WO1998051662A2 - Compounds and methods for the inhibition of the expression of vcam-1 - Google Patents
Compounds and methods for the inhibition of the expression of vcam-1 Download PDFInfo
- Publication number
- WO1998051662A2 WO1998051662A2 PCT/US1998/009781 US9809781W WO9851662A2 WO 1998051662 A2 WO1998051662 A2 WO 1998051662A2 US 9809781 W US9809781 W US 9809781W WO 9851662 A2 WO9851662 A2 WO 9851662A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- butyl
- substituted
- alkyl
- spacer
- aryl
- Prior art date
Links
- 0 ***c(c(*)c1*)c(*)c(*)c1O Chemical compound ***c(c(*)c1*)c(*)c(*)c1O 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/215—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids
- A61K31/22—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids of acyclic acids, e.g. pravastatin
- A61K31/222—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids of acyclic acids, e.g. pravastatin with compounds having aromatic groups, e.g. dipivefrine, ibopamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/215—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids
- A61K31/22—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids of acyclic acids, e.g. pravastatin
- A61K31/225—Polycarboxylic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/02—Muscle relaxants, e.g. for tetanus or cramps
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/02—Non-specific cardiovascular stimulants, e.g. drugs for syncope, antihypotensives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/14—Vasoprotectives; Antihaemorrhoidals; Drugs for varicose therapy; Capillary stabilisers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/10—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C323/18—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and singly-bound oxygen atoms bound to the same carbon skeleton having the sulfur atom of at least one of the thio groups bound to a carbon atom of a six-membered aromatic ring of the carbon skeleton
- C07C323/20—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and singly-bound oxygen atoms bound to the same carbon skeleton having the sulfur atom of at least one of the thio groups bound to a carbon atom of a six-membered aromatic ring of the carbon skeleton with singly-bound oxygen atoms bound to carbon atoms of the same non-condensed six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/10—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C323/18—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and singly-bound oxygen atoms bound to the same carbon skeleton having the sulfur atom of at least one of the thio groups bound to a carbon atom of a six-membered aromatic ring of the carbon skeleton
- C07C323/21—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and singly-bound oxygen atoms bound to the same carbon skeleton having the sulfur atom of at least one of the thio groups bound to a carbon atom of a six-membered aromatic ring of the carbon skeleton with the sulfur atom of the thio group bound to a carbon atom of a six-membered aromatic ring being part of a condensed ring system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/23—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C323/24—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/25—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/50—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton
- C07C323/51—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/56—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/64—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and sulfur atoms, not being part of thio groups, bound to the same carbon skeleton
- C07C323/66—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and sulfur atoms, not being part of thio groups, bound to the same carbon skeleton containing sulfur atoms of sulfo, esterified sulfo or halosulfonyl groups, bound to the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/64—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and sulfur atoms, not being part of thio groups, bound to the same carbon skeleton
- C07C323/67—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and sulfur atoms, not being part of thio groups, bound to the same carbon skeleton containing sulfur atoms of sulfonamide groups, bound to the carbon skeleton
Definitions
- This invention is in the area of methods and compositions for the inhibition of the expression of NCAM-1 and, in particular, for the treatment of diseases mediated by VCAM- 1, including cardiovascular and inflammatory diseases.
- Coronary heart disease remains the leading cause of death in the industrialized countries.
- the primary cause of CHD is atherosclerosis, a disease characterized by the deposition of lipids in the arterial vessel wall, resulting in a narrowing of the vessel passages and ultimately hardening the vascular system.
- Atherosclerosis as manifested in its major clinical complication, ischemic heart disease, continues to be a major cause of death in industrialized countries. It is now well accepted that atherosclerosis can begin with local injury to the arterial endothelium followed by proliferation of arterial smooth muscle cells from the medial layer to the intimal layer along the deposition of lipid and accumulation of foam cells in the lesion. As the atherosclerotic plaque develops it progressively occludes more and more of the affected blood vessel and can eventually lead to ischaemia or infarction. Therefore, it is desirable to provide methods of inhibiting the progression of atherosclerosis in patients in need thereof.
- Cardiovascular disease has been linked to several causative factors, which include hypercholesterolemia, hyperlipidemia, and the expression of VCAM-l in vascular endothelial cells.
- Adhesion of leukocytes to the endothelium represents a fundamental, early event in a wide variety of inflammatory conditions, including atherosclerosis, autoimmune disorders and bacterial and viral infections.
- Leukocyte recruitment to the endothelium is started when inducible adhesion molecule receptors on the surface of endothelial cells interact with counterreceptors on immune cells.
- Vascular endothelial cells determine which type of leukocytes (monocytes, lymphocytes, or neutrophils) are recruited, by selectively expressing specific adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM-1), intracellular adhesion molecule-1 (ICAM-1), and E-selectin.
- VCAM-1 vascular cell adhesion molecule-1
- IAM-1 intracellular adhesion molecule-1
- E-selectin E-selectin.
- VCAM-1 In the earliest stage of the atherosclerotic lesion, there is a localized endothelial expression of VCAM-1 and selective recruitment of mononuclear leukocytes that express the integrin counterreceptor VLA-4. Because of the selective expression of VLA-4 on monocytes and lymphocytes, but not neutrophils, VCAM-1 is important in mediating the selective adhesion of mononuclear leukocytes.
- VCAM-1 is a mediator in chronic inflammatory disorders such as asthma, rheumatoid arthritis and autoimmune diabetes.
- chronic inflammatory disorders such as asthma, rheumatoid arthritis and autoimmune diabetes.
- VCAM-1 and ICAM-1 are increased in asthmatics.
- blocking the integrin receptors for VCAM-1 and ICAM-1 suppressed both early and late phase responses in an ovalbumin-sensitized rat model of allergic airway responses. Rabb, 11.
- VCAM-1 is expressed by cells both as a membrane bound form and as a soluble form.
- VCAM-1 The soluble form of VCAM-1 has been shown to induce chemotaxis of vascular endothelial cells in vitro and stimulate an angiogenic response in rat cornea. Koch, A.F. et al., Nature 376, 517-519 (1995). Inhibitors of the expression of soluble VCAM-1 have potential therapeutic value in treating diseases with a strong angiogenic component, including tumor growth and metastasis. Folkman, J., and Shing, Y., Biol. Chem. 10931-10934 (1992).
- VCAM-1 is expressed in cultured human vascular endothelial cells after activation by lipopolysaccharide (LPS) and cytokines such as interleukin-1 (IL-1) and tumor necrosis factor (TNF-oc). These factors are not selective for activation of cell adhesion molecule expression.
- LPS lipopolysaccharide
- cytokines such as interleukin-1 (IL-1) and tumor necrosis factor (TNF-oc).
- linoleic acid, linolenic acid, arachidonic acid, linoleyl hydroperoxide (13-HPODE) and arachidonic hydroperoxide (15-HPETE) induce cell-surface gene expression of VCAM-1 but not ICAM-1 or E-selectin.
- Saturated fatty acids such as stearic acid
- monounsaturated fatty acids such as oleic acid
- the induction of VCAM-1 by PUFAs and their fatty acid hydroperoxides is suppressed by dithiocarbamates, including pyrrolidine dithiocarbamate (PDTC).
- the induction is mediated by an oxidized signal molecule, and that the induction is prevented when the oxidation of the molecule is blocked (i.e., the oxidation does not occur), reversed (i.e., the signal molecule is reduced), or when the redox modified signal is otherwise prevented from interacting with its regulatory target.
- Cells that are chronically exposed to higher than normal levels of polyunsaturated fatty acids or their oxidized counterparts can initiate an immune response that is not normal and which is out of proportion to the threat presented, leading to a diseased state.
- the oversensitization of vascular endothelial cells to PUFAs and ox-PUFAs can accelerate the formation, for example, of atherosclerotic plaque.
- WO95/30415 that includes the removal, decrease in the concentration of, or prevention of the formation of oxidized polyunsaturated fatty acids including but not limited to oxidized linoleic (C 18 ⁇ 9,12 ), linolenic (C 18 ⁇ 6,9 ' 12 ), arachidonic (C 20 ⁇ 5 1 U4 ) and eicosatrienoic (C 20 ⁇ 8 ' 1 4 ) acids.
- Nonlimiting examples of noncardiovascular inflammatory diseases that are mediated by VCAM-1 include rheumatoid and osteoarthritis, asthma, dermatitis, and multiple sclerosis.
- Serum lipoproteins are the carriers for lipids in the circulation. Lipoproteins are classified according to their density: chylomicrons, very low-density lipoproteins (VLDL), low density lipoproteins (LDL) and high-density lipoproteins (HDL). Chylomicrons primarily participate in transporting dietary triglycerides and cholesterol from the intestine to adipose tissue and liver. VLDL deliver endogenously synthesized triglycerides from liver to adipose and other tissues. LDL transports cholesterol to peripheral tissues and regulate endogenous cholesterol levels in those tissues. HDL transports cholesterol from peripheral tissues to the liver.
- VLDL very low-density lipoproteins
- LDL low density lipoproteins
- HDL high-density lipoproteins
- Oxidized LDL is a complex structure consisting of at least several chemically distinct oxidized materials, each of which, alone or in combination, may modulate cytokine-activated adhesion molecule gene expression.
- R fatty acid hydroperoxides such as linoleyl hydroperoxide (13- HPODE) are produced from free fatty acids by lipoxygenases and are an important component of oxidized LDL.
- Supplementation of saturated or monounsaturated fatty acids to cultured endothelial cells reduces their susceptibility to oxidant injury, whereas supplementation with polyunsaturated fatty acids (PUFA) enhances susceptibility to oxidant injury.
- PUFA polyunsaturated fatty acids
- LDL endothelial, smooth muscle, and/or inflammatory cells then convert LDL to ox- LDL.
- monocytes avidly take up ox-LDL through a "scavenger" receptor whose expression, unlike the LDL receptor, is not inhibited as the content of intracellular lipid rises.
- monocytes continue to take up ox-LDL and become lipid-engorged macrophage-foam cells that form the fatty streak.
- LDL functions in a way that directly results in deposition of the LDL cholesterol in the blood- vessel wall and that HDL functions in way that results in the HDL picking up cholesterol from the vessel wall and transporting it to the liver where it is metabolized
- LDL cholesterol levels correlate well with the risk of coronary heart disease whereas the HDL cholesterol levels are inversely associated with coronary heart disease [Patton et al., Clin. Chem. 29, 1980 (1983)]. It is generally accepted by those skilled in the art that reduction of abnormally high LDL cholesterol levels is effective therapy not only in the treatment of hypercholesterolemia but also in the treatment of atherosclerosis.
- LDL lipid such as the unsaturated fatty acid portions of LDL cholesteryl esters and phospholipids
- peroxidation of LDL lipid facilitates the accumulation of cholesterol in monocyte/macrophages which eventually are transformed into foam cells and become deposited in the sub-endothelial space of the vessel wall.
- the accumulation of foam cells in the vessel wall is recognized as an early event in the formation of an atherosclerotic plaque.
- peroxidation of LDL lipid is an important prerequisite to the facilitated accumulation of cholesterol in the vessel wall and the subsequent formation of an atherosclerotic plaque.
- monocyte/macrophages take up and degrade native LDL at relatively low rates and without marked accumulation of cholesterol.
- Elevated cholesterol levels are associated with a number of disease states, including restenosis, angina, cerebral atherosclerosis, and xanthoma. It is desirable to provide a method for reducing plasma cholesterol in patients with, or at risk of developing, restenosis, angina, cerebral arteriosclerosis, xanthoma, and other disease states associated with elevated cholesterol levels.
- HMG CoA reductase inhibitors have been termed statins or vastatins.
- Statins are among the most effective agents currently on the market for hypercholesterolemia, and include pravastatin (Pravchol, Bristol Myers Squibb), atorvastatin (Warner Lambert/Pfizer), simvastatin (Zocor, Merck), lovastatin (Mevacor, Merck), and fluvastatin (Lescol).
- LDL low density lipoprotein
- Probucol is chemically related to the widely used food additives 2,[3]-tert-butyl-4- hydroxyanisole (BHA) and 2,6-di-tert-butyl-4-methyl phenol (BHT). Its full chemical name is 4,4'-(isopropylidenedithio) bis(2,6-di-tert-butylphenol).
- Probucol is used primarily to lower serum cholesterol levels in hypercholesterolemic patients. Probucol is commonly administered in the form of tablets available under the trademark LorelcoTM. Unfortunately, probucol is almost insoluble in water and therefore cannot be injected intravenously. In fact, probucol is difficult for cells to absorb in vitro because of its poor miscibility in buffers and media for cell culture. Solid probucol is poorly absorbed into the blood, and is excreted in substantially unchanged form. Further, the tablet form of probucol is absorbed at significantly different rates and in different amounts by different patients.
- 5,262,439 to Parthasarathy discloses analogs of probucol with increased water solubility in which one or both of the hydroxyl groups are replaced with ester groups that increase the water solubility of the compound.
- the derivative is selected from the group consisting of a mono- or di- probucol ester of succinic acid, glutaric acid, adipic acid, seberic acid, sebacic acid, azelaic acid, or maleic acid.
- the probucol derivative is a mono- or di- ester in which the ester contains an alkyl or alkenyl group that contains a functionality selected from the group consisting of a carboxylic acid group, amine group, salt of an amine group, amide groups, amide groups, and aldehyde groups.
- probucol derivatives are hypocholesterolemic and hypohpemic agents: Fr 2168137 (bis 4-hydroxyphenylthioalkane esters); Fr 2140771 (tetralinyl phenoxy alkanoic esters of probucol); Fr 2140769 (benzofiiryloxyalkanoic acid derivatives of probucol); Fr 2134810 (bis-(3 -alky 1-5 -t-alky 1-4- thiazole-5-carboxy)phenylthio)alkanes; FR 2133024 (bis-(4- nicotinoyloxyphenylthio)propanes; and Fr 2130975 (bis(4-(phenoxyalkanoyloxy)- phenylthio)alkanes).
- U.S. Patent No. 5,155,250 to Parker, et al. discloses that 2,6-dialkyl-4-silylphenols are antiatherosclerotic agents. The same compounds are disclosed as serum cholesterol lowering agents in PCT Publication No. WO 95/15760, published on June 15, 1995.
- U.S. Patent No. 5,608,095 to Parker, et al. discloses that alkylated-4-silyl-phenols inhibit the peroxidation of LDL, lower plasma cholesterol, and inhibit the expression of VCAM-1, and thus are useful in the treatment of atherosclerosis.
- a series of European patent applications and to Shionogi Seiyaku Kabushiki Kaisha disclose phenolic thioethers for use in treating arteriosclerosis.
- European Patent Application No. 348 203 discloses phenolic thioethers which inhibit the denaturation of LDL and the incorporation of LDL by macrophages. The compounds are useful as anti-arteriosclerosis agents. Hydroxamic acid derivatives of these compounds are disclosed in European Patent Application No. 405 788 and are useful for the treatment of arteriosclerosis, ulcer, inflammation and allergy.
- Carbamoyl and cyano derivatives of the phenolic thioethers are disclosed in U. S. Patent No. 4,954,514 to Kita, et al.
- U. S. Patent No. 4,752,616 to Hall, et al. disclose arylthioalkylphenylcarboxylic acids for the treatment of thrombotic disease.
- the compounds disclosed are useful as platelet aggregation inhibitors for the treatment of coronary or cerebral thromboses and the inhibition of bronchoconstriction, among others.
- WO97/15546 to Nippon Shinyaku Co. Ltd. discloses carboxylic acid derivatives for the treatment of arterial sclerosis, ischemic heart diseases, cerebral infarction and post PTCA restenosis.
- the Dow Chemical Company is the assignee of patents to hypolipidemic 2-(3,5-di- tert-butyl-4-hydroxyphenyl)thio carboxamides.
- U. S. Patent Nos. 4,029,812
- cardiovascular disease is currently the leading cause of death in the United States, and ninety percent of cardiovascular disease is presently diagnosed as atherosclerosis, there is a strong need to identify new methods and pharmaceutical agents for its treatment.
- a more general goal is to identify selective methods for suppressing the expression of redox sensitive genes or activating redox sensitive genes that are suppressed.
- a redox sensitive gene for example MCP-1, IL-6 and thrombin receptor.
- the present invention provides a compound, composition and method for inhibiting the expression of VCAM-1, and thus can be used in the treatment of a disease mediated by VCAM-1, which includes administering a compound of formula (I) or (II), or a pharmaceutically acceptable salt thereof, optionally in a pharmaceutically acceptable carrier.
- the compounds of formula (I) are
- X is O, S, SO, SO 2 , CH 2 , or NH;
- Spacer is a group selected from the group consisting of -(CH 2 ) n -, -(CH 2 ) n -CO-, -(CH 2 ) n - N-,
- n 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10;
- Y is substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted alkyl, substituted or unsubstituted alkoxy, substituted or unsubstituted alkoxyalkyl, substituted or unsubstituted alkylthio, substituted or unsubstituted alkylthioalkyl, substituted or unsubstituted alkylsulfinyl, substituted or unsubstituted alkylsulfinylalkyl, substituted or unsubstituted alkylsulfonyl, substituted or unsubstituted alkylsulfonylalkyl, NH 2 , NHR, NR 2 , SO 2 -OH, OC(O)R, C(O)OH, C(O)OR, C(O)NH 2 , C(O)NHR, C(O)NR 2 , SO 2 NH 2 ,
- R is alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, alkyl-COOH, alkyl-COOalkyl, alkyl-COOaryl, heteroaryl, substituted heteroaryl, or when attached to a nitrogen atom, two adjacent R groups may combine to form a ring of 5 to 7 members;
- R 1 and R 2 are independently straight chained, branched, or cyclic alkyl which may be substituted, aryl, substituted aryl, heteroaryl, substituted heteroaryl, alkaryl, or aralkyl; and wherein substituents on the R 1 or R 2 groups are selected from the group consisting of hydrogen, halogen, alkyl, nitro, amino, alkylamino, dialkylamino, acyl, and acyloxy;
- R 3 and R 4 are independently any group that does not otherwise adversely affect the desired properties of the molecule, including H, halogen, or R 1 .
- the compound of formula (II) has the following structure
- R a , R b , R,., and Rj are independently any group that does not otherwise adversely affect the desired properties of the molecule, including hydrogen, straight chained, branched, or cyclic alkyl which may be substituted, aryl, substituted aryl, heteroaryl, substituted heteroaryl, alkaryl, substituted alkaryl, aralkyl or substituted aralkyl; substituents on the R ⁇ , R b , R,.
- R d groups are selected from the group consisting of hydrogen, halogen, alkyl, nitro, amino, haloalkyl, alkylamino, dialkylamino, acyl, and acyloxy;
- Z is selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, aralkyl, alkaryl, heteroaryl, heteroaralkyl, a carbohydrate group, -(CH,)-! ⁇ , -C(O)-R g , and -C(O)-(CH 2 ) n -R h , wherein (a) when each of R ⁇ R b , R,., and Rj are t-butyl, Z cannot be hydrogen and (b) when each of R a , R b , R e , and R d are t-butyl, Z cannot be the residue of
- R_. is selected from the group consisting of alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkyloxy, alkoxyalkyl, substituted alkoxyalkyl, NH 2 , NHR, NR 2 , mono- or polyhydroxy-substituted alkyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, acyloxy, substituted acyloxy, COOH, COOR, -CH(OH)R k , hydroxy, C(O)NH 2 , C(O)NHR, C(O)NR 2 ;
- R g is selected from the group consisting of alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkyloxy, alkoxyalkyl, substituted alkoxyalkyl, NH 2 , NHR, NR 2 , mono- or polyhydroxy-substituted alkyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl;
- R h is selected from the group consisting of alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkyloxy, alkoxyalkyl, substituted alkoxyalkyl, NH 2 , NHR, NR 2 , mono- or polyhydroxy-substituted alkyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, acyloxy, substituted acyloxy, COOH, COOR, -CH(OH)R k , hydroxy, O-phosphate, C(O)NH 2 , C(O)NHR, C(O)NR 2 and pharmaceutically acceptable salts thereof;
- R e , R g , and R h can independently be a substituent which improves the water solubility of the compound, including, but not limited to C(O)- spacer-SO 3 H, wherein spacer is as defined above, C(O)-spacer-SO 3 M, wherein M is a metal used to form a pharmaceutically acceptable salt, for example, sodium, C(O)-spacer-PO 3 H 2 , C(O)-spacer-PO 3 M 2 , C(O)-spacer-PO 3 HM, C(O)-spacer-PO 4 H, C(O)-spacer-PO 4 M, SO 3 M, - PO 3 H 2 -PO 3 M 2 , -PO 3 HM, cyclic phosphates, polyhydroxyalkyl, carbohydrate groups, C(O)- spacer- [O(C,_ 3 alkyl) p ] n , wherein n is as defined above and p is 1, 2, or 3, -
- the present invention further provides a method of inhibiting the peroxidation of LDL lipid in a patient in need thereof comprising administering to said patient an effective antioxidant amount of a compound of formula (I) or formula (II).
- a method for suppressing the expression of a redox-sensitive gene or activating a gene that is suppressed through a redox-sensitive pathway that includes administering an effective amount to prevent the oxidation of the oxidized signal, and typically, the oxidation of a PUFA of a compound of formula (I) or formula (II).
- Representative redox-sensitive genes that are involved in the presentation of an immune response include, but are not limited to, those expressing cytokines involved in initiating the immune response (e.g., IL-l ⁇ ), chemoattractants that promote the migration of inflammatory cells to a point of injury (e.g., MCP-1), growth factors (e.g., IL-6 and the thrombin receptor), and adhesion molecules (e.g., VCAM-1 and E-selectin).
- cytokines involved in initiating the immune response e.g., IL-l ⁇
- chemoattractants that promote the migration of inflammatory cells to a point of injury e.g., MCP-1
- growth factors e.g., IL-6 and the thrombin receptor
- adhesion molecules e.g., VCAM-1 and E-selectin
- alkyl refers to a saturated straight, branched, or cyclic, primary, secondary, or tertiary hydrocarbon of C, to C, 0 , and specifically includes methyl, ethyl, propyl, isopropyl, cyclopropyl, butyl, isobutyl, t-butyl, pentyl, cyclopentyl, isopentyl, neopentyl, hexyl, isohexyl, cyclohexyl, cyclohexylmethyl, 3- methylpentyl, 2,2-dimethylbutyl, and 2,3-dimethylbutyl.
- the alkyl group can be optionally substituted with one or more moieties selected from the group consisting of alkyl, halo, hydroxyl, carboxyl, acyl, acyloxy, amino, alkylamino, arylamino, alkoxy, aryloxy, nitro, cyano, sulfonic acid, sulfate, phosphonic acid, phosphate, or phosphonate, either unprotected, or protected as necessary, as known to those skilled in the art, for example, as taught in Greene, et al, Protective Groups in Organic Synthesis. John Wiley and Sons, Second Edition, 1991, hereby incorporated by reference.
- lower alkyl refers to a C, to C 5 saturated straight, branched, or if appropriate, a cyclic (for example, cyclopropyl) alkyl group.
- alkylene refers to a saturated hydrocarbyldiyl radical of straight or branched configuration made up of from one to ten carbon atoms. Included within the scope of this term are methylene, 1,2-ethane-diyl, 1,1-ethane-diyl, 1,3-propane-diyl, 1 ,2-propane-diyl, 1,3-butane-diyl, 1,4-butane-diyl and the like.
- the alkylene group can be optionally substituted with one or more moieties selected from the group consisting of alkyl, halo, hydroxyl, carboxyl, acyl, acyloxy, amino, alkylamino, arylamino, alkoxy, aryloxy, nitro, cyano, sulfonic acid, sulfate, phosphonic acid, phosphate, or phosphonate, either unprotected, or protected as necessary, as known to those skilled in the art, for example, as taught in Greene, et al, Protective Groups in Organic Synthesis. John Wiley and Sons, Second Edition, 1991, hereby incorporated by reference.
- -(CH 2 ) n - represents a saturated hydrocarbyldiyl radical of straight chain configuration.
- n is defined as 0-10.
- aryl refers to phenyl, biphenyl, or naphthyl, and preferably phenyl.
- aralkyl refers to an aryl group as defined above linked to the molecule through an alkyl group as defined above.
- alkaryl refers to an alkyl group as defind above linked to the molecule through an aryl group as defined above.
- the alkyl group can be optionally substituted as describe above and the aryl group can be optionally substituted with one or more moieties selected from the group consisting of alkyl, halo, hydroxyl, carboxyl, acyl, acyloxy, amino, alkylamino, arylamino, alkoxy, aryloxy, nitro, cyano, sulfonic acid, sulfate, phosphonic acid, phosphate, or phosphonate, either unprotected, or protected as necessary, as known to those skilled in the art, for example, as taught in Greene, et al, Protective Groups in Organic Synthesis. John Wiley and Sons, Second Edition, 1991.
- aryl phenyl; naphthyl; phenylmethyl; phenylethyl; 3,4,5-trihydroxyphenyl; 3,4,5-trimethoxyphenyl; 3,4,5-triethoxyphenyl; 4-chlorophenyl; 4-methylphenyl; 3,5-di- tertiarybutyl- 4-hydroxyphenyl; 4-fluorophenyl; 4-chloro-l -naphthyl; 2-methyl-l- naphthylmethyl; 2-naphthylmethyl; 4-chlorophenylmethyl; 4-tertiarybutylphenyl; 4- tertiarybutylphenylmethyl and the like.
- halo as used herein, includes chloro, bromo, iodo, and fluoro.
- alkoxy refers to a moiety of the structure -O-alkyl, wherein alkyl is as defined above.
- acyl refers to a group of the formula C(O)R', wherein R' is an alkyl, aryl, alkaryl or aralkyl group, or substituted alkyl, aryl, aralkyl or alkaryl, wherein these groups are as defined above.
- polyunsaturated fatty acid refers to a fatty acid (typically C 8 to C 2 ) that has at least two alkenyl bonds, and includes but is not limited to linoleic (C 18 ⁇ 9 - 12 ), linolenic (C 18 ⁇ 6 - 9 - 12 ), arachidonic (C 20 ⁇ 8 - 1 U4 ) acids.
- oxidized polyunsaturated fatty acid refers to an unsaturated fatty acid in which at least on of the alkenyl bonds has been converted to a hydroperoxide.
- Nonlimiting examples are 13-HPODE and 15-HPETE.
- salts or complexes refers to salts or complexes that retain the desired biological activity of the compounds of the present invention and exhibit minimal undesired toxicological effects.
- Nonlimiting examples of such salts are (a) acid addition salts formed with inorganic acids (for example, hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, nitric acid, and the like), and salts formed with organic acids such as acetic acid, oxalic acid, tartaric acid, succinic acid, malic acid, ascorbic acid, benzoic acid, tannic acid, pamoic acid, alginic acid, polyglutamic acid, naphthalenesulfonic acid, naphthalenedisulfonic acid, and polygalcturonic acid; (b) base addition salts formed with metal cations such as zinc, calcium, bismuth, barium, magnesium, aluminum, copper, cobalt, nickel, cadmium, sodium, potassium, and the like, or with a cturonic
- quaternary salts known by those skilled in the art, which specifically include the quaternary ammonium salt of the formula -NR + A " , wherien R is as defined above and A is a counterion, including chloride, bromide, iodide, -O-alkyl, toluenesulfonate, methylsulfonate, sulfonate, phosphate, or carboxylate (such as benzoate, succinate, acetate, glycolate, maleate, malate, citrate, tartrate, ascorbate, benzoate, cinnamoate, mandeloate, benzyloate, and diphenylacetate).
- A is a counterion, including chloride, bromide, iodide, -O-alkyl, toluenesulfonate, methylsulfonate, sulfonate, phosphate, or carboxylate (such as benzoate, succinate, acetate,
- VCAM-1 Diseases mediated by the VCAM-1 include, but are not limited to atherosclerosis, post- angioplasty restenosis, coronary artery disease, angina, small artery disease, and other cardiovascular diseases, as well as noncardiovascular inflammatory diseases such as rheumatoid arthritis, osteoarthritis, asthma, dermatitis, multiple sclerosis and psoriasis.
- the invention is a method for treating a disease mediated by the expression of VCAM-1 comprising administering a compound of the formula
- X is O, S, SO, SO 2 , CH 2 , or NH;
- Spacer is a group selected from the group consisting of -(CH 2 ) n -, -(CH 2 ) n -CO-, -(CH 2 ) n - N-, -(CH 2 ) n -O-, -(CH 2 ) n -S-, -(CH 2 O)-, -(OCH 2 )-, -(SCH 2 )-, -(CH 2 S-), -(aryl-O)-, -(O-aryl)-, -(alkyl- O)-, -(O-alkyl)-; n is O, 1, 2, 3, 4, 5, 6, 7, 8, 9, orlO;
- Y is substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted alkyl, NH 2 , NHR, NR 2 , SO 2 -OH, OC(O)R, C(O)OH, C(O)OR, C(O)NH 2 , C(O)NHR, C(O)NR 2 ;
- R is alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, substituted aryl, alkyl-COOH, alkyl-COOalkyl, alkyl-COOaryl, heteroaryl, substituted heteroaryl, or when attached to a nitrogen atom, two adjacent R groups may combine to form a ring of 5 to 7 members;
- R 1 and R 2 are independently straight chained, branched, or cyclic alkyl which may be substituted, aryl, substituted aryl, heteroaryl, substituted heteroaryl, alkaryl, or aralkyl; and wherein substituents on the R 1 or R 2 groups are selected from the group consisting of hydrogen, halogen, alkyl, nitro, amino, alkylamino, dialkylamino, acyl, and acyloxy;
- R 3 and R 4 are independently any group that does not otherwise adversely affect the desired properties of the molecule, including H, halogen, or R 1 .
- Preferred compounds of the present invention include compounds of formula (I) wherein
- X is S, SO or SO 2 ;
- Spacer is -(CH 2 ) n - or -(CH 2 ) n CO-; n is 0-10;
- Y is aryl, substituted aryl, heteroaryl, substituted heteroaryl, NH 2 , NHR, NR 2 , alkyl, substituted alkyl, acyloxy, and substituted acyloxy;
- R is alkyl, alkenyl, alkynyl, aryl, alkyl-COOH, alkyl-COOalkyl, alkyl- COOaryl, heteroaryl, or nitro substituted heteroaryl, or when attached to a nitrogen atom, two adjacent R groups may combine to form a ring of 5 to 7 members;
- R 1 and R 2 are independently straight chained, branched or cyclic C,_ 10 alkyl;
- R 3 and R 4 are independently hydrogen, halogen or R 1 .
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n - or -(CH 2 ) n -CO-; n is 0-10; Y is aryl, substituted aryl, heteroaryl, substituted heteroaryl, NH 2 , NHR, NR 2 , alkyl, substituted alkyl, acyloxy, and substituted acyloxy; R is alkyl, alkenyl, alkynyl, aryl, alkyl-COOH, alkyl- COOalkyl, alkyl-COOaryl, heteroaryl, or nitro substituted heteroaryl, or when attached to a nitrogen atom, two adjacent R groups may combine to form a ring of 5 to 7 members; R 1 and R 2 are independently straight chained, branched or cyclic C,. 5 alkyl; R 3 and R 4 are independently H.
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n - or -(CH 2 ) n -CO-; n is 0-10; Y is aryl; aryl which is mono- or polysubstituted by alkyl, alkenyl, alkynyl, halo, nitro, hydroxy, COOH, COOR, CONH 2 , CONHR, CONR 2 , -(CH 2 ) m -OH wherein m is 0- 10, haloalkyl, mono- or poly-hydroxysubstituted branched alkyl, a carbohydrate group, SO 2 OH, SO 2 NH 2 , SO 2 NHR, SO 2 NR 2 , or OCOR; heteroaryl; heteroaryl which is mono- or polysubstituted by alkyl, alkenyl, alkynyl, CH 2 NH 2 , CH 2
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n - or -(CH 2 ) n -CO-; n is 0-10; Y is aryl; aryl which is mono- or polysubstituted by alkyl, alkenyl, alkynyl, halo, nitro, hydroxy, COOH, COOR, CONH 2 , CONHR, CONR 2 , -(CH 2 ) m -OH wherein m is 0-10, haloalkyl, mono- or poly-hydroxysubstituted branched alkyl, a carbohydrate group, SO 2 OH, SO 2 NH 2 , SO 2 NHR, SO 2 NR 2 , or OCOR; R is alkyl, alkenyl, alkynyl, aryl, alkyl-COOH, alkyl- COOalkyl, alkyl-
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n - or -(CH 2 ) n -CO-; n is 0-10; Y is phenyl; phenyl which is mono- or polysubstituted by alkyl, alkenyl, alkynyl, halo, nitro, hydroxy, COOH, COOR, CONH 2 , CONHR, CONR 2 , -(CH 2 ) m -OH wherein m is 0-10, haloalkyl, mono- or poly-hydroxysubstituted branched alkyl, a carbohydrate group, SO 2 OH, SO 2 NH 2 , SO 2 NHR, SO 2 NR 2 , or OCOR; R is alkyl, alkenyl, alkynyl, alkyl-COOH, alkyl- COOalkyl, alkyl-COO
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n -; n is 0-10; Y is phenyl; phenyl which is mono- or polysubstituted by alkyl, alkenyl, alkynyl, halo, nitro, hydroxy, COOH, COOR, CONH 2 , CONHR, CONR 2 , -(CH 2 ) m -OH wherein m is 0-10, haloalkyl, mono- or poly-hydroxysubstituted branched alkyl, a carbohydrate group, SO 2 OH, SO 2 NH 2 , SO 2 NHR, SO 2 NR 2 , or OCOR; R is alkyl, alkenyl, alkynyl, alkyl-COOH, alkyl-COOalkyl, alkyl- COOaryl, aryl, heteroaryl or nitro-
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n -CO-; n is 0-10; Y is phenyl; phenyl which is mono- or polysubstituted by alkyl, alkenyl, alkynyl, halo, nitro, hydroxy, COOH, COOR, CONH 2 , CONHR, CONR 2 , -(CH 2 ) m -OH wherein m is 0-10, haloalkyl, mono- or poly-hydroxysubstituted branched alkyl, a carbohydrate group, SO 2 OH, SO 2 NH 2 , SO 2 NHR, SO 2 NR 2 , or OCOR; R is alkyl, alkenyl, alkynyl, alkyl-COOH, alkyl-COOalkyl, alkyl- COOaryl, aryl, heteroaryl or
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n - or -(CH 2 ) n -CO-; n is 0-10; Y is phenyl; phenyl which is mono- or polysubstituted by alkyl, halo, nitro, hydroxy, COOH, COOR, CONH 2 , CONHR, CONR 2 , -(CH 2 ) m -OH wherein m is 0-10, haloalkyl, mono- or poly-hydroxysubstituted branched alkyl, a carbohydrate group, SO 2 OH, SO 2 NH 2 , SO 2 NHR, SO 2 NR 2 , or OCOR; R is alkyl, alkyl-COOH, alkyl-COOalkyl, alkyl-COOaryl, aryl, heteroaryl or nitro-substituted heteroaryl
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n - or -(CH 2 ) n -CO-; n is 0-10; Y is phenyl; phenyl which is mono- or polysubstituted by alkyl, halo, nitro, hydroxy, COOH, COOR, CONH 2 , CONHR, CONR 2 , -(CH 2 ) m -OH wherein m is 0-10, haloalkyl, mono- or poly-hydroxysubstituted branched alkyl, a carbohydrate group, SO 2 OH, SO 2 NH 2 , SO 2 NHR, SO 2 NR 2 , or OCOR; R is alkyl, alkyl-COOH, alkyl-COOalkyl, alkyl-COOaryl, or nitro- substituted furanyl, or when attached to a nitrogen atom
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n - or -(CH 2 ) n -CO-; n is 0-10; Y is heteroaryl; heteroaryl which is mono- or polysubstituted by alkyl, alkenyl, alkynyl, CH 2 NH 2 , CH 2 NHR, CH 2 NR 2 , COOH, COOR; R is alkyl, alkenyl, alkynyl, aryl, alkyl-COOH, alkyl- COOalkyl, alkyl-COOaryl, heteroaryl, or nitro substituted heteroaryl, or when attached to a nitrogen atom, two adjacent R groups may combine to form a ring of 5 to 7 members; R 1 and R 2 are independently C ⁇ alkyl; R 3 and R 4 are independently H.
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n -; n is 0-10; Y is heteroaryl; heteroaryl which is mono- or polysubstituted by alkyl, alkenyl, alkynyl, CH 2 NH 2 , CH 2 NHR, CH 2 NR 2 , COOH, COOR; R is alkyl, alkenyl, alkynyl, aryl, alkyl-COOH, alkyl-COOalkyl, alkyl-COOaryl, heteroaryl, or nitro substituted heteroaryl, or when attached to a nitrogen atom, two adjacent R groups may combine to form a ring of 5 to 7 members; R 1 and R 2 are independently C,_ 5 alkyl; R 3 and R 4 are independently H.
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n -CO-; n is 0-10; Y is heteroaryl; heteroaryl which is mono- or polysubstituted by alkyl, alkenyl, alkynyl, CH 2 NH 2 , CH 2 NHR, CH 2 NR 2 , COOH, COOR; R is alkyl, alkenyl, alkynyl, aryl, alkyl-COOH, alkyl-COOalkyl, alkyl-COOaryl, heteroaryl, or nitro substituted heteroaryl, or when attached to a nitrogen atom, two adjacent R groups may combine to form a ring of 5 to 7 members; R 1 and R 2 are independently Cj. 5 alkyl; R 3 and R 4 are independently H.
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n - or -(CH 2 ) n -CO-; n is 0-10; Y is isoxazolyl or furanyl which may be optionally substituted by mono- or polysubstituted by alkyl, alkenyl, alkynyl, CH 2 NH 2 , CH 2 NHR, CH 2 NR 2 , COOH, COOR; R is alkyl, alkenyl, alkynyl, aryl, alkyl-COOH, alkyl-COOalkyl, alkyl-COOaryl, heteroaryl, or nitro substituted heteroaryl, or when attached to a nitrogen atom, two adjacent R groups may combine to form a ring of 5 to 7 members; R 1 and R 2 are independently Q.s alkyl; R 3 and R 4 are independently H.
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n - or -(CH 2 ) n -CO-; n is 0-10; Y is isoxazolyl which may be optionally substituted by mono- or polysubstituted by alkyl, alkenyl, alkynyl, CH 2 NH 2 , CH 2 NHR, CH 2 NR 2 , COOH, COOR; R is alkyl, alkenyl, alkynyl, aryl, alkyl-COOH, alkyl-COOalkyl, alkyl-COOaryl, heteroaryl, or nitro substituted heteroaryl, or when attached to a nitrogen atom, two adjacent R groups may combine to form a ring of 5 to 7 members; R 1 and R 2 are independently C]_ 5 alkyl; R 3 and R 4 are independently H.
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n - or -(CH 2 ) n -CO-; n is 0-10; Y is furanyl which may be optionally substituted by mono- or polysubstituted by alkyl, alkenyl, alkynyl, CH 2 NH 2 , CH 2 NHR, CH 2 NR 2 , COOH, COOR; R is alkyl, alkenyl, alkynyl, aryl, alkyl-COOH, alkyl-COOalkyl, alkyl-COOaryl, heteroaryl, or nitro substituted heteroaryl, or when attached to a nitrogen atom, two adjacent R groups may combine to form a ring of 5 to 7 members; R 1 and R 2 are independently C ⁇ alkyl; R 3 and R 4 are independently H.
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n - or -(CH 2 ) n -CO-; n is 0-10; Y is NH 2 , NHR or NR 2 ; R is alkyl, alkenyl, alkynyl, aryl, alkyl-COOH, alkyl-COOalkyl, alkyl- COOaryl, heteroaryl, or nitro substituted heteroaryl, or when attached to a nitrogen atom, two adjacent R groups may combine to form a ring of 5 to 7 members; R 1 and R 2 are independently C,_ 5 alkyl; R 3 and R 4 are independently H.
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n - or -(CH 2 ) n -CO-; n is 0-10; Y is NH 2 , NHR or NR 2 ; R is alkyl, or when attached to a nitrogen atom, two adjacent R groups may combine to form a ring of 5 to 7 members; R 1 and R 2 are independently C 5 alkyl; R 3 and R 4 are independently H.
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) ⁇ -; n is 0-10; Y is NH 2 , NHR or NR 2 ; R is alkyl, or when attached to a nitrogen atom, two adjacent R groups may combine to form a ring of 5 to 7 members; R 1 and R 2 are independently C,_ 5 alkyl; R 3 and R 4 are independently H.
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n -CO-; n is 0-10; Y is NH 2 , NHR or NR 2 ; R is alkyl, or when attached to a nitrogen atom, two adjacent R groups may combine to form a ring of 5 to 7 members; R 1 and R 2 are independently C ⁇ -5 alkyl; R 3 and R 4 are independently H.
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n - or -(CH 2 ) n -CO-; n is 0-10; Y is selected from the group consisting of straight chained, branched or cyclic alkyl; straight chained, branched, or cyclic alkyl substituted by OCOR, SO 2 OH, COOH or COOR; and OCOR; R is alkyl, alkenyl, alkynyl, and aryl, or when attached to a nitrogen atom, two adjacent R groups may combine to form a ring of 5 to 7 members; R 1 and R 2 are independently C 5 alkyl; R 3 and R 4 are independently H.
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n - or -(CH 2 ) n -CO-; n is 0-10; Y is selected from the group consisting of straight chained, branched or cyclic alkyl; straight chained, branched, or cyclic alkyl substituted by OCOR, SO 2 OH, COOH or COOR; and OCOR; R is alkyl or two adjacent R groups may combine to form a ring of 5 to 7 members; R 1 and R 2 are independently C ⁇ _ 5 alkyl; R 3 and R 4 are independently H.
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n -; n is 0-10; Y is selected from the group consisting of straight chained, branched or cyclic alkyl; straight chained, branched, or cyclic alkyl substituted by OCOR, SO 2 OH, COOH; or COOR; R is alkyl; R 1 and R 2 are independently C,_ 5 alkyl; R 3 and R 4 are independently H.
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n -CO-; n is 0-10; Y is selected from the group consisting of straight chained, branched or cyclic alkyl; straight chained, branched, or cyclic alkyl substituted by OCOR; R is alkyl; R 1 and R 2 are independently C 5 alkyl; R 3 and R 4 are independently H.
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n - or -(CH 2 ) n -CO-; n is 0-10; Y is OCOR; R is alkyl; R 1 and R 2 are independently C 1-5 alkyl; R 3 and R 4 are independently H.
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n -; n is 0-10; Y is OCOR; R is alkyl; R 1 and R 2 are independently C,_ 5 alkyl; R 3 and R 4 are independently H.
- Another preferred embodiment of the present invention includes compounds of formula (I) wherein X is S, SO, or SO 2 ; Spacer is -(CH 2 ) n -CO-; n is 0-10; Y is OCOR; R is alkyl; R 1 and R 2 are independently C,_ 5 alkyl; R 3 and R 4 are independently H.
- Examples of the present invention include compounds of formula (I) defined as follows:
- a compound of formula (II) and a method for treating a disease mediated by the expression of VCAM-1 comprising administering an effective amount of a compound of formula (II):
- R a , R b , R,., and Rj are independently hydrogen, straight chained, branched (for example, tert-butyl), or cyclic alkyl which may be substituted, aryl, substituted aryl, heteroaryl, substituted heteroaryl, alkaryl, substituted alkaryl, aralkyl or substituted aralkyl; substituents on the R ⁇ R,,, R j . and R ⁇ groups are selected from the group consisting of hydrogen, halogen, alkyl, nitro, amino, haloalkyl, alkylamino, dialkylamino, acyl, and acyloxy;
- Z is selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, aralkyl, alkaryl, heteroaryl, heteroaralkyl, a carbohydrate group, -(CH ⁇ -R e , -C(O)-R g , and -C(O)-(CH 2 ) n -R h , wherein (a) when each of R a , R b , R ⁇ ., and Rj are t-butyl, Z cannot be hydrogen and (b) when each of R a , R b , R,., and Rj are t-butyl, Z cannot be the residue of succinic acid;
- R e is selected from the group consisting of alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkoxy, alkoxyalkyl, substituted alkoxyalkyl, NH 2 , NHR, NR 2 , mono- or polyhydroxy-substituted alkyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, acyloxy, substituted acyloxy, COOH, COOR, -CH(OH)R k , hydroxy, C(O)NH 2 , C(O)NHR, C(O)NR 2 ;
- R g is selected from the group consisting of alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkoxy, alkoxyalkyl, substituted alkoxyalkyl, NH 2 , NHR, NR 2 , mono- or polyhydroxy-substituted alkyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl;
- R h is selected from the group consisting of alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, alkoxy, substituted alkoxy, alkoxyalkyl, substituted alkoxyalkyl, NH 2 , NHR, NR 2 , mono- or polyhydroxy-substituted alkyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, acyloxy, substituted acyloxy, COOH, COOR, -CH(OH)R k , hydroxy, O-phosphate, C(O)NH 2 , C(O)NHR, C(O)NR 2 and pharmaceutically acceptable salts thereof; or, in an alternative embodiment, R e , R g , and R h can independently be a substituent which improves the water solubility of the compound, including, but not limited to C(O)- spacer-SO 3 H, wherein spacer is as defined above,
- Substitutents on the groups defined above are selected from the group consisting of alkyl, alkenyl, alkynyl, hydroxy, halo, nitro, amino, alkylamino, dialkylamino, carboxy, aryl, heteroaryl, COOR, CONH 2 , CONHR, CONR 2 , haloalkyl, alkoxyalkyl, mono- or polyhydroxyalkyl, CH 2 -OR, CH 2 -OH, OCOR, O- ⁇ hosphate, SO 2 -NH 2 , SO 2 -NHR, SO 2 -NR 2 .
- a preferred embodiment of the present invention includes compounds of formula (II) wherein R ⁇ , R b , R,., and R ⁇ are independently hydrogen or straight chained, branched, or cyclic ., 0 alkyl; Z is selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, a carbohydrate group, -(CH 2 )- R e , -C(O)-R g , and -C(O)-(CH 2 ) n -R h , and pharmaceutically acceptable salts thereof.
- R a , R b , R restroom and Rj are independently hydrogen or straight chained, branched, or cyclic C,_ 5 alkyl;
- Z is selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, a carbohydrate group, -(CH 2 )-R e , -C(O)-R g , and -C(O)-(CH 2 ) n -R h , and pharmaceutically acceptable salts thereof.
- R ⁇ , b , R c , and Rj are independently hydrogen or straight chained, branched, or cyclic C,_ 5 alkyl;
- Z is selected from the group consisting of hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, -CH 2 -aryl substituted alkynyl, a carbohydrate group, -CH 2 -NR 2 , -CH 2 -alkoxy, -CH 2 -CHOH, -CH 2 -substituted aryl, -CH 2 - alkyl, -CH 2 -substituted alkyl, -CH 2 .-OCO-alkyl, -CH 2 -OCO-substituted alkyl, -CH 2 -COOR, - CH 2 -CH(OH)CH 2 NHCH 2 COOR, -CH 2 -CH
- R ⁇ R b , R ⁇ ., and Rj are independently hydrogen or straight chained, branched, or cyclic C 5 alkyl;
- Z is selected from the group consisting of hydrogen, alkyl, hydroxy alkyl, polyhydroxy alkyl, alkenyl, hydroxy alkenyl, acyl-substituted alkenyl, alkoxy alkyl, nitrophenylalkyl, aminophenylalkyl, alkylaminophenylalkyl, dialkylaminophenylalkyl, aminoalkyl, alkylaminoalkyl, dialkylaminoalkyl, carboxyalkyl, acyloxyalkyl, oxiranyl- substituted hydroxyalkyl, hydroxyalkyl-substituted oxiranylmethylene, oxiranyl-substituted hydroxyalkoxyalkyl oxiranylmethylene,
- Examples of the present invention include compounds of formula (II) wherein:
- R a t-butyl
- R b t-butyl
- R d t-butyl
- Z CO-(5-nitrofuran-2-yl)
- R a t-butyl
- R b t-butyl
- Z 3-carboxypropyl
- R a t-butyl
- R b t-butyl
- R c H
- R d H
- Z 4-aminobutyl
- R a t-butyl
- R b t-butyl
- Z CO-(2-carboxyethyl)
- R a t-butyl
- R b t-butyl
- R ⁇ . t-butyl
- Z CO-(2-methoxycarbonylethyl)
- R a t-butyl
- R b t-butyl
- R c t-butyl
- R d t-butyl
- Z 3-carboxypropyl
- R a t-butyl
- R b t-butyl
- R d t-butyl
- Z CO-2-carboxyethyl
- R a t-butyl
- R b t-butyl
- R ⁇ -butyl and R ⁇ -butyl
- Z CO-ammonium methyl (chloride)
- R a t-butyl
- R b t-butyl
- Z 3-(2-hydroxy-2-oxiranyl)ethoxyoxiran-
- R a t-butyl
- R b t-butyl
- R,. t-butyl
- R d t-butyl
- Z oxiranylmethyl
- R a t-butyl
- R b t-butyl
- R c t-butyl
- R d t-butyl
- Z 2,3-dihydroxypropyl
- R a t-butyl
- R b t-butyl
- Z ethyl
- R a t-butyl
- R b t-butyl
- R ⁇ t-butyl and R ⁇ -butyl
- Z CO-2-carboxyethyl
- R a t-butyl
- R b t-butyl
- R d t-butyl
- Z 3-methoxycarbonylpropyl
- R a t-butyl
- R b t-butyl
- R c t-butyl
- R d t-butyl
- Z 3-(N-N-diethylamino)propyl
- Z 2-ethoxycarbonylethenyl;
- R a t-butyl
- R b t-butyl
- Z carboxymethylaminocarbonylmethyl
- Z l,3- dicarboxypropylaminocarbonylmethyl;
- R a t-butyl
- R b t-butyl
- R ⁇ . t-butyl
- R d t-butyl
- Z 2-hydroxy-3-(l,3- diethoxycarbonyl)propylaminopropyl
- R a t-butyl
- R b t-butyl
- Z 3-hydroxypropenyl
- R a t-butyl
- R b t-butyl
- R,. t-butyl
- Z CH 2 CONH-(CH 2 )CH(NH 2 )COOH
- R a t-butyl
- R b t-butyl
- R c t-butyl
- R d t-butyl
- R a t-butyl
- R b t-butyl
- R c t-butyl
- R d t-butyl
- Z CO-2,2-dimethyl-3-hydroxypro ⁇ yl
- R a t-butyl
- R b t-butyl
- R c t-butyl
- R d t-butyl
- Z CH 2 CH(OH)CH 2 NH(2,3,4,5,6- pentahydroxyhexane.
- the compounds of formula (I) can be prepared by utilizing known procedures and techniques, or routine modifications thereof.
- a general synthetic scheme for preparing compounds of formula (I) is set forth in Scheme A, wherein all substituents, unless otherwise indicated, are previously defined.
- a quantity of the 4-mercapto-2,6-di-t-butylphenol is dissolved in ethanol to make a
- a phenol of structure (I) can be prepared by dissolving the appropriate 2,6- dialkyl-4-thiophenol (or suitably protected derivatives) in alcohol, preferably in ethanol, followed by addition of a halogenated aryl compound.
- the starting material, a 2,6-dialkyl-substituted thiophenol may be protected by any of the many protecting groups known to one of ordinary skill in the art.
- Suitable phenol protecting groups are ethers, such as methoxymethyl, 2-methoxyethoxymethyl, tetrahydropyranyl, t-butyl and benzyl; silyl ethers, such as trimethylsilyl and 1- butyldimethylsilyl; esters, such as acetate and benzoate; carbonates, such as methylcarbonate and benzyl carbonate; as well as sulfonates, such as methane sulfonate and toluene sulfonate.
- ethers such as methoxymethyl, 2-methoxyethoxymethyl, tetrahydropyranyl, t-butyl and benzyl
- silyl ethers such as trimethylsilyl and 1- butyldimethylsilyl
- esters such as acetate and benzoate
- carbonates such as methylcarbonate and benzyl carbonate
- sulfonates such as methane sulf
- 2,6-di-t-butyl-4-thiophenol (238 mg, 1 mmol) was dissolved in ethanol (0.7 mL) and cooled to 0° C. 5 N NaOH (0.6 mL, 3 mmol) was added followed by addition of 4- (bromomethyl)phenyl acetic acid (229 mg, 1 mmol). The reaction was warmed to room temperature and after 0.5 h the reaction was complete. The reaction was quenched with 1 N HCl (3.5 mL) and diluted with ether (25 mL). The ether layer was separated and washed with water (1 x 5 mL) and brine (1 x 5 mL), dried over MgSO 4 , filtered and concentrated.
- the aqueous layer was back-extracted with 2 x 2 mL of EtOAc; the combined organic layers were dried over anhydrous MgSO 4 .
- the drying agent was removed by filtration; solvent was removed by rotary evaporation to give a crude oil.
- the oil was purified by preparative thin-layer chromatography (pTLC) using 2 x 500 ⁇ plates and 1 : 1 hexanes- CH 2 C1 2 as eluant.
- the desired product (2,6-di-tert-butyl-4-thio(4'-nitrobenzyl)phenol) was obtained in 86% yield (90 mg).
- 2,6 di-t-butyl-4-thiophenol 180 mg, 0.755 mmol was dissolved in ethanol (1.5 mL) and then treated with 5 N NaOH (0.15 mL, 0.75 mmol). After 5 min, 4-(N,N- dimethylsulfonamide)benzyl bromide (210 mg, 0.755) in ethanol (1.5 mL) was added to the reaction. The resulting mixture was stirred at room temperature for 3 h. The reaction was quenched with IN HCl to pH 7, diluted with water (3 mL), extracted with ether (10 mL), separated and dried over MgSO 4 .
- 2,6-di-tert-butyl-4-thiophenol (0.84 mmol; 200 mg) was dissolved in 7.6 mL of DMF and treated with 1.1 mmol (46 mg) of NaH (60% dispersion in mineral oil) to give an orange mixture. After 15 min. , 0.76 mmol (0.12 mL) of 5-chloropentyl acetate was added. The mixture was stirred for 25 h then diluted with 20 mL of EtOAc and washed with H 2 O (25 mL). The organic layer was washed with brine then concentrated by rotary evaporation.
- the crude material was purified by radial chromatography (2 mm SiO 2 plate; 85 : 15 hexanes-EtOAc as eluant) to give 2,6-di-tert-butyl-4-thio((r- (acetoxy))pentyl)phenol (93 mg; 30% yield) as a light yellow, amorphous solid.
- a quantity of probucol (commercially available from Sigma Chemicals) in a 0.1 M solution of tetrahydrofuran is treated with 2 equivalents of sodium hydride and stirred at room temperature for 30 minutes.
- To the reaction mixture is added 3 equivalents of a primary alkyl bromide or iodide and the reaction stirred at room temperature for 16 hours.
- the reaction is quenched with 1 N aqueous HCl and diluted with ethyl acetate.
- the aqueous layer is removed and the ehtyl acetate layer is washed with water and then with an aqueous saturated sodium chloride solution.
- the ethyl acetate solution is dried over magnesium sulfate, gravity or vacuum filtered, and then concentrated.
- a second alternative method for the preparation of compounds of formula (II) wherein Z forms an ether group is the treatment of probucol with a primary alkyl bromide or iodide in acetonitrile in the presence of potassium fluoride absorbed on alumina according to the method of Ando et al. ⁇ Bull. Chem. Soc. Jpn., 55, 1982, 2504-2507).
- Phenol 4-[[l-[3,5-bis(l,l-dimethylethyl)4-[(4- nitrophenyl)methoxy]phenyl]thio]-l-methyIethyl]thio]2,6-bis(l,l- dimethylethyl)-
- Butanedioic acid mono [4-[[l-[[3,5-bis(l,l-dimethylethyl)-4- hydroxyphenyl]thio]-l-methylethyl]thio]2,6-bis(l,l-dimethylethyl)phenyl]ester
- Butanoic acid 4-[4-[[l-[[3,5-bis(l,l-dimethylethyl)-4-hydroxyphenyl]thio]-l- methylethyl]thio]2,6-dimethylphenoxy]-
- Phenol 4-[[l-[[4-(4-aminobutoxy)-3,5-bis(l,l-dimethylethyl)phenyl]thio]- l-methylethyl]thio]2,6-bis(l , 1-dimethylethyl)-
- Phenol 4-[[l-[[4-(4-aminobutoxy)-3,5-bis(l,l-dimethylethyl)phenyl]thio]-l- methylethyl]thio]2,6-bis(l,l-dimethylethyl)-
- Example 28 Propanoic acid, 2,2-dimethyl-, [4-[[l-[[3,5-bis(l,l-dimethylethyl)-4- hydroxyphenyl]thio]-l-methylethyl]thio]-2,6-bis(l,l- dimethylethyl)phenoxy]methyl ester
- acetonitrile 30 ml
- chloromethyl pivalate 6.0 g, 40 mmol
- potassium fluoride 8.0 g, 40% on alumina
- Phenol 4-[[l-[[4-(4-aminobutoxy)phenyl]thio]-l-methylethyl]thio]-2,6-bis(l,l- dimethylethyl)-
- Butanoic acid 4-[4-[[l-[[3,5-bis(l,l-dimethylethyl)-4-hydroxyphenyl]thio]-l- methylethyl]thio]phenoxy]-
- the resulting oil was chromatographed on silica gel eluting with 10% ethyl acetate/hexane followed by 20% ethyl acetate/hexane to yield 610 mg of the phthaloyl glycine ester.
- the phthaloyl glycine ester was taken up in DMF(8.6 mL) and hydrazine hydrate was added (0.136 mL, 2.34 mmol) and the reaction stirred overnight.
- IN HCl was added (5 mL) and the reaction stirred an additional 1 h.
- the reaction was diluted with ethyl acetate (25 mL) and washed with NaHCO3 (aq) (1X10 mL).
- Pentanedioic acid mono[4-[[l-[[3,5-bis(l,l-dimethylethyl)-4- hydroxyphenyl]thio]-l-methylethyl]thio]-2,6-bis(l , l-dimethylethyl)phenyl] ester
- Butanoic acid 4-[4-[[l-[[3,5-bis(l,l-dimethylethyI)-4-hydroxyphenyl]thio]-l- methylethyl]thio]-2,6-bis(l,l-dimethylethyl)phenoxy]-
- the ethyl acetate layer was dried over MgSO 4 , fileterd, concentrated and chromatographed on silia gel by elution with a concentration gradient of 10:90 methylene chloride/hexane to 60:40 methylene chloride hexane. The appropriate fractions were collected and concentrated to afford 442 mg of a white solid.
- the methyl ester was taken up in THF:MeOH:H 2 0 (4:1:1)(5 mL) and lithium hydroxide (63 mg, 1.5 mmol) was added. After 2.5 h the reaction was complete and quenched with 1 N HCl (3 mL) and extracted with ethyl acetate (15 mL).
- probucol 5.16 g, 10 mmol
- acetonitrile 50 ml
- potassium fluoride 2.9 g, 20 mmol
- Phenol 4-[[l-[[3,5-bis(l,l-dimethylethyl)-4-(oxiranylmethoxy)phenyl]thio]-l- methylethyl]thio]-2,6-bis(l,l-dimethylethyl)-
- Phenol 4-[[l-[[3,5-bis(l , l-dimethylethyl)-4-(3-ethoxy-2- hydroxypropoxy)phenyl]thio]-l-methylethyl]thio]-2,6-bis(l,l-dimethylethyl)-;
- Phenol 4-[[l-[[3,5-bis(l,l-dimethylethyl)-4-ethoxyphenyl]thio]-l- methylethyl]thio]-2,6-bis(l , 1-dimethylethyl)-
- Butanedioic acid mono[4-[[l-[[3,5-bis(l,l-dimethylethyI)-4- methoxyphenyl]thio]l-methylethyl]thio]-2,6-bis(l,l-dimethylethyl)phenyl] ester
- Example 22 The compound of Example 22 (1.13 g, 1.83 mmol) was taken up in DMF (3.6 mL) and 60% sodium hydride (183 mg, 4.6 mmol) was added followed 0.25 h later by methyl iodide (0.342 mL, 5.5 mmol). The reaction was allowed to stir overnight. The reaction was quenched with water (2 mL), diluted with ether (50 mL). The ether layer was washed with water (2X10 mL) and saturated aqueous sodium chloride (1X10 mL), dried over MgSO 4 ,filtere, and concentrated.
- Phenol 4-[[l-[[4-[2-[4-(dimethylamino)phenyl]ethoxy]-3,5-bis(l,l- dimethylethyl)phenyl]thio]-l-methylethyl]thio]-2,6-bis(l,l-dimethylethyl)-
- Example 46 ⁇ -D-Galactopyranose, 6-O-[4-[[l-[[3,5-bis(l,l-dimethylethyl)-4- hydroxyphenyl]thio]-l-methylethyI]thio]-2,6-bis(l,l-dimethylethyI)phenyl]- 1 ,2 :3,4-bis-O-(l -methylethylidene)
- Phenol 4-[[l-[[4-[3-(dimethylamino)propoxy]-3,5-bis(l,l- dimethylethyl)phenyI]thio]-l-methylethyl]thio]-2,6-bis(l,l-dimethylethyl)-
- Example 48 Glycine, N-[ [4- [ [1 - [ [3,5-bis(l ,1 -dimethylethyl)-4-hydroxyphenyl] thio]- l,methylethyl]thio]-2,6-bis(l,l-dimethylethyl)phenoxy]acetyl]-
- the reaction mixture was sti ⁇ ed overnight and the methylene chloride evaporated.
- the reaction was diluted with ether (10 mL) and washed with water (2X3 mL), dried over MgSO 4 , filtered, and concentrated.
- the crude mixture was purified by silica gel chromatography and elution with 50:50 ether/hexane to give 50 mg of the ethyl ester of the product.
- the ethyl ester dissolved in THF:H 2 0:MeOH (2:1:1)(1 mL) and LiOH-H20 (15 mg) was added and the reaction sti ⁇ ed for 1 h.
- the reaction mixture was sti ⁇ ed overnight and the methylene chloride evaporated.
- the reaction was diluted with ether(10 mL) and washed with water(2X3 mL), dried over MgSO , filtered, and concentrated.
- the crude mixture was purified by silica gel chromatography and elution with 50:50 ether/hexane to give 130 mg of the diethyl ester of the desired product.
- the diethyl ester was dissolved in THF:H 2 0:MeOH(2:l:l)(3 mL) and LiOH-H 2 0 (100 mg) was added and the reaction sti ⁇ ed for 1 h.
- Phenol 4-[[l-[[3,5-bis(l , l-dimethylethyl)-4-[[3-hydroxy-l- propenyl)oxy]phenyl]thio]-l-methylethyl]thio]-2,6-bis(l,l-dimethylethyl)-
- the reaction mixture was stirred overnight and the methylene chloride evaporated.
- the reaction was diluted with ether(10 mL) and washed with water (2X3 mL), dried over MgSO 4 , filtered, and concentrated.
- the crude mixture was purified by silica gel chromatography and elution with 50:50 ether/hexane followed by 70:30 ether/hexane to give 128 mg of the methyl ester of product.
- the methyl ester was dissolved in THF:H 2 0:MeOH(2: l:l)(3 mL) and LiOH-H 2 0 (50 mg) was added and the reaction stirred for 1 h.
- Butanoic acid 4-hydroxy-3,3-dimethyl-, 4-[[l-[[3,5-bis(l,l-dimethylethyl)-4- hydroxyphenyl]thio]-l-methylethyl]thio]-2,6-bis(l,l-dimethylethyl)phenyl ester
- the crude product mixture was dissolved in ether and chromatographed on silica gel with a concentration gradient of 70:30 hexane/ether to 0:100 hexane/ether. The appropriate fractions were combined and concentrated affording 700 mg of a white solid.
- the white solid (214 mg, 0.332 mmol)) was taken up in THF (6 mL) and borane-dimethylsulfide (2M in THF, 0.665 mL, 0.664 mmol) was added and the reaction sti ⁇ ed for 6 h. The reaction was qenched with concentrated HCl (0.100 mL) and the reaction sti ⁇ ed overnight.
- Butanoic acid 4-(suIfoxy)-, l-[4-[[3,5-bis(l,l-dimethylethyI)-4- hydroxyphenyl]thio]-l-methylethyl]thio]-2,6-bis(l,l-dimethylethyl)phenyl]ester
- Butanoic acid, 4-hydorxy-, 4-[[l-[[3,5-bis(l,l-dimethylethyl)-4- hydroxyphenyljthio]- 1 -methylethyl]thio]-2,6-bis( 1 , 1 -dimethylethyl)phenyl ester (12.5 g, 20.75 mmol) was dissolved in DMF (150 ml) and sulfur trioxide trimethylamine complex (12.5 g, 87.5 mmol) was added. The mixture was sti ⁇ ed at room temperature overnight. It was evaporated and the residue was dissolved in dichloromethane (100 ml), washed with water (2 x 50 ml).
- the present invention also includes the use of compounds of the formulas (I) and (II) in inhibiting the peroxidation of LDL lipid and in inhibiting the progression of atherosclerosis in patients in need thereof.
- patient refers to warm-blooded animals or mammals, and in particular humans, who are in need of the therapy described herein.
- HEPG2 cell was started in 10ml of MEM, 10% FBS, ImM Sodium Pyruvate. The cells were incubated in a tissue culture incubator. The cells were split into 4X96- wells plate in MEM, 10% FBS, ImM Sodium Pyruvate and allowed to grow to about
- the cells were treated with the desired concentration of compounds in 100 ⁇ l DMEM, 1%) RSA for 24 hours.
- the compounds are dissolved in DMSO.
- the range of concentration is lOuM - 40uM, with each concentration being done in triples.
- the coated plate is washed 3 times with IXPBS, pH 7.4, -0.05% Tween 20.
- IXPBS pH 7.4, -0.05% Tween 20
- Boehringer Mannheim Incubate at room temperature for 1 hour, rocking gently. Wash the coated plate 3X with IXPBS, pH 7.4, -0.05% Tween 20
- Example 63 VCAM-1 Assay Splitting the cells Two to four confluent PI 50 plates are trypsinized and the cells transfe ⁇ ed to a
- Water soluble compounds Compounds are initially screened at 50 ⁇ M and 10 ⁇ M. A 50 mM stock solution for each compound is prepared in culture medium. The stock solution is diluted to 5 mM and 1 mM. When 10 ⁇ L of the 5 mM solution is added to the well (1 mL medium/well), the final concentration will be 50 ⁇ M. Adding 10 ⁇ L of the 1 mM solution to the well will give a final concentration of 10 ⁇ M. Water insoluble compounds
- the compounds are added to the plate (each compound is done in duplicate). One plate is done for VCAM expression and one plate is done for ICAM expression.
- TNF is added to each well. 100 units/mL TNF is usually added to each well. Since each lot of TNF varies in the number of units, each new lot is titrated to determine the optimum concentration. Therefore this concentration will change. If 100 units/mL is bing used, dilute the TNF to 10 units/ ⁇ L and add 10 ⁇ L to each well.
- the plates are incubated at 37°C, 5% CO 2 overnight (approximately 16 hours). The next day the plates are checked under the microscope to see if there are any visual signs of toxicity. Records are made of any cell death, debris, or morphology changes, as well as insoluble compounds (particulate or turbity).
- the media 500 ⁇ L is saved and frozenat -70°C. Wash cells once with roughly 1 ml/well of Hanks Balance Salt Solution (HBSS) or PBS. Gently empty the wash solution and then tap the plate onto paper towels. Add either 250 ⁇ L/well of HBSS +5% FCCS to the plank (no primary antibody wells) or 250 ⁇ L/well of primary antibody diluted in HBSS +5% FCS. Incubate for 30 minutes at 37°C. Wash the wells twice with .5 mL/well HBSS or PBS and gently tap the plates onto paper towels after the last wash.
- HBSS Hanks Balance Salt Solution
- Substrate solution is made immediately prior to use and contains: water 10 mL
- TMB stock solution To 10 mg TMB, add 1 mL acetone. Store at 4°C protected from light.
- VCAM-1 Ab stock .1 ⁇ g/ ⁇ L final concentration 0.25 ⁇ g/mL
- ICAM-1 Ab stock .1 ⁇ g/ ⁇ L final concentration 0.25 ⁇ g/mL
- Butanedioic acid mono 5 6 23 65% at 15
- Butanedioic acid 40% at 100 mono[4-[[l-[[3,5- b ⁇ s( 1 , 1 -dimethylethyl)- 4- hydroxyphenyl]thio)- 1 -methylethyl ⁇ th ⁇ o-2,6- b ⁇ s(l ,l - d ⁇ methylethyl)phenyl methyl ester
- Pentanedioic acid (1 - NE 25 methylethyhdene)b ⁇ s(th ⁇ o ⁇ 2,6-b ⁇ s(l,l- d ⁇ methylethyl)-4, 1 - phenylene)] ester
- Pentanedioic acid 25 70% at 15 mono[4-[[l-[[3,5- b ⁇ s(l , l-d ⁇ methylethyl)-4- hydroxypheny 1] thio] - 1 - methylethyl]th ⁇ o]-2,6- b ⁇ s(l,l- d ⁇ methylethyl)phenyl] ester
- Butanedioic acid (1- NE 25 methylethyl ⁇ d ⁇ ne)b ⁇ s[th ⁇ o[2,6-b ⁇ (l,l- d ⁇ methylethyl)-4, 1 - phenylene ⁇ ⁇ ester,
- Mammals, and specifically humans, suffering from any of the above-described conditions can be treated by the topical, systemic or transdermal administration of a composition comprising an effective amount of the compound of formula (I) or formula (II) or a pharmaceutically acceptable salt thereof, optionally in a pharmaceutically acceptable carrier or diluent.
- the composition is administered subcutaneously, intravenously, intraperitoneally, intramuscularly, parenterally, orally, submucosally, by inhalation, transdermally via a slow release patch, or topically, in an effective dosage range to treat the target condition.
- An effective dose can be readily determined by the use of conventional techniques and by observing results obtained under analogous circumstances. In determining the effective dose, a number of factors are considered including, but not limited to: the species of patient; its size, age, and general health; the specific disease involved; the degree of involvement or the severity of the disease; the response of the individual patient; the particular compound administered; the mode of administration; the bioavailability characteristics of the preparation administered; the dose regimen selected; and the use of concomitant medication.
- Typical systemic dosages for all of the herein described conditions are those ranging from 0.1 mg/kg to 500 mg/kg of body weight per day as a single daily dose or divided daily doses. Prefe ⁇ ed dosages for the described conditions range from 5-1500 mg per day. A more particularly prefe ⁇ ed dosage for the desired conditions ranges from 25- 750 mg per day. Typical dosages for topical application are those ranging from 0.001 to 100%) by weight of the active compound.
- the compound is administered for a sufficient time period to alleviate the undesired symptoms and the clinical signs associated with the condition being treated.
- the active compound is included in the pharmaceutically acceptable carrier or diluent in an amount sufficient to deliver to a patient a therapeutic amount of compound in vivo in the absence of serious toxic effects.
- the concentration of active compound in the drug composition will depend on absorption, inactivation, and excretion rates of the drug as well as other factors known to those of skill in the art. It is to be noted that dosage values will also vary with the severity of the condition to be alleviated. It is to be further understood that for any particular subject, specific dosage regimens should be adjusted over time according to the individual need and the professional judgment of the person administering or supervising the administration of the compositions, and that the dosage ranges set forth herein are exemplary only and are not intended to limit the scope or practice of the claimed composition.
- the active ingredient may be administered at once, or may be divided into a number of smaller doses to be administered at varying intervals of time.
- a prefe ⁇ ed mode of administration of the active compound for systemic delivery is oral.
- Oral compositions will generally include an inert diluent or an edible carrier. They may be enclosed in gelatin capsules or compressed into tablets.
- the active compound can be incorporated with excipients and used in the form of tablets, troches, or capsules. Pharmaceutically compatible binding agents, and/or adjuvant materials can be included as part of the composition.
- the tablets, pills, capsules, troches and the like can contain any of the following ingredients, or compounds of a similar nature: a binder such as microcrystalline cellulose, gum tragacanth or gelatin; an excipient such as starch or lactose, a disintegrating agent such as alginic acid, Primogel, or corn starch; a lubricant such as magnesium stearate or Sterotes; a glidant such as colloidal silicon dioxide; a sweetening agent such as sucrose or saccharin; or a flavoring agent such as peppermint, methyl salicylate, or orange flavoring.
- a binder such as microcrystalline cellulose, gum tragacanth or gelatin
- an excipient such as starch or lactose, a disintegrating agent such as alginic acid, Primogel, or corn starch
- a lubricant such as magnesium stearate or Sterotes
- a glidant such as colloidal silicon dioxide
- dosage unit form When the dosage unit form is a capsule, it can contain, in addition to material of the above type, a liquid carrier such as a fatty oil.
- dosage unit forms can contain various other materials which modify the physical form of the dosage unit, for example, coatings of sugar, shellac, or other enteric agents.
- the compound or its salts can be administered as a component of an elixir, suspension, syrup, wafer, chewing gum or the like.
- a syrup may contain, in addition to the active compounds, sucrose as a sweetening agent and certain preservatives, dyes and colorings and flavors.
- the compound can also be mixed with other active materials which do not impair the desired action, or with materials that supplement the desired action.
- the active compounds can be administered in conjunction with other medications used in the treatment of cardiovascular disease, including lipid lowering agents such as probucol and nicotinic acid; platelet aggregation inhibitors such as aspirin; antithrombotic agents such as coumadin; calcium channel blockers such as varapamil, diltiazem, and nifedipine; angiotensin converting enzyme (ACE) inhibitors such as captopril and enalopril, and ⁇ -blockers such as propanalol, terbutalol, and labetalol.
- lipid lowering agents such as probucol and nicotinic acid
- platelet aggregation inhibitors such as aspirin
- antithrombotic agents such as coumadin
- calcium channel blockers such as varapamil, diltiazem, and nifedipine
- angiotensin converting enzyme (ACE) inhibitors such as captopril and enalopril
- the compounds can also be administered in combination with nonsteroidal antiinflammatories such as ibuprofen, indomethacin, fenoprofen, mefenamic acid, flufenamic acid, sulindac.
- nonsteroidal antiinflammatories such as ibuprofen, indomethacin, fenoprofen, mefenamic acid, flufenamic acid, sulindac.
- the compound can also be administered with corticosteriods.
- Solutions or suspensions used for parenteral, intradermal, subcutaneous, or topical application can include the following components: a sterile diluent such as water for injection, saline solution, fixed oils, polyethylene glycols, glycerine, propylene glycol or other synthetic solvents; antibacterial agents such as benzyl alcohol or methyl parabens; antioxidants such as ascorbic acid or sodium bisulfite; chelating agents such as ethylenediaminetetraacetic acid; buffers such as acetates, citrates or phosphates and agents for the adjustment of tonicity such as sodium chloride or dextrose. pH can be adjusted with acids or bases, such as hydrochloric acid or sodium hydroxide.
- the parenteral preparation can be enclosed in ampoules, disposable syringes or multiple dose vials made of glass or plastic.
- prefe ⁇ ed carriers are physiological saline, bacteriostatic water, Cremophor ELTM (BASF, Parsippany, NJ) or phosphate buffered saline (PBS).
- physiological saline bacteriostatic water
- Cremophor ELTM BASF, Parsippany, NJ
- PBS phosphate buffered saline
- the active compounds are prepared with carriers that will protect the compound against rapid elimination from the body, such as a controlled release formulation, including implants and microencapsulated delivery systems.
- a controlled release formulation including implants and microencapsulated delivery systems.
- Biodegradable, biocompatible polymers can be used, such as ethylene vinyl acetate, polyanhydrides, polyglycolic acid, collagen, polyorthoesters, and polylactic acid. Methods for preparation of such formulations will be apparent to those skilled in the art. The materials can also be obtained commercially from Alza Corporation and Nova Pharmaceuticals, Inc.
- Liposomal suspensions including liposomes targeted to infected cells with monoclonal antibodies to viral antigens
- liposome formulations may be prepared by dissolving appropriate lipid(s) (such as stearoyl phosphatidyl ethanolamine, stearoyl phosphatidyl choline, arachadoyl phosphatidyl choline, and cholesterol) in an inorganic solvent that is then evaporated, leaving behind a thin film of dried lipid on the surface of the container. An aqueous solution of the compound is then introduced into the container. The container is then swirled by hand to free lipid material from the sides of the container and to disperse lipid aggregates, thereby forming the liposomal suspension.
- appropriate lipid(s) such as stearoyl phosphatidyl ethanolamine, stearoyl phosphatidyl choline, arachadoyl phosphatidyl choline, and cholesterol
- Suitable vehicles or carriers for topical application can be prepared by conventional techniques, such as lotions, suspensions, ointments, creams, gels, tinctures, sprays, powders, pastes, slow-release transdermal patches, suppositories for application to rectal, vaginal, nasal or oral mucosa.
- thickening Oagents, emollients, and stabilizers can be used to prepare topical compositions.
- thickening agents include petrolatum, beeswax, xanthan gum, or polyethylene, humectants such as sorbitol, emollients such as mineral oil, lanolin and its derivatives, or squalene.
Abstract
Description



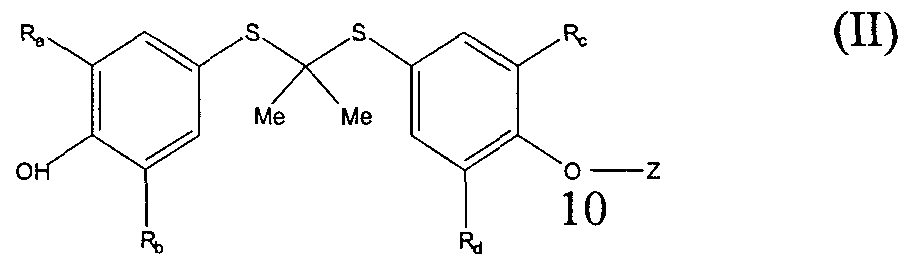







Claims






Priority Applications (23)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002289851A CA2289851C (en) | 1997-05-14 | 1998-05-14 | Compounds and methods for the inhibition of the expression of vcam-1 |
| PL343904A PL207885B1 (en) | 1997-05-14 | 1998-05-14 | Compounds and methods for the inhibition of the expression of vcam-1 |
| IL15707898A IL157078A (en) | 1997-05-14 | 1998-05-14 | Compounds and pharmaceutical compositions for the inhibition of the expression of vcam-1 |
| SI9830885T SI1464639T1 (en) | 1997-05-14 | 1998-05-14 | Succinic acid ester of probucol for the inhibition of the expression of VCAM-1 |
| IL15707798A IL157077A (en) | 1997-05-14 | 1998-05-14 | Compounds and pharmaceutical compositions for the inhibition of the expression of vcam-1 |
| SK1532-99A SK286392B6 (en) | 1997-05-14 | 1998-05-14 | Succinic acid ester of probucol for the inhibition of the expression of VCAM-1 and use for treatment cardiovascular and inflammatory diseases |
| IL13279798A IL132797A0 (en) | 1997-05-14 | 1998-05-14 | Compounds and methods for the inhibition of the expression of vcam-1 |
| DE69829966T DE69829966T2 (en) | 1997-05-14 | 1998-05-14 | A MONOETHER OF PROBUCOL AND METHODS FOR INHIBITING THE VCAM-1 EXPRESSION |
| BR9809819-5A BR9809819A (en) | 1997-05-14 | 1998-05-14 | Compounds and methods for inhibiting vcam-1 expression |
| SK50038-2007A SK286766B6 (en) | 1997-05-14 | 1998-05-14 | Composition and their use for treatment cardiovascular and inflammatory diseases |
| DK98922264T DK0994853T3 (en) | 1998-05-14 | 1998-05-14 | Monoether of probucol and methods for inhibiting expression of VCAM-1 |
| AT98922264T ATE294158T1 (en) | 1997-05-14 | 1998-05-14 | A MONOETHER OF PROBUCOL AND METHODS OF INHIBITING VCAM-1 EXPRESSION |
| HU0004592A HUP0004592A3 (en) | 1997-05-14 | 1998-05-14 | Compounds and methods for the inhibition of the expression of vcam-1 |
| EP98922264A EP0994853B1 (en) | 1997-05-14 | 1998-05-14 | A monoether of probucol and methods for the inhibition of the expression of vcam-1 |
| EA199901026A EA009370B1 (en) | 1997-05-14 | 1998-05-14 | Compound and pharmaceutical composition for the inhibition of the expression of vcam-1 |
| KR1019997010458A KR100882335B1 (en) | 1997-05-14 | 1998-05-14 | Compounds and methods for the inhibition of the expression of vcam-1 |
| AU74851/98A AU750041B2 (en) | 1997-05-14 | 1998-05-14 | Compounds and methods for the inhibition of the expression of VCAM-1 |
| IL16456898A IL164568A0 (en) | 1997-05-14 | 1998-05-14 | Compounds and methods for the inhibition of the expression of vcam-1 |
| JP54950298A JP3930056B2 (en) | 1997-05-14 | 1998-05-14 | Compounds and methods for inhibiting VCAM-1 expression |
| NO19995544A NO316221B1 (en) | 1997-05-14 | 1999-11-12 | Compounds for Inhibition of Expression of VCAM-1, Pharmaceutical Composition Including Same and Use of Same for Preparation of the Drug |
| HK00105042A HK1025947A1 (en) | 1997-05-14 | 2000-08-14 | A monoether of probucol and methods for the inhibition of the expression of vcam-1. |
| NO20032254A NO319855B1 (en) | 1997-05-14 | 2003-05-19 | Use of Compounds for the Preparation of Drug for Inhibition of Expression of VCAM-1. |
| IL164568A IL164568A (en) | 1997-05-14 | 2004-10-14 | Compounds and pharmaceutical compositions for treating diseases mediated by vcam-1 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US4702097P | 1997-05-14 | 1997-05-14 | |
| US60/047,020 | 1997-05-14 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO1998051662A2 true WO1998051662A2 (en) | 1998-11-19 |
| WO1998051662A3 WO1998051662A3 (en) | 2000-03-02 |
Family
ID=21946634
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1998/009773 WO1998051289A2 (en) | 1997-05-14 | 1998-05-14 | Monoesters of probucol for the treatment of cardiovascular and inflammatory disease |
| PCT/US1998/009781 WO1998051662A2 (en) | 1997-05-14 | 1998-05-14 | Compounds and methods for the inhibition of the expression of vcam-1 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1998/009773 WO1998051289A2 (en) | 1997-05-14 | 1998-05-14 | Monoesters of probucol for the treatment of cardiovascular and inflammatory disease |
Country Status (26)
| Country | Link |
|---|---|
| US (9) | US6147250A (en) |
| EP (2) | EP0994853B1 (en) |
| JP (4) | JP3930056B2 (en) |
| KR (5) | KR100882335B1 (en) |
| CN (7) | CN1977836A (en) |
| AT (3) | ATE356113T1 (en) |
| AU (2) | AU750041B2 (en) |
| BR (2) | BR9809819A (en) |
| CA (3) | CA2289851C (en) |
| CY (1) | CY1107645T1 (en) |
| CZ (4) | CZ301183B6 (en) |
| DE (3) | DE69829966T2 (en) |
| DK (2) | DK1464639T3 (en) |
| EA (5) | EA010183B1 (en) |
| ES (3) | ES2241139T3 (en) |
| HK (2) | HK1024629A1 (en) |
| HU (2) | HUP0004592A3 (en) |
| ID (2) | ID23877A (en) |
| IL (7) | IL164568A0 (en) |
| NO (3) | NO316221B1 (en) |
| NZ (2) | NZ501069A (en) |
| PL (2) | PL194329B1 (en) |
| PT (1) | PT1464639E (en) |
| SK (4) | SK286392B6 (en) |
| TR (2) | TR199902803T2 (en) |
| WO (2) | WO1998051289A2 (en) |
Cited By (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6121319A (en) * | 1997-05-14 | 2000-09-19 | Atherogenics, Inc. | Monoesters of probucol for the treatment of cardiovascular and inflammatory disease |
| WO2001070757A2 (en) * | 2000-03-21 | 2001-09-27 | Atherogenics, Inc. | Thioketals and thioethers for inhibiting the expression of vcam-1 |
| WO2001077072A2 (en) * | 2000-04-11 | 2001-10-18 | Atherogenics, Inc. | Compounds and methods to increase plasma hdl cholesterol levels and improve hdl functionality |
| US6323359B1 (en) * | 2000-05-02 | 2001-11-27 | Salsbury Chemicals, Inc. | Process for preparing probucol derivatives |
| WO2001098291A2 (en) * | 2000-06-20 | 2001-12-27 | Atherogenics, Inc. | 1,3-bis-(substituted-phenyl)-2-propen-1-ones and their use to treat vcam-1 mediated disorders |
| WO2002087556A2 (en) * | 2001-04-11 | 2002-11-07 | Atherogenics, Inc. | Probucol monoesters and their use to increase plasma hdl cholesterol levels and improve hdl functionality |
| WO2003039352A2 (en) * | 2001-11-09 | 2003-05-15 | Atherogenics, Inc. | Methods of reversing and preventing cardiovascular pathologies |
| US6852878B2 (en) | 1998-05-14 | 2005-02-08 | Atherogenics, Inc. | Thioketals and thioethers for inhibiting the expression of VCAM-1 |
| WO2005011594A2 (en) * | 2003-07-31 | 2005-02-10 | Epigenesis Pharmaceuticals Llc | Combination of dehydroepiandrosterone or dehydroepiandrosterone-sulfate with a tyrosine kinase inhibitor, delta opioid receptor antagonist, neurokinin receptor antagonist, or vcam inhibitor for treatment of asthma or chronic obstructive pulmonary disease |
| US6887712B1 (en) | 1998-11-09 | 2005-05-03 | Atherogenics, Inc. | Methods and compositions to lower plasma cholesterol levels |
| JP2005514344A (en) * | 2001-10-25 | 2005-05-19 | アセロジエニクス・インコーポレイテツド | Compounds and methods for transplant rejection therapy |
| WO2005051900A1 (en) * | 2003-11-25 | 2005-06-09 | Novo Nordisk A/S | Novel compounds for the treatment of obesity |
| WO2006063408A1 (en) * | 2004-12-17 | 2006-06-22 | Stocker Ronald O | Compositions and methods for treating cardiovascular disorders |
| US7071158B2 (en) | 1997-07-01 | 2006-07-04 | Atherogenics, Inc. | Antioxidant enhancement of therapy for hyperproliferative conditions |
| US7078431B2 (en) | 2000-06-20 | 2006-07-18 | Atherogenics, Inc. | 1,3-bis-(substituted-phenyl)-2-propen-1-ones and their use to treat VCAM-1 mediated disorders |
| JP2006188525A (en) * | 1999-05-28 | 2006-07-20 | Abbott Lab | Cell proliferation inhibitor |
| US7087645B2 (en) | 1997-05-14 | 2006-08-08 | Atherogenics, Inc. | Compounds and methods for treating transplant rejection |
| US7094801B2 (en) | 2001-12-19 | 2006-08-22 | Atherogenics, Inc. | Chalcone derivatives and their use to treat diseases |
| US7166605B2 (en) * | 2002-03-22 | 2007-01-23 | Nicox, S.A. | Probucol nitro-derivatives |
| US7173129B2 (en) | 2003-06-06 | 2007-02-06 | Athero Genics, Inc. | Sulfonamide-substituted chalcone derivatives and their use to treat diseases |
| US7196089B2 (en) | 2003-01-29 | 2007-03-27 | Asterand Uk Limited | EP4 receptor antagonists |
| US7271274B2 (en) | 2004-04-20 | 2007-09-18 | Ahterogenics, Inc. | Phenolic antioxidants for the treatment of disorders including arthritis, asthma and coronary artery disease |
| US7273948B2 (en) * | 2003-01-13 | 2007-09-25 | Atherogenics, Inc. | Process of preparing esters and ethers of probucol and derivatives thereof |
| US7294737B2 (en) | 2004-04-20 | 2007-11-13 | Atherogenics, Inc. | Process of preparing esters and ethers of probucol and derivatives thereof |
| US7417068B2 (en) | 2003-10-16 | 2008-08-26 | Asterand Uk Limited | EP4 receptor antagonists |
| WO2008118948A1 (en) | 2007-03-26 | 2008-10-02 | Atherogenics, Inc. | Methods and compositions of derivatives of probucol for the treatment of diabetes |
| US7897776B2 (en) | 2007-04-23 | 2011-03-01 | Salutria Pharmaceuticals Llc | Sulfonamide containing compounds for treatment of inflammatory disorders |
| US10882828B2 (en) | 2009-03-18 | 2021-01-05 | Resverlogix Corp. | Anti-inflammatory agents |
| US11649220B2 (en) | 2018-01-30 | 2023-05-16 | Demotech.Inc. | Probucol derivative, preparation method therefor and use thereof |
Families Citing this family (104)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001517617A (en) * | 1997-09-24 | 2001-10-09 | ノヴァ モレキュラー インク. | Methods for increasing APOE levels for the treatment of neurodegenerative diseases |
| US7361684B2 (en) * | 1999-06-28 | 2008-04-22 | Massachusetts Institute Of Technology | Screening of compounds for treatment of atherosclerosis and heart attack |
| US6403637B1 (en) | 1999-08-09 | 2002-06-11 | Univ Saint Louis | Methods of modulating matrix metalloproteinase activity and uses thereof |
| US6670355B2 (en) * | 2000-06-16 | 2003-12-30 | Wyeth | Method of treating cardiovascular disease |
| AUPQ872800A0 (en) * | 2000-07-12 | 2000-08-03 | Heart Research Institute, The | Compositions and methods for treating cardiovascular disorders |
| US6982251B2 (en) * | 2000-12-20 | 2006-01-03 | Schering Corporation | Substituted 2-azetidinones useful as hypocholesterolemic agents |
| EP1911462A3 (en) | 2001-01-26 | 2011-11-30 | Schering Corporation | Compositions comprising a sterol absorption inhibitor |
| US6939554B2 (en) * | 2002-02-05 | 2005-09-06 | Michigan Biotechnology Institute | Antimicrobial polymer |
| US7208467B2 (en) | 2002-06-07 | 2007-04-24 | Monty Krieger | Lipid-altering compositions for the treatment of infertility |
| US6960683B2 (en) * | 2002-07-12 | 2005-11-01 | Atherogenics, Inc. | Salt forms of poorly soluble probucol esters and ethers |
| US20050163821A1 (en) * | 2002-08-02 | 2005-07-28 | Hsing-Wen Sung | Drug-eluting Biodegradable Stent and Delivery Means |
| US20040110803A1 (en) * | 2002-09-13 | 2004-06-10 | Hossein Dovlatabadi | Methods and compositions for the use of D-malic acid to decrease serum triglyceride, cholesterol and lipoprotein levels |
| AU2003288925A1 (en) * | 2002-10-08 | 2004-05-04 | Massachusetts Institute Of Technology | Compounds for modulation of cholesterol transport |
| AU2002953533A0 (en) * | 2002-12-24 | 2003-01-16 | Arthron Limited | Fc receptor modulating compounds and compositions |
| ATE406364T1 (en) | 2003-03-07 | 2008-09-15 | Schering Corp | SUBSTITUTED AZETIDINONE DERIVATIVES, THEIR PHARMACEUTICAL FORMULATIONS AND THEIR USE IN THE TREATMENT OF HYPERCHOLESTEROLEMIA |
| DE602004018617D1 (en) | 2003-03-07 | 2009-02-05 | Schering Corp | SUBSTITUTED AZETIDINONE DERIVATIVES, THEIR PHARMACEUTICAL FORMULATIONS AND THEIR USE FOR THE TREATMENT OF HYPERCHOLESTEROLMIA |
| SI1603553T1 (en) * | 2003-03-17 | 2012-03-30 | Japan Tobacco Inc | Pharmaceutical compositions of cetp inhibitors |
| WO2005025492A2 (en) * | 2003-07-07 | 2005-03-24 | Emory University | Novel compositions, pharmaceutical compositions, and methods for the treatment and prevention of heart disease |
| US20050074443A1 (en) * | 2003-10-03 | 2005-04-07 | Treadwell Benjamin V. | Methods of attenuating autoimmune disease and compositions useful therefor |
| JP2007509054A (en) * | 2003-10-17 | 2007-04-12 | アミリン・ファーマシューティカルズ,インコーポレイテッド | Silylphenol promotes vascular health |
| EP1918000A2 (en) | 2003-11-05 | 2008-05-07 | Schering Corporation | Combinations of lipid modulating agents and substituted azetidinones and treatments for vascular conditions |
| US7744889B2 (en) * | 2003-11-14 | 2010-06-29 | Temple University - Of The Commonwealth System Of Higher Education | Alpha, beta-unsaturated sulfoxides for treating proliferative disorders |
| US20060025481A1 (en) * | 2004-02-09 | 2006-02-02 | Strange Matthew L | Process for preparation of probucol derivatives and polymorphic forms thereof |
| US7294736B2 (en) * | 2004-04-09 | 2007-11-13 | Cambrex Charles City, Inc. | Process for preparation of probucol derivatives |
| US20050282883A1 (en) * | 2004-04-29 | 2005-12-22 | John Griffin | Compositions and treatments for inhibiting kinase and/or HMG-CoA reductase |
| US7199126B2 (en) * | 2004-04-29 | 2007-04-03 | Pharmix Corporation | Compositions and treatments for inhibiting kinase and/or HMG-CoA reductase |
| US7183285B2 (en) * | 2004-04-29 | 2007-02-27 | Pharmix Corp. | Compositions and treatments for inhibiting kinase and/or HMG-CoA reductase |
| US20050272770A1 (en) * | 2004-04-29 | 2005-12-08 | John Griffin | Compositions and treatments for inhibiting kinase and/or HMG-CoA reductase |
| US20060063828A1 (en) * | 2004-06-28 | 2006-03-23 | Weingarten M D | 1,2-Bis-(substituted-phenyl)-2-propen-1-ones and pharmaceutical compositions thereof |
| EP1768660A4 (en) * | 2004-07-01 | 2009-05-13 | Atherogenics Inc | Compounds and methods for treating diabetic vascular diseases |
| US20060069080A1 (en) * | 2004-09-29 | 2006-03-30 | Veltri Enrico P | Combinations of substituted azetidinones and CB1 antagonists |
| US20060111436A1 (en) * | 2004-11-23 | 2006-05-25 | John Griffin | Compositions and treatments for modulating kinase and/or HMG-CoA reductase |
| NZ555320A (en) | 2004-12-03 | 2010-11-26 | Schering Corp | Substituted piperazines as CB1 antagonists |
| US7345191B2 (en) * | 2005-02-26 | 2008-03-18 | Cambrex Charles City, Inc. | Process for preparation of probucol derivatives |
| KR20080007373A (en) | 2005-04-21 | 2008-01-18 | 아테로제닉스, 인코포레이티드 | Process for the separation of probucol derivatives |
| US20070015779A1 (en) * | 2005-04-29 | 2007-01-18 | John Griffin | Compositions and treatments for inhibiting kinase and/or hmg-coa reductase |
| US7737155B2 (en) | 2005-05-17 | 2010-06-15 | Schering Corporation | Nitrogen-containing heterocyclic compounds and methods of use thereof |
| US20060269579A1 (en) * | 2005-05-25 | 2006-11-30 | Musculoskeletal Research Llc | Compositions for treating osteoarthritis |
| US7767710B2 (en) | 2005-05-25 | 2010-08-03 | Calosyn Pharma, Inc. | Method for treating osteoarthritis |
| JP2009528266A (en) | 2006-01-18 | 2009-08-06 | シェーリング コーポレイション | Cannabinoid receptor modifier |
| WO2007142581A1 (en) * | 2006-06-07 | 2007-12-13 | Astrazeneca Ab | Combination product for the treatment or prevention of dyslipidaemia |
| WO2008118946A1 (en) * | 2007-03-27 | 2008-10-02 | Atherogenics, Inc. | Methods and compositions using certain phenolic derivatives for the treatment of diabetes |
| WO2008130616A2 (en) * | 2007-04-19 | 2008-10-30 | Schering Corporation | Diaryl morpholines as cb1 modulators |
| JP2010531354A (en) * | 2007-06-26 | 2010-09-24 | アストラゼネカ アクチボラグ | Method for isolating a probucol derivative monosubstituted with a carboxy group |
| KR20100051625A (en) * | 2007-06-28 | 2010-05-17 | 인터벳 인터내셔널 비.브이. | Substituted piperazines as cb1 antagonists |
| CN101790521A (en) * | 2007-06-28 | 2010-07-28 | 英特维特国际股份有限公司 | Substituted-piperazinyl as the CB1 antagonist |
| WO2009042854A1 (en) * | 2007-09-26 | 2009-04-02 | Musculoskeletal Research Llc | Ion-channel regulator compositions and methods of using same |
| BRPI0817907B8 (en) | 2007-10-02 | 2021-06-22 | Lamodel Ltd | apparatus for administering a substance to an individual |
| US10420880B2 (en) | 2007-10-02 | 2019-09-24 | West Pharma. Services IL, Ltd. | Key for securing components of a drug delivery system during assembly and/or transport and methods of using same |
| US9345836B2 (en) | 2007-10-02 | 2016-05-24 | Medimop Medical Projects Ltd. | Disengagement resistant telescoping assembly and unidirectional method of assembly for such |
| US9656019B2 (en) | 2007-10-02 | 2017-05-23 | Medimop Medical Projects Ltd. | Apparatuses for securing components of a drug delivery system during transport and methods of using same |
| US7967795B1 (en) | 2010-01-19 | 2011-06-28 | Lamodel Ltd. | Cartridge interface assembly with driving plunger |
| AU2008317057B8 (en) | 2007-10-22 | 2014-02-13 | Merck Sharp & Dohme Corp. | Bicyclic Heterocycle Derivatives and their use as modulators of the activity of GPR119 |
| CN103251631A (en) * | 2008-05-13 | 2013-08-21 | 根梅迪卡治疗公司 | Salicylate conjugates useful for treating metabolic disorders |
| EP2297117B1 (en) | 2008-05-19 | 2012-10-31 | Merck Sharp & Dohme Corp. | Bicyclic heterocycle derivatives and use thereof as gpr119 modulators |
| EP2318404B1 (en) | 2008-07-16 | 2013-08-21 | Merck Sharp & Dohme Corp. | Bicyclic heterocycle derivatives and use thereof as gpr119 modulators |
| US9393369B2 (en) | 2008-09-15 | 2016-07-19 | Medimop Medical Projects Ltd. | Stabilized pen injector |
| US20100145305A1 (en) * | 2008-11-10 | 2010-06-10 | Ruth Alon | Low volume accurate injector |
| EP2379547A1 (en) | 2008-12-16 | 2011-10-26 | Schering Corporation | Pyridopyrimidine derivatives and methods of use thereof |
| WO2010075069A1 (en) | 2008-12-16 | 2010-07-01 | Schering Corporation | Bicyclic pyranone derivatives as nicotinic acid receptor agonists |
| WO2010075273A1 (en) | 2008-12-23 | 2010-07-01 | Schering Corporation | Bicyclic heterocycle derivatives and methods of use thereof |
| US8722882B2 (en) | 2008-12-23 | 2014-05-13 | Merck Sharp & Dohme Corp. | Pyrimidine derivatives as GPCR modulators for use in the treatment of obesity and diabetes |
| CA2747809A1 (en) | 2008-12-23 | 2010-07-01 | Joel M. Harris | Bicyclic heterocycle derivatives and methods of use thereof |
| US8152779B2 (en) * | 2008-12-30 | 2012-04-10 | Medimop Medical Projects Ltd. | Needle assembly for drug pump |
| WO2010106083A1 (en) * | 2009-03-16 | 2010-09-23 | Genmedica Therapeutics Sl | Combination therapies for treating metabolic disorders |
| JP2012520342A (en) * | 2009-03-16 | 2012-09-06 | ジェンメディカ・セラピューティックス・ソシエダッド・リミターダ | Anti-inflammatory and antioxidant conjugates useful for treating metabolic disorders |
| WO2010114957A1 (en) | 2009-04-03 | 2010-10-07 | Schering Corporation | Bicyclic piperidine and piperazine derivatives as gpcr modulators for the treatment of obesity, diabetes and other metabolic disorders |
| AR076024A1 (en) | 2009-04-03 | 2011-05-11 | Schering Corp | DERIVATIVES OF BRIDGED BICYCLIC HETEROCICLES AND METHODS OF USE OF THE SAME |
| HUE030253T2 (en) | 2009-05-18 | 2017-04-28 | Telomerase Activation Sciences Inc | Compositions and methods for increasing telomerase activity |
| US10071196B2 (en) | 2012-05-15 | 2018-09-11 | West Pharma. Services IL, Ltd. | Method for selectively powering a battery-operated drug-delivery device and device therefor |
| US8157769B2 (en) | 2009-09-15 | 2012-04-17 | Medimop Medical Projects Ltd. | Cartridge insertion assembly for drug delivery system |
| US10071198B2 (en) | 2012-11-02 | 2018-09-11 | West Pharma. Servicees IL, Ltd. | Adhesive structure for medical device |
| EP2493307B1 (en) | 2009-10-29 | 2016-04-27 | Merck Sharp & Dohme Corp. | Bridged bicyclic piperidine derivatives and methods of use thereof |
| US20120232073A1 (en) | 2009-11-23 | 2012-09-13 | Santhosh Francis Neelamkavil | Fused bicyclic pyrimidine derivatives and methods of use thereof |
| EP2503891B1 (en) | 2009-11-23 | 2016-08-03 | Merck Sharp & Dohme Corp. | Pyrimidine ether derivatives and methods of use thereof |
| EP2503887B1 (en) | 2009-11-24 | 2016-01-06 | Merck Sharp & Dohme Corp. | Substituted biaryl derivatives and methods of use thereof |
| US8348898B2 (en) | 2010-01-19 | 2013-01-08 | Medimop Medical Projects Ltd. | Automatic needle for drug pump |
| WO2011141907A1 (en) | 2010-05-10 | 2011-11-17 | Medimop Medical Projects Ltd. | Low volume accurate injector |
| US20130156720A1 (en) | 2010-08-27 | 2013-06-20 | Ironwood Pharmaceuticals, Inc. | Compositions and methods for treating or preventing metabolic syndrome and related diseases and disorders |
| US8466197B2 (en) | 2010-12-14 | 2013-06-18 | Genmedica Therapeutics Sl | Thiocarbonates as anti-inflammatory and antioxidant compounds useful for treating metabolic disorders |
| USD702834S1 (en) | 2011-03-22 | 2014-04-15 | Medimop Medical Projects Ltd. | Cartridge for use in injection device |
| US9072827B2 (en) | 2012-03-26 | 2015-07-07 | Medimop Medical Projects Ltd. | Fail safe point protector for needle safety flap |
| US9421323B2 (en) | 2013-01-03 | 2016-08-23 | Medimop Medical Projects Ltd. | Door and doorstop for portable one use drug delivery apparatus |
| KR20230050355A (en) | 2013-02-28 | 2023-04-14 | 프레지던트 앤드 펠로우즈 오브 하바드 칼리지 | Methods and compositions for mobilizing stem cells |
| US9011164B2 (en) | 2013-04-30 | 2015-04-21 | Medimop Medical Projects Ltd. | Clip contact for easy installation of printed circuit board PCB |
| US10293120B2 (en) | 2015-04-10 | 2019-05-21 | West Pharma. Services IL, Ltd. | Redundant injection device status indication |
| US10149943B2 (en) | 2015-05-29 | 2018-12-11 | West Pharma. Services IL, Ltd. | Linear rotation stabilizer for a telescoping syringe stopper driverdriving assembly |
| WO2016196934A1 (en) | 2015-06-04 | 2016-12-08 | Medimop Medical Projects Ltd. | Cartridge insertion for drug delivery device |
| US10576207B2 (en) | 2015-10-09 | 2020-03-03 | West Pharma. Services IL, Ltd. | Angled syringe patch injector |
| US9987432B2 (en) | 2015-09-22 | 2018-06-05 | West Pharma. Services IL, Ltd. | Rotation resistant friction adapter for plunger driver of drug delivery device |
| US10086145B2 (en) | 2015-09-22 | 2018-10-02 | West Pharma Services Il, Ltd. | Rotation resistant friction adapter for plunger driver of drug delivery device |
| CN112972833B (en) | 2015-10-09 | 2024-01-09 | 西医药服务以色列分公司 | Syringe needle cap remover |
| CN109219456B (en) | 2016-01-21 | 2020-05-15 | 西医药服务以色列有限公司 | Force containment in autoinjectors |
| CN109310816B (en) | 2016-01-21 | 2020-04-21 | 西医药服务以色列有限公司 | Needle insertion and retraction mechanism |
| JP6885960B2 (en) | 2016-01-21 | 2021-06-16 | ウェスト ファーマ サービシーズ イスラエル リミテッド | Drug delivery device with visual indicators |
| WO2017161076A1 (en) | 2016-03-16 | 2017-09-21 | Medimop Medical Projects Ltd. | Staged telescopic screw assembly having different visual indicators |
| US10376647B2 (en) | 2016-03-18 | 2019-08-13 | West Pharma. Services IL, Ltd. | Anti-rotation mechanism for telescopic screw assembly |
| JP6957525B2 (en) | 2016-06-02 | 2021-11-02 | ウェスト ファーマ サービシーズ イスラエル リミテッド | Needle evacuation by 3 positions |
| US9650332B1 (en) * | 2016-06-16 | 2017-05-16 | Yong Xu | Prodrug of probucol and method for preparing the same |
| EP3490635B1 (en) | 2016-08-01 | 2021-11-17 | West Pharma. Services Il, Ltd. | Partial door closure prevention spring |
| CN109562229B (en) | 2016-08-01 | 2021-07-13 | 西医药服务以色列有限公司 | Anti-rotation medicine barrel pin |
| CN106905208B (en) * | 2017-02-27 | 2018-09-07 | 江西瑞雅药业有限公司 | Probucol prodrug and preparation method thereof and pharmaceutical composition |
| CN110869072B (en) | 2017-05-30 | 2021-12-10 | 西部制药服务有限公司(以色列) | Modular drive mechanism for a wearable injector |
| US11857767B2 (en) | 2017-12-22 | 2024-01-02 | West Pharma. Services IL, Ltd. | Injector usable with different dimension cartridges |
Citations (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3179701A (en) * | 1962-05-14 | 1965-04-20 | Shell Oil Co | (3, 5-dialkyl-4-hydroxyphenyl) (3, 5-dialkyl-4-hydroxybenzyl) sulfides |
| GB1136539A (en) * | 1966-12-13 | 1968-12-11 | Uniroyal Inc | Phenolic sulphides |
| GB1148550A (en) * | 1967-12-02 | 1969-04-16 | Uniroyal Inc | Substituted benzylphenyl sulfides and their use as antioxidants |
| GB1199871A (en) * | 1967-05-11 | 1970-07-22 | Consolidation Coal Co | Improvements in or relating to Sulfur-Containing Bisphenols |
| DE2716125A1 (en) * | 1976-04-12 | 1977-10-27 | Yoshitomi Pharmaceutical | 3,5-DI-TERT.-BUTYL-4-HYDROXYPHENYLPYRIDINE COMPOUNDS, METHOD FOR THEIR MANUFACTURING AND PHARMACEUTICAL PREPARATIONS CONTAINING THEM |
| US4115590A (en) * | 1964-02-26 | 1978-09-19 | Ethyl Corporation | Binuclear phenols for reducing plasma lipid levels |
| EP0190682A2 (en) * | 1985-02-04 | 1986-08-13 | G.D. Searle & Co. | Novel disubstituted 4-hydroxyphenylthio anilides |
| US4755524A (en) * | 1986-01-31 | 1988-07-05 | G. D. Searle & Co. | Novel phenolic thioethers as inhibitors of 5-lipoxygenase |
| EP0292660A2 (en) * | 1987-03-17 | 1988-11-30 | Merrell Dow Pharmaceuticals Inc. | Alkylidenedithiobis (substituted) phenols for inhibiting interleukin-1 release and for alleviating interleukin-1 mediated conditions |
| EP0317165A1 (en) * | 1987-11-13 | 1989-05-24 | Riker Laboratories, Inc. | DI-t-butylphenols substituted by an alkoxy or benzyloxy group or a benzylthio group |
| EP0348203A1 (en) * | 1988-06-24 | 1989-12-27 | SHIONOGI SEIYAKU KABUSHIKI KAISHA trading under the name of SHIONOGI & CO. LTD. | Phenolic thioethers, their production and use |
| EP0405788A2 (en) * | 1989-06-29 | 1991-01-02 | SHIONOGI SEIYAKU KABUSHIKI KAISHA trading under the name of SHIONOGI & CO. LTD. | Di-tert-butyl(hydroxy)phenylthio substituted hydroxamic acid derivatives |
| US5262439A (en) * | 1992-04-30 | 1993-11-16 | The Regents Of The University Of California | Soluble analogs of probucol |
| EP0621255A1 (en) * | 1993-04-20 | 1994-10-26 | Adir Et Compagnie | Substituted phenoxy-isobutyric acids and esters |
| WO1995030415A1 (en) * | 1994-05-10 | 1995-11-16 | Emory University | Treatment for atherosclerosis and other cardiovascular and inflammatory diseases |
| WO1996012703A1 (en) * | 1994-10-25 | 1996-05-02 | G.D. Searle & Co. | Heteroalkyl and heteroarylthioalkyl thiophenolic compounds as 5-lipoxygenase inhibitors |
| WO1998022418A1 (en) * | 1996-11-20 | 1998-05-28 | Hoechst Marion Roussel, Inc. | Substituted phenols and thiophenols useful as antioxidant agents |
Family Cites Families (77)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3485843A (en) * | 1967-05-11 | 1969-12-23 | Dow Chemical Co | Piperazine adducts of ketone mercaptoles |
| US3479407A (en) * | 1968-11-06 | 1969-11-18 | Consolidation Coal Co | Sulfurization of 2,6-di-tert-butylphenol |
| US3576883A (en) * | 1969-06-30 | 1971-04-27 | Consolidation Coal Co | Alkylidenedithiobisphenols |
| DE2104524C3 (en) | 1970-09-16 | 1980-08-07 | Veb Kombinat Umformtechnik Herbert Warnke Erfurt, Ddr 5000 Erfurt | Hydraulic overload protection |
| FR2130975A5 (en) * | 1971-03-29 | 1972-11-10 | Aries Robert | Bis(4-(phenoxyalkanoyloxy)-phenylthio)alkanes - hypolipemics hypocholesterolemics |
| FR2133024A5 (en) * | 1971-04-06 | 1972-11-24 | Aries Robert | Bis-(4-nicotinoyloxyphenylthio) propanes - with hypocholesterolaemic and hypolipaemic activity |
| FR2134810A5 (en) * | 1971-04-21 | 1972-12-08 | Aries Robert | Bis-(3-alkyl-5-t-alkyl-4-(thiazole-5-carboxy)phenylthio) alcanes - - with hypocholesterolaemic and hypolipaemic activity |
| FR2140769A5 (en) * | 1971-06-07 | 1973-01-19 | Aries Robert | Benzofuryloxy alkanoic derivs of probucol - hypocholesterolemic and hypolipemic agents |
| FR2140771A5 (en) * | 1971-06-07 | 1973-01-19 | Aries Robert | Tetralinyl phenoxy alkanoic esters - of bis hydroxyphenylthio alkanes, hypolipemics etc |
| FR2168137A1 (en) * | 1972-01-17 | 1973-08-31 | Dynachim Sarl | Bis 4-hydroxyphenylthioalkane esters - with hypocholesterolaemic and hypolipaemic activities |
| JPS4975552A (en) * | 1972-11-20 | 1974-07-20 | ||
| US3952064A (en) * | 1973-03-12 | 1976-04-20 | Crown Zellerbach Corporation | Process for producing mercaptophenols |
| US4029812A (en) * | 1976-02-18 | 1977-06-14 | The Dow Chemical Company | Novel hypolipidemic 2-(3,5-di-tert-butyl-4-hydroxyphenyl)thio carboxamides |
| US4968514A (en) * | 1984-12-11 | 1990-11-06 | Forbes Polytech, Inc. | Beer bottle with fully reacted thermoplastic polyurethane crown capliner |
| US4679520A (en) | 1985-06-10 | 1987-07-14 | Harken Olaf T | Mainsail reefing and furling device and method |
| DE3530256A1 (en) | 1985-08-23 | 1987-02-26 | Merrell Dow Pharma | USE OF PROBUCOL IN THE TREATMENT OF HEART ARRYTHMY |
| DE3625279A1 (en) | 1986-07-25 | 1988-02-04 | Merrell Dow Pharma | USE OF PROBUCOL FOR THE PREVENTION AND TREATMENT OF HEART DISEASES |
| US4975467A (en) | 1987-03-17 | 1990-12-04 | Merrell Dow Pharmaceuticals Inc. | Method of inhibiting interleukin-1 release and alleviating interleukin-1 mediated conditions |
| US4752616A (en) * | 1987-06-29 | 1988-06-21 | E. R. Squibb & Sons, Inc. | Arylthioalkylphenyl carboxylic acids, compositions containing same and method of use |
| US4968710A (en) | 1987-11-13 | 1990-11-06 | Riker Laboratories, Inc. | Substituted di-t-butylphenols and anti-allergic use thereof |
| US5066822A (en) | 1987-11-13 | 1991-11-19 | Riker Laboratories, Inc | Di-t-butylphenols substituted by an alkoxy or benzyloxy group or a benzylthio group |
| CH675422A5 (en) | 1988-03-31 | 1990-09-28 | Symphar Sa | |
| JP2627003B2 (en) * | 1989-01-25 | 1997-07-02 | 塩野義製薬株式会社 | G-tert-butylhydroxyphenylthio derivative |
| US5527945A (en) | 1989-02-10 | 1996-06-18 | Basf Aktiengesellschaft | Diphenylheteroalkyl derivatives, the preparation thereof and drugs and cosmetics prepared therefrom |
| CA2017956A1 (en) * | 1989-07-06 | 1991-01-06 | Werner Bollag | Use of retinoids |
| DE3929913A1 (en) | 1989-09-08 | 1991-04-04 | Hoechst Ag | 4-HYDROXYTETRAHYDROPYRAN-2-ONE AND THE CORRESPONDING DIHYDROXYCARBONSAEUREDERIVATES, SALTS AND ESTERS, PROCESS FOR THEIR PREPARATION, THEIR USE AS A MEDICAMENT, PHARMACEUTICAL PREPARATES AND PREPARED PRODUCTS |
| US5115250A (en) * | 1990-01-12 | 1992-05-19 | Hewlett-Packard Company | Wiper for ink-jet printhead |
| US5112870A (en) | 1990-05-09 | 1992-05-12 | Merrell Dow Pharmaceuticals Inc. | Bis(alkyl-substituted-4-hydroxyphenylthio)alkane analogs as inhibitors of cataractogenesis |
| US5061734A (en) | 1990-05-09 | 1991-10-29 | Merrell Dow Pharmaceuticals Inc. | Bis(alkyl-substituted-4-hydroxyphenylthio)alkane analogs as inhibitors of cataractogenesis |
| US5298497A (en) * | 1990-05-15 | 1994-03-29 | E. R. Squibb & Sons, Inc. | Method for preventing onset of hypertension employing a cholesterol lowering drug |
| US5155250A (en) * | 1990-07-05 | 1992-10-13 | Merrell Dow Pharmaceuticals Inc. | 2,6-di-alkyl-4-silyl-phenols as antiatheroscerotic agents |
| US5085777A (en) * | 1990-08-31 | 1992-02-04 | E. I. Du Pont De Nemours And Company | Reverse osmosis membranes of polyamideurethane |
| FR2666583B1 (en) * | 1990-09-06 | 1994-09-09 | Adir | NOVEL DERIVATIVES OF SPIRO [4.5] DECANE, THEIR PREPARATION PROCESS AND THEIR PHARMACEUTICAL COMPOSITIONS CONTAINING THEM. |
| RU2024509C1 (en) * | 1991-05-07 | 1994-12-15 | Всероссийский научный центр по безопасности биологически активных веществ | Derivatives of 9-aminoacridine or their salts with organic or inorganic acids showing psychotropic, antiamnestic and lipid-regulating activity |
| GB9115951D0 (en) | 1991-07-24 | 1991-09-11 | Pfizer Ltd | Indoles |
| SK281016B6 (en) | 1992-02-05 | 2000-10-09 | Boehringer Ingelheim Kg | Amidine derivatives, method of their preparation, their use and pharmaceuticals them containing |
| US5310949A (en) | 1992-09-02 | 1994-05-10 | Merck & Co., Inc. | Cholesterol lowering compounds |
| GB9220571D0 (en) | 1992-09-30 | 1992-11-11 | Ici Plc | Quinazoline derivatives |
| US5821260A (en) * | 1992-10-30 | 1998-10-13 | Emory University | Treatment for atherosclerosis and other cardiovascular and inflammatory diseases |
| US5380747A (en) * | 1992-10-30 | 1995-01-10 | Emory University | Treatment for atherosclerosis and other cardiovascular and inflammatory diseases |
| US5662934A (en) * | 1993-01-05 | 1997-09-02 | Najarian; Thomas | Compositions and methods for lowering cholesterol while maintaining antioxidant levels |
| US5585235A (en) | 1993-04-13 | 1996-12-17 | Diagnescent Technologies, Inc. | Fluorescent assay and method that corrects for spectral interference |
| JPH06312978A (en) * | 1993-04-30 | 1994-11-08 | Japan Tobacco Inc | New phthalimide derivative having inhibiting activity against phospholipase a2 |
| US5411741A (en) | 1993-07-29 | 1995-05-02 | Zaias; Nardo | Method and composition for skin depigmentation |
| GB9320113D0 (en) | 1993-09-29 | 1993-11-17 | Zeneca Ltd | Tricyclic derivatives |
| ES2263094T3 (en) * | 1993-12-10 | 2006-12-01 | Aventis Inc. | USE OF 2,6-DIALQUIL-4-SILIL-PHENOLS TO TREAT XANTOMA. |
| US5426196A (en) | 1993-12-22 | 1995-06-20 | Glaxo Inc. | Synthesis of diaryl methanes |
| JPH07328425A (en) * | 1994-06-03 | 1995-12-19 | Toshiba Corp | Plasma chemical reaction device |
| CA2153553A1 (en) | 1994-07-13 | 1996-01-14 | Hidekazu Suzuki | Stable lipid emulsion |
| US5792787A (en) * | 1995-06-07 | 1998-08-11 | Emory University | Treatment for atherosclerosis and other cardiovascular and inflammatory diseases |
| JPH0959258A (en) * | 1995-08-11 | 1997-03-04 | Ono Pharmaceut Co Ltd | Guanidyl derivative |
| FR2738817B1 (en) * | 1995-09-14 | 1997-10-17 | Adir | NOVEL SUBSTITUTED ALKANOIC 2,2-DIMETHYL-OMEGA-PHENOXY ACIDS AND ESTERS, THEIR PREPARATION PROCESS AND THE PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| WO1997015546A1 (en) * | 1995-10-26 | 1997-05-01 | Nippon Shinyaku Co., Ltd. | Carboxylic acid derivatives and pharmaceutical compositions |
| KR100443893B1 (en) | 1996-01-15 | 2004-10-15 | 얀센 파마슈티카 엔.브이. | Angiogenesis inhibitor pyridazine amine |
| US5608095A (en) * | 1996-04-30 | 1997-03-04 | Hoechst Marion Roussel, Inc. | Alkyl-4-silyl-phenols and esters thereof as antiatherosclerotic agents |
| HUP9904567A3 (en) | 1996-06-20 | 2001-10-29 | Univ Texas | Use of azo, thioalkyl, thiocarbonyl derivatives substituted by fused heterocycles and/or phenyl group for the preparation of pharmaceutical compositions stimulating bone growth |
| WO1998030255A2 (en) | 1997-01-09 | 1998-07-16 | Localmed, Inc. | Localized intravascular delivery of antioxidant substances for inhibition of restenosis in recanalized blood vessels |
| GB9705502D0 (en) | 1997-03-17 | 1997-05-07 | Univ Wales Swansea The | Chlorination or aromatic compounds and catalysts therefor |
| AU6715098A (en) | 1997-03-24 | 1998-10-20 | Gilles Cote | Vascular remodeling agent |
| US6852878B2 (en) * | 1998-05-14 | 2005-02-08 | Atherogenics, Inc. | Thioketals and thioethers for inhibiting the expression of VCAM-1 |
| US6670398B2 (en) * | 1997-05-14 | 2003-12-30 | Atherogenics, Inc. | Compounds and methods for treating transplant rejection |
| AU750041B2 (en) * | 1997-05-14 | 2002-07-11 | Atherogenics, Inc. | Compounds and methods for the inhibition of the expression of VCAM-1 |
| KR20010020611A (en) * | 1997-07-01 | 2001-03-15 | 아테로제닉스, 인코포레이티드 | Antioxidant enhancement of therapy for hyperproliferative conditions |
| JP2001517617A (en) | 1997-09-24 | 2001-10-09 | ノヴァ モレキュラー インク. | Methods for increasing APOE levels for the treatment of neurodegenerative diseases |
| WO1999024400A1 (en) | 1997-11-10 | 1999-05-20 | Vyrex Corporation | Probucol esters and uses thereof |
| FR2785284B1 (en) | 1998-11-02 | 2000-12-01 | Galderma Res & Dev | VITAMIN D ANALOGS |
| DE19850532A1 (en) | 1998-11-03 | 2000-05-04 | Nematel Dr Rudolf Eidenschink | Bisphenylthio compounds |
| WO2000028332A1 (en) * | 1998-11-09 | 2000-05-18 | Atherogenics, Inc. | Methods and compositions to lower plasma cholesterol levels |
| EP1133483B1 (en) | 1998-11-23 | 2003-04-23 | Janssen Pharmaceutica N.V. | Il-5 inhibiting 6-azauracil derivatives |
| KR100979664B1 (en) | 1999-03-10 | 2010-09-02 | 유니버시티 오브 피츠버그 오브 더 커먼웰쓰 시스템 오브 하이어 에듀케이션 | Adipose-derived stem cells and lattices |
| BR0009507A (en) | 1999-03-30 | 2002-01-15 | Novartis Ag | Phthalazine derivatives for the treatment of inflammatory diseases |
| CA2403490A1 (en) | 2000-03-21 | 2001-10-25 | Atherogenics, Inc. | N-substituted dithiocarbamates for the treatment of biological disorders |
| EP1289944A2 (en) * | 2000-03-21 | 2003-03-12 | Atherogenics, Inc. | Thioketals and thioethers for inhibiting the expression of vcam-1 |
| US6323359B1 (en) | 2000-05-02 | 2001-11-27 | Salsbury Chemicals, Inc. | Process for preparing probucol derivatives |
| US20030064967A1 (en) * | 2001-04-11 | 2003-04-03 | Jayraz Luchoomun | Methods to increase plasma HDL cholesterol levels and improve HDL functionality with probucol monoesters |
| JP4017988B2 (en) | 2001-05-14 | 2007-12-05 | オムノバ ソリューソンズ インコーポレーティッド | Polymer surfactant derived from cyclic monomer having pendant fluorinated carbon group |
| US7187870B2 (en) * | 2003-10-15 | 2007-03-06 | Oewaves, Inc. | Tunable balanced opto-electronic filters and applications in opto-electronic oscillators |
-
1998
- 1998-05-14 AU AU74851/98A patent/AU750041B2/en not_active Ceased
- 1998-05-14 EA EA199901027A patent/EA010183B1/en not_active IP Right Cessation
- 1998-05-14 JP JP54950298A patent/JP3930056B2/en not_active Expired - Lifetime
- 1998-05-14 CN CNA2006101016892A patent/CN1977836A/en active Pending
- 1998-05-14 PL PL98336788A patent/PL194329B1/en not_active IP Right Cessation
- 1998-05-14 CA CA002289851A patent/CA2289851C/en not_active Expired - Fee Related
- 1998-05-14 EA EA200800375A patent/EA200800375A1/en unknown
- 1998-05-14 SK SK1532-99A patent/SK286392B6/en not_active IP Right Cessation
- 1998-05-14 CN CNB031530672A patent/CN1275596C/en not_active Expired - Fee Related
- 1998-05-14 BR BR9809819-5A patent/BR9809819A/en not_active Application Discontinuation
- 1998-05-14 US US09/079,213 patent/US6147250A/en not_active Expired - Lifetime
- 1998-05-14 CA CA002428130A patent/CA2428130A1/en not_active Abandoned
- 1998-05-14 NZ NZ501069A patent/NZ501069A/en unknown
- 1998-05-14 SK SK50038-2007A patent/SK286766B6/en not_active IP Right Cessation
- 1998-05-14 EP EP98922264A patent/EP0994853B1/en not_active Expired - Lifetime
- 1998-05-14 DE DE69829966T patent/DE69829966T2/en not_active Expired - Fee Related
- 1998-05-14 DK DK04075141T patent/DK1464639T3/en active
- 1998-05-14 EA EA199901026A patent/EA009370B1/en not_active IP Right Cessation
- 1998-05-14 AT AT04075141T patent/ATE356113T1/en active
- 1998-05-14 TR TR1999/02803T patent/TR199902803T2/en unknown
- 1998-05-14 HU HU0004592A patent/HUP0004592A3/en unknown
- 1998-05-14 ID IDW991584A patent/ID23877A/en unknown
- 1998-05-14 PT PT04075141T patent/PT1464639E/en unknown
- 1998-05-14 BR BR9809793-8A patent/BR9809793A/en not_active Application Discontinuation
- 1998-05-14 SK SK1531-99A patent/SK285695B6/en not_active IP Right Cessation
- 1998-05-14 KR KR1019997010458A patent/KR100882335B1/en not_active IP Right Cessation
- 1998-05-14 CN CNB988071711A patent/CN1200704C/en not_active Expired - Fee Related
- 1998-05-14 IL IL16456898A patent/IL164568A0/en unknown
- 1998-05-14 CN CN2006101018440A patent/CN1977837B/en not_active Expired - Fee Related
- 1998-05-14 US US09/078,935 patent/US6121319A/en not_active Expired - Lifetime
- 1998-05-14 AT AT98923411T patent/ATE304350T1/en not_active IP Right Cessation
- 1998-05-14 KR KR1020067027007A patent/KR20070008725A/en not_active Application Discontinuation
- 1998-05-14 WO PCT/US1998/009773 patent/WO1998051289A2/en active Application Filing
- 1998-05-14 DK DK98923411T patent/DK0981343T3/en active
- 1998-05-14 AU AU75711/98A patent/AU747801C/en not_active Ceased
- 1998-05-14 EA EA200601059A patent/EA012847B1/en not_active IP Right Cessation
- 1998-05-14 PL PL343904A patent/PL207885B1/en not_active IP Right Cessation
- 1998-05-14 NZ NZ528906A patent/NZ528906A/en not_active IP Right Cessation
- 1998-05-14 CZ CZ0402399A patent/CZ301183B6/en not_active IP Right Cessation
- 1998-05-14 CN CNB031530664A patent/CN1287783C/en not_active Expired - Fee Related
- 1998-05-14 CN CNA2007101939040A patent/CN101284808A/en active Pending
- 1998-05-14 IL IL13279898A patent/IL132798A0/en active IP Right Grant
- 1998-05-14 CZ CZ20060388A patent/CZ301313B6/en not_active IP Right Cessation
- 1998-05-14 AT AT98922264T patent/ATE294158T1/en not_active IP Right Cessation
- 1998-05-14 HU HU0004230A patent/HU226611B1/en not_active IP Right Cessation
- 1998-05-14 DE DE69837295T patent/DE69837295T2/en not_active Expired - Lifetime
- 1998-05-14 DE DE69831566T patent/DE69831566T2/en not_active Expired - Fee Related
- 1998-05-14 KR KR1019997010459A patent/KR20010012504A/en not_active Application Discontinuation
- 1998-05-14 EP EP98923411A patent/EP0981343B1/en not_active Expired - Lifetime
- 1998-05-14 JP JP54949898A patent/JP2001524986A/en not_active Withdrawn
- 1998-05-14 CZ CZ0402499A patent/CZ301302B6/en not_active IP Right Cessation
- 1998-05-14 WO PCT/US1998/009781 patent/WO1998051662A2/en active Application Filing
- 1998-05-14 TR TR1999/02802T patent/TR199902802T2/en unknown
- 1998-05-14 CA CA002292388A patent/CA2292388C/en not_active Expired - Fee Related
- 1998-05-14 ES ES98922264T patent/ES2241139T3/en not_active Expired - Lifetime
- 1998-05-14 CZ CZ20090366A patent/CZ301985B6/en not_active IP Right Cessation
- 1998-05-14 KR KR1020087015303A patent/KR100953990B1/en not_active IP Right Cessation
- 1998-05-14 ES ES04075141T patent/ES2283933T3/en not_active Expired - Lifetime
- 1998-05-14 SK SK5028-2006A patent/SK286674B6/en not_active IP Right Cessation
- 1998-05-14 EA EA200500249A patent/EA009987B1/en not_active IP Right Cessation
- 1998-05-14 ID IDW991582A patent/ID29158A/en unknown
- 1998-05-14 IL IL13279798A patent/IL132797A0/en not_active IP Right Cessation
- 1998-05-14 CN CNB98807169XA patent/CN100453530C/en not_active Expired - Fee Related
- 1998-05-14 ES ES98923411T patent/ES2248901T3/en not_active Expired - Lifetime
-
1999
- 1999-08-06 US US09/370,046 patent/US6548699B1/en not_active Expired - Fee Related
- 1999-11-08 IL IL132798A patent/IL132798A/en not_active IP Right Cessation
- 1999-11-12 NO NO19995544A patent/NO316221B1/en not_active IP Right Cessation
- 1999-11-12 NO NO19995543A patent/NO327603B1/en not_active IP Right Cessation
-
2000
- 2000-06-29 HK HK00103938A patent/HK1024629A1/en not_active IP Right Cessation
- 2000-08-14 HK HK00105042A patent/HK1025947A1/en not_active IP Right Cessation
-
2002
- 2002-01-30 US US10/060,734 patent/US6617352B2/en not_active Expired - Fee Related
- 2002-04-02 US US10/114,351 patent/US7375252B2/en not_active Expired - Fee Related
- 2002-04-02 US US10/115,206 patent/US6828447B2/en not_active Expired - Fee Related
- 2002-04-02 US US10/114,346 patent/US6602914B2/en not_active Expired - Fee Related
-
2003
- 2003-05-19 NO NO20032254A patent/NO319855B1/en not_active IP Right Cessation
- 2003-08-25 US US10/647,766 patent/US7189870B2/en not_active Expired - Fee Related
-
2004
- 2004-10-14 IL IL164568A patent/IL164568A/en not_active IP Right Cessation
-
2006
- 2006-04-19 JP JP2006115258A patent/JP2006232848A/en active Pending
- 2006-04-20 JP JP2006116322A patent/JP2006265257A/en active Pending
- 2006-04-24 IL IL175130A patent/IL175130A/en not_active IP Right Cessation
- 2006-09-13 IL IL178072A patent/IL178072A/en not_active IP Right Cessation
- 2006-12-19 KR KR1020067026799A patent/KR100919883B1/en not_active IP Right Cessation
-
2007
- 2007-06-04 CY CY20071100732T patent/CY1107645T1/en unknown
-
2008
- 2008-05-15 US US12/121,464 patent/US20080214660A1/en not_active Abandoned
Patent Citations (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3179701A (en) * | 1962-05-14 | 1965-04-20 | Shell Oil Co | (3, 5-dialkyl-4-hydroxyphenyl) (3, 5-dialkyl-4-hydroxybenzyl) sulfides |
| US4115590A (en) * | 1964-02-26 | 1978-09-19 | Ethyl Corporation | Binuclear phenols for reducing plasma lipid levels |
| GB1136539A (en) * | 1966-12-13 | 1968-12-11 | Uniroyal Inc | Phenolic sulphides |
| GB1199871A (en) * | 1967-05-11 | 1970-07-22 | Consolidation Coal Co | Improvements in or relating to Sulfur-Containing Bisphenols |
| GB1148550A (en) * | 1967-12-02 | 1969-04-16 | Uniroyal Inc | Substituted benzylphenyl sulfides and their use as antioxidants |
| DE2716125A1 (en) * | 1976-04-12 | 1977-10-27 | Yoshitomi Pharmaceutical | 3,5-DI-TERT.-BUTYL-4-HYDROXYPHENYLPYRIDINE COMPOUNDS, METHOD FOR THEIR MANUFACTURING AND PHARMACEUTICAL PREPARATIONS CONTAINING THEM |
| EP0190682A2 (en) * | 1985-02-04 | 1986-08-13 | G.D. Searle & Co. | Novel disubstituted 4-hydroxyphenylthio anilides |
| US4755524A (en) * | 1986-01-31 | 1988-07-05 | G. D. Searle & Co. | Novel phenolic thioethers as inhibitors of 5-lipoxygenase |
| EP0292660A2 (en) * | 1987-03-17 | 1988-11-30 | Merrell Dow Pharmaceuticals Inc. | Alkylidenedithiobis (substituted) phenols for inhibiting interleukin-1 release and for alleviating interleukin-1 mediated conditions |
| EP0317165A1 (en) * | 1987-11-13 | 1989-05-24 | Riker Laboratories, Inc. | DI-t-butylphenols substituted by an alkoxy or benzyloxy group or a benzylthio group |
| EP0348203A1 (en) * | 1988-06-24 | 1989-12-27 | SHIONOGI SEIYAKU KABUSHIKI KAISHA trading under the name of SHIONOGI & CO. LTD. | Phenolic thioethers, their production and use |
| EP0405788A2 (en) * | 1989-06-29 | 1991-01-02 | SHIONOGI SEIYAKU KABUSHIKI KAISHA trading under the name of SHIONOGI & CO. LTD. | Di-tert-butyl(hydroxy)phenylthio substituted hydroxamic acid derivatives |
| US5262439A (en) * | 1992-04-30 | 1993-11-16 | The Regents Of The University Of California | Soluble analogs of probucol |
| EP0621255A1 (en) * | 1993-04-20 | 1994-10-26 | Adir Et Compagnie | Substituted phenoxy-isobutyric acids and esters |
| WO1995030415A1 (en) * | 1994-05-10 | 1995-11-16 | Emory University | Treatment for atherosclerosis and other cardiovascular and inflammatory diseases |
| WO1996012703A1 (en) * | 1994-10-25 | 1996-05-02 | G.D. Searle & Co. | Heteroalkyl and heteroarylthioalkyl thiophenolic compounds as 5-lipoxygenase inhibitors |
| WO1998022418A1 (en) * | 1996-11-20 | 1998-05-28 | Hoechst Marion Roussel, Inc. | Substituted phenols and thiophenols useful as antioxidant agents |
Non-Patent Citations (12)
| Title |
|---|
| DATABASE CHEMABS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US STN, accession no. 110:212254, XP002115683 -& CHEMICAL ABSTRACTS, vol. 110, no. 23, 5 June 1989 (1989-06-05) Columbus, Ohio, US; abstract no. 212254a, XP002115682 & S.D. PASTOR ET AL: PHOSPHOROUS SULFUR, vol. 37, no. 3-4, 1988, pages 117-123, * |
| DATABASE CHEMABS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US STN, accession no. 122:187387, XP002115602 -& CHEMICAL ABSTRACTS, vol. 122, no. 15, 10 April 1995 (1995-04-10) Columbus, Ohio, US; abstract no. 187387, XP002115595 & JP 06 312978 A (NIPPON TOBACCO SANGYO ET AL) 8 November 1994 (1994-11-08) * |
| DATABASE CHEMABS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US STN, accession no. 124:146082, XP002115604 & V.I. KELAREV ET AL: KHIM. GETEROTSIKL. SOEDIN, no. 5, 1995, pages 667-673, * |
| DATABASE CHEMABS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US STN, accession no. 124:8690, XP002115603 -& CHEMICAL ABSTRACTS, vol. 124, no. 1, 1 January 1996 (1996-01-01) Columbus, Ohio, US; abstract no. 8690, XP002115596 & V.I. KELAREV ET AL: KHIM. GETEROTSIKL. SOEDIN, no. 4, 1995, pages 514-517, * |
| DATABASE CHEMABS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US STN, accession no. 126:277465, XP002115599 -& CHEMICAL ABSTRACTS, vol. 126, no. 21, 26 May 1997 (1997-05-26) Columbus, Ohio, US; abstract no. 277465, XP002115593 & JP 09 059258 A (ONO PHARMACEUTICAL CO) 4 March 1997 (1997-03-04) * |
| DATABASE CHEMABS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US STN, accession no. 127:75973, XP002115597 & V.Z. LANKIN ET AL: DOKL. AKAD. NAUK, vol. 351, no. 4, 1996, pages 554-557, * |
| DATABASE CHEMABS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US STN, accession no. 82:86196, XP002115598 -& CHEMICAL ABSTRACTS, vol. 82, no. 13, 31 March 1975 (1975-03-31) Columbus, Ohio, US; abstract no. 86196, XP002115592 & JP 49 075552 A (SAGAMI CHEMICAL RESEARCH CENTER) 20 July 1974 (1974-07-20) * |
| DATABASE CHEMABS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US STN, accession no. 86:5066, XP002115601 -& CHEMICAL ABSTRACTS, vol. 86, no. 1, 3 January 1977 (1977-01-03) Columbus, Ohio, US; abstract no. 5066, XP002115594 & A.I. MEDVEDEV ET AL: TEZISY DOKL. NAUCHN. SESS. KHIM. TEKHNOL. ORG. SOEDIN. SERY SERNISTYKH NEFTEI, 13TH,1974, pages 123-124, * |
| DATABASE CHEMABS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US STN, accession no. 94:30290, XP002115600 & CH.I. MAMEDOV: MATER. NAUCHN. KONF. ASPIR. AKAD. NAUK AZ. SSR, vol. 1, 1980, pages 127-131, * |
| DATABASE CHEMABS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US STN, CAPLUS accession no. 1970:445047, XP002124423 & M.B. NEUWORTH ET AL: J. MED. CHEM., vol. 13, no. 4, 1970, pages 722-725, * |
| DATABASE CHEMABS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US STN, CAPLUS accession no. 1986:28675, XP002124424 & CHEMICAL ABSTRACTS, vol. 104, no. 5, 3 February 1986 (1986-02-03) Columbus, Ohio, US; abstract no. 28675, & P. DE MEGLIO ET AL: FARMACO, ED. SCI., vol. 40, no. 11, 1985, pages 833-844, * |
| L. COMINACINI ET AL: FREE RADICAL BIOLOGY AND MEDICINE, vol. 22, no. 1-2, 1996, pages 117-127, XP002095164 * |
Cited By (48)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7087645B2 (en) | 1997-05-14 | 2006-08-08 | Atherogenics, Inc. | Compounds and methods for treating transplant rejection |
| US6828447B2 (en) * | 1997-05-14 | 2004-12-07 | Atherogenics, Inc. | Compounds and methods for the inhibition of the expression of VCAM-1 |
| US7189870B2 (en) | 1997-05-14 | 2007-03-13 | Atherogenic, Inc. | Compounds and methods for the inhibition of the expression of VCAM-1 |
| US6121319A (en) * | 1997-05-14 | 2000-09-19 | Atherogenics, Inc. | Monoesters of probucol for the treatment of cardiovascular and inflammatory disease |
| US7071158B2 (en) | 1997-07-01 | 2006-07-04 | Atherogenics, Inc. | Antioxidant enhancement of therapy for hyperproliferative conditions |
| US6852878B2 (en) | 1998-05-14 | 2005-02-08 | Atherogenics, Inc. | Thioketals and thioethers for inhibiting the expression of VCAM-1 |
| US6887712B1 (en) | 1998-11-09 | 2005-05-03 | Atherogenics, Inc. | Methods and compositions to lower plasma cholesterol levels |
| JP2006188525A (en) * | 1999-05-28 | 2006-07-20 | Abbott Lab | Cell proliferation inhibitor |
| AU2001247651B2 (en) * | 2000-03-21 | 2007-01-04 | Atherogenics, Inc | Thioketals and thioethers for inhibiting the expression of vcam-1 |
| WO2001070757A3 (en) * | 2000-03-21 | 2002-03-14 | Atherogenics Inc | Thioketals and thioethers for inhibiting the expression of vcam-1 |
| WO2001070757A2 (en) * | 2000-03-21 | 2001-09-27 | Atherogenics, Inc. | Thioketals and thioethers for inhibiting the expression of vcam-1 |
| WO2001077072A3 (en) * | 2000-04-11 | 2002-07-18 | Atherogenics Inc | Compounds and methods to increase plasma hdl cholesterol levels and improve hdl functionality |
| WO2001077072A2 (en) * | 2000-04-11 | 2001-10-18 | Atherogenics, Inc. | Compounds and methods to increase plasma hdl cholesterol levels and improve hdl functionality |
| US7183317B2 (en) | 2000-04-11 | 2007-02-27 | Atherogenics, Inc. | Compounds and methods to increase plasma HDL cholesterol levels and improve HDL functionality |
| US6881860B2 (en) | 2000-04-11 | 2005-04-19 | Atherogenics, Inc. | Compounds and methods to increase plasma HDL cholesterol levels and improve HDL functionality |
| US6323359B1 (en) * | 2000-05-02 | 2001-11-27 | Salsbury Chemicals, Inc. | Process for preparing probucol derivatives |
| WO2001098291A3 (en) * | 2000-06-20 | 2002-05-16 | Atherogenics Inc | 1,3-bis-(substituted-phenyl)-2-propen-1-ones and their use to treat vcam-1 mediated disorders |
| WO2001098291A2 (en) * | 2000-06-20 | 2001-12-27 | Atherogenics, Inc. | 1,3-bis-(substituted-phenyl)-2-propen-1-ones and their use to treat vcam-1 mediated disorders |
| US7078431B2 (en) | 2000-06-20 | 2006-07-18 | Atherogenics, Inc. | 1,3-bis-(substituted-phenyl)-2-propen-1-ones and their use to treat VCAM-1 mediated disorders |
| WO2002087556A2 (en) * | 2001-04-11 | 2002-11-07 | Atherogenics, Inc. | Probucol monoesters and their use to increase plasma hdl cholesterol levels and improve hdl functionality |
| WO2002087556A3 (en) * | 2001-04-11 | 2003-02-06 | Atherogenics Inc | Probucol monoesters and their use to increase plasma hdl cholesterol levels and improve hdl functionality |
| JP2005514344A (en) * | 2001-10-25 | 2005-05-19 | アセロジエニクス・インコーポレイテツド | Compounds and methods for transplant rejection therapy |
| EP1451138A4 (en) * | 2001-11-09 | 2005-06-15 | Atherogenics Inc | Methods of reversing and preventing cardiovascular pathologies |
| WO2003039352A3 (en) * | 2001-11-09 | 2003-10-23 | Atherogenics Inc | Methods of reversing and preventing cardiovascular pathologies |
| WO2003039352A2 (en) * | 2001-11-09 | 2003-05-15 | Atherogenics, Inc. | Methods of reversing and preventing cardiovascular pathologies |
| US7094801B2 (en) | 2001-12-19 | 2006-08-22 | Atherogenics, Inc. | Chalcone derivatives and their use to treat diseases |
| US7166605B2 (en) * | 2002-03-22 | 2007-01-23 | Nicox, S.A. | Probucol nitro-derivatives |
| US7273948B2 (en) * | 2003-01-13 | 2007-09-25 | Atherogenics, Inc. | Process of preparing esters and ethers of probucol and derivatives thereof |
| US7622604B2 (en) | 2003-01-13 | 2009-11-24 | Salutria Pharmaceuticals Llc | Process of preparing esters and ethers of probucol and derivatives thereof |
| US7528157B2 (en) | 2003-01-29 | 2009-05-05 | Asterand Uk Limited | EP4 receptor antagonists |
| US7858644B2 (en) | 2003-01-29 | 2010-12-28 | Asterand Uk Limited | EP4 receptor antagonists |
| US7196089B2 (en) | 2003-01-29 | 2007-03-27 | Asterand Uk Limited | EP4 receptor antagonists |
| US7507754B2 (en) | 2003-01-29 | 2009-03-24 | Asterand Uk Limited | EP4 receptor antagonists |
| US7173129B2 (en) | 2003-06-06 | 2007-02-06 | Athero Genics, Inc. | Sulfonamide-substituted chalcone derivatives and their use to treat diseases |
| WO2005011594A3 (en) * | 2003-07-31 | 2007-04-12 | Epigenesis Pharmaceuticals Llc | Combination of dehydroepiandrosterone or dehydroepiandrosterone-sulfate with a tyrosine kinase inhibitor, delta opioid receptor antagonist, neurokinin receptor antagonist, or vcam inhibitor for treatment of asthma or chronic obstructive pulmonary disease |
| WO2005011594A2 (en) * | 2003-07-31 | 2005-02-10 | Epigenesis Pharmaceuticals Llc | Combination of dehydroepiandrosterone or dehydroepiandrosterone-sulfate with a tyrosine kinase inhibitor, delta opioid receptor antagonist, neurokinin receptor antagonist, or vcam inhibitor for treatment of asthma or chronic obstructive pulmonary disease |
| US7417068B2 (en) | 2003-10-16 | 2008-08-26 | Asterand Uk Limited | EP4 receptor antagonists |
| US7569602B2 (en) | 2003-10-16 | 2009-08-04 | Asterand Uk Limited | Furan derivatives as EP4 receptor antagonists |
| WO2005051900A1 (en) * | 2003-11-25 | 2005-06-09 | Novo Nordisk A/S | Novel compounds for the treatment of obesity |
| US7271274B2 (en) | 2004-04-20 | 2007-09-18 | Ahterogenics, Inc. | Phenolic antioxidants for the treatment of disorders including arthritis, asthma and coronary artery disease |
| US7294737B2 (en) | 2004-04-20 | 2007-11-13 | Atherogenics, Inc. | Process of preparing esters and ethers of probucol and derivatives thereof |
| WO2006063408A1 (en) * | 2004-12-17 | 2006-06-22 | Stocker Ronald O | Compositions and methods for treating cardiovascular disorders |
| WO2008118948A1 (en) | 2007-03-26 | 2008-10-02 | Atherogenics, Inc. | Methods and compositions of derivatives of probucol for the treatment of diabetes |
| US8252840B2 (en) | 2007-03-26 | 2012-08-28 | Salutria Pharmaceuticals Llc | Methods of derivatives of probucol for the treatment of type II diabetes |
| US7897776B2 (en) | 2007-04-23 | 2011-03-01 | Salutria Pharmaceuticals Llc | Sulfonamide containing compounds for treatment of inflammatory disorders |
| US10882828B2 (en) | 2009-03-18 | 2021-01-05 | Resverlogix Corp. | Anti-inflammatory agents |
| US11407719B2 (en) | 2009-03-18 | 2022-08-09 | Resverlogix Corp. | Anti-inflammatory agents |
| US11649220B2 (en) | 2018-01-30 | 2023-05-16 | Demotech.Inc. | Probucol derivative, preparation method therefor and use thereof |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6602914B2 (en) | Compounds and methods for the inhibition of the expression of VCAM-1 | |
| CA2562992C (en) | Probucol analogues, and use thereof as anti-inflammatory inhibitors of vcam-1 | |
| AU2006202461B2 (en) | Compositions and methods for the inhibition of the expression of VCAM-1 | |
| AU2002300328B2 (en) | Compounds and methods for the inhibition of the expression of VCAM-1 | |
| EP1468989A2 (en) | Compounds and methods for the inhibition of the expression of VCAM-1 | |
| EP1726582A2 (en) | Compounds and methods for the inhibition of the expression of vcam-1 | |
| EP1695959A1 (en) | Compouds and methods for the inhibition of the expression of VCAM-1 | |
| MXPA99010402A (en) | Compounds and methods for the inhibition of the expression of vcam-1 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 132797 Country of ref document: IL Ref document number: 98807169.X Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH GM GW HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 500841 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 153299 Country of ref document: SK |
|
| ENP | Entry into the national phase |
Ref document number: 2289851 Country of ref document: CA Ref document number: 2289851 Country of ref document: CA Kind code of ref document: A Ref document number: 1998 549502 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/1999/010402 Country of ref document: MX Ref document number: PV1999-4023 Country of ref document: CZ Ref document number: 1019997010458 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1999/02803 Country of ref document: TR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998922264 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 74851/98 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 199901026 Country of ref document: EA |
|
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH GM GW HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998922264 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1019997010458 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: PV1999-4023 Country of ref document: CZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 74851/98 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 157077 Country of ref document: IL Ref document number: 157078 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 164568 Country of ref document: IL |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1998922264 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV2006-375 Country of ref document: CZ |
|
| ENP | Entry into the national phase |
Ref country code: SK Ref document number: SK A |