BROAD RANGE PCR AMPLIFICATION TECHNIQUES
Statement as to Federally Sponsored Research This invention was made with Government support from the
National Science Foundation under Grant No. MCB 9405961. The Government has certain rights in the invention.
Background of the Invention The invention relates to novel polymerase chain reaction (PCR) amplification techniques and their use, for example, for identifying single nucleotide polymorphisms.
Dense linkage maps are invaluable tools for genetic and genomic analysis. They facilitate high resolution genetic mapping, positional cloning of monogenic traits, genetic dissection of poly genie traits, fine-structure linkage disequilibrium studies, and the construction of genome- wide physical maps. Historically, genetic maps were constructed with visible markers, but it is difficult to examine many such markers in a single cross. The recognition that distantly related individuals differ in DNA sequence throughout their genome (Botstein et al., Am. J. Hum. Genet. 32: 314-331, 1980) led to the rapid incorporation of DNA markers into mapping strategies. Useful DNA markers have the following general characteristics: (1) they are inherited in a Mendelian fashion; (2) they are present in most individuals analyzed and recognize a sequence that is polymoφhic; (3) they correspond to a single site in the genome; (4) the probe used to recognize the marker hybridizes selectively and efficiently, even under conditions of low stringency; and (5) they can be distributed throughout a community, either as clones or as DNA sequences.
Until recently, the most commonly used DNA markers were restriction fragment length polymorphisms (RFLPs), anonymous single copy-number genomic clones that reveal a polymorphism in the length of a restriction fragment, typically by DNA blot hybridization. RFLP mapping is well-suited for determining the genetic location of any newly-cloned DNA sequence; the DNA fragment can be used as a hybridization probe (assuming it detects an RFLP) against the DNA filters used to construct the RFLP map. However, in many cases, new genes are identified by mutations, and mapping such a mutation onto an RFLP map can be a lengthy and arduous procedure.
Summary of the Invention
In general, the invention features a method for determining whether a nucleic acid sequence includes a particular allele of a polymoφhic sequence, involving:
(a) contacting a nucleic acid sequence, in the same or a separate reaction, with a first pair of PCR primers and a second pair of PCR primers under conditions that allow hybridization of the PCR primers to the nucleic acid sequence, the first pair of PCR primers hybridizing to opposite strands of the nucleic acid sequence and bordering the position of the polymoφhic sequence, and the second pair of PCR primers hybridizing to opposite strands of the nucleic acid sequence and bordering the position of the polymoφhic sequence, the PCR primers being characterized as follows:
(i) one of the first pair of PCR primers (a) being complementary at its 3'-terminal nucleotide to a first allele of the polymoφhic sequence (allele A), (b) being non-complementary at its 3'-terminal nucleotide to a second allele of the polymoφhic sequence (allele B), and (c) being non- complementary to the nucleic acid sequence at a single non-complementary
nucleotide in its 3'-terminal nucleotides 2-6; and
(ii) one of the second pair of PCR primers (a) being complementary at its 3 '-terminal nucleotide to the first allele of the polymoφhic sequence (allele A), (b) being non-complementary at its 3'- terminal nucleotide to the second allele of the polymoφhic sequence (allele B), and (c) being non-complementary to the nucleic acid sequence at one (and, preferably, two) or more nucleotides in its 3 '-terminal nucleotides 2-6;
(b) carrying out the amplification reactions; and
(c) detecting an amplification product as an indication of the presence, in the nucleic acid sequence, of the first allele of the polymoφhic sequence (allele A).
If desired, the method may involve the further steps of: (a) contacting the nucleic acid sequence, in the same or a separate reaction, with a third pair of PCR primers and a fourth pair of PCR primers under conditions that allow hybridization of the PCR primers to the nucleic acid sequence, the third pair of PCR primers hybridizing to opposite strands of the nucleic acid sequence and bordering the position of the polymoφhic sequence, and the fourth pair of PCR primers hybridizing to opposite strands of the nucleic acid sequence and bordering the position of the polymoφhic sequence, the PCR primers being characterized as follows:
(i) one of the third pair of PCR primers (a) being complementary at its 3'-terminal nucleotide to the second allele of the polymoφhic sequence (allele B), (b) being non-complementary at its 3'- terminal nucleotide to the first allele of the polymoφhic sequence (allele A), and (c) being non-complementary to the nucleic acid sequence at a single nucleotide in its 3'-terminal nucleotides 2-6; and
(ii) one of the fourth pair of PCR primers (a) being
complementary at its 3'-terminal nucleotide to the second allele of the polymoφhic sequence (allele B), (b) being non-complementary at its 3'- terminal nucleotide to the first allele of the polymoφhic sequence (allele A), and (c) being non-complementary to the nucleic acid sequence at one (and, preferably, two) or more nucleotides in its 3'-terminal nucleotides 2-6;
(b) carrying out the amplification reactions; and
(c) detecting an amplification product as an indication of the presence, in the nucleic acid sequence, of the second allele of the polymoφhic sequence (allele B). In a related aspect, the invention features kits for carrying out the method of the invention. One particular kit for determining whether a nucleic acid sequence includes a particular allele of a polymoφhic sequence includes (a) a first pair of PCR primers and a second pair of PCR primers, the first pair of PCR primers hybridizing to opposite strands of the nucleic acid sequence and bordering the position of the polymoφhic sequence, and the second pair of PCR primers hybridizing to opposite strands of the nucleic acid sequence and bordering the position of the polymoφhic sequence, the PCR primers being characterized as follows: (i) one of the first pair of PCR primers (a) being complementary at its 3 '-terminal nucleotide to a first allele of the polymoφhic sequence (allele A), (b) being non-complementary at its 3'-terminal nucleotide to a second allele of the polymoφhic sequence (allele B), and (c) being non- complementary to the nucleic acid sequence at a single non-complementary nucleotide in its 3'-terminal nucleotides 2-6; and (ii) one of the second pair of PCR primers (a) being complementary at its 3'-terminal nucleotide to the first allele of the polymoφhic sequence (allele A), (b) being non-complementary at its 3'-terminal nucleotide to the second allele of the polymoφhic sequence (allele B), and (c) being non-complementary to the nucleic acid sequence at one
(and, preferably, two) or more nucleotides in its 3 '-terminal nucleotides.
If desired, the kit may also include (a) a third pair of PCR primers and a fourth pair of PCR primers, the third pair of PCR primers hybridizing to opposite strands of said nucleic acid sequence and bordering the position of the polymoφhic sequence, and the fourth pair of PCR primers hybridizing to opposite strands of the nucleic acid sequence and bordering the position of the polymoφhic sequence, the PCR primers being characterized as follows: (i) one of the third pair of PCR primers (a) being complementary at its 3 '-terminal nucleotide to the second allele of said polymoφhic sequence (allele B), (b) being non-complementary at its 3 '-terminal nucleotide to the first allele of the polymoφhic sequence (allele A), and (c) being non-complementary to the nucleic acid sequence at a single nucleotide in its 3'-terminal nucleotides 2-6; and (ii) one of the fourth pair of PCR primers (a) being complementary at its 3'- terminal nucleotide to the second allele of the polymoφhic sequence (allele B), (b) being non-complementary at its 3 '-terminal nucleotide to the first allele of the polymoφhic sequence (allele A), and (c) being non-complementary to the nucleic acid sequence at one (and, preferably, two) or more nucleotides in its 3 '-terminal nucleotides 2-6.
In preferred embodiments of any of the above methods or kits, the amplification reaction involving the first pair of PCR primers and the amplification reaction involving the second pair of PCR primers have different ranges of specificity; have ranges of specificity that overlap; and together have a greater than 3000-fold, and preferably at least a 10,000-fold, range of specificity. In addition, the methods and kits are used to identify a single nucleotide polymoφhism; each of the primers of the first and the second primer pairs that includes a non-complementary nucleotide in 3 '-terminal nucleotides
2-6 may also include a unique hybridization tag and/or a universal primer binding site; the detection step is facilitated by the hybridization tag and/or the universal priming site; and the detection step is carried out on a solid support (for example, a chip) to which a binding partner for each hybridization tag is immobilized.
As used herein, by "polymoφhic sequence" is meant any nucleotide sequence capable of variation, and by "allele" is meant one such variation. Preferably, such a variation is common in a population of organisms and is inherited in a Mendelian fashion. Such alleles may or may not have associated phenotypes. A "single nucleotide polymoφhism" (or "SNP") is one type of "polymoφhic sequence" which is characterized by a sequence variation of only one nucleotide.
By "range of specificity" is meant the range of nucleic acid template:PCR primer ratios at which template sequences differing by at least one nucleotide may be discriminated by assaying for the presence of detectable PCR amplification product formation.
By "hybridization tag" is meant an oligonucleotide that differs sufficiently in sequence from a target nucleic acid (for example, a target nucleic acid to be amplified) that significant cross-hybridization does not occur. When multiple hybridization tags are utilized in a single reaction mixture, these tags also preferably differ in sequence from one another such that each has a unique binding partner.
As described more fully below, the technique described herein provides a significant advance over other PCR-based techniques, particularly for carrying out genomic mapping analyses. For example, one widely used, more conventional PCR-based approach involves the use of single, short PCR
primers of arbitrary sequence (called "RAPD" primers for "random amplified polymoφhic DNA;" Williams et al., Nucleic Acids Research 18: 6531-6535, 1990). In a given individual, amplification with a RAPD primer typically results in the synthesis of one or more DNA fragments, while in another individual, the primer fails to amplify the same set of fragments. Because RAPD markers are dominant, they do not allow heterozygotes to be reliably scored (see Botstein et al., 1980, supra). In addition, because RAPD primers typically have low melting temperatures, the amplification of a specific sequence or sequences using such a primer is highly sensitive to PCR conditions, including template concentration and annealing temperature. It is thus often difficult to correlate results obtained by different research groups (Devos and Gale, Theor. Appl. Genet. 84: 567-572, 1992). Finally, because RAPD primers frequently amplify more than one sequence, resulting in multiple bands, analysis of the results can be complicated (Riedy et al., PCR. Nucleic Acids Research 20: 918, 1992).
Similarly, another technique in current usage exploits "AFLPs," or "amplified fragment length polymoφhisms." In this method, DNAs from two polymoφhic individuals are cleaved with one or two restriction endonucleases and adapters are ligated to the ends of the cleaved fragments (Vos et al., Nucleic Acids Research 23: 4407-4414, 1995). The fragments are then amplified using primers that are homologous to the adapter(s) which contain a short stretch of random nucleotides at the 3' end. These random nucleotides limit the number of amplified fragments and reveal polymoφhisms between the two individuals which are detected by displaying the amplified products on an acrylamide sequencing gel. Although large numbers of AFLPs can be detected in a single lane in a sequencing gel, this technique is limited by its requirement for acrylamide gel detection, as well as by the fact that many fragments are
generally amplified in each lane, resulting in a complicated pattern that requires expensive, automated high-resolution imaging technology to reliably decipher. Finally, in yet another PCR technique, markers referred to as "simple sequence length polymoφhisms" or "SSLPs" are utilized. These makers are based on amplification across tandem repeats of one or a few nucleotides known as "microsatellites." Microsatellites occur randomly in most eukaryotic genomes and display a high degree of polymoφhism due to variations in the number of repeat units. Simple sequence repeats are very abundant in most mammalian genomes, and the most common simple sequence repeat is (CA)n (Dietrich et al., Proc. Natl. Acad. Sci. USA 92: 10849-10853, 1995). The repeat length varies among individuals in a species, apparently due to slippage during DNA replication (Dietrich et al., Genetics 131 : 423-447, 1992). One major advantage of SSLPs is that they are co-dominant markers. That is, different patterns are obtained for organisms that are homozygous and heterozygous for the paternal alleles. Another advantage of SSLPs is that, because they are highly polymoφhic at a given locus, randomly selected SSLPs are likely to be informative in any given mapping population, and are therefore especially useful for studying evolutionary relationships. However, like AFLPs, certain SSLP markers can only be assayed by acrylamide gel elecfrophoresis and currently available SSLP assay methods are not suited to high throughput analysis using micro DNA arrays (for example, displayed on DNA chips) (Fodor et al., Science 251 : 767-773, 1991; Chee et al., Science 274: 610-614, 1996; and Southern, Trends in Genetics 12: 110-115, 1996).
In contrast to the above techniques, the presently claimed approach provides a method for mapping polymoφhic alleles that combines a number of advantageous features into a single format. First, the present technique makes use of allele-specific markers that are co-dominant; this facilitates the
identification of polymoφhic markers in homozygotes as well as heterozygotes. In addition, the present PCR technique may be readily automated, making it a practical method for large scale mapping efforts. This automation feature stems from the fact that the technique makes use of two allele-specific primers for each particular allele having different and complementary ranges of specificity, a feature that results in an increase in the range of template DNA concentrations that may be reliably assayed. This aspect of the invention is particularly important because determinations of sample DNA concentrations need not be measured, allowing the present technique to be used in conjunction with increasingly popular solid state formats, such as DNA chip formats.
Other features and advantages of the invention will be apparent from the following detailed description, and from the claims.
Brief Description of the Drawings FIGURE 1 is a schematic representation of the allele-specific PCR method. In this figure, primer pairs specific for allele 1 (PI and P3) amplify allele 1, but should not (in theory) amplify allele 2. PI forms a mismatch at the 3' end when hybridized to allele 2.
FIGURES 2A and 2B are graphs indicating the increase in product yields of alleles A (target) and B (non-target) as functions of the number of PCR cycles when using different DNA template concentrations. In Figure 2A, 0.01 nanograms of template DNA are utilized, and, in Figure 2B, 40 nanograms of template DNA are used. The relative efficiencies of amplification used for the calculations were 0.001 for primer PI and 0.007 for primer P2 in a two primer system. PI and P2 are specific for allele A. The closed squares represent Allele A/Primer 1 ; the open squares represent Allele B/ Primer 1 ; the
closed circles represent Allele A/Primer 2; and the open circles represent Allele B/Primer 2.
FIGURE 3 is a graph illustrating the increase in product yield of target (closed squares) and non-target (open squares) as a function of DNA template concentration. Product yield was determined according to Ugozzoli and Wallace, 1991, infra.
FIGURE 4 is a graph illustrating the increase in product yield of allele A (target) and allele B (non-target) as a function of DNA template concentration in a two primer system. Values of product yield were calculated based on 35 cycles of amplification with primers PI and P2 (both specific for allele A). The closed squares represent Allele A Primer 1; the open squares represent Allele B/ Primer 1; the closed circles represent Allele A/Primer 2; and the open circles represent Allele B/Primer 2.
FIGURE 5 is series of photographs and graphs illustrating the hybridization pattern of alleles A and B amplified with allele-specific primers P1/P2 (specific for allele A) and P3/P4 (specific for allele B).
FIGURE 6 is a schematic representation of the use of the present method in a DNA chip format.
Detailed Description The present invention features an improved PCR amplification technique that makes use of two sets of PCR primers for each allele of a polymoφhic sequence that differ in their amplification efficiencies due to the presence of differing numbers of nucleotides that are mismatched relative to the target sequence to be amplified. This improvement increases the range of specificity for the amplification step and provides a technique useful for the reliable detection of single nucleotide (allele-specific) polymoφhisms. In so
doing, the present approach greatly facilitates the use of allele-specific markers in the construction of genetic linkage maps, the detection of mutations or alleles in many organisms, and the sub-species typing of individuals, strains, or varieties. This invention is of particular importance because it allows total automation of the single nucleotide polymoφhism detection process, for example, through the use of DNA chip technology, representing a significant advance in such detection procedures.
The present approach is now described in detail.
Allele-specific PCR Markers "Allele-specific PCR" is an application of PCR in which alleles that differ by one or more nucleotides can be distinguished on the basis of an amplification product (Ugozzoli and Wallace, Methods: A Companion to Methods in Enzymology 2: 42-48, 1991). As illustrated in Figure 1, the technique utilizes primers with specific mismatches at or near the 3' end that permit preferential amplification of one allele (the target allele) relative to another (the non-target allele) (Ugozzoli and Wallace, 1991, supra; and Cha et al., PCR Methods and Applications 2: 14-20, 1992). This procedure offers the possibility of generating single nucleotide polymoφhism (SNP)-based markers for the construction of linkage maps, and represents an excellent option for constructing dense maps composed entirely of these markers. Allele-specific PCR has been used previously in attempts to detect the presence or absence of one or more variant nucleotide sequences by amplification (European Patent Application No 89302331.7, Publication No 0332435), including attempts to detect point mutations associated with a variety of genetic diseases (Ugozzoli and Wallace, 1991, supra; Wenham et al., Clinical Chemistry 37: 241-244, 1991 ; and Chang et al, BioTechniques 22: 520-527, 1997).
Allele-specific markers are co-dominant (as long as primer pairs for specifically amplifying each of the two alleles are used), are very abundant, and are easily assayed on agarose gels. In their current usage, however, allele-specific markers have some general limitations. For example, one of the main limitations encountered in the past when using allele specific primers, and the most important obstacle for the use of these primers as markers for mapping puφoses, is their relatively poor range of specificity (briefly, "range of specificity" refers to the ability of the markers to discriminate between two alleles). Relatively poor specificity represents a major problem when using these markers at high template DNA concentrations, because of the possibility of obtaining false positive results (i.e., too much amplification of the non-target allele). Previous estimates of the sensitivity of allele-specific PCR (determined by ethidium bromide staining) established that the method can reliably detect a point mutation in genomic DNA samples occurring at a frequency of approximately 1 in 40 (mutant to wild type allele ratio) (Sarkar et al.,
Analytical Chemistry 186: 64-68, 1990). This indicates a rather poor sensitivity and consequently inadequate levels of specificity for the ready application of this methodology to the construction of linkage maps.
In contrast, the methods of the present invention make use of two allele-specific primers for the identification of each SNP allele. These two primers have different and complementary ranges of specificity, therefore increasing the range of template DNA concentrations that may be reliably assayed. One of the primers is specific at low template DNA concentrations, and the second one shows specificity at higher concentrations of template DNA, covering in total a larger range of concentration than single allele-specific primers cuπently in use.
Use of Allele-Specific Markers for the Construction of Linkage Maps
The allele-specific PCR procedure involves the detection of the presence or absence of one or more variant nucleotide sequences by amplification. The method relies on the presence of such nucleotide differences for the detection and analysis of genetic polymoφhisms (Ugozzoli and Wallace, 1991, supra). Specific primers containing a 3'-terminal mismatch are designed to preferentially amplify one allele relative to another, as mismatched 3 '-termini are PCR extended with much lower efficiencies than coπectly matched termini by DNA polymerases (Petruska et al., Proc. Natl. Acad. Sci. USA 85: 6252-6256, 1988).
Although efficiency of extension may be considerably reduced during the first cycle of amplification, once extension from a mismatched primer occurs, the resultant product is fully matched with both primers, and accumulates exponentially after it is formed. Therefore, primers with mismatches at or near the 3' end are still able to extend to some degree, and a PCR product is obtained from the amplification of both alleles at the end of the amplification. The degree of specificity of the primers is therefore determined by the difference in efficiency of extension observed when amplifying target and non- target alleles with mismatched primers. Consequently, allele-specific markers will show specificity only when the product yield from the target allele exceeds the threshold of detection for the system used, and the product yield from the non-target allele does not reach that detection level.
Reductions of up to 3, 000-fold in the efficiency of extension of mismatched primers compared to perfect match primers have been reported (Cha et al., 1992, supra). We calculated that for those values of relative efficiency of extension, allele-specific markers will show specificity over an approximate 2,000 to 3,000 fold range of DNA concentrations. These ranges
of specificity are generally acceptable when the detection system used to score the presence or absence of the PCR product is gel electrophoresis, and the concentration of the sample has been previously determined. However, for the use of solid state technologies, such as DNA chip technology, where multiplex PCR is employed and the template DNA concentrations is unknown, those ranges of specificity may be insufficient. In multiplex PCR, different mismatched primers compete for reactants. When the efficiencies of extension of the primers used in the same reaction are not similar, differences among primers are amplified in each round of PCR, modifying individual yields and creating an imbalance in the system (Ferrie et al., American Journal of Human Genetics 51 : 251-262, 1992). Under those circumstances the range of specificity of individual primers changes (Ferrie et al., 1992, supra) compromising the accuracy of the determinations. This difficulty is overcome by the present invention through an increase in the range of specificity of the markers and a resultant decrease in the possibility of error.
Broad Range PCR Amplification and its Use for Allele-Specific Markers
The present invention involves the use of two sets of allele-specific primers for the identification of each allele. Figure 2 illustrates the pattern of specificity observed for two alleles, A and B, when amplified with primers PI and P2, each of which preferentially amplifies allele A (shown in this figure is the amplification of only one allele for simplification puφoses). One of the primers is specific at low template DNA concentrations (Figure 2A), and the second one shows specificity at higher concentrations of template DNA (Figure 2B). Moreover, the range of specificity of the two primers used to identify each one of the alleles overlaps in order to cover the entire range of DNA template concentration of the sample (Figures 2 A and 2B).
According to the present technique, the two primers that are used to detect the same allele are designed to include one or two mismatches (but not a 3 '-terminal mismatch) near the 3' end, depending on the degree of specificity that each primer should possess. Mismatch combination, location, and number of mismatches determines the efficiency with which the mismatched primers are extended. Previous studies have shown that different mismatch combinations located at the 3' end are extended with different efficiencies by Taq polymerase (Newton et al., Nucleic Acids Research 17:2503-2516, 1989; Kwok et al., Nucleic Acids Research 18:999-1005, 1990; Li et al., Proc. Natl. Acad. Sci. USA 87:4580-4584, 1990; and Sommer et al., BioTechniques 12:82- 87,1992). However, the presence of a single mismatch at the 3'-terminus of the non-target allele is sometimes insufficient to generate the desired level of discrimination with respect to the target allele, especially for mismatch combinations with efficiencies of extension that are close to the perfect match. Under these circumstances, the addition of one, and even two, additional mismatches with the non- target allele may be used to destabilize the 3 '-end, providing greater differentiation with the target allele (Newton et al., 1989, supra; Cha et al., 1992, supra). For example, the addition of an extra mismatch within the last four bases of the primer may be coupled with the natural 3 '-terminal mismatch to reduce PCR product yield of the non- target allele compared to the 3'-end mismatch alone (Kwok et al., 1990, supra). On the other hand, single base mismatches located either one, two, or three bases from the 3 '-terminal nucleotide of the primer may be extended without significantly affecting the overall product yield of the target allele (which by definition is one that is perfectly matched with the primer at the 3'-terminus) (Kwok et al., 1990, supra).
Primers according to the present method are tailored to the particular
sequence to be amplified, rather than being part of a random (for example, degenerate) oligonucleotide pool. As noted above, for any particular polymoφhic sequence, the allele-specific primers for two particular alleles (A and B) of a polymoφhic sequence differ at their 3' terminal nucleotides, the primer designed to detect allele A being complementary to allele A at the 3'- terminal nucleotide position and the primer designed to detect allele B being complementary to allele B at the 3 '-terminal nucleotide position. The primer designed to detect allele A at low sample DNA concentration is generally designed by the addition of one introduced mismatch with respect to allele A which occurs within 6 nucleotides of the 3' end, but not at the 3 '-terminal nucleotide. Since the second primer designed to detect allele A needs to be specific at higher sample DNA concentrations, two or more (typically, 2 or 3) mismatches are introduced into this second primer with respect to allele A (but again not at the 3 '-terminal nucleotide) to decrease amplification efficiency of allele B to the required value. The mismatches in this second primer are positioned using the same general parameters, that is, within 6 nucleotides from the 3' end of the primer. Alternatively, the "high DNA concentration" primer for allele A may instead include only a single mismatch with allele A which causes a lower efficiency of extension than the "low DNA concentration" primer. By the above design, the two primers that detect allele A contain in addition to the "internal" mismatches described above, a 3'-terminal mismatch with allele B. For any of the above destabilizing mismatched nucleotides, the choice of a particular primer/template mismatch (i.e., A/A, T/T, C/C, G/G, A/C, C/A, A/G, G/A, T/C, C/T, T/G, or G/T) is dependent upon the mismatch combinations that are available and that which is appropriate for any given sequence context.
The length of the primers used as allele-specific primers in this
invention depend on the detection method used to identify the amplification products. In the case where gel elecfrophoresis is used to detect amplification products, for example, the allele-specific primers are in general between 18 and 30 nucleotides in length, and preferably between 24 and 26 nucleotides (with 24 nucleotides being the most preferred).
In the case where a DNA hybridization method to a solid support is used to identify the amplification products, the allele-specific primers contain the following elements. First, the primers include a sequence proximal to the 5' end of the primer that serves as a "forward" universal primer binding site (e.g., the sequence of the phage T3 binding site for RNA polymerase). Second, in the middle of the primer, it includes a so-called unique "tag" sequence composed of approximately 20 nucleotides that does not have a corresponding sequence in the target DNA to be amplified and which serves to bind the PCR product to a solid support that contains a sequence complementary to the tag. The length of the tag sequence can be varied as required depending on the method used to detect the PCR product. And third, the primer includes a sequence proximal to its 3' end that is approximately 20-24 nucleotides and that corresponds to the sequence flanking the polymoφhic sequence to be detected. The length of the reverse primers in this invention, irrespective of the detection method, are in general between 18 and 30 nucleotides in length, and preferably between 24 and 26 nucleotides (with 24 nucleotides being the most preferred). The reverse primers used in combination with the specificity primers may be chosen from any sequence complementary to the opposite nucleic acid strand and positioned on the opposite side of the allelic marker. These reverse primers are designed using standard PCR methodologies (see, for example, PCR Technology, Erlich, ed., Stockton Press, London, 1989; PCR Protocols: A Guide to Methods and Applications, Innis et al., eds., Academic
Press, Inc., New York, 1990; and Ausubel et al., Current Protocols in Molecular Biology, Wiley Interscience, New York, 1997).
Amplification reactions using the above primer sets are carried out by standard techniques (see references above), with the number of PCR cycles depending on the method of detection. In addition, the concentration of dNTPs may be used to modify primer specificity. For example, lower dNTP levels generally increase the stringency of the amplification (Kwok et al., A guide to the design and use of mismatched and degenerate primers, Manual Supplement, PCR Methods and Applications, S39-S47, 1994), because mismatch extension efficiency depends on the absolute concentration of the next correct nucleotide. In the experiments described herein, optimum dNTP concentration was approximately 125 mM. And the optimum values for primer concentration and Mg+2 concentration were 7.5 pmol per reaction and 1.5 mM, respectively.
The primers used in the present methods are preferably DNA, and can be synthesized using standard techniques and, when appropriate, detectably labeled using any desired standard method (Ausubel et al., supra). In one preferred method, PCR products are labeled using universal primers. By this technique, universal primer binding sites are included, for example, in the allele-specific primers used to amplify the polymoφhic sequences. The product of this initial amplification reaction is then further amplified using detectably labelled (for example, fluorescently labelled) universal primers (that are complementary to the universal primer binding sites) to generate detectably labelled amplification products. This universal primer technique is particularly useful in combination with a solid support (for example, a chip) format. In the methods of the invention, any detectable label may be used including, but not limited to, digoxigenin, fluorescent labels (e.g., fluorescein and rhodamine), enzymes (e.g., horseradish peroxidase and alkaline
phosphatase), biotin (which can be detected by anti-biotin specific antibodies or enzyme-conjugated avidin derivatives), radioactive labels (e.g., 32P and I25I), colorimetric reagents, and chemiluminescent reagents. The labels used are detected using standard methods. In addition, nucleic acid samples containing a polymoφhic sequence to be analyzed may be obtained from any source, e.g., a tissue homogenate, fluid, or culture, and these are also prepared using standard methods.
Moreover, as mentioned above, the present method may be carried out using solid support- type formats. The solid supports useful in the invention include, but are not limited to, agarose, acrylamide, and polystyrene beads; polystyrene microtiter plates (for use in, e.g., ELISA); and nylon and nitrocellulose membranes (for use in, e.g., dot or slot blot assays). In a preferred embodiment of the invention, the solid support contains an array of nucleic acid probes. In this case, solid supports made of materials such as glass (e.g., glass plates), silicon or silicon-glass (e.g., microchips), or gold (e.g., gold plates) can be used. Methods for attaching nucleic acid probes to precise regions on such solid surfaces, e.g., photolithographic methods, are well known in the art, and can be used to make solid supports for use in the invention. Examples of such techniques are described, for example, in Schena et al., Science 270:467-470, 1995; Kozal et al., Nature Medicine 2(7):753-759, 1996; Cheng et al., Nucleic Acids Research 24(2):380-385, 1996; Lipshutz et al., BioTechniques 19(3):442-447, 1995; Pease et al., Proc. Natl. Acad. Sci. USA 91 :5022-5026, 1994; Fodor et al., Nature 364:555-556, 1993; Pirrung et al., U.S. Patent No. 5,143,854; and Fodor et al., WO 92/10092. In practice, assaying a specific polymoφhic allele may involve four separate PCR reactions (two pairs of allele-specific primers for each one of the two target and non- target alleles). Depending on the technique used to assay
the PCR results, these reactions may be carried out separately (for example, if products are scored by a gel electrophoretic technique) or together (for example, if products are scored by hybridization to immobilized binding partners, such as those immobilized on a DNA chip). The actual results of the assay reflect the DNA concentration of the original template. Examples of all possible scoring alternatives are shown in Table 1. In this Table, P1/P2 preferentially amplify allele A, and P3/P4 are specific for and amplify allele B.
TABLE 1
Template DNA Genotype PI P2 P3 P4 Concentration
Low Allele A +
Medium Allele A + + - -
High Allele A + + + -
Low Allele B - - + -
Medium Allele B _ _ +
+
High Allele B + +
Low Heterozygous + +
Medium Heterozygous + + +
+
High Heterozygous + + +
+
Estimation of the Range of PCR Amplification Specificity
Figure 3 shows the range of specificity observed for a single allele-specific primer when the method of detection used is agarose gel
electrophoresis. The calculations for Figure 3 were made considering the lowest values of efficiency of extension that could be obtained on average from all different mismatch combinations. According to theoretical calculations and experimental data, we established that primers with an average relative efficiency of extension of 10"3 would maintain their specificity over an approximate 1,000 fold range of DNA concentrations.
On the other hand, Figure 4 shows the range of specificity obtained from the use of two allele-specific primers when the method of detection is also agarose gel elecfrophoresis. As shown in Figure 4 the range of specificity for the two marker system increased 10-fold compared to the method that used only one allele-specific marker (Figure 3). This analysis indicates that the use of two allele-specific primers allows the use of allele-specific markers in those cases in which the samples analyzed show up to 10,000-fold variations in their DNA concentrations. This increase in range of specificity is particularly useful for techniques in which DNA samples of different concentrations are utilized. For example, in fully automated DNA chip approaches, because samples are multiplexed and because it is not possible to adjust PCR conditions to take into account variations in DNA concentrations, this technique represents a significant improvement over standard methodologies.
Experimental Identification of Alleles Using Broad Range PCR Amplification Since the range of specificity covered by the primers used to identify each one of the alleles determines the specificity of the amplification step over an appropriate range of template concentrations, range of specificity constitutes an important parameter of the present technique. As deduced from Figures 3 and 4, the larger the ratio between the product yields obtained from the
amplification of target and non-target alleles with the mismatched primers, the larger the range of DNA concentrations within which the marker shows specificity.
In a series of experiments, we determined relative product yields between different mismatch primers and their respective perfect match primer to confirm that appropriate degrees of specificity were obtained for both primers in a set. The product yields obtained from amplification reactions using mismatch and perfect match primers were measured by standard Southern hybridization methods. The product yields obtained in all cases were quantified using a Phosphorlmager (Molecular Dynamics, Sunnyvale, CA) after exposing the hybridized blots to Phosphorlmager screens.
In these experiments, we first tested the effect of a single mismatch (in addition to the non- target 3 '-terminal mismatch) on marker specificity. Forty-five single nucleotide amplified polymoφhisms (or "SNAP") primers were generated with the required values of relative product yields for the markers to be assayed using agarose gel elecfrophoresis (some examples of the results obtained are shown in Table 2). In Table 2, the values indicate relative product yield for a variety of mismatch primers designed by using the addition of an extra mismatch at the 3'-terminus. Measurements of product yield and calculations of relative efficiencies of amplification were performed as described above. For the determination of specificity in agarose gels, a 400- fold range of template DNA concentration was used.
TABLE 2
The values obtained from these experiments showed that the addition of an extra mismatch near the 3 '-end of the primer considerably reduced PCR product yield of the non-target allele with respect to the values obtained for the 3 '-end mismatch alone (Table 3 includes a few examples).
TABLE 3
In some cases, values of relative product yield of up to 10"4 were obtained for
the amplification of non-target alleles relative to perfect match primers (Primers 22V2, 22V3, 29V9, 29V12, 41 VI 1, 46V4; Table 2). On the other hand, the presence of a single mismatch 2 or 3 bases from the 3' end did not have a significant effect at reducing the overall product yield of the target allele (Table 2). These experiments indicated that the relative efficiencies of extension between target and non-target alleles were reliably increased by primer design. Finally, we made designs for all 12 possible natural mismatch combinations that could be present in the SNP sequences, and, in all cases, reliably obtained the desired ranges of specificity for these primers (not shown).
Also, as shown in Table 2, the lowest value of relative product yield obtained from non-target alleles (compared to perfect match) was 2 x 10"4 when amplified with primers containing one extra mismatch near the 3' end (Primer 22 V2). This implied that the relative product yield obtained for the target alleles would have to be higher than 0.1 (with respect to the perfect match) in order to maintain the required 10"3 range of specificity. The use of primer combinations that decreased product yield of the target allele below values of 0.1 would only decrease specificity, since no further reduction on the product yield of non-target alleles would be obtained from the addition of such mismatch combinations.
In addition, in these experiments, other primers containing two extra mismatches (in addition to the 3 '-terminal non- target mismatch) were tested for their ability to increase marker specificity. These results are shown in Table 4. In this Table, the values indicate the relative product yield of non-target and target alleles (compared to perfect match) obtained after amplification with primers containing two additional mismatches near the 3 '-terminus.
TABLE 4
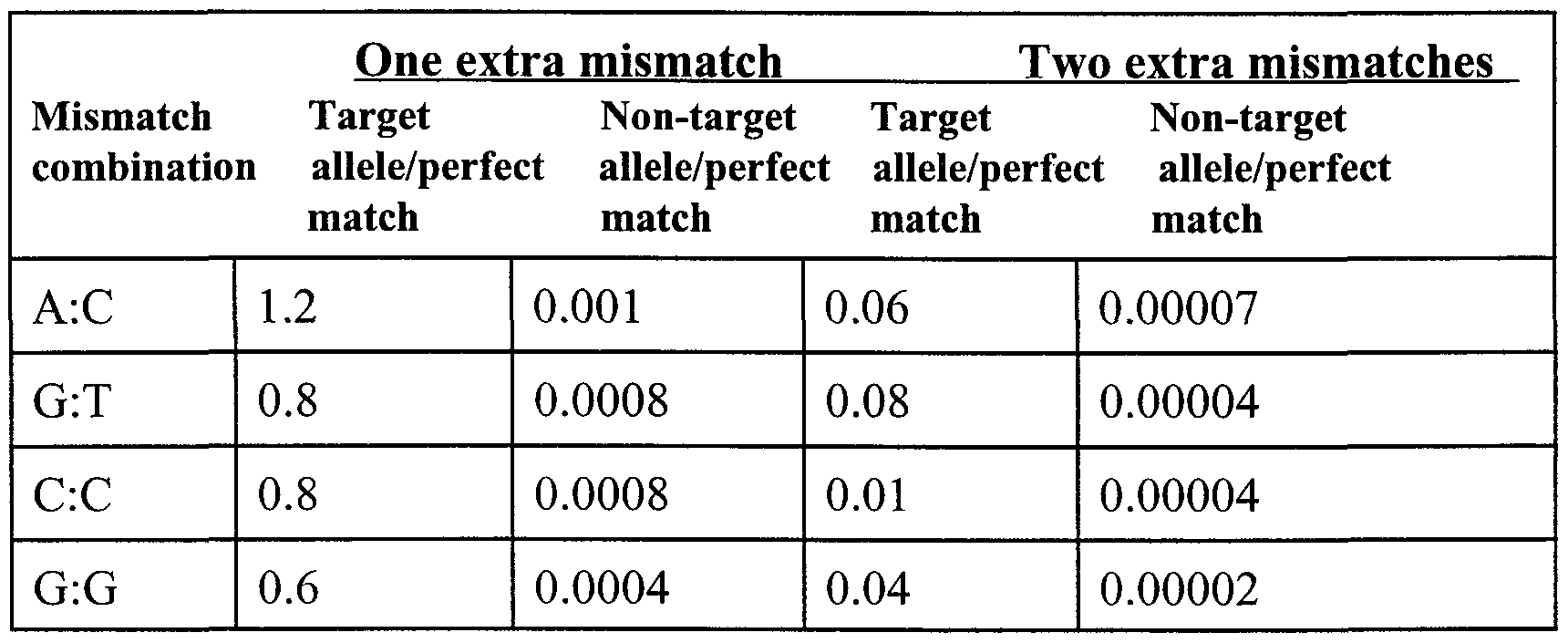
As shown in Table 4, relative product yields for non-target alleles of up to 2.3 x 10"5 (compared to the perfect match) were obtained when two additional mismatches were introduced to design the primers, indicating that specificity over higher ranges of DNA concentrations were obtained. In addition, in a parallel set of experiments, two primers with complementary ranges of specificity were generated by the introduction of one and two mismatches, respectively, near the 3' end, according to the required level of discrimination needed. In parallel reactions, PCR amplification was carried out using "perfect match" primers that contained sequences identical to those described above, but lacking the mismatched nucleotides. Table 5 shows the values of relative product yield obtained from the amplification of these allele-specific primers (values of product yield are relative to those obtained with perfect match primers). In this Table, "one extra mismatch" and "two extra mismatches" refers to the number of non-complementary nucleotides present in the primer, in addition to the 3 '-terminal mismatch of the primer with
respect to the non-target allele.
TABLE 5
As shown in Table 5, the addition of one extra mismatch (in addition to the 3 '-terminal non- target mismatch) at the 3' end resulted in a difference in relative product yield between target and non- target alleles of approximately 1, 000-fold (Table 5). In addition, the relative product yield of the target allele compared to the perfect match in these particular cases was relatively high (Table 5), allowing the marker to be specific under conditions of low sample concentration (Figure 2 A, Figure 4).
The addition of two extra mismatches (in addition to the 3 '-terminal non- target mismatch) near the 3' end decreased the product yield of non-target alleles to average levels of 10"5 compared to perfect match primers, making the markers specific at high sample DNA concentrations (Table 5, Figures 2B, and Figure 4). In all cases, ratios between product yield of the target allele amplified with the primer containing one extra mismatch and the non-target allele amplified with the primer containing two extra mismatches (extremes of the range) was at least 104 fold, a value that guarantees specificity over a 10,000-fold range of DNA concentrations (Figure 4). Finally, as shown in
Table 5, relative amplification efficiencies for the complementary primers overlapped in all cases, ensuring specificity over the entire range of DNA concentrations. These results indicated the feasibility of generating markers with ranges of specificity adequately high to cover large ranges of DNA concentration (approximately a 10,000-fold range).
In a final experiment the reliability of the method was tested by hybridization of the products amplified from two alleles, A and B, with allele-specific primers P1/P2 (specific for allele A), and P3/P4 (specific for allele B), holding the primer concentrations constant and using different concentrations of template DNA (ranging from 0.01 to 10 nanograms of DNA). Figure 5 shows that, at low template DNA concentration (0.01 nanograms), primers PI and P3 showed specificity for their respective alleles (A and B). At higher DNA concentrations (10 nanograms), in the case of allele B, primer P3 lost specificity, but primer P4 retained specificity. These results demonstrated the feasibility of the use of the two primer system in the construction of linkage maps in those cases where DNA concentrations vary over a 10,000-fold range.
Chip Based Approaches
Chip-based approaches, involving microarrays of DNA sequences as gene-specific hybridization targets, have been developed recently for the detection of single-nucleotide polymoφhisms and for the quantitative measurement of expression of genes in plants and humans (Schena et al., Science 270: 467-470, 1995; and Schena et al., Proc. Natl. Acad. Sci. USA 93: 10614-10619, 1996). The power of DNA chip technology for genome analysis resides in the large number of probes that can be tested using a single chip. The potential applications of this new technology are vast, and include use in mapping procedures. Although single-nucleotide polymoφhisms are quite
adaptable to chip-based assays (Jordan and Collins, Nature 380: 111-112, 1996), implementation of totally automated mapping systems using such markers has sometimes been problematic due to the lack of a robust methodology, particularly for monitoring single nucleotide polymoφhisms (Jordan and Collins, 1996, supra). In theory SNPs can be assayed directly on high density Affymetrix chips using so-called tiling procedures; however, these methods have generally not been sufficiently reproducible or sensitive to reliably assay most SNPs.
In contrast, the present technique is ideally suited to DNA chip applications. In particular, the capability of two (or more) sets of primers to maintain allele specificity within a broad range of DNA concentrations allows the use of allele-specific markers in this format, since reactions may be carried out without a requirement for previous determinations of sample DNA concentrations. By exploiting a combination of two allele-specific PCR primers with complementary ranges of specificity, a considerable increase is obtained in the overall range of DNA concentrations that may be reliably assayed compared to values obtained with single-allele specific primers.
As shown in Figure 6, the present technique facilitates the detection of allele-specific amplification products. In this figure, primers PI and P2 are specific for the allele associated with Arabidopsis thaliana ecotype Columbia, and primers P3 and P4 are specific for Arabidopsis ecotype Landsberg erecta. PI differs from P2, and P3 differs from P4, in the number of mismatches with the target sequence near the 3' end, resulting in primers having different but overlapping ranges of specificity. In addition, these primers each contain a multiplex oligonucleotide tag (a hybridization tag) that differs in sequence from the primer itself as well as the target sequence. Following amplification, the labelled PCR products (for example, radioactive or fluorescent PCR products)
are scored using DNA chips on which are immobilized (in discrete quadrants) binding partners for each of the multiplex tags. By carrying out hybridization to these tags, the presence of the allelic marker is determined, as well as a determination of whether the sample DNA was homozygous or heterozygous at that allele.
Any number of allelic markers may be simultaneously tested in this manner simply by including primer sets for each target marker in the PCR amplification reaction mixture, and assaying by hybridization to binding partners for each of those markers, for example, using unique multiplex tags immobilized on a solid support.
Other Embodiments The broad range PCR techniques described herein may be used in any appropriate context, although mapping represents a particularly useful application of the method. In addition, such mapping approaches find use in any number of organisms (including plants and animals) and are most useful for organisms having incomplete genomic sequence information. Other embodiments are within the claims.
What is claimed is: