WO1994018151A1 - Acylfulvene analogues as antitumor agents - Google Patents
Acylfulvene analogues as antitumor agents Download PDFInfo
- Publication number
- WO1994018151A1 WO1994018151A1 PCT/US1994/001232 US9401232W WO9418151A1 WO 1994018151 A1 WO1994018151 A1 WO 1994018151A1 US 9401232 W US9401232 W US 9401232W WO 9418151 A1 WO9418151 A1 WO 9418151A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- illudin
- cells
- tumor
- analog
- acylfulvene
- Prior art date
Links
- 0 C[C@@]1(C2=O)OC(C3)[C@]13C(C)=C1C2=CC(C)=C1* Chemical compound C[C@@]1(C2=O)OC(C3)[C@]13C(C)=C1C2=CC(C)=C1* 0.000 description 1
- GLUHRZJWBWGWFS-FUKGKQRISA-N C[C@@]1(C2=O)OC(C3)[C@]13C(C)=C1C2=CC(C)=C1Cc(cc1)ccc1O Chemical compound C[C@@]1(C2=O)OC(C3)[C@]13C(C)=C1C2=CC(C)=C1Cc(cc1)ccc1O GLUHRZJWBWGWFS-FUKGKQRISA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/12—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/12—Ketones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/215—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/215—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids
- A61K31/22—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids of acyclic acids, e.g. pravastatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/27—Esters, e.g. nitroglycerine, selenocyanates of carbamic or thiocarbamic acids, meprobamate, carbachol, neostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/45—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by condensation
- C07C45/455—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by condensation with carboxylic acids or their derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/45—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by condensation
- C07C45/46—Friedel-Crafts reactions
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/65—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by splitting-off hydrogen atoms or functional groups; by hydrogenolysis of functional groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
- C07C45/673—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by change of size of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/657—Unsaturated compounds containing a keto groups being part of a ring containing six-membered aromatic rings
- C07C49/665—Unsaturated compounds containing a keto groups being part of a ring containing six-membered aromatic rings a keto group being part of a condensed ring system
- C07C49/67—Unsaturated compounds containing a keto groups being part of a ring containing six-membered aromatic rings a keto group being part of a condensed ring system having two rings, e.g. tetralones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/657—Unsaturated compounds containing a keto groups being part of a ring containing six-membered aromatic rings
- C07C49/683—Unsaturated compounds containing a keto groups being part of a ring containing six-membered aromatic rings having unsaturation outside the aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/703—Unsaturated compounds containing a keto groups being part of a ring containing hydroxy groups
- C07C49/723—Unsaturated compounds containing a keto groups being part of a ring containing hydroxy groups polycyclic
- C07C49/727—Unsaturated compounds containing a keto groups being part of a ring containing hydroxy groups polycyclic a keto group being part of a condensed ring system
- C07C49/737—Unsaturated compounds containing a keto groups being part of a ring containing hydroxy groups polycyclic a keto group being part of a condensed ring system having three rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/703—Unsaturated compounds containing a keto groups being part of a ring containing hydroxy groups
- C07C49/747—Unsaturated compounds containing a keto groups being part of a ring containing hydroxy groups containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/753—Unsaturated compounds containing a keto groups being part of a ring containing ether groups, groups, groups, or groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/753—Unsaturated compounds containing a keto groups being part of a ring containing ether groups, groups, groups, or groups
- C07C49/755—Unsaturated compounds containing a keto groups being part of a ring containing ether groups, groups, groups, or groups a keto group being part of a condensed ring system with two or three rings, at least one ring being a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/007—Esters of unsaturated alcohols having the esterified hydroxy group bound to an acyclic carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2603/00—Systems containing at least three condensed rings
- C07C2603/93—Spiro compounds
- C07C2603/94—Spiro compounds containing "free" spiro atoms
Abstract
Description









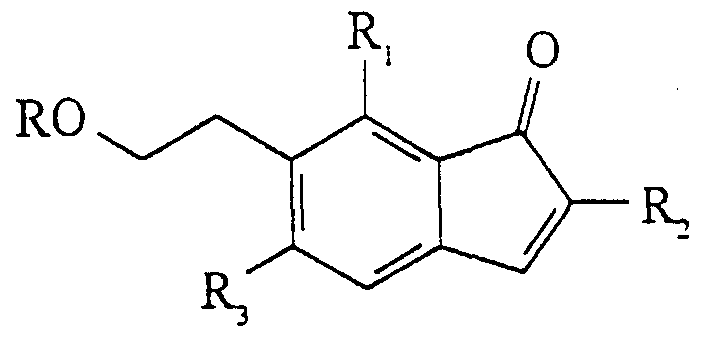




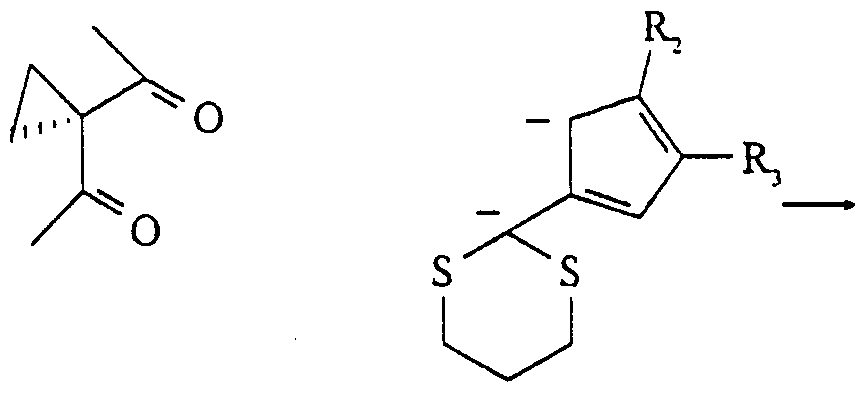


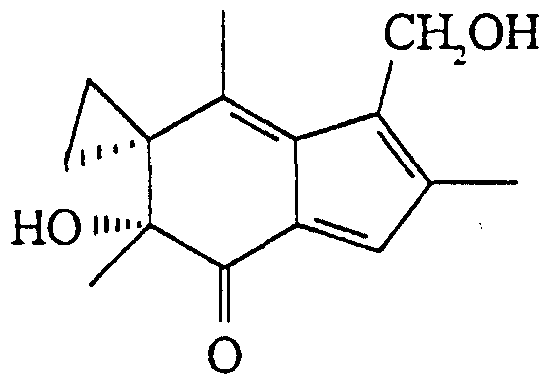




Claims




Priority Applications (15)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002155329A CA2155329C (en) | 1993-02-09 | 1994-02-02 | Acylfulvene analogues as antitumor agents |
| DK94908702T DK0683762T3 (en) | 1993-02-09 | 1994-02-02 | Acylfulven analogues as anti-tumor agents |
| KR1019950703280A KR100311327B1 (en) | 1993-02-09 | 1994-02-02 | Acylpulbene analogs as antitumor agents |
| AU61700/94A AU676889B2 (en) | 1993-02-09 | 1994-02-02 | Acylfulvene analogues as antitumor agents |
| DE69415507T DE69415507T2 (en) | 1993-02-09 | 1994-02-02 | ANALOGS OF ACYLFULVEN AS AN ANTITUARY AGENT |
| BR9405689A BR9405689A (en) | 1993-02-09 | 1994-02-02 | Process of inhibiting tumor growth in an individual composition capable of binding to a tumor antigen and therapeutic composition |
| MD96-0212A MD1418G2 (en) | 1993-02-09 | 1994-02-02 | Acylfulven analogues pharmaceutical composition on base thereof |
| TJ96000336A TJ266B (en) | 1993-02-09 | 1994-02-02 | Application of acylfulvene analogues and pharmaceutical composition on their basis |
| RU95121694A RU2145849C1 (en) | 1993-02-09 | 1994-02-02 | Use of acylfulven analogs and pharmaceutical composition based on thereof |
| JP51820894A JP3908270B2 (en) | 1993-02-09 | 1994-02-02 | Acylfulvene analogues and pharmaceutical compositions thereof |
| PL94310159A PL175024B1 (en) | 1993-02-09 | 1994-02-02 | Cylofulvenic analogues as antineoplastic agents |
| EP94908702A EP0683762B1 (en) | 1993-02-09 | 1994-02-02 | Acylfulvene analogues as antitumor agents |
| HU9502358A HU220059B (en) | 1993-02-09 | 1994-02-02 | Acylfulvene analogues, use of them for producing pharmaceutical compositions and pharmaceutical compositions containing them |
| NO19953099A NO318648B1 (en) | 1993-02-09 | 1995-08-07 | Use of 6-substituted acylfulven analogs having anti-tumor effect |
| GR990400821T GR3029736T3 (en) | 1993-02-09 | 1999-03-19 | Acylfulvene analogues as antitumor agents. |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/015,179 US5439936A (en) | 1989-10-03 | 1993-02-09 | Method of treating certain tumors using illudin analogs |
| US08/015,179 | 1993-02-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994018151A1 true WO1994018151A1 (en) | 1994-08-18 |
Family
ID=21769946
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1994/001232 WO1994018151A1 (en) | 1993-02-09 | 1994-02-02 | Acylfulvene analogues as antitumor agents |
Country Status (22)
| Country | Link |
|---|---|
| US (3) | US5439936A (en) |
| EP (1) | EP0683762B1 (en) |
| JP (1) | JP3908270B2 (en) |
| KR (1) | KR100311327B1 (en) |
| CN (1) | CN1046934C (en) |
| AT (1) | ATE174894T1 (en) |
| AU (1) | AU676889B2 (en) |
| BR (1) | BR9405689A (en) |
| CA (1) | CA2155329C (en) |
| CZ (1) | CZ288596B6 (en) |
| DE (1) | DE69415507T2 (en) |
| DK (1) | DK0683762T3 (en) |
| ES (1) | ES2125441T3 (en) |
| GR (1) | GR3029736T3 (en) |
| HU (1) | HU220059B (en) |
| MD (1) | MD1418G2 (en) |
| NO (1) | NO318648B1 (en) |
| NZ (1) | NZ262282A (en) |
| PL (1) | PL175024B1 (en) |
| RU (1) | RU2145849C1 (en) |
| TJ (1) | TJ266B (en) |
| WO (1) | WO1994018151A1 (en) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5856580A (en) * | 1996-08-08 | 1999-01-05 | The Regents Of University Of California | Total synthesis of antitumor acylfulvenes |
| US5932553A (en) * | 1996-07-18 | 1999-08-03 | The Regents Of The University Of California | Illudin analogs useful as antitumor agents |
| US6025328A (en) * | 1998-02-20 | 2000-02-15 | The Regents Of The University Of California | Antitumor agents |
| US7141603B2 (en) | 1999-02-19 | 2006-11-28 | The Regents Of The University California | Antitumor agents |
| EP1909783A2 (en) * | 2005-08-03 | 2008-04-16 | The Regents of the University of California | Illudin analogs useful as anticancer agents |
| EP2277595A2 (en) | 2004-06-24 | 2011-01-26 | Novartis Vaccines and Diagnostics, Inc. | Compounds for immunopotentiation |
| CN106083953A (en) * | 2016-06-15 | 2016-11-09 | 成都医学院 | A kind of from fiddlehead, extract former pteroside and the method preparing high purity raw pteroside |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6436916B1 (en) | 2000-10-12 | 2002-08-20 | Alvin Guttag | Ibuprofen-aspirin and hydroxymethylacylfulvene analogs |
| US7015247B2 (en) * | 2000-10-12 | 2006-03-21 | Alvin Guttag | Ibuprofen-aspirin, hydroxymethylacylfulvene analogs and L-sugar illudin analogs |
| TWI522109B (en) * | 2009-01-26 | 2016-02-21 | 臺北醫學大學 | Use of pterosin compounds for treating diabetes and obesity |
| US10285955B2 (en) | 2014-04-10 | 2019-05-14 | Af Chemicals, Llc | Affinity medicant conjugate |
| US11135182B2 (en) | 2014-04-10 | 2021-10-05 | Af Chemicals, Llc | Affinity medicant conjugates |
| JP2017518359A (en) | 2014-04-10 | 2017-07-06 | エイエフ ケミカルス, エルエルシーAf Chemicals, Llc | Affinity drug conjugate |
| US20210137850A1 (en) * | 2017-03-15 | 2021-05-13 | Memorial Sloan Kettering Cancer Center | Diagnosis & treatment of ercc3-mutant cancer |
| KR20210087435A (en) * | 2018-09-04 | 2021-07-12 | 랜턴 파마 인코포레이티드 | Iludin analogs, uses thereof, and methods for their synthesis |
| EP3667323A1 (en) | 2018-12-11 | 2020-06-17 | Kelner, Michael | Methods, compositions and devices for treating cancer with illudofulvenes |
| RU2738848C1 (en) * | 2019-10-24 | 2020-12-17 | Общество с ограниченной ответственностью "Научно-исследовательский институт ХимРар" (ООО "НИИ ХимРар") | Hepatitis b virus (hbv) inhibitor, which is a derivative of n-{3-[6-(dialkylamino)pyridazine-3-yl]phenyl}aryl sulphonamide derivatives and derivatives of n-{4-[6-(dialkylamino)pyridazine-3-yl]phenyl}aryl sulphonamide |
| US11591295B2 (en) | 2019-11-25 | 2023-02-28 | Af Chemicals Llc | Affinity illudofulvene conjugates |
| EP4035684A1 (en) | 2019-11-25 | 2022-08-03 | AF Chemical LLC | Affinity illudofulvene conjugates |
| CN112972443A (en) * | 2021-03-29 | 2021-06-18 | 杭州添帆生物科技有限公司 | Anticancer medicine and its application |
| WO2023239821A2 (en) * | 2022-06-07 | 2023-12-14 | Lantern Pharma Inc. | Treating cancers with combinations of acylfulvenes with ibrutinib or bortezomib |
| WO2024016014A2 (en) * | 2022-07-15 | 2024-01-18 | Lantern Pharma Inc. | Method for treating breast cancers and parp resistant breast cancers |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1991004754A2 (en) * | 1989-10-03 | 1991-04-18 | The Regents Of The University Of California | Illudin analogs as anti-tumor agents |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9017024D0 (en) * | 1990-08-03 | 1990-09-19 | Erba Carlo Spa | New linker for bioactive agents |
| FI85318C (en) * | 1990-08-14 | 1992-03-25 | Tecnomen Oy | COMPENSATION AV FELET I EN KLOCKAS GAONG. |
| JPH06223404A (en) * | 1993-01-25 | 1994-08-12 | Mitsubishi Kasei Corp | Optical information recording medium |
-
1993
- 1993-02-09 US US08/015,179 patent/US5439936A/en not_active Expired - Lifetime
-
1994
- 1994-02-02 WO PCT/US1994/001232 patent/WO1994018151A1/en active IP Right Grant
- 1994-02-02 AT AT94908702T patent/ATE174894T1/en not_active IP Right Cessation
- 1994-02-02 BR BR9405689A patent/BR9405689A/en not_active Application Discontinuation
- 1994-02-02 KR KR1019950703280A patent/KR100311327B1/en not_active IP Right Cessation
- 1994-02-02 CN CN94191560A patent/CN1046934C/en not_active Expired - Fee Related
- 1994-02-02 AU AU61700/94A patent/AU676889B2/en not_active Ceased
- 1994-02-02 JP JP51820894A patent/JP3908270B2/en not_active Expired - Fee Related
- 1994-02-02 PL PL94310159A patent/PL175024B1/en not_active IP Right Cessation
- 1994-02-02 TJ TJ96000336A patent/TJ266B/en unknown
- 1994-02-02 CA CA002155329A patent/CA2155329C/en not_active Expired - Fee Related
- 1994-02-02 ES ES94908702T patent/ES2125441T3/en not_active Expired - Lifetime
- 1994-02-02 NZ NZ262282A patent/NZ262282A/en not_active IP Right Cessation
- 1994-02-02 MD MD96-0212A patent/MD1418G2/en not_active IP Right Cessation
- 1994-02-02 CZ CZ19951986A patent/CZ288596B6/en not_active IP Right Cessation
- 1994-02-02 EP EP94908702A patent/EP0683762B1/en not_active Expired - Lifetime
- 1994-02-02 RU RU95121694A patent/RU2145849C1/en not_active IP Right Cessation
- 1994-02-02 DE DE69415507T patent/DE69415507T2/en not_active Expired - Lifetime
- 1994-02-02 DK DK94908702T patent/DK0683762T3/en active
- 1994-02-02 HU HU9502358A patent/HU220059B/en not_active IP Right Cessation
- 1994-07-15 US US08/276,169 patent/US5563176A/en not_active Expired - Lifetime
- 1994-11-01 US US08/332,940 patent/US5523490A/en not_active Expired - Lifetime
-
1995
- 1995-08-07 NO NO19953099A patent/NO318648B1/en not_active IP Right Cessation
-
1999
- 1999-03-19 GR GR990400821T patent/GR3029736T3/en unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1991004754A2 (en) * | 1989-10-03 | 1991-04-18 | The Regents Of The University Of California | Illudin analogs as anti-tumor agents |
Non-Patent Citations (1)
| Title |
|---|
| T.C. MCMORRIS ET AL.: "Structure and reactivity of illudins", TETRAHEDRON, vol. 45, no. 17, 1989, pages 5433 - 5440 * |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6380403B1 (en) | 1996-07-18 | 2002-04-30 | Regents Of The University Of California | Illudin analogs useful as antitumor agents |
| US5932553A (en) * | 1996-07-18 | 1999-08-03 | The Regents Of The University Of California | Illudin analogs useful as antitumor agents |
| US7713939B2 (en) | 1996-07-18 | 2010-05-11 | The Regents Of The University Of California | Illudin analogs useful as antitumor agents |
| US6069283A (en) * | 1996-07-18 | 2000-05-30 | The Regents Of The University Of California | Illudin analogs useful as antitumor agents |
| US7329759B2 (en) | 1996-07-18 | 2008-02-12 | The Regents Of The University Of California | Illudin analogs useful as antitumor agents |
| US6987193B2 (en) | 1996-07-18 | 2006-01-17 | The Regents Of The University Of California | Illudin analogs useful as antitumor agents |
| US6639105B2 (en) | 1996-07-18 | 2003-10-28 | The Regents Of The University Of California | Illudin analogs useful as antitumor agents |
| EP1454890A2 (en) * | 1996-08-08 | 2004-09-08 | The Regents of The University of California at San Diego | Anti-tumor Acylfulvenes |
| US6252093B1 (en) | 1996-08-08 | 2001-06-26 | The Regents Of The University Of California | Total synthesis of antitumor acylfulvenes |
| US6160184A (en) * | 1996-08-08 | 2000-12-12 | The Regents Of The University Of California | Total synthesis of antitumor acylfulvenes |
| US6469184B2 (en) | 1996-08-08 | 2002-10-22 | Regents Of The University Of California | Total synthesis of antitumor acylfulvenes |
| US6717017B2 (en) | 1996-08-08 | 2004-04-06 | The Regents Of The University Of California | Total synthesis of antitumor acylfulvenes |
| US5856580A (en) * | 1996-08-08 | 1999-01-05 | The Regents Of University Of California | Total synthesis of antitumor acylfulvenes |
| EP1454890A3 (en) * | 1996-08-08 | 2004-11-10 | The Regents Of The University Of California | Anti-tumor Acylfulvenes |
| US6855696B2 (en) | 1998-02-20 | 2005-02-15 | The Regents Of The University Of California | Antitumor agents |
| US6908918B2 (en) | 1998-02-20 | 2005-06-21 | Regents Of The University Of California | Antitumor agents |
| US6323181B1 (en) | 1998-02-20 | 2001-11-27 | The Regents Of The University Of California | Antitumor agents |
| US6548679B1 (en) | 1998-02-20 | 2003-04-15 | The Regents Of The University Of California | Antitumor agents |
| US7629380B2 (en) | 1998-02-20 | 2009-12-08 | The Regents Of The University Of California | Antitumor agents |
| US6025328A (en) * | 1998-02-20 | 2000-02-15 | The Regents Of The University Of California | Antitumor agents |
| US7141603B2 (en) | 1999-02-19 | 2006-11-28 | The Regents Of The University California | Antitumor agents |
| EP2277595A2 (en) | 2004-06-24 | 2011-01-26 | Novartis Vaccines and Diagnostics, Inc. | Compounds for immunopotentiation |
| EP1909783A2 (en) * | 2005-08-03 | 2008-04-16 | The Regents of the University of California | Illudin analogs useful as anticancer agents |
| US7655695B2 (en) | 2005-08-03 | 2010-02-02 | The Regents Of The University Of California | Illudin analogs useful as anticancer agents |
| EP1909783A4 (en) * | 2005-08-03 | 2010-08-04 | Univ California | Illudin analogs useful as anticancer agents |
| CN106083953A (en) * | 2016-06-15 | 2016-11-09 | 成都医学院 | A kind of from fiddlehead, extract former pteroside and the method preparing high purity raw pteroside |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU676889B2 (en) | Acylfulvene analogues as antitumor agents | |
| US5439942A (en) | Method of treating certain tumors using illudin analogs | |
| EP1454893B1 (en) | Illudin analogs useful as antitumor agents | |
| JPS59225150A (en) | Pesticidal fragrant compound, synthesis and intermediate therefor, drug containing same and use as medicine | |
| DE69907419T2 (en) | ANTITUMOR SUBSTANCES | |
| JPH0296523A (en) | Platinum chemical remedy product | |
| EP3988521A1 (en) | Cellular senescence-activating compounds | |
| DE60012750T2 (en) | NEW XANTHON DERIVATIVES, THEIR MANUFACTURE AND THEIR USE AS DRUGS | |
| US7405309B2 (en) | Pyranone derivatives useful for treating cancer | |
| AU2003297671A1 (en) | Diterpenoid compounds, compositions thereof and their use as anti-cancer or anti-fungal agents | |
| US5494930A (en) | Caribenolide I | |
| KR810001489B1 (en) | Process for preparing oxazolidinedione derivatives of vinca alkaloids | |
| US3934024A (en) | Method of producing analgesia and compositions useful therein | |
| JPS62500453A (en) | Use of pyrotin derivatives | |
| Kudinova et al. | Effect of the synthetic resorcinol derivative of caryophyllene on cholesterol biosynthesis and the functional activity of immune system cells | |
| Karekezi | Aspects of the biological activity of the schistosomicide oxamniquine | |
| WO1992000740A2 (en) | Bis-dioxopiperazines and their use as protection agents | |
| US20110306662A1 (en) | Steroid-Derived Cyclopamine Analogs and Methods for Using the Same in the Prevention or Treatment of Cancer |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 94191560.3 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AT AU BB BG BR BY CA CH CN CZ DE DK ES FI GB HU JP KP KR KZ LK LU LV MG MN MW NL NO NZ PL PT RO RU SD SE SK UA UZ VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| EX32 | Extension under rule 32 effected after completion of technical preparation for international publication | ||
| LE32 | Later election for international application filed prior to expiration of 19th month from priority date or according to rule 32.2 (b) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: PV1995-1986 Country of ref document: CZ Ref document number: 2155329 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1019950703280 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 262282 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1994908702 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1994908702 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: PV1995-1986 Country of ref document: CZ |
|
| EX32 | Extension under rule 32 effected after completion of technical preparation for international publication |
Free format text: AM+,KG+,MD+,TJ+,TM+ |
|
| LE32 | Later election for international application filed prior to expiration of 19th month from priority date or according to rule 32.2 (b) |
Free format text: AM*,KG*,MD*,TJ*,TM* |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 96-0212 Country of ref document: MD |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1994908702 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: PV1995-1986 Country of ref document: CZ |