WO1999010422A1 - Rheology modification of low density polyethylene - Google Patents
Rheology modification of low density polyethylene Download PDFInfo
- Publication number
- WO1999010422A1 WO1999010422A1 PCT/US1998/016214 US9816214W WO9910422A1 WO 1999010422 A1 WO1999010422 A1 WO 1999010422A1 US 9816214 W US9816214 W US 9816214W WO 9910422 A1 WO9910422 A1 WO 9910422A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polymer
- sulfonyl azide
- film
- poly
- ethylene
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G81/00—Macromolecular compounds obtained by interreacting polymers in the absence of monomers, e.g. block polymers
- C08G81/02—Macromolecular compounds obtained by interreacting polymers in the absence of monomers, e.g. block polymers at least one of the polymers being obtained by reactions involving only carbon-to-carbon unsaturated bonds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/36—Sulfur-, selenium-, or tellurium-containing compounds
- C08K5/43—Compounds containing sulfur bound to nitrogen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L23/04—Homopolymers or copolymers of ethene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L23/04—Homopolymers or copolymers of ethene
- C08L23/08—Copolymers of ethene
- C08L23/0807—Copolymers of ethene with unsaturated hydrocarbons only containing more than three carbon atoms
- C08L23/0815—Copolymers of ethene with aliphatic 1-olefins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/22—Mixtures comprising a continuous polymer matrix in which are dispersed crosslinked particles of another polymer
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L23/04—Homopolymers or copolymers of ethene
- C08L23/06—Polyethene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L23/04—Homopolymers or copolymers of ethene
- C08L23/08—Copolymers of ethene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L23/04—Homopolymers or copolymers of ethene
- C08L23/08—Copolymers of ethene
- C08L23/0807—Copolymers of ethene with unsaturated hydrocarbons only containing more than three carbon atoms
- C08L23/0838—Copolymers of ethene with aromatic monomers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L23/04—Homopolymers or copolymers of ethene
- C08L23/08—Copolymers of ethene
- C08L23/0846—Copolymers of ethene with unsaturated hydrocarbons containing other atoms than carbon or hydrogen atoms
- C08L23/0853—Vinylacetate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L23/16—Elastomeric ethene-propene or ethene-propene-diene copolymers, e.g. EPR and EPDM rubbers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2312/00—Crosslinking
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2314/00—Polymer mixtures characterised by way of preparation
- C08L2314/06—Metallocene or single site catalysts
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2666/00—Composition of polymers characterized by a further compound in the blend, being organic macromolecular compounds, natural resins, waxes or and bituminous materials, non-macromolecular organic substances, inorganic substances or characterized by their function in the composition
- C08L2666/02—Organic macromolecular compounds, natural resins, waxes or and bituminous materials
- C08L2666/04—Macromolecular compounds according to groups C08L7/00 - C08L49/00, or C08L55/00 - C08L57/00; Derivatives thereof
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/13—Hollow or container type article [e.g., tube, vase, etc.]
- Y10T428/1334—Nonself-supporting tubular film or bag [e.g., pouch, envelope, packet, etc.]
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/13—Hollow or container type article [e.g., tube, vase, etc.]
- Y10T428/1352—Polymer or resin containing [i.e., natural or synthetic]
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/13—Hollow or container type article [e.g., tube, vase, etc.]
- Y10T428/1352—Polymer or resin containing [i.e., natural or synthetic]
- Y10T428/1386—Natural or synthetic rubber or rubber-like compound containing
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/13—Hollow or container type article [e.g., tube, vase, etc.]
- Y10T428/1352—Polymer or resin containing [i.e., natural or synthetic]
- Y10T428/139—Open-ended, self-supporting conduit, cylinder, or tube-type article
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/31504—Composite [nonstructural laminate]
- Y10T428/31855—Of addition polymer from unsaturated monomers
- Y10T428/31909—Next to second addition polymer from unsaturated monomers
- Y10T428/31913—Monoolefin polymer
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/31504—Composite [nonstructural laminate]
- Y10T428/31855—Of addition polymer from unsaturated monomers
- Y10T428/31909—Next to second addition polymer from unsaturated monomers
- Y10T428/31913—Monoolefin polymer
- Y10T428/31917—Next to polyene polymer
Definitions
- This invention relates to coupling of polyolefms, more specifically coupling of polyolefms to form products suitable for films.
- the term "rheology modification” means change in melt viscosity of a polymer as determined by dynamic mechanical spectroscopy
- the melt strength increases while maintaining the high shear viscosity (that is viscosity measured at a shear of 100 rad/sec by DMS) so that a polymer exhibits more resistance to stretching during elongation of molten polymer at low shear conditions (that is viscosity measured at a shear of 0 1 rad/sec by DMS) and does not sacrifice the output at high shear conditions
- An increase in melt strength is typically observed when long chain branches or similar structures are introduced into a polymer
- Polyolefms are frequently rheology modified using nonselective chemistries involving free radicals generated for instance using peroxides or high energy radiation
- chemistries involving free radical generation at elevated temperatures also degrade the molecular weight, especially in polymers containing tertiary hydrogen such as polystyrene, polypropylene, polyethylene copolymers etc
- the reaction of polypropylene with peroxides and oentaeryth ⁇ tol t ⁇ acrylate is reported by Wang et al , Journal of Applied Polymer Science, Vol.
- US 3,058,944; 3,336,268; and 3,530,108 include the reaction of certain poly (sulfonyl azide) compounds with isotactic polypropylene or other polyolefms by nitrene insertion into C-H bonds.
- the product reported in US 3,058,944 is crosslinked.
- the product reported in US 3,530,108 is foamed and cured with cycloalkane- di (sulfonyl azide) of a given formula.
- bridged polymers because polymer chains are "bridged” with sulfonamide bridges.
- the disclosed process includes a mixing step such as milling or mixing of the sulfonylazide and polymer in solution or dispersion then a heating step where the temperature is sufficient to decompose the sulfonylazide (100°C to 225° depending on the azide decomposition temperature) .
- the starting polypropylene polymer for the claimed process has a molecular weight of at least 275,000.
- Blends taught in US 3,336,268 have up to 25 percent ethylene propylene elastomer.
- United States Patent 3,631,182 taught the use of azido formate for crosslinking polyolefms.
- United States Patent 3341418 taught the use of sulfonyl azide and azidoformate compounds to crosslink of thermoplastics material (PP (polypropylene), PS (polystyrene) , PVC (poly (vinyl chloride)) and their blends with rubbers (polyisobutene, EPM, etc. ) .
- PP polypropylene
- PS polystyrene
- PVC poly (vinyl chloride)
- Canadian patent 797,917 family member of NL 6,503,188
- teachings of Canadian patent 797,917 include rheology modification using from 0.001 to 0.075 weight percent poly (sulfonyl azide) to modify homopolymer polyethylene and its blends with, especially polyisobutylene .
- the polyethylene is referred to as linear polyethylene.
- Polyethylene having a density of 0.945 is exemplified. The product is said to be useful for thermoforming.
- polymers rheology modified rather than crosslinked that is having less than 10 percent gel as determined by xylene extraction specifically by ASTM 2765.
- medium and lower density polyethylene that is polymers having a density of from 0.94 g/cc to about 0.90 g/cc
- the polymers would desirably show a combination of processability improved over the starting material with retention or improvement of toughness, low heat seal initiation temperature, low haze, high gloss or hot tack properties characteristic of the starting material.
- LDPE low density polyethylene
- LLDPE linear low density polyethylene
- Thick polyethylene films are useful for instance as greenhouse films, mulch films, and agricultural films.
- the invention includes a process of preparing a coupled polymer characterized by heating an admixture containing (1) at least one ethylene polymer or blend of ethylene polymers having a density of at least 0.89 g/mL and less than 0.935 g/mL and a comonomer content between 0.5 and 50 weight percent of an alpha olefin having greater than 2 and less than 20 carbon atoms per molecule and (2) a coupling amount of at least one poly (sulfonyl azide) to at least the decomposition temperature of the poly(sulfonyl azide) for a period sufficient for decomposition of at least 80 weight percent of the poly (sulfonyl azide) and sufficient to result in a coupled polymer.
- the amount of poly (sulfonyl azide) is preferably from 0.01 to 5 weight percent of polymers in the admixture
- the invention also includes any composition which is the product of any of the processes of the invention and articles made from those compositions, particularly any film of any composition of the invention Additionally the invention includes a use of any composition of the invention in a process of blowing or calendaring a film. More particularly the invention includes articles of compositions of the invention which are trash bags, agricultural films, construction films, or geomembranes, grocery sacks, sealant layers, tie layers, produce bags, garment bags, shipping sacks, medical films, stretch films, shrink films, agricultural films, construction films, or stretch hooders .
- Polymers to which the practice of this invention is applicable include homopolymers and copolymers of ethylene (hereinafter ethylene polymers) with narrow and broad (including bimodal) molecular weight distribution.
- ethylene polymers ethylene polymers
- One type of preferred polymers for use in the practice of the invention are polymers prepared from ethylene in combination with other monomers polyme ⁇ zable therewith Such monomers include alpha olefms and other monomers having at least one double bond, preferably alpha olefms having greater than 2, more preferably greater than 5 carbons
- Another type of preferred polymer has long chain branches introduced in the polymerization thereof as discussed hereinafter.
- these ethylene polymers have long or short branches and therefore differ from the linearity of (high density) polyethylene homopolymer, some are referred to in the art as "LLDPE” or linear low density polyethylene or “SLEP” substantially linear ethylene polymers where the term “linear” came to be used historically to distinguish from earlier highly branched low density polyethylenes prepared using free radical polymerization
- such ethylene polymers having long chain branches have low density, that is preferably a density less than 0.935 g/mL, more preferably less than 0.93 g/mL, most preferably less than 0.92 g/mL.
- the density is at least 0.89 g/mL, more preferably at least 0.890 g/mL, most preferably at least 0.91 g/mL. It is understood by those skilled in the art that a blend of ethylene polymers, particularly a blend formed in a polymerization reactor (in reactor blend) is sometimes perceived as or referred to as an ethylene polymer.
- an ethylene polymer can be a bimodal blend of ethylene polymer components and have a density between 0.89 and 0.935 g/mL even though one or both of the components might have a density outside that range.
- the bimodal blend would, however, be considered to be preferred for the practice of this invention.
- Alpha olefins having more than 2 carbon atoms include propylene, 1-butene, 1-pentene, 1-hexene, 1-octene, 1-nonene, 1-decene, 1- unidecene, and 1-dodecene as well as 4 -methyl -l-pentene, 4-methyl-l- hexene, 5-methyl-l-hexene, and vinylcyclohexene .
- the ethylene polymers which may be rheology modified according to this invention may be any interpolymers of ethylene and at least one ⁇ -olefin. Suitable -olefins are represented by the following formula:
- Suitable ⁇ -olefins for use as comonomers in a solution, gas phase or slurry polymerization process or combinations thereof include 1-propylene, 1-butene, 1-isobutylene, l-pentene, 1- hexene, 4 -methyl -l-pentene, 1-heptene and 1-octene, as well as other monomer types such as tetrafluoroethylene, vinyl benzocyclobutane, and cycloalkenes, for example cyclopentene, cyclohexene, and cyclooctene.
- the ⁇ -olefin will be 1-butene, l-pentene, 4 -methyl -l-pentene, 1-hexene, 1-heptene, 1-octene, or mixtures thereof. More preferably, the ⁇ -olefin will be 1-hexene, 1-heptene, 1-octene, or mixtures thereof. Most preferably, the ⁇ -olefin will be 1-octene.
- the ethylene polymer rheology modified according to this invention is preferably a SLEP, that is a substantially linear ethylene polymer, defined hereinafter.
- the polyolefin is a homopolymer, copolymer, or interpolymer .
- the homo or copolymers contain ethylene repeating units.
- the comonomer content is greater than 1 weight percent as determined by 13 C NMR (carbon 13 nuclear magnetic resonance), preferably greater than 2, more preferably greater than 3, most preferably at least 5 weight percent of alpha olefin or cyclic olefin.
- the comonomer content is at least one comonomer polymerizable with ethylene, preferably less than 4 comonomers polymerizable with ethylene, more preferably less than 2 such comonomers .
- Polyolefms are formed by means within the skill in the art.
- the alpha olefin monomers and optionally other addition polymerizable monomers are polymerized under conditions within the skill in the art, Such conditions include those utilized in processes involving Ziegler- Natta catalysts such as those disclosed in U.S. Patent No. 4,076,698 (Anderson et al) ; 4,950,541 and the patents to which they refer, as well as 3,645,992 (Elston) as well as those processes utilizing metallocene and other single site catalysts such as exemplified by U.S. Patents 4,937,299 (Ewen et al . ) , 5,218,071 (Tsutsui et al . ) , 5,278,272, 5,324,800, 5,084,534, 5,405,922, 4,588,794, 5,204,419 and the processes subsequently discussed in more detail.
- Ziegler- Natta catalysts such as those disclosed in U.S. Patent No. 4,076,698 (Anderson et
- starting material polyolefins are preferably substantially linear ethylene polymers (SLEPs) .
- the substantially linear ethylene polymers (SLEPs) are homogeneous polymers having long chain branching. They are disclosed in U.S. Patent Nos. 5,272,236 and 5,278,272.
- SLEPs are available as polymers made by the InsiteTM Process and Catalyst Technology such as EngageTM polyolefin elastomers (POEs) commercially available from DuPont Dow Elastomers LLC and AffinityTM polyolefin plastomers (POPs) commercially available from The Dow Chemical Company.
- EngageTM polyolefin elastomers POEs
- POPs AffinityTM polyolefin plastomers
- SLEPs can be prepared via the solution, slurry, or gas phase, preferably solution phase, polymerization of ethylene and one or more optional ⁇ -olefin comonomers in the presence of a constrained geometry catalyst, such as is disclosed in European Patent Application 416,815-B.
- the substantially linear ethylene/ ⁇ -olefin polymers are made by a continuous process using suitable constrained geometry catalysts, preferably constrained geometry catalysts as disclosed in U.S. Patent No. 5,132,380.
- suitable constrained geometry catalysts preferably constrained geometry catalysts as disclosed in U.S. Patent No. 5,132,380.
- the monocyclopentadienyl transition metal olefin polymerization catalysts taught in USP 5,026,798, are also suitable for use in preparing the polymers of the present invention, so long as the reaction conditions are as specified below.
- Suitable cocatalysts for use herein include but are not limited to, for example, polymeric or oligomeric aluminoxanes , especially methyl aluminoxane, as well as inert, compatible, noncoordinating, ion forming compounds.
- Preferred cocatalysts are inert, noncoordinating, boron compounds.
- continuous process means a process in which reactants are continuously added and product is continuously withdrawn such that an approximation of a steady state (that is substantially constant concentration of reactants and product while carrying out the process) is achieved.
- the polymerization conditions for manufacturing the substantially linear ethylene/ ⁇ -olefin polymers of the present invention are generally those useful in the solution polymerization process, although the application of the present invention is not limited thereto. Slurry and gas phase polymerization processes are also believed to be useful, provided the proper catalysts and polymerization conditions are employed.
- substantially linear olefin polymers and copolymers can also be used in making the substantially linear olefin polymers and copolymers to be rheologically modified according to the present invention, such as those disclosed in USP 3,914,342.
- the multiple reactors can be operated in series or in parallel, with at least one constrained geometry catalyst employed in one of the reactors .
- substantially linear means that, in addition to the short chain branches attributable to homogeneous comonomer incorporation, the ethylene polymer is further characterized as having long chain branches in that the polymer backbone is substituted with an average of 0.01 to 3 long chain branches/1000 carbons.
- Preferred substantially linear polymers for use in the invention are substituted with from 0.01 long chain branch/1000 carbons to 1 long chain branch/1000 carbons, and more preferably from 0.05 long chain branch/1000 carbons to 1 long chain branch/1000 carbons .
- linear means that the polymer lacks measurable or demonstrable long chain branches, that is, the polymer is substituted with an average of less than 0.01 long chain branch/1000 carbons.
- long chain branching means a chain length longer than the short chain branch that results from the incorporation of the ⁇ -olefin (s) into the polymer backbone.
- Each long chain branch has the same comonomer distribution as the polymer backbone and can be as long as the polymer backbone to which it is attached.
- the empirical effect of the presence of long chain branching in the substantial linear ethylene/ ⁇ -olefin interpolymers used in the invention is manifested in its enhanced rheological properties which are quantified and expressed herein in terms of gas extrusion rheometry (GER) results or a combination thereof melt flow, I 10 /I 2 , increases .
- GER gas extrusion rheometry
- the presence of short chain branching of up to 6 carbon atoms in length can be determined in ethylene polymers by using 13 C nuclear magnetic resonance (NMR) spectroscopy and is quantified using the method described by Randall (Rev. Macromol . Chem. Phys . , C.29, V. 2&3, p. 285-297) .
- deGroot and Chum found that the presence of octene does not change the hydrodynamic volume of the polyethylene samples in solution and, as such, one can account for the molecular weight increase attributable to octene short chain branches by knowing the mole percent octene in the sample. By deconvoluting the contribution to molecular weight increase attributable to 1-octene short chain branches, deGroot and Chum showed that GPC-DV may be used to quantify the level of long chain branches in substantially linear ethylene/octene copolymers .
- SLEPs are further characterized as having: (a) a melt flow ratio, I 10 /I 2 > . 5.63, (b) a molecular weight distribution, M w /M n as determined by gel permeation chromatography and defined by the equation: (M w /M n ) ⁇ (I 10 /I 2 ) - 4.63, (c) a critical shear stress at the onset of gross melt fracture, as determined by gas extrusion rheometry, of greater than 4 x 10 6 dynes/cm 2 or a gas extrusion rheology such that the critical shear rate at onset of surface melt fracture for the SLEP is at least 50 percent greater than the critical shear rate at the onset of surface melt fracture for a linear ethylene polymer, the linear ethylene polymer has an I 2 , M ⁇ M,, and, preferably density, which are each within ten percent of the SLEP and wherein the respective critical shear rates of the SLEP and the linear ethylene
- the I10/I2 ratio indicates the degree of long chain branching, that is, the higher the I10/I2 ratio, the more long chain branching in the polymer.
- the I10/I2 ratio of the substantially linear ethylene/ ⁇ -ole in polymers is at least 5.63, preferably at least 7, especially at least 8 or above, and as high as 25.
- the melt index as measured by ASTM D-1238 (190°C/2.16 Kg) for the substantially linear olefin polymers useful herein is preferably at least 0.1 grams/10 minutes (g/10 min), more preferably at least 0.5 g/10 min and especially at least 1 g/10 min up to preferably 100 g/10 min, more preferably up to 50 g/10 min, and especially up to 20 g/10 min.
- Determination of the critical shear rate and critical shear stress in regards to melt fracture as well as other rheology properties such as rheological processing index (PI) is performed using a gas extrusion rheometer (GER) .
- the gas extrusion rheometer is described by M. Shida, R.N. Shroff and L.V. Cancio in Polymer Engineering Science, Vol. 17, No. 11, p. 770 (1977), and in
- the SLEPs for use in the invention includes ethylene interpolymers and have a PI in the range of 0.01 kpoise to 50 kpoise, preferably 15 kpoise or less.
- the SLEPs used herein have a PI less than or equal to 70 percent of the PI of a linear ethylene polymer (either a Ziegler polymerized polymer or a linear uniformly branched polymer as described by Elston in US Patent 3,645,992) having an I 2 , M w /M r- and density, each within ten percent of the SLEPs.
- the rheological behavior of SLEPs can also be characterized by the Dow Rheology Index (DRI), which expresses a polymer's "normalized relaxation time as the result of long chain branching.”
- DRI Dow Rheology Index
- SLEP INSITETM Technology Polyolefms
- DRI values range from 0 for polymers which do not have any measurable long chain branching (for example, TafmerTM products available from Mitsui Petrochemical Industries and ExactTM products available from Exxon Chemical Company) to 15 and are independent of melt index.
- DRI provides improved correlations to melt elasticity and high shear flowability relative to correlations of the same attempted with melt flow ratios.
- DRI is preferably at least 0.1, and especially at least 0.5, and most especially at least 0.8.
- DRI can be calculated from the equation:
- DRI (3652879 * ⁇ ⁇ ⁇ 649 / ⁇ -1)/10
- ⁇ is the characteristic relaxation time of the material and ⁇ is the zero shear viscosity of the material.
- Baseline determination of viscosity and shear rate data are obtained using a Rheometric Mechanical Spectrometer (RMS-800) under dynamic sweep mode from 0.1 to 100 radians/second at 190 C and a Gas Extrusion Rheometer (GER) at extrusion pressures from 1,000 psi to 5,000 psi (6.89 to 34.5 MPa) , which corresponds to shear stress from 0.086 to 0.43 MPa, using a 0.0754 mm diameter, 20:1 L/D die at 190 C.
- Specific material determinations can be performed from 140 to 190 C as required to accommodate melt index variations.
- the onset of surface melt fracture is defined as the loss of extrudate gloss .
- the loss of extrudate gloss is the point at which the surface roughness of the extrudate can only be detected by a 40X magnification.
- the critical shear rate at the onset of surface melt fracture for the SLEPs is at least 50 percent greater than the critical shear rate at the onset of surface melt fracture of a linear ethylene polymer having essentially the same I 2 and M w /M n .
- Gross melt fracture occurs at unsteady extrusion flow conditions and ranges in detail from regular (alternating rough and smooth, helical, etc.) to random distortions. For commercial acceptability to maximize the performance properties of films, coatings and moldings, surface defects should be minimal, if not absent.
- the critical shear stress at the onset of gross melt fracture for the SLEPs, especially those having a density >0.910 g/cc, used in the invention is greater than 4 x 10 ⁇ dynes/cm 2 .
- the critical shear rate at the onset of surface melt fracture (OSMF) and the onset of gross melt fracture (OGMF) will be used herein based on the changes of surface roughness and configurations of the extrudates extruded by a GER.
- the SLEPs used in the invention are also preferably characterized by a single DSC melting peak.
- the single melting peak is determined using a differential scanning calorimeter standardized with indium and deionized water. The method involves 3-7 mg sample sizes, a "first heat" to 180 C which is held for 4 minutes, a cool down at 10 C/min. to -30 C which is held for 3 minutes, and heat up at 10 C/min. to 140 C for the "second heat” .
- the single melting peak is taken from the "second heat” heat flow vs. temperature curve. Total heat of fusion of the polymer is calculated from the area under the curve .
- the single melting peak may show, depending on equipment sensitivity, a "shoulder or a "hump" on the low melting side that constitutes less than 12 percent, typically, less than 9 percent, and more typically less than 6 percent of the total heat of fusion of the polymer.
- Such an artifact is observable for other homogeneously branched polymers such as ExactTM resins and is discerned on the basis of the slope of the single melting peak varying monotonically through the melting region of the artifact.
- Such an artifact occurs within 34 C, typically within 27 C, and more typically within 20 C of the melting point of the single melting peak.
- the heat of fusion attributable to an artifact can be separately determined by specific integration of its associated area under the heat flow vs. temperature curve.
- the molecular weight distributions of ethylene ⁇ -olefin polymers are determined by gel permeation chromatography (GPC) on a Waters 150C high temperature chromatographic unit equipped with a differential refractometer and three columns of mixed porosity. The columns are supplied by Polymer Laboratories and are commonly packed with pore sizes of 10 ⁇ 2 and 10 '1 mm) .
- the solvent is 1, 2 , 4-trichlorobenzene, from which about 0.3 percent by weight solutions of the samples are prepared for injection.
- the flow rate is about 1.0 milliliters/minute, unit operating temperature is about 140° C and the injection size is about 100 microliters.
- the molecular weight determination with respect to the polymer backbone is deduced by using narrow molecular weight distribution polystyrene standards (from Polymer Laboratories) in conjunction with their elution volumes.
- the equivalent polyethylene molecular weights are determined by using appropriate Mark-Houwink coefficients for polyethylene and polystyrene (as described by Williams and Ward in Journal of Polymer Science, Polymer Letters, Vol. 6, p. 621, 1968) to derive the following equation:
- the polymer is reacted with a polyfunctional compound capable of insertion reactions into C-H bonds.
- a polyfunctional compound capable of insertion reactions into C-H bonds.
- Such polyfunctional compounds have at least two, preferably 2, functional groups capable of C-H insertion reactions.
- Those skilled in the art are familiar with C-H insertion reactions and functional groups capable of such reactions.
- coupling agents Compounds having at least two functional groups capable of C-H insertion under reaction conditions are referred to herein as coupling agents.
- Such coupling agents include alkyl and aryl azides (R-N 3 ) , acyl azides (R-C(0)N 3 ), azidoformates (R-O-C (O) -N 3 ) , phosphoryl azides ( (RO) _- (PO) -N 3 ) , phosphinic azides (R 2 -P (0) -N 3 ) and silyl azides (R 3 -Si- N 3 ) .
- Polyfunctional compounds capable of insertions into C-H bonds also include poly (sulfonyl azide) s.
- the poly (sulfonyl azide) is any compound having at least two sulfonyl azide groups (-S0 2 N 3 ) reactive with the polyolefin.
- the poly(sulfonyl azide) s have a structure X-R-X wherein each X is S0 2 N 3 and R represents an unsubstituted or inertly substituted hydrocarbyl, hydrocarbyl ether or silicon-containing group, preferably having sufficient carbon, oxygen or silicon, preferably carbon, atoms to separate the sulfonyl azide groups sufficiently to permit a facile reaction between the polyolefin and the sulfonyl azide, more preferably at least 1, more preferably at least 2, most preferably at least 3 carbon, oxygen or silicon, preferably carbon, atoms between functional groups.
- each R advantageously has at least one carbon or silicon atom between X' s and preferably has less than 50, more preferably less than 30, most preferably less than 20 carbon, oxygen or silicon atoms. Within these limits, larger is better for reasons including thermal and shock stability.
- R is straight- chain alkyl hydrocarbon, there are preferably less than 4 carbon atoms between the sulfonyl azide groups to reduce the propensity of the nitrene to bend back and react with itself.
- Silicon containing groups include silanes and siloxanes, preferably siloxanes.
- inertly substituted refers to substitution with atoms or groups which do not undesirably interfere with the desired reaction (s) or desired properties of the resulting coupled polymers.
- groups include fluorine, aliphatic or aromatic ether, siloxane as well as sulfonyl azide groups when more than two polyolefin chains are to be joined.
- Suitable structures include R as aryl, alkyl, aryl alkaryl, arylalkyl silane, siloxane or heterocyclic, groups and other groups which are inert and separate the sulfonyl azide groups as described.
- R includes at least one aryl group between the sulfonyl groups, most preferably at least two aryl groups (such as when R is 4,4' diphenylether or 4 , 4' -biphenyl) .
- R is one aryl group, it is preferred that the group have more than one ring, as in the case of naphthylene bis(sulfonyl azides).
- Poly (sulfonyl) azides include such compounds as 1, 5-pentane bis(sulfonyl azide), 1,8-octane bis(sulfonyl azide), 1,10-decane bis (sulfonyl azide), 1, 10-octadecane bis (sulfonyl azide), l-octyl-2 , 4 , 6-benzene t ⁇ s (sulfonyl azide), 4 , 4' -diphenyl ether b ⁇ s(sulfonyl azide), 1, 6-b ⁇ s (4' -sulfonazidophenyl) hexane, 2,7- naphthalene bis (sulfonyl azide), and mixed sulfonyl azides of chlorinated aliphatic hydrocarbons containing an average of from 1 to 8 chlorine atoms and from 2 to 5 sulfonyl azide groups per molecule, and mixtures thereof.
- Preferred poly (sulfonyl azide) s include oxy- bis (4-sulfonylaz ⁇ dobenzene) , 2 , 7-naphthalene b ⁇ s(sulfonyl azido) , 4 , 4' -bis (sulfonyl azido) biphenyl , 4 , 4 ' -diphenyl ether bis (sulfonyl azide) and bis (4 -sulfonyl azidophenyl) methane, and mixtures thereof.
- Sulfonyl azides are conveniently prepared by the reaction of sodium azide with the corresponding sulfonyl chloride, although oxidation of sulfonyl hydrazmes with various reagents (nitrous acid, dmitrogen tetroxide, nitrosonium tetrafluoroborate) has been used
- Polyfunctional compounds capable of insertions into C-H bonds also include carbene-formmg compounds such as salts of alkyl and aryl hydrazones and diazo compounds, and nitrene- forming compounds such as alkyl and aryl azides (R-N,) , acyl azides (R-C(O)N-,), azidoformates (R-O-C(O) -N 3 ) , sulfonyl azides (R-SO -NJ , phosphoryl azides ((RO) 2 - (PO) -N 3 ) , phosphmic azides (R 2
- Some of the coupling agents of the invention are preferred because of their propensity to form a greater abundance of carbon-hydrogen insertion products.
- Such compounds as the salts of hydrazones, diazo compounds, azidoformates, sulfonyl azides, phosphoryl azides, and silyl azides are preferred because they form stable s glet-state electron products (carbenes and nitrenes) which carry out efficient carbon-hydrogen insertion reactions, rather than substantially 1) rearranging via such mechanisms as the Curtius-type rearrangement, as is the case with acyl azides and phosphinic azides, or 2) rapidly converting to the triplet-state electron configuration which preferentially undergoes hydrogen atom abstraction reactions, which is the case with alkyl and aryl azides
- selection from among the preferred coupling agents is conveniently possible because of the differences in the temperatures at which the different classes of coupling agents are converted to the active carbene or nitrene products.
- carbenes are formed from diazo compounds efficiently at temperatures less than 100°C, while salts of hydrazones, azidoformates and the sulfonyl azide compounds react at a convenient rate at temperatures above 100°C, up to temperatures of 200°C.
- salts of hydrazones, azidoformates and the sulfonyl azide compounds react at a convenient rate at temperatures above 100°C, up to temperatures of 200°C.
- convenient rates it is meant that the compounds react at a rate that is fast enough to make commercial processing possible, while reacting slowly enough to allow adequate mixing and compounding to result in a final product with the coupling agent adequately dispersed and located substantially in the desired position in the final product.
- Phosphoryl azides may be reacted at temperatures in excess of 180°C up to 300°C, while silyl azides react preferentially at temperatures of from 250°C to 400°C.
- the poly (sulfonyl azide) is used in a rheology modifying amount, that is an amount effective to increase the low-shear viscosity (at 0.1 rad/sec) of the polymer preferably at least 5 percent as compared with the starting material polymer, but less than a crosslinking amount, that is an amount sufficient to result in less than 1 weight percent of gel as measured by ASTM D2765-procedure A.
- the amount of azide sufficient to increase the low shear viscosity and result in less than 1 weight percent gel will depend on molecular weight of the azide used and polymer, the amount is preferably less than 5 percent, more preferably less than 2 percent, most preferably less than l weight percent poly (sulfonyl azide) based on total weight of polymer when the poly (sulfonyl azide) has a molecular weight of from 200 to 2000.
- the amount of poly (sulfonyl azide) is preferably at least 0.01 weight percent, more preferably at least 0.05 weight percent, most preferably at least 0.10 weight percent based on total polymer.
- the sulfonyl azide is admixed with the polymer and heated to at least the decomposition temperature of the sulfonyl azide.
- decomposition temperature of the azide it is meant that temperature at which the azide converts to the sulfonyl nitrene, eliminating nitrogen and heat in the process, as determined by differential scanning calorimetry (DSC).
- DSC differential scanning calorimetry
- the poly (sulfonyl azide) begins to react at a kinetically significant rate (convenient for use in the practice of the invention) at temperatures of 130°C and is almost completely reacted at 160°C in a DSC (scanning at 10°C/min) .
- ARC scanning at 2°C/ hr
- Extent of reaction is a function of time and temperature. At the low levels of azide used in the practice of the invention, the optimal properties are not reached until the azide is essentially fully reacted.
- Temperatures for use in the practice of the invention are also determined by the softening or melt temperatures of the polymer starting materials. For these reasons, the temperature is advantageously greater than 90°C, preferably greater than 120°C, more preferably greater than 150°C, most preferably greater than 180°C.
- Preferred times at the desired decomposition temperatures are times that are sufficient to result in reaction of the coupling agent with the polymer (s) without undesirable thermal degradation of the polymer matrix.
- Preferred reaction times in terms of the half life of the coupling agent that is the time required for half of the agent to be reacted at a preselected temperature, which half life is determinable by DSC is 5 half lives of the coupling agent.
- the reaction time is preferably at least 4 minutes at 200 °C.
- Admixing of the polymer and coupling agent is conveniently accomplished by any means within the skill in the art. Desired distribution is different in many cases, depending on what rheological properties are to be modified. In a homopolymer or copolymer it is desirable to have as homogeneous a distribution as possible, preferably achieving solubility of the azide in the polymer melt .
- Preferred processes include at least one of (a) dry blending the coupling agent with the polymer, preferably to form a substantially uniform admixture and adding this mixture to melt processing equipment, for example a melt extruder to achieve the coupling reaction, at a temperature at least the decomposition temperature of the coupling agent; (b) introducing, for example by injection, a coupling agent in liquid form, for example dissolved in a solvent therefor or in a slurry of coupling agent in a liquid, into a device containing polymer, preferably softened, molten or melted polymer, but alternatively in particulate form, in solution or dispersion, more preferably in melt processing equipment; (c) forming a first admixture of a first amount of a first polymer and a coupling agent, advantageously at a temperature less than the decomposition temperature of the coupling agent, preferably by melt blending, and then forming a second admixture of the first admixture with a second amount of a second polymer
- process (c) is conveniently used to make a concentrate with a first polymer composition having a lower melting temperature, advantageously at a temperature below the decomposition temperature of the coupling agent, and the concentrate is melt blended into a second polymer composition having a higher melting temperature to complete the coupling reaction.
- Concentrates are especially preferred when temperatures are sufficiently high to result in loss of coupling agent by evaporation or decomposition not leading to reaction with the polymer, or other conditions would result that effect.
- some coupling occurs during the blending of the first polymer and coupling agent, but some of the coupling agent remains unreacted until the concentrate is blended into the second polymer composition.
- Each polymer or polymer composition includes at least one homopolymer, copolymer, terpolymer, or interpolymer and optionally includes additives within the skill in the art.
- the coupling agent is added in a dry form it is preferred to mix the agent and polymer in a softened or molten state below the decomposition temperature of the coupling agent then to heat the resulting admixture to a temperature at least equal to the decomposition temperature of the coupling agent.
- melt processing is used to mean any process in which the polymer is softened or melted, such as extrusion, pelletizing, film blowing and casting, thermoforming, and compounding in polymer melt form, .
- the polyolefin (s) and coupling agent are suitably combined in any manner which results in desired reaction thereof, preferably by mixing the coupling agent with the polymer (s) under conditions which allow sufficient mixing before reaction to avoid uneven amounts of localized reaction then subjecting the resulting admixture to heat sufficient for reaction.
- a substantially uniform admixture of coupling agent and polymer is formed before exposure to conditions in which chain coupling takes place.
- a substantially uniform admixture is one in which the distribution of coupling agent in the polymer is sufficiently homogeneous to be evidenced by a polymer having a melt viscosity after treatment according to the practice of the invention either higher at low angular frequency (for example 0.1 rad/sec) or lower at higher angular frequency (for example 100 rad/sec) than that of the same polymer which has not been treated with the coupling agent but has been subjected to the same shear and thermal history.
- decomposition of the coupling agent occurs after mixing sufficient to result in a substantially uniform admixture of coupling agent and polymer.
- This mixing is preferably attained with the polymer in a molten or melted state, that is above the crystalline melt temperature, or in a dissolved or finely dispersed condition rather than in a solid mass or particulate form.
- the molten or melted form is more preferred to insure homogeniety rather than localized concentrations at the surface.
- any equipment is suitably used, preferably equipment which provides sufficient mixing and temperature control in the same equipment, but advantageously practice of the invention takes place in such devices as an extruder or a static polymer mixing devise such as a Brabender blender.
- extruder is used for its broadest meaning to include such devices as a device which extrudes pellets or pelletizer.
- at least one step of the process of the invention takes place in the melt extrusion step.
- reaction take place in a solvent or other medium
- reaction it is preferred that the reaction be n a bulk phase to avoid later steps for removal of the solvent or other medium.
- a polymer above the crystalline melt temperature is advantageous for even mixing and for reaching a reaction temperature (the decomposition temperature of the sulfonyl azide) .
- the process of the present invention takes place in a single vessel, that is mixing of the coupling agent and polymer takes place in the same vessel as heating to the decomposition temperature of the coupling agent
- the vessel is preferably a twin-screw extruder, but is also advantageously a single- screw extruder or a batch mixer
- the reaction vessel more preferably has at least two zones of different temperatures into which a reaction mixture would pass, the first zone advantageously being at a temperature at least the crystalline melt temperature or the softening temperature of the polymer (s) and preferably less than the decomposition temperature of the coupling agents and the second zone being at a temperature sufficient for decomposition of the coupling agent.
- the first zone is preferably at a temperature sufficiently high to soften the polymer and allow it to combine with the coupling agent through distributive mixing to a substantially uniform admixture .
- the preferred embodiment for incorporation of coupling agent is to solution blend the coupling agent in solution or admixture into the polymer, to allow the polymer to imbibe (absorb or adsorb at least some of the coupling agent) , and then to evaporate the solvent After evaporation, the resulting mixture is extruded.
- the solvent is preferably a solvent for the coupling agent, and more preferably also for the polymer when the polymer is soluble such as in the case of polycarbonate
- solvents include polar solvents such as acetone, THF (tetrahydrofuran) and chlorinated hydrocarbons such as methylene chloride
- polar solvents such as acetone, THF (tetrahydrofuran) and chlorinated hydrocarbons such as methylene chloride
- other non-polar compounds such as mineral oils in which the coupling agent is sufficiently miscible to disperse the coupling agent in a polymer, are used.
- the coupling agent be added to the post-reactor area of a polymer processing plant.
- the coupling agent is added in either powder or liquid form to the powdered polyethylene after the solvent is removed by decantation and prior to the drying and densification extrusion process.
- the coupling agent is preferably added in either powder or liquid form to the powdered polyethylene before the densification extrusion.
- the coupling agent is preferably added to the polymer solution prior to the densification extrusion process.
- rheology modified or chain coupled polymers that is the polymers which have sulfonamide, amine, alkyl-substituted or aryl- substituted carboxamide, alkyl-substituted or aryl-substituted phosphoramide, alkyl-substituted or aryl-substituted methylene coupling between different polymer chains.
- Resulting compounds advantageously show higher low shear viscosity than the original polymer due to coupling of long polymer chains to polymer backbones.
- Rheology modified polymers are especially useful as blown film for better bubble stability as measured by low shear viscosity.
- Polymers rheology modified according to the practice of the invention are superior to the corresponding unmodified polymer starting materials for these applications due to the elevation of viscosity, of preferably at least 5 percent at low shear rates (0.1 rad/sec) , sufficiently high melt strengths to avoid deformation during thermal processing or to achieve bubble strength during blow molding, and sufficiently low high shear rate viscosities to facilitate molding and extrusion.
- Advantageously toughness and tensile strength of the starting material is maintained or improved.
- Polymers resulting from the practice of the invention are different from those resulting from practice of prior art processes such as shown in CA 797,917.
- At least one mechanical property of tear strength, puncture resistance or low haze (as measured by ASTM D1003) a film prepared from a composition of the invention is superior to that of a film of the linear polyethylene modified as taught in CA 797,917.
- a film made from the material taught in the reference would have a puncture resistance of less than 15 in- lb (17.25 cm/kg), machine direction Elmendorf tear strength of less than 500 g, and haze of more than 35 percent.
- the puncture resistance is measured at room temperature using an instrument for the purpose available from Instron Inc.
- Model 4201 under the trade designation Model 4201 with a hardware upgrade commercially available from Sintech Inc. under the trade designation MTS Sintech ReNew testing frame with the Sintech (Version 3.08) Testing Software, film with dimensions of 6"x 6" (15 cm x 15 cm) , a round specimen holder measuring 12.56" square (78.5 cm 2 ), a puncture probe of polished stainless steel ball measuring 1/2" (1.25 cm), with 7.5" (18.75 cm) maximum travel and travel speed of 10"/min (25.4 cm /min) , to measure the energy required to break the film.
- Film and film structures particularly benefit from this invention and can be made using conventional blown film fabrication techniques or other, preferably biaxial, orientation processes such as tenter frames or double bubble processes.
- Conventional blown film processes are described, for example, in The Encyclopedia of Chemical Technology, Kirk-Othmer, Third Edition, John Wiley & Sons, New York, 1981, vol. 16, pp. 416-417 and Vol . 18, pp. 191-192.
- Biaxial orientation film manufacturing process such as described in a "double bubble" process as in U.S. Patent 3,456,044 (Pahlke) , and the processes described in U.S. Patent 4,352,849 (Mueller), U.S. Patent 4,597,920 (Golike) , U.S.
- Patent 4,820,557 (Warren), U.S. Patent 4,837,084 (Warren), U.S. Patent 4,865,902 (Golike et al.), U.S. Patent 4,927,708 (Herran et al . ) , U.S. Patent 4,952,451 (Mueller), U.S. Patent 4,963,419 (Lustig et al . ) , and U.S. Patent 5,059,481 (Lustig et al . ) , can also be used to make film structures from the novel compositions described herein.
- the film structures can also be made as described in a tenter-frame technique, such as that used for oriented polypropylene.
- the films may be monolayer or multilayer films.
- the film made using this invention can also be coextruded with the other layer (s) or the film can be laminated onto another layer (s) in a secondary operation, such as that described in Packaging Foods With Plastics, by Wilmer A. Jenkins and James P. Harrington (1991) or that described in "Coextrusion For Barrier Packaging” by W.J.
- a monolayer film is produced via tubular film (that is, blown film techniques) or flat die (that is, cast film) as described by K.R. Osborn and W.A. Jenkins in "Plastic Films, Technology and Packaging Applications” (Technomic Publishing Co., Inc., 1992), then the film must go through an additional post- extrusion step of adhesive or extrusion lamination to other packaging material layers to form a multilayer structure.
- the film may still be laminated to additional layers of packaging materials, depending on the other physical requirements of the final film.
- "Laminations vs. Coextrusion" by D. Dumbleton (Converting Magazine (September 1992) also discusses lamination versus coextrusion.
- Monolayer and coextruded films can also go through other post extrusion techniques, such as a biaxial orientation process .
- Extrusion coating is yet another technique for producing multilayer film structures using the novel compositions described herein.
- the novel compositions comprise at least one layer of the film structure. Similar to cast film, extrusion coating is a flat die technique.
- a sealant can be extrusion coated onto a substrate either in the form of a monolayer or a coextruded extrudate.
- the novel compositions described herein comprise at least one layer of the total multilayer film structure
- Other layers of the multilayer structure include but are not limited to barrier layers, tie layers, structural layers or a combination thereof
- Various materials can be used for these layers, with some of them being used as more than one layer m the same film structure.
- the multilayer film structures comprise from 2 to 7 layers
- the rheology-modifled polymers and intermediates used to make rheology-modifled polymers may be used alone or in combination with one or more additional polymers in a polymer blend When additional polymers are present, they may be selected from any of the modified or unmodified homogeneous polymers described for this invention any modified or unmodified heterogeneous poly
- compositions of the invention are particularly useful in thick films such as are particularly useful in agricultural films, for example useful as mulch, greenhouse films, and geomembranes.
- the term "thick films” is used to designate films having a thickness greater than 6 mils (greater than 15 x 10E-02 mmeters) .
- Such films are preferably prepared by blown film processes within the skill in the art.
- compositions of the invention are preferably capable of blow up ratios (ratio of square area of bubble to square area of die diameter) of at least 2, more preferably at least 2.5 with an adequate melt strength to produce a stable bubble at an extruder pressure less than 2000 psi (13,784kPa) .
- the starting material ethylene polymer preferably has a density less than 0.935, more preferably up to 0.930, most preferably up to 0.920; and preferably at least 0.89, more preferably at least 0.890, most preferably at least 0.900.
- the polymer modified according to the practice of the invention preferably has an 12 as measured by the procedures of ASTM 1238 Procedure A at 190°C and 2.16 Kg of less than 5 g/10 min, more preferably less than lg/lOmin to avoid an unstable bubble which results when the melt index is too high.
- Heterogeneous polyethylenes that are optionally combined with the rheology-modified polymers according to this invention fall into two broad categories, those prepared with a free radical initiator at high temperature and high pressure, and those prepared with a coordination catalyst at high temperature and relatively low pressure.
- the former are generally known as low density polyethylenes (LDPE) and are characterized by branched chains of polymerized monomer units pendant from the polymer backbone.
- LDPE polymers generally have a density between 0.910 and 0.935 g/cc.
- Ethylene polymers and copolymers prepared by the use of a coordination catalyst, such as a Ziegler or Phillips catalyst, are generally known as linear polymers because of the substantial absence of branch chains of polymerized monomer units pendant from the backbone.
- High density polyethylene generally having a density of 0.941 to 0.965 g/cc, is typically a homopolymer of ethylene, and it contains relatively few branch chains relative to the various linear copolymers of ethylene and an ⁇ -olefin.
- HDPE is well known, commercially available in various grades, and may be used in this invention.
- the density of a linear ethylene/ ⁇ -olefin copolymer is a function of both the length of the ⁇ -olefin and the amount of such monomer in the copolymer relative to the amount of ethylene, the greater the length of the ⁇ -olefin and the greater the amount of ⁇ -olefin present, the lower the density of the copolymer.
- LLCPE is typically a copolymer of ethylene and an ⁇ -olefin of 3 to 12 carbon atoms, preferably 4 to 8 carbon atoms (for example, 1- butene, 1-octene, etc.), that has sufficient ⁇ -olefin content to reduce the density of the copolymer to that of LDPE.
- ⁇ -olefin 3 to 12 carbon atoms, preferably 4 to 8 carbon atoms (for example, 1- butene, 1-octene, etc.), that has sufficient ⁇ -olefin content to reduce the density of the copolymer to that of LDPE.
- ULDPE ultra low density polyethylene
- VLDPE very low density polyethylene
- the densities (according to ASTM D-792) of these linear polymers advantageously range from 0.87 to 0.91 g/cc.
- Heterogeneous linear ethylene polymers are available from The Dow Chemical Company as DowlexTM LLDPE and as AttaneTM ULDPE resins. Heterogeneous linear ethylene polymers can be prepared via the solution, slurry or gas phase polymerization of ethylene and one or more optional ⁇ -olefin comonomers in the presence of a Ziegler Natta catalyst, by processes such as are disclosed in U.S. Patent No. 4,076,698 to /Anderson et al .
- heterogeneous ethylene polymers are typically characterized as having molecular weight distributions, M w /M n , in the range of from 3.5 to 4.1. Relevant discussions of both of these classes of materials, and their methods of preparation are found in U.S. Patent No. 4,950,541 and the patents to which it refers .
- compositions of the invention and compositions produced by practice of the invention are particularly useful because of their surprising properties.
- the preferred medium density polyethylenes and ethylene copolymers (density 0.90 g/mL, comonomer content 0.5 - 5 mole percent) of the invention are particularly useful as blown films such as in trash bags, grocery sacks, sealant layers, tie layers, produce bags, garment bags, shipping sacks, medical films, stretch film, shrink film, agricultural film, construction film, geomembranes, stretch hooders, green house films, and mulch films, preferably trash bags, agricultural film, greenhouse film, construction film, and geomembranes.
- medium density preferred embodiments are useful in cast films such as are useful in stretch films, diaper backsheets, industrial wrap, produce wrap, meat wrap, consumer wrap, and shrink film elastic film, preferably as elastic film.
- the low density preferred embodiments are also particularly useful for calendaring to form such materials as sheeting, packaging films, and non-packaging films.
- the strain was determined to be within the linear viscoelastic regime by performing a strain sweep at 0.1 rad/s and 190 C, by strain sweep from 2 to 30 percent strain m 2 percent steps to determine the minimum required strain to produce torques within the specification of the transducer; another strain sweep at 100 rad/s and 190 C was used to determine the maximum strain before nonlinearity occurred according to the procedure disclosed by J. M. Dealy and K. F. Wissbrun, "Melt Rheology and Its Role m Plastics Processing", Van Nostrand, New York (1990) . All testing was performed in a nitrogen purge to minimize oxidative degradation.
- the melt index was measured according to ASTM D-1238 condition
- Samples were prepared using either a HaakeBuchler Rheomix 600 mixer with roller style blades, attached to a HaakeBuchler Rheocord 9000 Torque rheometer, or using a Brabender mixer (Type R.E E No. A- 19/S B) with a 50 g mixing bowl
- Example 2 the procedure of Example 1 was repeated but using 0.1 weight percent 4 , 4' -oxybis (benzenesulfonyl azide).
- Rheological properties were measured for each sample plus an unmodified control (Comparative Sample A) at 190°C over a frequency range of 0.1 to 100 rad/second using a Rheometrics mechanical spectrometer equipped with parallel 25 mm diameter plates according to manufacture's directions.
- the low shear viscosity was the viscosity measured at the lowest frequency.
- the high shear viscosity was determined NSC at 100 rad/sec.
- Dowlex 2045 containing 1250 ppm calcium stearate, 200 ppm hindered polyphenol antioxidant commercially available from Ciba Geigy Corporation under the trade designation Irganox 1010 with 0, 0.05, and 0.1 weight percent of 4 , 4' -oxybis (benzenesulfonyl azide) CAS# [7456- 68-0] for C.S. C, Example 5, and Example 6, respectively.
- Rheology modification efficiency was surprisingly influenced by the molecular weight distribution, molecular weight, type and amount of comonomer.
- the efficiency (based on percent change in viscosity at 0.1 rad shear rate) of the polymers tested decreased in the following order: Ex. 3&4, Ex. 5&6, Ex. 7&8, Ex. 1&2.
- DOWLEX 2035 containing 200 ppm of Irganox 1010 antioxidant (previously identified) and 750 ppm of synthetic dihydrotalcite commercially available from Kyowa under the trade designation DHT 4A stabilizer referred to hereinafter by the trade designation.
- the amount of azide used was based on the final predicted melt index.
- DOWLEX is a trademark of The Dow Chemical Company.
- the extruder measured temperature was 130°C, 175°C, 215°C, 221°C, and 221°C for zones 1, 2, 3, 4, and 5, respectively.
- the temperature was measured using thermocouples that are placed to the body of the barrel of the extruder.
- the distances of the thermocouples from the center of the feed zone were 8.8, 38.8, 56.2, 66.3, 78.8, and 88.8 cm from the feed to the discharge (die) of the extruder for Zones 1, 2, 3, 4 , nd 5, respectively.
- the melt temperature and die temperatures were 230°C and 220° C, respectively.
- the melt-extruded resin ran through a water cooling bath (at 19° C) before it was pelletized.
- the output rate for this process was 30 pounds/hr (13.6 kg/hr) .
- a total of 300 pounds (136.2 kg) of the coupled resin was collected for further study.
- the final resin (after treatment) had a measured 1.0 g/10 min melt index and 0.919 g/cc density.
- the film was made directly from unmodified pellets of a homopolymer of ethylene having MI of 0.22 g/10 min, density of 0.921 g/cc commercially available from The Dow Chemical Co. under the trade designation LDPE-132I which designation is used hereinafter for the polyethylene.
- the film was prepared directly from the dry blend of pellets of 30 weight percent low density polyethylene (0.47 g/10 min MI, 0.9190 g/cc) commercially available from The Dow Chemical Co. under the trade designation LDPE-662I (containing 300 ppm Irganox 1010 antioxidant) which designation is used hereinafter for the polyethylene and 70 weight percent of a linear low density ethylene/octene copolymer (1.00 g/lOmin MI, 0.920 g/cc density) commercially available from The Dow Chemical Co. under the trade designation DOWLEX LLDPE 2045 (containing the additives identified in Ex. 6) which designation was used hereinafter for the polyethylene.
- DOWLEX LLDPE 2045 containing the additives identified in Ex. 6 which designation was used hereinafter for the polyethylene.
- the resulting blend was used to provide a better balance of properties such as tear strength and melt strength than would be achieved by using LDPE 1321 resin as one of the blend components.
- Die gap 70 mil (1.75 mm) Die type: Sano Die diameter: 6 inches (15.2 cm) Screw type : Barr ET Output rate: 188 lb/h (85.1 kg/hr)
- Cooling Air yes Blow up ratio: 2.0 & 2.9 Film gauge: 6 .0 mil (0.15 mm) Shear at the die: -106 /s (metric unit)
- Puncture at room temperature was measured using an instrument for the purposed commercially available from Instron Inc. under the trade designation Instron Model 4201 with a hardware upgrade commercially available from Sintech Inc. and a testing frame commercially available from Sintech Inc. under the trade designation MTS Sintech ReNew testing frame along with software commercially available from Sintech Inc. under the trade designation Sintech (Version 3.08) Testing Software.
- Four samples of each film with dimensions of 6"x 6" (15x15 cm) were measured using a round specimen holder 12.56" (31.9 cm) square.
- a puncture probe was a 1/2" (1.27 cm) polished stainless steel ball with 7.5" (18.75 cm) maximum travel and travel speed of 10' "/min (25.4 cm/min) . The energy required to break the film was measured.
- Elmendorf Tear Strength was measured at 23 °C according to the procedures of ASTM D1922. MD (Machine Direction) Ult (ultimate) Tensile Strength and CD (Cross Direction) Ult Tensile Strength were measured according to the procedures of ASTM D638.
- CD and MD were Cross Direction and Machine Direction orientation of the film
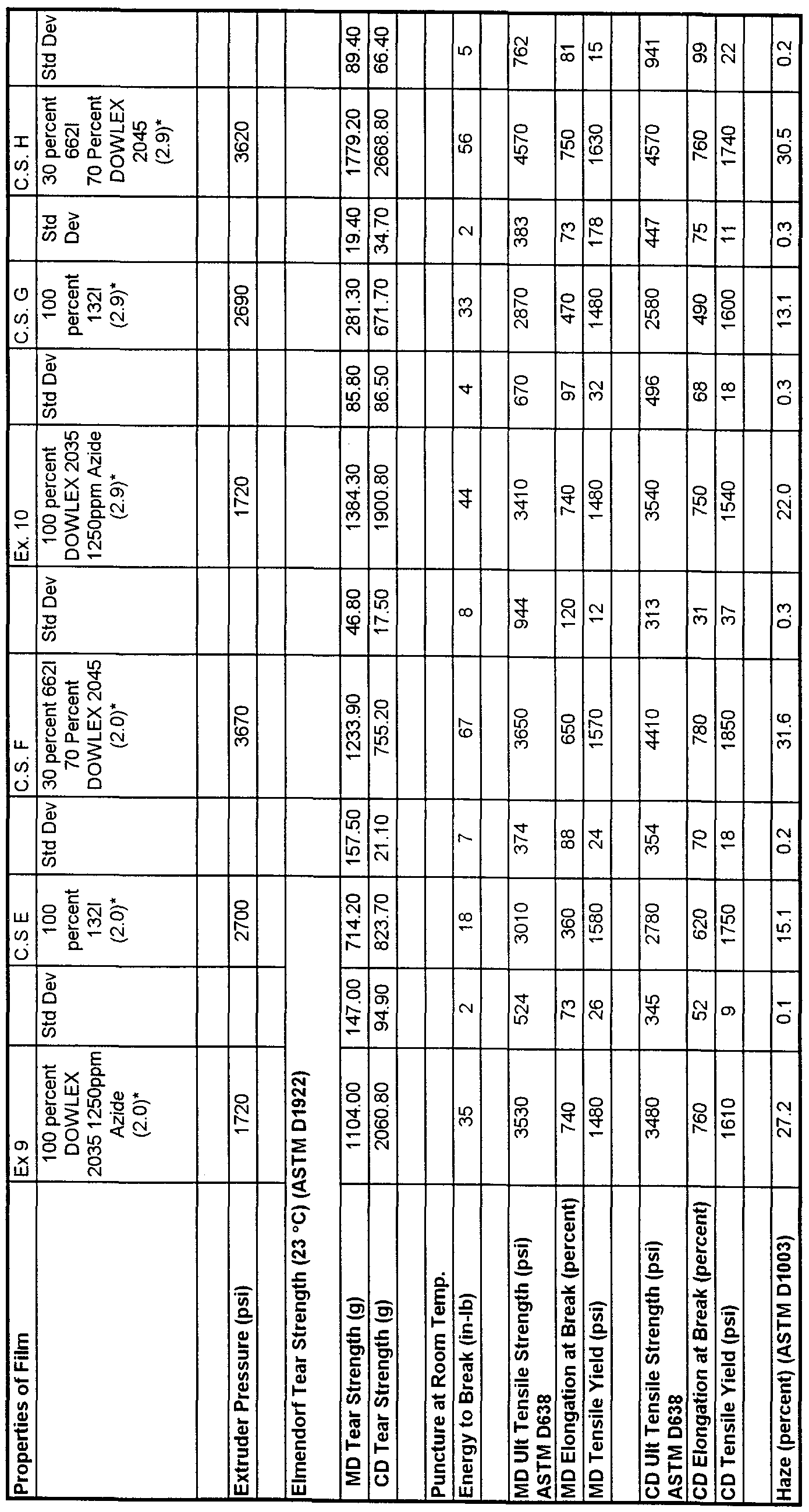
- the film of Example 9 As compared to the film of Comparative Sample E, the film of Example 9 exhibited much better mechanical properties such as tear strength and puncture resistance. It also had acceptable processability as shown by the lower extruder pressures as compared with that of Comparative Sample E. Good bubble stability was observed as was good quality film as indicated by visual observation. (A stable bubble does not waver or pump up and down causing film thickness variation.
- Comparative Sample F was the blend of LDPE and LLDPE to achieve better physical properties; while this blend provides better mechanical performance compared to a single resin alone, as indicated by tear strength and energy to break, the processability was decreased by the additional of the LLDPE (that is it requires very high extruder pressures, 3670 psi (25300 kPa) ) . This is not advantageous to production of thick blown films.
- Example 10 shows that the coupled Dowlex 2035 LLDPE resin can be blown at very high blow up ratios (2.9) . This is significant in that it allows the film to be made into very wide lay flats for greenhouse and agricultural films.
- the physical properties of the film of Example 9 compared to that of Comparative Sample G were far superior in tear strength and puncture resistance.
- the film of Example 10 had lower die pressure than Comparative Sample H indicating that Example 10 should have higher output than Comparative Sample H.
Abstract
Description




Claims
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP98938337A EP1007586B1 (en) | 1997-08-27 | 1998-08-05 | Rheology modification of low density polyethylene |
| AT98938337T ATE306517T1 (en) | 1997-08-27 | 1998-08-05 | RHEOLOGY CHANGE IN LOW DENSITY POLYETHYLENE |
| KR1020007002009A KR100562615B1 (en) | 1997-08-27 | 1998-08-05 | Rheology modification of low density polyethylene |
| CA 2301644 CA2301644C (en) | 1997-08-27 | 1998-08-05 | Rheology modification of low density polyethylene |
| AU86882/98A AU8688298A (en) | 1997-08-27 | 1998-08-05 | Rheology modification of low density polyethylene |
| JP2000507741A JP2001514288A (en) | 1997-08-27 | 1998-08-05 | Rheological modification of low density polyethylene |
| DE1998631871 DE69831871T2 (en) | 1997-08-27 | 1998-08-05 | RHEOLOGY CHANGE IN POLYETHYLENE LOW DENSITY |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US5758297P | 1997-08-27 | 1997-08-27 | |
| US60/057,582 | 1997-08-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1999010422A1 true WO1999010422A1 (en) | 1999-03-04 |
Family
ID=22011502
Family Applications (5)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1998/016214 WO1999010422A1 (en) | 1997-08-27 | 1998-08-05 | Rheology modification of low density polyethylene |
| PCT/US1998/016213 WO1999010421A1 (en) | 1997-08-27 | 1998-08-05 | Rheology modification of interpolymers of alpha-olefins and vinyl aromatic monomers |
| PCT/US1998/016215 WO1999010423A1 (en) | 1997-08-27 | 1998-08-05 | Rheology modification of elastomers |
| PCT/US1998/016810 WO1999010415A1 (en) | 1997-08-27 | 1998-08-13 | Process of rheology modification of polymers |
| PCT/US1998/017685 WO1999010427A1 (en) | 1997-08-27 | 1998-08-26 | Rheology modification of polymers prepared using metallocenes |
Family Applications After (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1998/016213 WO1999010421A1 (en) | 1997-08-27 | 1998-08-05 | Rheology modification of interpolymers of alpha-olefins and vinyl aromatic monomers |
| PCT/US1998/016215 WO1999010423A1 (en) | 1997-08-27 | 1998-08-05 | Rheology modification of elastomers |
| PCT/US1998/016810 WO1999010415A1 (en) | 1997-08-27 | 1998-08-13 | Process of rheology modification of polymers |
| PCT/US1998/017685 WO1999010427A1 (en) | 1997-08-27 | 1998-08-26 | Rheology modification of polymers prepared using metallocenes |
Country Status (21)
| Country | Link |
|---|---|
| US (9) | US6211302B1 (en) |
| EP (5) | EP1007586B1 (en) |
| JP (5) | JP4344471B2 (en) |
| KR (5) | KR100562615B1 (en) |
| CN (3) | CN1273592A (en) |
| AR (5) | AR016885A1 (en) |
| AT (3) | ATE322519T1 (en) |
| AU (5) | AU8688398A (en) |
| BR (3) | BR9814447A (en) |
| CA (5) | CA2301644C (en) |
| DE (3) | DE69831871T2 (en) |
| ES (2) | ES2247709T3 (en) |
| HU (1) | HUP0002756A3 (en) |
| ID (1) | ID25634A (en) |
| MY (4) | MY128986A (en) |
| NO (1) | NO322848B1 (en) |
| PL (1) | PL339083A1 (en) |
| TR (2) | TR200000905T2 (en) |
| TW (5) | TW591070B (en) |
| WO (5) | WO1999010422A1 (en) |
| ZA (5) | ZA987750B (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001083605A3 (en) * | 2000-05-04 | 2002-01-31 | Dow Chemical Co | Molecular melt and methods for making and using the molecular melt |
| US7399737B2 (en) | 2005-06-13 | 2008-07-15 | Exxonmobil Chemical Patents Inc. | Lube additives |
| WO2011062961A1 (en) * | 2009-11-23 | 2011-05-26 | Dow Global Technologies Inc. | Compositions, films and methods of preparing the same |
Families Citing this family (131)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6506842B1 (en) * | 1997-01-29 | 2003-01-14 | Dupont Dow Elastomers L.L.C. | Rheology-modified thermoplastic elastomer compositions and articles fabricated therefrom |
| AU742775B2 (en) * | 1997-03-10 | 2002-01-10 | Schoeller Arca Systems Ab | A process for the manufacturing of thermoplastic products with high creep strain resistance |
| US6211302B1 (en) * | 1997-08-27 | 2001-04-03 | The Dow Chemical Company | Rheology modification of interpolymers of alpha-olefins and vinylidene aromatic monomers |
| US6709742B2 (en) | 1998-05-18 | 2004-03-23 | Dow Global Technologies Inc. | Crosslinked elastic fibers |
| ATE495205T1 (en) * | 1998-11-02 | 2011-01-15 | Dow Global Technologies Inc | SHEER-FLUFFILIZABLE ETHYLENE/ALPHA-OLEFIN/DIENE POLYMERS AND THEIR PRODUCTION |
| US6277916B1 (en) | 1999-02-25 | 2001-08-21 | The Dow Chemical Company | Process for preparing thermoplastic vulcanizates |
| US6365693B1 (en) * | 1999-05-07 | 2002-04-02 | Dupont Dow Elastomers, L.L.C. | Fluoroalkanesulfonyl azide copolymers |
| AU5884000A (en) * | 1999-06-24 | 2001-01-09 | Dow Chemical Company, The | Polyolefin composition with improved impact properties |
| EP1457518A1 (en) * | 1999-12-03 | 2004-09-15 | Dow Global Technologies, Inc. | Grafted thermoplastic compositions and fabricated articles therefrom |
| EP1254191A2 (en) * | 1999-12-03 | 2002-11-06 | The Dow Chemical Company | Grafted thermoplastic compositions and fabricated articles therefrom |
| US6291618B1 (en) | 1999-12-23 | 2001-09-18 | The Dow Chemical Company | Phosphazene azide coupling agents and their use in rheology modification of polymers |
| US6593005B2 (en) | 2000-01-24 | 2003-07-15 | Dow Global Technologies Inc. | Composition and films thereof |
| US6939919B2 (en) * | 2000-05-26 | 2005-09-06 | Dow Global Technologies Inc. | Polyethylene rich/polypropylene blends and their uses |
| US6548600B2 (en) | 2000-09-22 | 2003-04-15 | Dupont Dow Elastomers L.L.C. | Thermoplastic elastomer compositions rheology-modified using peroxides and free radical coagents |
| US20040041347A1 (en) * | 2000-11-30 | 2004-03-04 | Beach Jefferey Kurtis | Permanently lubricated gasket |
| US7469905B2 (en) * | 2000-11-30 | 2008-12-30 | Springseal Inc. | Permanently lubricated film gasket and method of manufacture |
| DE60108173T2 (en) | 2000-12-22 | 2005-06-02 | Dow Global Technologies, Inc., Midland | propylene copolymer |
| CA2460009A1 (en) * | 2001-10-01 | 2003-04-10 | Dow Global Technologies Inc. | Blow moldable propylene polymer compositions |
| US20050070673A1 (en) * | 2001-10-01 | 2005-03-31 | Novak Leo R. | Thermoformable propylene polymer compositions |
| US20030138627A1 (en) * | 2001-11-02 | 2003-07-24 | Finlayson Malcolm F. | Molecular melt and methods for making and using the molecular melt |
| DE60313305T2 (en) * | 2002-03-11 | 2007-12-27 | Dow Global Technologies, Inc., Midland | REVERSIBLE, THERMOFIXED, ELASTIC FIBERS, PRODUCTION METHOD AND ARTICLES PRODUCED THEREFROM |
| EP1860214B1 (en) | 2002-03-11 | 2009-04-29 | Dow Global Technologies Inc. | Reversible, heat-set, elastic fibers, and method of making and articles made from same |
| US6921563B2 (en) * | 2002-05-24 | 2005-07-26 | Ticona Gmbh | Multi-layer laminate, packaging material and packages made therefrom |
| US6734253B2 (en) * | 2002-07-19 | 2004-05-11 | Dow Global Technologies, Inc. | Scratch and mar resistant propylene polymer composition |
| US6864195B2 (en) * | 2002-08-15 | 2005-03-08 | Bfs Diversified Products, Llc | Heat weldable roofing membrane |
| US6846884B2 (en) * | 2002-09-27 | 2005-01-25 | Union Carbide Chemicals & Plastics Technology Corporation | Control of resin properties |
| US7338994B2 (en) * | 2002-10-01 | 2008-03-04 | Dow Global Technologies Inc. | Rheology-modified thermoplastic elastomer compositions for extruded profiles |
| US6908687B2 (en) | 2002-12-30 | 2005-06-21 | Exxonmobil Oil Corporation | Heat-shrinkable polymeric films |
| US7101628B2 (en) * | 2003-03-20 | 2006-09-05 | Bfs Diversified Products, Llc | Thermoplastic vulcanizate and membrane for covering a roof made therefrom |
| US6930148B2 (en) * | 2003-04-08 | 2005-08-16 | Texas Petrochemicals Lp | Enhanced polyisobutylene modified hot melt adhesive formulation |
| AU2004247669B2 (en) * | 2003-06-10 | 2010-03-25 | Dow Global Technologies Llc | Film layers made from ethylene polymer blends |
| WO2005010896A1 (en) * | 2003-07-24 | 2005-02-03 | Union Carbide Chemicals & Plastic Technology Corporation | Cable insulation system with flexibility, high temperature deformation resistance, and reduced degree of stickiness |
| US7632086B2 (en) * | 2003-10-03 | 2009-12-15 | Exxonmobil Chemical Patents Inc. | Melt fracture reduction |
| CN100545201C (en) * | 2003-10-22 | 2009-09-30 | 宝洁公司 | Composition with the extrusion blow molded thermoplastic package combination |
| US7906586B2 (en) * | 2003-12-09 | 2011-03-15 | Dow Global Technologies Llc | Thermoplastic olefinic compositions |
| PL1574772T3 (en) * | 2004-03-12 | 2008-12-31 | Borealis Tech Oy | A crosslinked ethylene polymer pressure pipe and a method for the preparation thereof |
| US8129009B2 (en) * | 2004-04-13 | 2012-03-06 | E. I. Du Pont De Nemours And Company | Composition comprising ethylene copolymer |
| JP2005320469A (en) * | 2004-05-11 | 2005-11-17 | Idemitsu Kosan Co Ltd | Polycarbonate resin composition and molded article |
| WO2005123829A1 (en) * | 2004-06-14 | 2005-12-29 | Polyone Corporation | Durable and low friction thermoplastic elastomer compositions |
| US20060020063A1 (en) * | 2004-07-22 | 2006-01-26 | Guenther Gerhard K | Controlled finishes for free surface polyethylene resins |
| WO2006063338A1 (en) * | 2004-12-08 | 2006-06-15 | Mark Knapp | Molded gasket and method of making |
| AU2005316788B2 (en) | 2004-12-17 | 2012-04-05 | Dow Global Technologies Llc | Rheology modified polyethylene compositions |
| EP1674504A1 (en) * | 2004-12-22 | 2006-06-28 | Total Petrochemicals Research Feluy | Geo-membrane applications |
| CA2592833A1 (en) * | 2005-01-03 | 2006-07-13 | Dow Global Technologies Inc. | Elastomeric resin compositions with improved resistance to draw resonance |
| US7858723B2 (en) * | 2005-01-31 | 2010-12-28 | Mitsui Chemicals, Inc. | Ethylene-based resin and molded object obtained therefrom |
| AU2006220811A1 (en) * | 2005-03-04 | 2006-09-14 | Dow Global Technologies Inc. | Improved polyethylene resin compositions having low mi and high melt strength |
| US7642330B2 (en) * | 2005-03-31 | 2010-01-05 | Exxonmobil Chemical Patents Inc. | Method of selecting polyolefins based on rheological properties |
| US20060222796A1 (en) * | 2005-04-04 | 2006-10-05 | Morris Barry A | Structure comprising metallized film and ethylene copolymer |
| JP5318567B2 (en) * | 2005-05-12 | 2013-10-16 | ダウ グローバル テクノロジーズ エルエルシー | Thermoformed extruded sheet material with low gloss |
| DE102005023040A1 (en) * | 2005-05-13 | 2006-11-16 | Basell Polyolefine Gmbh | Polyolefinic molding composition for producing pipes with improved resistance comprises thermoplastic polyolefin, and organic polyoxy compound and/or organic polyhydroxy compound |
| US20070045904A1 (en) * | 2005-08-31 | 2007-03-01 | Ulcej John A | Blow molding extrusion apparatus and method |
| US7498282B2 (en) * | 2005-10-26 | 2009-03-03 | Dow Global Technology Inc. | Multi-layer, elastic articles |
| US7491761B2 (en) * | 2005-11-01 | 2009-02-17 | Solutia Incorporated | Poly(vinyl butyral) pellets |
| EP1954730B2 (en) | 2005-11-23 | 2019-10-23 | Dow Global Technologies LLC | Heterogeneous, compositionally phase separated, ethylene alpha-olefin interpolymers |
| US8153243B2 (en) | 2005-12-09 | 2012-04-10 | Dow Global Technologies Llc | Interpolymers suitable for multilayer films |
| US20070182106A1 (en) * | 2006-02-06 | 2007-08-09 | Biesenberger Jeffrey J | Lubeless pipe gasket and method of fabrication |
| WO2007106417A1 (en) | 2006-03-10 | 2007-09-20 | Dow Global Technologies Inc. | Polyethylene resins for sheet and thermoforming applications |
| US7462391B2 (en) * | 2006-05-17 | 2008-12-09 | The Goodyear Tire & Rubber Company | Multi-layered veneer for a tire sidewall |
| JP2010504390A (en) | 2006-09-21 | 2010-02-12 | ユニオン カーバイド ケミカルズ アンド プラスティックス テクノロジー エルエルシー | Method for controlling characteristics in multimodal systems |
| JP5508016B2 (en) * | 2006-10-06 | 2014-05-28 | 株式会社ブリヂストン | Branched polymer and method for synthesis and use thereof |
| US8474830B2 (en) | 2007-06-07 | 2013-07-02 | Springseal, Inc. | Gasket |
| US20090018253A1 (en) * | 2007-07-09 | 2009-01-15 | Eastman Chemical Company | Perfromance additives for thermoplastic elastomers |
| DE102008050219B4 (en) | 2007-10-05 | 2020-06-10 | The Yokohama Rubber Co., Ltd. | Rubber composition for tires, manufacture and use of the like and vulcanized composition |
| US8404773B2 (en) | 2008-03-17 | 2013-03-26 | Dow Global Technologies Llc | Coating composition, method of producing the same, articles made therefrom, and method of making such articles |
| CA2726090C (en) | 2008-05-27 | 2017-11-28 | Springseal, Inc. | Pipe coupling assembly |
| US20110230670A1 (en) * | 2008-07-31 | 2011-09-22 | 3M Innovation Properties Company | Azide compositions and methods of making and using thereof |
| US8772414B2 (en) * | 2008-09-16 | 2014-07-08 | Dow Global Technologies Llc | Polymeric compositions and foams, methods of making the same, and articles prepared from the same |
| US8119549B2 (en) * | 2009-01-27 | 2012-02-21 | Milliken & Company | Consolidated fibrous structure |
| US8147957B2 (en) * | 2009-01-27 | 2012-04-03 | Milliken & Company | Consolidated fibrous structure |
| US8114507B2 (en) * | 2009-01-27 | 2012-02-14 | Milliken & Company | Multi-layered fiber |
| US7960024B2 (en) * | 2009-01-27 | 2011-06-14 | Milliken & Company | Multi-layered fiber |
| US8029633B2 (en) * | 2009-01-27 | 2011-10-04 | Milliken & Company | Method of forming a consolidated fibrous structure |
| US20100239796A1 (en) * | 2009-03-23 | 2010-09-23 | Gagne Joseph Donald | Lap sealable laminate and packaging made therefrom |
| US9265633B2 (en) | 2009-05-20 | 2016-02-23 | 480 Biomedical, Inc. | Drug-eluting medical implants |
| US20110319987A1 (en) * | 2009-05-20 | 2011-12-29 | Arsenal Medical | Medical implant |
| US8888840B2 (en) * | 2009-05-20 | 2014-11-18 | Boston Scientific Scimed, Inc. | Drug eluting medical implant |
| EP2432425B1 (en) * | 2009-05-20 | 2018-08-08 | 480 Biomedical, Inc. | Medical implant |
| RU2012102056A (en) * | 2009-06-23 | 2013-07-27 | ДАУ ГЛОБАЛ ТЕКНОЛОДЖИЗ ЭлЭлСи. | POLYPROPYLENE WITH CONTROLLED RHEOLOGY |
| WO2011002998A1 (en) * | 2009-07-01 | 2011-01-06 | Dow Global Technologies Inc. | Ethylenic polymer and its use |
| US8431651B2 (en) | 2009-07-14 | 2013-04-30 | Dow Global Technologies Llc | Vacuum thermoformed, extruded sheeting with improved reduced gloss |
| DE202009013150U1 (en) * | 2009-10-01 | 2011-02-17 | Peguform Gmbh | Mounting system for mounting a bumper cover |
| WO2011059994A2 (en) | 2009-11-11 | 2011-05-19 | 3M Innovative Properties Company | Polymeric compositions and method of making and articles thereof |
| US20130075409A1 (en) | 2010-04-14 | 2013-03-28 | Mridula Kapur | Fuel containers made from polyethylene compositions with improved creep resistance |
| CN102985476B (en) * | 2010-05-27 | 2015-08-19 | 陶氏环球技术有限责任公司 | Polymer composition, its preparation method and the goods prepared by said composition |
| US8269107B2 (en) | 2010-05-28 | 2012-09-18 | General Cable Technologies Corporation | Halogen-free flame retardant polyolefin |
| CN103328565B (en) | 2010-09-15 | 2015-08-26 | 陶氏环球技术有限责任公司 | There is the propylene-alpha-olefin copolymers composition of the foaming window of improvement |
| WO2012068727A1 (en) * | 2010-11-24 | 2012-05-31 | Dow Global Technologies Llc | Composition comprising propylene-alpha-olefin copolymer, olefin block copolymer and dpo-bsa molecular melt |
| BRPI1107080A2 (en) | 2010-12-30 | 2013-04-24 | Braskem Sa | article forming by blowing and compression |
| US8801049B2 (en) | 2011-04-29 | 2014-08-12 | Springseal, Inc. | Pipe coupling system and method |
| KR102048446B1 (en) * | 2012-08-29 | 2019-11-25 | 다우 글로벌 테크놀로지스 엘엘씨 | Ethylene-based polymer compositions and foams |
| JP5711713B2 (en) * | 2012-10-01 | 2015-05-07 | 住友電気工業株式会社 | Multi-layer heat recovery article |
| US9115274B2 (en) | 2012-12-13 | 2015-08-25 | General Cable Technologies Corporation | Fire and water resistant cable cover |
| US9087629B2 (en) | 2012-12-13 | 2015-07-21 | General Cable Technologies Corporation | Fire and water resistant cable |
| JP6002027B2 (en) | 2012-12-20 | 2016-10-05 | 住友電気工業株式会社 | Multi-layer heat recovery article, wire splice and wire harness |
| JP5651161B2 (en) | 2012-12-20 | 2015-01-07 | 住友電気工業株式会社 | Multi-layer heat recovery article, wire splice and wire harness |
| EP2945994B1 (en) | 2013-01-18 | 2018-07-11 | Basf Se | Acrylic dispersion-based coating compositions |
| KR102367286B1 (en) * | 2013-01-18 | 2022-02-24 | 셀가드 엘엘씨 | Surface modifying agents, modified materials and methods |
| EP2956505B1 (en) | 2013-02-14 | 2017-01-04 | Dow Global Technologies LLC | Ethylene-based polymer compositions with improved processibility |
| US10577440B2 (en) | 2013-03-13 | 2020-03-03 | Chevron Phillips Chemical Company Lp | Radically coupled resins and methods of making and using same |
| US10654948B2 (en) | 2013-03-13 | 2020-05-19 | Chevron Phillips Chemical Company Lp | Radically coupled resins and methods of making and using same |
| JP6527133B2 (en) * | 2013-03-15 | 2019-06-05 | ダウ グローバル テクノロジーズ エルエルシー | EPDM packaging system and process |
| JP6405366B2 (en) * | 2013-05-02 | 2018-10-17 | ダウ グローバル テクノロジーズ エルエルシー | Polyethylene composition and articles made therefrom |
| US10102979B2 (en) | 2013-05-17 | 2018-10-16 | Miltec Corporation | Actinic and electron beam radiation curable water based electrode binders and electrodes incorporating same |
| US9475928B2 (en) | 2013-06-24 | 2016-10-25 | Dow Global Technologies Llc | Reinforced polypropylene composition |
| KR102034340B1 (en) | 2013-06-24 | 2019-10-18 | 다우 글로벌 테크놀로지스 엘엘씨 | Reinforced polypropylene composition |
| EP2883890A4 (en) | 2013-06-28 | 2016-07-20 | Lg Chemical Ltd | Trinary elastic copolymer comprising diene and method for preparing same |
| CN104797610A (en) | 2013-06-28 | 2015-07-22 | 株式会社Lg化学 | Trinary elastic copolymer comprising diene and method for preparing same |
| IN2015DN02232A (en) | 2013-06-28 | 2015-08-21 | Lg Chemical Ltd | |
| KR101585204B1 (en) | 2013-06-28 | 2016-01-13 | 주식회사 엘지화학 | Elastic diene terpolymer and preparation method thereof |
| KR101684648B1 (en) * | 2014-04-21 | 2016-12-08 | 주식회사 엘지화학 | Elastic diene terpolymer and preparation method thereof |
| AR098696A1 (en) | 2013-12-10 | 2016-06-08 | General Cable Tech Corp | THERMALLY CONDUCTIVE COMPOSITIONS AND CABLES OF THESE |
| MX2016009988A (en) | 2014-02-07 | 2016-11-15 | General Cable Tech Corp | Methods of forming cables with improved coverings. |
| EP3110885B1 (en) | 2014-02-24 | 2018-10-03 | Dow Global Technologies LLC | Thermoplastic polyolefin with reduced gloss for non-carpeted flooring |
| KR101783897B1 (en) | 2014-12-10 | 2017-10-10 | 주식회사 엘지화학 | Polyolefin pellet for preparing fiber and fiber comprising the same |
| WO2016109628A1 (en) * | 2014-12-30 | 2016-07-07 | Dow Global Technologies Llc | Sulfonylazide derivative for tie layer |
| EP3241222A4 (en) | 2014-12-30 | 2018-07-18 | General Cable Technologies Corporation | Multi-layer cables |
| US10183924B2 (en) | 2015-12-30 | 2019-01-22 | Dow Global Technologies Llc | Sulfonylazide derivative for diene polymer |
| TWI750182B (en) * | 2016-06-23 | 2021-12-21 | 美商陶氏全球科技有限責任公司 | Aliphatic sulfonyl azide anhydride for tie layer |
| TWI746568B (en) | 2016-06-23 | 2021-11-21 | 美商陶氏全球科技有限責任公司 | Production of sulfonyl azide anhydride |
| DE102016121554A1 (en) * | 2016-11-10 | 2018-05-17 | Wobben Properties Gmbh | Multilayer composite component |
| US11028193B2 (en) | 2017-04-13 | 2021-06-08 | Braskem America, Inc. | Azide-modified olefin as polymeric coupling agent |
| WO2018195756A1 (en) * | 2017-04-25 | 2018-11-01 | 南通大学 | Test tube stand and receiving plate unit with magnetic base used for super clean bench |
| JP7305642B2 (en) | 2017-12-29 | 2023-07-10 | ダウ グローバル テクノロジーズ エルエルシー | Polyolefin-acrylic particles |
| US11584844B2 (en) | 2017-12-29 | 2023-02-21 | Dow Global Technologies Llc | Process for preparing crosslinked polyolefin particles |
| CN111417682B (en) | 2017-12-29 | 2023-01-10 | 陶氏环球技术有限责任公司 | Method for modifying polycarbonate blends |
| KR102295069B1 (en) | 2018-08-17 | 2021-08-26 | 주식회사 엘지화학 | Separator for electrochemical device and manufacturing method thereof |
| KR102474477B1 (en) * | 2018-11-29 | 2022-12-05 | 롯데케미칼 주식회사 | Polyolefin composition having imporoved stress resistance |
| US11073214B2 (en) * | 2019-03-29 | 2021-07-27 | Rapak, Llc | Duckbill valve and method for making a duckbill valve |
| SG11202112115XA (en) | 2019-05-30 | 2021-12-30 | Dow Global Technologies Llc | Method of extruding linear low-density polyethylene (lldpe) without surface fractures in the melt |
| FR3099766B1 (en) | 2019-08-07 | 2021-07-30 | Michelin & Cie | Ethylene-rich diene block polymer having a statistical block and a polyethylene block. |
| FR3116536B1 (en) | 2020-11-26 | 2022-11-25 | Michelin & Cie | An ethylene-rich diene triblock polymer having a random block and two polyethylene terminal blocks. |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3203936A (en) * | 1961-03-15 | 1965-08-31 | Hercules Powder Co Ltd | Cross-linking ethylene polymers |
| DE1569025A1 (en) * | 1964-03-13 | 1970-07-09 | Hercules Inc | Improvements in or relating to polymers containing bridge bonds |
| US5118531A (en) * | 1987-12-18 | 1992-06-02 | Uniroyal Chemical Company, Inc. | Pumpability sealant composition |
| EP0702032A2 (en) * | 1994-09-19 | 1996-03-20 | Sentinel Products Corp. | Cross-linked foam structures of essentially linear polyolefines and process for manufacture |
Family Cites Families (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA797917A (en) | 1968-10-29 | Hercules Incorporated | Bridged polymers | |
| US3203937A (en) | 1961-03-15 | 1965-08-31 | Hercules Powder Co Ltd | Cross-linking ethylene polymers |
| BE622066A (en) | 1961-03-15 | |||
| US3282864A (en) * | 1963-05-31 | 1966-11-01 | Phillips Petroleum Co | Di(sulfonylazides) as crosslinking agents for hydrocarbon polymers |
| US3336268A (en) | 1963-10-14 | 1967-08-15 | Hercules Inc | Bridged polypropylene |
| US3336258A (en) | 1963-11-26 | 1967-08-15 | Du Pont | Aromatic polyamide-acids cross-linked with ditertiary diamines |
| US3530108A (en) | 1964-06-12 | 1970-09-22 | Hercules Inc | Elastic polypropylene film and process |
| US3341480A (en) | 1964-07-02 | 1967-09-12 | Hercules Inc | Cellular polyolefin |
| FR1478692A (en) | 1965-04-15 | 1967-07-12 | ||
| US3298975A (en) | 1966-03-18 | 1967-01-17 | Hercules Inc | Expandable partially-foamed and cured polypropylene composition |
| US3855184A (en) * | 1971-01-27 | 1974-12-17 | E Bostick | Crosslinking of aromatic polymers with organic polysulfonazides |
| NL7207689A (en) | 1972-06-07 | 1973-12-11 | ||
| FR2245702B1 (en) * | 1973-08-24 | 1976-05-07 | Rhone Poulenc Ind | |
| JPS5744456B2 (en) | 1974-04-10 | 1982-09-21 | ||
| DE2911719C2 (en) | 1979-03-24 | 1982-07-01 | Fa. Carl Freudenberg, 6940 Weinheim | Process for the production of lightweight foams from thermoplastic crystalline plastics |
| US4579905A (en) | 1983-08-15 | 1986-04-01 | General Electric Company | Graftlinked polymers and process for making |
| US4714716A (en) | 1984-11-16 | 1987-12-22 | The Dow Chemical Company | Lightly crosslinked linear olefinic polymer foams and process for making |
| US4694025A (en) | 1984-11-16 | 1987-09-15 | The Dow Chemical Company | Alcohol control of lightly crosslinked foamed polymer production |
| GB2205103A (en) | 1987-05-21 | 1988-11-30 | Shell Int Research | Modified block copolymers and process for the preparation thereof |
| US5037895A (en) | 1988-08-04 | 1991-08-06 | Phillips Petroleum Company | Process for improving thermoplastic hydrocarbon polymers with polysulfonazide and free radical catalyst |
| US5594078A (en) | 1991-07-23 | 1997-01-14 | Phillips Petroleum Company | Process for producing broad molecular weight polyolefin |
| US5498581A (en) | 1994-06-01 | 1996-03-12 | Phillips Petroleum Company | Method for making and using a supported metallocene catalyst system |
| BR9509193A (en) | 1994-09-02 | 1997-10-21 | Dow Chemical Co | Thermoset elastomer process for preparing a thermoplastic vulcanized thermoset elastomer process for making a thermoplastic vulcanized and fabricated part |
| US5869591A (en) * | 1994-09-02 | 1999-02-09 | The Dow Chemical Company | Thermoset interpolymers and foams |
| US5654454A (en) | 1995-05-30 | 1997-08-05 | Phillips Petroleum Company | Metallocene preparation and use |
| EP1007589B1 (en) * | 1997-08-27 | 2003-10-01 | The Dow Chemical Company | Coupling of blends of alpha-olefin/vinyl aromatic monomer or hindered aliphatic vinyl monomer interpolymers with polyolefins |
| US6211302B1 (en) * | 1997-08-27 | 2001-04-03 | The Dow Chemical Company | Rheology modification of interpolymers of alpha-olefins and vinylidene aromatic monomers |
| DE19941841A1 (en) | 1999-09-02 | 2001-03-08 | Claas Selbstfahr Erntemasch | Self-propelled agricultural machine |
| US6291618B1 (en) * | 1999-12-23 | 2001-09-18 | The Dow Chemical Company | Phosphazene azide coupling agents and their use in rheology modification of polymers |
-
1998
- 1998-08-05 US US09/129,155 patent/US6211302B1/en not_active Expired - Fee Related
- 1998-08-05 DE DE1998631871 patent/DE69831871T2/en not_active Expired - Lifetime
- 1998-08-05 WO PCT/US1998/016214 patent/WO1999010422A1/en active IP Right Grant
- 1998-08-05 AT AT98938338T patent/ATE322519T1/en not_active IP Right Cessation
- 1998-08-05 EP EP98938337A patent/EP1007586B1/en not_active Expired - Lifetime
- 1998-08-05 US US09/129,161 patent/US6376623B1/en not_active Expired - Lifetime
- 1998-08-05 BR BR9814447A patent/BR9814447A/en not_active Application Discontinuation
- 1998-08-05 AU AU86883/98A patent/AU8688398A/en not_active Abandoned
- 1998-08-05 AT AT98938337T patent/ATE306517T1/en active
- 1998-08-05 KR KR1020007002009A patent/KR100562615B1/en not_active IP Right Cessation
- 1998-08-05 WO PCT/US1998/016213 patent/WO1999010421A1/en not_active Application Discontinuation
- 1998-08-05 WO PCT/US1998/016215 patent/WO1999010423A1/en active IP Right Grant
- 1998-08-05 CA CA 2301644 patent/CA2301644C/en not_active Expired - Fee Related
- 1998-08-05 ES ES98938337T patent/ES2247709T3/en not_active Expired - Lifetime
- 1998-08-05 EP EP19980938336 patent/EP1007585A1/en not_active Withdrawn
- 1998-08-05 KR KR1020007002005A patent/KR100562616B1/en not_active IP Right Cessation
- 1998-08-05 JP JP2000507742A patent/JP4344471B2/en not_active Expired - Fee Related
- 1998-08-05 CA CA 2301836 patent/CA2301836C/en not_active Expired - Fee Related
- 1998-08-05 DE DE1998634123 patent/DE69834123T2/en not_active Expired - Lifetime
- 1998-08-05 JP JP2000507740A patent/JP2001514287A/en active Pending
- 1998-08-05 KR KR1020007002004A patent/KR20010023366A/en not_active Application Discontinuation
- 1998-08-05 AU AU86882/98A patent/AU8688298A/en not_active Abandoned
- 1998-08-05 EP EP19980938338 patent/EP1007587B1/en not_active Expired - Lifetime
- 1998-08-05 AU AU86881/98A patent/AU743240B2/en not_active Ceased
- 1998-08-05 CA CA 2301602 patent/CA2301602A1/en not_active Abandoned
- 1998-08-05 CN CN98809860A patent/CN1273592A/en active Pending
- 1998-08-05 JP JP2000507741A patent/JP2001514288A/en active Pending
- 1998-08-05 TR TR200000905T patent/TR200000905T2/en unknown
- 1998-08-05 US US09/129,163 patent/US6521306B1/en not_active Expired - Lifetime
- 1998-08-13 AT AT98939934T patent/ATE239048T1/en not_active IP Right Cessation
- 1998-08-13 ES ES98939934T patent/ES2192783T3/en not_active Expired - Lifetime
- 1998-08-13 CA CA 2301527 patent/CA2301527A1/en not_active Abandoned
- 1998-08-13 BR BR9814441A patent/BR9814441A/en not_active IP Right Cessation
- 1998-08-13 KR KR1020007001979A patent/KR20010023341A/en not_active Application Discontinuation
- 1998-08-13 AU AU88283/98A patent/AU8828398A/en not_active Abandoned
- 1998-08-13 US US09/133,244 patent/US6143829A/en not_active Expired - Lifetime
- 1998-08-13 WO PCT/US1998/016810 patent/WO1999010415A1/en not_active Application Discontinuation
- 1998-08-13 DE DE1998614098 patent/DE69814098T2/en not_active Expired - Lifetime
- 1998-08-13 CN CN98810324A patent/CN1276811A/en active Pending
- 1998-08-13 EP EP19980939934 patent/EP1007583B1/en not_active Expired - Lifetime
- 1998-08-13 JP JP2000507736A patent/JP2001514284A/en active Pending
- 1998-08-25 MY MYPI98003868A patent/MY128986A/en unknown
- 1998-08-25 MY MYPI98003863A patent/MY133100A/en unknown
- 1998-08-25 MY MYPI98003864A patent/MY121755A/en unknown
- 1998-08-26 HU HU0002756A patent/HUP0002756A3/en unknown
- 1998-08-26 ZA ZA9807750A patent/ZA987750B/en unknown
- 1998-08-26 PL PL33908398A patent/PL339083A1/en unknown
- 1998-08-26 KR KR10-2000-7001910A patent/KR100535582B1/en not_active IP Right Cessation
- 1998-08-26 ZA ZA9807746A patent/ZA987746B/en unknown
- 1998-08-26 WO PCT/US1998/017685 patent/WO1999010427A1/en active IP Right Grant
- 1998-08-26 ZA ZA9807745A patent/ZA987745B/en unknown
- 1998-08-26 EP EP19980941064 patent/EP1007590A1/en not_active Withdrawn
- 1998-08-26 CN CNB988094347A patent/CN1166730C/en not_active Expired - Lifetime
- 1998-08-26 US US09/140,603 patent/US6528136B1/en not_active Expired - Lifetime
- 1998-08-26 AR ARP980104237 patent/AR016885A1/en unknown
- 1998-08-26 TR TR200001175T patent/TR200001175T2/en unknown
- 1998-08-26 BR BR9814442A patent/BR9814442A/en not_active Application Discontinuation
- 1998-08-26 ID ID20000351A patent/ID25634A/en unknown
- 1998-08-26 AR ARP980104238 patent/AR016886A1/en unknown
- 1998-08-26 AR ARP980104239 patent/AR016887A1/en unknown
- 1998-08-26 AR ARP980104241 patent/AR016889A1/en unknown
- 1998-08-26 CA CA 2301835 patent/CA2301835C/en not_active Expired - Lifetime
- 1998-08-26 MY MYPI98003897A patent/MY126503A/en unknown
- 1998-08-26 ZA ZA9807752A patent/ZA987752B/en unknown
- 1998-08-26 AU AU89210/98A patent/AU743302B2/en not_active Ceased
- 1998-08-26 JP JP2000507746A patent/JP4342721B2/en not_active Expired - Lifetime
- 1998-08-26 ZA ZA9807753A patent/ZA987753B/en unknown
- 1998-08-26 AR ARP980104240 patent/AR016888A1/en unknown
- 1998-09-29 TW TW87114112A patent/TW591070B/en active
- 1998-09-29 TW TW87114107A patent/TW584641B/en active
- 1998-09-29 TW TW87114109A patent/TW588059B/en active
- 1998-10-01 TW TW87114111A patent/TW584652B/en active
-
2000
- 2000-02-25 NO NO20000966A patent/NO322848B1/en unknown
- 2000-09-29 TW TW91132924A patent/TW200300428A/en unknown
- 2000-10-23 US US09/694,649 patent/US6359073B1/en not_active Expired - Lifetime
-
2001
- 2001-10-29 US US10/021,134 patent/US6506848B2/en not_active Expired - Lifetime
- 2001-12-10 US US10/013,152 patent/US6552129B2/en not_active Expired - Lifetime
-
2003
- 2003-01-21 US US10/347,713 patent/US6777502B2/en not_active Expired - Lifetime
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3203936A (en) * | 1961-03-15 | 1965-08-31 | Hercules Powder Co Ltd | Cross-linking ethylene polymers |
| DE1569025A1 (en) * | 1964-03-13 | 1970-07-09 | Hercules Inc | Improvements in or relating to polymers containing bridge bonds |
| US5118531A (en) * | 1987-12-18 | 1992-06-02 | Uniroyal Chemical Company, Inc. | Pumpability sealant composition |
| EP0702032A2 (en) * | 1994-09-19 | 1996-03-20 | Sentinel Products Corp. | Cross-linked foam structures of essentially linear polyolefines and process for manufacture |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001083605A3 (en) * | 2000-05-04 | 2002-01-31 | Dow Chemical Co | Molecular melt and methods for making and using the molecular melt |
| JP2003531939A (en) * | 2000-05-04 | 2003-10-28 | ダウ グローバル テクノロジーズ インコーポレイテッド | Molecular melt and method of making and using molecular melt |
| US7399737B2 (en) | 2005-06-13 | 2008-07-15 | Exxonmobil Chemical Patents Inc. | Lube additives |
| WO2011062961A1 (en) * | 2009-11-23 | 2011-05-26 | Dow Global Technologies Inc. | Compositions, films and methods of preparing the same |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1007586B1 (en) | Rheology modification of low density polyethylene | |
| EP0717759A1 (en) | Batch inclusion packages | |
| MXPA00002017A (en) | Rheology modification of polymers prepared using metallocenes | |
| CZ2000710A3 (en) | Modification process of polymer rheology that are prepared by employing metallocenes | |
| MXPA00002008A (en) | Rheology modification of interpolymers of alpha-olefins and vinyl aromatic monomers |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH GM HR HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2301644 Country of ref document: CA Ref country code: CA Ref document number: 2301644 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020007002009 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998938337 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998938337 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020007002009 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1998938337 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1020007002009 Country of ref document: KR |