229910000497 Amalgam Inorganic materials 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 1
- 229910052686 Californium Inorganic materials 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 238000007445 Chromatographic isolation Methods 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229910052685 Curium Inorganic materials 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- OXQKEKGBFMQTML-UHFFFAOYSA-N D-glycero-D-gluco-heptitol Natural products OCC(O)C(O)C(O)C(O)C(O)CO OXQKEKGBFMQTML-UHFFFAOYSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 239000004386 Erythritol Substances 0.000 description 1
- UNXHWFMMPAWVPI-UHFFFAOYSA-N Erythritol Natural products OCC(O)C(O)CO UNXHWFMMPAWVPI-UHFFFAOYSA-N 0.000 description 1
- 102100032865 General transcription factor IIH subunit 5 Human genes 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 101000655402 Homo sapiens General transcription factor IIH subunit 5 Proteins 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 1
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 1
- 239000004166 Lanolin Substances 0.000 description 1
- 238000005684 Liebig rearrangement reaction Methods 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 description 1
- WAEMQWOKJMHJLA-UHFFFAOYSA-N Manganese(2+) Chemical compound [Mn+2] WAEMQWOKJMHJLA-UHFFFAOYSA-N 0.000 description 1
- 229910015463 Mo3S4 Inorganic materials 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 229910004878 Na2S2O4 Inorganic materials 0.000 description 1
- OXQKEKGBFMQTML-WAHCGKIUSA-N Perseitol Natural products OC[C@H](O)[C@H](O)C(O)[C@H](O)[C@H](O)CO OXQKEKGBFMQTML-WAHCGKIUSA-N 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical class OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- 229910052778 Plutonium Inorganic materials 0.000 description 1
- RVGRUAULSDPKGF-UHFFFAOYSA-N Poloxamer Chemical compound C1CO1.CC1CO1 RVGRUAULSDPKGF-UHFFFAOYSA-N 0.000 description 1
- JVWLUVNSQYXYBE-UHFFFAOYSA-N Ribitol Natural products OCC(C)C(O)C(O)CO JVWLUVNSQYXYBE-UHFFFAOYSA-N 0.000 description 1
- 239000002262 Schiff base Substances 0.000 description 1
- 150000004753 Schiff bases Chemical class 0.000 description 1
- 244000000231 Sesamum indicum Species 0.000 description 1
- 235000003434 Sesamum indicum Nutrition 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 229910052771 Terbium Inorganic materials 0.000 description 1
- WDLRUFUQRNWCPK-UHFFFAOYSA-N Tetraxetan Chemical compound OC(=O)CN1CCN(CC(O)=O)CCN(CC(O)=O)CCN(CC(O)=O)CC1 WDLRUFUQRNWCPK-UHFFFAOYSA-N 0.000 description 1
- 229910052775 Thulium Inorganic materials 0.000 description 1
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- RUSUZAGBORAKPY-UHFFFAOYSA-N acetic acid;n'-[2-(2-aminoethylamino)ethyl]ethane-1,2-diamine Chemical compound CC(O)=O.CC(O)=O.CC(O)=O.CC(O)=O.CC(O)=O.CC(O)=O.NCCNCCNCCN RUSUZAGBORAKPY-UHFFFAOYSA-N 0.000 description 1
- 239000012445 acidic reagent Substances 0.000 description 1
- 229910052768 actinide Inorganic materials 0.000 description 1
- 150000001255 actinides Chemical class 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 208000021825 aldosterone-producing adrenal cortex adenoma Diseases 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910001413 alkali metal ion Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 229910001420 alkaline earth metal ion Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000005210 alkyl ammonium group Chemical group 0.000 description 1
- 125000005600 alkyl phosphonate group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- SNAAJJQQZSMGQD-UHFFFAOYSA-N aluminum magnesium Chemical compound [Mg].[Al] SNAAJJQQZSMGQD-UHFFFAOYSA-N 0.000 description 1
- 238000010640 amide synthesis reaction Methods 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 239000003957 anion exchange resin Substances 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 230000000890 antigenic effect Effects 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- ANUAIBBBDSEVKN-UHFFFAOYSA-N benzene-1,2,4,5-tetramine Chemical compound NC1=CC(N)=C(N)C=C1N ANUAIBBBDSEVKN-UHFFFAOYSA-N 0.000 description 1
- 229910052790 beryllium Inorganic materials 0.000 description 1
- 210000000746 body region Anatomy 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- QXDMQSPYEZFLGF-UHFFFAOYSA-L calcium oxalate Chemical compound [Ca+2].[O-]C(=O)C([O-])=O QXDMQSPYEZFLGF-UHFFFAOYSA-L 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 239000006229 carbon black Substances 0.000 description 1
- 125000004181 carboxyalkyl group Chemical group 0.000 description 1
- 150000007942 carboxylates Chemical group 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000004203 carnauba wax Substances 0.000 description 1
- 235000013869 carnauba wax Nutrition 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 229940082500 cetostearyl alcohol Drugs 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 229920001429 chelating resin Polymers 0.000 description 1
- 230000009920 chelation Effects 0.000 description 1
- SERARPRVBWDEBA-GXDHUFHOSA-N chembl1994738 Chemical compound OC1=CC=CC=C1\C=N\NC1=CC=CC=C1 SERARPRVBWDEBA-GXDHUFHOSA-N 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 229910001914 chlorine tetroxide Inorganic materials 0.000 description 1
- 201000001883 cholelithiasis Diseases 0.000 description 1
- 238000011097 chromatography purification Methods 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 239000008139 complexing agent Substances 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 150000004696 coordination complex Chemical class 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 150000001924 cycloalkanes Chemical class 0.000 description 1
- 229940097362 cyclodextrins Drugs 0.000 description 1
- 125000000058 cyclopentadienyl group Chemical group C1(=CC=CC1)* 0.000 description 1
- 230000009849 deactivation Effects 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 238000001784 detoxification Methods 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 235000015872 dietary supplement Nutrition 0.000 description 1
- LJSQFQKUNVCTIA-UHFFFAOYSA-N diethyl sulfide Chemical compound CCSCC LJSQFQKUNVCTIA-UHFFFAOYSA-N 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- KBQHZAAAGSGFKK-UHFFFAOYSA-N dysprosium atom Chemical compound [Dy] KBQHZAAAGSGFKK-UHFFFAOYSA-N 0.000 description 1
- 239000008157 edible vegetable oil Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000008387 emulsifying waxe Substances 0.000 description 1
- ZSWFCLXCOIISFI-UHFFFAOYSA-N endo-cyclopentadiene Natural products C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 description 1
- UYAHIZSMUZPPFV-UHFFFAOYSA-N erbium Chemical compound [Er] UYAHIZSMUZPPFV-UHFFFAOYSA-N 0.000 description 1
- UNXHWFMMPAWVPI-ZXZARUISSA-N erythritol Chemical compound OC[C@H](O)[C@H](O)CO UNXHWFMMPAWVPI-ZXZARUISSA-N 0.000 description 1
- 235000019414 erythritol Nutrition 0.000 description 1
- 229940009714 erythritol Drugs 0.000 description 1
- IDGUHHHQCWSQLU-UHFFFAOYSA-N ethanol;hydrate Chemical compound O.CCO IDGUHHHQCWSQLU-UHFFFAOYSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- IFQUWYZCAGRUJN-UHFFFAOYSA-N ethylenediaminediacetic acid Chemical compound OC(=O)CNCCNCC(O)=O IFQUWYZCAGRUJN-UHFFFAOYSA-N 0.000 description 1
- OGPBJKLSAFTDLK-UHFFFAOYSA-N europium atom Chemical compound [Eu] OGPBJKLSAFTDLK-UHFFFAOYSA-N 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 230000005294 ferromagnetic effect Effects 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 229950004545 gadopenamide Drugs 0.000 description 1
- FBPFZTCFMRRESA-GUCUJZIJSA-N galactitol Chemical compound OC[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-GUCUJZIJSA-N 0.000 description 1
- 208000001130 gallstones Diseases 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 229910052735 hafnium Inorganic materials 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- MWSXXXZZOZFTPR-UHFFFAOYSA-N hex-3-ene-1,6-diol Chemical group OCCC=CCCO MWSXXXZZOZFTPR-UHFFFAOYSA-N 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- KJZYNXUDTRRSPN-UHFFFAOYSA-N holmium atom Chemical compound [Ho] KJZYNXUDTRRSPN-UHFFFAOYSA-N 0.000 description 1
- 239000012456 homogeneous solution Substances 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical compound [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- NTHXOOBQLCIOLC-UHFFFAOYSA-N iohexol Chemical compound OCC(O)CN(C(=O)C)C1=C(I)C(C(=O)NCC(O)CO)=C(I)C(C(=O)NCC(O)CO)=C1I NTHXOOBQLCIOLC-UHFFFAOYSA-N 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 235000019388 lanolin Nutrition 0.000 description 1
- 229940039717 lanolin Drugs 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- MMIPFLVOWGHZQD-UHFFFAOYSA-N manganese(3+) Chemical compound [Mn+3] MMIPFLVOWGHZQD-UHFFFAOYSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 125000005358 mercaptoalkyl group Chemical group 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 229910044991 metal oxide Inorganic materials 0.000 description 1
- 150000004706 metal oxides Chemical class 0.000 description 1
- OJURWUUOVGOHJZ-UHFFFAOYSA-N methyl 2-[(2-acetyloxyphenyl)methyl-[2-[(2-acetyloxyphenyl)methyl-(2-methoxy-2-oxoethyl)amino]ethyl]amino]acetate Chemical compound C=1C=CC=C(OC(C)=O)C=1CN(CC(=O)OC)CCN(CC(=O)OC)CC1=CC=CC=C1OC(C)=O OJURWUUOVGOHJZ-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 229960000554 metrizamide Drugs 0.000 description 1
- 239000000693 micelle Substances 0.000 description 1
- 239000013081 microcrystal Substances 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- MEFBJEMVZONFCJ-UHFFFAOYSA-N molybdate Chemical compound [O-][Mo]([O-])(=O)=O MEFBJEMVZONFCJ-UHFFFAOYSA-N 0.000 description 1
- 150000002763 monocarboxylic acids Chemical class 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- VLBJGMYFMCNIRG-UHFFFAOYSA-N n-(2-bromoethyl)-n-(3,3-dimethylbutanoyl)-3,3-dimethylbutanamide Chemical compound CC(C)(C)CC(=O)N(CCBr)C(=O)CC(C)(C)C VLBJGMYFMCNIRG-UHFFFAOYSA-N 0.000 description 1
- 150000004767 nitrides Chemical class 0.000 description 1
- 150000002843 nonmetals Chemical group 0.000 description 1
- 231100000956 nontoxicity Toxicity 0.000 description 1
- GLDOVTGHNKAZLK-UHFFFAOYSA-N octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCCCO GLDOVTGHNKAZLK-UHFFFAOYSA-N 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 150000004028 organic sulfates Chemical class 0.000 description 1
- 229910052762 osmium Inorganic materials 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 150000002926 oxygen Chemical class 0.000 description 1
- 239000003002 pH adjusting agent Substances 0.000 description 1
- 125000000913 palmityl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 150000004686 pentahydrates Chemical class 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-M perchlorate Chemical compound [O-]Cl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-M 0.000 description 1
- 150000004965 peroxy acids Chemical class 0.000 description 1
- OXQKEKGBFMQTML-BIVRFLNRSA-N perseitol Chemical compound OC[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO OXQKEKGBFMQTML-BIVRFLNRSA-N 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- AQSJGOWTSHOLKH-UHFFFAOYSA-N phosphite(3-) Chemical class [O-]P([O-])[O-] AQSJGOWTSHOLKH-UHFFFAOYSA-N 0.000 description 1
- 150000003017 phosphorus Chemical class 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 238000006303 photolysis reaction Methods 0.000 description 1
- 230000015843 photosynthesis, light reaction Effects 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 229960000540 polacrilin potassium Drugs 0.000 description 1
- 229960000502 poloxamer Drugs 0.000 description 1
- 229920001983 poloxamer Polymers 0.000 description 1
- 229920000729 poly(L-lysine) polymer Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- WVWZXTJUCNEUAE-UHFFFAOYSA-M potassium;1,2-bis(ethenyl)benzene;2-methylprop-2-enoate Chemical compound [K+].CC(=C)C([O-])=O.C=CC1=CC=CC=C1C=C WVWZXTJUCNEUAE-UHFFFAOYSA-M 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 239000005297 pyrex Substances 0.000 description 1
- YVVONORJTFWCED-UHFFFAOYSA-N pyridine-2-carboxylic acid pyridine-3-carboxylic acid Chemical compound OC(=O)C1=CC=CN=C1.OC(=O)C1=CC=CC=N1 YVVONORJTFWCED-UHFFFAOYSA-N 0.000 description 1
- 150000003222 pyridines Chemical class 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 1
- 239000010948 rhodium Substances 0.000 description 1
- HEBKCHPVOIAQTA-ZXFHETKHSA-N ribitol Chemical compound OC[C@H](O)[C@H](O)[C@H](O)CO HEBKCHPVOIAQTA-ZXFHETKHSA-N 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 150000003346 selenoethers Chemical class 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- JVBXVOWTABLYPX-UHFFFAOYSA-L sodium dithionite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])=O JVBXVOWTABLYPX-UHFFFAOYSA-L 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 229910052979 sodium sulfide Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 229960002920 sorbitol Drugs 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 229910052712 strontium Inorganic materials 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 125000003107 substituted aryl group Chemical group 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 150000003463 sulfur Chemical class 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 150000004772 tellurides Chemical class 0.000 description 1
- WMOVHXAZOJBABW-UHFFFAOYSA-N tert-butyl acetate Chemical compound CC(=O)OC(C)(C)C WMOVHXAZOJBABW-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- CIHOLLKRGTVIJN-UHFFFAOYSA-N tert‐butyl hydroperoxide Chemical compound CC(C)(C)OO CIHOLLKRGTVIJN-UHFFFAOYSA-N 0.000 description 1
- RAOIDOHSFRTOEL-UHFFFAOYSA-N tetrahydrothiophene Chemical compound C1CCSC1 RAOIDOHSFRTOEL-UHFFFAOYSA-N 0.000 description 1
- OULAJFUGPPVRBK-UHFFFAOYSA-N tetratriacontyl alcohol Natural products CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCO OULAJFUGPPVRBK-UHFFFAOYSA-N 0.000 description 1
- 150000007970 thio esters Chemical class 0.000 description 1
- 150000003568 thioethers Chemical class 0.000 description 1
- 150000003573 thiols Chemical group 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 239000013638 trimer Substances 0.000 description 1
- FYZFRYWTMMVDLR-UHFFFAOYSA-M trimethyl(3-trimethoxysilylpropyl)azanium;chloride Chemical compound [Cl-].CO[Si](OC)(OC)CCC[N+](C)(C)C FYZFRYWTMMVDLR-UHFFFAOYSA-M 0.000 description 1
- XFNJVJPLKCPIBV-UHFFFAOYSA-N trimethylenediamine Chemical compound NCCCN XFNJVJPLKCPIBV-UHFFFAOYSA-N 0.000 description 1
- JBWKIWSBJXDJDT-UHFFFAOYSA-N triphenylmethyl chloride Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(Cl)C1=CC=CC=C1 JBWKIWSBJXDJDT-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- SWGJCIMEBVHMTA-UHFFFAOYSA-K trisodium;6-oxido-4-sulfo-5-[(4-sulfonatonaphthalen-1-yl)diazenyl]naphthalene-2-sulfonate Chemical compound [Na+].[Na+].[Na+].C1=CC=C2C(N=NC3=C4C(=CC(=CC4=CC=C3O)S([O-])(=O)=O)S([O-])(=O)=O)=CC=C(S([O-])(=O)=O)C2=C1 SWGJCIMEBVHMTA-UHFFFAOYSA-K 0.000 description 1
- 150000003657 tungsten Chemical class 0.000 description 1
- IWIACDUBSJLWEJ-UHFFFAOYSA-N tungsten(5+) Chemical compound [W+5] IWIACDUBSJLWEJ-UHFFFAOYSA-N 0.000 description 1
- 210000004291 uterus Anatomy 0.000 description 1
- GPPXJZIENCGNKB-UHFFFAOYSA-N vanadium Chemical compound [V]#[V] GPPXJZIENCGNKB-UHFFFAOYSA-N 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000004034 viscosity adjusting agent Substances 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 238000002424 x-ray crystallography Methods 0.000 description 1
- 238000004846 x-ray emission Methods 0.000 description 1
- 239000000811 xylitol Substances 0.000 description 1
- 235000010447 xylitol Nutrition 0.000 description 1
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 1
- 229960002675 xylitol Drugs 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/0002—General or multifunctional contrast agents, e.g. chelated agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/06—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/0474—Organic compounds complexes or complex-forming compounds, i.e. wherein a radioactive metal (e.g. 111In3+) is complexed or chelated by, e.g. a N2S2, N3S, NS3, N4 chelating group
- A61K51/0482—Organic compounds complexes or complex-forming compounds, i.e. wherein a radioactive metal (e.g. 111In3+) is complexed or chelated by, e.g. a N2S2, N3S, NS3, N4 chelating group chelates from cyclic ligands, e.g. DOTA
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F11/00—Compounds containing elements of Groups 6 or 16 of the Periodic System
- C07F11/005—Compounds containing elements of Groups 6 or 16 of the Periodic System compounds without a metal-carbon linkage
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F15/00—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic System
- C07F15/02—Iron compounds
- C07F15/025—Iron compounds without a metal-carbon linkage
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2123/00—Preparations for testing in vivo
Definitions
- the present invention relates to the use in
- diagnostic imaging in particular X-ray, ultrasound and scintigraphy of contrast agents comprising complexes of multinuclear moieties, and to contrast media containing such complexes.
- All diagnostic imaging is based on the achievement of different signal levels from different structures within the body.
- X-ray imaging for example, for a given body structure to be visible in the image, the X-ray attenuation by that structure must differ from that of the surrounding tissues.
- the difference in signal between the body structure and its surroundings is frequently termed contrast and much effort has been devoted to means of enhancing contrast in diagnostic imaging since the greater the contrast between a body structure and its surroundings the higher the quality of the images and the greater their value to the physician performing the diagnosis.
- the greater the contrast the smaller the body structures that may be visualized in the imaging procedure i.e. increased contrast can lead to increased spatial resolution.
- the diagnostic quality of images is strongly dependent on the inherent noise level in the imaging procedure - and the ratio of the contrast level to the noise level can thus be seen to represent an effective diagnostic quality factor for diagnostic images.
- contrast agents were insoluble inorganic barium salts which enhanced X-ray attenuation in the body zones into which they distributed. More recently the field of X- ray contrast agents has been dominated by soluble iodine containing compounds such as those marketed by Nycomed AS under the trade names Omnipaque and Amipaque.
- APCA aminopolycarboxylic acid
- contrast enhancement may be achieved particularly effectively by the use of multinuclear complexes, that is complexes wherein the complexed moiety itself comprises two or more contrast enhancing atoms.
- the complex would comprise two or more heavy metal atoms and for MRI the complex would contain two or more metal atoms with paramagnetic properties.
- the word "atom” is used to refer to ionic and covalently bonded forms and not simply to isolated uncharged atoms.
- the complexed moiety while it is polynuclear, will not generally be so large as to be considered to be a particle itself. Thus it will generally have maximum dimensions 500 ⁇ or less, e.g. of 80 ⁇ or less, especially 40 ⁇ or less. More particularly, the multinuclear entity will have a distinct,
- the invention provides a diagnostic imaging contrast medium comprising a physiologically tolerable multinuclear complex of formula I
- M n B u A v is a multinuclear entity; each M which may be the same or different is a contrast enhancing metal atom covalently bonded to at least one atom B where u is non-zero; each B which may be the same or different is a non-metal bridging atom covalently bonded to at least two metal atoms M and optionally to further atoms; each A which may be the same or different is a non-metal non-bridging atom covalently bonded to a metal atom M; each L which may be the same or different is a ligand co-ordinately bonded to at least one metal atom M; n is a positive integer of value 2 or greater at least one metal atom M being tungsten or a lanthanide where n represents 2; u is zero or a positive integer, u being at least 2 unless n is at least 5 or at least one M is a lanthanide; x is a positive integer; and v and v
- physiologically tolerable salt thereof together with at least one pharmaceutical carrier or excipient.
- the invention provides the use of a multinuclear complex for the manufacture of a contrast medium as defined above for use in imaging of the human or non-human animal body.
- the invention provides a method of generating an image of a human or non-human animal, preferably mammalian, body which method comprises administering to said body a
- physiologically tolerable contrast enhancing amount of a multinuclear complex as defined above and generating an image of at least part of said body into which said agent distributes, e.g. by X-ray, MRI, ultrasound, or scintigraphy.
- the invention also provides a multinuclear complex, especially a complex of group Ib, IIb, IIIb, IVb, Vb, VIb, VIIb or VIII (CAS) metals, or lanthanides or actinides more especially group Vb, VIb, VIIb or VIII metals, particularly
- the invention also provides a diagnostic imaging contrast medium comprising a multinuclear complex as defined above together with at least one sterile pharmaceutical carrier or excipient.
- Multinuclear complexes have particular potential as contrast agents since, relative to mononuclear complexes such as the paramagnetic metal ion APCA chelates and polychelates conventionally proposed for use as X-ray contrast agents, the increase in the contrast enhancing atom content of the molecule is achieved with relatively little increase in the volume occupied by the contrast agent complexes, that is to say the use of multinuclear complexes enables a high ratio of contrast enhancing atom to overall complex volume to be achieved.
- the total quantity of the contrast agent necessary in order to achieve the same contrast effect may be reduced and thus problems associated with contrast agent solubility or toxicity or with contrast medium viscosity may also be reduced.
- the complexed moiety should comprise two or more contrast enhancing metal atoms (preferably in the form of a molecular ion).
- the multinuclear moiety also contains further atoms which may have little or no contrast enhancing effect but which may for example function as bridging atoms bonding the contrast
- bridging atoms and groups include the atoms of groups IVa, Va, VIa and VIIa (CAS), e.g. oxygen, sulphur, selenium, tellurium, halogen atoms, nitrides, and bridging groups such as hydroxyl, carboxylates, alkoxy, aryloxy, phosphorus containing compounds and substituted nitrogen and phosphorus atoms, e.g. alkyl substituted nitrogen, phosphorus or P-oxide phosphorus atoms.
- groups IVa, Va, VIa and VIIa e.g. oxygen, sulphur, selenium, tellurium, halogen atoms, nitrides
- bridging groups such as hydroxyl, carboxylates, alkoxy, aryloxy, phosphorus containing compounds and substituted nitrogen and phosphorus atoms, e.g. alkyl substituted nitrogen, phosphorus or P-oxide phosphorus atoms.
- Carbonyl bridging groups are not favourable due to their inherent toxic effect in vivo and, advantageously the multinuclear complex will not contain such groups.
- the complexed multinuclear moiety will contain at least 2, for example up to 100 (or even greater), especially up to 60, especially up to 50, for example up to 30, such as 2-15, especially 2 to 6, preferably 3 to 5 contrast enhancing metal atoms, particularly preferably 2, 3 or 4.
- the appropriate nature, e.g. the element, the isotope or the oxidation state, of the contrast enhancing metal atoms is of course dependent on the imaging technique in which the multinuclear complex is intended to function as a contrast agent. While generally speaking the
- multinuclear moiety will preferably contain at least two group Vb, VIb, VIIb or VIII atoms it may additionally contain other metal, especially transition metal, atoms and for X-ray imaging the multinuclear moiety will conveniently contain two or preferably three or more contrast enhancing atoms having atomic numbers of at least 37, preferably at least 50, while for scintigraphy the moiety will conveniently contain contrast enhancing atoms which are radioactive isotopes, e.g. radioactive metal ions, and for MRI the complex preferably contains paramagnetic metal atoms, especially lannhanide or first or second row transition metal atoms.
- a ferromagnetically coupled paramagnetic complex useful for MRI contrast enhancement may thus take the form of a multinuclear complex containing two or more paramagnetic metal atoms selected from chromium, iron, nickel, manganese, cobalt, vanadium, molybdenum,
- the metal ions include chromium, manganese, gadolinium, and iron, and the preferred external complexing substances includes organosulfates and their derivatives,
- the ferromagnetically coupled, paramagnetic complex preferably has a net nuclear spin of greater than about 1/2 and has more than about 5 unpaired electrons. It should also preferably contain labile water groups.
- the multinuclear moiety should contain two or more heavy metal atoms, e.g.
- the multinuclear complex comprises at least one metal ion selected from W, Mo, Re, Rh, Tc, V, Nb, Ru and Re, however Mo. and W are particularly preferred.
- the choice of heavy metal used in the multinuclear complexes will be determined by a variety of factors including the toxicity of the overall complex and the X-ray absorption characteristics of the heavy atom. In this regard it should be noted that while the X-ray absorption cross section for atoms generally increases with increasing atomic number, the absorption cross section is itself dependent on the X-ray
- the multinuclear complexes according to the invention will each have optimum photon energy ranges making them particularly suitable for operation with X-ray imaging apparatus utilizing X-rays having such photon energy ranges.
- multinuclear complexes containing atoms of more than one heavy element one may create X-ray contrast agents having optimal performance in more than one photon energy band or over a broader band.
- the complexes used according to the present invention are thus particularly attractive since they can be selected so as to match their X-ray attenuation profiles with the X-ray emission profiles of particular X-ray sources - in effect the invention provides "tunable" X-ray contrast media.
- n, u and v are preferably 2 to
- n and also preferably u are at least 3 , particularly at least 4;
- x is preferably 1 to 20, especially 1 to 10, and
- w depends on the size and identity of the ligand - nonetheless w is preferably 1 or 2, especially 1.
- Non-chelant complexing agents such as amines and mono carboxylic acids, e.g. acetic acid and amino acids, optionally substituted aromatic amines (such as pyrazole or pyridine), phosphines (e.g. alkyl and/or aryl
- phosphines phosphites, phosphates, phosphonates and phosphinates are known and may be used in the formation of the multinuclear complexes of the invention.
- multinuclear complexes should be as high as possible and accordingly it is particularly preferred that the multinuclear moiety should be bound in a chelate
- L may conveniently represent a linear, branched or cyclic polyamino, polyaminocarboxylic or polycarboxylic acid.
- L may be represented by the formula
- R 1 which may be the same or different represents
- R 2 C 1-4 hydroxyalkyl, carboxy-C 1-4 alkyl or amino-C 1-4 alkyl groups or together both R 1 groups represent a group
- R 3 is an R 2 group or a C 1-4 alkyl group optionally substituted by hydroxyl, carboxyl, aryl or amino groups,
- each R 2 independently represents a hydrogen atom or an optionally amidated or esterified carboxy C 1-4 alkyl group, wherein any amine nitrogen is substituted by group selected from hydrogen atoms and optionally hydroxylated C 1-4 alkyl groups.
- L may be selected from
- the multinuclear complex used according to the invention may be ionic or, more preferably, may carry no net charge; most preferably the complex is non-ionic. Moreover it may be water-soluble or, less preferably, water-insoluble. Any necessary counterions should of course most preferably also be physiologically
- Suitable countercations include for example
- protons alkali and alkaline earth metal ions, e.g.
- ammonium and organic cations e.g. organic amine cations, quaternary ammonium, pyridinium, meglumine, alkylammonium,
- Suitable counteranions include for example halide (e.g. chloride, bromide, iodide, I 3 Other suitable chelant moieties will be discussed in greater detail later.
- JACS 112 7402-7403 (1990); JACS 110:
- each L' which may be the same or different is a ligand, either a molecule, an ion or one liganding moiety of a multidentate ligand; and W are each zero or positive integers).
- L' groups can of course be provided by one chelant (L) and the metal atoms may be covalently bound to further atoms (generally designated by the letter A in the formulae referred to herein) not indicated by L, L or B and which function neither as ligands nor as bridges.
- the M-B bonds to the bridging atoms B of formula II may be dative rather than covalent bonds.
- Examples of such complexes thus include the macrocyclic binuclear chelates such as those of formula III
- each R which may be same or different is hydrogen or an organic group and each M is as defined above, for example a metal atom or ion, e.g. Ni, Pb(II) or Cu(II).
- multinuclear complexes wherein the metal atoms M are not linked by covalent bonds does fall within the scope of the invention, it is particularly preferred that the multinuclear complexes be of the bridged type wherein the metal atoms M are covalently linked via bridging atoms or atom pairs (e.g. S 2 bridges) or bridging ligands (eg. acetate, hydroxo etc).
- bridging atoms or atom pairs e.g. S 2 bridges
- bridging ligands eg. acetate, hydroxo etc.
- analogues are known and they and complexes thereof are useful according to the invention, especially as X-ray contrast agents.
- Many such complexes are known and typical exemplary structures include the bi-, tri-, tetra- and hexa- nuclear structures of formulae II, IIIa, IV and V
- each B, M, L' and W' are defined above and where other non-bridging atoms covalently bonded to metals M are omitted for the sake of clarity).
- M may represent Mo, W, Re, Tc, V, Nb, Ta, Ru or Fe, but it is also possible for one (or more) of the metals M to be replaced by a transition or other metal, e.g. Hg, besides those metals specifically listed above in such a case however the substitute metal should be in a minority.
- a transition or other metal e.g. Hg
- M 2-50 clusters e.g. M 2 , M 3 , M 4 , M 5 , M 6 , M 7 , M 8 , M 9 , M 10 , M 11 , M 12 , M 17 , M 18 , M 36 and M 48 clusters, are known from the
- the complexes above may be electrically charged or neutral - for administration as contrast agents they may preferably be complexed with ligands/chelating agents which serve to improve water solubility and to reduce toxicity and to leave unaffected, to only slightly increase or, most preferably, to reduce the magnitude of the overall electronic charge carried by the complex and stabilize redox sites of clusters.
- the structural formulae can conveniently be written M 2 L q ( ⁇ 2 B) 2 and M 3 L r ( ⁇ 3 B) ( ⁇ 2 B) 3 ,M 4 L s ( ⁇ 3 B) 4 and M 6 L 1 ( ⁇ 3 B) 8 respectively ( ⁇ 3 B indicating that the B is a bridging atom bonded to 3 metals, and q, r, s and t respectively being integers identifying the total number of
- the multinuclear complexes be chelate complexes and it is especially preferred that a single multidentate chelant be used to coordinate at least two and preferably all of the liganded centres.
- a multidentate chelant L coordinating for example three metals would be referred to in these formulae as ( ⁇ 3 L).
- a multinuclear entity of formula I can comprise a unit of formula M 3 ( ⁇ 3 B) ( ⁇ 2 B) 3 , M 4 ( ⁇ 3 B) 4 or M 6 ( ⁇ 3 B) 8 (wherein M and B are defined above, but
- each M may indpendently represent Mo or W, and each B may independently represent O, S, Se or Te or a nitrogen or phosphorus atom covalently bonded to a proton or an organic group).
- molybdenum and/or tungsten e.g. APCA chelates of multinuclear entities of formula
- M 2 Z 2 ( ⁇ 2 Z) 2 i.e. M 2 Z 2 ( ⁇ 2 Z) 2
- each M is independently W or Mo and each Z is independently O, S, Se, Te, CI, Br or I, e.g.
- Mo 4 ( ⁇ 3 Se) 4 Mo 2 O 2 ( ⁇ 2 Se) 2 , Mo 3 ( ⁇ 3 O) ( ⁇ ,O) 3 , W 6 ( ⁇ 3 S) 8 ,
- Particularly preferred multinuclear clusters of type M 2 B 2 include those of formula:
- M preferably represents Mo, W or Re, whilst B and A preferably each represent O, S, Se or Te.
- multinuclear cluster of this type include:
- each R 50 may represent a hydrogen atom or a straight chained or branched alkyl group optionally substituted by hydroxy, thiol, alkylthiol groups or by a carbonyl group which may itself be optionally alkyl substituted on the amide nitrogen.
- R 50 represents a C 1-6 alkyl, optionally substituted as
- R 50 may represent an alkyl group of formula
- M preferably represents Mo, W or Re
- a and B each preferably represent O, S, Se or Te.
- multinuclear clusters of this formula include
- the multinuclear clusters described above are preferably chelated by ligands which are carbohydrates, for example sugar alcohols, especially those with a backbone chain containing of from 4 to 7 carbon atoms.
- ligands which are carbohydrates, for example sugar alcohols, especially those with a backbone chain containing of from 4 to 7 carbon atoms.
- sugar alcohols include perseitol, galactitol, D-mannitol, erythritol, D-threitol, p-arabinatol, xylitol, ribitol and D-glucitol.
- Preferred M 3 B 4 multinuclear clusters may be any suitable M 3 B 4 multinuclear clusters.
- each M is preferably Mo, W or Re and each B is preferably O, S, Se or Te.
- Examples of such clusters include
- Preferred ligands for the M 3 B 4 clusters include those of formula
- each Z may represent a nitrogen or phosphorous atom
- each Y may represent a nitrogen, phosphorous, oxygen or sulphur atom or a group NR 50 or PR 50 (where R 50 is as defined above) and each group R 51 may represent a group
- the multinuclear entities useful according to the invention thus include many known polynuclear inorganic molecules and ions and complexes thereof (including complexes with organic species), including those known under the general term polyoxoanions.
- Such multinuclear entities can be described by the following general formula:
- each D which may be the same or different represents a non-metal atom, molecule or ion within the multinuclear entity that is bonded to one or more metals M,
- each G which may be the same or different represents an atom, molecule, ion or metal complex coordinatively bound to the M ⁇ D ⁇ entity to yield the charged or
- each J which may be the same or different, is a physiologicaly tolerable counterion (e.g. an alkali metal, alkaline earth metal, ammonium, quaternary ammonium, organic amine etc cation where the cluster is anionic),
- a physiologicaly tolerable counterion e.g. an alkali metal, alkaline earth metal, ammonium, quaternary ammonium, organic amine etc cation where the cluster is anionic
- ⁇ and ⁇ are numbers having a value of at least 2, e.g. 2-100, ⁇ is a positive number, and 5 and k are each zero or postive numbers.
- At least two (and preferably the majority) of the M atoms will generally be group Vb, VIb, VIIb or VIII elements (e.g. V, Fe, Nb, Mo, Tc, Ru, Rh, Ta, W and Re, especially Nb, Ta, Re, Mo, V, W, particularly Mo and W) while other (minority) M atoms or M atom complex may be selected for example from a secondary group comprising other transition metals, group Ila elements and
- lanthanides as well as other diverse metals such as Be, Al, Si, Ge, Sn, etc. for example as mentioned by
- X a is selected from B, Al, Si, Ge, P, As, Se, Te, S, I, Co, Mn, Cu, alkylphosphonate, arylphosphonate
- M a , M b , M c are independently selected from W, Mo, V, Nb, Ta and Re; ⁇ , ⁇ , ⁇ , ⁇ , and ⁇ are zero or positive numbers; ⁇ is zero for isopolyoxoanions and mixed isopolyoxoanions or is a positive integer for
- the counter-cations for polyoxoanions can be protons, organic cations (e.g. Ph + , CH 3 + , as in AR Seidle et al, JACS 108: 6430-6431 (1986)), alkali metal
- Quaternary ammonium salts of polyoxoanions can be developed which display remarkable hydrolytic stability and are highly soluble in organic media.
- Organo-soluble salts i.e. lipophilic
- Organo-soluble salts exist as an ion pair whereby the polyoxoanion is effectively coated with the greasy quaternary ammonium salt.
- Lipophilic polyoxoanions and chelant complexes can be dissolved in suitable oils, placed inside
- cyclodextrins incorporated into liposomes or micelles.
- An oil solution can be combined with water and emulsion stabilizers to form an emulsion which can be utilized as a GI x-ray contrast agent.
- Liposomal formulations are interesting as liver agents. Cyclodextrin formulations are potential intravascular agents.
- reaction may be used as an analytical method for the separation and identification of sugars. Recently, there has been interest in understanding the structural and physical inorganic aspects of these complexes.
- polyalcohol ligand provides hydroxide coordinating groups attached to four adjacent carbon atoms in the ligand backbone as shown here for a generic polyalcohol ligand: (where R 52 is a straight chained or branched alkyl group)
- the complex is formed by a condensation process involving acid:
- complexes are colorless. Due to both the fact that the complex is a salt and the polyhydroxy nature of the ligand, these metal-ligand complexes exhibit very high water solubility.
- coordinating ligands are biocompatible. Higher cluster complexes may also be formed.
- modified polyoxoanions may be formed for example by the reaction of a reactive metal-organic complex with "defect" (substitutionally active)
- polyoxoanions such that the charge of the polyoxoanion, its solubility, or other desired physical/chemical properties can be systematically varied. Such complexes may obviate the need for separate counter-cations.
- M' Proper choice of M' can lead to stable substituted polyoxometalates while proper choice of R can lead to changes in overall complex charge (osmolality), solubility, etc
- R when R is a monoquarternary ammonium, then an electrically neutral polyoxoanion [SiW 11 O 4 ⁇ (Si (CH 2 ) n (NR 3 ) ) 2 ]° can be prepared.
- modified polyoxyanions may be attached to polychelants, for example the polychelants disclosed in WO-A-90/12050, WO-A-91/05762 and in Angew. Chem. Int. Ed. Engl. 29, pages 138-175 (1990) of Tomalia et al.
- a modified polyoxoanion may comprise one or more heteroatoms which displace the original metal atoms in the cluster so altering the characteristics of the heteropolyanions as discussed above.
- heteroatoms include Be, Y, La, Ti, Zr, Hi, V, Nb, Ta, Cr, Mo, W, Mn, Re, Co, Rh, Ni, Pt, Cu, Zn, B, Al, Ga, In, Tl, C, Si, Ge, Sn, P, As, Sb, S, Se, Te, I, Ce, Pr, Nd, Sm, Eu, Gd, Ho, Er, Yb, Th, U, Np and Am.
- Preferred heteroatoms include W, Mo, V, Ti, Sn, Nb, Rh, Ge, Re, Sb, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Al, Ga, In, Tl, Rh, Ru, Os, Ce, Pr, Nd, Sm, Eu, Ho, Th, U, Pr, Tb, Pu, Np, Am, Cm, Cf, La, Sr, Ba and Si.
- Metal clusters especially HPA's, which contain a mixture of heavy metals (which are contrast enhancing in X-ray imaging) and paramagnetic metals (which are
- contrast enhancing in MRI are especially attractive.
- Clusters containing for example 10 to 20 heavy metals (such as Mo or W) and 1 to 4 paramagnetic metals (such as Gd, Mn, Fe or Dy) are especially interesting.
- EDTA EDTA
- other chelants are suitable for the preparation of the multinuclear chelate complexes used according to the invention.
- bifunctional chelants and chelate-based contrast agents e.g. those described in WO-A-89/00557 (Berg) and the documents mentioned therein and in the search report appended thereto, US-A-4647447 (Gries), US-A-4826673 (Dean), EP-A-230893 (Felder), EP-A-217577 (Frincke), US-A-4652519 (Warshawsky), US-A-4687659 (Quay), and numerous other recent patent publications of Nycomed AS, Salutar Inc, Schering AG, Squibb, Bracco, Mallinckrodt, Dow and Guerbet.
- the chelants useful for complexing the multinuclear moiety and which form coordinate bonds directly with the metals of the multinuclear moiety can be selected from a wide range of structures. Many of the most useful chelants are of general formula XII
- a is an integer of from 2 to 12, preferably 2 to 10, e.g. 2, 3, or 4;
- b is an integer of from 1 to 8, preferably 2, 3 or 4;
- each R independently is hydrogen, a hydrophilic or linking group (e.g. a hydroxyalkyl group) or two groups R 1 , or one R 1 and one group Z 3 , together represent a saturated or unsaturated heterocyclic or carbocyclic ring, preferably with 5-7 ring atoms;
- a hydrophilic or linking group e.g. a hydroxyalkyl group
- two groups R 1 , or one R 1 and one group Z 3 together represent a saturated or unsaturated heterocyclic or carbocyclic ring, preferably with 5-7 ring atoms
- each X independently is O, S, NZ 3 or PZ 3 ,
- each Z 3 indpendently is hydrogen, hydroxyalkyl
- mercaptoalkyl carboxyalkyl (or an amide or ester derivative thereof e.g. -CH 2 CONHCH 3 ) or optionally hydroxy or mercapto substituted acyl, or is a side chain ((CHR 1 ) a X * ) c Z * (where c is 1 to 4 and X * and Z * are as defined for X and Z 3 but do not represent any group containing a X* or Z * group) or two groups Z 3 together form a briding group ((CHR 1 ) a X * ) c (CHR 1 ) a ) or are salts thereof).
- polyamines and polyethers especially linear or cyclic polyamines and polyethers, such as
- ethylenediamine, 1,4,7-triazacyclononane and cyclen can be used as chelants, in general APCAs and starburst dendrimers (see e.g. Tomalia, Angew. Chem. Engl. Ed. 29: 138-175 (1990)) are preferred, particularly DTPA, EDTA, TTHA and derivatives thereof and other cyclic and non-cyclic APCAs as defined in WO-A-89/00557 and APCAs of formula XIII
- each R 1 is independently hydrogen or an optionally hydroxylated and/or alkoxylated alkyl group or an organic side chain adapted for the attachment of or attached to a macromolecule;
- d and e each is an integer having a value of 1, 2 or 3; each X 10 is independently a carboxyl (ie COOH) or
- each Y is independently a group X 10 , SR 1 , OR, or N(R 3 ) 2 ;
- E is a group (CHR 2 ) f (X" (CHR 2 ) f ) g
- f is an integer of from 2 to 5, preferably 2 or 3 , g is zero, 1 or 2, preferably zero or 1, each f
- X" is O, S or N(CHR 1 ) d Y, preferably O or S,
- each R 2 is independently R 1 or, when the carbon to which it is attached is not bonded to a nitrogen, hydroxyl, or two R 2 groups, especially where f is 2, may together with the intervening carbons form a cycloalkyl group
- R 3 is independently a group R 1 or N(R 3 ) 2 represents a preferably saturated heterocyclic group preferably having 5 or 6 ring members, optionally containing as a further heteroatom a nitrogen or oxygen and optionally substituted by R 1 groups. It is also possible for the multinuclear moiety to be chelated by a chelant which is then itself attached to form part of an oligomer or polymer, such as a starburst dendrimer.
- any alkyl moiety preferably has a carbon atom content of up to 8
- any cycloalkyl group preferably is a C 3-8 , especially C 5-7
- any carboxyl derivative is preferably a CON(R 3 ) 2 or CON(OH) R 1 group.
- Suitable chelants include the pyrazole for example Prog. I1.
- R 20 and R 21 which may be the same different, represent hydrogen atoms or an optionally hydroxylated, optionally alkoxylated alkyl group (e.g. C 1-8 alkyl), or a pyrazolyl group PZ of formula MtR 24 3 or
- R 22 and R 23 are groups as defined for R 20 and R 21 respectively but do not represent PZ groups, or one may represent bond or an alkylene chain (e.g. C 1-10 n-alkylene) linked to a group R 22 or R 23 of a further group of formula XIV (i.e. to form a dimer).
- Mt is B, Al or Ge
- R 24 is hydrogen, alkyl, amine, APCA or aryl (e.g. 5 to 8 ring membered homo or heteroaryl such as phenyl or pyrazolyl (e.g. PZ) group.
- Z is zero or a positive integer, e.g. 1-10.
- oligomers such as trimers, tetramers or polymers may also be formed in a similar fashion.
- the trispyrazolylborate ligands have several features to recommend them for metal-ion cluster
- the molecule functions as a tridentate coordinating ligand that presents aromatic amine
- cluster/ligand complex This may be especially useful where use of the carboxylate ion is undesirable.
- the poly(tris-pyrazolylborate) ligands moreover can form a single coordinating molecule capable of binding to more than one metal ion:
- Suitable chelants include compounds of formulae:
- X 2 O, S or NR 41
- R 40 CH 2 COOH, CH 2 CH 2 SH, CH 2 CH 2 OH, CH 2 PO 3 H 2 ,
- R 41 CH(CH 2 X 4 ) 2 CH 2 CHX 4 CH 2 X 4 , CH 2 CHX 4 CHX 4 CH 2 X 4 ,
- R 42 alkyl, eg. C 1-6 alkyl, or substituted C 4-12 homo or heteroaryls
- substituted aryl means subsituted by one or more groups selected from carboxyl (and derivatives thereof e.g. salts, amides and esters), hydroxyl, mercapto, phosphates, amines and phosphines).
- X 6 NR 40 , PR 40 , N(CH 2 ) 2 X 3 or one X 6 may provide an
- alkylene chain (eg. CH 2 CH 2 ) linking the macrocycle to a similar macrocycle
- M 6 B 8 complexes e.g. W 6 ( ⁇ 3 S) 8
- chelants such as (xvi) to (xx) are of particular interest.
- M 2 B 2 complexes e.g.
- W 2 O 2 ( ⁇ 2 O) 2 2+ chelants such as NTA, IDA, EDTA, HEDTA, DTPA, DTPA-BMA, HEDDA, TTDA, EDTA-BMA, TBEDDA, MEEDDA, TTHA, EDDA, EHPG, PDTA, CHDTA, HPDTA, TACN, TTHA, CMPA-BMPA, DTPA-BMPA and triazacyclononane monoacetic acid,
- TTHA are of particular interest.
- Particularly preferred chelants include cyclen, TTHA, EDTA, DTPA, DOTA, D03A, HP-D03A, the 6-oxa and 6-thia analogues of DTPA and amides thereof, e.g. DTPA-BMA and DTPA-BMO (6-carboxymethyl-3,9-bis(morpholinocarbonyl-methyl)-3,6,9-triazaundecanedioic acid - the Gd(III) chelate whereof is sometimes referred to as gadopenamide).
- DTPA-BMA and DTPA-BMO 6-carboxymethyl-3,9-bis(morpholinocarbonyl-methyl)-3,6,9-triazaundecanedioic acid - the Gd(III) chelate whereof is sometimes referred to as gadopenamide.
- polyoxa/polyaza cycloalkane macrocyclic chelants e.g. such as are described in US-A-3860611, US-A-3952015 and JOC 22: 1029 (1957), JOC 39: 2351
- a macromolecule conveniently be any tissue, organ or cell targeting macromolecule, for example a
- biomolecule such as a protein, an antibody or antibody fragment, or alternatively a biologically relatively inert material such as a polysaccharide or poly-sugar alcohol, e.g. dextran, starch, cyclodextrin etc).
- a biologically relatively inert material such as a polysaccharide or poly-sugar alcohol, e.g. dextran, starch, cyclodextrin etc.
- introduced may, in intermediate or final stages, be subject to reduction or deprotection steps.
- LCO.E.CO.L (XIX) may be aminated with or without a subsequent reduction step according to the following schemes:
- the polyamine starting materials are either
- polyamines include NH 2 (CH 2 ) 2-5 NH 2 , NH 2 (CH 2 ) 2 O(CH 2 ) 2 NH 2 ,
- substituted polyamines may also be prepared by methods described in or analogous to those of EP-A-287465
- Derivatization of the polyamines may be effected using alkylation agents such as those described by EP-A-230893 (Felder), e.g. HalCH 2 COL", HalCH(COOH) CH 2 O Benzyl, or HalCH(COOH) 2 (where Hal is CI or Br and L" is OH, NHAlkyl or NAlkyl 2 (e.g NHCH 3 or N(CH 3 ) 2 ) or HalCH 2 NAlkyl 2 (e.g. ClCH 2 N(CH 3 ) 2 ), followed where necessary by
- reaction may be of many of the same or similar
- thioesters could equally be produced by reaction of an aminocarboxylic acid reagent with a chloroalkylsulphide, e.g.
- the chelants of formula (XIII) can also be produced by amination of polyfunctional reagents.
- One example of this procedure is given by Huber et al. J. Chem. Soc. Chem. Comm. (1989) 879, i.e.
- Amine intermediates can also be produced by
- the resulting polyamine can then be converted to a compound of formula XIII by reaction with X 9 CH 2 CN (where X 9 is OH or, prferably, a halogen) followed by
- polyfunctional acids may be reacted with appropriate amines if necessary after activation of the acid groups, reduction of the amide and alkylation will yield chelants of formula XIII.
- E 3 is -CHOHCH 2 CH 2 - , -(CHOH) 2 -,
- the chelant may be provided with a reactive side chain (e.g. described by Meares et al. Anal. Biochem. 142: 68(1984), etc).
- a reactive side chain e.g. described by Meares et al. Anal. Biochem. 142: 68(1984), etc.
- attachment can be resumed for example using the methods developed by Salutar Inc. (See for example WO-A-90/12050 and Sieving et al., Bioconjugate Chem. 1: 65-71 (1990)) or the mixed anhydride or cyclic anhydride methods of Krejcarek et al Biochemical and Biophysical Research Comm. 77 : 881 (1977) or Hnatowich et al. Science 220: 613 (1983) etc.
- Attachment of the chelant may be either directly to the macromolecule or, preferably, to an intermediate polymer, e.g. poly-L-lysine or polyethylene-imine, onto which a plurality of chelants may be loaded, e.g. as discussed in EP-A-331616 (Deutsch).
- multinuclear complexes used according to the invention may be prepared by the methods suggested in the literature or by analogous methods.
- novel complexes may be prepared from known complexes by ligand interchange.
- oxalatotungstate(V) may be used as a starting material and ligand exchange
- [M 3 ( ⁇ 3 B) ( ⁇ 2 B) 3 (H 2 O) 9 ] 4+ (where M is Mo or W and B is O or S) can be prepared by methods known from the literature. The co-ordinated waters in these complexes can readily be replaced by chelants xvi to xxiii to reduce toxicity. Single or mixed ligand combinations may be used to produce ionic or non-ionic complexes.
- the chelated molybdenum and tungsten M 3 complexes can also be prepared by reaction of chelants (xvi) to
- M Mo or W) or [M 3 ( ⁇ 3 -S) ( ⁇ 2 -S 2 ) (S 2 ) 3 ] 2-
- the coordinated water in the tetranuclear aqua complexes may be substituted by ligands such as chelants i to vii to reduce toxicity. Selected examples are shown below.
- M 1 is hydrogen, alkali metal (e.g. Na, Li, K), ammonium, quaternary ammonium, etc,
- This reaction is conveniently performed in a solvent such as THF, THT, DMF etc under an inert atmosphere.
- the product (XXV) is then treated with water to produce W 3 S 4 Cl 4 (H 2 O) 9 (compound (XXVI)) which can be used to produce larger clusters.
- reaction is effected using photolysis in solvents and under an inert atmosphere as discussed under (M) above.
- the oxidizing agent ([o]) is conveniently hydrogen peroxide, OCl-, TBHP, O 2 , O 3 or a peracid.
- the oxidation is suitably carried out in a solvent such as described under (M) above and can be performed to yield a family of W(VI) mixed sulfur/oxygen clusters.
- the clusters produced according to processes (M) - (Q) may be reacted to replace L 3 ligands by water-soluble chelaring agents, e.g. such as those discussed above, to produce more soluble products.
- the multinuclear complexes of the lanthanides, of iron and of manganese are particularly attractive.
- Gd or Dy or La complexes such as Ln 4 Cu 4 , Ln 2 Cu 4 , Ln 8 Cu 12 , Ln 2 Cu 2 and Ln 3 Cu and also of the
- the multinuclear complexes will conveniently be formulated together with pharmaceutical or veterinary carriers or excipient.
- the contrast media of the invention may conveniently contain pharmaceutical or veterinary formulation aids, for example stabilizers, antioxidants, osmolality adjusting agents, buffers, pH adjusting agents, colorants, flavours, viscosity adjusting agents and the like. They may be in forms suitable for
- parenteral or enteral administration for example, injection or infusion or administration directly into a body cavity having an external voidance duct, for example the gastrointestinal tract, the bladder and the uterus.
- the media of the invention may be in conventional pharmaceutical adminstration forms such as tablets, coated tablets, capsules, powders, solutions, suspensions, dispersions, syrups, suppositories,
- emulsions emulsions, liposomes, etc; solutions, suspensions and dispersions in physiologically acceptable carrier media, e.g. water for injections, will however generally be preferred.
- physiologically acceptable carrier media e.g. water for injections
- media for parenteral administration will preferably contain small quantities, e.g. 0.01 to 10 mole percent relative to the multinuclear complex of free chelants or of weak chelate complexes with physiologically tolerable chelated species (e.g. Ca 2+ ); small additions of sodium or calcium salts may also advantageously be made.
- physiologically tolerable chelated species e.g. Ca 2+
- small additions of sodium or calcium salts may also advantageously be made.
- the media of the invention should generally have a heavy atom content of 1 millimole/l to 5 mole/1, preferably 0.1 to 2 mole/l. Dosages of from 0.05 to 2.0 mmoles/Kg, e.g. 0.5 to 1.5 mmoles/kg will generally be sufficient to provide adequate contrast although dosages of 0.8 to 1.2
- mmoles/kg will normally be preferred.
- M n clusters (where M is a therapeutically or contrast effective metal and n is 2 or greater) complexed by a chelating ligand L (such as APCA, PZ etc), may be administered therapeutically.
- the medium to be administered will contain a
- therapeutically required metal which may be the same as or replace an image enhancing metal in the multi-nuclear complexes of the present invention.
- therapeutic metals include Sb, Ti, Mo, Pd, W.
- polyoxyanions - especially HPA's (heteropolyoxyanions) - are preferred ligands.
- the clusters of the present invention may be used advantageously in bioanalytical applications.
- a further aspect of the present invention includes therapeutic and bioanalytical uses of multi-nuclear complexes of formula I.
- the present invention provides a particularly effective means by which contrast media efficiency may be enhanced by increasing the relative proportion of molecular volume that is occupied by the contrast enhancing heavy or paramagnetic metal atom.
- contrast media efficiency may be enhanced by increasing the relative proportion of molecular volume that is occupied by the contrast enhancing heavy or paramagnetic metal atom.
- this also enables higher K-edge value atoms than the iodine of the now conventional X-ray contrast media to be utilized
- the title compound was prepared by a slightly modified version of the procedure described in Cotton, F.A., et. al., Polyhedron 5; 907 (1986).
- 1.0 g W(CO) 6 was refluxed with 0.8 g anhydrous Na 2 S in 100 mL of acetate anhydride for 12 hours under N 2 .
- the product was dissolved in 0.3 M HCl.
- the solution was loaded onto a AG50W-X8 cationexchange column. Elution with 0.3M HCl yielded an orange band (1st band), peak at 458 nm, identified as W 3 O 3 S(H 2 O) 9 CL 4 (e 349 M -1 cm -1 ).
- the isolated band was purified on a second cation-exchange column and
- the reagent K 3 [W 2 Cl 9 ] can be prepared as described by Shibahara et al., Inorg. Chim Acta 127: L39 (1987).
- the reagent K 2 [WCl 6 ] can be prepared as described by Kennedy, et. al., J. Chem. Soc. (1963) 3392.
- TTHA-BDHA Ligand a Triethylenetetramine hexaceticacid (5.0 g, 10.1 mmol) and acetic anhydride (9.55 mL, 101 mmol) were dissolved in dry pyridine and heated to 65°C with stirring for 18 hours. To the resulting dark brown slurry was added diethyl ether, the solution was
- N,N,N"',N"'-tetra(t-butyloxycarbonylmethyl)-N',N"-ditrifluoroacetylaminomethyl-triethylenetetramine N,N,N"',N"'-tetra(t-butyloxycarbonylmethyl)-N',N"-ditrifluoroacetylaminomethyl-triethylenetetramine is dissolved in a methanol/water solution of potassium carbonate (20 equivalents) and heated to 60°C with stirring for 6 hours. The solution is evaporated to dryness and extracted several times with chloroform.
- N,N,N"',N"'-tetra(t-butyloxycarbonylmethyl)-N',N"-di(2-aminoethyl)-triethylenetetramine is evaporated to give N,N,N"',N"'-tetra(t-butyloxycarbonylmethyl)-N',N"-di(2-aminoethyl)-triethylenetetramine.
- N,N,N"',N"'-tetra(t-butyloxycarbonylmethyl)-N',N"-di(2-aminoethyl)-triethylenetetramine is dissolved in acetonitrile with an excess of potassium carbonate, and a solution of bromoethanol (3 equivalents) in
- N,N,N"',N"'-tetra(t-butyloxycarbonylmethyl)-N',N"-di(N,N-bis(2-hydroxyethyl)-2-aminoethyl)-triethylenetetramine is dissolved in methylene chloride and trifluoroacetic acid is added dropwise. The solution is stirred for one hour and evaporated to dryness, and the hydrloysis is repeated three core times, to give the title compound.
- N,N'-di(N,N-di- acetyl)aminoethyl TACN is dissolved in aqueous NaOH at pH 11 and bromoethanol is added dropwise with stirring. The solution is then heated to 40°C for 18 hours and the resulting solution is passed through an ion exchange column to give the title
- TACN (0.129 g, 0.001 mol) in water containing a trace of sodium iodide was treated with N-(2-chloroethyl)iminodiacetic acid hydrochloride (0.765 g, 0.0033 mol) at 40°C and pH 11.5-12 (NaOH). After 3 hours, the solution was passed through a column of Amberlite IR 120 ion exchange resin (acid form) and evaporated to dryness. The solid residue was recrystallized from
- step (e) The oil obtained in step (e) was used without any further purification.
- the oil was dissolved in 20 mL of methylene chloride and 20 mL of trifluoroacetic acid were added dropwise. The reaction was stirred for 24 hours at 25°C. The solvent was then stripped off and the resulting oil taken up in 30 mL of methanol. A catalytic amount of palladium was added and the reaction allowed to stir at 25°C for 24 hours under a hydrogen atmosphere. The reaction mixture was then filtered through charcoal to remove the catalyst and concentrated to yield the title compound as a yellow solid. The title compound was further purified by ion-exchange chromatography.
- the ligands of Intermediate Examples 6 to 11 may be used in the subsequent Examples in place of the ligands actually specified, eg. DTPA, EGTA, EDTA, etc.
- Example 1
- the potassium salt (1.61 g, 1 mmol) of the oxalato complex of tungsten (V) (prepared according to Baba et al., Mem. Fac. Tech. Tokyo Metropolitan Univ. 32: 3207-3220 (1982)) and 1 ,2-diaminopropane-N,N,N',N'-tetraacetic acid (0.612 g, 2 mmol) were dissolved under nitrogen in a mixture of 50 ml oxygen-free water and sodium acetate-solution (1M, 8 ml) and heated to 100°C. Calcium acetate-solution (1M, 10 ml) was added with stirring and the mixture allowed to cool. After
- the title compound is prepared by dissolving the barium salt of Example 1 in warm water. After addition of a stoichiometric amount of a 1M sodium sulfate solution the mixture is allowed to cool, filtered and the
- the dimethylammonium salt of W 3 ( ⁇ 3 -S) ( ⁇ 2 -S) 3 (TTHA) was dissolved in a minimum amount of water and loaded onto a column containing BioRad AG50W-X8 resin in the proton form.
- the dimethyl ammonium cation remains on the column and the product is eluted from the column with water as H 2 [W 3 ( ⁇ 3 -S) ( ⁇ 2 -S) 3 (TTHA)].
- the purple effluent is reduced in volume by rotary evaporation and carefully neutralized with 1N NaOH.
- the purple solution at pH 7 is loaded onto a column containing Sephadex G-15 resin.
- the second band containing Na 2 [W 3 ( ⁇ 3 -S) ( ⁇ 2 - S) 3 (TTHA)] is eluted with water.
- the yield was 7.3 g.
- Positive ion FAB-MS spectral data showed a molecular ion at 1216, corresponding to the mass of Na 2 [W 3 ( ⁇ 3 -S) ( ⁇ 2 - S) 3 (TTHA)]+H + .
- HPLC chromatography indicates a single tungsten containing peak (monitored at 570 nm).
- Example 4A The salt from Example 4A (3.00 g, 2.469 mmol) was dissolved in water (24 mL). The pH was adjusted to 6.9 by careful addition of 1 M hydrochloric acid. Water was added to 24.60 mL. The osmolality (254 mOsm/Kg) was adjusted to 337 mOsm/Kg by the addition of 73 mg of NaCl. The solution was passed through a 0.22 ⁇ M sterile filter and placed in one 30 mL vial. The solution contained 0.10 mmol of the disodium salt of Example 4A per mL. The LD 50 in mice was found to be greater than 5 mmol/Kg.
- the resulting orange solution is concentrated to 25 ml under reduced pressure to precipitate the excess salts.
- the orange filtrate is placed on a sephadex G-15 column.
- the orange eluant with water is treated with Na 2 SO 4 to convert the barium complex into the sodium one.
- the insoluble BaSO 4 is filtered off and the orange filtrate is reduced to a small volume, 15 ml.
- the concentrated orange filtrate is mixed with ethanol to precipitate the title product.
- the orange precipitate is collected by filtration, washed with acetone, and dried in the air.
- the title compound was prepared by refluxing the mixture of W 3 ( ⁇ 3 -5) ( ⁇ 2 -S) 3 (H 2 O) 9 Cl 4 (Intermediate Example 1) and W(CO) 6 in pyridine. 50 mg of W 3 ( ⁇ 3 -S) ( ⁇ 2 -S) 3 (H 2 O) 9 Cl 4 were dissolved in 3 mL of N 2 saturated pyridine giving a dark green solution, followed by the addition of 20 mg of W(CO) 6 . The resulting green suspension was refluxed under N 2 for an hour. During the reflux, the W(CO) 6 was dissolved into the green solution, which eventually became a brown suspension. The brown suspension was mixed with 2-3 ml of ether to precipitate more product.
- Cetomacrogal Emulsifying Wax A non-ionic emulsifying agent which contains cetostearyl alcohol and
- cetomacrogol considered non-toxic; forms a pourable product using 5% wax; recommended for use with salts of polyvalent metals and medicaments based on nitrogenous compounds.
- Cetyl Alchohol CH 3 (CH 2 ) 14 CH 2 OH; used as an emulsifier in 2-5% concentrations; commonly used for suppositories.
- Cholesterol C 24 H 46 O; can be used as an emulsifying agent; some reported complicity of cholesterol in
- Corn oil Composed of 44% oleic acid; 39% linoleic acid; 7% palmitic acid; 3% stearic acid, and other components; used as an oral nutritional supplement 67% as an
- Cottonseed oil Composed of 39% linoleic acid; 33% oleic acid; 19% palmitic acid; 2% stearic acid, and other components; used as an IV emulsifying agent in 10-15% w/v concentration; some side effects of IV use reported; Reference: J. Am. Med. Assoc., 166, 1042 (1958).
- Glycerol monostearate C 24 H 40 O 4 ; non-toxic and edible; used as an emulsifying aid; useful with quat salts;
- Sesame seed oil Composed of 45% oleic acid; 40%
- Emulsion stabilizers Bentonite; Calcium Stearate;
- Magnesium aluminum silicate mineral oil; lanolin oil; polacrilin potassium; propylene glycol; poloxamer.
Abstract
Description

































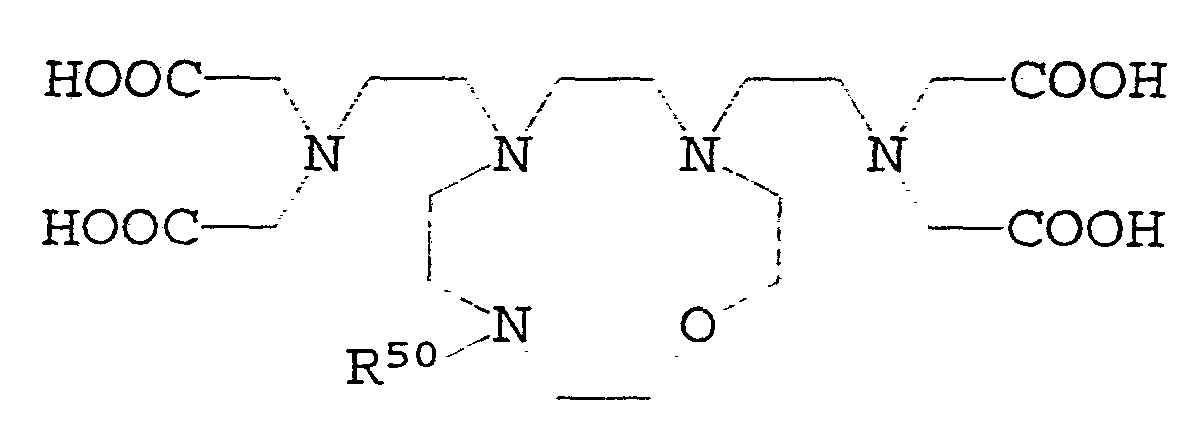





















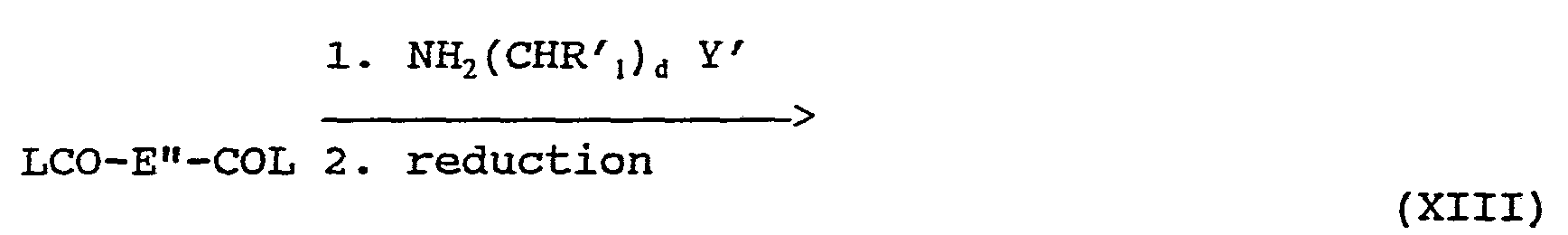






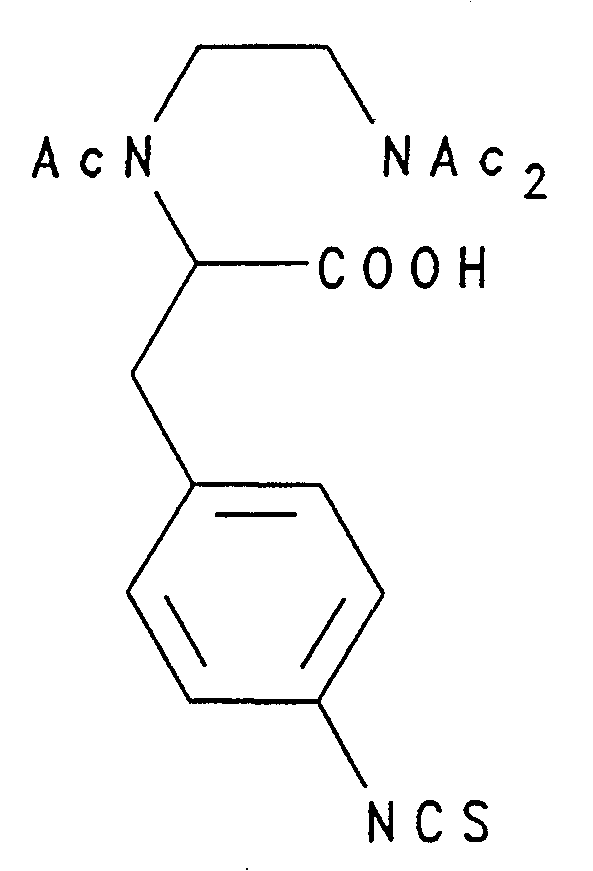
























Claims

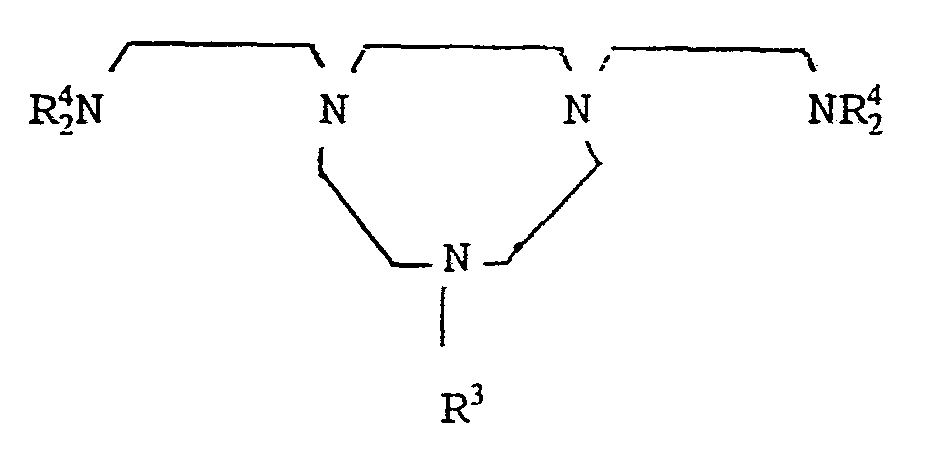
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/122,461 US5482699A (en) | 1991-03-27 | 1992-03-27 | Multinuclear complexes for x-ray imaging |
| JP4506847A JPH06506449A (en) | 1991-03-27 | 1992-03-27 | contrast medium |
| EP92907484A EP0577675B1 (en) | 1991-03-27 | 1992-03-27 | Contrast media |
| DE69230400T DE69230400T2 (en) | 1991-03-27 | 1992-03-27 | Contrast medium |
| AU14338/92A AU660013B2 (en) | 1991-03-27 | 1992-03-27 | Contrast media |
| FI934194A FI934194A (en) | 1991-03-27 | 1993-09-24 | KONTRASTMEDEL |
| NO933421A NO933421L (en) | 1991-03-27 | 1993-09-24 | The contrast agent |
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB909006977A GB9006977D0 (en) | 1990-03-28 | 1990-03-28 | Compositions |
| GB919106579A GB9106579D0 (en) | 1991-03-27 | 1991-03-27 | Contrast media |
| GB9106579.7 | 1991-03-27 | ||
| GB919120507A GB9120507D0 (en) | 1991-03-27 | 1991-09-26 | Contrast media |
| GB9120507.0 | 1991-09-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1992017215A1 true WO1992017215A1 (en) | 1992-10-15 |
Family
ID=27265012
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1992/000698 WO1992017215A1 (en) | 1990-03-28 | 1992-03-27 | Contrast media |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO1992017215A1 (en) |
Cited By (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993019788A1 (en) * | 1992-04-03 | 1993-10-14 | Mallinckrodt Medical, Inc. | Methods and compositions for magnetic resonance imaging |
| US5364953A (en) * | 1992-11-05 | 1994-11-15 | Mallinckrodt Medical, Inc. | High relaxivity, paramagnetic, metal clusters for magnetic resonance imaging |
| EP0739337A1 (en) * | 1994-01-14 | 1996-10-30 | Mallinckrodt Medical, Inc. | Functionalized macrocyclic ligands for imaging applications |
| WO1996040287A2 (en) * | 1995-06-07 | 1996-12-19 | Nycomed Salutar, Inc. | Preparation and use of contrast agents |
| WO1997026921A2 (en) * | 1996-01-23 | 1997-07-31 | Nycomed Salutar Inc. | Contrast media |
| US5817289A (en) * | 1995-01-26 | 1998-10-06 | Nycomed Imaging As | Non-cluster type bismuth compounds |
| EP0871637A1 (en) * | 1995-07-20 | 1998-10-21 | Mallinckrodt Medical, Inc. | Tungsten clusters |
| US6056939A (en) * | 1998-08-28 | 2000-05-02 | Desreux; Jean F. | Self-assembling heteropolymetallic chelates as imaging agents and radiopharmaceuticals |
| US6117412A (en) * | 1995-01-26 | 2000-09-12 | Nycomed Imaging As | Non-cluster type bismuth compounds |
| US6511648B2 (en) | 1998-12-18 | 2003-01-28 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6511649B1 (en) | 1998-12-18 | 2003-01-28 | Thomas D. Harris | Vitronectin receptor antagonist pharmaceuticals |
| US6524553B2 (en) | 1998-03-31 | 2003-02-25 | Bristol-Myers Squibb Pharma Company | Quinolone vitronectin receptor antagonist pharmaceuticals |
| US6537520B1 (en) | 1998-03-31 | 2003-03-25 | Bristol-Myers Squibb Pharma Company | Pharmaceuticals for the imaging of angiogenic disorders |
| US6548663B1 (en) | 1998-03-31 | 2003-04-15 | Bristol-Myers Squibb Pharma Company | Benzodiazepine vitronectin receptor antagonist pharmaceuticals |
| US6558649B1 (en) | 1998-12-18 | 2003-05-06 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6569402B1 (en) | 1998-12-18 | 2003-05-27 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6770259B2 (en) | 2000-11-03 | 2004-08-03 | Bristol-Myers Squibb Pharma Company | Simultaneous dual isotope imaging of cardiac perfusion and cardiac inflammation |
| US6794518B1 (en) | 1998-12-18 | 2004-09-21 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6838074B2 (en) | 2001-08-08 | 2005-01-04 | Bristol-Myers Squibb Company | Simultaneous imaging of cardiac perfusion and a vitronectin receptor targeted imaging agent |
| US6878363B2 (en) | 2000-05-17 | 2005-04-12 | Bristol-Myers Squibb Pharma Company | Use of small molecule radioligands to discover inhibitors of amyloid-beta peptide production and for diagnostic imaging |
| US7060248B2 (en) | 2000-02-15 | 2006-06-13 | Bristol-Myers Squibb Pharma Company | Matrix metalloproteinase inhibitors |
| WO2006064451A2 (en) * | 2004-12-17 | 2006-06-22 | Koninklijke Philips Electronics N.V. | Targeting contrast agents or targeting therapeutic agents for molecular imaging and therapy |
| US7138104B2 (en) | 2001-08-08 | 2006-11-21 | Bristol-Myers Squibb Company | Simultaneous imaging of cardiac perfusion and a vitronectin receptor targeted imaging agent |
| WO2007088051A2 (en) | 2006-01-31 | 2007-08-09 | Bayer Schering Pharma Aktiengesellschaft | Modulation of mdl-1 activity for treatment of inflammatory disease |
| US7344702B2 (en) | 2004-02-13 | 2008-03-18 | Bristol-Myers Squibb Pharma Company | Contrast agents for myocardial perfusion imaging |
| US7354914B2 (en) | 2000-06-01 | 2008-04-08 | Bristol-Myers Squibb Pharma Company | Lactams substituted by cyclic succinates as inhibitors of Aβ protein production |
| US7485283B2 (en) | 2004-04-28 | 2009-02-03 | Lantheus Medical Imaging | Contrast agents for myocardial perfusion imaging |
| US7824659B2 (en) | 2005-08-10 | 2010-11-02 | Lantheus Medical Imaging, Inc. | Methods of making radiolabeled tracers and precursors thereof |
| WO2011035140A1 (en) | 2009-09-18 | 2011-03-24 | Paka Pulmonary Pharmaceuticals, Inc. | Methods and compositions for delivery of contrast moieties to the lungs |
| EP2733148A1 (en) * | 2012-11-15 | 2014-05-21 | Bayer Pharma Aktiengesellschaft | Tungsten complexes with di-dentate ligands |
| WO2014122225A1 (en) * | 2013-02-06 | 2014-08-14 | Iticon Gmbh | Heteropolyoxometalates |
| WO2014144708A1 (en) | 2013-03-15 | 2014-09-18 | The Regents Of The University Of California | Peptides having reduced toxicity that stimulate cholesterol efflux |
| US8936777B2 (en) | 2010-02-08 | 2015-01-20 | Lantheus Medical Imaging, Inc. | Methods and apparatus for synthesizing imaging agents, and intermediates thereof |
| US9408927B2 (en) | 2008-02-29 | 2016-08-09 | Lantheus Medical Imaging, Inc. | Contrast agents for applications including perfusion imaging |
| CN106631996A (en) * | 2016-12-20 | 2017-05-10 | 新疆大学 | Preparation method of organic-inorganic hybrid polyoxometallate nanorod |
| US9687571B2 (en) | 2009-04-15 | 2017-06-27 | Lantheus Medical Imaging, Inc. | Stabilization of radiopharmaceutical compositions using ascorbic acid |
| US9713651B2 (en) | 2012-08-10 | 2017-07-25 | Lantheus Medical Imaging, Inc. | Compositions, methods, and systems for the synthesis and use of imaging agents |
Citations (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3952015A (en) | 1973-11-12 | 1976-04-20 | Carl George Krespan | Macrocyclic compounds having oxa and aza linkages in the ring and containing spirooxetane groups |
| FR2348699A1 (en) * | 1976-04-21 | 1977-11-18 | Hoffmann La Roche | X-RAY CONTRAST SUBSTANCE |
| WO1986002352A1 (en) * | 1984-10-18 | 1986-04-24 | Board Of Regents, The University Of Texas System | Gadolinium chelates as nmr contrast agents |
| US4647447A (en) | 1981-07-24 | 1987-03-03 | Schering Aktiengesellschaft | Diagnostic media |
| US4652519A (en) | 1983-02-03 | 1987-03-24 | Yeda Research And Development Company Limited | Bifunctional chelating agents and process for their production |
| EP0217577A2 (en) | 1985-09-12 | 1987-04-08 | Hybritech Incorporated | Antibody complexes of hapten-modified diagnostic or therapeutic agents |
| EP0230893A2 (en) | 1986-01-30 | 1987-08-05 | BRACCO INDUSTRIA CHIMICA Società per Azioni | Paramagnetic chelates |
| EP0130934B1 (en) * | 1983-07-01 | 1987-08-05 | Schering Aktiengesellschaft | Complexing agents, complexes and complex salts |
| US4687659A (en) | 1984-11-13 | 1987-08-18 | Salutar, Inc. | Diamide-DTPA-paramagnetic contrast agents for MR imaging |
| EP0263059A2 (en) * | 1986-09-26 | 1988-04-06 | Schering Aktiengesellschaft | Complexes amides |
| EP0287465A1 (en) | 1987-04-14 | 1988-10-19 | Guerbet S.A. | Cyclic ligands containing nitrogen, metal complexes formed by these ligands, diagnostic compositions containing them and process for their preparation |
| WO1989000557A1 (en) | 1987-07-16 | 1989-01-26 | Cockbain, Julian, Roderick, Michaelson | Aminopolycarboxylic acids and derivatives thereof |
| US4826673A (en) | 1985-01-09 | 1989-05-02 | Mallinckrodt, Inc. | Methods and compositions for enhancing magnetic resonance imaging |
| EP0331616A2 (en) | 1988-02-29 | 1989-09-06 | Schering Aktiengesellschaft | Polymer-bonded complexe builders, their complexes, process for their preparation and pharmaceutical agents containing the same |
| WO1989010372A1 (en) * | 1988-04-22 | 1989-11-02 | Centre National De La Recherche Scientifique (Cnrs | Cold marking of molecules of peptidic or proteinic nature |
| EP0361960A2 (en) * | 1988-09-29 | 1990-04-04 | RANNEY, David F. | Methods and compositions for magnetic resonance imaging |
| WO1990012050A1 (en) | 1989-04-07 | 1990-10-18 | Salutar, Inc. | Chelants |
| WO1991014460A1 (en) | 1990-03-28 | 1991-10-03 | Cockbain, Julian, Roderick, Michaelson | Contrast media |
| WO1991015467A1 (en) * | 1990-04-09 | 1991-10-17 | Cockbain, Julian, Roderick, Michaelson | Aminopolycarboxylic acid chelating agents |
-
1992
- 1992-03-27 WO PCT/EP1992/000698 patent/WO1992017215A1/en active IP Right Grant
Patent Citations (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3952015A (en) | 1973-11-12 | 1976-04-20 | Carl George Krespan | Macrocyclic compounds having oxa and aza linkages in the ring and containing spirooxetane groups |
| FR2348699A1 (en) * | 1976-04-21 | 1977-11-18 | Hoffmann La Roche | X-RAY CONTRAST SUBSTANCE |
| US4647447A (en) | 1981-07-24 | 1987-03-03 | Schering Aktiengesellschaft | Diagnostic media |
| US4652519A (en) | 1983-02-03 | 1987-03-24 | Yeda Research And Development Company Limited | Bifunctional chelating agents and process for their production |
| EP0130934B1 (en) * | 1983-07-01 | 1987-08-05 | Schering Aktiengesellschaft | Complexing agents, complexes and complex salts |
| WO1986002352A1 (en) * | 1984-10-18 | 1986-04-24 | Board Of Regents, The University Of Texas System | Gadolinium chelates as nmr contrast agents |
| US4687659A (en) | 1984-11-13 | 1987-08-18 | Salutar, Inc. | Diamide-DTPA-paramagnetic contrast agents for MR imaging |
| US4826673A (en) | 1985-01-09 | 1989-05-02 | Mallinckrodt, Inc. | Methods and compositions for enhancing magnetic resonance imaging |
| EP0217577A2 (en) | 1985-09-12 | 1987-04-08 | Hybritech Incorporated | Antibody complexes of hapten-modified diagnostic or therapeutic agents |
| EP0230893A2 (en) | 1986-01-30 | 1987-08-05 | BRACCO INDUSTRIA CHIMICA Società per Azioni | Paramagnetic chelates |
| EP0263059A2 (en) * | 1986-09-26 | 1988-04-06 | Schering Aktiengesellschaft | Complexes amides |
| EP0287465A1 (en) | 1987-04-14 | 1988-10-19 | Guerbet S.A. | Cyclic ligands containing nitrogen, metal complexes formed by these ligands, diagnostic compositions containing them and process for their preparation |
| WO1989000557A1 (en) | 1987-07-16 | 1989-01-26 | Cockbain, Julian, Roderick, Michaelson | Aminopolycarboxylic acids and derivatives thereof |
| EP0331616A2 (en) | 1988-02-29 | 1989-09-06 | Schering Aktiengesellschaft | Polymer-bonded complexe builders, their complexes, process for their preparation and pharmaceutical agents containing the same |
| WO1989010372A1 (en) * | 1988-04-22 | 1989-11-02 | Centre National De La Recherche Scientifique (Cnrs | Cold marking of molecules of peptidic or proteinic nature |
| EP0361960A2 (en) * | 1988-09-29 | 1990-04-04 | RANNEY, David F. | Methods and compositions for magnetic resonance imaging |
| WO1990012050A1 (en) | 1989-04-07 | 1990-10-18 | Salutar, Inc. | Chelants |
| WO1991014460A1 (en) | 1990-03-28 | 1991-10-03 | Cockbain, Julian, Roderick, Michaelson | Contrast media |
| WO1991015467A1 (en) * | 1990-04-09 | 1991-10-17 | Cockbain, Julian, Roderick, Michaelson | Aminopolycarboxylic acid chelating agents |
Non-Patent Citations (30)
| Title |
|---|
| AM. J HOSP. PHARM., vol. 24, 1967, pages 143 |
| AM. PROF. PHARMACY, vol. 16, 1950, pages 874 |
| ASPINALL ET AL., JACS, vol. 63, 1941, pages 852 |
| BABA ET AL., MEM. FAC. TECH. TOKYO METROPOLITAN UNIV., vol. 32, 1982, pages 3207 |
| BABA ET AL., MEM. FAC. TECH. TOKYO METROPOLITAN UNIV., vol. 32, 1982, pages 3207 - 3220 |
| BEDELL ET AL., INORG. CHEM., vol. 21, 1982, pages 874 |
| BRECHBIEL ET AL., INORG. CHEM., vol. 25, 1986, pages 2772 |
| COLLENBERG, Z. ANORG. ALLG. CHEM., vol. 102, 1918, pages 247 - 276 |
| HOLM ET AL., JACS, vol. 112, 1990, pages 8015 - 8023 |
| HUBER ET AL., J. CHEM. SOC. CHEM. COMM., 1989, pages 879 |
| Inorganic Chemistry, vol. 16, no. 10, 1977, (Washington, DC, US), V.R. OTT et al.: "Di-mu-oxo, mu-oxo-mu-sulfido, and di-mu-sulfido complexes of molybdenum(V) with EDTA, cysteine, and cysteine ester ligands. Preparation and electrochemical and spectral properties", pages 2538-2545, see the whole article (cited in the application) * |
| J. Am. Chem. Soc., vol. 108, 1986, (Washington, DC, US), T. SHIBAHARA et al.: "Preparation of triangular tungsten(IV) aqua ion, W3S44+, and X-ray structure of (bpyH)5[W3S4(NCS)9]3H2O", pages 2757-2758, see the whole article (cited in the application) * |
| J. Am. Chem. Soc., vol. 110, 1988, (Washington, DC, US), T. SAITO et al.: "Synthesis of [Mo6S8(PEt3)6] by reductive dimerization of a trinuclear molybdenum chloro sulfido cluster complex coordinated with triethylphosphine and methanol: a molecular model for superconducting Chevrel phases", pages 1646-1647, see the whole article (cited in the application) * |
| J. AM. MED. ASSOC., vol. 166, 1958, pages 1042 |
| J. Inorg. Nucl. Chem., vol. 36, 1974, (Oxford, GB), J. NOVAK et al.: "Tungsten(V) complexes of ethylenediaminetetraacetic acid", pages 1061-1065, see the whole article (cited in the application) * |
| J. ORG. CHEM., vol. 49, 1984, pages 133 |
| J. PHARM SCI., vol. 71, no. 2, 1982, pages 182 |
| J. PHARM SCI., vol. 71, no. 3, 1982, pages 370 |
| J. PHARM SCI., vol. 71, no. 5, 1982, pages 495 |
| JACS, vol. 108, 1986, pages 2757 - 2758 |
| JOC, vol. 39, 1974, pages 2351 |
| JOC, vol. 45, 1980, pages 1177 |
| KASINA ET AL., J. MED. CHEM., vol. 29, 1986, pages 1933 |
| KREJCAREK ET AL., BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMM., vol. 77, 1977, pages 881 |
| MEARES ET AL., ANAL. BIOCHEM., vol. 142, 1984, pages 68 |
| NORDLANDER ET AL., TETR. LETT., 1978, pages 4987 |
| SHIBAHARA ET AL., 37TH NAT. CONF. COORD. CHEM., TOKYO |
| TOMALIA, ANGEW. CHEM. ENGL. ED., vol. 29, 1990, pages 138 - 175 |
| V6GTLE ET AL., LIEBIGS ANN. CHEM., 1977, pages 1344 |
| YEH ET AL., ANALYTICAL BIOCHEM., vol. 100, 1979, pages 152 |
Cited By (70)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993019788A1 (en) * | 1992-04-03 | 1993-10-14 | Mallinckrodt Medical, Inc. | Methods and compositions for magnetic resonance imaging |
| US5364953A (en) * | 1992-11-05 | 1994-11-15 | Mallinckrodt Medical, Inc. | High relaxivity, paramagnetic, metal clusters for magnetic resonance imaging |
| EP0739337A1 (en) * | 1994-01-14 | 1996-10-30 | Mallinckrodt Medical, Inc. | Functionalized macrocyclic ligands for imaging applications |
| EP0739337A4 (en) * | 1994-01-14 | 1997-03-26 | Mallinckrodt Medical Inc | Functionalized macrocyclic ligands for imaging applications |
| US6303101B1 (en) | 1995-01-26 | 2001-10-16 | Nycomed Imaging As | Bismuth compounds |
| US6117412A (en) * | 1995-01-26 | 2000-09-12 | Nycomed Imaging As | Non-cluster type bismuth compounds |
| US5817289A (en) * | 1995-01-26 | 1998-10-06 | Nycomed Imaging As | Non-cluster type bismuth compounds |
| WO1996040287A2 (en) * | 1995-06-07 | 1996-12-19 | Nycomed Salutar, Inc. | Preparation and use of contrast agents |
| WO1996040287A3 (en) * | 1995-06-07 | 1997-02-27 | Nycomed Salutar Inc | Preparation and use of contrast agents |
| EP0871637A1 (en) * | 1995-07-20 | 1998-10-21 | Mallinckrodt Medical, Inc. | Tungsten clusters |
| EP0871637A4 (en) * | 1995-07-20 | 2000-01-05 | Mallinckrodt Medical Inc | Tungsten clusters |
| WO1997026921A3 (en) * | 1996-01-23 | 1997-10-23 | Nycomed Salutar Inc | Contrast media |
| WO1997026921A2 (en) * | 1996-01-23 | 1997-07-31 | Nycomed Salutar Inc. | Contrast media |
| US6548663B1 (en) | 1998-03-31 | 2003-04-15 | Bristol-Myers Squibb Pharma Company | Benzodiazepine vitronectin receptor antagonist pharmaceuticals |
| US7052673B2 (en) | 1998-03-31 | 2006-05-30 | Bristol-Myers Squibb Pharma Company | Pharmaceuticals for the imaging of angiogenic disorders |
| US6524553B2 (en) | 1998-03-31 | 2003-02-25 | Bristol-Myers Squibb Pharma Company | Quinolone vitronectin receptor antagonist pharmaceuticals |
| US6537520B1 (en) | 1998-03-31 | 2003-03-25 | Bristol-Myers Squibb Pharma Company | Pharmaceuticals for the imaging of angiogenic disorders |
| US6056939A (en) * | 1998-08-28 | 2000-05-02 | Desreux; Jean F. | Self-assembling heteropolymetallic chelates as imaging agents and radiopharmaceuticals |
| US7018611B2 (en) | 1998-12-18 | 2006-03-28 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6558649B1 (en) | 1998-12-18 | 2003-05-06 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6569402B1 (en) | 1998-12-18 | 2003-05-27 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6683163B2 (en) | 1998-12-18 | 2004-01-27 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6689337B2 (en) | 1998-12-18 | 2004-02-10 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6743412B2 (en) | 1998-12-18 | 2004-06-01 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US7332149B1 (en) | 1998-12-18 | 2008-02-19 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6794518B1 (en) | 1998-12-18 | 2004-09-21 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6818201B2 (en) | 1998-12-18 | 2004-11-16 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6511649B1 (en) | 1998-12-18 | 2003-01-28 | Thomas D. Harris | Vitronectin receptor antagonist pharmaceuticals |
| US7090828B2 (en) | 1998-12-18 | 2006-08-15 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6511648B2 (en) | 1998-12-18 | 2003-01-28 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US7321045B2 (en) | 1998-12-18 | 2008-01-22 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US7060248B2 (en) | 2000-02-15 | 2006-06-13 | Bristol-Myers Squibb Pharma Company | Matrix metalloproteinase inhibitors |
| US6878363B2 (en) | 2000-05-17 | 2005-04-12 | Bristol-Myers Squibb Pharma Company | Use of small molecule radioligands to discover inhibitors of amyloid-beta peptide production and for diagnostic imaging |
| US7354914B2 (en) | 2000-06-01 | 2008-04-08 | Bristol-Myers Squibb Pharma Company | Lactams substituted by cyclic succinates as inhibitors of Aβ protein production |
| US7456278B2 (en) | 2000-06-01 | 2008-11-25 | Bristol-Myers Squibb Pharma Corporation | Lactams substituted by cyclic succinates as inhibitors of Aβ protein production |
| US6770259B2 (en) | 2000-11-03 | 2004-08-03 | Bristol-Myers Squibb Pharma Company | Simultaneous dual isotope imaging of cardiac perfusion and cardiac inflammation |
| US7138104B2 (en) | 2001-08-08 | 2006-11-21 | Bristol-Myers Squibb Company | Simultaneous imaging of cardiac perfusion and a vitronectin receptor targeted imaging agent |
| US6838074B2 (en) | 2001-08-08 | 2005-01-04 | Bristol-Myers Squibb Company | Simultaneous imaging of cardiac perfusion and a vitronectin receptor targeted imaging agent |
| US9161997B2 (en) | 2001-08-16 | 2015-10-20 | David S. Casebier | Contrast agents for myocardial perfusion imaging |
| US10889550B2 (en) | 2004-02-13 | 2021-01-12 | Lantheus Medical Imaging, Inc. | Contrast agents for myocardial perfusion imaging |
| US10125106B2 (en) | 2004-02-13 | 2018-11-13 | Lantheus Medical Imaging, Inc. | Contrast agents for myocardial perfusion imaging |
| US7344702B2 (en) | 2004-02-13 | 2008-03-18 | Bristol-Myers Squibb Pharma Company | Contrast agents for myocardial perfusion imaging |
| EP3385253A1 (en) | 2004-02-13 | 2018-10-10 | Lantheus Medical Imaging, Inc. | Contrast agents for myocardial perfusion imaging |
| US9718786B2 (en) | 2004-02-13 | 2017-08-01 | Lantheus Medical Imaging, Inc. | Contrast agents for myocardial perfusion imaging |
| US8226929B2 (en) | 2004-02-13 | 2012-07-24 | Lantheus Medical Imaging, Inc. | Contrast agents for myocardial perfusion imaging |
| US7485283B2 (en) | 2004-04-28 | 2009-02-03 | Lantheus Medical Imaging | Contrast agents for myocardial perfusion imaging |
| EP2253333A2 (en) | 2004-04-28 | 2010-11-24 | Lantheus Medical Imaging, Inc. | Contrast Agents for Myocardial Perfusion Imaging |
| US8263042B2 (en) | 2004-04-28 | 2012-09-11 | Lantheus Medical Imaging, Inc. | Contrast agents for myocardial perfusion imaging |
| WO2006064451A2 (en) * | 2004-12-17 | 2006-06-22 | Koninklijke Philips Electronics N.V. | Targeting contrast agents or targeting therapeutic agents for molecular imaging and therapy |
| WO2006064451A3 (en) * | 2004-12-17 | 2007-03-01 | Koninkl Philips Electronics Nv | Targeting contrast agents or targeting therapeutic agents for molecular imaging and therapy |
| US7824659B2 (en) | 2005-08-10 | 2010-11-02 | Lantheus Medical Imaging, Inc. | Methods of making radiolabeled tracers and precursors thereof |
| WO2007088051A2 (en) | 2006-01-31 | 2007-08-09 | Bayer Schering Pharma Aktiengesellschaft | Modulation of mdl-1 activity for treatment of inflammatory disease |
| US10245332B2 (en) | 2008-02-29 | 2019-04-02 | Lantheus Medical Imaging, Inc. | Contrast agents for applications including perfusion imaging |
| US9408927B2 (en) | 2008-02-29 | 2016-08-09 | Lantheus Medical Imaging, Inc. | Contrast agents for applications including perfusion imaging |
| US9687571B2 (en) | 2009-04-15 | 2017-06-27 | Lantheus Medical Imaging, Inc. | Stabilization of radiopharmaceutical compositions using ascorbic acid |
| WO2011035140A1 (en) | 2009-09-18 | 2011-03-24 | Paka Pulmonary Pharmaceuticals, Inc. | Methods and compositions for delivery of contrast moieties to the lungs |
| US10022462B2 (en) | 2010-02-08 | 2018-07-17 | Lantheus Medical Imaging, Inc. | Methods and apparatus for synthesizing imaging agents, and intermediates thereof |
| US10842892B2 (en) | 2010-02-08 | 2020-11-24 | Lantheus Medical Imaging, Inc. | Methods and apparatus for synthesizing imaging agents, and intermediates thereof |
| US9603951B2 (en) | 2010-02-08 | 2017-03-28 | Lantheus Medical Imaging, Inc. | Methods and apparatus for synthesizing imaging agents, and intermediates thereof |
| US8936777B2 (en) | 2010-02-08 | 2015-01-20 | Lantheus Medical Imaging, Inc. | Methods and apparatus for synthesizing imaging agents, and intermediates thereof |
| US9919064B2 (en) | 2012-08-10 | 2018-03-20 | Lantheus Medical Imaging, Inc. | Compositions, methods, and systems for the synthesis and use of imaging agents |
| US10500293B2 (en) | 2012-08-10 | 2019-12-10 | Lantheus Medical Imaging, Inc. | Compositions, methods, and systems for the synthesis and use of imaging agents |
| US9713651B2 (en) | 2012-08-10 | 2017-07-25 | Lantheus Medical Imaging, Inc. | Compositions, methods, and systems for the synthesis and use of imaging agents |
| US11744906B2 (en) | 2012-08-10 | 2023-09-05 | Lantheus Medical Imaging, Inc. | Compositions, methods, and systems for the synthesis and use of imaging agents |
| EP2733148A1 (en) * | 2012-11-15 | 2014-05-21 | Bayer Pharma Aktiengesellschaft | Tungsten complexes with di-dentate ligands |
| RU2667894C2 (en) * | 2013-02-06 | 2018-09-25 | Пом Патентфервальтгунгс Гбр | Heteropolyoxometalates |
| WO2014122225A1 (en) * | 2013-02-06 | 2014-08-14 | Iticon Gmbh | Heteropolyoxometalates |
| US10531665B2 (en) | 2013-02-06 | 2020-01-14 | Pom Patentverwaltungs Gbr | Heteropolyoxometalates |
| WO2014144708A1 (en) | 2013-03-15 | 2014-09-18 | The Regents Of The University Of California | Peptides having reduced toxicity that stimulate cholesterol efflux |
| CN106631996A (en) * | 2016-12-20 | 2017-05-10 | 新疆大学 | Preparation method of organic-inorganic hybrid polyoxometallate nanorod |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1992017215A1 (en) | Contrast media | |
| US5482699A (en) | Multinuclear complexes for x-ray imaging | |
| US5458869A (en) | Multinuclear complexes for X-ray imaging | |
| US5804161A (en) | Contrast agents | |
| US5804163A (en) | Contrast agents for magnetic resonance imaging aminosaccharide | |
| US4957939A (en) | Sterile pharmaceutical compositions of gadolinium chelates useful enhancing NMR imaging | |
| US4647447A (en) | Diagnostic media | |
| US5560903A (en) | Method of enhancing paramagnetism in chelates for MRI | |
| WO1995019185A1 (en) | Functionalized aza-macrobicyclic ligands for imaging applications | |
| NZ236267A (en) | 10-(2'-hydroxy-3'-polyoxaalkyl)-1,4,7-triscarboxymethyl-1,4,7,10-tetraazacyclododecane | |
| CA1230342A (en) | Tc99m-phenida, radioscintigraphic agent for diagnosis of hepatobiliary disease | |
| EP0876161B1 (en) | Contrast media | |
| US5717121A (en) | Preparation and use of contrast agents | |
| EP0517765A1 (en) | Preparations for mr diagnosis | |
| MXPA98009385A (en) | Agents against |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU CA FI HU JP KR NO PL RO RU US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IT LU MC NL SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| EX32 | Extension under rule 32 effected after completion of technical preparation for international publication | ||
| LE32 | Later election for international application filed prior to expiration of 19th month from priority date or according to rule 32.2 (b) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2106976 Country of ref document: CA Ref document number: 1992907484 Country of ref document: EP Ref document number: 08122461 Country of ref document: US Ref document number: 934194 Country of ref document: FI |
|
| WWP | Wipo information: published in national office |
Ref document number: 1992907484 Country of ref document: EP |
|
| LE32 | Later election for international application filed prior to expiration of 19th month from priority date or according to rule 32.2 (b) | ||
| EX32 | Extension under rule 32 effected after completion of technical preparation for international publication | ||
| LE32 | Later election for international application filed prior to expiration of 19th month from priority date or according to rule 32.2 (b) | ||
| LE32 | Later election for international application filed prior to expiration of 19th month from priority date or according to rule 32.2 (b) | ||
| LE32 | Later election for international application filed prior to expiration of 19th month from priority date or according to rule 32.2 (b) | ||
| LE32 | Later election for international application filed prior to expiration of 19th month from priority date or according to rule 32.2 (b) | ||
| EX32 | Extension under rule 32 effected after completion of technical preparation for international publication | ||
| LE32 | Later election for international application filed prior to expiration of 19th month from priority date or according to rule 32.2 (b) | ||
| ENP | Entry into the national phase |
Ref document number: 1995 473574 Country of ref document: US Date of ref document: 19950607 Kind code of ref document: A |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1992907484 Country of ref document: EP |