WO2005076783A2 - Sulfone substituted imidazo ring ethers - Google Patents
Sulfone substituted imidazo ring ethers Download PDFInfo
- Publication number
- WO2005076783A2 WO2005076783A2 PCT/US2004/040383 US2004040383W WO2005076783A2 WO 2005076783 A2 WO2005076783 A2 WO 2005076783A2 US 2004040383 W US2004040383 W US 2004040383W WO 2005076783 A2 WO2005076783 A2 WO 2005076783A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- alkyl
- heterocyclyl
- alkylenyl
- heteroaryl
- Prior art date
Links
- 0 CC1(*)C=Cc2ncc3nc(*)[n](*)c3c2C=C1 Chemical compound CC1(*)C=Cc2ncc3nc(*)[n](*)c3c2C=C1 0.000 description 16
- JYYSVVKAKJIJBG-UHFFFAOYSA-N CC(C)(C[n]1c2c(cccc3)c3nc(N)c2nc1COC)OCCS(C)(=O)=O Chemical compound CC(C)(C[n]1c2c(cccc3)c3nc(N)c2nc1COC)OCCS(C)(=O)=O JYYSVVKAKJIJBG-UHFFFAOYSA-N 0.000 description 1
- YWDGOMOQAZENKS-UHFFFAOYSA-N CCCCc1nc2c(N)nc(CCCC3)c3c2[n]1CC(C)(C)OCCS(C)(=O)=O Chemical compound CCCCc1nc2c(N)nc(CCCC3)c3c2[n]1CC(C)(C)OCCS(C)(=O)=O YWDGOMOQAZENKS-UHFFFAOYSA-N 0.000 description 1
- FJKBNCRNYQIRQI-UHFFFAOYSA-N CCOCc1nc2c(N)nc(cccc3)c3c2[n]1CCOCCS(C)(=O)=O Chemical compound CCOCc1nc2c(N)nc(cccc3)c3c2[n]1CCOCCS(C)(=O)=O FJKBNCRNYQIRQI-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/14—Ortho-condensed systems
Definitions
- l-[2-(4-piperidyl)ethyl]-lH- imidazo[4,5-c]quinoline was synthesized as a possible anticonvulsant and cardiovascular agent.
- 2-oxoimidazo[4,5-c]quinolines have been reported.
- Certain lH-imidazo[4,5-c]quinolin-4-amines and 1- and 2-substituted derivatives thereof were later found to be useful as antiviral agents, bronchodilators and immunomodulators.
- the present invention provides a new class of compounds that are useful in inducing cytokine biosynthesis in animals.
- Such compounds are of the following Formula
- the compounds of Formulas I and la are useful as immune response modifiers due to their ability to induce cytokine biosynthesis (e.g., induces the synthesis of at least one cytokine) and otherwise modulate the immune response when administered to animals. This makes the compounds useful in the treatment of a variety of conditions such as viral diseases and tumors that are responsive to such changes in the immune response.
- the invention further provides pharmaceutical compositions containing an effective amount of a compound of Formula I or Formula la and methods of inducing cytokine biosynthesis in an animal, treating a viral infection and/or treating a neoplastic disease in an animal by administering an effective amount of a compound of Formula I to the animal.
- methods of synthesizing compounds of Formula I and Formula la and intermediates useful in the synthesis of these compounds are provided.
- “a,” “an,” “the,” “at least one,” and “one or more” are used interchangeably.
- the terms “comprises” and variations thereof do not have a limiting meaning where these terms appear in the description and claims. The above summary of the present invention is not intended to describe each disclosed embodiment or every implementation of the present invention.
- Xi-i and X ⁇ - 2 are independently selected from the group consisting of
- Z is selected from the group consisting of -S-, -S(O)-, and -S(O) 2 -;
- Ri is selected from the group consisting of: C MO alkyl, C 2 - ⁇ o alkenyl, C 2 - ⁇ 0 alkynyl, aryl, aryl-Ci-io alkylenyl, aryloxy-Ci-io alkylenyl, C O alkylarylenyl, heteroaryl, heteroaryl-Ci-io alkylenyl, heteroaryloxy-Ci-io alkylenyl, Ci-io alkylheteroarylenyl, heterocyclyl, heterocyclyl-Ci-io alkyl
- X M and X ⁇ - 2 are independently selected from the group consisting of C MO alkylene, C 4 - ⁇ o alkenylene, and C 4 - ⁇ o alkynylene; wherein the terminal carbon atoms of alkenylene and alkynylene are tetrahedral;
- Z is selected from the group consisting of-S-, -S(O)-, and — S(O) 2 -;
- Ri is selected from the group consisting of: C MO alkyl, C 2 - ⁇ o alkenyl, C 2 - ⁇ o alkynyl, aryl, aryl-Ci-io alkylenyl, aryloxy-Ci-io alkylenyl, CM O alkylarylenyl, heteroaryl, heteroaryl-Ci-io alkylenyl, heteroaryloxy-Ci-io alkylenyl, C O alkylheteroarylenyl, heterocyclyl
- R 4 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, hydroxyalkyl, haloalkyl, haloalkoxy, halogen,
- A is selected from the group consisting of -O-, -C(O)-, -S(O)o- 2 -, -CH 2 -, and -N(R4)-;
- Q is selected from the group consisting of a bond, -C(R 6 )-, -C(R 6 )-C(R 6 )-, -S(O) 2 -, -C(R 6 )-N(R 8 )- -, -S(O) 2 -N(R 8 )-, -C(R 6 )-O-, and -C(R 6 )-N(OR 9 )-;
- V is selected from the group consisting of -C(R 6 )-, -O-C(R 6 )-, -N(R 8 )-C(R 6 )-, and -S(O) 2 -;
- W is selected from the group consisting of a bond, -C(O)-, and
- X M and X ⁇ - 2 are independently selected from the group consisting of C MO alkylene, C 4 - ⁇ o alkenylene, and C 4 - ⁇ o alkynylene; wherein the terminal carbon atoms of alkenylene and alkynylene are tetrahedral;
- Z is selected from the group consisting of-S-, -S(O)-, and -S(O) 2 -;
- Ri is selected from the group consisting of: C MO alkyl, C - ⁇ o alkenyl, C 2 - ⁇ o alkynyl, aryl, aryl-Ci-io alkylenyl, aryloxy-Ci-io alkylenyl, C MO alkylarylenyl, heteroaryl, heteroaryl-Ci-io alkylenyl, heteroaryloxy-Ci-io alkylenyl, C MO alkylheteroarylenyl, heterocyclyl, hetero
- Xi-i and X ⁇ - are independently selected from the group consisting of C O alkylene, C 4 - ⁇ o alkenylene, and C - ⁇ o alkynylene; wherein the terminal carbon atoms of alkenylene and alkynylene are tetrahedral;
- Z is selected from the group consisting of -S-, -S(O)-, and -S(O) -;
- Ri is selected from the group consisting of: C MO alkyl, C 2 - ⁇ o alkenyl, C 2 - ⁇ o alkynyl, aryl, aryl-Ci-io alkylenyl, aryloxy-C ⁇ _ ⁇ o alkylenyl, C MO alkylarylenyl, heteroaryl, heteroaryl-Ci-io alkylenyl, heteroaryloxy-Ci-io alkylenyl, C MO alkylheteroarylenyl, heterocyclyl,
- R 4 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, hydroxyalkyl, haloalkyl, haloalkoxy, halogen,
- R is C 2 - 7 alkylene;
- R 8 is selected from the group consisting of hydrogen, alkyl, alkoxyalkylenyl, and arylalkylenyl;
- R 9 is selected from the group consisting of hydrogen and alkyl;
- Rio is C 3 - 8 alkylene;
- A is selected from the group consisting of -O-, -C(O)-, -S(O)o- 2 -, -CH 2 -, and
- Q is selected from the group consisting of a bond, -C(R 6 )-, -C(R 6 )-C(R 6 )-, -S(O) 2 -, -C(R 6 )-N(R 8 )-W-, -S(O) 2 -N(R 8 )-, -C(R 6 )-O-, and -C(R 6 )-N(OR 9 )-; V is selected from the group consisting of -CCReK -O-C(R 6 )-, -N(R 8 )-C(R 6 )-, and -S(O) 2 -; is selected from the group consisting of a bond, -C(O)-, and -S(O) 2 -; and a and b are independently integers from 1 to 6 with the proviso that a + b is ⁇ 7; or a pharmaceutically acceptable salt thereof.
- the present invention is selected from the group consisting of
- Xi-i and X ⁇ - 2 are independently selected from the group consisting of
- Z is selected from the group consisting of-S-, -S(O)-, and-S(O) 2 -;
- Ri is selected from the group consisting of: C O alkyl, C 2 - ⁇ o alkenyl, C2-10 alkynyl, aryl, aryl-C ⁇ -10 alkylenyl, aryloxy-C 1 -1 0 alkylenyl, C MO alkylarylenyl, heteroaryl.
- 1 ⁇ is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, hydroxyalkyl, haloalkyl, haloalkoxy, halogen,
- R 5 is selected from the group consisting of:
- R 7 is C 2 . 7 alkylene;
- R 8 is selected from the group consisting of hydrogen, alkyl, alkoxyalkylenyl, and arylalkylenyl;
- R 9 is selected from the group consisting of hydrogen and alkyl;
- Rio is C - 8 alkylene;
- A is selected from the group consisting of -O-, -C(O)-, -S(O) 0 - 2 -, -CH 2 -, and
- Q is selected from the group consisting of a bond, -C(R 6 )-, -C(R )-C(R 6 )-, -S(O) 2 -, -C(R 6 )-N(R 8 )-W-, -S(O) 2 -N(R 8 )-, -C(R 6 )-O-, and -C(R 6 )-N(OR 9 )-; V is selected from the group consisting of -C(R 6 )-, -O-C(R 6 )-, -N(R 8 )-C(R 6 )-, and -S(O) 2 -; W is selected from the group consisting of a bond, -C(O)-, and -S(O) 2 -; and a and b are independently integers from 1 to 6 with the proviso that a + b is ⁇ 7; or a pharmaceutically acceptable salt thereof.
- the present invention provides a
- Xi-i and X ⁇ - 2 are independently selected from the group consisting of C MO alkylene, C 4 - ⁇ o alkenylene, and C 4 - ⁇ o alkynylene; wherein the terminal carbon atoms of alkenylene and alkynylene are tetrahedral;
- Z is selected from the group consisting of-S-, -S(O)-, and -S(O) 2 -;
- Ri is selected from the group consisting of: C MO alkyl, C2-10 alkenyl, C2- 10 alkynyl, aryl, aryl-C ⁇ -10 alkylenyl, aryloxy-Ci-io alkylenyl, CM O alkylarylenyl, heteroaryl, heteroaryl-Ci-io alkylenyl, heteroaryloxy-Ci-io alkylenyl, C MO alkylheteroarylenyl, heterocyclyl, heterocyclyl-
- R A is selected from the group consisting of: halogen, hydroxy, alkyl, alkenyl, haloalkyl, alkoxy, alkylthio, and -N(R 9 ) 2 ; n is 0 to 4; and
- R" is hydrogen or a non-interfering substituent; or a pharmaceutically acceptable salt thereof.
- the present invention provides a compound of Formula ma:
- X M and X ⁇ - 2 are independently selected from the group consisting of C MO alkylene, C 4 - ⁇ o alkenylene, and C 4 - ⁇ o alkynylene; wherein the terminal carbon atoms of alkenylene and alkynylene are tetrahedral;
- Z is selected from the group consisting of-S-, -S(O)-, and -S(O) 2 -;
- R] is selected from the group consisting of: C MO alkyl, C 2 - ⁇ o alkenyl, C 2 - ⁇ o alkynyl, aryl, aryl-Ci-io alkylenyl, aryloxy-Ci-io alkylenyl, C O alkylarylenyl, heteroaryl, heteroaryl-Ci-io alkylenyl, heteroaryloxy-Ci-io alkylenyl, C MO alkylheteroarylenyl, heterocyclyl
- R is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, hydroxyalkyl, haloalkyl, haloalkoxy, halogen,
- R 7 is C 2 . 7 alkylene;
- R 8 is selected from the group consisting of hydrogen, alkyl, alkoxyalkylenyl, and arylalkylenyl;
- R is selected from the group consisting of hydrogen and alkyl;
- Rio is C 3 .
- A is selected from the group consisting of -O-, -C(O)-, -S(O)o- 2 - 5 -CH 2 -, and -N(R4>;
- Q is selected from the group consisting of a bond, -C(R 6 )-, -C(R 6 )-C(R 6 )-, -S(O) 2 -, -C(R 6 )-N(R 8 )-W-, -S(O) 2 -N(R 8 )-, -C(R 6 )-O-, and -C(R 6 )-N(OR 9 )-;
- V is selected from the group consisting of -C(R 6 )-, -O-C(R 6 )-,
- the present invention provides a compound of Formula ma:
- X M and X 1 - 2 are independently selected from the group consisting of CM O alkylene, C4.-1 0 alkenylene, and C - ⁇ 0 alkynylene; wherein the terminal carbon atoms of alkenylene and alkynylene are tetrahedral;
- Z is selected from the group consisting of -S-, -S(O)-, and -S(O) 2 -;
- Ri is selected from the group consisting of: CM O alkyl, C2-10 alkenyl, C2-10 alkynyl, aryl, aryl-Ci-io alkylenyl, aryloxy-Ci-10 alkylenyl, C MO alkylarylenyl, heteroaryl, heteroaryl-Ci-10 alkylenyl, heteroaryloxy-C ⁇ -10 alkylenyl, C MO alkylheteroarylenyl, heterocyclyl, heterocyclyl-C ⁇ -10 al
- R is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, hydroxyalkyl, haloalkyl, haloalkoxy, halogen,
- R is C 2 - alkylene;
- R 8 is selected from the group consisting of hydrogen, alkyl, alkoxyalkylenyl, and arylalkylenyl;
- R is selected from the group consisting of hydrogen and alkyl;
- Rio is C - 8 alkylene;
- A is selected from the group consisting of -O-, -C(O)-, -S(O) 0 . 2 -, -CH 2 -, and
- Q is selected from the group consisting of a bond, -C(R 6 )-, -C(R 6 )-C(R 6 )-, -S(O) 2 -, -C(R 6 )-N(R 8 )-W-, -S(O) 2 -N(R 8 )-, -C(R 6 )-O-, and -C(R 6 )-N(OR 9 )-;
- V is selected from the group consisting of -C(R 6 )-, -O-C(R 6 )-,
- the present invention provides a compound of Formula IV:
- X M and X ⁇ - 2 are independently selected from the group consisting of C MO alkylene, C 4 - ⁇ o alkenylene, and C 4 - ⁇ o alkynylene; wherein the terminal carbon atoms of alkenylene and alkynylene are tetrahedral;
- Z is selected from the group consisting of-S-, -S(O)-, and -S(O) 2 -;
- Ri is selected from the group consisting of: C MO alkyl, C 2 - ⁇ o alkenyl, C2-10 alkynyl, aryl, aryl-Ci-io alkylenyl, aryloxy-Ci-1 0 alkylenyl, C MO alkylarylenyl, heteroaryl, heteroaryl-Ci-10 alkylenyl, heteroaryloxy-Ci-io alkylenyl, CM O alkylheteroarylenyl, heterocyclyl, heterocyclyl
- X M and X 1 - 2 are independently selected from the group consisting of C MO alkylene, C - ⁇ o alkenylene, and C 4 - ⁇ o alkynylene; wherein the terminal carbon atoms of alkenylene and alkynylene are tetrahedral;
- Z is selected from the group consisting of -S-, -S(O)-, and -S(O) 2 -;
- Ri is selected from the group consisting of: C MO alkyl, C - ⁇ o alkenyl, C 2 - ⁇ o alkynyl, ' aryl, aryl-C ⁇ -10 alkylenyl, aryloxy-C M 0 alkylenyl, C MO alkylarylenyl, heteroaryl, heteroaryl-Ci-10 alkylenyl, heteroaryloxy-C ⁇ -10 alkylenyl, C MO alkylheteroarylenyl, heterocyclyl, heterocyclyl-
- R 4 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, hydroxyalkyl, haloalkyl, haloalkoxy, halogen,
- R 5 is selected from the group consisting of:
- Q is selected from the group consisting of a bond, -C(R 6 )-, -C(R 6 )-C(R 6 )-, -S(O) 2 -, -C(R 6 )-N(R 8 )-W-, -S(O) 2 -N(R 8 )-, -C(R 6 )-O-, and -C(R 6 )-N(OR 9 )-;
- V is selected from the group consisting of -C(R 6 )-, -O-C(R 6 )-,
- the present invention also provides intermediate compounds, which are useful, for example, in preparing compounds of Formulas I-Ifla.
- the present invention provides a compound of Formula V:
- T is -NH 2 or -NO 2 ;
- Xi-i and X 1 - 2 are independently selected from the group consisting of
- Ri is selected from the group consisting of: C MO alkyl, C 2 - 10 alkenyl, C2-10 alkynyl, aryl, aryl-C ⁇ -10 alkylenyl, aryloxy-Ci-10 alkylenyl, C ⁇ -10 alkylarylenyl, heteroaryl, heteroaryl-Ci-10 alkylenyl, heteroaryloxy-C ⁇ -10 alkylenyl, CM O alkylheteroarylenyl, heterocyclyl, heterocyclyl-C ⁇ -10 alkylenyl, and C M O alkyl, C 2 - ⁇ 0 alkenyl, C2-10 alkynyl, aryl, aryl-C ⁇ - 10 alkylenyl, ary
- T is -NH 2 or -NO 2 ;
- XM and X ⁇ - 2 are independently selected from the group consisting of
- the present invention provides a compound of Formula VIE:
- X and X ⁇ - 2 are independently selected from the group consisting of
- Ri is selected from the group consisting of: C MO alkyl, C 2 - ⁇ o alkenyl, C 2 - ⁇ o alkynyl, aryl, aryl-Ci-io alkylenyl, aryloxy-Ci-10 alkylenyl, C MO alkylarylenyl, heteroaryl, heteroaryl-Ci-io alkylenyl, heteroaryloxy-C ⁇ -10 alkylenyl, C MO alkylheteroarylenyl, heterocyclyl, heterocyclyl-Ci-io alkylenyl, and CMO alkyl, C2-10 alkenyl, C 2 . ⁇ o alkynyl, aryl, aryl-Cno alkylen
- R 7 is C 2 . 7 alkylene;
- R 8 is selected from the group consisting of hydrogen, alkyl, alkoxyalkylenyl, and arylalkylenyl;
- R is selected from the group consisting of hydrogen and alkyl;
- Rio is C 3 . 8 alkylene;
- A is selected from the group consisting of -O-, -C(O)-, -S(O)o- 2 -, -CH 2 -, and
- Q is selected from the group consisting of a bond, -C(R 6 )-, -C(R 6 )-C(R 6 )-,
- V is selected from the group consisting of -C(R 6 )-, -O-C(R 6 )-, -N(R 8 )-C(R 6 )-, and -S(O) 2 -;
- W is selected from the group consisting of a bond, -C(O)-, and -S(O) 2 -; and a and b are independently integers from 1 to 6 with the proviso that a + b is ⁇ 7; or a pharmaceutically acceptable salt thereof.
- the present invention provides a compound of Formula IX:
- X M and X ⁇ _ 2 are independently selected from the group consisting of C MO alkylene, C 4 - ⁇ o alkenylene, and C 4 - ⁇ o alkynylene; wherein the terminal carbon atoms of alkenylene and alkynylene are tetrahedral; Ri is selected from the group consisting of: C MO alkyl, C 2 - ⁇ o alkenyl, C2-10 alkynyl, aryl, aryl-Ci-io alkylenyl, aryloxy-Ci-1 0 alkylenyl, CM O alkylarylenyl, heteroaryl, heteroaryl-Ci-1 0 alkylenyl, heteroaryloxy-Ci-io alkylenyl, C MO alkylheteroarylenyl, heterocyclyl, heterocyclyl-Ci-io alkylenyl, and C O alkyl, C2-10 alkenyl, C2-
- R- t is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, hydroxyalkyl, haloalkyl, haloalkoxy, hal
- R 7 is C 2 - 7 alkylene;
- R 8 is selected from the group consisting of hydrogen, alkyl, alkoxyalkylenyl, and arylalkylenyl;
- R 9 is selected from the group consisting of hydrogen and alkyl;
- Rio is C 3 .
- A is selected from the group consisting of -O-, -C(O)-, -S(O)o-2- 5 -CH 2 -, and -N(R4>;
- Q is selected from the group consisting of a bond, -C(R 6 )-, -C(R 6 )-C(R 6 )-, -S(O) 2 -, -C(R 6 )-N(R 8 )-W-, -S(O) 2 -N(R 8 )-, -C(R 6 )-O-, and -C(R 6 )-N(OR 9 )-;
- V is selected from the group consisting of -C(R 6 )-, -O-C(R 6 ) ⁇ ,
- the present invention provides a compound of Formula
- X M and X ⁇ - 2 are independently selected from the group consisting of C MO alkylene, C 4 _ ⁇ o alkenylene, and C 4 -1 0 alkynylene; wherein the terminal carbon atoms of alkenylene and alkynylene are tetrahedral;
- R is selected from the group consisting of alkyl, alkoxy, hydroxy, fluoro, and trifluoromethyl;
- n is 0 to 4;
- R 2 is selected from the group consisting of -X-R 4 , -X-Y-R 4 , and -X-R 5 ;
- X is selected from the group consisting of alkylene; alkenylene, alkynylene, arylene, heteroarylene, and heterocyclylene wherein the alkylene, alkenylene, and alkynylene groups can be optionally interrupted or terminated with arylene, heteroarylene, or heterocyclylene, and optionally interrupted by one or more -O- groups;
- R 4 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, hydroxyalkyl, haloalkyl, haloalkoxy, halogen,
- R 7 is C2- 7 alkylene;
- R 8 is selected from the group consisting of hydrogen, alkyl, alkoxyalkylenyl, and arylalkylenyl;
- R 9 is selected from the group consisting of hydrogen and alkyl;
- Rio is C 3 . 8 alkylene;
- A is selected from the group consisting of -O-, -C(O)-, -S(O)o-2-, -CH 2 -, and
- Q is selected from the group consisting of a bond, -C(R 6 )-, -C(R 6 )-C(R 6 )-, -S(O) 2 -, -C(R 6 )-N(R 8 )-W-, -S(O) 2 -N(R 8 )-, -C(R 6 )-O-, and -C(R 6 )-N(OR 9 >; V is selected from the group consisting of -C(R 6 )-, -O-C(R 6 )-, -N(R 8 )-C(R 6 )-, and -S(O) 2 -; W is selected from the group consisting of a bond, -C(O)-, and -S(O) 2 -; and a and b are independently integers from 1 to 6 with the proviso that a + b is ⁇ 7; or a pharmaceutically acceptable salt thereof.
- the present invention provides a compound
- X M and X ⁇ - 2 are independently selected from the group consisting of C O alkylene, C - ⁇ o alkenylene, and C 4 - ⁇ o alkynylene; wherein the terminal carbon atoms of alkenylene and alkynylene are tetrahedral;
- R is selected from the group consisting of alkyl, alkoxy, hydroxy, fluoro, and trifluoromethyl;
- n is 0 to 4;
- R 2 is selected from the group consisting of -R4, -X-R 4 , -X-Y-R4, and
- X is selected from the group consisting of alkylene, alkenylene, alkynylene, arylene, heteroarylene, and heterocyclylene wherein the alkylene, alkenylene, and alkynylene groups can be optionally interrupted or terminated with arylene, heteroarylene, or heterocyclylene, and optionally interrupted by one or more -O- groups;
- Y is selected from
- R4 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, hydroxyalkyl, haloalkyl, haloalkoxy, halogen,
- V is selected from the group consisting of -C(R 6 )-, -O-C(R 6 )-, -N(R 8 )-C(R 6 >, and -S(O) 2 -;
- W is selected from the group consisting of a bond, -C(O)-, and -S(O) 2 -; and a and b are independently integers from 1 to 6 with the proviso that a + b is ⁇ 7.
- the present invention provides a compound of Formula XXINa:
- X 1 - 1 and X ⁇ - 2 are independently selected from the group consisting of C MO alkylene, C 4 - ⁇ o alkenylene, and C 4 - ⁇ o alkynylene; wherein the terminal carbon atoms of alkenylene and alkynylene are tetrahedral;
- R is selected from the group consisting of alkyl, alkoxy, hydroxy, fluoro, and trifluoromethyl;
- n is 0 to 4;
- R 2 is selected from the group consisting of -R 4 , -X-R 4 , -X-Y-R4, and -X-R 5 ;
- X is selected from the group consisting of alkylene, alkenylene, alkynylene, arylene, heteroarylene, and heterocyclylene wherein the alkylene, alkenylene, and alkynylene groups can be optionally interrupted or terminated with arylene, heteroarylene, or heterocyclylene, and optionally interrupted by one or
- R 4 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, hydroxyalkyl, haloalkyl, haloalkoxy, halogen,
- R 5 is selected from the group consisting of:
- R 7 is C2- 7 alkylene;
- R 8 is selected from the group consisting of hydrogen, alkyl, alkoxyalkylenyl, and arylalkylenyl;
- R is selected from the group consisting of hydrogen and alkyl;
- Rio is C 3 - 8 alkylene;
- A is selected from the group consisting of -O-, -C(O)-, -S(O)o-2-, -CH2-, and -NCR,)-;
- Q is selected from the group consisting of a bond, -C(R 6 )-, -C(R 6 )-C(R 6 )-, -S(O) 2 -, -C(R 6 )-N(R 8 )-W-, -S(O) 2 -N(R 8 )-, -C(R 6 )-O-, and -C(R 6 )
- W is selected from the group consisting of a bond, -C(O)-, and -S(O)2-; and a and b are independently integers from 1 to 6 with the proviso that a + b is ⁇ 7; or a pharmaceutically acceptable salt thereof.
- non-interfering means that the ability of thte compound or salt, which includes a non-interfering substituent, to modulate the biosynthesis of one or more cytokines is not destroyed by the non-interfering substitutent.
- R is hydrogen or a non-interfering substituent.
- Illustrative mon-interfering R" groups include those described above for R .
- a carbon atom which is tetrahedral is a carbon atom which is bonded to four atoms wherein each of the four bonds is a sirxgle bond.
- alkyl As used herein, the terms "alkyl,” “alkenyl,” “alkynyl” and the prefix “alk-” are inclusive of both straight chain and branched chain groups and of cyclic groups, i.e. cycloalkyl and cycloalkenyl, as well as combinations thereof. Unless otherwise specified, these groups contain from 1 to 20 carbon atoms, with alkenyi groups containing from 2 to 20 carbon atoms, and alkynyl groups containing from 2 to 20 carbon atoms. In some embodiments, these groups have a total of up to 10 carbon atoms, up to 8 carbon atoms, up to 6 carbon atoms, or up to 4 carbon atoms.
- Cyclic groups can be monocyclic or polycyclic and preferably have from 3 to 10 ring carbon atoms.
- Exemplary cyclic groups include cyclopropyl, cyclopropylmethyl, cyclopentyl, cyclohexyl, adamantyl, and substituted and unsubstituted bornyl, norbornyl, and norbornenyl.
- the term "-cyclic(CH 2 ) 3 . 6 -” represents the divalent fo m of cycloalkyl groups of three to six carbon atoms.
- "-cyclic(CH 3 . 6 -" is — C / ⁇ H 2 C CH 2 (c H 2 ) p ⁇ wherein p is an integer of 0 to 3.
- alkylene alkenylene
- -and “alkynylene” are the divalent forms of the "alkyl,” “alkenyl,” and “alkynyl” groups defined above.
- alkylenyl alkenylenyl
- alkynylenyl are use when “alkylene,” “alkenylene,” and “alkynylene,” respectively, are substituted.
- an aaylalkylenyl group comprises an alkylene moiety to which an aryl group is attached.
- haloalkyl is inclusive of groups that are substituted by one or more halogen atoms, including perfluorinated groups. This is also true of other groups that include the prefix “halo-.” Examples of suitable haloalkyl groups are chloromethyl, trifluoromethyl, and the like.
- aryl as used herein includes carbocyclic aromatic rings or ring systems.
- aryl groups include phenyl, naphthyl, biphenyl, fluorenyl and indenyl.
- heteroatom refers to the atoms O, S, or N.
- heteroaryl includes aromatic rings or ring systems that contain at least one ring heteroatom (e.g., O, S, N).
- Suitable heteroaryl groups include furyl, thienyl, pyridyl, quinolinyl, isoquinolinyl, indolyl, isoindolyl, triazolyl, pynolyl, tetrazolyl, imidazolyl, pyrazolyl, oxazolyl, thiazolyl, benzofuranyl, benzothiophenyl, carbazolyl, benzoxazolyl, pyrimidinyl, benzimidazolyl, quinoxalinyl, benzothiazolyl, naphthyridinyl, isoxazolyl, isothiazolyl, purinyl, quinazolinyl, pyrazinyl, 1-oxidopyridyl, pyridazinyl, triazinyl, tetrazinyl, oxadiazolyl, thiadiazolyl, and so on.
- heterocyclyl includes non-aromatic rings or ring systems that contain at least one ring heteroatom (e.g., O, S, N) and includes all of the fully saturated and partially unsaturated derivatives of the above mentioned heteroaryl groups.
- heterocyclic groups include pyrrolidinyl, tetraliydrofuranyl, morpholinyl, thiomorpholinyl, piperidinyl, piperazinyl, thiazolidinyl, imidazolidinyl, isothiazolidinyl, tetrahydropyranyl, quinuclidinyl, homopiperidinyl (azepanyl), homopiperazinyl (diazepanyl), 1,3-dioxolanyl, aziridinyl, dihydroisoquinolin-(lH)-yl, octahydroisoquinolin-(lH)-yl, dihydroquinolin- (2H)-yl, octahydroquinolin-(2H)-yl, dihydro-lH-imidazolyl, and the like.
- heterocyclyl contains a nitrogen atom
- the point of attachment of the heterocyclyl group may be the nitrogen atom.
- arylene “heteroarylene,” and “heterocyclylene” are the divalent forms of the “aryl,” “heteroaryl,” and “heterocyclyl” groups defined above.
- arylenyl “heteroarylenyl,” and “heterocyclylenyl” are used when “arylene,” “heteroarylene,” and “heterocyclylene,” respectively, are substituted.
- an alkylarylenyl group comprises an arylene moiety to which an alkyl group is attached.
- fused aryl ring includes fused carbocyclic aromatic rings or ring systems. Examples of fused aryl rings include benzo, naphtho, fluoreno, and indeno. In certain embodiments, the fused aryl ring is benzo.
- fused heteroaryl ring includes the fused forms of 5 or 6 membered aromatic rings that contain one heteroatom selected from S and N. In certain embodiments, the fused heteroaryl ring is pyrido or thieno. In certain embodiments, the
- fused heteroaryl ring is pyrido.
- the pyrido ring is wherein the highlighted bond indicates the position where the ring is fused.
- the term "fused 5 to 7 membered saturated ring" includes rings which are fully saturated except for the bond where the ring is fused.
- the ring is a cyclohexene ring.
- the ring is tetrahydropyrido or dihydrothieno. hi certain embodiments, the ring is
- each group (or substituent or variable) is independently selected, whether explicitly stated or not.
- each R 9 group is independently selected, hi another example, when an -N(R 8 )-C(R 6 )-N(R 8 )- group is present, each R 8 group is independently selected.
- the invention is inclusive of the compounds described herein in any of their pharmaceutically acceptable forms, including isomers (e.g., diastereomers and enantiomers), salts, solvates, polymorphs, and the like, hi particular, if a compound is optically active, the invention specifically includes each of the compound's enantiomers as well as racemic mixtures of the enantiomers. It should be understood that the term “compound” includes any or all of such forms, whether explicitly stated or not (although at times, “salts" are explicitly stated).
- each one of the following variables e.g., A”, R", i, R 2 , R, R A , XM, X1-2, Z, Q, T, m, and n, and so on
- each one of the following variables e.g., A”, R", i, R 2 , R, R A , XM, X1-2, Z, Q, T, m, and n, and so on
- each one of the following variables e.g., A”, R", i, R 2 , R, R A , XM, X1-2, Z, Q, T, m, and n, and so on
- each of the resulting combinations of variables is an embodiment of the present invention.
- A" is a fused aryl ring or heteroaryl ring containing one heteroatom selected from the group consisting of N and S, wherein the aryl or heteroaryl ring is unsubstituted or substituted by one or more R groups, or a fused 5 to 7 membered saturated ring, optionally containing one heteroatom selected from the group consisting of N and S, and unsubstituted or substituted by one or more R A groups.
- A" is a fused aryl ring which is unsubstituted or substituted by one or more R groups. In some embodiments the fused aryl ring is unsubstituted.
- A" is a fused heteroaryl ring containing one heteroatom selected from the group consisting of N and S, and unsubstituted or substituted by one or more R groups. In some embodiments the fused heteroaryl ring is unsubstituted. In some embodiments, A" is a fused 5 to 7 membered saturated ring which is unsubstituted or substituted by one or more RA groups. In some embodiments, the 5 to 7 membered ring is unsubstituted. In some embodiments, A" is a fused 5 to 7 membered saturated ring containing one heteroatom selected from the group consisting of N and S, and unsubstituted or substituted by one or more R A groups.
- each R is independently selected from the group consisting of alkyl, alkoxy, hydroxy, fluoro, and trifluoromethyl.
- each R A is independently selected from the group consisting of halogen, hydroxy, alkyl, alkenyl, haloalkyl, alkoxy, alkylthio, and -N(R 9 ) 2 .
- Ri is selected from the group consisting of C MO alkyl, C 2 - 10 alkenyl, C2-10 alkynyl, aryl, aryl-C ⁇ -1 0 alkylenyl, aryloxy-Ci- 1 0 alkylenyl, C MO alkylarylenyl, heteroaryl, heteroaryl-Ci-10 alkylenyl, heteroaryloxy-C ⁇ -10 alkylenyl, C O alkylheteroarylenyl, heterocyclyl, heterocyclyl-C ⁇ - 10 alkylenyl, and CM O alkyl, C 2 - ⁇ o alkenyl, C2-10 alkynyl, aryl, aryl-Ci-10 alkylenyl, aryloxy-Ci-io alkylenyl, C MO alkylarylenyl, heteroaryl, heteroaryl-Ci-10 alkylenyl, heteroaryloxy-C ⁇ -10 alkyleny
- Ri is linear or branched C ⁇ - 4 alkyl, aryl, or 5 to 10 membered heteroaryl containing one or two heteroatoms, wherein the alkyl, aryl, or heteroaryl group may be unsubstituted or substituted with one or more substituents independently selected from the group consisting of C MO alkyl, C MO alkoxy, hydroxy-Ci-io alkyl, halo-Ci-io alkyl, halo-Ci-io alkoxy, halogen, nitro, hydroxy, cyano, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclyl, amino, C MO alkylamino, di(C ⁇ - ⁇ o alkyl)amino, and in the case of C MO alkyl, C 2 - ⁇ o alkenyl, C2- 10 alkyn
- Ri is methyl, ethyl, 1 -propyl, 2-propyl, 2-methylpropyl, 2- hydroxy-2-methylpropyl, phenyl, 4-chlorophenyl, or 4-fluorophenyl.
- Ri is methyl, ethyl, 2-propyl, 2-methylpropyl, 2-hydroxy-2- methylpropyl, or phenyl.
- R" is hydrogen or a non-interfering substituent.
- R" is selected from the group consisting of -R 4 , -X-R 4 , In some embodiments, R" is hydrogen, alkyl, hydroxyalkylenyl, or alkoxyalkylenyl. In some embodiments, R" is hydrogen, C ⁇ - alkyl, hydroxyC ⁇ - 4 alkylenyl, or
- R" is alkyl, hydroxyalkylenyl, or alkoxyalkylenyl. In some embodiments, R" is C ⁇ - 4 alkyl, hydroxyC ⁇ - alkylenyl, or Ci_ 4 alkoxyC ⁇ _ alkylenyl. In some embodiments, R" is hydrogen, methyl, ethyl, propyl, butyl, 2-hydroxyethyl, hydroxymethyl, 2-methoxyethyl, or ethoxymethyl. In some embodiments, R" is methyl, ethyl, propyl, butyl, 2-methoxyethyl, or ethoxymethyl.
- R 2 is selected from the group consisting of -R 4 , -X-R 4 , -X-Y-R4, and -X-R 5 .
- R 2 is hydrogen, alkyl, hydroxyalkylenyl, or alkoxyalkylenyl- .
- R 2 is hydrogen, C ⁇ 4 alkyl, hydroxyC ⁇ - 4 alkylenyl, or C ⁇ - 4 alkoxyC ⁇ - 4 alkylenyl.
- R 2 is alkyl, or alkoxyalkylenyl.
- R 2 is C alkyl, hydroxyC ⁇ - alkylenyl, or C ⁇ - 4 alkoxyC ⁇ - 4 alkylenyl. hi some embodiments, R 2 is C ⁇ - alkyl, or C ⁇ - 4 alkoxyC ⁇ - alkylenyl. In some embodiments, R 2 is hydrogen, methyl, ethyl, propyl, butyl, 2-hydroxyethyl-, hydroxymethyl, 2-methoxyethyl, or ethoxymethyl. In some embodiments, R 2 is methyl, ethyl, propyl, butyl, 2-methoxyethyl, or ethoxymethyl. In some embodiments, XM and X ⁇ .
- X M and X ⁇ - 2 are independently selected from C 2 . 4 alkylene groups.
- X M is -(CH 2 ) 2 - 4 -, -CH 2 -C(CH 3 ) 2 -, or
- -CH 2 -cyclic(CH 2 ) 3 - 6 - is
- X M is -(CH 2 )2-4-, or -CH2-C(CH 3 )2-.
- X1- 1 is
- X ⁇ - 2 is -(CH 2 ) 2 - or -(CH 2 ) 3 -.
- Z is selected from the group consisting of-S-, -S(O)-, and ⁇ S(O) 2 -. hi some embodiments, Z is -S(O) 2 -. In some embodiments, Z is -S(O)-. In some embodiments, Z is -S-. In some embodiments, m is an integer of 0 to 3. In some embodiments, m is 0. In some embodiments, n is an integer of 0 to 4. In some embodiments, n is 0. In some embodiments, A is selected from the group consisting of -O-, -C(O)-, -
- A is -O-.
- Q is selected from the group consisting of a bond, -C(R 6 )-, -C(R 6 )-C(R 6 )-, -S(O) 2 -, -C(R 6 )-N(R 8 )-W-, -S(O) 2 -N(R 8 )-, -C(R 6 )-O-, and -C(R 6 )-N(OR 9 )-.
- Q is -C(R 6 )-, -S(O) 2 -, or -C(R 6 )-N(R 8 )-W-.
- T is -NH 2 or -NO 2 .
- T is -NH2.
- V is selected from the group consisting of -C(R 6 )-, -O-C(R 6 )-, -N(R 8 )-C(R 6 )-, and -S(O) 2 -.
- W is selected from the group consisting of a bond, -C(O)-, and -S(O) 2 -.
- W is a bond.
- a and b are independently integers from 1 to 6 with the proviso that a + b is ⁇ 7.
- a and b are each the integer 2.
- X is selected from the group consisting of alkylene, alkenylene, alkynylene, arylene, heteroarylene, and heterocyclylene wherem the alkylene, alkenylene, and alkynylene groups can be optionally interrupted or terminated with arylene, heteroarylene, or heterocyclylene, and optionally interrupted by one or more -O- groups.
- X is alkylene.
- X is -(CH2) ⁇ - -.
- Y is selected from the group consisting of -O-, -S(O)o-2-, -S(O) 2 -N(R 8 )-, -C(R 6 )-, -C(R 6 )-O-, -O-C(R 6 )-, -O-C(O)-O-, -N(R 8 )-Q-, -C(R 6 )-N(R 8 )-, -O-C(R 6 )-N(R 8 )-, -C(R 6 )-N(OR 9 )-,
- Y is selected from the group consisting of -S(O)o- 2 -, -S(O) 2 -N(R 8 )-, -C(R 6 )-, -C(R 6 )-O-, -O-C(R 6 )-, -O-C(O)-O-, -N(R 8 )-Q-, -C(R 6 )-N(R 8 )-, -O-C(R 6 )-N(R 8 )-, -C(R 6 )-N(OR 9 )-,
- 1 ⁇ is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, hydroxyalkyl, haloalkyl, haloalkoxy,
- X and X ⁇ - 2 are independently selected from C 2 - 7 alkylene groups; and in certain embodiments X M and X ⁇ - 2 are independently selected from C 2 - 4 alkylene groups.
- Xi-i is -(CH 2 ) _ 4 -, -CH 2 -
- X M is -(CH2)2- 4 - or - CH 2 -C(CH 3 ) 2 -.
- X ⁇ - 2 is -(CH 2 ) 2 -, or -(CH 2 ) 3 -.
- Z is -S(O) 2 -.
- Fonnulas I, la, ⁇ , Ila, in, ma, rV, orIVa, Z is -S(O)-.
- Rj is linear or branched C M alkyl, aryl, or 5 to 10 membered heteroaryl containing one or two heteroatoms, wherein the alkyl, aryl, or heteroaryl group may be unsubstituted or substituted with one or more substituents.
- Ri is methyl, ethyl, 1 -propyl, 2-propyl, 2-methylpropyl, 2-hydroxy-2-methylpropyl, phenyl, 4-chlorophenyl, or 4-fluorophenyl. In certain embodiments Ri is methyl, ethyl, 2-propyl, 2-methylpropyl, 2-hydroxy-2-methylpropyl, or phenyl. In some embodiments, particularly embodiments of Formulas la, Ila, ma, IVa, Vm, IX, XXma, XXIVa, or XXV, X is -(CH 2 ) ⁇ - 3 -.
- R" is hydrogen, alkyl, hydroxyalkylenyl, or alkoxyalkylenyl.
- R" is hydrogen, methyl, ethyl, propyl, butyl, 2-hydroxyethyl, hydroxymethyl, 2-methoxyethyl, or ethoxymethyl.
- particularly embodiments of Formulas la Ila, ma, INa, Nm,
- R 2 is hydrogen, alkyl, hydroxyalkylenyl, or alkoxyalkylenyl. h certain embodiments R 2 is hydrogen, methyl, ethyl, propyl, butyl, 2-hydroxyethyl, hydroxymethyl, 2-methoxyethyl, or ethoxymethyl. In some embodiments, particularly embodiments of Fom ⁇ ulas la, Ha, a, IVa, VEH, IX, XXHIa, XXIVa, or XXV, R 2 is alkyl or alkoxyalkylenyl.
- R 2 is methyl, ethyl, propyl, butyl, 2-methoxyethyl, or ethoxymethyl.
- particularly embodiments of Formulas ⁇ , Ha, DI, ma, V, VI, Vm, IX, XXffia, XXINa, or XXN, n is 0.
- particularly embodiments of Formulas IN or IVa m is 0.
- the compound or salt induces the biosynthesis of one or more cytokines.
- Step (1) a 4-chloro-3- nitroquinoline of Formula XXVII is reacted with an amine of the formula R I -S(O) 2 -X I - 2 -O-X M - ⁇ H 2 or a salt thereof to provide a 3-nitroquinolin-4-amine of Formula XXVHI.
- the reaction can be carried out by adding the 4-chloro-3-nitroquinoline to a solution of an amine of the formula R I -S(O)2-X I - 2 -O-X M -NH 2 or salt thereof in a suitable solvent such as anhydrous dichloromethane in the presence of a base such as triethylamine.
- a suitable solvent such as anhydrous dichloromethane
- the reaction can be run at ambient temperature.
- the product or a pharmaceutically acceptable salt thereof can be isolated by conventional methods.
- Many 4-chloro-3-nitroquinolines of Formula XXVII are known or can be prepared using known synthetic methods, see for example, U.S. Patent Nos. 4,689,338; 5,175,296; 5,367,076; and 5,389,640; and the references cited therein.
- Amines of the formula R I -S(O) 2 -X I - 2 -O-X M -NH 2 or salts thereof can be prepared using known synthetic methods.
- the hydrochloride salt of CH 3 -S(O) 2 -CH 2 - CH2-O-CH2-CH2-NH2 can be prepared by reacting sodium thiomethoxide with 2- ⁇ 2-[(tert- butoxycarbonyl)amino]ethoxy ⁇ ethyl methanesulfonate followed by oxidation of the sulfur atom and removal of the tert-butoxycarbonyl group as described in Parts A-C of Example 1 infra.
- step (2) of Reaction Scheme I a 3-nitroquinolin-4-amine of Formula XX VHI is reduced to provide a quinoline-3,4-diamine of Formula VII.
- the reduction can be carried out using a conventional heterogeneous hydrogenation catalyst such as platinum on carbon or palladium on carbon.
- the reaction can be conveniently carried out in a Parr vessel in a suitable solvent such as acetonitrile, toluene and/or isopropanol.
- the product or a pharmaceutically acceptable salt thereof can be isolated by conventional methods.
- a quinoline-3,4-diamine of Formula VII is reacted with a carboxylic acid or an equivalent thereof to provide a lH-imidazo[4,5-c]quinoline of Formula VHI.
- Suitable equivalents to a carboxylic acid include orthoesters, and 1,1- dialkoxyalkyl alkanoates.
- the carboxylic acid or equivalent is selected such that it will provide the desired R 2 substituent in a compound of Formula Vm.
- tri ethyl orthoformate will provide a compound with hydrogen at the 2-position
- trimethyl orthovalerate will provide a compound with butyl at the 2-position.
- the reaction can be run in the absence of solvent or in an inert solvent such as toluene.
- step (3) can be carried out by (i) reacting a compound of Formula VII with an acyl halide of formula R 2 -C(O)Cl or R 2 -C(O)Br and then (ii) cyclizing.
- the acyl halide is added to a solution of a compound of Formula VII in an inert solvent such as acetonitrile, pyridine or dichloromethane.
- the reaction can be carried out at ambient temperature.
- the product of part (i) is heated in pyridine or alternatively in an alcohol such as ethanol with a tertiary amine such as triethylamine.
- the two steps can be combined into a single step in solvents such as pyridine, dichloromethane, and dichloroethane.
- a lH-imidazo[4,5-c]quinoline of Formula NUI is oxidized to provide an N-oxide of Formula IX using a conventional oxidizing agent that is capable of forming N-oxides.
- reaction can be conveniently carried out by treating a solution of a compound of Formula Nm in a suitable solvent such as chloroform or dichloromethane with 3-chloroperoxybenzoic acid at ambient temperature.
- a suitable solvent such as chloroform or dichloromethane
- 3-chloroperoxybenzoic acid at ambient temperature.
- an ⁇ -oxide of Formula IX is aminated to provide a lH-imidazo[4,5- c]quinolin-4-amine of Formula fla-1, which is a subgenus of Formulas I, la, II, Ha, and ub.
- a compound of Formula IX is reacted with an acylating agent.
- Suitable acylating agents include alkyl- or arylsulfonyl chorides (e.g., benzenesulfonyl choride, methanesulfonyl choride, orj3-toluenesulfonyl chloride).
- the product of part (i) is reacted with an excess of an aminating agent.
- Suitable aminating agents include ammonia (e.g.
- ammonium hydroxide in the form of ammonium hydroxide
- ammonium salts e.g., ammonium carbonate, ammonium bicarbonate, ammonium phosphate
- the reaction can be carried out by dissolving a compound of Formula EX in a suitable solvent such as dichloromethane or chloroform, adding ammonium hydroxide to the solution, and then adding / ?-toluenesulfonyl chloride.
- a suitable solvent such as dichloromethane or chloroform
- step (4) the oxidation of step (4a) and the amination of step (4b) can be carried out without isolating the product of the oxidation to provide a 1H- imidazo[4,5-c]quinolin-4-amine of Formula IIa-1.
- step (4) after the lH-imidazo[4,5- ejquinoline of Formula NHI is consumed by reaction with 3-chloroperoxybenzoic acid as described in step (4a), the aminating and acylating agents are added to the reaction mixture as described in step (4b) above.
- the product or a pharmaceutically acceptable salt thereof can be isolated using conventional methods.
- Formula X is reduced to provide a quinoline-3,4-diamine of Formula XL
- the reaction can be carried out as in step (2) of Reaction Scheme I.
- the product or a pharmaceutically acceptable salt thereof can be isolated by conventional methods.
- Many 3-nitroquinolin-4- amines of Formula X are known or can be prepared using known synthetic methods, see for example, U.S. Patent Nos. 4,689,338; 5,175,296; and 5,389,640; and the references cited therein.
- step (2) of Reaction Scheme ⁇ a quinoline-3,4-diamine of Formula XI is reacted with a carboxylic acid or an equivalent thereof to provide a lH-imidazo[4,5-c]quinoline of Formula -XII.
- step (3) of Reaction Scheme I The reaction can be conveniently carried out as described in step (3) of Reaction Scheme I.
- the product or a pharmaceutically acceptable salt thereof can be isolated by conventional methods.
- a suitable solvent such DMF or tetrahydrofuran.
- the reaction can be run at ambient temperature.
- the product or a pharmaceutically acceptable salt thereof can be isolated by conventional methods. Many vinyl sulfones are commercially available or can be prepared using known synthetic methods.
- step (4) of Reaction Scheme ⁇ a lH-imidazo[4,5-c]quinoline of Formula Xm is oxidized to provide an N-oxide of Formula XIV.
- the reaction can be conveniently carried out as in step (4a) of Reaction scheme I.
- step (5) of Reaction Scheme ⁇ an N-oxide of Formula XIV is aminated to provide a lH-imidazo[4,5-c]quinolin-4-amine of Formula IIa-2, which is a subgenus of
- n is as defined above;
- Ria and R 2a are a subset of Ri and R , respectively, as defined above, which do not include those groups that one skilled in the art would recognize as being susceptible to reduction or decomposition under the mildly acidic conditions in step (1).
- susceptible groups include, for example, alkenyl, alkynyl, and aryl groups, and groups bearing nitro and -S- substitutents.
- R, Ri, R 2 , X , and n are as defined above;
- P is a protecting group such as, for example, tert-butoxycarbonyl; and Hal is chloro, bromo, or iodo.
- h step (1) the amino group of an amino alcohol of Formula XN is protected with a removable protecting group such as an alkoxycarbonyl group (e.g., tert-butoxycarbonyl) to provide a protected amine of Formula XNI.
- the reaction can be conveniently carried out by adding a base, such as aqueous sodium hydroxide, to a solution of the hydroxy amine of Formula XN in a suitable solvent such as tetrahydrofuran, and then adding tert-butyl dicarbonate.
- a base such as aqueous sodium hydroxide
- a suitable solvent such as tetrahydrofuran
- tert-butyl dicarbonate tert-butyl dicarbonate.
- the product can be isolated by conventional methods.
- step (2) of Reaction Scheme IN the hydroxy group of the protected amine of Formula XVI is alkylated with an allyl halide to provide an allyloxy compound of Formula XVH.
- the reaction can be conveniently carried out by combining the protected amine of Formula XVI with allyl bromide in a biphasic mixture of aqueous 50% sodium hydroxide in an inert solvent such as dichloromethane in the presence of a phase transfer catalyst such as benzyltrimethylammonium chloride.
- the reaction can be carried out at ambient temperature.
- the product can be isolated by conventional methods.
- step (3) of Reaction Scheme IV the allyloxy compound of Formula XV ⁇ is hydroborated and oxidized to provide a hydroxypropoxy compound of Formula XVIH.
- the reaction is carried out by first adding 9-borabicyclo[3.3.1 ]nonane, dissolved in a suitable solvent such as tetrahydrofuran, to an allyloxy compound of Formula XVH, then adding water followed by aqueous sodium hydroxide, and then adding an excess of hydrogen peroxide. After completion of the reaction, excess peroxide can be neutralized with aqueous sodium metabisulfite.
- the product can be isolated by conventional methods.
- step (4) of Reaction Scheme IV the amine protecting group on the hydroxypropoxy compound of Formula XVm is removed to provide a hydroxypropoxy amine of Formula XIX.
- Removal of the ⁇ ert-butoxycarbonyl protecting group can be conveniently carried out by adding hydrochloric acid in a solvent such as dioxane to the hydroxypropoxy compound of Formula XVIII.
- a hydroxypropoxy amine of Formula XIX or a salt thereof is reacted with a 4-chloro-3-nitroquinoline of Formula XXVH to provide a 3- nitroquinolin-4-amine of Formula XX.
- the reaction can be carried out by adding a 4- chloro-3-nitroquinoline to a solution of a hydroxpropoxy amine of Formula XIX or a salt thereof in a suitable solvent such as anhydrous dichloromethane in the presence of a base such as triethylamine.
- a suitable solvent such as anhydrous dichloromethane
- the reaction can be run at ambient temperature.
- the product or a pharmaceutically acceptable salt thereof can be isolated by conventional methods.
- step (6) of Reaction Scheme TV the hydroxy group of a 3-nitroquinolin-4-amine of Formula XX is replaced with a halogen to provide a 3-nitroquinolin-4-amine of Fonnula XXI.
- the reaction can be carried out by adding thionyl chloride to a 3- nitroquinolin-4-amine of Formula XX in a suitable solvent such as dichloromethane.
- the reaction can be run at an elevated temperature, for example, at reflux.
- the product or a pharmaceutically acceptable salt thereof can be isolated by conventional methods.
- step (7) of Reaction Scheme IV a 3-nitroquinolin-4-amine of Formula XXI is reduced to provide a quinoline-3,4-diamine of Formula XXH.
- the reduction can be carried out as in step (2) of Reaction Scheme I.
- the product or a pharmaceutically acceptable salt thereof can be isolated by conventional methods.
- step (8) of Reaction Scheme IN a quinoline-3,4-diamine of Formula XX ⁇ is reacted with a carboxylic acid or an equivalent thereof to provide a lH-imidazo[4,5- cjquinoline of Formula XXJTI.
- the reaction can be carried out as in step (3) of Reaction Scheme I.
- the product or a pharmaceutically acceptable salt thereof can be isolated by conventional methods.
- step (9) of Reaction Scheme IV a lH-imidazo[4,5-e]quinoline of Formula
- XX ⁇ I is oxidized to a 5- ⁇ -oxide and then animated to provide a lH-imidazo[4,5- c]quinolin-4-amine of Formula XXIV.
- the reaction can be conveniently carried out as in step (4) or steps (4a) and (4b) of Reaction Scheme I.
- the product or a pharmaceutically acceptable salt thereof can be isolated using conventional methods.
- step (10) of Reaction Scheme IV a lH-imidazo[4,5-c]quinolin-4-amine of Formula XXIV is reacted with a sodium thiolate of formula Na + S ⁇ -R ⁇ to provide a 1H- imidazo[4,5-c]quinolin-4-amine of Formula IJa-4, which is a subgenus of Formulas I, la, ⁇ , ⁇ a, and lib.
- the reaction can be carried out by adding a lH-imidazo[4,5-c]quinolin-4- amine of Formula XXIV to a sodium thiolate of formula Na + S " -R ⁇ in a suitable solvent such as DMF.
- the reaction can be run at ambient temperature.
- the product or a phannaceutically acceptable salt thereof can be isolated using conventional methods.
- Sodium thiolates of formula Na S " -R ⁇ are commercially available or can be readily prepared by adding a thiol of formula ⁇ S-Ri to a suspension of sodium hydride in a suitable solvent such as DMF.
- a suitable solvent such as DMF.
- the sulfide moiety of a lH-imidazo[4,5- c]quinolin-4-amine of Formula ⁇ a-4 can be oxidized to a sulfinyl or sulfonyl moiety to provide a lH-imidazo[4,5-c]quinolin-4-amine of Formula ⁇ a-5, which is a subgenus of Formulas I, la, H, Ha, and Hb.
- the reaction can be canied out by treating a solution of a lH-imidazo[4,5-c]quinolin-4-amine of Formula ⁇ a-4 in a suitable solvent such as dichloromethane or chloroform with 3-chloroperoxybenzoic acid at ambient temperature.
- a suitable solvent such as dichloromethane or chloroform
- the degree of oxidation is controlled by adjusting the amount of 3-chloroperoxybenzoic acid used in the reaction.
- Using approximately one equivalent will provide the sulfinyl moiety, and using two equivalents will provide the sulfonyl moiety.
- the product or a pharmaceutically acceptable salt thereof can be isolated using conventional methods.
- Step (1) a lH-imidazo[4,5- c]quinolin-4-amine of Formula XXIVb is reduced to provide a 6,7,8,9-tetrahydro-lH- imidazo[4,5-c]quinolin-4-amine of Formula XXVI.
- the reaction can be carried out as in step (1) of Reaction Scheme HI.
- the product or a pharmaceutically acceptable salt thereof can be isolated using conventional methods.
- step (2) of Reaction Scheme V a 6,7,8,9-tetrahydro-lJz r -imidazo[4,5-e]quinolin- 4-amine of Formula XXVI is reacted with a sodium thiolate of .formula Na + S " -R ⁇ to provide a 6,7,8,9-tetrahydro-lH-imidazo[4,5-c]quinolin-4-amine of Formula ma-2, which is a subgenus of Formulas I, la, IH, ma, and Hlb.
- the reaction can be carried out and sodium thiolates obtained as described in step (10) of Reaction Scheme IV.
- the product or a pharmaceutically acceptable salt thereof can be isolated using conventional methods.
- step (3) of Reaction Scheme V a 6,7,8,9-tefrahydro-l_/J-imidazo[4,5-c]quinolin- 4-amine of Formula IHa-2 can be oxidized to provide a 6,7,8,9-tetrahydro-lH-imidazo[4,5- c]quinolin-4-amine of Formula I ⁇ a-3, which is a subgenus of Formulas I, la, HI, ma, and Hlb.
- the reaction can be carried out as described in step (11) of Reaction Scheme TV.
- the product or a pharmaceutically acceptable salt thereof can be isolated using conventional methods.
- Reaction Scheme VI where R, Ri, R 2 , XM, X ⁇ - 2 , and m are as defined above.
- Reaction Scheme VI begins with a 4-chloro-3-nitro[l,5]naphthyridine of Formula XXIX.
- Compounds of Formula XXIX and their preparation are known; see for example U.S. Patent No. 6,194,425 and the references cited therein.
- Steps (1) through (4) of Reaction Scheme VI can be carried out as described for the corresponding steps (1) through (4) of Reaction Scheme I to provide a lH-imidazo[4,5-e][l,5]naphthyridine of Formula IVa-1, which is a subgenus of Formulas I, la, TV, and IVa.
- the product or a pharmaceutically acceptable salt thereof can be isolated using conventional methods.
- Reaction Scheme VH where R, R b R 2 , X M , X ⁇ - 2 , and m are as defined above and Ph is phenyl.
- Reaction Scheme VH begins with a 5-chloro-4-nitrotetrazolo[l,5- ][l,7]naphthyridine of Formula XXXrV.
- Compounds of Formula XXXIV can be prepared using the synthetic methods described in U.S. Patent No. 6,194,425 and the references cited therein.
- Steps (1) through (3) of Reaction Scheme VH can be carried out as described for the corresponding steps (1) through (3) of Reaction Scheme I to provide a lH-tetrazolo[l,5- ⁇ ]imidazo[4,5- c][l,7]naphthyridine of FonnulaXXXV.
- a lH-tetrazolo[l,5- ⁇ ]imidazo[4,5- c][l,7]naphthyridine of Formula XXXV is reacted with triphenylphosphine to form a N- triphenylphosphinyl intermediate of Formula XXXVI.
- the reaction can be carried out by combining a compound of Fonnula XXXV with triphenylphosphine under a nitrogen atmosphere in a suitable solvent such as toluene or 1,2-dichlorobenzene and heating at reflux.
- a suitable solvent such as toluene or 1,2-dichlorobenzene and heating at reflux.
- N-triphenylphosphinyl intermediate of Formula XXXNI is hydrolyzed to provide a lH-imidazo[4,5-c][l,7]naphthyridine of Formula XXXV ⁇ , which is a subgenus of Formulas I and la.
- the hydrolysis can be carried out by general methods well known to those skilled in the art, for example, by heating in a lower alkanol in the presence of an acid.
- the product or a pharmaceutically acceptable salt thereof can be isolated using conventional methods.
- Reaction Scheme VIE where R, Ri, R 2 , Xi-i, X ⁇ - 2 , and m are as defined above, Ph is phenyl, and -Otf is a trifluoromethanesulfonate group.
- Reaction Scheme VHI begins with a 4- nitrotetrazolo[l,5- ⁇ ][l,8]naphthyridine of Formula XXXVIH.
- Compounds of Formula XXXVm can be prepared using the synthetic methods described in U.S. Patent No. 6,194,425 and the references cited therein.
- Steps (1) through (5) of Reaction Scheme Nm can be carried out as described for the conesponding steps (1) through (5) of Reaction Scheme VH to provide a lH-imidazo[4,5-c][l,8]naphthyridine of Formula XLI, which is a subgenus of Formulas I and la.
- the product or a pharmaceutically acceptable salt thereof can be isolated using conventional methods.
- Compounds of the invention can also be prepared using variations of the synthetic routes shown in Reaction Schemes I through NHL
- the reduction method described in Reaction Scheme H for the preparation of tetrahydroquinolines can also be used to prepared tetrahydronaphthyridines and the synthetic route shown in Reaction Scheme IN for the preparation of quinolines can be used to prepare [l,5]naphthyridines by using a 4-chloro-3-nitro[l,5]naphthyridine in lieu of a 4-chloro-3-nitroquinoline.
- Compounds of the invention can also be prepared using the synthetic routes described in the EXAMPLES below.
- compositions of the invention contain a therapeutically effective amount of a compound or salt of the invention as described above in combination with a pharmaceutically acceptable carrier.
- a therapeutically effective amount and “effective amount” mean an amount of the compound or salt sufficient to induce a therapeutic or prophylactic effect, such as cytokine induction, immunomodulation, antitumor activity, and/or antiviral activity.
- compositions of the invention will contain sufficient active ingredient to provide a dose of about 100 nanograms per kilogram (ng kg) to about 50 milligrams per kilogram (mg/kg), preferably about 10 micrograms per kilogram ( ⁇ g/kg) to about 5 mg/kg, of the compound or salt to the subject.
- a variety of dosage forms maybe used, such as tablets, lozenges, capsules, parenteral formulations, syrups, creams, ointments, aerosol formulations, transdermal patches, transmucosal patches and the like.
- the compounds or salts of the invention can be administered as the single therapeutic agent in the treatment regimen, or the compounds or salts of the invention may be administered in combination with one another or with other active agents, including additional immune response modifiers, antivirals, antibiotics, antibodies, proteins, peptides, oligonucleotides, etc.
- Compounds or salts of the invention have been shown to induce the production of certain cytokines in experiments performed according to the test set forth below.
- Cytokines whose production may be induced by the administration of compounds or salts of the invention generally include interferon- ⁇ (IFN- ⁇ ) and/or tumor necrosis factor- ⁇ (TNF- ⁇ ) as well as certain interleukins (IL). Cytokines whose biosynthesis may be induced by compounds or salts of the invention include IFN- ⁇ , TNF- ⁇ , IL-1, IL-6, IL-10 and IL-12, and a variety of other cytokines.
- the invention provides a method of inducing cytokine biosynthesis in an animal comprising administering an effective amount of a compound or salt or composition of the invention to the animal.
- the animal to which the compound or salt or composition is administered for induction of cytokine biosynthesis may have a disease as described infra, for example a viral disease or a neoplastic disease, and administration of the compound or salt may provide therapeutic treatment.
- the compound or salt may be administered to the animal prior to the animal acquiring the disease so that administration of the compound or salt may provide a prophylactic treatment.
- compounds or salts of the invention can affect other aspects of the innate immune response. For example, natural killer cell activity may be stimulated, an effect that may be due to cytokine induction.
- the compounds or salts may also activate macrophages, which in turn stimulate secretion of nitric oxide and the production of additional cytokines. Further, the compounds or salts may cause proliferation and differentiation of B-lymphocytes.
- Compounds or salts of the invention can also have an effect on the acquired immune response.
- T helper type 1 cytokine IFN- ⁇
- T helper type 2 T H 2
- IL-4, IL-5 and IL-13 T helper type 2
- the compound or salt or composition may be administered alone or in combination with one or more active components as in, for example, a vaccine adjuvant.
- the compound or salt and other component or components may be administered separately; together but independently such as in a solution; or together and associated with one another such as (a) covalently linked or (b) non-covalently associated, e.g., in a colloidal suspension.
- Conditions for which compounds or salts identified herein may be used as treatments include, but are not limited to: (a) viral diseases such as, for example, diseases resulting from infection by an adenoviras, a herpesvirus (e.g., HSN-I, HSN-H, CMN, or VZV), a poxvirus (e.g., an orthopoxvirus such as variola or vaccinia, or molluscum contagiosum), a picornavirus (e.g., rhinovirus or enterovirus), an orthomyxovirus (e.g., influenzavirus), a paramyxovirus (e.g., parainfluenzavirus, mumps virus, measles virus, and respiratory syncytial virus (RSV)), a coronavirus (e.g., SARS), a papovavirus (e.g., papillomaviruses, such as those that cause genital warts, common warts, or plantar war
- Streptococcus Chlamydia, Mycoplasma, Pneumococcus, Neisseria, Clostridium, Bacillus, Corynebacterium, Mycobacterium, Campylobacter, Vibrio, Senatia, Providencia, Chromobacterium, Brucella, Yersinia, Haemophilus, or Bordetella;
- other infectious diseases such chlamydia, fungal diseases including but not limited to candidiasis, aspergillosis, histoplasmosis, cryptococcal meningitis, or parasitic diseases including but not limited to malaria, pneumocystis carnii pneumonia, leishmaniasis, cryptosporidiosis, toxoplasmosis, and trypanosome infection
- neoplastic diseases such as intraepithelial neoplasias, cervical dysplasia, actinic keratosis, basal cell carcinoma, squam
- compounds or salts of the present invention may be useful as a vaccine adjuvant for use in conjunction with any material that raises either humoral and/or cell mediated immune response, such as, for example, live viral, bacterial, or parasitic immunogens; inactivated viral, tumor-derived, protozoal, organism-derived, fungal, or bacterial immunogens, toxoids, toxins; self-antigens; polysaccharides; proteins; glycoproteins; peptides; cellular vaccines; DNA vaccines; autologous vaccines; recombinant proteins; and the like, for use in connection with, for example, BCG, cholera, plague, typhoid, hepatitis A, hepatitis B, hepatitis C, influenza A, influenza B, parainfluenza, polio, rabies, measles, mumps, rubella, yellow fever, tetanus, diphtheria, hemophilus influenza b, tuberculosis, meningo
- Compounds or salts of the present invention may be particularly helpful in individuals having compromised immune function.
- compounds or salts may be used for treating the opportunistic infections and tumors that occur after suppression of cell mediated immunity in, for example, transplant patients, cancer patients and HIV patients.
- one or more of the above diseases or types of diseases for example, a viral disease or a neoplastic disease maybe treated in an animal in need thereof (having the disease) by administering a therapeutically effective amount of a compound or salt of the invention to the animal.
- An amount of a compound or salt effective to induce cytokine biosynthesis is an amount sufficient to cause one or more cell types, such as monocytes, macrophages, dendritic cells and B-cells to produce an amount of one or more cytokines such as, for example, IFN- ⁇ , TNF- ⁇ , IL-1, IL-6, IL-10 and IL-12 that is increased (induced) over a background level of such cytokines.
- the precise amount will vary according to factors known in the art but is expected to be a dose of about 100 ng/kg to about 50 mg/kg, preferably about 10 ⁇ g/kg to about 5 mg/kg.
- the invention also provides a method of • treating a viral infection in an animal and a method of treating a neoplastic disease in an animal comprising administering an effective amount of a compound or salt or composition of the invention to the animal.
- An amount effective to treat or inhibit a viral infection is an amount that will cause a reduction in one or more of the manifestations of viral infection, such as viral lesions, viral load, rate of virus production, and mortality as compared to untreated control animals.
- the precise amount that is effective for such treatment will vary according to factors known in the art but is expected to be a dose of about 100 ng/kg to about 50 mg/kg, preferably about 10 ⁇ g/kg to about 5 mg/kg.
- An amount of a compound or salt effective to treat a neoplastic condition is an amount that will cause a reduction in tumor size or in the number of tumor foci. Again, the precise amount will vary according to factors known in the art but is expected to be a dose of about 100 ng/kg to about 50 mg/kg, preferably about 10 ⁇ g/kg to about 5 mg/kg. Objects and advantages of this invention are further illustrated by the following examples, but the particular materials and amounts thereof recited in these examples, as well as other conditions and details, should not be construed to unduly limit this invention.
- Part B A solution of the material from Part A in chloroform (478 mL) was placed in a cold water bath. Solid 3-chloroperoxybenzoic acid (45 g of -60%) was added in portions over a period of 20 minutes. The reaction mixture was partitioned between chloroform (50 mL) and saturated aqueous sodium carbonate (100 mL). The organic layer was washed sequentially with saturated aqueous sodium carbonate (100 mL) and brine (100 mL), dried over anhydrous sodium sulfate, filtered, and then concentrated under reduced pressure to provide the crude product as a clear oil. The oil was purified by column chromatography
- Part C A solution of the material from Part B in methanol (22 mL) was chilled in an ice/water bath. Hydrochloric acid (94 mL of a 4M solution in dioxane) was added dropwise over a period of 22 minutes. The ice bath was removed and the reaction mixture was allowed to stir at ambient temperature for 45 minutes and then it was concentrated under reduced pressure. The residue was twice dissolved in methanol and then reconcentrated to provide 2-[2-(methylsulfonyl)ethoxy]ethaneamine hydrochloride as a clear oil.
- Part G 3-Chloroperoxybenzoic acid (1.5 g of -60%, 5.3 mmol) was added in portions over a period of 8 minutes to a solution of the material from Part F (5.3 mmol) in chloroform (53 mL).
- the reaction mixture was stirred at ambient temperature for 20 minutes during which time a precipitate formed.
- the suspension was diluted with chloroform (100 mL) and saturated aqueous sodium carbonate (50 mL) and then filtered to provide 2-methyl-l- ⁇ 2-[2-(methylsulfonyl)ethoxy]ethyl ⁇ -lH-imidazo[4,5-c]quinoline 5-oxide as a white solid.
- the crude wet material was carried on to the next step.
- Part H The material from Part G was suspended in dichloromethane (26 mL). Ammonium hydroxide (9 mL, 28%> solution in water) was added. P ⁇ ra-toluenesulfonyl chloride (1.0 g, 5.3 mmol) was added. After stirring at ambient temperature for 30 minutes the reaction mixture was filtered. Analysis (H NMR) of the isolated solid showed a 2: 1 mixture of desired product to N-oxide. The crude material was subjected to additional amination using the same reaction conditions.
- Trimethyl orthovalerate (1.3 g, 8.1 mmol) was added to a solution of N 4 - ⁇ 2-[2- (methylsulfonyl)ethoxy]ethyl ⁇ quinoline-3,4-diamine (-2.3 g, -7.4 mmol) in acetonitrile (37 mL). Pyridine hydrochloride (-100 mg) was added and the reaction mixture was heated to reflux with the volatiles being collected in a Dean Stark trap. After 15 minutes analysis by TLC indicated that the starting material was consumed.
- reaction mixture was stined at ambient temperature for 30 minutes then additional 3-chloroperoxybenzoic acid (0.4 g) was added.
- the reaction mixture was stined at ambient temperature for 15 minutes and then partitioned between dichloromethane (100 mL) and saturated aqueous sodium carbonate (50 mL).
- the organic layer was washed sequentially with saturated aqueous sodium carbonate (50 mL) and brine (50 mL), dried over anhydrous sodium sulfate, filtered, and then concentrated under reduced pressure to provide 2-butyl-l- ⁇ 2-[2- (methylsulfonyl)ethoxy]ethyl ⁇ -lH-imidazo[4,5-c]quinoline 5-oxide as an orange foam.
- Part C Aqueous ammonium hydroxide (12 mL, 30% solution in water) was added to a rapidly stined solution of the material from Part B in dichloromethane (36 mL). Para- toluenesulfonyl chloride (1.4 g, 7.2 mmol) was added. The reaction mixture was stined at ambient temperature until analysis by TLC showed that the starting material had been consumed. The reaction mixture was partitioned between chloroform (100 mL) and saturated aqueous sodium carbonate (50 mL).
- Part B 3-Chloroperoxybenzoic acid (2.15 g of 60%>, 7.47 mmol) was added over a period of 10 minutes to a suspension of the material from Part A (6.79 mmol) in chloroform (34 mL). After 20 minutes analysis by TLC indicated that the starting material had been consumed. Ammonium hydroxide (34 mL, 28%o solution in water) was added. The resulting biphasic mixture was homogenized by stirring for several minutes. Para- toluenesulfonyl chloride (1.4 g, 7.5 mmol) was added in a single portion. The reaction mixture was stined at ambient temperature for 15 minutes. The reaction mixture was poured into a separatory funnel and the layers were separated.
- Part B 3-Chloroperoxybenzoic acid (3.24 g of 60%, 11.3 mmol) was added in portions to a solution of the material from Part A (10.2 mmol) in chloroform (51 mL) over a period of 8 minutes. After 20 minutes analysis by TLC indicated that the starting material was consumed. The reaction mixture was partitioned between chloroform (100 mL) and saturated aqueous sodium carbonate (100 mL). The aqueous layer was extracted with chloroform (50 mL).
- Triethyl orthopropionate (2.5 mL,12 mmol) was added to a stined solution of N 4 - ⁇ 2-[2-(methylsulfonyl)ethoxy]ethyl ⁇ quinoline-3,4-diamine (3.2 g, 10 mmol) in acetonitrile (50 mL). Pyridine hydrochloride (0.3 g) was added and the reaction mixture was heated to reflux with the volatiles being collected in a Dean Stark trap. After 40 minutes analysis by TLC indicated that the starting material was consumed.
- reaction mixture was concentrated under reduced pressure to provide 3.2 g of 2-ethyl-l- ⁇ 2-[2- (methylsulfonyl)ethoxy]ethyl ⁇ -lH-imidazo[4,5-c]quinoline as an oil which slowly solidified.
- Part B Using the method of Example 4 Part B, the material from Part A was oxidized to provide 2-ethyl- 1 - ⁇ 2-[2-(methylsulfonyl)ethoxy]ethyl ⁇ - lH-imidazo[4,5-c]quinoline 5- Oxide as an orange solid.
- Part C Using the method of Example 4 Part C the material from Part B was aminated and purified to provide 0.8 g of 2-ethyl-l- ⁇ 2-[2-(methylsulfonyl)ethoxy]ethyl ⁇ -lH- imidazo[4,5-c]quinolin-4-amine as a white powder, mp 160-163 °C.
- Part B A solution of the material from Part A (3.33 g, 8.42 mmol) in ethanol (42 mL) and triethylamine (3.5 mL, 25.3 mmol) was heated at reflux for 2 hours. Analysis by TLC showed that the starting material had been consumed. The reaction mixture was allowed to cool and then it was concentrated under reduced pressure. The residue was combined with water (50 mL) and then extracted with dichloromethane (2 X 100 mL). The combined extracts were washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to provide crude product as a light yellow oil. This material was purified by column chromatography (silica gel eluting with
- Part C 3-Chloroperoxybenzoic acid (2.2 g of 60%, 7.6 mmol) was added in portions to a solution of the material from Part B (6.9 mmol) in chloroform (34 mL) over a period of 8 minutes. After 20 minutes analysis by TLC indicated that the starting material was consumed. Ammonium hydroxide (34 mL, 30% solution in water) was added. The resulting biphasic mixture was stined until both phases were clear and red. Solid para- toluenesulfonyl chloride (1.3 g, 6.6 mmol) was added in several portions. The reaction mixture was stined at ambient temperature. After 10 minutes analysis by TLC indicated that the starting material was consumed.
- the reaction mixture was partitioned between dichloromethane (100 mL) and saturated aqueous sodium carbonate (50 mL). The organic layer was washed sequentially with saturated aqueous sodium carbonate (50 mL) and with brine (50 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to provide crude product.
- the crude product was purified by column chromatography (silica gel eluting with 95/5 dichloromethane/methanol) to provide a light brown foam. This material was dissolved in hot methanol (50 mL). The solution was treated with activated charcoal (1 g of DARCO) and then filtered though a layer of
- Part E Using the method of Example 4 Part B, the material from Part D was oxidized to provide 2-ethoxymethyl- 1 - ⁇ 2-[2-(phenylsulfonyl)ethoxy] -2-methylpropyl ⁇ - 1H- imidazo[4,5-c]quinoline 5-oxide as an orange foam.
- Part B Using the method of Example 4 Part B, the material from Part A was oxidized to provide l- ⁇ 2-[2-(methylsulfonyl)ethoxy]-2-methylpropyl ⁇ -2-ethoxymethyl-lH- imidazo[4,5-c]quinoline 5-oxide as an orange foam.
- the pH of the solution was adjusted to pH 12 by the addition of aqueous 50% sodium hydroxide and then the solution was extracted with dichloromethane (2 x 100 mL). The combined extracts were washed with brine (100 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to provide crude product as a white foam. This material was recrystallized from ethyl acetate.
- the slurry was cooled in an ice bath.
- the pH was adjusted to pH 2 by the addition of sulfuric acid (1 L of 1M in water).
- the resulting solution was extracted with ethyl acetate (4 x 500 mL).
- the combined extracts were washed sequentially with water (3 x 500 mL) and brine (2 x 500 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to give a first lot of crude product. Analysis of the aqueous and brine washes indicated that they contained product so they were extracted with ethyl acetate.
- the combined extracts were dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to provide a second lot of crude product. The two lots were combined to provide 263.8 g of tert-butyl (2-hydroxyethyl)carbamate as a colorless oil.
- Part D The crude product from Part C was combined with hydrochloric acid (1.242 L of 4M in dioxane) and then stined under a nitrogen atmosphere for -42 hours.
- the reaction mixture was concentrated under reduced pressure, triturated with dichloromethane/methanol and then concentrated under reduced pressure to provide an oil.
- the oil was triturated with diethyl ether four times and then dried under vacuum to provide crade 3-(2-aminoethoxy)propan-l-ol hydrochloride.
- This material was purified by column chromatography (1.5 Kg of silica gel eluting with 98/2 dichloromethane/methanol) to provide 98.1 g of 3- ⁇ 2-[(3-nitroquinolin-4-yl)amino]ethoxy ⁇ propan-l-ol as a yellow solid.
- Part G A slurry of the material from Part F in toluene (75 mL) was added to a Parr vessel containing 5%> platinum on carbon (0.50 g) and toluene (25 mL). The vessel was placed on a shaker and then pressurized with hydrogen to 50 psi (3.4 X 10 5 Pa). After 16 hours analysis by TLC indicated that the reaction was not complete. Additional catalyst (0.50 g) was added and the reaction was pressurized with hydrogen to 50 psi (3.4 X 10 5 Pa). After
- reaction was refluxed under a nitrogen atmosphere for 19 hours at which time analysis by HPLC indicated that all of the starting material had been consumed.
- reaction mixture was concentrated under reduced pressure to provide 10.5 g of crude l-[2-(3- chloropropoxy)ethyl]-2-propyl-lH-imidazo[4,5-c]quinoline as a black oil.
- Part i 3-Chloroperoxybenzoic acid (12.74 g of 75%, 55.4 mmol) was added in a single portion to a solution of the material from Part H in dichloromethane (100 mL).
- the reaction mixture was stined at ambient temperature for 30 minutes at which time analysis by HPLC indicated that all of the starting material had been consumed.
- Ammonium hydroxide 50 mL of 27%) was added followed by the slow addition of tosyl chloride (6.64 g, 34.8 mmol). After the addition was complete the reaction mixture was stined at ambient temperature for 30 minutes at which time analysis by HPLC indicated that all of the starting material had been consumed.
- reaction mixture was diluted with dichloromethane, washed sequentially with aqueous potassium carbonate, water, and brine, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to prove 8.5 g of crude product as a gray solid.
- This material was purified by column chromatography (93 g of silica gel eluting with ethyl acetate) to provide 4.5 g of product.
- Part B 3-Chloroperoxybenzoic acid (2.79 g of 75%, 12.1 mmol) was added to a solution of the material from Part A (2.34 g, 6.06 mmol) in dichloromethane (25 mL). After 15 minutes analysis by ⁇ PLC indicated that all of the starting material had been consumed. The reaction mixture was diluted with dichloromethane (50 mL), washed sequentially with aqueous saturated potassium carbonate, water (3 x 50 mL), and brine (2 x 50 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to provide crude product as a brown oil.
- This material was purified by column chromatography (300 g of silica gel eluting with 98/2 dichloromethane/methanol) followed by recrystallization from acetonitrile to provide 1.53 g of l-(2- ⁇ 3-[(l- methylethyl)sulfonyl]propoxy ⁇ ethyl)-2-propyl-lH-imidazo[4,5-c]quinolin-4-amine as a white crystalline solid, mp 176.0-177.0 °C.
- Example 15 1 -(2- ⁇ 3-[(2-Methylphenyl)sulfonyl]propoxy ⁇ ethyl)-2-propyl- lH-imidazo[4,5-e]quinolin-4-amine
- reaction mixture was poured into water (250 mL) with rapid stirring and the resulting mixture was extracted with dichloromethane (50 mL). The organic layer was washed sequentially with aqueous saturated potassium carbonate and brine, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to provide 3.68 g of crude l-(2- ⁇ 3-[(2-methylphenyl)thio]propoxy ⁇ ethyl)-2- propyl-lH-imidazo[4,5-c]quinolin-4-amine as a brown gummy solid.
- Part B 3-Chloroperoxybenzoic acid (4.10 g of 75%>, 17.6 mmol) was added to a solution of the material from Part A in dichloromethane (30 mL). After 15 minutes analysis by ⁇ PLC indicated that all of the starting material had been consumed.
- the reaction mixture was diluted with IN potassium hydroxide (100 mL) and dichloromethane (50 mL) and stined for 10 minutes. The organic layer was separated then washed sequentially with IN potassium hydroxide (2 x 50 mL), water (1 x 50 L) and brine (2 x 50 mL), dried over magnesium sulfate, filtered, and concentrated under reduced pressure to provide 3.61 g of a brown solid.
- This material was triturated with ethyl acetate containing a small amount of dichloromethane to provide 2.1 g of a brown solid.
- This material was purified by column chromatography (silica gel eluting sequentially with ethyl acetate, 95/5 ethyl acetate/methanol, and 9/1 ethyl acetate/methanol) to provide 1.2 g of 2-propyl-l-(2- ⁇ 3- [(pyridine-2-yl)sulfonyl]propoxy ⁇ ethyl)- lH-imidazo[4,5-c]quinolin-4-amine as a white powder, mp 150.0-151.0 °C. !
- Example 17 1 - ⁇ 2-[3-(Methylthio)propoxy]ethyl ⁇ -2-propyl- lH-imidazo[4,5-c]quinolin-4-amine
- Example 20 1 - ⁇ 2-[3-(Decane- 1 -sulfonyl)propoxy]ethyl ⁇ -2-propyl- lH-imidazo[4,5-c]quinolin-4-amine
- Part B The material from Part B was oxidized using the method of Example 19.
- the crude product was purified by column chromatography (silica gel eluting with 95/5 dichloromethane/methanol) to provide 1.3 g of a dark brown oil.
- the oil was dissolved in methanol (100 mL).
- the solution was heated gently then combined with activated charcoal (1 g of DARCO) and stined for 10 minutes.
- the mixture was filtered through a layer of CELITE filter aid and the filter cake was washed with methanol.
- the filtrate was concentrated under reduced pressure to provide a colorless oil.
- the oil was triturated with diethyl ether to provide a solid.
- Part B A suspension of 4-chloro-2-methyl-l- ⁇ 2-methyl-2-[2- (methylsulfonyl)ethoxy]propyl ⁇ -lH-imidazo[4,5-c]quinoline (1.80 g, 4.55 mmol) in a solution of ammonia in methanol (7 M, 18 mL) in a high pressure vessel was heated at 150 °C for 14 hours. The vessel was allowed to cool to room temperature. A solid was isolated by filtration, washed with methanol, and purified by flash chromatography (silica gel, eluted with 10% methanol in dichloromethane) to provide a white solid.
- the crude product was subjected to flash chromatography (silica gel, eluted with 5%> methanol in dichloromethane) to yield a mixture of compounds that were combined and resubjected to the reaction conditions.
- the mixture was allowed to cool to room temperature and was concentrated under reduced pressure to afford l-(2-propyl-lH-imidazo[4,5-c]quinolin-l-yl)-2-methylpropan-2-ol as a white solid that was used in the next step without purification.
- Part B Sodium metal (0.03 g, 1.3 mmol) was added to a solution of the material from Part A (1.7 g, 6.0 mmol) in tefraliydrofuran (24 mL). The reaction mixture was sonicated for 1 hour, then was heated at reflux for 2 hours. The reaction mixture was allowed to cool to room temperature and methyl vinyl sulfone (1.3 g, 12 mmol) was added dropwise. The reaction mixture was stined at room temperature for 17 hours, then water (50 mL) was added. The mixture was exfracted with ethyl acetate (3 x 50 mL).
- Part C Using the method of Example 4 Part B, the material from Part B was oxidized to provide l- ⁇ 2-methyl-2-[2-(methylsulfonyl)ethoxy]propyl ⁇ -2-propyl-lH-imidazo[4,5- cjquinoline 5-oxide as a pale orange oil.
- Part B A mixture of l-(nitromethyl)cyclopentanol (8.3 g, 57.2 mmol) and 20%> palladium hydroxide on carbon (0.6 g) in ethanol (150 mL) was hydrogenated at 35 psi (2.4 x 10 5 Pa) hydrogen pressure on a Pan apparatus for 1 day. After workup, the reaction was not complete and was subjected to the reaction conditions again for 8 days with fresh catalyst.
- the mixture was filtered through CELITE filter agent and the filtrate was concentrated to yield an oil that contained a 13:1 ratio of the desired amine product, 1- (aminomethyl)cyclopentanol, to the conesponding hydroxylamine.
- the oil was concentrated from toluene to remove the ethanol and used in the next experiment without further purification.
- Part D A mixture of l- ⁇ [(3-nitroquinolin-4-yl)amino]methyl ⁇ cyclopentanol (8.26 g, 28.8 mmol) and 5% platinum on carbon (0.9 g) in ethanol (200 mL) was hydrogenated on a Pan apparatus overnight. The mixture was filtered through CELITE filter agent and the filtrate was concentrated. The product l- ⁇ [(3-aminoquinolin-4-yl)amino]methyl ⁇ -l- cyclopentanol was concentrated from toluene and dichloromethane to remove residual ethanol, then used immediately in the next step.
- Part G Using the method of Example 4 Part B, the material from Part F was oxidized to provide 2-(ethoxymethyl)- 1 -( ⁇ 1 -[2-(methylsulfonyl)ethoxy] cyclopentyl ⁇ methyl)- 1H- imidazo[4,5-c]quinoline 5-oxide as a pale orange oil, which was used in the next step without purification.
- Part B A mixture of the l- ⁇ [(3-nitroquinolin-4-yl)amino]methyl ⁇ cyclohexanol prepared above (87%> purity, 7.00 g, 20.3 mmol) and 5% platinum on carbon (0.60 g) in toluene (160 mL) and ethanol (20 mL) was hydrogenated at 20-30 psi (1.4 x 10 5 to 2.1 x 10 5 Pa) on a Pan apparatus for 2 hours. The mixture was filtered through CELITE filter agent, which was rinsed with toluene. The filtrate was concentrated to an oil and the oil was concentrated twice from toluene. To the oil was added CH 2 C1 2 (200 mL).
- Part D Using the method of Example 4 Part B, the material from Part C was oxidized to provide 2-(ethoxymethyl)- 1 -( ⁇ 1 -[2-(methylsulfonyl)ethoxy] cyclohexyl ⁇ methyl)- 1H- imidazo[4,5-e] quinoline 5-oxide as an orange oil, which was used in the next step without purification.
- Part E Using the method of Example 4 Part C, the material from Part D was aminated.
- Part B The material from Part A was combined with ethanol (20 mL) and triethylamine (2.5 mL) to yield a solution that was heated at reflux for 10 hours. The solution was allowed to cool to room temperature, then was concentrated under reduced pressure to afford a brown solid. The solid was purified by flash chromatography (silica gel, 5%> methanol in dichloromethane) to provide 2.15 g of 2-hexyl-l- ⁇ 2-[2- (methylsulfonyl)ethoxy]ethyl ⁇ -lH-imidazo[4,5-c]quinoline as an off-white solid.
- Part C Using the method of Example 4 Part B, the material from Part B was oxidized to provide 2-hexyl-l- ⁇ 2-[2-(methylsulfonyl)ethoxy]ethyl ⁇ -lH-imidazo[4,5-c]quinoline 5- oxide as an orange solid, which was used in the next step without purification.
- Part D Using the method of Example 4 Part C, the material from Part C was aminated.
- the crude product was purified by flash chromatography (silica gel, 5% methanol in dichloromethane) to provide a powder that was crystallized from acetonitrile to provide 1.3 g of 2-hexyl-l- ⁇ 2-[2-(methylsulfonyl)ethoxy]ethyl ⁇ -lH-imidazo[4,5-c]quinolin-4- amine as crystalline plates, mp 158-160 °C.
- Part B A mixture of l-(nitromethyl)cyclobutanol (8.2 g, 62.5 mmol) and 20% palladium hydroxide on carbon (1.0 g) in ethanol (200 mL) was hydrogenated at 40 psi (2.8 x 10 5 Pa) on a Pan apparatus for 6 days. More 20% palladium hydroxide on carbon (1.2 g) was added and the mixture was hydrogenated at 40 psi (2.8 x 10 5 Pa) for an additional 5 days. The mixture was filtered through CELITE filter agent, which was rinsed with ethanol. The filtrate was concentrated to an oil that was concentrated from dichloromethane and chloroform to remove residual ethanol. l-(Aminomethyl)cyclobutanol was obtained as an off white solid (6.15 g) that was used without further purification in the next step.
- Part D A mixture of l- ⁇ [(3-nitroquinolin-4-yl)amino]methyl ⁇ cyclobutanol (6.32 g, 23.1 mmol) and 20%> palladium hydroxide on carbon (0.60 g) in ethanol (100 mL) was hydrogenated overnight on a Pan apparatus at 40 psi (2.8 x 10 5 Pa). The mixture was filtered through CELITE filter agent, which was rinsed several times with ethanol, and the filtrate was concentrated to provide 5.66 g of l- ⁇ [(3-aminoquinolin-4- yl)amino]methyl ⁇ cyclobutanol as a pale yellow solid. The solid was concentrated from toluene and chloroform to remove ethanol before using directly in the next reaction.
- Part B A suspension of 4-chloro- 1 - ⁇ 2-methyl-2-[2-(methylsulfonyl)ethoxy]propyl ⁇ - ⁇ H- imidazo[4,5-c]quinoline (4.30 g, 11.3 mmol) in a solution of ammonia in methanol (7 M, 43 mL) in a high pressure vessel was heated at 150 °C for 12 hours. The vessel was allowed to cool to room temperature. The mixture was concentrated under reduced pressure to afford a solid that was slurried in water (50 mL), saturated aqueous sodium carbonate, and methanol.
- the solid was isolated by filtration, washed with water, and purified by flash chromatography (silica gel, eluted with 10% methanol in dichloromethane) to provide a white solid.
- the solid was heated in methanol (200 mL) at reflux, isolated by filtration, washed with methanol, and dried to yield 2.2 g of l- ⁇ 2- methyl-2-[2-(methylsulfonyl)ethoxy]propyl ⁇ -lH-imidazo[4,5-c]quinolin-4-arnine as a white powder, mp 261-264 °C.
- Part B Sodium hydride (60%» dispersion in oil, 55 mg, 1.38 mmol) was added to a stined solution of l-(2-butyl-lH-imidazo[4,5-c]quinolin-l-yl)-2-methylpropan-2-ol (4.10 g, 13.8 mmol) in tetrahydrofuran (55 mL). After 5 minutes, methyl vinyl sulfone (2.90 g, 27.6 mmol) was added dropwise. The reaction mixture was stined at room temperature for 1 hour. Water (50 mL) and a small amount of brine were added and the mixture was extracted with ethyl acetate (2 x 50 mL).
- Part D Using the method of Example 4 Part C, the material from Part C was aminated.
- Part B A mixture of l- ⁇ [(3-nitroquinolin-4-yl)amino]methyl ⁇ cyclopropanol (3.50 g, 13.5 mmol) and 5%> platinum on carbon (350 mg) in ethyl acetate (70 mL) and methanol (7 mL) was hydrogenated under 35 psi (2.4 x 10 5 Pa) on a Pan apparatus for 3 hours. The mixture was filtered through CELITE filter agent, which was rinsed with ethyl acetate.
- Part D The material from Part C was dissolved in ethanol (50 mL) and triethylamine (5.5 mL) was added. The reaction mixture was stined at 60 °C for 5 hours. The reaction mixture was allowed to cool to room temperature and was concentrated under reduced pressure. The residue was dissolved in dichloromethane (70 mL) and washed with saturated aqueous sodium bicarbonate (50 mL). The aqueous layer was back-extracted with dichloromethane (25 mL). The combined organic layers were dried over magnesium sulfate, filtered, and concentrated under reduced pressure to yield an oil. The oil was triturated with acetonitrile.
- Part F Using the method of Example 4 Part B, the material from Part E was oxidized to provide 2-(ethoxymethyl)- 1 -( ⁇ 1 -[2-(methylsulfonyl)ethoxy]cyclopropyl ⁇ methyl)- 1H- imidazo[4,5-c]quinoline 5-oxide as an orange solid, which was used in the next step without purification.
- Part G Using the method of Example 4 Part C, the material from Part F was aminated.
- Part B Sodium hydride (60% dispersion in oil, 30 mg, 0.74 mmol) was added to a stined solution of l-(2-ethyl-lH-imidazo[4,5-c]quinolin-l-yl)-2-methylpropan-2-ol (2.00 g, 7.43 mmol) in tetrahydrofuran (30 mL). After five minutes, methyl vinyl sulfone (1.60 g, 14.9 mmol) was added dropwise. The reaction mixture was stined at room temperature for 1.5 hours and a solid formed.
- Part C Using the method of Example 4 Part B, the material from Part B was oxidized to provide 2-ethyl-l- ⁇ 2-methyl-2-[2-(methylsulfonyl)ethoxy]propyl ⁇ -lH-imidazo[4,5- cjquinoline 5-oxide as an off-white foam, which was used in the next step without purification.
- Part D Using the method of Example 4 Part C, the material from Part C was aminated.
- the crude product was purified by flash chromatography (silica gel, eluted with 5%> methanol in dichloromethane) to provide 2.3 g of 2-ethyl-l- ⁇ 2-methyl-2-[2- (methylsulfonyl)ethoxy]propyl ⁇ -lH-imidazo[4,5-c]quinolin-4-amine as a white foam.
- a portion of the material (1.2 g) was crystallized from acetonitrile to provide 2-ethyl-l- ⁇ 2- methyl-2-[2-(methylsulfonyl)ethoxy]propyl ⁇ -lH-imidazo[4,5-c]quinolin-4-amine as tan, crystalline plates, mp 206-209 °C.
- Part B A solution of potassium carbonate (3.4 g, 24.3 mmol) in water (9 mL) was added to a stined suspension of N- ⁇ 4-[(2-hydroxy-2-methylpropyl)amino]quinolin-3-yl ⁇ -2- methoxyacetamide hydrochloride (5.5 g, 16.2 mmol) in ethanol (23 mL). The reaction mixture was heated at reflux for 1.5 hours, then was allowed to cool to room temperature. The mixture was concentrated under reduced pressure to yield an aqueous slurry that was diluted with water (50 mL) and a small amount of brine. The mixture was extracted with dichloromethane (2 x 50 mL).
- Part F A suspension of l-(4-amino-2-ethoxymethyl-lH-imidazo[4,5-c][l,5]naphthyridin- l-yl)-2-methylpropan-2-ol (2.87 g, 9.10 mmol), di-tert-butyl di-carbonate (5 g, 22.75 mmol), triethylamine (2.3 g, 22.75 mmol), 4-(dimethylamino)pyridine (111 mg, 0.91 mmol) and acetonitrile (91 mL) was heated to reflux at which time a solution was obtained. The solution was heated at reflux for 2 hours and then concentrated under reduced pressure.
- the resulting oil was partitioned between dichloromethane (200 mL) and saturated aqueous ammonium chloride (100 mL). The organic layer was separated, washed with saturated aqueous sodium bicarbonate (100 mL), dried over sodium sulfate, filtered, and then concentrated under reduced pressure to provide an orange oil.
- the oil was purified by column chromatography (silica gel eluted with 7/3 ethyl acetate/hexanes) to provide 4 g of NN-(bis tert-butoxycarbonyl)- l-(4-amino-2-ethoxymethyl-lH- imidazo[4,5-c][l,5]naphthyridin-l-yl)-2-methylpropan-2-ol as a peach oil which solidified under vacuum.
- Examples 41 - 4.8 The examples in the table below can be prepared by treating the indicated starting material with boron tribromide.
- the reaction can be carried out by adding a solution of boron tribromide (2.5 equivalents of 1 M in dichloromethane) to a suspension or solution of the staring material (1 equivalent) in dichloromethane at 0 °C.
- the reaction mixture is maintained at 0 °C until complete and then quenched with methanol.
- the product can be isolated using conventional methods.
- Examples 49 - 50 The examples in the table below can be prepared using the method described for Examples 41 - 48.
- exemplary compounds have the following Formula (Ia-6) wherein X M , X ⁇ - 2 , Ri, and R are defined in the table below, wherein each line of the table represents a specific compound.
- Certain exemplary compounds including some of those described above in the Examples, have the following Formulas (IIa-6, HIa-4, IVa-2, and XLII) and the following substituents (X M , X ⁇ - , R I , and R 2 ) wherein each line of the table is matched with each Formula to represent a specific embodiment of the invention and wherein
- CYTOKINE ⁇ NDUCTION IN HUMAN CELLS Compounds of the invention have been found to induce cytokine biosynthesis when tested using the method described below.
- An in vitro human blood cell system is used to assess cytokine induction. Activity is based on the measurement of interferon ( ⁇ ) and tumor necrosis factor ( ⁇ ) (IFN- ⁇ and TNF- ⁇ , respectively) secreted into culture media as described by Testerman et. al. in "Cytokine Induction by the Immunomodulators Imiquimod and S-27609", Journal of Leukocyte Biology, 58, 365-372 (September, 1995). Blood Cell Preparation for Culture Whole blood from healthy human donors is collected by venipuncture into EDTA vacutainer tubes.
- PBMC Peripheral blood mononuclear cells
- HISTOPAQUE-1077 HISTOPAQUE-1077. Blood is diluted 1:1 with Dulbecco's Phosphate Buffered Saline (DPBS) or Hank's Balanced Salts Solution
- DPBS Dulbecco's Phosphate Buffered Saline
- Hank's Balanced Salts Solution DPBS
- HBSS HBSS
- DPBS DPBS
- HBSS HBSS
- RPMI complete 4 x 10 6 cells/mL in RPMI complete.
- the PBMC suspension is added to 48 well flat bottom sterile tissue culture plates (Costar, Cambridge, MA or Becton Dickinson Labware, Lincoln Park, NJ) containing an equal volume of RPMI complete media containing test compound.
- the compounds are solubilized in dimethyl sulfoxide (DMSO).
- DMSO concentration should not exceed a final concentration of 1% for addition to the culture wells.
- the compounds are generally tested at concentrations ranging from 30-0.014 ⁇ M.
- test compound is added at 60 ⁇ M to the first well containing RPMI complete and serial 3 fold dilutions are made in the wells.
- the PBMC suspension is then added to the wells in an equal volume, bringing the test compound concentrations to the desired range (30-0.014 ⁇ M).
- the final concentration of PBMC suspension is 2 x 10 ⁇ cells/mL.
- the plates are covered with sterile plastic lids, mixed gently and then incubated for 18 to 24 hours at 37°C in a 5% carbon dioxide atmosphere.
- the plates are centrifuged for 10 minutes at 1000 rpm (approximately 200 x g) at 4°C.
- the cell-free culture supernatant is removed with a sterile polypropylene pipet and fransfened to sterile polypropylene tubes.
- Samples are maintained at -30 to -70°C until analysis.
- the samples are analyzed for interferon ( ⁇ ) by ELISA and for tumor necrosis factor ( ⁇ ) by ELISA or IGEN Assay.
- Interferon ( ⁇ ) and Tumor Necrosis Factor ( ⁇ ) Analysis by ELISA Interferon ( ⁇ ) concentration is determined by ELISA using a Human Multi-Species kit from PBL Biomedical Laboratories, New Brunswick, NJ.
- TNF Tumor necrosis factor
- ⁇ Tumor necrosis factor
- ELISA kits available from Biosource hitemational, Camarillo, CA.
- the TNF concentration can be determined by ORIGEN M-Series Immunoassay and read on an IGEN M-8 analyzer from IGEN International, Gaithersburg, MD.
- the immunoassay uses a human TNF capture and detection antibody pair from Biosource International, Camarillo, CA. Results are expressed in pg/mL.
Abstract
Description































































































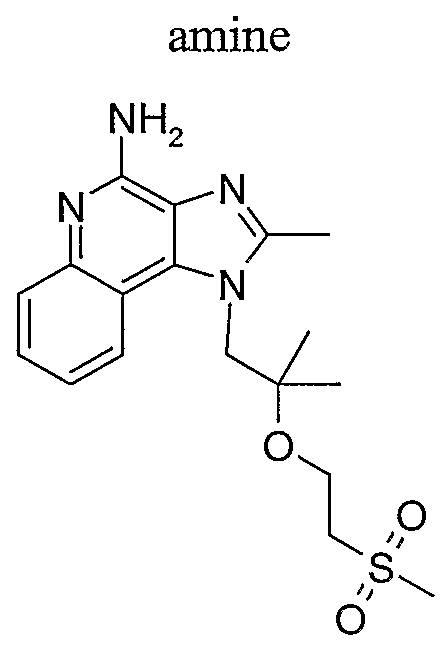


















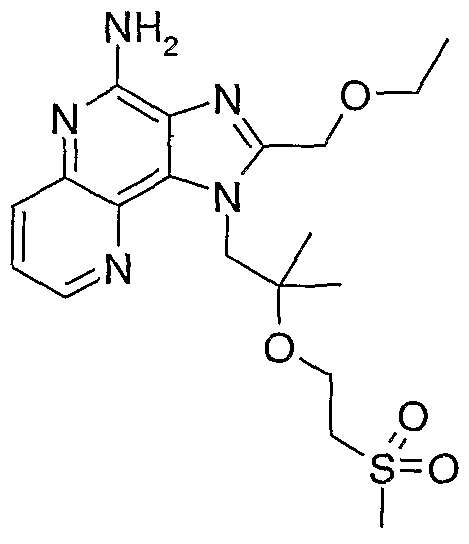

















Claims





























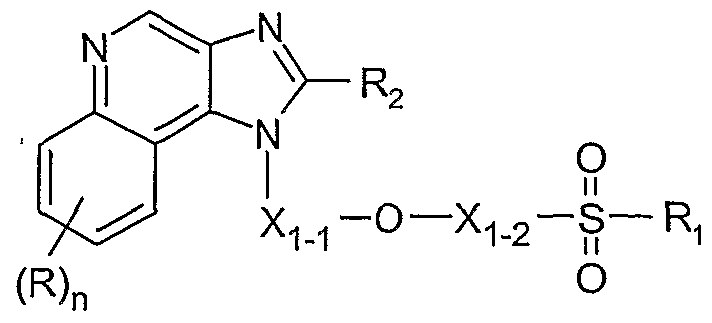










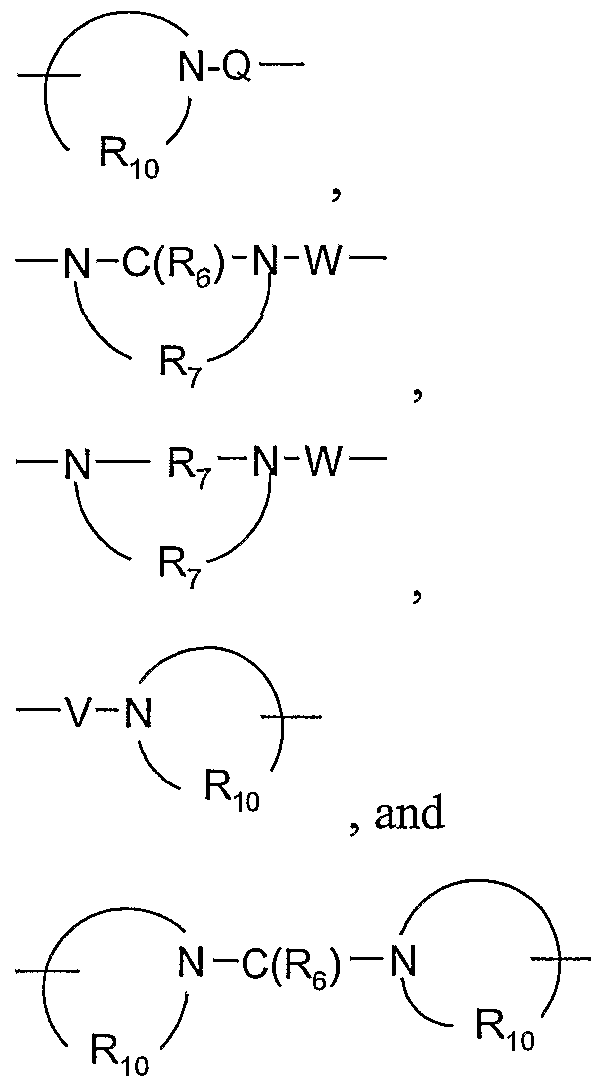





Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002549216A CA2549216A1 (en) | 2003-12-04 | 2004-12-03 | Sulfone substituted imidazo ring ethers |
| AU2004315771A AU2004315771A1 (en) | 2003-12-04 | 2004-12-03 | Sulfone substituted imidazo ring ethers |
| US10/596,117 US7939526B2 (en) | 2003-12-04 | 2004-12-03 | Sulfone substituted imidazo ring ethers |
| EP04821353A EP1694674A4 (en) | 2003-12-04 | 2004-12-03 | Sulfone substituted imidazo ring ethers |
| JP2006542750A JP2007513170A (en) | 2003-12-04 | 2004-12-03 | Sulfone substituted imidazo ring ether |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US52677203P | 2003-12-04 | 2003-12-04 | |
| US60/526,772 | 2003-12-04 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2005076783A2 true WO2005076783A2 (en) | 2005-08-25 |
| WO2005076783A3 WO2005076783A3 (en) | 2005-12-29 |
Family
ID=34860172
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2004/040383 WO2005076783A2 (en) | 2003-12-04 | 2004-12-03 | Sulfone substituted imidazo ring ethers |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US7939526B2 (en) |
| EP (1) | EP1694674A4 (en) |
| JP (1) | JP2007513170A (en) |
| CN (1) | CN1914203A (en) |
| AR (1) | AR048289A1 (en) |
| AU (1) | AU2004315771A1 (en) |
| CA (1) | CA2549216A1 (en) |
| TW (1) | TW200533352A (en) |
| WO (1) | WO2005076783A2 (en) |
Cited By (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007075468A1 (en) * | 2005-12-16 | 2007-07-05 | Coley Pharmaceutical Group, Inc. | Substituted imidazoquinolines, imidazonaphthyridines, and imidazopyridines, compositions, and methods |
| JP2009509971A (en) * | 2005-09-23 | 2009-03-12 | コーリー ファーマシューティカル グループ,インコーポレイテッド | Process for 1H-imidazo [4,5-c] pyridine and analogs thereof |
| US7879849B2 (en) | 2003-10-03 | 2011-02-01 | 3M Innovative Properties Company | Pyrazolopyridines and analogs thereof |
| US7906506B2 (en) | 2006-07-12 | 2011-03-15 | 3M Innovative Properties Company | Substituted chiral fused [1,2] imidazo [4,5-c] ring compounds and methods |
| US7915281B2 (en) | 2004-06-18 | 2011-03-29 | 3M Innovative Properties Company | Isoxazole, dihydroisoxazole, and oxadiazole substituted imidazo ring compounds and method |
| US7923429B2 (en) | 2003-09-05 | 2011-04-12 | 3M Innovative Properties Company | Treatment for CD5+ B cell lymphoma |
| US7943610B2 (en) | 2005-04-01 | 2011-05-17 | 3M Innovative Properties Company | Pyrazolopyridine-1,4-diamines and analogs thereof |
| US7943636B2 (en) | 2005-04-01 | 2011-05-17 | 3M Innovative Properties Company | 1-substituted pyrazolo (3,4-C) ring compounds as modulators of cytokine biosynthesis for the treatment of viral infections and neoplastic diseases |
| US7943609B2 (en) | 2004-12-30 | 2011-05-17 | 3M Innovative Proprerties Company | Chiral fused [1,2]imidazo[4,5-C] ring compounds |
| US7968563B2 (en) | 2005-02-11 | 2011-06-28 | 3M Innovative Properties Company | Oxime and hydroxylamine substituted imidazo[4,5-c] ring compounds and methods |
| US8034938B2 (en) | 2004-12-30 | 2011-10-11 | 3M Innovative Properties Company | Substituted chiral fused [1,2]imidazo[4,5-c] ring compounds |
| EP2394650A1 (en) | 2004-12-30 | 2011-12-14 | 3M Innovative Properties Co. | Use of resiquimod for the treatment of cutaneous metastases |
| US8221771B2 (en) | 2003-08-05 | 2012-07-17 | 3M Innovative Properties Company | Formulations containing an immune response modifier |
| WO2014107663A2 (en) | 2013-01-07 | 2014-07-10 | The Trustees Of The University Of Pennsylvania | Compositions and methods for treating cutaneous t cell lymphoma |
| US8871782B2 (en) | 2003-10-03 | 2014-10-28 | 3M Innovative Properties Company | Alkoxy substituted imidazoquinolines |
| US8951528B2 (en) | 2006-02-22 | 2015-02-10 | 3M Innovative Properties Company | Immune response modifier conjugates |
| WO2015162075A1 (en) * | 2014-04-22 | 2015-10-29 | F. Hoffmann-La Roche Ag | 4-amino-imidazoquinoline compounds |
| US9248127B2 (en) | 2005-02-04 | 2016-02-02 | 3M Innovative Properties Company | Aqueous gel formulations containing immune response modifiers |
| US9475775B2 (en) | 2015-03-06 | 2016-10-25 | Hoffmann-La Roche Inc. | Benzazepine dicarboxamide compounds |
| EP3153180A1 (en) | 2011-06-03 | 2017-04-12 | 3M Innovative Properties Company | Heterobifunctional linkers with polyethylene glycol segments and immune response modifier conjugates made therefrom |
| US10005772B2 (en) | 2006-12-22 | 2018-06-26 | 3M Innovative Properties Company | Immune response modifier compositions and methods |
| US10370338B2 (en) | 2016-05-23 | 2019-08-06 | Hoffmann-La Roche Inc. | Benzazepine dicarboxamide compounds with tertiary amide function |
| WO2019166946A1 (en) | 2018-02-28 | 2019-09-06 | Pfizer Inc. | Il-15 variants and uses thereof |
| WO2019224715A1 (en) | 2018-05-23 | 2019-11-28 | Pfizer Inc. | Antibodies specific for cd3 and uses thereof |
| WO2019224716A2 (en) | 2018-05-23 | 2019-11-28 | Pfizer Inc. | Antibodies specific for gucy2c and uses thereof |
| US10526309B2 (en) | 2015-10-02 | 2020-01-07 | The University Of North Carolina At Chapel Hill | Pan-TAM inhibitors and Mer/Axl dual inhibitors |
| US10533007B2 (en) | 2016-04-19 | 2020-01-14 | Innate Tumor Immunity, Inc. | NLRP3 modulators |
| US10533005B2 (en) | 2017-02-17 | 2020-01-14 | Innate Tumor Immunity, Inc. | NLRP3 modulators |
| US10544102B2 (en) | 2016-05-23 | 2020-01-28 | Hoffmann-La Roche Inc. | Benzazepine dicarboxamide compounds with secondary amide function |
| US10556903B2 (en) | 2016-04-19 | 2020-02-11 | Innate Tumor Immunity, Inc. | NLRP3 modulators |
| US10590112B2 (en) | 2016-06-12 | 2020-03-17 | Hoffmann-La Roche Inc. | Dihydropyrimidinyl benzazepine carboxamide compounds |
| WO2020128893A1 (en) | 2018-12-21 | 2020-06-25 | Pfizer Inc. | Combination treatments of cancer comprising a tlr agonist |
| WO2021124073A1 (en) | 2019-12-17 | 2021-06-24 | Pfizer Inc. | Antibodies specific for cd47, pd-l1, and uses thereof |
| WO2022013775A1 (en) | 2020-07-17 | 2022-01-20 | Pfizer Inc. | Therapeutic antibodies and their uses |
Families Citing this family (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040265351A1 (en) | 2003-04-10 | 2004-12-30 | Miller Richard L. | Methods and compositions for enhancing immune response |
| AR045260A1 (en) | 2003-08-12 | 2005-10-19 | 3M Innovative Properties Co | COMPOUNDS CONTAINING IMIDAZO-OXIMA REPLACED |
| MXPA06002199A (en) | 2003-08-27 | 2006-05-22 | 3M Innovative Properties Co | Aryloxy and arylalkyleneoxy substituted imidazoquinolines. |
| WO2005079195A2 (en) | 2003-10-03 | 2005-09-01 | 3M Innovative Properties Company | Pyrazolopyridines and analogs thereof |
| US8598192B2 (en) | 2003-11-14 | 2013-12-03 | 3M Innovative Properties Company | Hydroxylamine substituted imidazoquinolines |
| CA2545774A1 (en) | 2003-11-14 | 2005-06-02 | 3M Innovative Properties Company | Oxime substituted imidazo ring compounds |
| CA2547020C (en) | 2003-11-25 | 2014-03-25 | 3M Innovative Properties Company | 1h-imidazo[4,5-c]pyridine-4-amine derivatives as immune response modifier |
| WO2005066170A1 (en) | 2003-12-29 | 2005-07-21 | 3M Innovative Properties Company | Arylalkenyl and arylalkynyl substituted imidazoquinolines |
| JP2007517044A (en) | 2003-12-30 | 2007-06-28 | スリーエム イノベイティブ プロパティズ カンパニー | Imidazoquinolinyl, imidazopyridinyl, and imidazonaphthyridinylsulfonamide |
| WO2005094531A2 (en) | 2004-03-24 | 2005-10-13 | 3M Innovative Properties Company | Amide substituted imidazopyridines, imidazoquinolines, and imidazonaphthyridines |
| US8017779B2 (en) | 2004-06-15 | 2011-09-13 | 3M Innovative Properties Company | Nitrogen containing heterocyclyl substituted imidazoquinolines and imidazonaphthyridines |
| US8541438B2 (en) | 2004-06-18 | 2013-09-24 | 3M Innovative Properties Company | Substituted imidazoquinolines, imidazopyridines, and imidazonaphthyridines |
| US8026366B2 (en) | 2004-06-18 | 2011-09-27 | 3M Innovative Properties Company | Aryloxy and arylalkyleneoxy substituted thiazoloquinolines and thiazolonaphthyridines |
| WO2006038923A2 (en) | 2004-06-18 | 2006-04-13 | 3M Innovative Properties Company | Aryl substituted imidazonaphthyridines |
| US8378102B2 (en) | 2005-02-09 | 2013-02-19 | 3M Innovative Properties Company | Oxime and hydroxylamine substituted thiazolo[4,5-c] ring compounds and methods |
| WO2006086449A2 (en) | 2005-02-09 | 2006-08-17 | Coley Pharmaceutical Group, Inc. | Alkyloxy substituted thiazoloquinolines and thiazolonaphthyridines |
| US8658666B2 (en) | 2005-02-11 | 2014-02-25 | 3M Innovative Properties Company | Substituted imidazoquinolines and imidazonaphthyridines |
| EP1851224A2 (en) | 2005-02-23 | 2007-11-07 | 3M Innovative Properties Company | Hydroxyalkyl substituted imidazoquinolines |
| CA2598639A1 (en) | 2005-02-23 | 2006-08-31 | Coley Pharmaceutical Group, Inc. | Hydroxyalkyl substituted imidazonaphthyridines |
| WO2006091567A2 (en) | 2005-02-23 | 2006-08-31 | Coley Pharmaceutical Group, Inc. | Hydroxyalkyl substituted imidazoquinoline compounds and methods |
| CA2598437A1 (en) | 2005-02-23 | 2006-08-31 | Coley Pharmaceutical Group, Inc. | Method of preferentially inducing the biosynthesis of interferon |
| ZA200803029B (en) | 2005-09-09 | 2009-02-25 | Coley Pharm Group Inc | Amide and carbamate derivatives of alkyl substituted /V-[4-(4-amino-1H-imidazo[4,5-c] quinolin-1-yl)butyl] methane-sulfonamides and methods |
| EP1922317A4 (en) | 2005-09-09 | 2009-04-15 | Coley Pharm Group Inc | Amide and carbamate derivatives of n-{2-ý4-amino-2- (ethoxymethyl)-1h-imidazoý4,5-c¨quinolin-1-yl¨-1,1-dimethylethyl}methanesulfonamide and methods |
| US8088790B2 (en) | 2005-11-04 | 2012-01-03 | 3M Innovative Properties Company | Hydroxy and alkoxy substituted 1H-imidazoquinolines and methods |
| US8329721B2 (en) | 2006-03-15 | 2012-12-11 | 3M Innovative Properties Company | Hydroxy and alkoxy substituted 1H-imidazonaphthyridines and methods |
| WO2008030511A2 (en) | 2006-09-06 | 2008-03-13 | Coley Pharmaceuticial Group, Inc. | Substituted 3,4,6,7-tetrahydro-5h, 1,2a,4a,8-tetraazacyclopenta[cd]phenalenes |
| MX359517B (en) | 2010-08-17 | 2018-10-01 | 3M Innovative Properties Company Star | Lipidated immune response modifier compound compositions, formulations, and methods. |
| US9475804B2 (en) | 2011-06-03 | 2016-10-25 | 3M Innovative Properties Company | Heterobifunctional linkers with polyethylene glycol segments and immune response modifier conjugates made therefrom |
| WO2016057931A1 (en) | 2014-10-10 | 2016-04-14 | The Research Foundation For The State University Of New York | Trifluoromethoxylation of arenes via intramolecular trifluoromethoxy group migration |
| CA3086439A1 (en) | 2017-12-20 | 2019-06-27 | 3M Innovative Properties Company | Amide substitued imidazo[4,5-c]quinoline compounds with a branched chain linking group for use as an immune response modifier |
| GB202213166D0 (en) | 2022-09-08 | 2022-10-26 | Cambridge Entpr Ltd | Novel compounds, compositions and therapeutic uses thereof |
Family Cites Families (98)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US664260A (en) * | 1897-09-29 | 1900-12-18 | William Hamlin | Manufacture of starch. |
| US660747A (en) * | 1900-05-31 | 1900-10-30 | Bausch & Lomb | Objective. |
| US3314941A (en) * | 1964-06-23 | 1967-04-18 | American Cyanamid Co | Novel substituted pyridodiazepins |
| ZA848968B (en) * | 1983-11-18 | 1986-06-25 | Riker Laboratories Inc | 1h-imidazo(4,5-c)quinolines and 1h-imidazo(4,5-c)quinolin-4-amines |
| IL73534A (en) * | 1983-11-18 | 1990-12-23 | Riker Laboratories Inc | 1h-imidazo(4,5-c)quinoline-4-amines,their preparation and pharmaceutical compositions containing certain such compounds |
| US5238944A (en) * | 1988-12-15 | 1993-08-24 | Riker Laboratories, Inc. | Topical formulations and transdermal delivery systems containing 1-isobutyl-1H-imidazo[4,5-c]quinolin-4-amine |
| US5756747A (en) * | 1989-02-27 | 1998-05-26 | Riker Laboratories, Inc. | 1H-imidazo 4,5-c!quinolin-4-amines |
| US5037986A (en) * | 1989-03-23 | 1991-08-06 | Minnesota Mining And Manufacturing Company | Olefinic 1H-imidazo[4,5-c]quinolin-4-amines |
| US4929624A (en) * | 1989-03-23 | 1990-05-29 | Minnesota Mining And Manufacturing Company | Olefinic 1H-imidazo(4,5-c)quinolin-4-amines |
| NZ232740A (en) | 1989-04-20 | 1992-06-25 | Riker Laboratories Inc | Solution for parenteral administration comprising a 1h-imidazo(4,5-c) quinolin-4-amine derivative, an acid and a tonicity adjuster |
| US4988815A (en) * | 1989-10-26 | 1991-01-29 | Riker Laboratories, Inc. | 3-Amino or 3-nitro quinoline compounds which are intermediates in preparing 1H-imidazo[4,5-c]quinolines |
| CA2093132C (en) * | 1990-10-05 | 2002-02-26 | John F. Gerster | Process for the preparation of imidazo[4,5-c]quinolin-4-amines |
| US5175296A (en) * | 1991-03-01 | 1992-12-29 | Minnesota Mining And Manufacturing Company | Imidazo[4,5-c]quinolin-4-amines and processes for their preparation |
| US5389640A (en) * | 1991-03-01 | 1995-02-14 | Minnesota Mining And Manufacturing Company | 1-substituted, 2-substituted 1H-imidazo[4,5-c]quinolin-4-amines |
| US5268376A (en) * | 1991-09-04 | 1993-12-07 | Minnesota Mining And Manufacturing Company | 1-substituted 1H-imidazo[4,5-c]quinolin-4-amines |
| US5266575A (en) * | 1991-11-06 | 1993-11-30 | Minnesota Mining And Manufacturing Company | 2-ethyl 1H-imidazo[4,5-ciquinolin-4-amines |
| IL105325A (en) * | 1992-04-16 | 1996-11-14 | Minnesota Mining & Mfg | Immunogen/vaccine adjuvant composition |
| US5395937A (en) * | 1993-01-29 | 1995-03-07 | Minnesota Mining And Manufacturing Company | Process for preparing quinoline amines |
| US5352784A (en) * | 1993-07-15 | 1994-10-04 | Minnesota Mining And Manufacturing Company | Fused cycloalkylimidazopyridines |
| DE69425661T2 (en) | 1993-07-15 | 2001-04-19 | Minnesota Mining & Mfg | IMIDAZO [4,5-c] PYRIDINE-4-AMINE |
| US5482936A (en) * | 1995-01-12 | 1996-01-09 | Minnesota Mining And Manufacturing Company | Imidazo[4,5-C]quinoline amines |
| JPH09208584A (en) | 1996-01-29 | 1997-08-12 | Terumo Corp | Amide derivative, pharmaceutical preparation containing the same, and intermediate for synthesizing the same |
| US5741908A (en) * | 1996-06-21 | 1998-04-21 | Minnesota Mining And Manufacturing Company | Process for reparing imidazoquinolinamines |
| US5693811A (en) * | 1996-06-21 | 1997-12-02 | Minnesota Mining And Manufacturing Company | Process for preparing tetrahdroimidazoquinolinamines |
| KR100518903B1 (en) * | 1996-10-25 | 2005-10-06 | 미네소타 마이닝 앤드 매뉴팩춰링 캄파니 | Immune response modifier compounds for treatment of the th2 mediated and related diseases |
| US5939090A (en) * | 1996-12-03 | 1999-08-17 | 3M Innovative Properties Company | Gel formulations for topical drug delivery |
| JP4101302B2 (en) * | 1997-01-09 | 2008-06-18 | テルモ株式会社 | Novel amide derivatives and synthetic intermediates |
| US6446153B2 (en) * | 1997-03-14 | 2002-09-03 | Intel Corporation | Shared embedded microcontroller interface |
| JPH1180156A (en) | 1997-09-04 | 1999-03-26 | Hokuriku Seiyaku Co Ltd | 1-(substitutedaryl)alkyl-1h-imidazopyridin-4-amine derivative |
| UA67760C2 (en) * | 1997-12-11 | 2004-07-15 | Міннесота Майнінг Енд Мануфакчурінг Компані | Imidazonaphthyridines and use thereof to induce the biosynthesis of cytokines |
| JPH11222432A (en) | 1998-02-03 | 1999-08-17 | Terumo Corp | Preparation for external use containing amide derivative inducing interferon |
| US6569104B2 (en) * | 1998-07-16 | 2003-05-27 | Canon Kabushiki Kaisha | Blood vessel detecting apparatus |
| US6110929A (en) * | 1998-07-28 | 2000-08-29 | 3M Innovative Properties Company | Oxazolo, thiazolo and selenazolo [4,5-c]-quinolin-4-amines and analogs thereof |
| JP2000119271A (en) | 1998-08-12 | 2000-04-25 | Hokuriku Seiyaku Co Ltd | 1h-imidazopyridine derivative |
| US20020058674A1 (en) * | 1999-01-08 | 2002-05-16 | Hedenstrom John C. | Systems and methods for treating a mucosal surface |
| SK287112B6 (en) * | 1999-01-08 | 2009-12-07 | 3M Innovative Properties Company | Use of immune response modifiying compound for cervical dysplasias treatment |
| US6558951B1 (en) * | 1999-02-11 | 2003-05-06 | 3M Innovative Properties Company | Maturation of dendritic cells with immune response modifying compounds |
| JP2000247884A (en) | 1999-03-01 | 2000-09-12 | Sumitomo Pharmaceut Co Ltd | Arachidonic acid-induced skin disease-treating agent |
| US6573273B1 (en) * | 1999-06-10 | 2003-06-03 | 3M Innovative Properties Company | Urea substituted imidazoquinolines |
| US6756382B2 (en) * | 1999-06-10 | 2004-06-29 | 3M Innovative Properties Company | Amide substituted imidazoquinolines |
| US6451810B1 (en) * | 1999-06-10 | 2002-09-17 | 3M Innovative Properties Company | Amide substituted imidazoquinolines |
| US6541485B1 (en) * | 1999-06-10 | 2003-04-01 | 3M Innovative Properties Company | Urea substituted imidazoquinolines |
| US6331539B1 (en) * | 1999-06-10 | 2001-12-18 | 3M Innovative Properties Company | Sulfonamide and sulfamide substituted imidazoquinolines |
| US6376669B1 (en) * | 1999-11-05 | 2002-04-23 | 3M Innovative Properties Company | Dye labeled imidazoquinoline compounds |
| US6894060B2 (en) * | 2000-03-30 | 2005-05-17 | 3M Innovative Properties Company | Method for the treatment of dermal lesions caused by envenomation |
| CA2306170A1 (en) * | 2000-04-18 | 2001-10-18 | Kenneth Curry | Novel amino, carboxy derivatives of barbituric acid |
| US20020055517A1 (en) * | 2000-09-15 | 2002-05-09 | 3M Innovative Properties Company | Methods for delaying recurrence of herpes virus symptoms |
| JP2002145777A (en) | 2000-11-06 | 2002-05-22 | Sumitomo Pharmaceut Co Ltd | Therapeutic agent for arachidonic acid-induced dermatosis |
| US6677347B2 (en) * | 2000-12-08 | 2004-01-13 | 3M Innovative Properties Company | Sulfonamido ether substituted imidazoquinolines |
| US6660747B2 (en) * | 2000-12-08 | 2003-12-09 | 3M Innovative Properties Company | Amido ether substituted imidazoquinolines |
| US6660735B2 (en) * | 2000-12-08 | 2003-12-09 | 3M Innovative Properties Company | Urea substituted imidazoquinoline ethers |
| UA74852C2 (en) * | 2000-12-08 | 2006-02-15 | 3M Innovative Properties Co | Urea-substituted imidazoquinoline ethers |
| US6545017B1 (en) * | 2000-12-08 | 2003-04-08 | 3M Innovative Properties Company | Urea substituted imidazopyridines |
| EP1360486A2 (en) * | 2000-12-08 | 2003-11-12 | 3M Innovative Properties Company | Screening method for identifying compounds that selectively induce interferon alpha |
| US6664264B2 (en) * | 2000-12-08 | 2003-12-16 | 3M Innovative Properties Company | Thioether substituted imidazoquinolines |
| US6525064B1 (en) * | 2000-12-08 | 2003-02-25 | 3M Innovative Properties Company | Sulfonamido substituted imidazopyridines |
| US20020107262A1 (en) * | 2000-12-08 | 2002-08-08 | 3M Innovative Properties Company | Substituted imidazopyridines |
| US6664265B2 (en) * | 2000-12-08 | 2003-12-16 | 3M Innovative Properties Company | Amido ether substituted imidazoquinolines |
| US6667312B2 (en) * | 2000-12-08 | 2003-12-23 | 3M Innovative Properties Company | Thioether substituted imidazoquinolines |
| US6664260B2 (en) | 2000-12-08 | 2003-12-16 | 3M Innovative Properties Company | Heterocyclic ether substituted imidazoquinolines |
| US6545016B1 (en) * | 2000-12-08 | 2003-04-08 | 3M Innovative Properties Company | Amide substituted imidazopyridines |
| US6677348B2 (en) * | 2000-12-08 | 2004-01-13 | 3M Innovative Properties Company | Aryl ether substituted imidazoquinolines |
| US7226928B2 (en) * | 2001-06-15 | 2007-06-05 | 3M Innovative Properties Company | Methods for the treatment of periodontal disease |
| US20030133913A1 (en) * | 2001-08-30 | 2003-07-17 | 3M Innovative Properties Company | Methods of maturing plasmacytoid dendritic cells using immune response modifier molecules |
| WO2003030902A1 (en) * | 2001-10-09 | 2003-04-17 | Tularik Inc. | Imidazole derivates as anti-inflammatory agents |
| AU2002360278A1 (en) * | 2001-10-12 | 2003-11-11 | Coley Pharmaceutical Gmbh | Methods and products for enhancing immune responses using imidazoquinoline compounds |
| DE60230340D1 (en) * | 2001-11-16 | 2009-01-22 | 3M Innovative Properties Co | N-Ä4- (4-amino-2-ethyl-1H-imidazoÄ4,5-quinolin-1-yl) -butyl-methanesulfonamide, pharmaceutical composition containing the same and their use |
| KR100962751B1 (en) | 2001-11-29 | 2010-06-09 | 쓰리엠 이노베이티브 프로퍼티즈 컴파니 | Pharmaceutical Formulations Comprising an Immune Response Modifier |
| US6677349B1 (en) * | 2001-12-21 | 2004-01-13 | 3M Innovative Properties Company | Sulfonamide and sulfamide substituted imidazoquinolines |
| BR0307788A (en) * | 2002-02-22 | 2006-04-04 | 3M Innovative Properties Co | reduction method and treatment of uv-b-induced immunosuppression |
| GB0211649D0 (en) * | 2002-05-21 | 2002-07-03 | Novartis Ag | Organic compounds |
| US6743920B2 (en) * | 2002-05-29 | 2004-06-01 | 3M Innovative Properties Company | Process for imidazo[4,5-c]pyridin-4-amines |
| MXPA04012199A (en) * | 2002-06-07 | 2005-02-25 | 3M Innovative Properties Co | Ether substituted imidazopyridines. |
| DK1543002T3 (en) * | 2002-07-23 | 2006-12-27 | Teva Gyogyszergyar Zartkoeruee | Preparation of 1H-imidazo (4,5-c) quinoline-4-amines via 1H-imidazo (4,5-c) quinoline-4-phthalimide intermediates |
| CA2493158A1 (en) * | 2002-07-26 | 2004-02-05 | Biogal Gyogyszergyar Rt. | Preparation of 1h-imidazo [4,5-c] quinolin-4-amines via novel 1h-imidazo [4,5-c] quinolin-4-cyano and 1h-imidazo [4,5-c] quinolin-4-carboxamide intermediates |
| MXPA05001647A (en) * | 2002-08-15 | 2005-04-19 | 3M Innovative Properties Co | Immunostimulatory compositions and methods of stimulating an immune response. |
| EP1542688A4 (en) * | 2002-09-26 | 2010-06-02 | 3M Innovative Properties Co | 1h-imidazo dimers |
| AU2003287324A1 (en) * | 2002-12-11 | 2004-06-30 | 3M Innovative Properties Company | Gene expression systems and recombinant cell lines |
| AU2003287316A1 (en) * | 2002-12-11 | 2004-06-30 | 3M Innovative Properties Company | Assays relating to toll-like receptor activity |
| CA2510375A1 (en) * | 2002-12-20 | 2004-07-15 | 3M Innovative Properties Company | Aryl / hetaryl substituted imidazoquinolines |
| AU2003300184B8 (en) * | 2002-12-30 | 2009-12-03 | 3M Innovative Properties Company | Immunostimulatory combinations |
| EP1592302A4 (en) * | 2003-02-13 | 2007-04-25 | 3M Innovative Properties Co | Methods and compositions related to irm compounds and toll-like receptor 8 |
| WO2004075865A2 (en) * | 2003-02-27 | 2004-09-10 | 3M Innovative Properties Company | Selective modulation of tlr-mediated biological activity |
| WO2004078138A2 (en) * | 2003-03-04 | 2004-09-16 | 3M Innovative Properties Company | Prophylactic treatment of uv-induced epidermal neoplasia |
| EP1605943A4 (en) * | 2003-03-07 | 2008-01-16 | 3M Innovative Properties Co | 1-amino 1h-imidazoquinolines |
| ATE556711T1 (en) * | 2003-03-13 | 2012-05-15 | 3M Innovative Properties Co | METHOD FOR IMPROVING SKIN QUALITY |
| WO2004080292A2 (en) * | 2003-03-13 | 2004-09-23 | 3M Innovative Properties Company | Method of tattoo removal |
| EP1608282A4 (en) * | 2003-03-13 | 2010-12-08 | 3M Innovative Properties Co | Methods for diagnosing skin lesions |
| JP4139251B2 (en) * | 2003-03-17 | 2008-08-27 | 本田技研工業株式会社 | Electric management machine |
| US20040192585A1 (en) | 2003-03-25 | 2004-09-30 | 3M Innovative Properties Company | Treatment for basal cell carcinoma |
| EP1613956A2 (en) * | 2003-03-25 | 2006-01-11 | 3M Innovative Properties Company | Selective activation of cellular activities mediated through a common toll-like receptor |
| WO2004091500A2 (en) * | 2003-04-10 | 2004-10-28 | 3M Innovative Properties Company | Delivery of immune response modifier compounds |
| EP1617845A4 (en) * | 2003-04-28 | 2006-09-20 | 3M Innovative Properties Co | Compositions and methods for induction of opioid receptors |
| AR045260A1 (en) | 2003-08-12 | 2005-10-19 | 3M Innovative Properties Co | COMPOUNDS CONTAINING IMIDAZO-OXIMA REPLACED |
| PL1653959T3 (en) | 2003-08-14 | 2015-10-30 | 3M Innovative Properties Co | Lipid-modified immune response modifiers |
| NZ545536A (en) * | 2003-09-05 | 2010-04-30 | Anadys Pharmaceuticals Inc | TLR7 ligands for the treatment of hepatitis C |
| US7687628B2 (en) * | 2003-10-01 | 2010-03-30 | Taro Pharmaceuticals U.S.A., Inc. | Method of preparing 4-amino-1H-imidazo(4,5-c)quinolines and acid addition salts thereof |
| ITMI20032121A1 (en) * | 2003-11-04 | 2005-05-05 | Dinamite Dipharma Spa In Forma Abbr Eviata Dipharm | PROCEDURE FOR THE PREPARATION OF IMIQUIMOD AND ITS INTERMEDIATES |
-
2004
- 2004-12-03 US US10/596,117 patent/US7939526B2/en not_active Expired - Fee Related
- 2004-12-03 AR ARP040104518A patent/AR048289A1/en unknown
- 2004-12-03 CN CNA2004800414001A patent/CN1914203A/en active Pending
- 2004-12-03 CA CA002549216A patent/CA2549216A1/en not_active Abandoned
- 2004-12-03 AU AU2004315771A patent/AU2004315771A1/en not_active Abandoned
- 2004-12-03 EP EP04821353A patent/EP1694674A4/en not_active Withdrawn
- 2004-12-03 JP JP2006542750A patent/JP2007513170A/en active Pending
- 2004-12-03 TW TW093137541A patent/TW200533352A/en unknown
- 2004-12-03 WO PCT/US2004/040383 patent/WO2005076783A2/en active Application Filing
Non-Patent Citations (1)
| Title |
|---|
| See references of EP1694674A4 * |
Cited By (48)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8221771B2 (en) | 2003-08-05 | 2012-07-17 | 3M Innovative Properties Company | Formulations containing an immune response modifier |
| US7923429B2 (en) | 2003-09-05 | 2011-04-12 | 3M Innovative Properties Company | Treatment for CD5+ B cell lymphoma |
| US7879849B2 (en) | 2003-10-03 | 2011-02-01 | 3M Innovative Properties Company | Pyrazolopyridines and analogs thereof |
| US8871782B2 (en) | 2003-10-03 | 2014-10-28 | 3M Innovative Properties Company | Alkoxy substituted imidazoquinolines |
| US7915281B2 (en) | 2004-06-18 | 2011-03-29 | 3M Innovative Properties Company | Isoxazole, dihydroisoxazole, and oxadiazole substituted imidazo ring compounds and method |
| EP2394650A1 (en) | 2004-12-30 | 2011-12-14 | 3M Innovative Properties Co. | Use of resiquimod for the treatment of cutaneous metastases |
| US7943609B2 (en) | 2004-12-30 | 2011-05-17 | 3M Innovative Proprerties Company | Chiral fused [1,2]imidazo[4,5-C] ring compounds |
| US8034938B2 (en) | 2004-12-30 | 2011-10-11 | 3M Innovative Properties Company | Substituted chiral fused [1,2]imidazo[4,5-c] ring compounds |
| US10071156B2 (en) | 2005-02-04 | 2018-09-11 | 3M Innovative Properties Company | Aqueous gel formulations containing immune response modifiers |
| US9248127B2 (en) | 2005-02-04 | 2016-02-02 | 3M Innovative Properties Company | Aqueous gel formulations containing immune response modifiers |
| US7968563B2 (en) | 2005-02-11 | 2011-06-28 | 3M Innovative Properties Company | Oxime and hydroxylamine substituted imidazo[4,5-c] ring compounds and methods |
| US7943636B2 (en) | 2005-04-01 | 2011-05-17 | 3M Innovative Properties Company | 1-substituted pyrazolo (3,4-C) ring compounds as modulators of cytokine biosynthesis for the treatment of viral infections and neoplastic diseases |
| US7943610B2 (en) | 2005-04-01 | 2011-05-17 | 3M Innovative Properties Company | Pyrazolopyridine-1,4-diamines and analogs thereof |
| JP2009509971A (en) * | 2005-09-23 | 2009-03-12 | コーリー ファーマシューティカル グループ,インコーポレイテッド | Process for 1H-imidazo [4,5-c] pyridine and analogs thereof |
| WO2007075468A1 (en) * | 2005-12-16 | 2007-07-05 | Coley Pharmaceutical Group, Inc. | Substituted imidazoquinolines, imidazonaphthyridines, and imidazopyridines, compositions, and methods |
| EP3085373A1 (en) | 2006-02-22 | 2016-10-26 | 3M Innovative Properties Company | Immune response modifier conjugates |
| US8951528B2 (en) | 2006-02-22 | 2015-02-10 | 3M Innovative Properties Company | Immune response modifier conjugates |
| US7906506B2 (en) | 2006-07-12 | 2011-03-15 | 3M Innovative Properties Company | Substituted chiral fused [1,2] imidazo [4,5-c] ring compounds and methods |
| US10005772B2 (en) | 2006-12-22 | 2018-06-26 | 3M Innovative Properties Company | Immune response modifier compositions and methods |
| US10144735B2 (en) | 2006-12-22 | 2018-12-04 | 3M Innovative Properties Company | Immune response modifier compositions and methods |
| EP3153180A1 (en) | 2011-06-03 | 2017-04-12 | 3M Innovative Properties Company | Heterobifunctional linkers with polyethylene glycol segments and immune response modifier conjugates made therefrom |
| EP3756669A1 (en) | 2013-01-07 | 2020-12-30 | The Trustees of the University of Pennsylvania | Compositions for use for treating cutaneous t cell lymphoma |
| WO2014107663A2 (en) | 2013-01-07 | 2014-07-10 | The Trustees Of The University Of Pennsylvania | Compositions and methods for treating cutaneous t cell lymphoma |
| RU2698902C2 (en) * | 2014-04-22 | 2019-09-02 | Ф. Хоффманн-Ля Рош Аг | 4-amino-imidazoquinoline derivatives, useful in treating diseases mediated by tlr7 and/or tlr8 agonists |
| US9447097B2 (en) | 2014-04-22 | 2016-09-20 | Hoffmann-La Roche Inc. | 4-amino-imidazoquinoline compounds |
| US9334268B2 (en) | 2014-04-22 | 2016-05-10 | Hoffman-La Roche Inc. | 4-amino-imidazoquinoline compounds |
| TWI607007B (en) * | 2014-04-22 | 2017-12-01 | 赫孚孟拉羅股份公司 | 4-amino-imidazoquinoline compounds |
| WO2015162075A1 (en) * | 2014-04-22 | 2015-10-29 | F. Hoffmann-La Roche Ag | 4-amino-imidazoquinoline compounds |
| KR101905292B1 (en) | 2014-04-22 | 2018-11-21 | 에프. 호프만-라 로슈 아게 | 4-amino-imidazoquinoline compounds |
| US9822065B1 (en) | 2015-03-06 | 2017-11-21 | Hoffmann-La Roche Inc. | Benzazepine dicarboxamide compounds |
| US9597333B2 (en) | 2015-03-06 | 2017-03-21 | Hoffmann-La Roche Inc. | Benzazepine dicarboxamide compounds |
| US9475775B2 (en) | 2015-03-06 | 2016-10-25 | Hoffmann-La Roche Inc. | Benzazepine dicarboxamide compounds |
| US10526309B2 (en) | 2015-10-02 | 2020-01-07 | The University Of North Carolina At Chapel Hill | Pan-TAM inhibitors and Mer/Axl dual inhibitors |
| US10556903B2 (en) | 2016-04-19 | 2020-02-11 | Innate Tumor Immunity, Inc. | NLRP3 modulators |
| US10533007B2 (en) | 2016-04-19 | 2020-01-14 | Innate Tumor Immunity, Inc. | NLRP3 modulators |
| US10544102B2 (en) | 2016-05-23 | 2020-01-28 | Hoffmann-La Roche Inc. | Benzazepine dicarboxamide compounds with secondary amide function |
| US10370338B2 (en) | 2016-05-23 | 2019-08-06 | Hoffmann-La Roche Inc. | Benzazepine dicarboxamide compounds with tertiary amide function |
| US10590112B2 (en) | 2016-06-12 | 2020-03-17 | Hoffmann-La Roche Inc. | Dihydropyrimidinyl benzazepine carboxamide compounds |
| US11046671B2 (en) | 2016-06-12 | 2021-06-29 | Hoffmann-La Roche Inc. | Dihydropyrimidinyl benzazepine carboxamide compounds |
| US10533005B2 (en) | 2017-02-17 | 2020-01-14 | Innate Tumor Immunity, Inc. | NLRP3 modulators |
| US11827632B2 (en) | 2017-02-17 | 2023-11-28 | Innate Tumor Immunity, Inc | NLRP3 modulators |
| WO2019166946A1 (en) | 2018-02-28 | 2019-09-06 | Pfizer Inc. | Il-15 variants and uses thereof |
| WO2019224716A2 (en) | 2018-05-23 | 2019-11-28 | Pfizer Inc. | Antibodies specific for gucy2c and uses thereof |
| WO2019224715A1 (en) | 2018-05-23 | 2019-11-28 | Pfizer Inc. | Antibodies specific for cd3 and uses thereof |
| US11434292B2 (en) | 2018-05-23 | 2022-09-06 | Pfizer Inc. | Antibodies specific for CD3 and uses thereof |
| WO2020128893A1 (en) | 2018-12-21 | 2020-06-25 | Pfizer Inc. | Combination treatments of cancer comprising a tlr agonist |
| WO2021124073A1 (en) | 2019-12-17 | 2021-06-24 | Pfizer Inc. | Antibodies specific for cd47, pd-l1, and uses thereof |
| WO2022013775A1 (en) | 2020-07-17 | 2022-01-20 | Pfizer Inc. | Therapeutic antibodies and their uses |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2007513170A (en) | 2007-05-24 |
| TW200533352A (en) | 2005-10-16 |
| AR048289A1 (en) | 2006-04-19 |
| CN1914203A (en) | 2007-02-14 |
| US7939526B2 (en) | 2011-05-10 |
| WO2005076783A3 (en) | 2005-12-29 |
| AU2004315771A1 (en) | 2005-08-25 |
| EP1694674A2 (en) | 2006-08-30 |
| US20070155767A1 (en) | 2007-07-05 |
| CA2549216A1 (en) | 2005-08-25 |
| EP1694674A4 (en) | 2010-07-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7939526B2 (en) | Sulfone substituted imidazo ring ethers | |
| AU2005283085B2 (en) | Substituted imidazoquinolines, imidazopyridines, and imidazonaphthyridines | |
| US8802853B2 (en) | Arylalkenyl and arylalkynyl substituted imidazoquinolines | |
| WO2006091568A2 (en) | Hydroxyalkyl substituted imidazonaphthyridines | |
| EP1730143A2 (en) | Amide substituted imidazopyridines, imidazoquinolines, and imidazonaphthyridines | |
| AU2004312508A1 (en) | Imidazoquinolinyl, imidazopyridinyl, and imidazonaphthyridinyl sulfonamides | |
| WO2005123080A2 (en) | Nitrogen-containing heterocyclyl substituted imidazoquinolines and imidazonaphthyridines | |
| WO2005123079A2 (en) | Urea substituted imidazopyridines, imidazoquinolines, and imidazonaphthyridines | |
| WO2006029115A2 (en) | 2-amino 1h imidazo ring systems and methods | |
| EP1846405A2 (en) | Oxime and hydroxylamine substituted imidazo 4,5-c ring compounds and methods | |
| WO2007056112A2 (en) | Hydroxy and alkoxy substituted 1h-imidazoquinolines and methods | |
| WO2006093514A2 (en) | Aryl and arylalkylenyl substituted thiazoloquinolines and thiazolonaphthyridines | |
| EP1682544A2 (en) | Hydroxylamine substituted imidazo ring compounds | |
| EP1831221A2 (en) | Substituted chiral fused 1,2 imidazo 4,5-c ring compounds | |
| US9938275B2 (en) | Substituted imidazoquinolines, imidazopyridines, and imidazonaphthyridines |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10596117 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2549216 Country of ref document: CA Ref document number: 1966/CHENP/2006 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004315771 Country of ref document: AU Ref document number: 2006542750 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004821353 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2004315771 Country of ref document: AU Date of ref document: 20041203 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004315771 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200480041400.1 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004821353 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10596117 Country of ref document: US |