WO1995006031A1 - Inhibitors of tnf-alpha secretion - Google Patents
Inhibitors of tnf-alpha secretion Download PDFInfo
- Publication number
- WO1995006031A1 WO1995006031A1 PCT/US1994/009343 US9409343W WO9506031A1 WO 1995006031 A1 WO1995006031 A1 WO 1995006031A1 US 9409343 W US9409343 W US 9409343W WO 9506031 A1 WO9506031 A1 WO 9506031A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- alkyl
- tnf
- compound according
- hydrogen
- Prior art date
Links
- 0 *NCC(OC1CC1)=O Chemical compound *NCC(OC1CC1)=O 0.000 description 3
- KXYAVSFOJVUIHT-UHFFFAOYSA-N C=Cc1cc(cccc2)c2cc1 Chemical compound C=Cc1cc(cccc2)c2cc1 KXYAVSFOJVUIHT-UHFFFAOYSA-N 0.000 description 1
- YUFGIUWXYHMZSZ-NDOUWSIVSA-N CC(C)(C)OC(NC(/C=C/C1=CC=C2C=CC=CC2C1)C(N[C@@H](C[O](C)C(c1ccccc1)=O)C(N)=O)=O)=O Chemical compound CC(C)(C)OC(NC(/C=C/C1=CC=C2C=CC=CC2C1)C(N[C@@H](C[O](C)C(c1ccccc1)=O)C(N)=O)=O)=O YUFGIUWXYHMZSZ-NDOUWSIVSA-N 0.000 description 1
- CEEVUNYNDBMBDP-UHFFFAOYSA-N CC(COC(c1ccccc1)=O)(C(N)=O)NC(CN)=O Chemical compound CC(COC(c1ccccc1)=O)(C(N)=O)NC(CN)=O CEEVUNYNDBMBDP-UHFFFAOYSA-N 0.000 description 1
- MLPCQFIWRGHDAE-CJESRSHOSA-N CCCC(CC(NO)=O)C(N[C@@H](Cc1cc(cccc2)c2cc1)C(N[C@@H](C)C(NN)=O)=O)=O Chemical compound CCCC(CC(NO)=O)C(N[C@@H](Cc1cc(cccc2)c2cc1)C(N[C@@H](C)C(NN)=O)=O)=O MLPCQFIWRGHDAE-CJESRSHOSA-N 0.000 description 1
- CAYVDSAGVMAGEX-GVFCDGLHSA-N CCCC(CC(NO)=O)C(N[C@@H](Cc1ccc(cccc2)c2c1)C(N[C@@H](C)C(NNOC(c1ccccc1)=O)=O)=O)=O Chemical compound CCCC(CC(NO)=O)C(N[C@@H](Cc1ccc(cccc2)c2c1)C(N[C@@H](C)C(NNOC(c1ccccc1)=O)=O)=O)=O CAYVDSAGVMAGEX-GVFCDGLHSA-N 0.000 description 1
- MITVTFSYSXVCCT-NSVYTFCXSA-N CCCC(CC(OC)=O)C(N[C@@H](Cc1cc2ccccc2cc1)C(N[C@@H](C)C(NNOC(c1ccccc1)=O)=O)=O)=O Chemical compound CCCC(CC(OC)=O)C(N[C@@H](Cc1cc2ccccc2cc1)C(N[C@@H](C)C(NNOC(c1ccccc1)=O)=O)=O)=O MITVTFSYSXVCCT-NSVYTFCXSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06086—Dipeptides with the first amino acid being basic
- C07K5/06095—Arg-amino acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C259/00—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups
- C07C259/04—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups without replacement of the other oxygen atom of the carboxyl group, e.g. hydroxamic acids
- C07C259/06—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups without replacement of the other oxygen atom of the carboxyl group, e.g. hydroxamic acids having carbon atoms of hydroxamic groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/02—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link
- C07K5/0202—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link containing the structure -NH-X-X-C(=0)-, X being an optionally substituted carbon atom or a heteroatom, e.g. beta-amino acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06034—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/0606—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing heteroatoms not provided for by C07K5/06086 - C07K5/06139, e.g. Ser, Met, Cys, Thr
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06078—Dipeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06086—Dipeptides with the first amino acid being basic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- TNF- ⁇ is a principal mediator of the host response to gram-negative bacteria.
- Lipopolysaccharide LPS, also called endotoxin
- endotoxin derived from the cell wall of gram-negative bacteria
- TNF- ⁇ secreted has only been recently elucidated. Kriegler et al. Cell, 53, 45-53, (1988) conjectured that TNF- ⁇ "secretion" is due to the cleaving of the 26 kD membrane-bound molecule by a proteolytic enzyme or protease. Scuderi et. al., J. Immunology, 143, 168-173 (1989), suggested that the release of TNF- ⁇ from human leukocyte cells is dependent on one or more serine proteases, e.g., a leukocyte elastase or trypsin.
- serine proteases e.g., a leukocyte elastase or trypsin.
- protease which causes the cleavage of the TNF- ⁇ molecule into the 17 kD protein is, in fact, a metalloprotease which is believed to reside in the plasma membrane of cells producing TNF- ⁇ .
- the physicochemical characteristics of the enzyme have not been published.
- Cardiovascular Disease which includes:
- Neurologic which includes:
- Metabolic/Idiopathic which includes:
- Inhibitors of TACE would prevent the cleavage of cell-bound TNF- ⁇ thereby reducing the level of TNF- ⁇ in serum and tissues. Such inhibitors would be of significant clinical utility and could be potential therapeutics for treating the above TNF- ⁇ -related disorders.
- the invention relates to compounds of formula I:
- n 0, 1 or 2;
- B is unsubstituted or substituted C 2 to C 8 alkylene
- R 1 , R 2 and R 3 each independent of the other is hydrogen, alkylene(cycloalkyl), OR 4 , SR 4 , N(R 4 )(R 5 ), halogen, substituted or unsubstituted C 1 to C 8 alkyl, C 1 to
- n 0, 1 or 2;
- Y is hydrogen, unsubstituted or substituted C 1 to C 8 alkyl, alkylene(cycloalkyl), the group -R 8 -COOR 9 or the group -R 10 N(R 1 1 )(R 12 ); wherein R 8 is C 1 to C 8 alkylene; R 9 is hydrogen or C 1 to C 8 alkyl; R 10 is unsubstituted or substituted C 1 to C 8 alkylene; and R 11 and R 12 are each, independent of the other, hydrogen or C 1 to C 8 alkyl;
- A is a protected or an unprotected ⁇ -amino acid radical
- A is the same or different protected or unprotected ⁇ -amino acid radical; and the pharmaceutically acceptable salts thereof;
- the compound is capable of reducing serum TNF- ⁇ levels by at least 80% when administered at 25mg/kg in a murine model of LPS-induced sepsis syndrome; and a pharmaceutically acceptable carrier.
- the invention is directed to a compound of formula I:
- X is hydroxamic acid, thiol, phosphoryl or carboxyl
- n 0, 1 or 2;
- R 1 , R 2 and R 3 each independent of the other is hydrogen, alkylene(cycloalkyl), OR 4 , SR 4 , N(R 4 )(R 5 ), halogen, substituted or unsubstituted C 1 to C 8 alkyl, C 1 to
- A is a protected or an unprotected ⁇ -amino acid radical; provided that when n is 1 , A is a protected or an unprotected ⁇ -amino acid radical; when n is 2, A is the same or different protected or unprotected ⁇ -amino acid radical;
- B is unsubstituted or substituted C 2 to C 8 alkylene
- the compounds of formula I are useful as inhibitors of TNF- ⁇ secretion, and particularly useful as inhibitors of the TNF- ⁇ converting enzyme (TACE).
- TACE TNF- ⁇ converting enzyme
- the invention also relates to a method for treating a mammal having a condition or a disease characterized by overproduction or unregulated production of TNF- ⁇ , comprising administering to the mammal a composition comprising an effective amount of a biologically active compound of formula II:
- X is hydroxamic acid, thiol, phosphoryl or carboxyl
- n 0, 1 or 2;
- Y is hydrogen, unsubstituted or substituted C 1 to C 8 alkyl, alkylene(cycloalkyl), the group -R 8 -COOR 9 or the group -R 10 N(R 1 1 )(R 12 ); wherein R 8 is C 1 to C 8 alkylene; R 9 is hydrogen or C 1 to C 8 alkyl; R 10 is unsubstituted or substituted C 1 to
- R 11 and R 12 are each, independent of the other, hydrogen or C 1 to
- A is a protected or an unprotected ⁇ -amino acid radical; and when n is 2, A is the same or different protected or unprotected ⁇ -amino acid radical; and the pharmaceutically acceptable salts thereof;
- the compound is capable of reducing serum TNF levels by at least 80% when administered at 25mg/kg in a murine model of LPS-induced sepsis syndrome; and a pharmaceutically acceptable carrier.
- the invention includes pharmaceutical compositions containing a compound according to formula I as the active component.
- pharmaceutical compositions comprising a compound according to formula II and a protein which binds TNF are described.
- An example of a protein which binds TNF is an anti-TNF antibody or a soluble TNF receptor which is described in EPA 0418014, assigned to the assignee of the instant application. The disclosure of EPA 0418014 is incorporated herein by reference.
- Alkyl means a straight or branched, univalent, saturated or unsaturated hydrocarbon group of 1 to 8 carbon atoms. Alkyl groups include the straight-chain groups methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, vinyl, allyl, butenyl, pentenyl, hexenyl, heptenyl and octenyl as well as the branched isomers thereof. "Substituted alkyl” means an alkyl group substituted with one or more of hydroxy, amino, halogen, or thiol.
- Alkylene means a bivalent alkyl group as defined above.
- substituted alkylene means an alkylene group substituted with one or more of hydroxy, amino, halogen or thiol groups.
- Aryl means an aromatic or heteroaromatic group, including for example, phenyl, naphthyl, pyridyl, quinolyl, thienyl, furyl and the like, optionally substituted with one or more of C 1 to C 8 alkyl, hydroxy, amino, halogen, thiol or alkyl groups.
- Alkylene(cycloalkyl) refers to groups of the structure -R 13 -R 14 wherein R 13 is an alkylene as defined above, and R 14 is a univalent cyclic alkane radical, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and the like.
- Alkylenearyl means the group -R 15 -R 16 , wherein R 15 is a substituted or unsubstituted alkylene group as defined above, and R 16 is a substituted or unsubstituted aryl group as defined above.
- Biologically active as used in defining certain compounds of formula II, designates a compound capable of (a) inhibiting secretion of TNF- ⁇ ; (b) preventing cleavage of membrane-bound TNF- ⁇ by TACE; or (c) reducing serum TNF levels by at least 80% when administered at 25 mg/kg in a standard murine model of LPS-induced sepsis syndrome.
- preferred radicals for X are hydroxamic acid, thiol and phosphoryl. More preferred X radicals are hydroxamic acid and thiol, while the most preferred radical is hydroxamic acid.
- the preferred value for m is 1.
- R 1 or R 2 radicals are hydrogen, C 1 to C 8 alkyl and C 1 to C 8 alkylenearyl.
- R 1 or R 2 is alkyl, preferred is C 1 to C 6 alkyl and most preferred is C 1 to C 4 alkyl.
- R 1 or R 2 is alkylenearyl, preferred alkylene groups are C 1 to C 6 alkylene, and more preferred is C 1 to C 4 alkylene; and preferred aryl groups are phenyl and substituted phenyl.
- the most preferred alkylenearyl group for R 1 or R 2 is C 1 to C 4 alkylenephenyl.
- the most preferred group for R 1 is hydrogen and the most preferred group for R 2 is isobutyl.
- R 3 radicals are substituted and unsubstituted C 1 to C 8 alkyl and C 1 to C 8 alkylenearyl.
- R 3 is alkyl, preferred is C 1 to C 6 alkyl and more preferred is C 1 to C 4 alkyl, with t- butyl being most preferred.
- R 3 is C 1 to C 8 alkylenearyl
- preferred alkylene groups are C 1 to C 6 alkylene, and more preferred is C 1 to C4 alkylene
- preferred aryl groups are phenyl, naphthyl, and thienyl, each optionally substituted with hydroxy, amino, halogen, thiol or alkyl groups.
- R 3 Preferred groups for R 3 are therefore C 1 to C 4 alkylenephenyl, C 1 to C 4 alkylenenaphthyl, and C 1 to C 4 alkylenethienyl. More preferred is C 1 to C 4 alkylenenaphthyl, with methylenenaphthyl being most preferred.
- R 3 is a protected or unprotected side chain of a naturally occurring ⁇ -amino acid
- R 3 preferably is an arginine, lysine, tryptophan or tyrosine side chain.
- the most preferred radicals for R 3 are t-buyl, methylene(cyclohexyl) and methylene-(2'naphthyl).
- Y is preferably hydrogen, unsubstituted or substituted C 1 to C 8 alkyl or the group -R 10 N(R 1 1 )(R 1 2 ).
- R 10 preferably being unsubstituted or substituted C 1 to C 6 alkylene
- R 1 1 and R 12 preferably are each independently hydrogen or C 1 to C 6 alkyl.
- More preferred R 10 radicals are unsubstituted or substituted C 1 to C 4 alkylene, with dimethylene being most preferred.
- More preferred radicals for R 10 and R 11 are hydrogen or C 1 to C 4 alkyl, with hydrogen being most preferred.
- a hydroxylamine reagent described above can be hydroxylamine or alternatively, it can be an O-protected hydroxylamine such as commercially available O-trimethylsilyl hydroxylamine, O-tert-butylhydroxylamine, or O-benzylhydroxylamine.
- precursor compound (Io) may be carried out by condensing the dicarboxylate compound (Ie), with the amine (In), wherein R" is an activating group
- the benzyl ester compound (lb) is treated with a Wittig reagent, typically methyl or tert-butyl triphenylphosphoranylidene acetate, to form the alkene (Ic), as a mixture of E- and Z- isomers.
- a Wittig reagent typically methyl or tert-butyl triphenylphosphoranylidene acetate
- Reduction of the alkene compound (Ic) is carried out with hydrogen, in the presence of an appropriate catalyst (typically palladium on activated charcoal), to both hydrogenate the double bond and to remove the benzyl ester, giving the mono-ester compound (Id) as a enantiomeric mixture.
- Compound (Ie) is obtained by treating the mono-ester compound (Id) using any of a variety of conventional carboxylate activation procedures.
- the preparation of the amine compound (In) is achieved by condensing the compound (II) with the amine compound (Ik), wherein P' is an amine protective group other than P, and R" is an activating group such as an active ester, anhydride or other group that causes condensation with the amine terminus of (Ik) to occur with formation of a peptide bond, to give compound (Im). Removal of P is accomplished under appropriate conditions (Bodanszky, M.; Bodanszky, A., "The Practice of Peptide Synthesis”; Springer-Verlag: Berlin, 1984; Chapter III) to produce compound (In), either as corresponding amine or the amine salt.
- compound (If) is available commercially and others can be easily synthesized by classical methods.
- the amine-nitrile (If) is protected with an appropriate protective group reagent to produce the protected amine-nitrile (Ig).
- P is typically CBZ, BOC or FMOC groups, but can be any other suitable group.
- the protected amine-nitrile (Ig) undergoes reduction with a reagent such as borane-methyl sulfide complex or sodium borohydride/cobalt (II) chloride, to give the mono-protected diamine (Ih) which can be isolated as its amine salt.
- the compounds of formula II may be administered orally, parenterally, via inhalation, transdermally, intra-nasally, intra-ocularlly, mucosally, rectally and topically. Such administration may be in dosage unit formulations containing conventional adjuvants and carrier materials.
- parenteral as used herein includes subcutaneous injections, intravenous, intramuscular, intracisternal injection or infusion techniques.
- the diastereomers of (A) were separated and purified by reverse phase HPLC using a C 1 8 column, eluting with water containing 0.1% trifluoracetic acid with a gradient of acetonitrile (0-60% in 30 minutes) and also containing 0.1% trifluoroacetic acid, ("Method A"), to give a purified early eluting diastereomer and a purified late eluting diastereomer, which had retention times of 21 and 23 minutes respectively.
- TLC Rf 0.13 (chloroform-methanol 9:l)
- N-CBZ-aminoacetonitrile (1e) was dissolved in anhydrous tetrahydrofuran (32 ml).
- the solution was stirred and 64 ml of borane-methylsulfide complex (2M in tetrahydrofuran) was added via syringe.
- the mixture was heated to reflux and stirred overnight.
- the mixture was cooled with an ice bath as 5 ml of water was added slowly, with vigorous stirring. The stirring was continued for ca. 5 minutes, then 75 ml of 6M HCl was slowly added.
- the non- homogeneous solution was transferred to a flask containing 100 ml of absolute ethanol, and heated until it became homogeneous.
- the hot solution was dried over a small amount of anhydrous sodium sulfate, filtered, and concentrated in vacuo to obtain a solid.
- the solid was triturated with cold 1 :3 ethyl acetatehexane and collected by filtration to give 1.46g (71% yield) of L-3-(2'-naphthyl)alanyl-L-alanine, 2-(benzyloxy-carbonyl-amino)-ethyl amide (lj) as a white solid.
- the diastereomers of (1) were separated by reverse phase HPLC using a C 1 8 column and eluting with water containing 0.1% trifluoroacetic acid with a gradient of acetonitrile (0-60% in 30 minutes) also containing 0.1% trifluoroacetic acid (hereinafter "Method A”).
- the purified diastereomers (1n) and (1o) had retention times of 20 and 22 minutes, respectively.
- Diastereomer (1n) showed the following NMR data.
- 4-methylpentanoyl chloride 12(b) was prepared by adding dropwise with stirring, 38ml (0.52 mol) of thionyl chloride to 50g (0.43 mol) of 4-methylvaleric acid over 30 minutes. The mixture was heated during the addition, leading to vigorous HCl gas evolution. When the thionyl chloride addition was completed, the reaction mixture was refluxed for 1 hour. The reaction mixture was distilled, with collection of the distillate between 135 and 148 °C.
- reaction mixture was cooled to -78 °C and 34.6ml (0.25 mol) of 12(b) was added over 10 minutes. Stirring was continued at -78 °C for one hour, then the reaction mixture was allowed to stir at room temperature overnight. The tetrahydrofuran was removed in vacuo by rotary evaporation to produce an orange residue.
- reaction mixture was stirred at -5 °C for 20 minutes, then a solution of 25.0g (0.0934 mol) of 12(d) dissolved in 380 ml of anhydrous tetrahydrofuran (pre-cooled to -5°C) was added.
- the reaction was stirred at -5 °C for 2 hours, then water (50ml) was added.
- the reaction was allowed to warm to room temperature.
- the tetrahydrofuran was removed by rotary evaporation to produce a residue.
- the residue was dissolved in ethyl acetate (250ml) and washed with water (125ml) and brine (125ml).
- the reaction was filtered to remove the solid, using 500ml of dichloromethane and 250ml of water to rinse the solid collected.
- the filtrate was tranferred to a separatory funnel and the layers were separated.
- the lower(dichloromethane) layer was filtered and concentrated in vacuo by rotary evaporation to produce a dark oil.
- the oil was purified with two successive flash chromatography columns [each column: 500 grams of silica gel 60, eluted with 1900ml of 1:4 ethyl acetate: hexane, and 1000 ml of ethyl acetate] to produce 26.6 (65% yield) of 12(f) as a viscous oil.
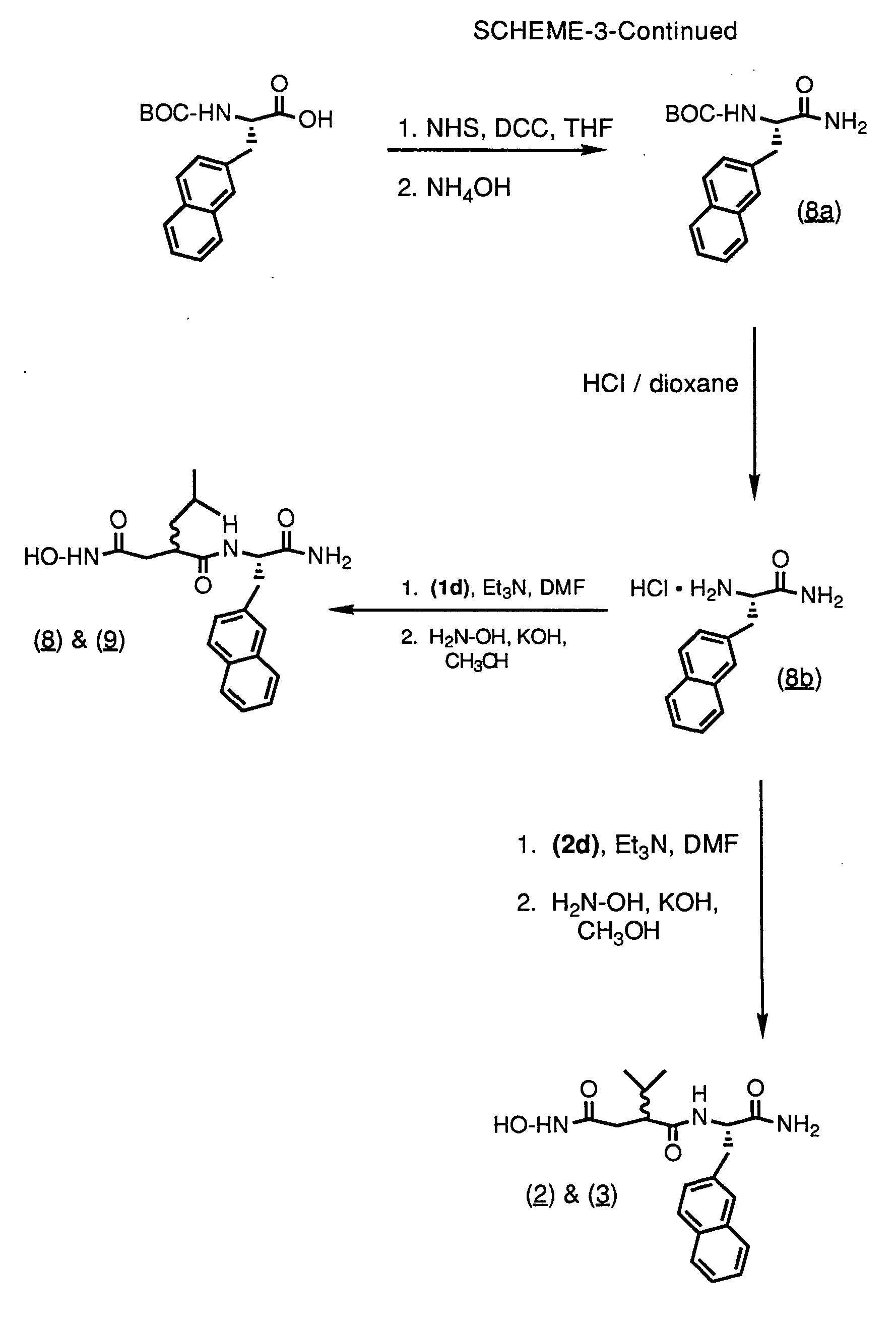
- the diastereomers (2) and (3) can be made from L-3-(2'-naphthyl)alanine amide hydrochloride (8b) and compound (2d), using the sequence of reactions used to prepare Compound (1) from Compounds (1j) and (1d).
- Compounds (2) and (3) were separated by reverse phase HPLC as described above.
- Compound (5b) was prepared from Compound (5a) in 87% yield, by the method used to prepare Compound (lj).
- TLC Rf 0.11 (chloroform-isopropanol 9:1); 1 H NMR (CDCl 3 ) ⁇ 1.28(d,3H), 1.43(m,1H), 1.70(m,4H), 3.30(m,6H), 3.91(m,2H) 4.34(m,1H), 5.03(s,2H), 5.11(s,2H), 5.22(s,2H), 5.50(m,1H) , 7.01(m,1H) , 7.33(bmJ5H), 7.76(d,1H), 9.25(m,1H), 9.41(m,1H); 13 C NMR (CDCl 3 ) ⁇ 17.7, 24.5, 31.1, 40.3, 40.6, 44.1, 48.6, 54.1, 66.7, 66.9, 68.9, 127.9, 128.0, 128.1, 128.
- Hydroxamate (5d) was prepared from Compound (5c) in 78% yield as a mixture of diastereomers.
- Compound (6b) was prepared from Compounds (6a) and (1d) in 69% yield using the method previously described to prepare Compound (A 2 ).
- TLC Rf 0.21 and 0.29 (chloroform-isopropanol 9: 1);
- 1 H NMR (d 6 -DMSO; mixture of diastereomers) ⁇ 0.81(m,3H), 0.88(m,3H), 1.17 & 1.23(d,3H), 1.40(bm,8H), 2.46(m,3H), 2.78(m,1H), 2.98(m,2H), 3.54 & 3.56(s, 3H), 4.08(m,1H), 4.16(m,1H), 5.00(s,2H), 7.04(m,1H), 7.23(t,1H), 7.34(m,6H), 7.58 & 7.68(d,1H), 8.10 & 8.42(d,1H).
- Compound (7c) was prepared from Compound (7b) in 48% yield with the method used to prepare Compound (6c). A single diastereomer of Compound (7c) was isolated by
- the dicyclohexylurea by-product was removed by filtration, and the filtrate was transferred to a flask containing 1.5 ml (0.022 mol) of concentrated NH4OH. After the mixture had stirred at room temperature for 1 hour, the solvent was removed in vacuo to give a residue. The residue was dissolved in ethyl acetate (350 ml) and washed with water (100 ml), 1M HCL (100 ml), water (100 ml), saturated sodium bicarbonate solution (100 ml) and finally with brine (100 ml). After drying over anhydrous magnesium sulfate, the solution was filtered and concentrated in vacuo to produce a solid.
- the diastereomers (8) and (9) can be made from L-3-(2'-naphthyl)alanine amide hydrochloride (8b) and (1d), using the sequence of reactions used to prepare Compound (1) from Compounds (1j) and (1d).
- N-BOC-L-3-(2'-naphthyl)alanyl-L-(O-benzyl)serine amide (10a) was prepared from N-BOC-L-3-(2'-naphthyl)alanine and L-(O-benzyl)serine amide in 80% yield with the method used to prepare (7a).
- Compound (10d) was prepared from Compound (10c) in 74% yield with the method used to prepare Compound (lm). TLC: Rf 0.12 (chloroform-isopropanol 9:1).
- Compound (10) was prepared from Compound (10d) in 84% yield with the method used to prepare Compound (1n). HPLC retention times: 25.2 and 27.1 minutes (method A).
- Compound (11b) was prepared from Compounds (11a) and (1d), in 86% yield using the method previously described to prepare Compound (A 2 ).
- the following example demonstrates the selective in vitro inhibition of T-cell TNF- ⁇ secretion, as compared to TNF-ß and IFN- ⁇ secretion, by Compound 1.
- Human peripheral blood T-cells were purified from peripheral blood mononuclear cells by rosetting with 2-aminoethylisothiouronium bromide hydrobromide-treated sheep erythrocytes. After hypotonic lysis of sheep erythrocytes, monocytes were depleted by plastic adherence for one hour at 37 °C.
- the peripheral blood T-cells were stimulated with anti-CD3 antibody (OKT3) which was immobilized on the culture wells at 10 ⁇ g/ml in PBS plus 10 ng/ml of the phorbol ester, PMA.
- Culture medium comprised RPMI 1640 medium containing 10% fetal bovine serum, 50 U/ml penicillin, and 50 ⁇ g/ml streptomycin. The stimulation was performed in the presence or absence of the inhibitor Compound 1 (200 ⁇ M), and TNF- ⁇ in the medium was assayed by ELISA. Results are shown in Table I.
- Compound 1 inhibited TNF- ⁇ release by 72% and 63%, respectively, while there was no inhibitory effect on the release of TNF- ⁇ or interferon- ⁇ .
- Compound 1 clearly demonstrates selective inhibition of TNF- ⁇ secretion and has no effect on either
- TNF- ⁇ or interferon- ⁇ secretion are TNF- ⁇ or interferon- ⁇ secretion.
- T-cells which have been stimulated by PMA and ionomycin.
- the alloreactive human T-cell clone, PL-1 does not express cell surface TNF- ⁇ in the absence of stimulation.
- cell surface TNF- ⁇ as well as the ligands for CD40 and 41 BB, are rapidly induced on the cell surface.
- Detection of cell surface TNF- ⁇ was performed by staining with an Fc fusion protein consisting of an Fc portion of a human IgG1 molecule (1gGFc) coupled with an extracellular domain of TNF receptor (p80).
- Detection of cell surface ligands for 41BB and CD40 was performed by staining with analogous Fc fusion proteins consisting of IgGFc and extracellular domains of 41BB and CD40, respectively.
- a fusion molecule consisting of IgGFc and the extracellular portion of the IL-4 receptor (IL-4R:Fc) was utilized as a negative control for staining, since PL-1 cells do not express cell-surface IL-4 in response to PMA stimulation.
- IL-4R:Fc IL-4 receptor
- mice Female Balb/c mice (18-20g) were injected i.v. with 400 ⁇ g of LPS. Simultaneously, the mice were injected subcutaneously with 500 ⁇ g of Compound A or Compound 1 in 0.5 ml of saline containing 0.02% DMSO. Control mice received LPS intravenously and saline/DMSO subcutaneously. Two hours following the LPS injection, serum was obtained and pooled from two mice in each treatment group. TNF- ⁇ levels were determined by ELISA and are shown in the following Table III.
- Compound 1 inhibits the secretion of TNF- ⁇ at least by 80%, and essentially by 100%, as the TNF- ⁇ levels were undetectable. Comparatively, Compound A reduced serum TNF- ⁇ levels by approximately 60% as compared to the saline/DMSO control.
- mice were injected i.v. with
- mice were injected subcutaneously with 500 ⁇ g
- Compound 1 As compared to mice that received LPS + saline, Compound 1 reduced serum TNF- ⁇ levels by 84%. Overall, Compound 1 reduced serum TNF- ⁇ levels by 85.4 ⁇ 2.98% as compared to TNF- ⁇ levels in mice that received LPS only. From Tables III and IV, Compound 1 effectively reduces serum TNF- ⁇ levels by at least 80% when administered at 25 mg/kg in a murine model of LPS-induced sepsis syndrome. 250 ⁇ g Compound A versus Compound 1 versus control
- mice Female Balb/c mice (18-20g) were injected i.v. with 450 ⁇ g of LPS. Simultaneously, the mice were injected subcutaneously with 250 ⁇ g of Compound A or Compound 1 in 0.25 ml of saline containing 0.02% DMSO. Control mice received LPS intravenously and saline/DMSO subcutaneously. Two hours following the LPS injection, serum was obtained from three mice in each treatment group. TNF- ⁇ levels were determined by ELISA. The results are expressed as the mean optical density (OD) obtained in the ELISA from each treatment group, and are shown in Table V. The background OD of the control sample was 0.162 ⁇ 0.003. TABLE V
- Table V illustrates the effect of Compound 1 and Compound A on inhibiting serum
- TNF- ⁇ release in LPS -stimulated mice Compound 1 reduced serum TNF- ⁇ levels to those of the control, thereby indicating a complete inhibition of TNF- ⁇ secretion at 250 ⁇ g/ml.
- Each of Compound 1 and Compound A was diluted to 50 ⁇ M in normal mouse serum and incubated at 37°C. At various times, aliquots were withdrawn, diluted 100-fold into ice-cold PBS, and tested for inhibitory efficacy against purified TACE. After 40 minutes, Compound A showed a decrease in inhibitory effect corresponding to a 3-4 fold loss in concentration of the compound, and Compound 1 showed no decrease in inhibitory effect.
Abstract
Description































Claims


Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| NZ271893A NZ271893A (en) | 1993-08-23 | 1994-08-19 | Inhibitors of tnf-alpha secretion |
| CA002170158A CA2170158A1 (en) | 1993-08-23 | 1994-08-19 | Inhibitors of tnf-alpha secretion |
| EP94925940A EP0715619A4 (en) | 1993-08-23 | 1994-08-19 | Inhibitors of tnf-alpha secretion |
| JP7507668A JPH09503201A (en) | 1993-08-23 | 1994-08-19 | Inhibitor of TNF-alpha secretion |
| AU75694/94A AU687436B2 (en) | 1993-08-23 | 1994-08-19 | Inhibitors of TNF-alpha secretion |
| FI960803A FI960803A (en) | 1993-08-23 | 1996-02-22 | TNF-alpha secretion inhibitors |
| NO960723A NO960723L (en) | 1993-08-23 | 1996-02-23 | Inhibitors of TNF-alpha secretion |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11060193A | 1993-08-23 | 1993-08-23 | |
| US18301994A | 1994-01-18 | 1994-01-18 | |
| US08/110,601 | 1994-01-18 | ||
| US08/183,019 | 1994-01-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1995006031A1 true WO1995006031A1 (en) | 1995-03-02 |
Family
ID=26808203
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1994/009343 WO1995006031A1 (en) | 1993-08-23 | 1994-08-19 | Inhibitors of tnf-alpha secretion |
Country Status (8)
| Country | Link |
|---|---|
| EP (1) | EP0715619A4 (en) |
| JP (1) | JPH09503201A (en) |
| AU (2) | AU687436B2 (en) |
| CA (1) | CA2170158A1 (en) |
| FI (1) | FI960803A (en) |
| NO (1) | NO960723L (en) |
| NZ (1) | NZ271893A (en) |
| WO (1) | WO1995006031A1 (en) |
Cited By (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996035714A1 (en) * | 1995-05-10 | 1996-11-14 | Chiroscience Limited | Peptide compounds which inhibit metalloproteinase and tnf liberation and their therapeutic uses |
| WO1996035712A1 (en) * | 1995-05-10 | 1996-11-14 | Chiroscience Limited | Peptidyl compounds which inhibit metalloproteinase and tnf liberation and their therapeutic use |
| WO1996035711A1 (en) * | 1995-05-10 | 1996-11-14 | Chiroscience Limited | Peptide compounds which inhibit metalloproteinase and tnf liberation, and their therapeutic use |
| WO1997012902A1 (en) * | 1995-10-05 | 1997-04-10 | Darwin Discovery Limited | Thio-substituted peptides as inhibitors for metalloproteinases and tnf liberation |
| WO1997019053A1 (en) | 1995-11-23 | 1997-05-29 | British Biotech Pharmaceuticals Limited | Metalloproteinase inhibitors |
| US5665777A (en) * | 1995-11-14 | 1997-09-09 | Abbott Laboratories | Biphenyl hydroxamate inhibitors of matrix metalloproteinases |
| WO1997038007A1 (en) * | 1996-04-04 | 1997-10-16 | Darwin Discovery Limited | Peptidyl compounds having mmp and tnf- liberation inhibitory activity |
| WO1998030541A1 (en) * | 1997-01-07 | 1998-07-16 | Abbott Laboratories | C-terminal ketone hydroxamic acid inhibitors of matrix metalloproteinases and tnfa secretion |
| WO1998057657A1 (en) * | 1997-06-18 | 1998-12-23 | The Rockefeller University | Methods for identifying antibodies and peptides useful in the treatment of septic shock and experimental arthritis and uses thereof |
| US5883241A (en) * | 1995-09-05 | 1999-03-16 | Celltech Therapeutics Limited | DNA sequences coding for a human metalloproteinase and variants thereof |
| WO1999018942A1 (en) * | 1997-10-10 | 1999-04-22 | Imperial College Innovations Ltd. | Use of csaidtm compounds for the management of uterine contractions |
| US5952320A (en) * | 1997-01-07 | 1999-09-14 | Abbott Laboratories | Macrocyclic inhibitors of matrix metalloproteinases and TNFα secretion |
| US5985911A (en) * | 1997-01-07 | 1999-11-16 | Abbott Laboratories | C-terminal ketone inhibitors of matrix metalloproteinases and TNFα secretion |
| US5990293A (en) * | 1996-09-05 | 1999-11-23 | Celltech Therapeutics Limited | Human metalloproteinase, variants thereof and DNA sequence coding therefor |
| WO1999059568A1 (en) * | 1998-05-16 | 1999-11-25 | British Biotech Pharmaceuticals Limited | Hydroxamic acid derivatives as antibacterials |
| US5994312A (en) * | 1994-10-05 | 1999-11-30 | Darwin Discovery, Ltd. | Peptidyl compounds |
| WO2000018358A2 (en) * | 1998-09-30 | 2000-04-06 | The Procter & Gamble Company | Method of treating hair loss using ketoamides |
| WO2000018361A2 (en) * | 1998-09-30 | 2000-04-06 | The Procter & Gamble Company | Method of treating hair loss using sulfonamides |
| WO2000041473A2 (en) * | 1999-01-13 | 2000-07-20 | Jomaa Pharmaka Gmbh | Use of 3-isoxazolidinones and hydroxylamine acids for the treatment of infections |
| US6169075B1 (en) | 1996-09-10 | 2001-01-02 | British Biotech Pharmaceuticals Limited | Cytostatic agents |
| US6172064B1 (en) | 1998-08-26 | 2001-01-09 | Glaxo Wellcome Inc. | Formamides as therapeutic agents |
| US6191150B1 (en) | 1998-08-26 | 2001-02-20 | Glaxo Wellcome Inc. | Formamide compounds as therapeutic agents |
| WO2001022952A2 (en) * | 1999-09-25 | 2001-04-05 | Smithkline Beecham P.L.C. | Use of tace inhibitors |
| US6288261B1 (en) | 1998-12-18 | 2001-09-11 | Abbott Laboratories | Inhibitors of matrix metalloproteinases |
| US6300341B1 (en) | 1998-09-30 | 2001-10-09 | The Procter & Gamble Co. | 2-substituted heterocyclic sulfonamides |
| US6307049B1 (en) | 1998-09-30 | 2001-10-23 | The Procter & Gamble Co. | Heterocyclic 2-substituted ketoamides |
| US6329550B1 (en) | 1998-12-31 | 2001-12-11 | Aventis Pharmaceuticals Inc. | Amidomalonamides useful as inhibitors of MMP of matrix metalloproteinase |
| US6329400B1 (en) | 1998-08-26 | 2001-12-11 | Glaxo Wellcome Inc. | Formamide compounds as therapeutic agents |
| WO2002006227A1 (en) * | 2000-07-18 | 2002-01-24 | Chugai Seiyaku Kabushiki Kaisha | Matrix metalloprotease inhibitors |
| WO2002028829A2 (en) * | 2000-09-25 | 2002-04-11 | Questcor Pharmaceuticals, Inc. | Peptide deformylase inhibitors |
| US6406877B2 (en) | 1995-06-08 | 2002-06-18 | Immunex Corporation | TNF-α converting enzyme |
| US6462023B1 (en) | 1996-09-10 | 2002-10-08 | British Biotech Pharmaceuticals, Ltd. | Cytostatic agents |
| US6838466B2 (en) | 2001-12-20 | 2005-01-04 | Schering Corporation | Compounds for the treatment of inflammatory disorders |
| US7915225B2 (en) | 1999-04-19 | 2011-03-29 | Immunex Corporation | Soluble tumor necrosis factor receptor treatment of medical disorders |
| WO2020070239A1 (en) | 2018-10-04 | 2020-04-09 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Egfr inhibitors for treating keratodermas |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1986003747A1 (en) * | 1984-12-19 | 1986-07-03 | Ciba-Geigy Ag | New carbonic acid esters |
| EP0274453A2 (en) * | 1987-01-06 | 1988-07-13 | Société anonyme dite: LABORATOIRE ROGER BELLON | Collagenase inhibitor derivatives, their preparation and pharmaceutical compositions containing them |
| EP0498665A1 (en) * | 1991-02-07 | 1992-08-12 | British Biotech Pharmaceuticals Limited | Hydroxamic acids derivatives, process for their preparation and use thereof |
| WO1992022523A2 (en) * | 1991-06-14 | 1992-12-23 | Research Corporation Technologies, Inc. | Peptide derivatives of collagenase inhibitor |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9215665D0 (en) * | 1992-07-23 | 1992-09-09 | British Bio Technology | Compounds |
| GB9323165D0 (en) * | 1993-11-10 | 1994-01-05 | Chiros Ltd | Compounds |
| GB9411088D0 (en) * | 1994-06-03 | 1994-07-27 | Hoffmann La Roche | Hydroxylamine derivatives |
-
1994
- 1994-08-19 EP EP94925940A patent/EP0715619A4/en not_active Ceased
- 1994-08-19 CA CA002170158A patent/CA2170158A1/en not_active Abandoned
- 1994-08-19 AU AU75694/94A patent/AU687436B2/en not_active Ceased
- 1994-08-19 WO PCT/US1994/009343 patent/WO1995006031A1/en not_active Application Discontinuation
- 1994-08-19 JP JP7507668A patent/JPH09503201A/en active Pending
- 1994-08-19 NZ NZ271893A patent/NZ271893A/en unknown
-
1996
- 1996-02-22 FI FI960803A patent/FI960803A/en unknown
- 1996-02-23 NO NO960723A patent/NO960723L/en unknown
-
1998
- 1998-01-06 AU AU50302/98A patent/AU5030298A/en not_active Abandoned
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1986003747A1 (en) * | 1984-12-19 | 1986-07-03 | Ciba-Geigy Ag | New carbonic acid esters |
| EP0274453A2 (en) * | 1987-01-06 | 1988-07-13 | Société anonyme dite: LABORATOIRE ROGER BELLON | Collagenase inhibitor derivatives, their preparation and pharmaceutical compositions containing them |
| US4918105A (en) * | 1987-01-06 | 1990-04-17 | Sa Laboratoire Roger Bellon | Novel compounds with collagenase-inhibiting activity, a process for their preparation and pharmaceutical compositions in which these compounds are present |
| EP0498665A1 (en) * | 1991-02-07 | 1992-08-12 | British Biotech Pharmaceuticals Limited | Hydroxamic acids derivatives, process for their preparation and use thereof |
| WO1992022523A2 (en) * | 1991-06-14 | 1992-12-23 | Research Corporation Technologies, Inc. | Peptide derivatives of collagenase inhibitor |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP0715619A4 * |
Cited By (62)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6180611B1 (en) | 1994-10-05 | 2001-01-30 | Darwin Discovery, Ltd. | Peptidyl compounds |
| US5994312A (en) * | 1994-10-05 | 1999-11-30 | Darwin Discovery, Ltd. | Peptidyl compounds |
| US5994293A (en) * | 1995-05-10 | 1999-11-30 | Darwin Discovery Ltd. | Peptidyl compounds and their therapeutic use |
| US6110896A (en) * | 1995-05-10 | 2000-08-29 | Darwin Discovery, Ltd. | Peptidyl compounds and their therapeutic use |
| WO1996035711A1 (en) * | 1995-05-10 | 1996-11-14 | Chiroscience Limited | Peptide compounds which inhibit metalloproteinase and tnf liberation, and their therapeutic use |
| WO1996035712A1 (en) * | 1995-05-10 | 1996-11-14 | Chiroscience Limited | Peptidyl compounds which inhibit metalloproteinase and tnf liberation and their therapeutic use |
| WO1996035714A1 (en) * | 1995-05-10 | 1996-11-14 | Chiroscience Limited | Peptide compounds which inhibit metalloproteinase and tnf liberation and their therapeutic uses |
| AU710502B2 (en) * | 1995-05-10 | 1999-09-23 | Darwin Discovery Limited | Peptidyl compounds which inhibit metalloproteinase and TNF liberation and their therapeutic use |
| US5981491A (en) * | 1995-05-10 | 1999-11-09 | Darwin Discovery Limited | Peptidyl compounds and their therapeutic use |
| US6406901B1 (en) | 1995-06-08 | 2002-06-18 | Immunex Corporation | TNF-a converting enzyme |
| US7695948B2 (en) | 1995-06-08 | 2010-04-13 | Immunex Corporation | Antibodies that bind TNF-α converting enzyme |
| US7199224B2 (en) | 1995-06-08 | 2007-04-03 | Immunex Corporation | Antibodies that bind TNF-α converting enzyme |
| US6406877B2 (en) | 1995-06-08 | 2002-06-18 | Immunex Corporation | TNF-α converting enzyme |
| US6555354B2 (en) | 1995-06-08 | 2003-04-29 | Immunex Corporation | TNF-α converting enzyme |
| US5883241A (en) * | 1995-09-05 | 1999-03-16 | Celltech Therapeutics Limited | DNA sequences coding for a human metalloproteinase and variants thereof |
| US5981490A (en) * | 1995-10-05 | 1999-11-09 | Darwin Discovery, Ltd. | Peptidyl compounds |
| WO1997012902A1 (en) * | 1995-10-05 | 1997-04-10 | Darwin Discovery Limited | Thio-substituted peptides as inhibitors for metalloproteinases and tnf liberation |
| US5665777A (en) * | 1995-11-14 | 1997-09-09 | Abbott Laboratories | Biphenyl hydroxamate inhibitors of matrix metalloproteinases |
| WO1997019053A1 (en) | 1995-11-23 | 1997-05-29 | British Biotech Pharmaceuticals Limited | Metalloproteinase inhibitors |
| AU720239B2 (en) * | 1996-04-04 | 2000-05-25 | Darwin Discovery Limited | Peptidyl compounds having MMP and TNF- liberation inhibitory activity |
| US5872146A (en) * | 1996-04-04 | 1999-02-16 | Chiroscience Limited | Mercapto alkyl peptidyl compounds having MMP and TNF inhibitory activity |
| WO1997038007A1 (en) * | 1996-04-04 | 1997-10-16 | Darwin Discovery Limited | Peptidyl compounds having mmp and tnf- liberation inhibitory activity |
| US5990293A (en) * | 1996-09-05 | 1999-11-23 | Celltech Therapeutics Limited | Human metalloproteinase, variants thereof and DNA sequence coding therefor |
| US6462023B1 (en) | 1996-09-10 | 2002-10-08 | British Biotech Pharmaceuticals, Ltd. | Cytostatic agents |
| US6169075B1 (en) | 1996-09-10 | 2001-01-02 | British Biotech Pharmaceuticals Limited | Cytostatic agents |
| WO1998030541A1 (en) * | 1997-01-07 | 1998-07-16 | Abbott Laboratories | C-terminal ketone hydroxamic acid inhibitors of matrix metalloproteinases and tnfa secretion |
| US5952320A (en) * | 1997-01-07 | 1999-09-14 | Abbott Laboratories | Macrocyclic inhibitors of matrix metalloproteinases and TNFα secretion |
| US5985911A (en) * | 1997-01-07 | 1999-11-16 | Abbott Laboratories | C-terminal ketone inhibitors of matrix metalloproteinases and TNFα secretion |
| AU730308B2 (en) * | 1997-06-18 | 2001-03-01 | Rockefeller University, The | Methods for identifying antibodies and peptides useful in the treatment of septic shock and experimental arthritis and uses thereof |
| WO1998057657A1 (en) * | 1997-06-18 | 1998-12-23 | The Rockefeller University | Methods for identifying antibodies and peptides useful in the treatment of septic shock and experimental arthritis and uses thereof |
| WO1999018942A1 (en) * | 1997-10-10 | 1999-04-22 | Imperial College Innovations Ltd. | Use of csaidtm compounds for the management of uterine contractions |
| US6992190B2 (en) | 1998-05-16 | 2006-01-31 | British Biotech Pharmaceuticals Ltd. | Hydroxamic acid derivatives as antibacterials |
| WO1999059568A1 (en) * | 1998-05-16 | 1999-11-25 | British Biotech Pharmaceuticals Limited | Hydroxamic acid derivatives as antibacterials |
| GB2353708A (en) * | 1998-05-16 | 2001-03-07 | British Biotech Pharm | Hydroxamic acid derivatives as antibacterials |
| US7323563B2 (en) | 1998-05-16 | 2008-01-29 | British Biotech Pharmaceuticals Ltd. | Hydroxamic acid derivatives as antibacterials |
| EP1754477A3 (en) * | 1998-05-16 | 2007-03-14 | Vernalis (Oxford) Limited | Hydroxamic acid derivatives as antibacterials |
| EP1754477A2 (en) * | 1998-05-16 | 2007-02-21 | Vernalis (Oxford) Limited | Hydroxamic acid derivatives as antibacterials |
| US6441042B1 (en) | 1998-05-16 | 2002-08-27 | British Biotech Pharmaceuticals Limited | Hydroxamic acid derivatives as antibacterials |
| US6172064B1 (en) | 1998-08-26 | 2001-01-09 | Glaxo Wellcome Inc. | Formamides as therapeutic agents |
| US6329400B1 (en) | 1998-08-26 | 2001-12-11 | Glaxo Wellcome Inc. | Formamide compounds as therapeutic agents |
| US6191150B1 (en) | 1998-08-26 | 2001-02-20 | Glaxo Wellcome Inc. | Formamide compounds as therapeutic agents |
| WO2000018361A2 (en) * | 1998-09-30 | 2000-04-06 | The Procter & Gamble Company | Method of treating hair loss using sulfonamides |
| WO2000018358A3 (en) * | 1998-09-30 | 2000-07-27 | Procter & Gamble | Method of treating hair loss using ketoamides |
| US6300341B1 (en) | 1998-09-30 | 2001-10-09 | The Procter & Gamble Co. | 2-substituted heterocyclic sulfonamides |
| WO2000018361A3 (en) * | 1998-09-30 | 2000-05-25 | Procter & Gamble | Method of treating hair loss using sulfonamides |
| WO2000018358A2 (en) * | 1998-09-30 | 2000-04-06 | The Procter & Gamble Company | Method of treating hair loss using ketoamides |
| US6307049B1 (en) | 1998-09-30 | 2001-10-23 | The Procter & Gamble Co. | Heterocyclic 2-substituted ketoamides |
| US6288261B1 (en) | 1998-12-18 | 2001-09-11 | Abbott Laboratories | Inhibitors of matrix metalloproteinases |
| US6329550B1 (en) | 1998-12-31 | 2001-12-11 | Aventis Pharmaceuticals Inc. | Amidomalonamides useful as inhibitors of MMP of matrix metalloproteinase |
| WO2000041473A3 (en) * | 1999-01-13 | 2001-11-29 | Jomaa Pharmaka Gmbh | Use of 3-isoxazolidinones and hydroxylamine acids for the treatment of infections |
| WO2000041473A2 (en) * | 1999-01-13 | 2000-07-20 | Jomaa Pharmaka Gmbh | Use of 3-isoxazolidinones and hydroxylamine acids for the treatment of infections |
| US7915225B2 (en) | 1999-04-19 | 2011-03-29 | Immunex Corporation | Soluble tumor necrosis factor receptor treatment of medical disorders |
| US8119605B2 (en) | 1999-04-19 | 2012-02-21 | Immunex Corporation | Soluble tumor necrosis factor receptor treatment of medical disorders |
| WO2001022952A3 (en) * | 1999-09-25 | 2003-02-20 | Smithkline Beecham Plc | Use of tace inhibitors |
| WO2001022952A2 (en) * | 1999-09-25 | 2001-04-05 | Smithkline Beecham P.L.C. | Use of tace inhibitors |
| WO2002006227A1 (en) * | 2000-07-18 | 2002-01-24 | Chugai Seiyaku Kabushiki Kaisha | Matrix metalloprotease inhibitors |
| WO2002028829A2 (en) * | 2000-09-25 | 2002-04-11 | Questcor Pharmaceuticals, Inc. | Peptide deformylase inhibitors |
| WO2002028829A3 (en) * | 2000-09-25 | 2003-12-24 | Questcor Pharmaceuticals Inc | Peptide deformylase inhibitors |
| US6838466B2 (en) | 2001-12-20 | 2005-01-04 | Schering Corporation | Compounds for the treatment of inflammatory disorders |
| US7034057B2 (en) | 2001-12-20 | 2006-04-25 | Schering Corporation | Compounds for the treatment of inflammatory disorders |
| US7598242B2 (en) | 2001-12-20 | 2009-10-06 | Schering Corporation | Compounds for the treatment of inflammatory disorders |
| WO2020070239A1 (en) | 2018-10-04 | 2020-04-09 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Egfr inhibitors for treating keratodermas |
Also Published As
| Publication number | Publication date |
|---|---|
| AU687436B2 (en) | 1998-02-26 |
| CA2170158A1 (en) | 1995-03-02 |
| NO960723D0 (en) | 1996-02-23 |
| AU5030298A (en) | 1998-03-05 |
| AU7569494A (en) | 1995-03-21 |
| NO960723L (en) | 1996-02-23 |
| EP0715619A1 (en) | 1996-06-12 |
| FI960803A (en) | 1996-04-22 |
| FI960803A0 (en) | 1996-02-22 |
| NZ271893A (en) | 1997-11-24 |
| EP0715619A4 (en) | 1997-03-19 |
| JPH09503201A (en) | 1997-03-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5594106A (en) | Inhibitors of TNF-α secretion | |
| AU687436B2 (en) | Inhibitors of TNF-alpha secretion | |
| JP3372260B2 (en) | Antiviral peptide derivatives | |
| US4691007A (en) | Proline derivatives | |
| KR890003603B1 (en) | Process for preparing renin inhibitors containing 5-amino-2,5-disubstituted-4-hydroxypentanoic acid residues | |
| JP3205558B2 (en) | New peptide derivatives | |
| EP0468339B1 (en) | Alpha-keto-amide derivatives having protease inhibiting activity | |
| US6777443B2 (en) | Dipeptide derivatives | |
| EP0618223A2 (en) | Peptides inhibiting interleukin 1-bêta release useful as antiinflammatory agents | |
| US6235717B1 (en) | Pharmaceutical compounds | |
| US6995177B1 (en) | HCV NS3 protease inhibitors | |
| JPH0832720B2 (en) | Tuftsin analogs, process for their preparation and pharmaceutical compositions | |
| CA2217857A1 (en) | Peptide compounds which inhibit metalloproteinase and tnf liberation and their therapeutic uses | |
| WO1988003022A1 (en) | Relatively low molecular weight polypeptides as renin inhibitors | |
| EP0218688A1 (en) | Dihalo-statine substituted renin inhibitors. | |
| JPS60136595A (en) | Substituted ethylenediamine derivative, manufacture and medicine | |
| US5218089A (en) | Retro-inverso analogues of thymopentin and the method for their synthesis | |
| JPH10182613A (en) | Tetraphydroisoquinoline derivative | |
| JPH0665175A (en) | Radiotherapeutic or irradiation therapeutic agent and preparation of pharmaceutical containing same new derivative of beta-amino acid | |
| CN85107088A (en) | The preparation of the heterocyclic immunostimulants that peptide replaces | |
| JPH0647599B2 (en) | Heptanoyl-Glu-Asp-Ala-amino acid immunostimulant | |
| AU600549B2 (en) | Novel compounds | |
| AU740954B2 (en) | (3R)-3-amino-4-carboxybutyraldehyde derivatives inhibiting the release of interleukin-1/beta | |
| US6180759B1 (en) | Process for the preparation of azacycloalkylakanoyl pseudotetrapeptides | |
| US5091510A (en) | Retro-inverso analogues of thymopentin, and their use in the preparation of pharmaceutical compositions |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AM AT AU BB BG BR BY CA CH CN CZ DE DK ES FI GB GE HU JP KE KG KP KR KZ LK LT LU LV MD MG MN MW NL NO NZ PL PT RO RU SD SE SI SK TJ TT UA UZ VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): KE MW SD AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2170158 Country of ref document: CA Ref document number: 271893 Country of ref document: NZ Ref document number: 960803 Country of ref document: FI |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1994925940 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1994925940 Country of ref document: EP |
|
| WWR | Wipo information: refused in national office |
Ref document number: 1994925940 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1994925940 Country of ref document: EP |