WO2005121348A1 - Lipid encapsulated interfering rna - Google Patents
Lipid encapsulated interfering rnaInfo
- Publication number
- WO2005121348A1 WO2005121348A1 PCT/CA2005/000886 CA2005000886W WO2005121348A1 WO 2005121348 A1 WO2005121348 A1 WO 2005121348A1 CA 2005000886 W CA2005000886 W CA 2005000886W WO 2005121348 A1 WO2005121348 A1 WO 2005121348A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- lipid
- peg
- nucleic acid
- particle
- interfering rna
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
- A61K31/7105—Natural ribonucleic acids, i.e. containing only riboses attached to adenine, guanine, cytosine or uracil and having 3'-5' phosphodiester links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Liposomes
- A61K9/1271—Non-conventional liposomes, e.g. PEGylated liposomes, liposomes coated with polymers
- A61K9/1272—Non-conventional liposomes, e.g. PEGylated liposomes, liposomes coated with polymers with substantial amounts of non-phosphatidyl, i.e. non-acylglycerophosphate, surfactants as bilayer-forming substances, e.g. cationic lipids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/111—General methods applicable to biologically active non-coding nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/88—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation using microencapsulation, e.g. using amphiphile liposome vesicle
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/14—Type of nucleic acid interfering N.A.
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/35—Nature of the modification
- C12N2310/351—Conjugate
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2320/00—Applications; Uses
- C12N2320/30—Special therapeutic applications
- C12N2320/32—Special delivery means, e.g. tissue-specific
Definitions
- the present invention relates to compositions and methods for the therapeutic delivery of a nucleic acid comprising a serum-stable lipid delivery vehicle encapsulating a nucleic acid to provide efficient RNA interference (RNAi) in a cell or mammal. More particularly, the present invention is directed to using a small interfering RNA (siRNA) encapsulated in a serum-stable lipid particle having a small diameter suitable for systemic delivery.
- siRNA small interfering RNA
- RNA interference is an evolutionarily conserved, sequence specific mechanism triggered by double stranded RNA (dsRNA) that induces degradation of complementary target single stranded mRNA and "silencing" of the corresponding translated sequences (McManus and Sharp, Nature Rev. Genet. 3:737 (2002)).
- RNAi functions by enzymatic cleavage of longer dsRNA strands into biologically active "short- interfering RNA" (siRNA) sequences of about 21-23 nucleotides in length (Elbashir, et al. , Genes Dev. 15:188 (2001)).
- siRNA can be used downregulate or silence the transcription and translation of a gene product of interest. For example, it is desirable to downregulate genes associated with liver diseases and disorders such as hepatitis. In particular, it is desirable to downregulate genes associated with hepatitis viral infection and survival.
- RNA nucleic acid delivery system
- Viral vectors are relatively efficient gene delivery systems, but suffer from a variety of limitations, such as the potential for reversion to the wild type as well as immune response concerns.
- nonviral gene delivery systems are receiving increasing attention (Worgall, et al, Human Gene Tlierapy 8:37 (1997); Peeters, et al, Human Gene Therapy 7:1693 (1996); Yei, et al, Gene Tlierapy 1: 192 (1994); Hope, et al, Molecular Membrane Biology 15:1 (1998)).
- viral systems are rapidly cleared from the circulation, limiting transfection to "first-pass" organs such as the lungs, liver, and spleen. In addition, these systems induce immune responses that compromise delivery with subsequent injections.
- Plasmid DNA-cationic liposome complexes are currently the most commonly employed nonviral gene delivery vehicles (Feigner, Scientific American 276:102 (1997); Chonn, et al, Current Opinion in Biotechnology 6:698 (1995)).
- cationic liposome complexes made of an amphipathic compound, a neutral lipid, and a detergent for transfecting insect cells are disclosed in U.S. Patent No. 6,458,382.
- Cationic liposome complexes are also disclosed in U.S. Patent Publication No. 2003/0073640.
- liposomal delivery systems include, for example, the use of reverse micelles, anionic and polymer liposomes.
- Reverse micelles are disclosed in U.S. Patent No. 6,429,200.
- Anionic liposomes are disclosed in U.S. Patent Application No. 2003/0026831.
- Polymer liposomes, that incorporate dextrin or glycerol-phosphocholine polymers, are disclosed in U.S. Patent Application Nos. 2002/0081736 and 2003/0082103, respectively.
- a gene delivery system containing an encapsulated nucleic acid for systemic delivery should be small (i.e., less than about 100 nm diameter) and should remain intact in the circulation for an extended period of time in order to achieve delivery to affected tissues.
- This requires a highly stable, serum-resistant nucleic acid-containing particle that does not interact with cells and other components of the vascular compartment.
- the particle should also readily interact with target cells at a disease site in order to facilitate intracellular delivery of a desired nucleic acid.
- nucleic acids can be encapsulated in small (about 70 nm diameter) "stabilized nucleic acid-lipid particles” (SNALP) that consist of a single plasmid encapsulated within a bilayer lipid vesicle (Wheeler, et al, Gene Therapy 6:271 (1999)).
- SNALPs typically contain the "fusogenic" lipid dioleoylphosphatidylethanolamine (DOPE), low levels of cationic lipid, and are stabilized in aqueous media by the presence of a poly(ethylene glycol) (PEG) coating.
- DOPE lipid dioleoylphosphatidylethanolamine
- PEG poly(ethylene glycol)
- SNALP have systemic application as they exhibit extended circulation lifetimes following intravenous (i.v.) injection, accumulate preferentially at distal tumor sites due to the enhanced vascular permeability in such regions, and can mediate transgene expression at these tumor sites.
- the levels of transgene expression observed at the tumor site following i.v. injection of SPLP containing the luciferase marker gene are superior to the levels that can be achieved employing plasmid DNA-cationic liposome complexes (lipoplexes) or naked DNA.
- the present invention comprises novel, stable nucleic acid-lipid particles (SNALP) encapsulating one or more interfering RNA molecules, methods of making the SNALP.
- SNALP stable nucleic acid-lipid particles
- SNALPs and methods of delivering and/or administering the SNALPs.
- the invention provides for a nucleic acid-lipid particle comprising an interfering RNA and a cationic lipid of Formula I or II and having the following structures:
- R 1 and R 2 are independently selected from the group consisting of: H and -C 3 alkyls; and R 3 and R 4 are independently selected from the group consisting of alkyl groups having from about 10 to about 20 carbon atoms, wherein at least one of R 3 and R 4 comprises at least two sites of unsaturation.
- that cationic lipid is selected from l,2-DiLinoleyloxy-N,N-dimethylaminopropane (DLinDMA) and 1,2- Dilinolenyloxy-N,N-dimethylaminopropane (DLenDMA).
- the interfering RNA molecule is fully encapsulated within the lipid bilayer of the nucleic acid- lipid particle such that the nucleic acid in the nucleic acid-lipid particle is resistant in aqueous solution to degradation by a nuclease.
- the nucleic acid particle is substantially non-toxic to mammals.
- the nucleic acid lipid particles may further comprise a non-cationic lipid, a bilayer stabilizing component (i.e., a conjugated lipid that prevents aggregation of particles, a cationic polymer lipid, a sterol (e.g., cholesterol) and combinations thereof.
- the interfering RNA is a small-interfering RNA molecule that is less than about 60 nucleotides in length or a double-stranded RNA greater than about 25 nucleotides in length.
- the interfering RNA is transcribed from a plasmid, in particular a plasmid comprising a DNA template of a target sequence.
- the non-cationic lipid is selected from distearoylphosphatidylcholine (DSPC), dioleoylphosphatidylcholine (DOPC), dipalmitoylphosphatidylcholine (DPPC), dioleoylphosphatidylglycerol (DOPG), dipalmitoylphosphatidylglycerol (DPPG), dioleoyl-phosphatidylethanolamine (DOPE), palmitoyloleoylphosphatidylcholine (POPC), palmitoyloleoyl- phosphatidylethanolamine (POPE) and dioleoyl- phosphatidylethanolamine 4-(N-maleimidomethyl)-cyclohexane-l- carboxylate (DOPE-mal), dipalmitoyl phosphatidyl ethanolamine (DPPE), dimyristoylphosphoethaholamine (DMPE), distearoyl-phosphati
- the conjugated lipid that inhibits aggregation of particles is one or more of a polyethyleneglycol (PEG)-lipid conjugate, a polyamide (ATTA)-lipid conjugate, and a mixture thereof.
- PEG-lipid conjugate is one or more of a PEG-dialkyloxypropyl (DAA), a PEG-diacylglycerol (DAG), a PEG-phospholipid, a PEG-ceramide, and a mixture thereof.
- the PEG-DAG conjugate is one or more of a PEG-dilauroylglycerol (C ⁇ 2 ), a PEG-dimyristoylglycerol (C 14 ), a PEG- dipalmitoylglycerol (C 16 ), and a PEG-distearoylglycerol (C 18 ).
- the PEG- DAA conjugate is one or more of a PEG-dilauryloxypropyl (C 12 ), a PEG- dimyristyloxypropyl (C 14 ), a PEG-dipalmityloxypropyl (C 16 ), and a PEG- distearyloxypropyl (C 18 ).
- the nucleic acid-lipid particles of the present invention are useful for the therapeutic delivery of nucleic acids comprising an interfering RNA sequence.
- an interfering RNA is formulated into a nucleic acid-lipid particle, and the particles are administered to patients requiring such treatment.
- cells are removed from a patient, the interfering RNA delivered in vitro, and reinjected into the patient.
- the present invention provides for a method of introducing a nucleic acid into a cell by contacting a cell with a nucleic acid-lipid particle comprised of a cationic lipid, a non-cationic lipid, a conjugated lipid that inhibits aggregation, and an interfering RNA.
- At least about 5%, 10%, 15%, 20%, or 25% of the total injected dose of the nucleic acid-lipid particles is present in plasma about 8, 12, 24, 36, or 48 hours after injection, other embodiments, more than 20%, 30%, 40% and as much as 60%, 70% or 80% of the total injected dose of the nucleic acid-lipid particles is present in plasma about 8, 12, 24, 36, or 48 hours after injection.
- the presence of an interfering RNA in cells of the lung, liver, tumor or at a site of inflammation is detectable at about 8, 12, 24, 36, 48, 60, 72 or 96 hours after administration.
- downregulation of expression of the target sequence is detectable at about 8, 12, 24, 36, 48, 60, 72 or 96 hours after administration.
- downregulation of expression of the target sequence occurs preferentially in tumor cells or in cells at a site of inflammation.
- the presence of an interfering RNA in cells at a site distal to the site of administration is detectable at least four days after intravenous injection of the nucleic acid-lipid particle.
- the presence of an interfering RNA in of cells in the lung, liver or a tumor is detectable at least four days after injection of the nucleic acid-lipid particle.
- the nucleic acid-lipid particle is administered parenterally or intraperitoneally.
- the particles are suitable for use in intravenous nucleic acid transfer as they are stable in circulation, of a size required for pharmacodynamic behavior resulting in access to extravascular sites and target cell populations.
- the invention also provides for pharmaceutically acceptable compositions comprising a nucleic acid-lipid particle.
- Another embodiment of the present invention provides methods for in vivo delivery of interfering RNA.
- a nucleic acid-lipid particle comprising a cationic lipid, a non-cationic lipid, a conjugated lipid that inhibits aggregation of particles, and interfering RNA is administered (e.g., intravenously) to a subject (e.g., a mammal such as a human).
- the invention provides methods for in vivo delivery of interfering RNA to the liver of a mammalian subject.
- a further embodiment of the present invention provides a method of treating a disease or disorder in a mammalian subject.
- a therapeutically effective amount of a nucleic acid-lipid particle comprising a cationic lipid, a non-cationic lipid, a conjugated lipid that inhibits aggregation of particles, and interfering RNA is administered to the mammalian subject (e.g., a rodent such as a mouse, a primate such as a human or a monkey).
- the disease or disorder is associated with expression and/or overexpression of a gene and expression or overexpression of the gene is reduced by the interfering RNA.
- Figure 1 illustrates the structures of two exemplary cationic lipids of the invention: l,2-DiLinoleyloxy-N,N-dimethylaminopropane (DLinDMA) and 1,2- Dilinolenyloxy-N,N-dimethylaminopropane (DLenDM A) .
- Figure 2 illustrates the synthetic scheme for DLinDMA.
- Figure 3 illustrates the synthetic scheme for DLenDMA.
- Figure 4 illustrates downregulating ⁇ -galactosidase expression in CT26.CL25 cells via in vitro delivery of encapsulated anti- ⁇ -galactosidase siRNA in DSPC:Cholesterol:DODMA:PEG-DMG liposomes.
- Figure 5 illustrates that clearance studies with LUVs showed that SNALPs containing PEG-DAGs were comparable to SNALPs containing PEG-CeramideC20.
- Figure 6 illustrates the pharmacokinetic properties of SNALPs containing PEG- DAGs.
- Figure 7 illustrates the biodistribution properties of SNALPs containing PEG- DAGs.
- Figure 8 illustrates the luciferase gene expression 24 hrs post IV administration of SPLPs containing PEG-CeramideC 2 o versus PEG-DAGs in Neuro-2a Tumor Bearing Male A/J Mice.
- Figure 9 illustrates the luciferase gene expression 48 hrs post IV administration of SPLPs containing PEG-CeramideC 2 o versus PEG-DAGs in Neuro-2a Tumor Bearing Male A/J Mice.
- Figure 10 illustrates the luciferase gene expression 72 hrs post IV administration of SPLPs containing PEG-CeramideC 2 o versus PEG-DAGs in Neuro-2a Tumor Bearing Male A/J Mice.
- Figure 11 illustrates data showing luciferase gene expression in tumors 48 hours after intravenous administration of SPLP comprising PEG-DAA conjugates and PEG- DAG conjugates.
- Figure 12 illustrates data showing luciferase gene expression in liver, lung, spleen, heart, and tumor following intravenous administration of SPLP comprising PEG- DAA conjugates and PEG-DAG conjugates.
- Figure 13 illustrates data from clearance studies in Neuro-2a tumor bearing male A/J mice after administration of SPLPs comprising a PEG-DAA conjugate and containing a plasmid encoding luciferase under the control of the CMV promoter and SNALPs comprising a PEG-DAA conjugate and containing anti-luciferase siRNA.
- Figure 14 illustrates data from studies of the pharmacokinetic properties of SPLPs comprising a PEG-DAA conjugate and containing a plasmid encoding luciferase under the control of the CMV promoter and SNALPs comprising a PEG-DAA conjugate and containing anti-luciferase siRNA in Neuro-2a tumor bearing male A J mice.
- Figure 15 illustrates data from clearance studies in Neuro-2a tumor bearing male A/J mice after administration of SPLPs comprising a PEG-DAA conjugate or a PEG-DAG conjugate and containing a plasmid encoding luciferase under the control of the CMV promoter, pSPLPs comprising a PEG-DAG conjugate and containing a plasmid encoding luciferase under the control of the CMV promoter and SNALPs comprising a PEG-DAA conjugate and containing anti-luciferase siRNA.
- Figure 16 illustrates data from studies of the pharmacokinetic properties of SPLPs comprising a PEG-DAA conjugate or a PEG-DAG conjugate and containing a plasmid encoding luciferase under the control of the CMV promoter, pSPLPs comprising a PEG-DAG conjugate and containing a plasmid encoding luciferase under the control of the CMV promoter and SNALPs comprising a PEG-DAA conjugate and containing anti- luciferase siRNA in Neuro-2a tumor bearing male A/J mice.
- Figure 17 illustrates in vitro data demonstrating silencing of luciferase expression in luciferase expressing cells treated with SPLPs comprising a PEG-lipid conjugate and containing a plasmid encoding luciferase under the control of the CMV promoter and SNALPs comprising a PEG-lipid conjugate and containing anti-luciferase siRNA.
- Figure 18 illustrates in vivo data demonstrating silencing of luciferase expression in Neuro-2a tumor bearing male A/J mice treated with SPLPs comprising a PEG-DAA conjugate and containing a plasmid encoding luciferase under the control of the CMV promoter and SNALPs comprising a PEG-DAA conjugate and containing anti-luciferase siRNA.
- Figure 19 illustrates in vivo data demonstrating silencing of luciferase expression in Neuro-2a tumor bearing male A/J mice treated with SPLPs comprising a PEG-DAA conjugate and containing a plasmid encoding luciferase under the control of the CMV promoter and SNALPs comprising a PEG-DAA conjugate and containing anti-luciferase siRNA.
- Figure 20 illustrates in vivo data demonstrating silencing of luciferase expression in Neuro-2a tumor bearing male A/J mice treated with SPLPs comprising a PEG-DAA conjugate and containing a plasmid encoding luciferase under the control of the CMV promoter and SNALPs comprising a PEG-DAA conjugate and containing anti-luciferase siRNA.
- Figure 21 illustrates in vivo data demonstrating silencing of luciferase expression in Neuro-2a tumor bearing male A/J mice treated with SPLPs comprising a PEG-DAA conjugate and containing a plasmid encoding luciferase under the control of the CMV promoter and SNALPs comprising a PEG-DAA conjugate and containing anti-luciferase siRNA.
- Figure 22 illustrates in vivo data demonstrating silencing of luciferase expression in Neuro-2a tumor bearing male A/J mice treated with SPLPs comprising a PEG-DAA conjugate and containing a plasmid encoding luciferase under the control of the CMV promoter and SNALPs comprising a PEG-DAA conjugate and containing anti-luciferase siRNA.
- Figure 23 illustrates data showing silencing of gene expression following in vitro transfection of Neuro2a cells stably expressing luciferase by an SPLP (i.e., SNALP) comprising DODAC, DODMA, or DLinDMA and encapsulating an anti-luciferase siRNA sequence.
- Figure 24 illustrates data showing SNALP-mediated gene silencing in vitro.
- Figure 25 illustrates data showing luciferase gene expression in tumors 48 hours following intravenous delivery of SPLP encapsulating a plasmid encoding luciferase.
- the SPLP comprised PEG-C-DMA conjugates and either DODMA or DLinDMA.
- the PEG moieties had molecular weight of either 2000 or 750.
- Figure 26 illustrates data showing luciferase gene expression in Neuro2A tumor bearing male A/J mice 48 hours after intravenous administration of SPLP encapsulating a plasmid encoding luciferase.
- the SPLP comprised varying percentages (Le., 15%, 10%, 5% or 2.5 %) of PEG-C-DMA and either DODMA or DLinDMA.
- Figure 27 illustrates data showing the percentage of the injected dose of SPLP, SNALP, or empty vesicles remaining in plasma of male A/J mice following a single intravenous administration of 3 H-CHE-labeled SPLP or SNALP, or empty vesicles, containing various percentages (Le., 2%, 5%, 10%, or 15%) of PEG-C-DMA.
- Figure 28 illustrates data showing the biodistribution SPLP, SNALP or empty vesicles in Neuro-2A tumor-bearing male A/J mice 48 hours after a single intravenous administration of H-CHE-labelled formulations comprising varying percentages of PEG- C-DMA.
- FIG. 29 illustrates data showing silencing of luciferase expression in distal, stable Neuro2A-G tumors in A/J mice 48 hours after intravenous administration of SNALP comprising DLinDMA.
- Figure 30 illustrates data showing silencing of luciferase expression in Neuro2A- G cells following delivery of SNALP formulations comprising DLinDMA and encapsulating anti-luciferase siRNA.
- Figure 31 illustrates data showing silencing of luciferase expression in Neuro2A- G cells following delivery of SNALP formulations comprising DLinDMA and encapsulating anti-luciferase siRNA. Delivery of the SNALP formulations was performed in the absence or presence of chloroquine. DETAILED DESCRIPTION OF THE INVENTION
- the present invention demonstrates the unexpected success of encapsulating short interfering RNA (siRNA) molecules in SNALPs comprising cationic lipids of Formula I, II, or mixture thereof.
- siRNA short interfering RNA
- the SNALPs described herein can be used to deliver an siRNA to a cell to silence a target sequence of interest.
- SNALP comprising any of a broad range of concentrations of additional cationic lipids, non-cationic lipids, and other lipids can be used to practice the present invention.
- the SNALP can be prepared with any nucleic acid comprising an interfering RNA sequence, from any source and comprising any polynucleotide sequence, and can be prepared using any of a large number of methods.
- lipid refers to a group of organic compounds that include, but are not limited to, esters of fatty acids and are characterized by being insoluble in water, but soluble in many organic solvents. They are usually divided into at least three classes: (1) “simple lipids” which include fats and oils as well as waxes; (2) “compound lipids” which include phospholipids and glycolipids; (3) “derived lipids” such as steroids.
- lipid encapsulated can refer to a lipid formulation that provides a compound with full encapsulation, partial encapsulation, or both.
- the nucleic acid is fully encapsulated in the lipid formulation (e.g., to form an SPLP, pSPLP, or other SNALP).
- SNALP refers to a stable nucleic acid lipid particle, including SPLP.
- a SNALP represents a vesicle of lipids coating a reduced aqueous interior comprising a nucleic acid (e.g., ssDNA, dsDNA, ssRNA, micro RNA (miRNA), short hairpin RNA (shRNA), dsRNA, siRNA, or a plasmid, including plasmids from which an interfering RNA is transcribed).
- a nucleic acid e.g., ssDNA, dsDNA, ssRNA, micro RNA (miRNA), short hairpin RNA (shRNA), dsRNA, siRNA, or a plasmid, including plasmids from which an interfering RNA is transcribed.
- SPLP refers to a nucleic acid lipid particle comprising a nucleic acid (e.g., a plasmid) encapsulated within a lipid vesicle.
- SNALPs and SPLPs typically contain a cationic lipid, a non-cationic lipid, and a lipid that prevents aggregation of the particle (e.g. , a PEG-lipid conjugate).
- SNALPs and SPLPs have systemic application as they exhibit extended circulation lifetimes following intravenous (i.v.) injection, accumulate at distal sites (e.g., sites physically separated from the administration site and can mediate expression of the transfected gene at these distal sites.
- SPLPs include "pSPLP" which comprise an encapsulated condensing agent-nucleic acid complex as set forth in WO 00/03683.
- vesicle-forming lipid is intended to include any amphipathic lipid having a hydrophobic moiety and a polar head group, and which by itself can form spontaneously into bilayer vesicles in water, as exemplified by most phospholipids.
- vesicle-adopting lipid is intended to include any amphipathic lipid that is stably incorporated into lipid bilayers in combination with other amphipathic lipids, with its hydrophobic moiety in contact with the interior, hydrophobic region of the bilayer membrane, and its polar head group moiety oriented toward the exterior, polar surface of the membrane.
- Vesicle-adopting lipids include lipids that on their own tend to adopt a nonlamellar phase, yet which are capable of assuming a bilayer structure in the presence of a bilayer-stabilizing component.
- a typical example is DOPE
- Bilayer stabilizing components include, but are not limited to, conjugated lipids that inhibit aggregation of the SNALPs, polyamide oligomers (e.g., ATTA-lipid derivatives), peptides, proteins, detergents, lipid-derivatives, PEG-lipid derivatives such as PEG coupled to dialkyloxypropyls, PEG coupled to diacylglycerols, PEG coupled to phosphatidyl-ethanolamines, and PEG conjugated to ceramides as described in U.S. Patent No. 5,885,613.
- conjugated lipids that inhibit aggregation of the SNALPs include, but are not limited to, conjugated lipids that inhibit aggregation of the SNALPs, polyamide oligomers (e.g., ATTA-lipid derivatives), peptides, proteins, detergents, lipid-derivatives, PEG-lipid derivatives such as PEG coupled to dialkyloxypropyls, PEG coupled to diacylgly
- amphipathic lipid refers, in part, to any suitable material wherein the hydrophobic portion of the lipid material orients into a hydrophobic phase, while the hydrophilic portion orients toward the aqueous phase.
- Amphipathic lipids are usually the major component of a lipid vesicle. Hydrophilic characteristics derive from the presence of polar or charged groups such as carbohydrates, phosphate, carboxylic, sulfato, amino, sulfhydryl, nitro, hydroxy and other like groups.
- Hydrophobicity can be conferred by the inclusion of apolar groups that include, but are not limited to, long chain saturated and unsaturated aliphatic hydrocarbon groups and such groups substituted by one or more aromatic, cycloaliphatic or heterocyclic group(s).
- apolar groups that include, but are not limited to, long chain saturated and unsaturated aliphatic hydrocarbon groups and such groups substituted by one or more aromatic, cycloaliphatic or heterocyclic group(s).
- amphipathic compounds include, but are not limited to, phospholipids, aminolipids and sphingolipids.
- phospholipids include, but are not limited to, phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, phosphatidylinositol, phosphatidic acid, palmitoyloleoyl phosphatidylcholine, lysophosphatidylcholine, lysophosphatidylethanolamine, dipalmitoylphosphatidylcholine, dioleoylphosphatidylcholine, distearoylphosphatidylcholine or dilinoleoylphosphatidylcholine.
- amphipathic lipids Other compounds lacking in phosphorus, such as sphingolipid, glycosphingolipid families, diacylglycerols and ⁇ -acyloxyacids, are also within the group designated as amphipathic lipids. Additionally, the amphipathic lipid described above can be mixed with other lipids including triglycerides and sterols.
- neutral lipid refers to any of a number of lipid species that exist either in an uncharged or neutral zwitterionic form at a selected pH.
- such lipids include, for example, diacylphosphatidylcholine, diacylphosphatidylethanolamine, ceramide, sphingomyelin, cephalin, cholesterol, cerebrosides and diacylglycerols.
- noncationic lipid refers to any neutral lipid as described above as well as anionic lipids.
- Non-cationic lipids include, e.g., distearoylphosphatidylcholine (DSPC), dioleoylphosphatidylcholine (DOPC), dipalmitoylphosphatidylcholine (DPPC), dioleoylphosphatidylglycerol (DOPG), dipalmitoylphosphatidylglycerol (DPPG), dioleoyl-phosphatidylethanolamine (DOPE), palmitoyloleoylphosphatidylcholine (POPC), palmitoyloleoyl- phosphatidylethanolamine (POPE) and dioleoyl- phosphatidylethanolamine 4-(N-maleimidomethyl)-cyclohexane-l-carboxylate (DOPE- mal), dipalmitoyl phosphatidyl phosphatidy
- anionic lipid refers to any lipid that is negatively charged at physiological pH. These lipids include, but are not limited to, phosphatidylglycerol, cardiolipin, diacylphosphatidylserine, diacylphosphatidic acid, N-dodecanoyl phosphatidylethanolamines, N-succinyl phosphatidylethanolamines, N- glutarylphosphatidylethanolamines, lysylphosphatidylglycerols, palmitoyloleyolphosphatidylglycerol (POPG), and other anionic modifying groups joined to neutral lipids.
- phosphatidylglycerol cardiolipin
- diacylphosphatidylserine diacylphosphatidic acid
- N-dodecanoyl phosphatidylethanolamines N-succinyl phosphatidylethanolamines
- N- glutarylphosphatidylethanolamines N
- cationic lipid refers to any of a number of lipid species that carry a net positive charge at a selected pH, such as physiological pH.
- lipids include, but are not limited to: l,2-DiLinoleyloxy-N,N-dimethylaminopropane (DLinDMA) and 1,2- Dilinolenyloxy-N,N-dimethylaminopropane (DLenDMA), N,N-dioleyl-N,N- dimethylammonium chloride (DODAC); N-(2,3-dioleyloxy)propyl)-N,N,N- trimethylammonium chloride (DOTMA); N,N-distearyl-N,N-dimethylammonium bromide (DDAB); N-(2,3-dioleoyloxy)pro ⁇ yl)-N,N,N-trimethylammonium chloride (DOTAP); 3 - (N-(N',N'
- hydrophobic lipid refers to compounds having apolar groups that include, but are not limited to, long chain saturated and unsaturated aliphatic hydrocarbon groups and such groups optionally substituted by one or more aromatic, cycloaliphatic or heterocyclic group(s). Suitable examples include, but are not limited to, diacylglycerol, dialkylglycerol, N-N-dialkylamino, l,2-diacyloxy-3-aminopropane and l,2-dialkyl-3- aminopropane.
- the term "fusogenic” refers to the ability of a liposome, an SNALP or other drug delivery system to fuse with membranes of a cell.
- the membranes can be either the plasma membrane or membranes surrounding organelles, e.g., endosome, nucleus, etc.
- diacylglycerol refers to a compound having 2-fatty acyl chains, R 1 and R 2 , both of which have independently between 2 and 30 carbons bonded to the 1- and 2-position of glycerol by ester linkages.
- the acyl groups can be saturated or have varying degrees of unsaturation.
- Diacylglycerols have the following general formula:
- dialkyloxypropyl refers to a compound having 2-alkyl chains, R 1 and R 2 , both of which have independently between 2 and 30 carbons.
- the alkyl groups can be saturated or have varying degrees of unsaturation.
- Dialkyloxypropyls have the following general formula:
- ATTA or "polyamide” refers to, but is not limited to, compounds disclosed in U.S. Patent Nos. 6,320,017 and 6,586,559. These compounds include a compound having the formula
- R is a member selected from the group consisting of hydrogen, alkyl and acyl
- R 1 is a member selected from the group consisting of hydrogen and alkyl; or optionally, R and R and the nitrogen to which they are bound form an azido moiety
- R is a member of the group selected from hydrogen, optionally substituted alkyl, optionally substituted aryl and a side chain of an amino acid
- R is a member selected from the group consisting of hydrogen, halogen, hydroxy, alkoxy, mercapto, hydrazino, amino and NR 4 R 5 , wherein R 4 and R 5 are independently hydrogen or alkyl
- n is 4 to 80
- m is 2 to 6
- p is 1 to 4
- q is 0 or 1.
- polypeptide polypeptide
- peptide protein
- polypeptide polypeptide
- protein protein
- amino acid polymers in which one or more amino acid residue is an artificial chemical mimetic of a corresponding naturally occurring amino acid, as well as to naturally occurring amino acid polymers and non-naturally occurring amino acid polymers.
- the terms encompass amino acid chains of any length, including full-length proteins (i.e., antigens), wherein the amino acid residues are linked by covalent peptide bonds.
- amino acid refers to naturally occurring and synthetic amino acids, as well as amino acid analogs and amino acid mimetics that function in a manner similar to the naturally occurring amino acids.
- basic amino acid refers to naturally- occurring amino acids as well as synthetic amino acids and/or or amino acid mimetics having a net positive charge at a selected pH, such as physiological pH. This group includes, but is not limited to, lysine, arginine, asparagine, glutamine, histidine and the like.
- Naturally occurring amino acids are those encoded by the genetic code, as well as those amino acids that are later modified, e.g., hydroxyproline, ⁇ -carboxyglutamate, and O-phosphoserine.
- Amino acid analogs refers to compounds that have the same basic chemical structure as a naturally occurring amino acid, i.e., an ⁇ carbon that is bound to a hydrogen, a carboxyl group, an amino group, and an R group, e.g., homoserine, norleucine, methionine sulf oxide, methionine methyl sulfondum. Such analogs have modified R groups (e.g., norleucine) or modified peptide backbones, but retain the same basic chemical structure as a naturally occurring amino acid.
- Amino acid mimetics refers to chemical compounds that have a structure that is different from the general chemical structure of an amino acid, but that functions in a manner similar to a naturally occurring amino acid.
- nucleic acid or “polynucleotide” refers to a polymer containing at least two deoxyribonucleotides or ribonucleotides in either single- or double-stranded form.
- nucleic acids containing known analogues of natural nucleotides that have similar binding properties as the reference nucleic acid and are metabolized in a manner similar to naturally occurring nucleotides.
- a particular nucleic acid sequence also implicitly encompasses conservatively modified variants thereof (e.g., degenerate codon substitutions), alleles, orthologs, SNPs, and complementary sequences as well as the sequence explicitly indicated.
- degenerate codon substitutions may be achieved by generating sequences in which the third position of one or more selected (or all) codons is substituted with mixed-base and/or deoxyinosine residues (Batzer et al., Nucleic Acid Res.
- Nucleotides contain a sugar deoxyribose (DNA) or ribose (RNA), a base, and a phosphate group. Nucleotides are linked together through the phosphate groups.
- Bases include purines and pyrimidines, which further include natural compounds adenine, thymine, guanine, cytosine, uracil, inosine, and natural analogs, and synthetic derivatives of purines and pyrimidines, which include, but are not limited to, modifications which place new reactive groups such as, but not limited to, amines, alcohols, thiols, carboxylates, and alkylhalides.
- DNA may be in the form of antisense, plasmid DNA, parts of a plasmid DNA, pre-condensed DNA, product of a polymerase chain reaction (PCR), vectors (PI, PAC, BAC, YAC, artificial chromosomes), expression cassettes, chimeric sequences, chromosomal DNA, or derivatives of these groups.
- PCR polymerase chain reaction
- PI polymerase chain reaction
- PAC PAC
- BAC BAC
- YAC artificial chromosomes
- expression cassettes chimeric sequences
- chromosomal DNA or derivatives of these groups.
- nucleic acid is used interchangeably with gene, cDNA, mRNA encoded by a gene, and an interfering RNA molecule.
- Constantly modified variants applies to both amino acid and nucleic acid sequences. With respect to particular nucleic acid sequences, “conservatively modified variants” refers to those nucleic acids that encode identical or essentially identical amino acid sequences, or where the nucleic acid does not encode an amino acid sequence, to essentially identical sequences. Because of the degeneracy of the genetic code, a large number of functionally identical nucleic acids encode any given protein. For instance, the codons GCA, GCC, GCG and GCU all encode the amino acid alanine. Thus, at every position where an alanine is specified by a codon, the codon can be altered to any of the corresponding codons described without altering the encoded polypeptide.
- nucleic acid variations are "silent variations," which are one species of conservatively modified variations. Every nucleic acid sequence herein that encodes a polypeptide also describes every possible silent variation of the nucleic acid.
- each codon in a nucleic acid except AUG, which is ordinarily the only codon for methionine, and TGG, which is ordinarily the only codon for tryptophan
- TGG which is ordinarily the only codon for tryptophan
- DNA or RNA sequence that comprises partial length or entire length coding sequences necessary for the production of a polypeptide or precursor (e.g., hepatitis virus A, B, C, D, E, or G; or herpes simplex virus).
- a polypeptide or precursor e.g., hepatitis virus A, B, C, D, E, or G; or herpes simplex virus.
- Gene product refers to a product of a gene such as an RNA transcript.
- RNA transcript refers to a product of a gene such as an RNA transcript.
- interfering RNA or "RNAi” or “interfering RNA sequence” refers to double-stranded RNA that results in the degradation of specific mRNAs and can be used to interfere with translation from a desired mRNA target transcript.
- RNAi Short RNAi that is about 15-30 nucleotides in length is referred to as "small-interfering RNA” or “siRNA.” Longer RNAi is generally referred to as “double-stranded RNA” or “dsRNA.”
- dsRNA double-stranded RNA
- a DNA molecule that transcribes dsRNA or siRNA also provides RNAi.
- DNA molecules for transcribing dsRNA are disclosed in U.S. Patent No. 6,573,099, and in U.S. Patent Publication Nos. 20020160393 and 20030027783. DNA molecules for transcribing siRNA are reviewed in Tuschl and Borkhardt, Molecular Interventions, 2:158 (2002).
- RNAi RNA-binding protein
- a detectable decrease can be as small as about 5 % or 10 %, or as great as about 80 %, 90 % or 100 %. More typically, a detectable decrease is about 20 %, 30 %, 40 %, 50 %, 60 %, or 70 %.
- aqueous solution refers to a composition comprising in whole, or in part, water.
- organic lipid solution refers to a composition comprising in whole, or in part, an organic solvent having a lipid.
- distal site refers to a physically separated site, which is not limited to an adjacent capillary bed, but includes sites broadly distributed throughout an organism.
- Serum-stable in relation to nucleic acid-lipid particles means that the particle is not significantly degraded after exposure to a serum or nuclease assay that would significantly degrade free DNA.
- Systemic delivery refers to delivery that leads to a broad biodistribution of a compound within an organism. Some techniques of administration can lead to the systemic delivery of certain compounds, but not others. Systemic delivery means that a useful, preferably therapeutic, amount of a compound is exposed to most parts of the body. To obtain broad biodistribution generally requires a blood lifetime such that the compound is not rapidly degraded or cleared (such as by first pass organs (liver, lung, etc.) or by rapid, nonspecific cell binding) before reaching a disease site distal to the site of administration.
- Systemic delivery of nucleic acid-lipid particules can be by any means known in the art including, for example, intravenous, subcutaneous, intraperitoneal, In a preferred embodiment, systemic delivery of nucleic acid-lipid particles is by intravenous delivery.
- SNALPs Stable Nucleic Acid-Lipid Particles
- the stable nucleic acid-lipid particles or, alternatively, SNALPs typically comprise cationic lipid (i.e., a cationic lipid of Formula I or II) and nucleic acids.
- Such SNALPs also preferably comprise noncationic lipid and a bilayer stabilizing component (i.e., a conjugated lipid that inhibits aggregation of the SNALPs).
- the SNALPs of the present invention typically have a mean diameter of about 50 nm to about 150 nm, more typically about 100 nm to about 130 nm, most typically about 110 nm to about 115 nm, and are substantially nontoxic.
- the nucleic acids present in the SNALPs of the present invention are resistant in aqueous solution to degradation with a nuclease.
- the present invention provides stabilized nucleic acid-lipid particles (SPLPs or SNALPs) and other lipid-based carrier systems (e.g., a liposome, a micelle, a virosome, a lipid-nucleic acid particle, a nucleic acid complex and mixtures thereof) containing cationic lipids of the present invention, i.e., cationic lipids of Formula I, Formula II, or a combination thereof.
- the lipid-nucleic acid particles of the present invention typically comprise a nucleic acid, a cationic lipid of Formula I or Formula II, a non-cationic lipid and a PEG-lipid conjugate.
- the cationic lipid of Formula I or Formula II typically comprises from about 2% to about 60%, from about 5% to about 50%, from about 10% to about 45%, from about 20% to about 40%, or about 30% of the total lipid present in said particle.
- the non-cationic lipid typically comprises from about 5% to about 90%, from about 10% to about 85%, from about 20% to about 80%, from about 30% to about 70%, from about 40% to about 60% or about 48% of the total lipid present in said particle.
- the PEG-lipid conjugate typically comprises from about 1% to about 20%, from about 1.5% to about 18%, from about 4% to about 15%, from about 5% to about 12%, or about 2% of the total lipid present in said particle.
- the nucleic acid-lipid particles of the present invention may further comprise cholesterol.
- the cholesterol typically comprises from about 10% to about 60%, from about 12% to about 58%, from about 20% to about 55%, or about 48% of the total lipid present in said particle.
- the proportions of the components of the nucleic acid-lipid particles may be varied, e.g., using the ERP assay described herein.
- the cationic lipid may comprise from about 5% to about 15% of the total lipid present in said particle and for local or regional delivery, the cationic lipid comprises from about 40% to about 50% of the total lipid present in said particle.
- Cationic lipids of Formula I and II may be used in the present invention, either alone or in combination with one or more other cationic lipid species or non-cationic lipid species.
- Cationic lipids of Formula I and II have the following structures:
- R 1 and R 2 are independently selected and are H or -C 3 alkyls.
- R 3 and R 4 are independently selected and are alkyl groups having from about 10 to about 20 carbon atoms; at least one of R and R comprises at least two sites of unsaturation.
- R 3 and R 4 are both the same, Le., R 3 and ' R 4 are both linoleyl (C18), etc.
- R 3 and R 4 are different, i.e., R 3 is myristyl (C14) and R 4 is linoleyl (C18).
- the cationic lipids of the present invention are symmetrical, Le., R and R are both the same.
- both R and R 4 comprise at least two sites of unsaturation.
- R 3 and R 4 are independently selected from dodecadienyl, tetradecadienyl, hexadecadienyl, linoleyl, and icosadienyl.
- R 3 and R 4 are both linoleyl.
- R and R comprise at least three sites of unsaturation and are independently selected from, e.g., dodecatrienyl, tetradectrienyl, hexadecatrienyl, linolenyl, and icosatrienyl.
- the cationic lipids of Formula I and Formula II described herein typically carry a net positive charge at a selected pH, such as physiological pH. It has been surprisingly found that cationic lipids comprising alkyl chains with multiple sites of unsaturation, e.g., at least two or three sites of unsaturation, are particularly useful for forming lipid-nucleic acid particles with increased membrane fluidity. A number of cationic lipids and related analogs, which are also useful in the present invention, have been described in co-pending USSN 08/316,399; U.S. Patent Nos. 5,208,036, 5,264,618, 5,279,833 and 5,283,185, and WO 96/10390.
- Additional suitable cationic lipids include, e.g., dioctadecyldimethylammonium (“DODMA”), Distearyldimethylammonium (“DSDMA”), N,N-dioleyl-N,N- dimethylammonium chloride (“DODAC”); N-(2,3-dioleyloxy)propyl)-N,N,N- trimethylammonium chloride (“DOTMA”); N,N-distearyl-N,N-dimethylammonium bromide (“DDAB”); N-(2,3-dioleoyloxy)propyl)-N,N,N-trimethylammonium chloride (“DOTAP”); 3 -(N-(N',N , -dimethylaminoethane)-carbamoyl)cholesterol (“DC-Chol”) and N-( 1 ,2-dimyristyloxyprop-3 -yl)-N,N-
- the noncationic lipids used in the present invention can be any of a variety of neutral uncharged, zwitterionic or anionic lipids capable of producing a stable complex. They are preferably neutral, although they can alternatively be positively or negatively charged.
- noncationic lipids useful in the present invention include: phospholipid-related materials, such as lecithin, phosphatidylethanolamine, lysolecithin, lysophosphatidylethanolamine, phosphatidylserine, phosphatidylinositol, sphingomyelin, cephalin, cardiolipin, phosphatidic acid, cerebrosides, dicetylphosphate, distearoylphosphatidylcholine (DSPC), dioleoylphosphatidylcholine (DOPC), dipalmitoylphosphatidylcholine (DPPC), dioleoylphosphatidylglycerol (DOPG), dipalmitoylphosphatidylglycerol (DPPG), dioleoyl-phosphatidylethanolamine (DOPE), palmitoyloleoylphosphatidylcholine (POPC), palmitoyloleoyl- phosphatid
- Noncationic lipids or sterols such as cholesterol may be present.
- Additional nonphosphorous containing lipids are, e.g., stearylamine, dodecylamine, hexadecylamine, acetyl palmitate, glycerolricinoleate, hexadecyl stereate, isopropyl myristate, amphoteric acrylic polymers, triethanolamine-lauryl sulfate, alkyl- aryl sulfate polyethyloxylated fatty acid amides, dioctadecyldimethyl ammonium bromide and the like, diacylphosphatidylcholine, diacylphosphatidylethanolamine, ceramide, sphingomyelin, cephalin, and cerebrosides.
- Noncationic lipids such as lysophosphatidylcholine and lysophosphatidylethanolamine may be present.
- Noncationic lipids also include polyethylene glycol-based polymers such as PEG 2000, PEG 5000 and polyethylene glycol conjugated to phospholipids or to ceramides (referred to as PEG-Cer), as described in co-pending USSN 08/316,429.
- the noncationic lipids are diacylphosphatidylcholine (e.g.
- distearoylphosphatidylcholine dioleoylphosphatidylcholine, dipalmitoylphosphatidylcholine and dilinoleoylphosphatidylcholine
- diacylphosphatidylethanolamine e.g., dioleoylphosphatidylethanolamine and palmitoyloleoylphosphatidylethanolamine
- ceramide or sphingomyelin e.g., dioleoylphosphatidylethanolamine and palmitoyloleoylphosphatidylethanolamine
- ceramide or sphingomyelin sphingomyelin.
- the acyl groups in these lipids are preferably acyl groups derived from fatty acids having o-C ⁇ carbon chains. More preferably the acyl groups are lauroyl, myristoyl, palmitoyl, stearoyl or oleoyl.
- the SPLPs of the present invention comprise bilayer stabilizing component (BSC) such as an ATTA-lipid or a PEG-lipid, such as PEG coupled to dialkyloxypropyls (PEG-DAA) as described in, e.g., WO 05/026372, PEG coupled to diacylglycerol (PEG-DAG) as described in, e.g., U.S. Patent Publication Nos. 20030077829 and 2005008689), PEG coupled to phosphatidylethanolamine (PE) (PEG-PE), or PEG conjugated to ceramides, or a mixture thereof (see, U.S. Patent No.
- BSC bilayer stabilizing component
- an ATTA-lipid or a PEG-lipid such as PEG coupled to dialkyloxypropyls (PEG-DAA) as described in, e.g., WO 05/026372, PEG coupled to diacylglycerol (PEG-DAG) as described in,
- the BSC is a conjugated lipid that inhibits aggregation of the SPLPs.
- Suitable conjugated lipids include, but are not limited to PEG-lipid conjugates, ATTA-lipid conjugates, cationic- polymer-lipid conjugates (CPLs) or mixtures thereof.
- the SPLPs comprise either a PEG-lipid conjugate or an ATTA-lipid conjugate together with a CPL.
- PEG is a polyethylene glycol, a linear, water-soluble polymer of ethylene PEG repeating units with two terminal hydroxyl groups.
- PEGs are classified by their molecular weights; for example, PEG 2000 has an average molecular weight of about 2,000 daltons, and PEG 5000 has an average molecular weight of about 5,000 daltons. PEGs are commercially available from Sigma Chemical Co.
- MePEG-OH monomethoxypolyethylene glycol
- MePEG-S monomethoxypolyethylene glycol-succinate
- MePEG-S-NHS monomethoxypolyethylene glycol-succinimidyl succinate
- MePEG-NH 2 monomethoxypolyethylene glycol-amine
- MePEG-TRES monomethoxypolyethylene glycol-imidazolyl-carbonyl
- the PEG has an average molecular weight of from about 550 daltons to about 10,000 daltons, more preferably of about 750 daltons to about 5,000 daltons, more preferably of about 1,000 daltons to about 5,000 daltons, more preferably of about 1,500 daltons to about 3,000 daltons and, even more preferably, of about 2,000 daltons, or about 750 daltons.
- the PEG can be optionally substituted by an alkyl, alkoxy, acyl or aryl group.
- PEG can be conjugated directly to the lipid or may be linked to the lipid via a linker moiety.
- Any linker moiety suitable for coupling the PEG to a lipid can be used including, e.g. , non-ester containing linker moieties and ester- containing linker moieties.
- the linker moiety is a non-ester containing linker moiety.
- non-ester containing linker moiety refers to a linker moiety that does not contain a carboxylic ester bond (-OC(O)-).
- Suitable non-ester containing linker moieties include, but are not limited to, amido (-C(O)NH-), amino (-NR-), carbonyl (-C(O)-), carbamate (-NHC(O)O-), urea (-NHC(O)NH-), disulphide (-S-S-), ether (-O-), succinyl (-(O)CCH 2 CH 2 C(O)-), succinamidyl (- NHC(O)CH CH C(O)NH-), ether, disulphide, etc. as well as combinations thereof (such as a linker containing both a carbamate linker moiety and an amido linker moiety).
- a carbamate linker is used to couple the PEG to the lipid.
- an ester containing linker moiety is used to couple the PEG to the lipid.
- Suitable ester containing linker moieties include, e.g., carbonate (- OC(O)O-), succinoyl, phosphate esters (-O-(O)POH-O-), sulfonate esters, and combinations thereof.
- Phosphatidylemanolamines having a variety of acyl chain groups of varying chain lengths and degrees of saturation can be conjugated to polyethyleneglycol to form the bilayer stabilizing component.
- phosphatidylethanolamines are commercially available, or can be isolated or synthesized using conventional techniques known to those of skilled in the art. Phosphatidylethanolamines containing saturated or unsaturated fatty acids with carbon chain lengths in the range of do to C 2 o are preferred. Phosphatidylethanolamines with mono- or diunsaturated fatty acids and mixtures of saturated and unsaturated fatty acids can also be used.
- Suitable phosphatidylethanolamines include, but are not limited to, the following: dimyristoylphosphatidylethanolamine (DMPE), dipalmitoylphosphatidylethanolamine (DPPE), dioleoylphosphatidylethanolamine (DOPE) and distearoylphosphatidylethanolamine
- DMPE dimyristoylphosphatidylethanolamine
- DPPE dipalmitoylphosphatidylethanolamine
- DOPE dioleoylphosphatidylethanolamine
- distearoylphosphatidylethanolamine distearoylphosphatidylethanolamine
- ATTA or "polyamide” refers to, but is not limited to, compounds disclosed in U.S. Patent Nos. 6,320,017 and 6,586,559. These compounds include a compound having the formula
- R is a member selected from the group consisting of hydrogen, alkyl and acyl
- R 1 is a member selected from the group consisting of hydrogen and alkyl; or optionally, R 1 9 and R and the nitrogen to which they are bound form an azido moiety
- R is a member of the group selected from hydrogen, optionally substituted alkyl, optionally substituted aryl and a side chain of an amino acid
- R is a member selected from the group consisting of hydrogen, halogen, hydroxy, alkoxy, mercapto, hydrazino, amino and NR 4 R 5 , wherein R 4 and R 5 are independently hydrogen or alkyl
- n is 4 to 80
- m is 2 to 6
- p is 1 to 4
- q is 0 or 1.
- diacylglycerol refers to a compound having 2-fatty acyl chains, R 1 and R 2 , both of which have independently between 2 and 30 carbons bonded to the 1- and 2-position of glycerol by ester linkages.
- the acyl groups can be saturated or have varying degrees of unsaturation.
- Diacylglycerols have the following general formula: (IV)
- dialkyloxypropyl refers to a compound having 2-alkyl chains, R 1 and R 2 , both of which have independently between 2 and 30 carbons.
- the alkyl groups can be saturated or have varying degrees of unsaturation.
- Dialkyloxypropyls have the following general formula:
- the PEG-lipid is a PEG-DAA conjugate has the following formula:
- R and R are independently selected and are long-chain alkyl groups having from about 10 to about 22 carbon atoms.
- the long-chain alkyl groups can be saturated or unsaturated.
- Suitable alkyl groups include, but are not limited to, lauryl (C12), myristyl (C14), palmityl (C16), stearyl (C18) and icosyl (C20).
- R and R are the same, Le., R and R are both myristyl (z.e., dimyristyi), R 1 and R 2 are both stearyl (Le., distearyl), etc.
- R 1 and R 2 are independently selected and are alkyl groups having from about 10 to about 20 carbon atoms; PEG is a polyethyleneglycol; and L is a non-ester-containing linker moiety as described above.
- Suitable alkyl groups include, but are not limited to, lauryl (C12), myristyl (C14), palmityl (C16), stearyl (C18) and icosyl (C20).
- R 1 and R 2 are the same, Le., they are both myristyl (C14) or both palmityl (C16) or both stearyl (C18).
- the alkyl groups are saturated.
- PEG is a polyethylene glycol having an average molecular weight ranging of about 550 daltons to about 10,000 daltons, more preferably of about 750 daltons to about 5,000 daltons, more preferably of about 1,000 daltons to about 5,000 daltons, more preferably of about 1,500 daltons to about 3,000 daltons and, even more preferably, of about 2,000 daltons, or about 750 daltons.
- the PEG can be optionally substituted with alkyl, alkoxy, acyl or aryl.
- the terminal hydroxyl group is substituted with a methoxy or methyl group.
- L is a non-ester containing linker moiety or an ester containing linker moiety.
- L is a non-ester containing linker moiety.
- Suitable non-ester containing linkers include, but are not limited to, an amido linker moiety, an amino linker moiety, a carbonyl linker moiety, a carbamate linker moiety, a urea linker moiety, an ether linker moiety, a disulphide linker moiety, a succinamidyl linker moiety and combinations thereof.
- the non-ester containing linker moiety is a carbamate linker moiety (i.e., a PEG-C-DAA conjugate). In another preferred embodiment, the non-ester containing linker moiety is an amido linker moiety (Le., a PEG-A-DAA conjugate). In a preferred embodiment, the non- ester containing linker moiety is a succinamidyl linker moiety (i.e., a PEG-S-DAA conjugate).
- the PEG-DAA conjugates are synthesized using standard techniques and reagents known to those of skill in the art. It will be recognized that the PEG-DAA conjugates will contain various amide, amine, ether, thio, carbamate and urea linkages. T hose of skill in the art will recognize that methods and reagents for forming these bonds are well known and readily available. See, e.g., March, ADVANCED ORGANIC CHEMISTRY (Wiley 1992), Larock, COMPREHENSIVE ORGANIC TRANSFORMATIONS (VCH 1989); and Furniss, VOGEL'S TEXTBOOK OF PRACTICAL ORGANIC CHEMISTRY 5th ed. (Longman 1989).
- the PEG-DAA conjugate is a dilauryloxypropyl (C12)-PEG conjugate, dimyristyloxypropyl (C14)-PEG conjugate, a dipalmitoyloxypropyl (C16)-PEG conjugate or a disteryloxypropyl (C18)-PEG conjugate.
- C12 dimyristyloxypropyl
- C14 dimyristyloxypropyl
- C18 disteryloxypropyl
- hydrophilic polymers can be used in place of PEG.
- suitable polymers include, but are not limited to, polyvinylpyrrolidone, polymethyloxazoline, polyethyloxazoline, polyhydroxypropyl methacrylamide, polymethacrylamide and polydimethylacrylamide, polylactic acid, polyglycolic acid, and derivatized celluloses, such as hydroxymethylcellulose or hydroxyethylcellulose.
- the SNALPs and SPLPs of the present invention can further comprise cationic poly(ethylene glycol) (PEG) lipids, or CPLs, that have been designed for insertion into lipid bilayers to impart a positive charge ⁇ ee, Chen, et al, Bioconj. Chem. 11:433-437 (2000)).
- PEG poly(ethylene glycol)
- CPLs cationic poly(ethylene glycol) lipids
- Suitable SPLPs and SPLP-CPLs for use in the present invention, and methods of making and using SPLPs and SPLP-CPLs, are disclosed, e.g., in U.S. Patent No. 6,852,334 and WO 00/62813.
- Cationic polymer lipids useful in the present invention have the following architectural features: (1) a lipid anchor, such as a hydrophobic lipid, for incorporating the CPLs into the lipid bilayer; (2) a hydrophilic spacer, such as a polyethylene glycol, for linking the lipid anchor to a cationic head group; and (3) a polycationic moiety, such as a naturally occurring amino acid, to produce a protonizable cationic head group.
- Suitable CPL include compounds of Formula VII: A-W-Y (VII) wherein A, W and Y are as described below.
- A is a lipid moiety such as an amphipathic lipid, a neutral lipid or a hydrophobic lipid that acts as a lipid anchor.
- Suitable lipid examples include vesicle-forming lipids or vesicle adopting lipids and include, but are not limited to, diacylglycerolyls, dialkylglycerolyls, N-N-dialkylaminos, l,2-diacyloxy-3- aminopropanes and l,2-dialkyl-3-aminopropanes.
- W is a polymer or an oligomer, such as a hydrophilic polymer or oligomer.
- the hydrophilic polymer is a biocompatible polymer that is nonimmunogenic or possesses low inherent immunogenicity.
- the hydrophilic polymer can be weakly antigenic if used with appropriate adjuvants.
- Suitable nonimmunogenic polymers include, but are not limited to, PEG, polyamides, polylactic acid, polyglycolic acid, polylactic acid/polyglycolic acid copolymers and combinations thereof.
- the polymer has a molecular weight of about 250 to about 7000 daltons.
- "Y" is a polycationic moiety.
- polycationic moiety refers to a compound, derivative, or functional group having a positive charge, preferably at least 2 positive charges at a selected pH, preferably physiological pH.
- Suitable polycationic moieties include basic amino acids and their derivatives such as arginine, asparagine, glutamine, lysine and histidine; spermine; spermidine; cationic dendrimers; polyamines; polyamine sugars; and amino polysaccharides.
- the polycationic moieties can be linear, such as linear tetralysine, branched or dendrimeric in structure.
- Polycationic moieties have between about 2 to about 15 positive charges, preferably between about 2 to about 12 positive charges, and more preferably between about 2 to about 8 positive charges at selected pH values.
- the selection of which polycationic moiety to employ may be determined by the type of liposome application which is desired.
- the charges on the polycationic moieties can be either distributed around the entire liposome moiety, or alternatively, they can be a discrete concentration of charge density in one particular area of the liposome moiety e.g. , a charge spike. If the charge density is distributed on the liposome, the charge density can be equally distributed or unequally distributed. All variations of charge distribution of the polycationic moiety are encompassed by the present invention.
- the lipid "A,” and the nonimmunogenic polymer “W,” can be attached by various methods and preferably, by covalent attachment. Methods known to those of skill in the art can be used for the covalent attachment of "A” and “W.” Suitable linkages include, but are not limited to, amide, amine, carboxyl, carbonate, carbamate, ester and hydrazone linkages. It will be apparent to those skilled in the art that "A” and “W” must have complementary functional groups to effectuate the linkage. The reaction of these two groups, one on the lipid and the other on the polymer, will provide the desired linkage.
- the polycationic moiety can have a ligand attached, such as a targeting ligand or a chelating moiety for complexing calcium.
- the cationic moiety maintains a positive charge.
- the ligand that is attached has a positive charge.
- Suitable ligands include, but are not limited to, a compound or device with a reactive functional group and include lipids, amphipathic lipids, carrier compounds, bioaffinity compounds, biomaterials, biopolymers, biomedical devices, analytically detectable compounds, therapeutically active compounds, enzymes, peptides, proteins, antibodies, immune stimulators, radiolabels, fluorogens, biotin, drugs, haptens, DNA, RNA, polysaccharides, liposomes, virosomes, micelles, immunoglobulins, functional groups, other targeting moieties, or toxins.
- the nucleic acid component of the present invention comprises an interfering RNA that silences (e.g., partially or completely inhibits) expression of a gene of interest.
- An interfering RNA can be provided in several forms.
- an interfering RNA can be provided as one or more isolated small-interfering RNA (siRNA) duplexes, longer double-stranded RNA (dsRNA) or as siRNA or dsRNA transcribed from a transcriptional cassette in a DNA plasmid.
- siRNA small-interfering RNA
- dsRNA double-stranded RNA
- siRNA siRNA or dsRNA transcribed from a transcriptional cassette in a DNA plasmid.
- the interfering RNA can be administered alone or in combination with the administration of conventional agents used to treat the disease or disorder associated with the gene of interest.
- Genes of interest include, but are not limited to, genes associated with viral infection and survival, genes associated with liver and kidney diseases and disorders, genes associated with tumorigenesis and cell transformation, angiogenic genes, immunomodulator genes, such as those associated with inflammatory and autoimmune responses, ligand receptor genes, and genes associated with neurodegenerative disorders. 1. Selecting siRNA sequences
- Suitable siRNA sequences can be identified using any means known in the art. Typically, the methods described in Elbashir, et al, Nature 411:494-498 (2001) and Elbashir, et al, EMBO J 20: 6877-6888 (2001) are combined with rational design rules set forth in Reynolds et al, Nature Biotech. 22(3):326-330 (2004).
- the sequence within about 50 to about 100 nucleotides 3 ' of the AUG start codon of a transcript from the target gene of interest is scanned for dinucleotide sequences (e.g., AA, CC, GG, or UU) (see, e.g., Elbashir, et al, EMBO J 20: 6877-6888 (2001)).
- the nucleotides immediately 3' to the dinucleotide sequences are identified as potential siRNA target sequences.
- the 19, 21, 23, 25, 27, 29, 31, 33, 35 or more nucleotides immediately 3' to the dinucleotide sequences are identified as potential siRNA target sites.
- the dinucleotide sequence is an AA sequence and the 19 nucleotides immediately 3' to the AA dinucleotide are identified as a potential siRNA target site.
- siRNA target sites are spaced at different positions along the length of the target gene.
- potential siRNA target sites may be further analyzed to identify sites that do not contain regions of homology to other coding sequences. For example, a suitable siRNA target site of about 21 base pairs typically will not have more than 16-17 contiguous base pairs of homology to other coding sequences. If the siRNA sequences are to be expressed from an RNA Pol III promoter, siRNA target sequences lacking more than 4 contiguous A's or T's are selected.
- siRNA sequences complementary to the siRNA target sites may be designed.
- the siRNA sequences may also be analyzed by a rational design algorithm to identify sequences that have one or more of the following features: (1) G/C content of about 25% to about 60% G/C; (2) at least 3 A/Us at positions 15-19 of the sense strand; (3) no internal repeats; (4) an A at position 19 of the sense strand; (5) an A at position 3 of the sense strand; (6) a U at position 10 of the sense strand; (7) no G/C at position 19 of the sense strand; and (8) no G at position 13 of the sense strand.
- siRNA design tools that incorporate algorithms that assign suitable values of each of these features and are useful for selection of siRNA can be found at, e.g., http://boz094.ust.hk/RNAi/siRNA.
- the sequence is analyzed for the presence or absence of immunostimulatory motifs (e.g., GU- rich motifs) as described in, e.g., co-pending U.S. Provisional Patent Application Nos. 60/585301, filed July 2, 2004; 60/589363, filed July 19, 2004; 60/627326, filed November 12, 2004; and 60/665297, filed March 25, 2005.
- the immunostimulatory siRNA molecules can be modified to increase or decrease their immunostimulatory properties and the non-immunostimulatory molecules can be modified so that they possess immunostimulatory properties Generating siRNA
- siRNA can be provided in several forms including, e.g. , as one or more isolated small-interfering RNA (siRNA) duplexes, longer double-stranded RNA (dsRNA) or as siRNA or dsRNA transcribed from a transcriptional cassette in a DNA plasmid.
- siRNA may also be chemically synthesized.
- the synthesized or transcribed siRNA have 3' overhangs of about 1-4 nucleotides, preferably of about 2-3 nucleotides and 5' phosphate termini.
- the siRNA sequences may have overhangs (e.g., 3' or 5' overhangs as described in (Elbashir, et al, Genes Dev.
- RNA population can be used to provide long precursor RNAs, or long precursor RNAs that have substantial or complete identity to a selected target sequence can be used to make the siRNA.
- the RNAs can be isolated from cells or tissue, synthesized, and/or cloned according to methods well known to those of skill in the art.
- the RNA can be a mixed population (obtained from cells or tissue, transcribed from cDNA, subtracted, selected, etc.), or can represent a single target sequence.
- RNA can be naturally occurring (e.g., isolated from tissue or cell samples), synthesized in vitro (e.g., using T7 or SP6 polymerase and PCR products or a cloned cDNA); or chemically synthesized.
- the complement is also transcribed in vitro and hybridized to form a dsRNA.
- the RNA complements are also provided (e.g., to form dsRNA for digestion by E. coli RNAse III or Dicer), e.g., by transcribing cDNAs corresponding to the RNA population, or by using RNA polymerases.
- the precursor RNAs are then hybridized to form double stranded RNAs for digestion.
- the dsRNAs can be directly administered to a subject or can be digested in vitro prior to administration.
- siRNA can be transcribed as sequences that automatically fold into duplexes with hairpin loops from DNA templates in plasmids having RNA polymerase III transcriptional units, for example, based on the naturally occurring transcription units for small nuclear RNA U6 or human RNase P RNA HI (see,
- a transcriptional unit or cassette will contain an RNA transcript promoter sequence, such as an HI -RNA or a U6 promoter, operably linked to a template for transcription of a desired siRNA sequence and a termination sequence, comprised of 2-3 uridine residues and a polythymidine (T5) sequence (polyadenylation signal) (Brummelkamp, Science, supra).
- an RNA transcript promoter sequence such as an HI -RNA or a U6 promoter
- the selected promoter can provide for constitutive or inducible transcription.
- Compositions and methods for DNA-directed transcription of RNA interference molecules is described in detail in U.S. Patent No. 6,573,099.
- the transcriptional unit is incorporated into a plasmid or DNA vector from which the interfering RNA is transcribed. Plasmids suitable for in vivo delivery of genetic material for therapeutic purposes are described in detail in U.S. Patent Nos. 5,962,428 and 5,910,488.
- the selected plasmid can provide for transient or stable delivery of a target cell. It will be apparent to those of skill in the art that plasmids originally designed to express desired gene sequences can be modified to contain a transcriptional unit cassette for transcription of siRNA.
- RNA, synthesizing RNA, hybridizing nucleic acids, making and screening cDNA libraries, and performing PCR are well known in the art (see, e.g., Gubler & Hoffman, Gene 25:263-269 (1983); Sambrook et al, supra; Ausubel et al, supra), as are PCR methods (see U.S. Patents 4,683,195 and 4,683,202; PCR Protocols: A Guide to Methods and Applications (Innis et al., eds, 1990)).
- Expression libraries are also well known to those of skill in the art.
- a suitable plasmid is engineered to contain, in expressible form, a template sequence that encodes a partial length sequence or an entire length sequence of a gene product of interest. Template sequences can also be used for providing isolated or synthesized siRNA and dsRNA. Generally, it is desired to downregulate or silence the transcription and translation of a gene product of interest.
- Genes of interest include, but are not limited to, genes associated with viral infection and survival, genes associated with metabolic diseases and disorders (e.g., liver diseases and disorders), genes associated with tumorigenesis and cell transformation, angiogenic genes, immunomodulator genes, such as those associated with inflammatory and autoimmune responses, ligand receptor genes, and genes associated with neurodegenerative disorders.
- Genes associated with viral infection and survival include those expressed by a virus in order to bind, enter and replicate in a cell.
- viral sequences associated with chronic viral diseases include sequences of Hepatitis viruses (Hamasaki, et al, FEBS Lett. 543:51 (2003); Yokota, et al, EMBO Rep. 4:602 (2003); Schlomai, et al, Hepatology 37:764 (2003); Wilson, et al, Proc. Natl. Acad. Sci. 100:2783 (2003); Kapadia, et al, Proc. Natl. Acad. Sci. 100:2014 (2003); and FIELDS VIROLOGY (Knipe et al. eds. 2001)), Human
- HIV Immunodeficiency Virus
- Herpes viruses Jia, et al, J. Virol. 77:3301 (2003)
- HPV Human Papilloma Viruses
- Exemplary hepatitis viral nucleic acid sequences that can be silenced include, but are not limited to: nucleic acid sequences involved in transcription and translation (e.g., Enl, En2, X, P), nucleic acid sequences encoding structural proteins (e.g., core proteins including C and C-related proteins; capsid and envelope proteins including S, M, and/or L proteins, or fragments thereof) (see, e.g., FIELDS VIROLOGY, 2001, supra).
- Exemplary Hepatitis C nucleic acid sequences that can be silenced include, but are not limited to: serine proteases (e.g., NS3/NS4), helicases (e.g.
- Hepatitis A nucleic acid sequences are set forth in e.g., Genbank Accession No. NC_001489 ; Hepatitis B nucleic acid sequences are set forth in, e.g., Genbank Accession No. NC_003977; Hepatitis C nucleic acid sequences are set forth in, e.g., Genbank Accession No. NC_004102; Hepatitis D nucleic acid sequence are set forth in, e.g., Genbank Accession No.
- NC_001653 Hepatitis E nucleic acid sequences are set forth in e.g., Genbank Accession No. NC_001434;. and Hepatitis G nucleic acid sequences are set forth in e.g., Genbank Accession No. NC_001710.
- Genes associated with metabolic diseases and disorders include, for example genes expressed in, for example, dyslipidemia (e.g., liver X receptors (e.g., LXR ⁇ and LXR ⁇ Genbank Accession No. NM_007121), farnesoid X receptors (FXR) (Genbank Accession No.
- dyslipidemia e.g., liver X receptors (e.g., LXR ⁇ and LXR ⁇ Genbank Accession No. NM_007121)
- FXR farnesoid X receptors
- NM_005123 sterol-regulatory element binding protein (SREBP), Site-1 protease (S IP), 3-hydroxy-3-methylglutaryl coenzyme-A reductase (HMG coenzyme-A reductase), Apolipoprotein (ApoB), and Apolipoprotein (ApoE)) and diabetes (e.g., Glucose 6- phosphatase) (see, e.g., Forman et al, Cell 81:687 (1995); Seol et al, Mol. Endocrinol.
- genes associated with metabolic diseases and disorders include genes that are expressed in the liver itself as well as and genes expressed in other organs and tissues.
- Examples of gene sequences associated with tumorigenesis and cell transformation include translocation sequences such as MLL fusion genes, BCR-ABL (Wilda, et al, Oncogene, 21:5716 (2002); Scherr, et al, Blood 101:1566), TEL-AML1, EWS-FLI1, TLS-FUS, PAX3-FKHR, BCL-2, AML1-ETO and AML1-MTG8
- Angiogenic genes are able to promote the formation of new vessels. Of particular interest is Vascular Endothelial Growth Factor (VEGF) (Reich, et al, Mol. Vis. 9:210 (2003)) or VEGFr. siRNA sequences that target VEGFr are set forth in, e.g., GB 2396864; U.S. Patent Publication No. 20040142895; and CA2456444.
- VEGF Vascular Endothelial Growth Factor
- Anti-angiogenic genes are able to inhibit neovascularization. These genes are particularly useful for treating those cancers in which angiogenesis plays a role in the pathological development of the disease.
- anti-angiogenic genes include, but are not limited to, endostatin (see e.g., U.S. Patent No. 6,174,861), angiostatin (see, e.g., U.S. Patent No. 5,639,725), and VEGF-R2 (see e.g., Decaussin et al. (1999) J. Pathol 188(4): 369-737).
- endostatin see e.g., U.S. Patent No. 6,174,861
- angiostatin see, e.g., U.S. Patent No. 5,639,725
- VEGF-R2 see e.g., Decaussin et al. (1999) J. Pathol 188(4): 369-737.
- Immunomodulator genes are genes that modulate one or more immune responses.
- immunomodulator genes include cytokines such as growth factors (e.g., TGF- ⁇ ., TGF- ⁇ , EGF, FGF, IGF, NGF, PDGF, CGF, GM-CSF, SCF, etc.), interleukins (e.g., IL-2, IL-3, IL-4, IL-6, IL-7, IL-10, IL-12, IL-15, E -20, etc.), interferons (e.g. , IFN- ⁇ , IFN- ⁇ , IFN- ⁇ , etc.), TNF (e.g. , TNF- ), and Flt3-Ligand.
- growth factors e.g., TGF- ⁇ ., TGF- ⁇ , EGF, FGF, IGF, NGF, PDGF, CGF, GM-CSF, SCF, etc.
- interleukins e.g., IL-2, IL-3, IL-4
- Fas and Fas Ligand genes are also immunomodulator target sequences of interest (Song, et al, Nat. Med. 9:347 (2003)).
- Genes encoding secondary signaling molecules in hematopoietic and lymphoid cells are also included in the present invention, for example, Tec family kinases, such as Bruton's tyrosine kinase (Btk) (Heinonen, et al, FEBS Lett. 527:274 (2002)).
- Btk Bruton's tyrosine kinase
- Cell receptor ligands include ligands that are able to bind to cell surface receptors (e.g., insulin receptor, EPO receptor, G-protein coupled receptors, receptors with tyrosine kinase activity, cytokine receptors, growth factor receptors, etc.), to modulate (e.g,. inhibit, activate, etc.) the physiological pathway that the receptor is involved in (e.g., glucose level modulation, blood cell development, mitogenesis, etc.).
- cell receptor ligands include cytokines, growth factors, interleukins, interferons, erythropoietin (EPO), insulin, glucagon, G-protein coupled receptor ligands, etc.).
- Templates coding for an expansion of trinucleotide repeats find use in silencing pathogenic sequences in neurodegenerative disorders caused by the expansion of trinucleotide repeats, such as spinobulbular muscular atrophy and Huntington's Disease (Caplen, et al, Hum. Mol. Genet. 11:175 (2002)).
- Tumor suppressor genes are genes that are able to inhibit the growth of a cell, particularly tumor cells. Thus, delivery of these genes to tumor cells is useful in the treatment of cancers.
- Tumor suppressor genes include, but are not limited to, p53 (Lamb et al, Mol. Cell Biol 6:1379-1385 (1986), Ewen et al, Science 255:85-87 (1992), Ewen etal. (1991) Cell 66:1155-1164, and Hu et al, EMBO J. 9:1147-1155 (1990)), RBI (Toguchida etal. (1993) Genomics 17:535-543), WT1 (Hastie, N. D., Curr. Opin. Genet. Dev. 3:408-413 (1993)), NF1 (Trofatter et al, Cell 72:791-800 (1993), Cawthon et al,
- VHL Longif et al, Science 260:1317-1320 (1993)
- APC Gaden et al, Cell 66:589-600 (1991)
- DAP kinase see e.g., Diess et al (1995) Genes Dev. 9: 15-30
- pl6 see e.g., Marx (1994) Science 264(5167): 1846
- ARF see e.g., Jo et al. (1995) Cell 83(6): 993-1000
- Neurofibromin see e.g., Huynh et al (1992) Neurosci. Lett. 143(1-2): 233-236
- PTEN see e.g., Li et al. (1997) Science 275(5308): 1943-1947).
- the present invention provides a method of preparing serum-stable nucleic acid- lipid particles in which the plasmid or other nucleic acid is encapsulated in a lipid bilayer and is protected from degradation.
- the particles made by the methods of this invention typically have a size of about 50 nm to about 150 nm, more typically about 100 nm to about 130 nm, most typically about 110 nm to about 115 nm.
- the particles can be formed by any method known in the art including, but not limited to: a continuous mixing method, a detergent dialysis method, or a modification of a reverse-phase method which utilizes organic solvents to provide a single phase during mixing of the components.
- the cationic lipids are lipids of Formula I and II or combinations thereof.
- the noncationic lipids are ESM, DOPE, DOPC, DPPE, DMPE, 16:0 Monomethyl Phosphatidylethanolamine, 16:0 Dimethyl Phosphatidylethanolamine, 18:1 Trans Phosphatidylethanolamine, 18:0 18:1 Phosphatidylethanolamine (SOPE), 16:0 18:1 Phosphatidylethanolamine, DSPE, polyethylene glycol-based polymers (e.g., PEG 2000, PEG 5000, PEG-modified diacylglycerols, or PEG-modified dialkyloxypropyls), distearoylphosphatidylcholine (DSPC), cholesterol, or combinations thereof.
- polyethylene glycol-based polymers e.g., PEG 2000, PEG 5000, PEG-modified diacylglycerols, or PEG-modified
- the organic solvents are methanol, chloroform, methylene chloride, ethanol, diethyl ether or combinations thereof.
- the nucleic acid is a plasmid; the cationic lipid is a lipid of Formula I or II or combinations thereof; the noncationic lipid is ESM, DOPE, PEG-DAAs, distearoylphosphatidylcholine (DSPC), cholesterol, or combinations thereof (e.g. DSPC and PEG-DAAs); and the organic solvent is methanol, chloroform, methylene chloride, ethanol, diethyl ether or combinations thereof.
- the present invention provides for nucleic acid-lipid particles produced via a continuous mixing method, e.g., process that includes providing an aqueous solution comprising a nucleic acid such as an siRNA or a plasmid, in a first reservoir, and providing an organic lipid solution in a second reservoir, and mixing the aqueous solution with the organic lipid solution such that the organic lipid solution mixes with the aqueous solution so as to substantially instantaneously produce a liposome encapsulating the nucleic acid (e.g., siRNA).
- a continuous mixing method e.g., process that includes providing an aqueous solution comprising a nucleic acid such as an siRNA or a plasmid, in a first reservoir, and providing an organic lipid solution in a second reservoir, and mixing the aqueous solution with the organic lipid solution such that the organic lipid solution mixes with the aqueous solution so as to substantially instantaneously produce a liposome encapsulating the nucleic acid (e
- the serum-stable nucleic acid-lipid particles formed using the continuous mixing method typically have a size of from about 50 nm to about 150 nm, more typically about 100 nm to about 130 nm, most typically about 110 nm to about 115 nm.
- the particles thus formed do not aggregate and are optionally sized to achieve a uniform particle size.
- the particles are formed using detergent dialysis.
- a plasmid or other nucleic acid e.g., siRNA
- a detergent solution of cationic lipids to form a coated nucleic acid complex.
- coated nucleic acids can aggregate and precipitate.
- the presence of a detergent reduces this aggregation and allows the coated nucleic acids to react with excess lipids (typically, non-cationic lipids) to form particles in which the plasmid or other nucleic acid is encapsulated in a lipid bilayer.
- the present invention provides a method for the preparation of serum-stable nucleic acid-lipid particles, comprising: (a) combining a nucleic acid with cationic lipids in a detergent solution to form a coated nucleic acid-lipid complex; (b) contacting non-cationic lipids with the coated nucleic acid-lipid complex to form a detergent solution comprising a nucleic acid-lipid complex and non-cationic lipids; and (c) dialyzing the detergent solution of step (b) to provide a solution of serum- stable nucleic acid-lipid particles, wherein the nucleic acid is encapsulated in a lipid bilayer and the particles are serum-stable and have a size of from about 50 to about 150 nm.
- An initial solution of coated nucleic acid-lipid complexes is formed by combining the nucleic acid with the cationic lipids in a detergent solution.
- the detergent solution is preferably an aqueous solution of a neutral detergent having a critical micelle concentration of 15-300 mM, more preferably 20-50 mM.
- suitable detergents include, for example, N,N'-((octanoylimino)- bis-(trimethylene))-bis-(D-gluconamide) (BIGCHAP); BRIJ 35; Deoxy-BIGCHAP; dodecylpoly(ethylene glycol) ether; Tween 20; Tween 40; Tween 60; Tween 80; Tween 85; Mega 8; Mega 9; Zwittergent® 3-08; Zwittergent® 3-10; Triton X-405; hexyl-, heptyl-, octyl- and nonyl- ⁇ -D-glucopyranoside; and heptylthioglucopyranoside; with octyl ⁇ -D-glucopyranoside and Tween-20 being the most preferred.
- the concentration of detergent in the detergent solution is typically about 100 mM to about 2 M, preferably from about 200 mM to about 1.5 M.
- the cationic lipids and nucleic acids will typically be combined to produce a charge ratio (+/-) of about 1:1 to about 20:1, preferably in a ratio of about 1:1 to about 12:1, and more preferably in a ratio of about 2:1 to about 6:1. Additionally, the overall concentration of nucleic acid in solution will typically be from about 25 ⁇ g/mL to about 1 mg/mL, preferably from about 25 ⁇ g/mL to about 200 ⁇ g/mL, and more preferably from about 50 ⁇ g/mL to about 100 ⁇ g/mL.
- the combination of nucleic acids and cationic lipids in detergent solution is kept, typically at room temperature, for a period of time which is sufficient for the coated complexes to form.
- the nucleic acids and cationic lipids can be combined in the detergent solution and warmed to temperatures of up to about 37°C.
- the coated complexes can be formed at lower temperatures, typically down to about 4°C.
- the nucleic acid to lipid ratios (mass/mass ratios) in a formed nucleic acid-lipid particle will range from about 0.01 to about 0.08. The ratio of the starting materials also falls within this range because the purification step typically removes the unencapsulated nucleic acid as well as the empty liposomes.
- the nucleic acid-lipid particle preparation uses about 400 ⁇ g nucleic acid per 10 mg total lipid or a nucleic acid to lipid ratio of about 0.01 to about 0.08 and, more preferably, about 0.04, which corresponds to 1.25 mg of total lipid per 50 ⁇ g of nucleic acid.
- the detergent solution of the coated nucleic acid-lipid complexes is then contacted with non-cationic lipids to provide a detergent solution of nucleic acid-lipid complexes and non-cationic lipids.
- the non-cationic lipids which are useful in this step include, diacylphosphatidylcholine, diacylphosphatidylethanolamine, ceramide, sphingomyelin, cephalin, cardiolipin, and cerebrosides.
- the non-cationic lipids are diacylphosphatidylcholine, diacylphosphatidylethanolamine, ceramide or sphingomyelin.
- the acyl groups in these lipids are preferably acyl groups derived from fatty acids having C10-C24 carbon chains. More preferably the acyl groups are lauroyl, myristoyl, palmitoyl, stearoyl or oleoyl.
- the non-cationic lipid will be 1,2-sr ⁇ -dioleoylphosphatidylemanolamine (DOPE), palmitoyl oleoyl phosphatidylcholine (POPC), egg phosphatidylcholine (EPC), distearoylphosphatidylcholine (DSPC), cholesterol, or a mixture thereof.
- DOPE 1,2-sr ⁇ -dioleoylphosphatidylemanolamine
- POPC palmitoyl oleoyl phosphatidylcholine
- EPC egg phosphatidylcholine
- DSPC distearoylphosphatidylcholine
- cholesterol or a mixture thereof.
- the nucleic acid-lipid particles will be fusogenic particles with enhanced properties in vivo and the non-cationic lipid will be DSPC or DOPE.
- the nucleic acid-lipid particles of the present invention may further comprise cholesterol.
- the non-cationic lipids will further comprise polyethylene glycol-based polymers such as PEG 2000, PEG 5000 and polyethylene glycol conjugated to a diacylglycerol, a ceramide or a phospholipid, as described in U.S. Patent No. 5,820,873 and U.S. Patent Publication No. 20030077829.
- the non-cationic lipids will further comprise polyethylene glycol-based polymers such as PEG 2000, PEG 5000 and polyethylene glycol conjugated to a dialkyloxypropyl.
- the amount of non-cationic lipid which is used in the present methods is typically about 2 to about 20 mg of total lipids to 50 ⁇ g of nucleic acid. Preferably the amount of total lipid is from about 5 to about 10 mg per 50 ⁇ g of nucleic acid.
- the detergent is removed, preferably by dialysis.
- the removal of the detergent results in the formation of a lipid-bilayer which surrounds the nucleic acid providing serum-stable nucleic acid-lipid particles which have a size of from about 50 nm to about 150 nm, more typically about 100 nm to about 130 nm, most typically about 110 nm to about 115 nm.
- the particles thus formed do not aggregate and are optionally sized to achieve a uniform particle size.
- the serum-stable nucleic acid-lipid particles can be sized by any of the methods available for sizing liposomes. The sizing may be conducted in order to achieve a desired size range and relatively narrow distribution of particle sizes.
- Extrusion of the particles through a small-pore polycarbonate membrane or an asymmetric ceramic membrane is also an effective method for reducing particle sizes to a relatively well-defined size distribution.
- the suspension is cycled through the membrane one or more times until the desired particle size distribution is achieved.
- the particles may be extruded through successively smaller-pore membranes, to achieve a gradual reduction in size.
- the present invention provides a method for the preparation of serum-stable nucleic acid-lipid particles, comprising: (a) preparing a mixture comprising cationic lipids and non-cationic lipids in an organic solvent; (b) contacting an aqueous solution of nucleic acid with said mixture in step (a) to provide a clear single phase; and (c) removing said organic solvent to provide a suspension of nucleic acid-lipid particles, wherein said nucleic acid is encapsulated in a lipid bilayer, and said particles are stable in serum and have a size of from about 50 to about 150 nm.
- nucleic acids or plasmids
- cationic lipids or non-cationic lipids which are useful in this group of embodiments are as described for the detergent dialysis methods above.
- organic solvent which is also used as a solubilizing agent, is in an amount sufficient to provide a clear single phase mixture of nucleic acid and lipids.
- Suitable solvents include, but are not limited to, chloroform, dichloromethane, diethylether, cyclohexane, cyclopentane, benzene, toluene, methanol, or other aliphatic alcohols such as propanol, isopropanol, butanol, tert-butanol, iso-butanol, pentanol and hexanol.
- Combinations of two or more solvents may also be used in the present invention.
- Contacting the nucleic acid with the organic solution of cationic and non-cationic lipids is accomplished by mixing together a first solution of nucleic acid, which is typically an aqueous solution, and a second organic solution of the lipids.
- a first solution of nucleic acid which is typically an aqueous solution
- a second organic solution of the lipids One of skill in the art will understand that this mixing can take place by any number of methods, for example by mechanical means such as by using vortex mixers.
- the organic solvent is removed, thus forming an aqueous suspension of serum-stable nucleic acid-lipid particles.
- the methods used to remove the organic solvent will typically involve evaporation at reduced pressures or blowing a stream of inert gas (e.g., nitrogen or argon) across the mixture.
- the serum-stable nucleic acid-lipid particles thus formed will typically be sized from about 50 nm to about 150 nm, more typically about 100 nm to about 130 nm, most typically about 110 nm to about 115 nm. To achieve further size reduction or homogeneity of size in the particles, sizing can be conducted as described above.
- the methods will further comprise adding nonlipid polycations which are useful to effect the delivery to cells using the present compositions.
- nonlipid polycations examples include, but are limited to, hexadimethrine bromide (sold under the brand name POLYBRENE®, from Aldrich Chemical Co.,
- Suitable polycations include, for example, salts of poly-L-ornithine, poly-L-arginine, poly-L-lysine, poly-D-lysine, polyallylamine and polyethyleneimine.
- the formation of the nucleic acid-lipid particles can be carried out either in a mono-phase system (e.g., a Bligh and Dyer monophase or similar mixture of aqueous and organic solvents) or in a two-phase system with suitable mixing.
- a mono-phase system e.g., a Bligh and Dyer monophase or similar mixture of aqueous and organic solvents
- a two-phase system with suitable mixing.
- the cationic lipids and nucleic acids are each dissolved in a volume of the mono-phase mixture. Combination of the two solutions provides a single mixture in which the complexes form.
- the complexes can form in two-phase mixtures in which the cationic lipids bind to the nucleic acid (which is present in the aqueous phase), and
- the present invention provides a method for the preparation of nucleic acid-lipid particles, comprising: (a) contacting nucleic acids with a solution comprising non-cationic lipids and a detergent to form a nucleic acid-lipid mixture; (b) contacting cationic lipids with the nucleic acid-lipid mixture to neutralize a portion of the negative charge of the nucleic acids and form a charge-neutralized mixture of nucleic acids and lipids; and (c) removing the detergent from the charge-neutralized mixture to provide the nucleic acid-lipid particles in which the nucleic acids are protected from degradation.
- the solution of non-cationic lipids and detergent is an aqueous solution.
- Contacting the nucleic acids with the solution of non-cationic lipids and detergent is typically accomplished by mixing together a first solution of nucleic acids and a second solution of the lipids and detergent.
- this mixing can take place by any number of methods, for example, by mechanical means such as by using vortex mixers.
- the nucleic acid solution is also a detergent solution.
- the amount of non-cationic lipid which is used in the present method is typically determined based on the amount of cationic lipid used, and is typically of from about 0.2 to 5 times the amount of cationic lipid, preferably from about 0.5 to about 2 times the amount of cationic lipid used.
- the nucleic acids are precondensed as described in, e.g., U.S. Patent Application No. 09/744,103.
- the nucleic acid-lipid mixture thus formed is contacted with cationic lipids to neutralize a portion of the negative charge which is associated with the nucleic acids (or other polyanionic materials) present.
- the amount of cationic lipids used will typically be sufficient to neutralize at least 50 % of the negative charge of the nucleic acid.
- the negative charge will be at least 70 % neutralized, more preferably at least 90 % neutralized.
- Cationic lipids which are useful in the present invention include, for example, DLinDMA and, DLenDMA.
- lipids and related analogs have been described in U.S. Provisional Patent Application Nos. 60/578,075, filed June 7, 2004; 60/610,746, filed September 17, 2004; and 60/679,427, filed May 9, 2005.
- Contacting the cationic lipids with the nucleic acid-lipid mixture can be accomplished by any of a number of techniques, preferably by mixing together a solution of the cationic lipid and a solution containing the nucleic acid-lipid mixture. Upon mixing the two solutions (or contacting in any other manner), a portion of the negative charge associated with the nucleic acid is neutralized. Nevertheless, the nucleic acid remains in an uncondensed state and acquires hydrophilic characteristics.
- the detergent (or combination of detergent and organic solvent) is removed, thus forming the nucleic acid-lipid particles.
- the methods used to remove the detergent will typically involve dialysis.
- organic solvents are present, removal is typically accomplished by evaporation at reduced pressures or by blowing a stream of inert gas (e.g. , nitrogen or argon) across the mixture.
- inert gas e.g. , nitrogen or argon
- the particles thus formed will typically be sized from about 50 nm to several microns, more typically about 50 nm to about 150 nm, even more typically about 100 nm to about 130 nm, most typically about 110 nm to about 115 nm.
- the nucleic acid-lipid particles can be sonicated, filtered or subjected to other sizing techniques which are used in liposomal formulations and are known to those of skill in the art.
- the methods will further comprise adding nonlipid polycations which are useful to effect the lipofection of cells using the present compositions.
- suitable nonlipid polycations include, hexadimethrine bromide (sold under the brand name POLYBRENE®, from Aldrich Chemical Co., Milwaukee, Wisconsin, USA) or other salts of hexadimethrine.
- suitable polycations include, for example, salts of poly-L-ornithine, poly-L-arginine, poly-L-lysine, poly-D-lysine, polyallylamine and polyethyleneimine. Addition of these salts is preferably after the particles have been formed.
- the present invention provides methods for the preparation of nucleic acid-lipid particles, comprising: (a) contacting an amount of cationic lipids with nucleic acids in a solution; the solution comprising from about 15-35 % water and about 65-85 % organic solvent and the amount of cationic lipids being sufficient to produce a +/- charge ratio of from about 0.85 to about 2.0, to provide a hydrophobic nucleic acid-lipid complex; (b)contacting the hydrophobic, nucleic acid-lipid complex in solution with noncationic lipids, to provide a nucleic acid-lipid mixture; and (c)removing the organic solvents from the nucleic acid-lipid mixture to provide nucleic acid-lipid particles in which the nucleic acids are protected from degradation.
- nucleic acids, non-cationic lipids, cationic lipids and organic solvents which are useful in this aspect of the invention are the same as those described for the methods above which used detergents.
- the solution of step (a) is a mono-phase. In another group of embodiments, the solution of step (a) is two-phase.
- the non-cationic lipids are ESM, DOPE, DOPC, polyethylene glycol-based polymers (e.g., PEG 2000, PEG 5000, PEG-modified diacylglycerols, or PEG-modified dialkyloxypropyls), distearoylphosphatidylcholine (DSPC), DPPE, DMPE, 16:0 Monomethyl Phosphatidylethanolamine, 16:0 Dimethyl Phosphatidylethanolamine, 18:1 Trans Phosphatidylethanolamine, 18:0 18:1 Phosphatidylethanolamine (SOPE), 16:0 18:1 Phosphatidylethanolamine, DSPE, cholesterol, or combinations thereof.
- the organic solvents are methanol, chloroform, methylene chloride, ethanol, diethyl ether or combinations thereof.
- the nucleic acid is a plasmid from which an interfering RNA is transcribed;
- the cationic lipid is DLindMA, DLenDMA, DODAC, DDAB, DOTMA, DOSPA, DMRIE, DOGS or combinations thereof;
- the non-cationic lipid is ESM, DOPE, DAG-PEGs, distearoylphosphatidylcholine (DSPC), DPPE, DMPE, 16:0 Monomethyl Phosphatidylethanolamine, 16:0 Dimethyl Phosphatidylethanolamine, 18:1 Trans Phosphatidylethanolamine, 18:0 18:1 Phosphatidylethanolamine (SOPE), 16:0 18:1 Phosphatidylethanolamine DSPE, cholesterol, or combinations thereof (e.g. DSPC and PEG-DAA); and the organic solvent is methanol, chloroform, methylene chloride, ethanol, diethyl
- contacting the nucleic acids with the cationic lipids is typically accomplished by mixing together a first solution of nucleic acids and a second solution of the lipids, preferably by mechanical means such as by using vortex mixers.
- the resulting mixture contains complexes as described above.
- These complexes are then converted to particles by the addition of non-cationic lipids and the removal of the organic solvent.
- the addition of the non-cationic lipids is typically accomplished by simply adding a solution of the non-cationic lipids to the mixture containing the complexes. A reverse addition can also be used. Subsequent removal of organic solvents can be accomplished by methods known to those of skill in the art and also described above.
- the amount of non-cationic lipids which is used in this aspect of the invention is typically an amount of from about 0.2 to about 15 times the amount (on a mole basis) of cationic lipids which was used to provide the charge-neutralized nucleic acid-lipid complex. Preferably, the amount is from about 0.5 to about 9 times the amount of cationic lipids used.
- the present invention provides nucleic acid-lipid particles which are prepared by the methods described above.
- the nucleic acid-lipid particles are either net charge neutral or carry an overall charge which provides the particles with greater gene lipofection activity.
- the nucleic acid component of the particles is a nucleic acid which interferes with the production of an undesired protein.
- the nucleic acid comprises an interfering RNA, the non-cationic lipid is egg sphingomyelin and the cationic lipid is DLinDMA or DLenDMA.
- the nucleic acid comprises an interfering RNA
- the noncationic lipid is a mixture of DSPC and cholesterol
- the cationic lipid is DLinDMA or DLenDMA.
- the non-cationic lipid may further comprise cholesterol.
- the post-insertion technique results in SNALPs having CPLs mainly in the external face of the SNALP bilayer membrane, whereas standard techniques provide SNALPs having CPLs on both internal and external faces.
- the method is especially useful for vesicles made from phospholipids (which can contain cholesterol) and also for vesicles containing PEG-lipids (such as PEG-DAAs and PEG-DAGs).
- PEG-lipids such as PEG-DAAs and PEG-DAGs.
- the serum-stable nucleic acid-lipid particles of the present invention are useful for the introduction of nucleic acids into cells. Accordingly, the present invention also provides methods for introducing a nucleic acids (e.g., a plasmid or and siRNA) into a cell. The methods are carried out in vitro or in vivo by first forming the particles as described above and then contacting the particles with the cells for a period of time sufficient for delivery of the nucleic acid to the cell to occur. [0178]
- the nucleic acid-lipid particles of the present invention can be adsorbed to almost any cell type with which they are mixed or contacted.
- the particles can either be endocytosed by a portion of the cells, exchange lipids with cell membranes, or fuse with the cells. Transfer or incorporation of the nucleic acid portion of the particle can take place via any one of these pathways. In particular, when fusion takes place, the particle membrane is integrated into the cell membrane and the contents of the particle combine with the intracellular fluid.
- the nucleic acid-lipid particles of the present invention can be administered either alone or in mixture with a physiologically-acceptable carrier (such as physiological saline or phosphate buffer) selected in accordance with the route of administration and standard pharmaceutical practice.
- a physiologically-acceptable carrier such as physiological saline or phosphate buffer
- physiological saline will be employed as the pharmaceutically acceptable carrier.
- suitable carriers include, e.g., water, buffered water, 0.4% saline, 0.3% glycine, and the like, including glycoproteins for enhanced stability, such as albumin, lipoprotein, globulin, etc.
- the pharmaceutical carrier is generally added following particle formation. Thus, after the particle is formed, the particle can be diluted into pharmaceutically acceptable carriers such as normal saline.
- the concentration of particles in the pharmaceutical formulations can vary widely, i.e., from less than about 0.05%, usually at or at least about 2-5% to as much as 10 to 30% by weight and will be selected primarily by fluid volumes, viscosities, etc., in accordance with the particular mode of administration selected.
- the concentration may be increased to lower the fluid load associated with treatment. This may be particularly desirable in patients having atherosclerosis-associated congestive heart failure or severe hypertension.
- particles composed of irritating lipids may be diluted to low concentrations to lessen inflammation at the site of administration.
- compositions of the present invention may be sterilized by conventional, well known sterilization techniques.
- Aqueous solutions can be packaged for use or filtered under aseptic conditions and lyophilized, the lyophilized preparation being combined with a sterile aqueous solution prior to administration.
- the compositions can contain pharmaceutically acceptable auxiliary substances as required to approximate physiological conditions, such as pH adjusting and buffering agents, tonicity adjusting agents and the like, for example, sodium acetate, sodium lactate, sodium chloride, potassium chloride, and calcium chloride.
- the particle suspension may include lipid-protective agents which protect lipids against free-radical and lipid- peroxidative damages on storage.
- nucleic acid-lipid particles can be incorporated into a broad range of topical dosage forms including, but not limited to, gels, oils, emulsions, topical creams, pastes, ointments, lotions and the like.
- nucleic acid-lipid particles such as those disclosed in WO 96/40964, U.S. Patent Nos. 5,705,385, 5,976,567, 5,981,501, and 6,410,328.
- This latter format provides a fully encapsulated nucleic acid-lipid particle that protects the nucleic acid from nuclease degradation in serum, is nonimmunogenic, is small in size and is suitable for repeat dosing.
- administration can be in any manner known in the art, ' e.g., by injection, oral administration, inhalation, transdermal application, or rectal administration. Administration can be accomplished via single or divided doses.
- the pharmaceutical compositions are preferably administered parenterally, i.e., intraarticularly, intravenously, intraperitoneally, subcutaneously, or intramuscularly. More preferably, the pharmaceutical compositions are administered intravenously or intraperitoneally by a bolus injection (see, e.g., Stadler, et al, U.S. Patent No. 5,286,634).
- Intracellular nucleic acid delivery has also been discussed in Straubringer, et al, Methods Enzy ol, Academic Press, New York. 101:512 (1983); Mannino, et al, Biotechniques 6:682 (1988); Nicolau, et al, Crit. Rev. Ther. Drug Carrier Syst. 6:239 (1989), and Behr, Ace. Chem. Res. 26:274 (1993). Still other methods of administering lipid based therapeutics are described in, for example, Rahman et al, U.S. Patent No. 3,993,754; Sears, U.S. Patent No. 4,145,410; Papahadjopoulos et al, U.S. Patent No.
- the lipid nucleic acid particles can be administered by direct injection at the site of disease or by injection at a site distal from the site of disease (see, e.g., Culver, HUMAN GENE THERAPY, MaryAnn Liebert, Inc., Publishers, New York. pp.70-71(1994)).
- compositions of the present invention can be made into aerosol formulations (i.e., they can be "nebulized") to be administered via inhalation (see, Brigham, et al, Am. J. Sci. 298(4):278 (1989)). Aerosol formulations can be placed into pressurized acceptable propellants, such as dichlorodifluoromethane, propane, nitrogen, and the like.
- Formulations suitable for parenteral administration include aqueous and non-aqueous, isotonic sterile injection solutions, which can contain antioxidants, buffers, bacteriostats, and solutes that render the formulation isotonic with the blood of the intended recipient, and aqueous and non- aqueous sterile suspensions that can include suspending agents, solubilizers, thickening agents, stabilizers, and preservatives, hi the practice of this invention, compositions can be administered, for example, by intravenous infusion, orally, topically, intraperitoneally, intravesically or intrathecally.
- Formulations suitable for oral administration can consist of (a) liquid solutions, such as an effective amount of the packaged nucleic acid suspended in diluents, such as water, saline or PEG 400; (b) capsules, sachets or tablets, each containing a predetermined amount of the active ingredient, as liquids, solids, granules or gelatin; (c) suspensions in an appropriate liquid; and (d) suitable emulsions.
- liquid solutions such as an effective amount of the packaged nucleic acid suspended in diluents, such as water, saline or PEG 400
- capsules, sachets or tablets each containing a predetermined amount of the active ingredient, as liquids, solids, granules or gelatin
- suspensions in an appropriate liquid such as water, saline or PEG 400
- Tablet forms can include one or more of lactose, sucrose, mannitol, sorbitol, calcium phosphates, corn starch, potato starch, microcrystalline cellulose, gelatin, colloidal silicon dioxide, talc, magnesium stearate, stearic acid, and other excipients, colorants, fillers, binders, diluents, buffering agents, moistening agents, preservatives, flavoring agents, dyes, disintegrating agents, and pharmaceutically compatible carriers.
- Lozenge forms can comprise the active ingredient in a flavor, e.g., sucrose, as well as pastilles comprising the active ingredient in an inert base, such as gelatin and glycerin or sucrose and acacia emulsions, gels, and the like containing, in addition to the active ingredient, carriers known in the art.
- a flavor e.g., sucrose
- an inert base such as gelatin and glycerin or sucrose and acacia emulsions, gels, and the like
- the nucleic acid-lipid formulations are formulated with a suitable pharmaceutical carrier.
- Many pharmaceutically acceptable carriers may be employed in the compositions and methods of the present invention. Suitable formulations for use in the present invention are found, for example, in REMINGTON'S PHARMACEUTICAL SCIENCES, Mack Publishing Company, Philadelphia, PA, 17th ed. (1985).
- aqueous carriers may be used, for example, water, buffered water, 0.4% saline, 0.3% glycine, and the like, and may include glycoproteins for enhanced stability, such as albumin, lipoprotein, globulin, etc.
- glycoproteins for enhanced stability such as albumin, lipoprotein, globulin, etc.
- normal buffered saline (135-150 mM NaCl) will be employed as the pharmaceutically acceptable carrier, but other suitable carriers will suffice.
- These compositions can be sterilized by conventional liposomal sterilization techniques, such as filtration.
- compositions may contain pharmaceutically acceptable auxiliary substances as required to approximate physiological conditions, such as pH adjusting and buffering agents, tonicity adjusting agents, wetting agents and the like, for example, sodium acetate, sodium lactate, sodium chloride, potassium chloride, calcium chloride, sorbitan monolaurate, triethanolamine oleate, etc.
- auxiliary substances such as pH adjusting and buffering agents, tonicity adjusting agents, wetting agents and the like, for example, sodium acetate, sodium lactate, sodium chloride, potassium chloride, calcium chloride, sorbitan monolaurate, triethanolamine oleate, etc.
- the methods of the present invention may be practiced in a variety of hosts.
- Preferred hosts include mammalian species, such as avian (e.g., ducks), primates (e.g., humans and chimpanzees as well as other nonhuman primates), canines, felines, equines, bovines, ovines, caprines, rodents (e.g., rats and mice), lagomorphs, and swine.
- the amount of particles administered will depend upon the ratio of nucleic acid to lipid; the particular nucleic acid used, the disease state being diagnosed; the age, weight, and condition of the patient and the judgment of the clinician; but will generally be between about 0.01 and about 50 mg per kilogram of body weight; preferably between about 0.1 and about 5 mg/kg of body weight or about 10 8 -10 10 particles per injection.
- compositions and methods of the present invention are used to treat a wide variety of cell types, in vivo and in vitro.
- Suitable cells include, e.g., hematopoietic precursor (stem) cells, fibroblasts, keratinocytes, hepatocytes, endothelial cells, skeletal and smooth muscle cells, osteoblasts, neurons, quiescent lymphocytes, terminally differentiated cells, slow or noncycling primary cells, parenchymal cells, lymphoid cells, epithelial cells, bone cells, and the like.
- nucleic acid lipid particles encapsulating an interfering RNA is particularly suited for targeting tumor cells of any cell type.
- SNALP's accumulate at tumor sites and predominantly transfect tumor cells. See, Fenske, et al, Methods Enzymol, Academic Press, New York 346:36 (2002).
- the methods and compositions can be employed with cells of a wide variety of vertebrates, including mammals, and especially those of veterinary importance, e.g, canine, feline, equine, bovine, ovine, caprine, rodent, lagomorph, swine, etc., in addition to human cell populations.
- the nucleic acid-lipid particles are detectable in the subject 8, 12, 24, 48, 60, 72, or 96 hours after administration of the particles.
- the presence of the particles can be detected in the cells, tissues, or other biological samples from the subject.
- the particles by be detected, e.g., by direct detection of the particles, detection of the interfering RNA sequence, detection of the target sequence of interest (Le., by detecting expression or reduced expression of the sequence of interest), or a combination thereof. 1. Detection of Particles
- Nucleic acid-lipid particles are detected herein using any methods known in the art.
- a label can be coupled directly or indirectly to a component of the SNALP or other lipid-based carrier system using methods well known in the art.
- a wide variety of labels can be used, with the choice of label depending on sensitivity required, ease of conjugation with the SNALP component, stability requirements, and available instrumentation and disposal provisions.
- Suitable labels include, but are not limited to, spectral labels, such as fluorescent dyes (e.g., fluorescein and derivatives, such as fluorescein isothiocyanate (FITC) and Oregon GreenTM; rhodamine and derivatives, such Texas red, tetrarhodimine isothiocynate (TRITC), etc., digoxigenin, biotin, phycoerythrin, AMCA, CyDyesTM, and the like; radiolabels, such as 3 H, 125 1, 35 S, 14 C, 32 P, 33 P, etc.; enzymes, such as horse radish peroxidase, alkaline phosphatase, etc.; spectral colorimetric labels, such as colloidal gold or colored glass or plastic beads, such as polystyrene, polypropylene, latex, etc. The label can be detected using any means known in the art. 2. Detection of Nucleic Acids
- fluorescent dyes e.g., flu
- Nucleic acids are detected and quantified herein by any of a number of means well known to those of skill in the art.
- the detection of nucleic acids proceeds by well known methods such as Southern analysis, northern analysis, gel electrophoresis, PCR, radiolabeling, scintillation counting, and affinity chromatography. Additional analytic biochemical methods such as spectrophotometry, radiography, electrophoresis, capillary electrophoresis, high performance liquid chromatography (HPLC), thin layer chromatography (TLC), hyperdiffusion chromatography, may also be employed
- nucleic acid hybridization format is not critical.
- a variety of nucleic acid hybridization formats are known to those skilled in the art.
- common formats include sandwich assays and competition or displacement assays.
- Hybridization techniques are generally described in "Nucleic Acid Hybridization, A Practical Approach,” Ed. Hames, B.D. and Higgins, S.J., IRL Press, 1985.
- the sensitivity of the hybridization assays may be enhanced through use of a nucleic acid amplification system which multiplies the target nucleic acid being detected.
- a nucleic acid amplification system which multiplies the target nucleic acid being detected.
- In vitro amplification techniques suitable for amplifying sequences for use as molecular probes or for generating nucleic acid fragments for subsequent subcloning are known.
- RNA polymerase mediated techniques e.g., NASBATM
- PCR polymerase chain reaction
- LCR ligase chain reaction
- NASBATM RNA polymerase mediated techniques
- the select sequences can be generally amplified using, for example, nonspecific PCR primers and the amplified target region later probed for a specific sequence indicative of a mutation.
- Oligonucleotides for use as probes e.g., in in vitro amplification methods, for use as gene probes, or as inhibitor components are typically synthesized chemically according to the solid phase phosphoramidite triester method described by Beaucage and Caruthers, Tetrahedron Letts., 22(20):1859 1862 (1981), e.g., using an automated synthesizer, as described in Needham VanDevanter et al, Nucleic Acids Res., 12:6159
- oligonucleotides where necessary, is typically performed by either native acrylamide gel electrophoresis or by anion exchange HPLC as described in Pearson and Regnier, J. Chrom., 255:137 149 (1983).
- sequence of the synthetic oligonucleotides can be verified using the chemical degradation method of Maxam and Gilbert (1980) in Grossman and Moldave (eds.) Academic Press, New York, Methods in Enzymology, 65:499.
- In situ hybridization assays are well known and are generally described in Angerer et al, Methods EnzymoL, 152:649 (1987).
- in situ hybridization assay cells are fixed to a solid support, typically a glass slide. If DNA is to be probed, the cells are denatured with heat or alkali. The cells are then contacted with a hybridization solution at a moderate temperature to permit annealing of specific probes that are labeled. The probes are preferably labeled with radioisotopes or fluorescent reporters.
- the transfection efficiency of the nucleic acid-lipid particles described herein can be optimized using an ERP assay.
- the ERP assay can be used to distinguish the effect of various cationic lipids, non-cationic lipids, and bilayer stabilizing components of the SNALPs based on their relative effect on binding/uptake or fusion with/destabilization of the endosomal membrane.
- This assay allows one to determine quantitatively how each component of the SNALPs affects transfection efficacy, thereby optimizing the SNALPs.
- the Endosomal Release Parameter or, alternatively, ERP is defined as: REPORTER GENE EXPRESSION/CELL SNALP UPTAKE/CELL
- any reporter gene e.g. , luciferase, ⁇ -galactosidase, green fluorescent protein, etc.
- the lipid component or, alternatively, any component of the SNALP or lipid-based formulation
- any detectable label provided the does inhibit or interfere with uptake into the cell.
- the ERP assay of the present invention can assess the impact of the various lipid components (e.g., cationic lipid of Formula I or II, non-cationic lipid, PEG-lipid derivative, PEG-DAA conjugate, ATTA-lipid derivative, calcium, CPLs, cholesterol, etc.) on cell uptake and transfection efficiencies, thereby optimizing the SPLP or other lipid-based carrier system.
- the ERPs for each of the various SPLPs or other lipid-based formulations one can readily determine the optimized system, e.g., the SPLP or other lipid-based formulation that has the greatest uptake in the cell coupled with the greatest transfection efficiency.
- Suitable labels for carrying out the ERP assay of the present invention include, but are not limited to, spectral labels, such as fluorescent dyes (e.g., fluorescein and derivatives, such as fluorescein isothiocyanate (FITC) and Oregon Green ; rhodamine and derivatives, such Texas red, tetrarhodimine isothiocynate (TRITC), etc., digoxigenin, biotin, phycoerythrin, AMCA, CyDyes d , and the like; radiolabels, such as 3 H, 125 1, 35 S, 14 C, 32 P, 33 P, etc.
- fluorescent dyes e.g., fluorescein and derivatives, such as fluorescein isothiocyanate (FITC) and Oregon Green
- rhodamine and derivatives such Texas red, tetrarhodimine isothiocynate (TRITC), etc.
- digoxigenin biotin, phyco
- spectral colorimetric labels such as colloidal gold or colored glass or plastic beads, such as polystyrene, polypropylene, latex, etc.
- the label can be coupled directly or indirectly to a component of the SNALP using methods well known in the art. As indicated above, a wide variety of labels can be used, with the choice of label depending on sensitivity required, ease of conjugation with the SNALP component, stability requirements, and available instrumentation and disposal provisions.
- DPPS l,2-Distearoyl ⁇ sft-glycero-3-phosphocholine
- DSPC l,2-Distearoyl ⁇ sft-glycero-3-phosphocholine
- TNS was obtained from Sigma-Aldrich Canada (Oakville, ON).
- RiboGreen was obtained from Molecular Probes (Eugene, OR).
- the alkyl mesylates were purchased from Nu-Chek Prep, Inc. (Elysian, MN, USA).
- siRNA anti-luciferase and mismatch control was purchased from Dharmacon (Lafayette, CO, USA).
- the anti-luciferase sense sequence was 5'- G.A.U.U.A.U.G.U.C.C.G.G.U.A.U.G.U.A.U.G.U.A.U.U.U.U.U.U-3'.
- the anti-luciferase antisense sequence was 5'-U.A.C.A.U.A.A.C.C.G.G.A.C.A.U.A.A.U.C.U.C.U.C.U.U.C.U.U-3'. All other chemicals were purchased from Sigma-Aldrich (Oakville, ON, Canada).
- DSDMA and DODMA were synthesized using the respective alkyl bromides with methodology derived from that of a DOTMA precursor (Feigner et al, PNAS USA, 84, 7413-7417 (1987)).
- 3-(Dimethylamino)-l,2- propanediol (714 mg, 6 mmol) and 95% sodium hydride (NaH, 1.26 g, 50 mmol) were stirred in benzene (30 mL) under argon for 30 minutes.
- the correct (either oleyl or stearyl) alkyl bromide (5.0 g, 15 mmol) was added and the reaction refluxed under argon for 18 hours.
- the reaction mixture was then cooled in an ice bath while quenching via the slow addition of ethanol. Following dilution with a further 150 mL of benzene, the mixture was washed with distilled water (2 x 150 mL) and brine (150 mL), using ethanol ( ⁇ 20 mL) to aid phase separation if necessary. The organic phase was dried over magnesium sulphate and evaporated. The crude product was purified on a silica gel (Kiesel Gel 60) column eluted with chloroform containing 0-5% methanol.
- reaction mixture is then cooled in an ice bath while quenching via the slow addition of ethanol. Following dilution with a further 150 mL of benzene, the mixture is washed with distilled water (2 x 150 mL) and brine (150 mL). The organic phase is dried over magnesium sulphate and evaporated to give the crude product.
- TLC thin layer chromatography
- R f 0.5
- Decolorization and further purification of DLinDMA is effected with a second column, this time eiuting with 20 - 50% ethyl acetate in hexane.
- PEG-C-DMA was synthesized as follows. In brief, a C 1 lipid anchor was prepared by first alkylating the hydroxyl groups of 3- allyloxypropane-l,2-diol with myristyl bromide. The allyl group was subsequently removed via palladium catalysis, resulting in the C 14 hydroxyl lipid. The hydroxyl group was converted to the primary amine by mesylation and amination to yield 1,2- dimyristyloxypropyl-3 -amine, the lipid anchor.
- PEG-C-DMA SNALP Preparation: S ⁇ ALP with a lipid composition of DSPC: Choi :PEG-C- DMA:Cationic Lipid (20:48:2:30 molar percent) were prepared using the spontaneous vesicle formation by ethanol dilution method [Jeffs et al., Pharm. Res. In Press (2005)].
- the sample's were diafiltered against 100 mL of PBS (20 wash volumes) using a cross flow ultrafiltration cartridge (Amersham Biosciences, Piscataway, ⁇ J) and sterile filtered through Acrodisc 0.2 ⁇ m Posidyne filters (Pall Corp., Ann Arbor, MI).
- the siR ⁇ A concentration of final samples was determined using the RiboGreen assay and a siR ⁇ A standard curve. Particle size and polydispersity was determined using a Malvern Instruments Zetasizer 3000HSA (Malvern, UK). Nucleic acid encapsulation was determined using a RiboGreen assay, comparing fluorescence in the presence and absence of Triton X-100.
- TNS Assay 20 ⁇ M of SNALP lipid and 6 ⁇ M of TNS were mixed in a fluorescence cuvette in 2mL of 20 mM sodium phosphate, 25 mM citrate, 20 mM ammonium acetate and 150 mM NaCl, at a pH that was varied from 4.5 to 9.5.
- the pK a values are the point at which 50% of the molecules present are charged. By assuming that minimum fluorescence represents zero charge, and maximum fluorescence represents 100% charge, pK a can be estimated by measuring the pH at the point exactly half way between the values of minimum and maximum charge.
- Multilamellar vesicles were prepared comprising DPPS and cationic lipid at a molar ratio of 1:1. This was accomplished by drying the lipids from chloroform solution, transferring to 10 mm NMR tubes, and hydrating in 1.5 mL of 10 mM sodium citrate, pH 4. Free induction decays (FIDs) corresponding to 1000 scans were obtained with a 3.0 ⁇ s, 60o pulse with a 1 s interpulse delay and a spectral width of 25000 Hz. A gated two-level proton decoupling was used to ensure sufficient decoupling with minimum sample heating.
- FIDs Free induction decays
- the luminescence for each was measured using a Berthold MicroLumatPlus LB96V plate luminometer.
- the resulting luciferase activity was then normalized for the amount of protein using the Micro BCA assay kit (Pierce). Luciferase knockdown relative to a control was then determined for each system.
- SNALP were prepared incorporating the non-exchangeable tritium-labeled lipid cholesteryl hexadecyl ether (3H-CHE) (11.1 ⁇ Ci ⁇ mol total lipid) [Bally et al., in Liposome Technology, Vol. Ill, pp. 27-41, CRC Press (1993)].
- Neuro2A cells ATCC, VA, USA
- SNALP Uptake of SNALP Containing Cy 3 -labeled siRNA: SNALP were formulated as previously described, but using siRNA labelled with the fluorophore Cy3 (Cy3-siRNA was a gift of Sirna Therapeutics Inc, Boulder, CO). The encapsulation, siRNA concentration, and particle size were determined as described.
- Cy3 fluorescence within the cells was visualized using a rhodamine cube set (Microgen Optics, Redding, CA) and the DAPI fluorescence was visualized using a DAPI cube set (Carsen Group, Markham, ON). Digital pictures were captured using an Olympus DP70 camera system. Pictures of the cells were taken at exposure times of 1/4 sec when examining Cy3 fluorescence and 1/80 sec when examining DAPI fluorescence.
- Example 2 SNALP formulations encapsulating siRNA [0221] This example demonstrates encapsulating siRNA in SNALP formulated with either short- or long-chain PEG-DAG and produced by continuously mixing organic lipid and aqueous buffer solutions.
- C M PEG-DMG
- PEG-distearylglycerol C 18
- ⁇ - gal siRNA encapsulated in DSPC:Cholesterol:DODMA:PEG-DMG/PEG-DSG SNALP by this method resulted in > 90 % encapsulation (Ribogreen Assay) and -120 nm particle size (Malvern sizer).
- the preparations had the following characteristics:
- Example 3 Downregulation of intracellular expression in cells by delivering in vitro an SNALP formulation encapsulating siRNA.
- Lipid/therapeutic nucleic acid particles formulated according to the above noted techniques can be assayed for serum stability by a variety of methods.
- 1 ⁇ g of DNA encapsulated in the particle of interest is incubated in a total volume of 100 ⁇ L of 5 mM HEPES, 150 mM NaCl, 10.0 mM MgCl 2 pH 7.4.
- DNase treated samples are treated with either 100 or 10 U of DNase I (Gibco - BRL).
- 1.0 % Triton X-100 can be added in control experiments to ensure that lipid formulations are not directly inactivating the enzyme.
- Samples are incubated at 37°C for 30 min after which time the DNA is isolated by addition of 500 ⁇ L of DNAZOL followed by 1.0 mL of ethanol.
- the samples are centrifuged for 30 min at 15,000 rpm in a tabletop microfuge. The supernatant is decanted and the resulting DNA pellet is washed twice with 80% ethanol and dried.
- This DNA is resuspended in 30 ⁇ L of TE buffer. 20 ⁇ L of this sample is loaded on a 1.0% agarose gel and subjected to electrophoresis in TAE buffer.
- SNALP small nucleic acid-lipid particles
- PEG poly(ethyleneglycol)
- SNALP with long circulation times accumulated to levels corresponding to five to ten percent of the total injected dose per gram of tumor or greater than 1000 copies of plasmid DNA per cell, giving rise to levels of gene expression that were more than two orders of magnitude greater than those observed in any other tissue.
- the liver accumulated 20-30 % of the total injected dose, very low levels of gene expression were observed in the liver. This is thought to be due to the limited hepatocellular uptake of the PEG-ylated SNALP.
- CPL cationic PEG lipid
- CPL-SNALP yielded 10 5 -fold more in vitro gene expression than native SNALP.
- CPL-SNALP When CPL-SNALP were administered intravenously they yielded a substantial (250 fold) increase in hepatic gene expression compared to native SNALP.
- the increase in CPL-SNALP potency was specific to the liver.
- the levels of gene expression measured in the lung, kidney, spleen or heart remained unchanged, contributing to more than two orders of magnitude differential in the gene expression measured in the liver vs. other organs.
- These results illustrate the potential for modulating the delivery properties of PEG-lipid containing systems while retaining the stability and small uniform size required to achieve systemic gene delivery. In particular they demonstrate that disease site targeting and tissue specific gene expression can be re-programmed by altering the lipid composition of non- viral gene delivery systems.
- Example 6 SNALPs containing PEG-DAG conjugates [0233] This example demonstrates the preparation of a series of PEG-diacylglycerol lipids (PEG-DAG) SNALPs.
- the encapsulated nucleic acid is a plasmid.
- PEG-DAG SNALP were prepared incorporating 10 mol percent PEG- dilaurylglycerol (C 12 ), PEG-dimyristylglycerol (C 1 ), PEG-dipalmitoylglycerol (C 16 ) or PEG-disterylglycerol (C 18 ) and evaluated for in vitro transfection activity, pharmacokinetics and the biodistribution of gene expression resulting from systemic administration in tumor bearing mice.
- PEG-DAG lipid containing SNALP demonstrated a similar relationship between acyl chain length and in vitro transfection activity to those containing PEG-ceramides.
- Shorter acyl chain anchors (dimyristyl (C 1 ) and dipalmitoyl (C 16 )) resulted in SNALP particles that were less stable but have higher transfection activity in vitro than those incorporating longer acyl chain anchors (disteryl (C 18 )). Evaluation of the pharmacokinetics of PEG-DAG containing SNALP confirmed a correlation between the stability of the PEG lipid component and the circulation lifetime of SNALP.
- SNALP containing PEG-dimyristylglycerol (C 14 ), PEG-dipalmitoylglycerol (C 16 ) and PEG-disterylglycerol (C 18 ) demonstrated circulation half-lives of 0.75, 7 and 15 hours respectively. Extended circulation lifetime in turn correlates with an increase in tumor delivery and concomitant gene expression.
- PEG-disterylglycerol (C 18 ) containing SNALP bypass so-called 'first pass' organs, including the lung, and elicited gene expression in distal tumor tissue.
- the level of reporter gene expression observed in tumors represents a 100 to 1000-fold differential over that observed in any other tissue.
- HEPES HEPES
- OGP OGP
- 3 H-cholesteryl hexadecyl ether were obtained from a number of different commercial sources.
- DOPE:DODAC:PEG-Diacylglycerols 82.5:7.5: 10) large unilamellar vesicles were prepared via detergent dialysis in Hepes Buffered Saline (150 mM NaCl and 10 mM HEPES) for 48 hours.
- Lipid stock solutions were prepared in ethanol and then dried down to create a lipid film which was reconstituted in final 200mM OGP.
- LUVs were labeled with H-cholesteryl hexadecyl ether at luCi/lmg lipid.
- Particle sizes were determined by nicomp analysis. Radioactivity was determined by scintillation counting with Picofluor20.
- SNALP containing PEG-Diacyglycerols were formulated via detergent dialysis by varying the salt concentration to maximize the percent of DNA encapsulation. Optimal salt concentration was chosen for the 48 hour detergent dialysis. Empty vesicles were removed by one step sucrose centrifugation. 3.5 % sucrose was used to separate out the empty particles from the plasmid-containing PEG-Diacylglycerol formulations except for PEG-Dimyristylglycerol containing SNALP which used 5.0 % sucrose. Empty vesicles migrated to the top of the tube which were fractioned out and removed. In vitro Transfection
- the lipids were present in the SPLP in the following molar ratios (20:55:15:10).
- the following formulations were made: A PBS sterile filtered, 5 mL.
- mice 1.5xl0 6 Neuro2A cells were administered to each mouse on day 0.
- mice were randomized and treated with one dose of an SPLP formulation or PBS by intravenous (IV) injection. Dose amounts are based on body weight measurements taken on the day of dosing. 48 hours after SPLP administration, the mice were sacrificed, their blood was collected, and the following tissues were collected weighed, immediately frozen and stored at -80°C until further analysis: tumor, liver (cut in 2 halves), lungs, spleen & heart.
- Gene expression in collected tissues was determined by assaying for enzymatic activity of expressed luciferase reporter protein. The results are shown in Figures 11 and 12.
- SPLP comprising PEG-dialkyloxypropyls can conveniently be used to transfect distal tumor to substantially the same extent as SPLP comprising PEG-diacylglycerols.
- the transfection levels seen with SPLP containing PEG-dialkyloxypropyl are similar to those seen with SPLP containing PEG-diacylglycerols (e.g. PEG-DSG). It was also shown that similar to the PEG- diacylglycerol system, very little transfection occurred in non-tumor tissues.
- the SPLP comprising PEG-dialkyloxypropyls exhibit reduced toxicity compared to other SPLP formulations.
- Example 8 SNALPs containing PEG-dialkyloxypropyl conjugates
- This example described experiments analyzing the biodistribution (local and systemic) and pharmacokinetics of a series of PEG-dialkyloxypropyl lipids SNALPs (i.e., SPLP containing encapsulated siRNA. Local Biodistribution
- SPLP PEG-dialkyloxypropyl lipids SNALPs
- mice were randomized and treated with one dose of an SNALP formulation comprising 100 ⁇ g siRNA or PBS by intravenous (IV) injection in a total volume of 230 ⁇ l. Dose amounts are based on body weight measurements taken on the day of dosing. 24 hours after SPLP administration, the mice were sacrificed, their blood was collected, and the following tissues were collected weighed, immediately frozen and stored at -80C until further analysis: tumor, liver (cut in 2 halves), lungs, spleen & heart.
- IV intravenous
- mice (Jackson Laboratories) were seeded subcutaneously with Neuro 2 A cells at a dose of 1.5 x 10 cells in a total volume of 50 ⁇ L phosphate buffered saline on day zero. After tumors reached appropriate size (typically on day 9 or later), 200 ⁇ l (100 ⁇ g nucleic acid) of the SPLP or SNALP preparations described above, were administered intravenously. 0.25, 1, 2, 4, and 8 hours after administration of SPLP or SNALP, mice were weighed and blood (typically 25 ⁇ L) was collected by tail nick. 24 hours after administration of SPLP or SNALP, mice were sacrificed, blood was collected and assayed for clearance of [ H]CHE.
- SPLP containing PEG-DSG had the highest tumor accumulation at 10.9 % inject dose per gram tissue.
- the SiRNA SNALP had slightly more tumor accumulation than an SPLP sample with the same PEG-lipid at 7.3%, which also correlates relatively well with the plasma half-life for this SNALP.
- the pSPLP formulation had tumor accumulation at 7.5%, which is lower than the comparable PEG-DSG SPLP.
- This example illustrates silencing of gene expression in Neuro 2A tumor bearing mice after co-administration of SPLPs containing a plasmid encoding luciferase under the control of the CMV promoter and SNALPs containing anti-luciferase siRNA.
- mice 36 male A/J mice (Jackson Laboratories) were seeded subcutaneously with Neuro 2 A cells at a dose of 1.5 x 10 6 cells in a total volume of 50 ⁇ L phosphate buffered saline on day zero. Once tumors reached appropriate size (typically on day 9 or later), 200-240 ⁇ l PBS, SPLP, or SNALP formulations (100 ⁇ g nucleic acid total) prepared as described in Example 6 above, were administered intravenously. 24, 48, or 72 after administration of PBS, SPLP or a mixture of SPLP and SNALP, mice were sacrificed and organs (e.g., liver, lung, spleen, kidney, heart) and tumors were collected and evaluated for luciferase activity.
- organs e.g., liver, lung, spleen, kidney, heart
- SNALP were prepared containing siRNA duplex directed against the ⁇ - Galactosidase reporter gene and applied to the ⁇ -galactosidase expressing stable cell line: CT26CL25, plated at 2xl0 4 cells/well at a concentration of l.O ⁇ g/mL siRNA. Cells were exposed to SNALP for 24 hours and ⁇ -galactosidase activity was determined after 96 hours. Silencing was observed in 90% of the cells in culture which correlates with silencing of a target protein in 40% of cells in vivo.
- Example 11 Liver distribution of Rhodamine labeled SNALP Following a Single Intravenous Administration
- SNALP were prepared containing siRNA duplex directed against the ⁇ - Galactosidase reporter gene using and administered to A/J mice intravenously, through the tail vein. Tissues were collected at 24 hours, snap frozen and sectioned for visualization of SNALP dissemination. Cells were stained with rhodamine and counterstained with DAPI, which stains nuclei. The in vivo biodistribution of the SNALP favors the liver, with as much as 50% of the administered SNALP material delivered to the liver. The SNALP delivered to the liver is found in a diffuse pattern, distributed throughout the liver.
- Example 12 Silencing of gene expression following delivery of siRNA encapsulated in SPLP comprising cationic lipids
- This example describes experiments comparing expression of nucleic acids following in vitro transfection of Neuro2A cells with SNALP comprising: (1) DODAC, DODMA, or DLinDMA; (2) PEG-C-DMA; and (3) an siRNA duplex directed against luciferase encapsulated in the SNALP (Le., siRNA comprising the following sequence: GAUUAUGUCCGGUUAUGUAUU and targeting the DNA sequence complementary to: GATTATGTCCGGTTATGTATT).
- Neuro2A cells were stably transfected with a plasmid encoding luciferase under the control of the CMV promoter (pLO55).
- the stably transfected cells were then transfected with SNALP comprising: 15, 20, 25, 30, 35, or 40% of DODAC, DODMA, or DLinDMA; 2% PEG-C-DMA, and an siRNA duplex directed against luciferase encapsulated in the SNALP.
- Luciferase protein expression was measured 48 hours after transfection with SNALP.
- SNALP comprising 30% DLinDMA was more effective in reducing luciferase expression in the Neuro2A cells than SNALP comprising DODAC or DODMA were.
- DLinDMA the most fusogenic lipid with the lowest apparent phase transition temperature, yielded the greatest knockdown when incorporated in SNALP, with luciferase expression only 21% that of the untreated control. This was followed by the DLenDMA formulation (32%), and DODMA (54%). The close correspondence between knockdown efficiency and the H ⁇ phase forming ability of the cationic lipid as observed suggests that the two parameters are linked.
- Example 13 SNALP Containing Unsaturated Cationic Lipids Show Increased Gene- Silencing Activity
- This example describes experiments demonstrating in vivo transfection of organs with that SPLP comprising 15% DLinDMA can be used SPLP encapsulating a plasmid encoding luciferase under the control of the CMV promoter were administered to Neuro2A tumor bearing male A J mice.
- the SPLP had the following formulations:
- Luciferase gene expression was assessed in liver, lung, spleen, heart and tumors 48 hours after intravenous administration of the SPLP. The results are shown in Figure 25.
- Example 15 In Vivo Transfection of Tumor by Additional SPLP Formulations [0271] This example describes experiments demonstrating in vivo transfection of organs with that SPLP comprising DLinDMA or DODMA and varying percentages (15%, 10%, 5%, or 2.5%) of PEG-C-DMA. SPLP encapsulating a plasmid encoding luciferase were administered to Neuro2A tumor bearing male A/J mice. The SPLP had the following formulations:
- Example 17 Biodistribution of Lipid Vesicles Comprising PEG-C-DMA
- the example describes experiments conducted to assess the biodistribution of lipid vesicles comprising various percentages of PEG-C-DMA.
- a single intravenous dose of 3 H-CHE-labeled SPLP, SNALP, or empty vesicles was administered to Neuro 2A tumor bearing male A/J mice.
- SPLP comprised the cationic lipid DODMA
- SNALP comprised the cationic lipid DLinDMA.
- the lipid vesicles had the following formulations:
- Example 18 Silencing of Gene Expression at a Distal Tumor
- SNALP comprising DLinDMA and encapsulating an anti-luciferase siRNA sequence.
- Neuro 2A cells were stably transfected with a plasmid encoding luciferase under the control of the CMV promoter (pLO55) to generate Neuro 2A-G cells.
- the SNALP encapsulating the anti-luciferase siRNA sequence i.e., siRNA comprising the following sequence: GAUUAUGUCCGGUUAUGUAUU and targeting the DNA sequence complementary to: GATTATGTCCGGTTATGTATT
- siRNA comprising the following sequence: GAUUAUGUCCGGUUAUGUAUU and targeting the DNA sequence complementary to: GATTATGTCCGGTTATGTATT
- the SNALP formulations were as follows:
- Luciferase gene expression was measured 48 hours following administration of SNALP comprising DLinDMA and encapsulating an anti-luciferase siRNA sequence. The results are shown in Figure 29.
- Example 19 Silencing of Gene Expression in Neuro2A-G Tumor Cells in vitro
- This example describes experiments demonstrating gene silencing in mammalian cells following contact with SNALP comprising DLinDMA and encapsulating an anti- luciferase siRNA sequence described in Example 3 above.
- Neuro 2A cells were stably transfected with a plasmid encoding luciferase as described in Example 3 above to generate Neuro 2A-G cells.
- the Neuro 2A-G cell were contacted with SNALP formulations for 24 or 48 hours.
- the SNALP formulations comprised either PEG-C-DLA (C 12 ) or PEG-C-DMA (C 14 ) and are as follows:
- Luciferase gene expression was measured 24 or 48 hours following contacting the Neuro 2A-G cells with SNALP encapsulating an anti-luciferase siRNA sequence. The results are shown in Figure 30.
- Example 20 Silencing of Gene Expression in Neuro2A-G Tumor Cells in vitro
- This example describes experiments demonstrating gene silencing in mammalian cells following contact with SNALP comprising DLinDMA and encapsulating an anti- luciferase siRNA sequence described in Example 3 above.
- Neuro 2A cells were stably transfected with a plasmid encoding luciferase as described in Example 3 above to generate Neuro 2A-G cells.
- the Neuro 2A-G cells were contacted with SNALP formulations for 48 hours in the presence and absence of chloroquine.
- the SNALP formulations contained varying percentages of PEG-C-DMA (C 1 ) and either DODMA or DLinDMA.
- the formulation were as follows:
Abstract
Description

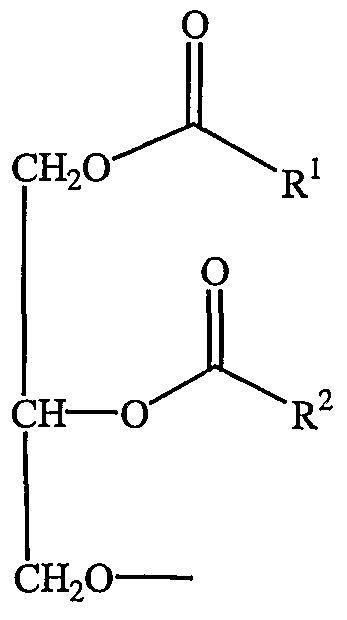
















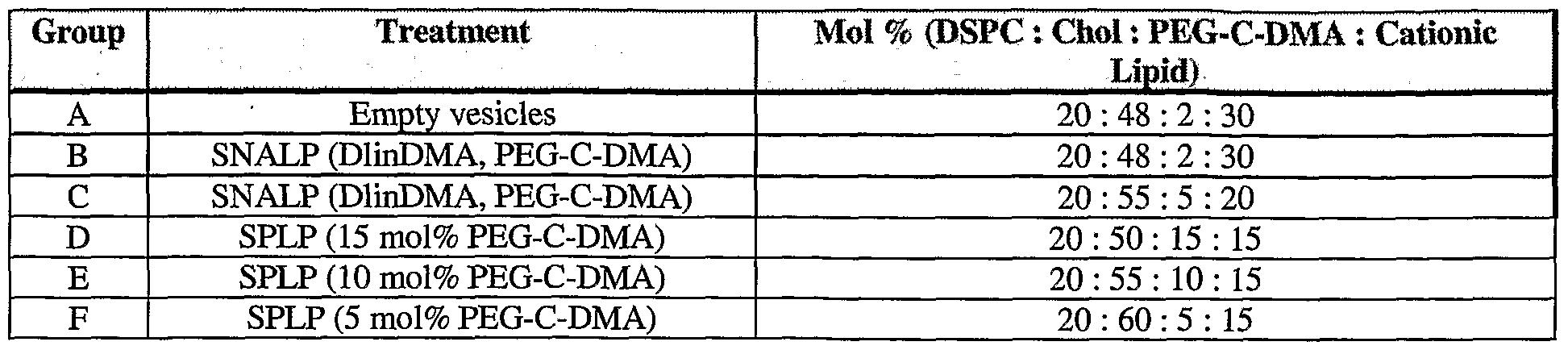




Claims



Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AT05757651T ATE536418T1 (en) | 2004-06-07 | 2005-06-07 | LIPID ENCAPSULATED INTERFERENCE RNA |
| CN2005800225822A CN1981044B (en) | 2004-06-07 | 2005-06-07 | Lipid encapsulated interfering RNA |
| AU2005252273A AU2005252273B2 (en) | 2004-06-07 | 2005-06-07 | Lipid encapsulated interfering RNA |
| EP05757651A EP1766035B1 (en) | 2004-06-07 | 2005-06-07 | Lipid encapsulated interfering rna |
| CA2569664A CA2569664C (en) | 2004-06-07 | 2005-06-07 | Lipid encapsulated interfering rna |
| JP2007526139A JP4796062B2 (en) | 2004-06-07 | 2005-06-07 | Lipid-encapsulating interfering RNA |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US57807504P | 2004-06-07 | 2004-06-07 | |
| US57796104P | 2004-06-07 | 2004-06-07 | |
| US60/578,075 | 2004-06-07 | ||
| US60/577,961 | 2004-06-07 | ||
| US61074604P | 2004-09-17 | 2004-09-17 | |
| US60/610,746 | 2004-09-17 | ||
| US67942705P | 2005-05-09 | 2005-05-09 | |
| US60/679,427 | 2005-05-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2005121348A1 true WO2005121348A1 (en) | 2005-12-22 |
Family
ID=35503071
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CA2005/000886 WO2005121348A1 (en) | 2004-06-07 | 2005-06-07 | Lipid encapsulated interfering rna |
Country Status (7)
| Country | Link |
|---|---|
| US (4) | US7799565B2 (en) |
| EP (1) | EP1766035B1 (en) |
| JP (1) | JP4796062B2 (en) |
| AT (1) | ATE536418T1 (en) |
| AU (1) | AU2005252273B2 (en) |
| CA (1) | CA2569664C (en) |
| WO (1) | WO2005121348A1 (en) |
Cited By (189)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006016097A2 (en) * | 2004-08-13 | 2006-02-16 | Ic Vec Limited | Vector comprising polymer modified sirna liposomes |
| WO2006053430A1 (en) * | 2004-11-17 | 2006-05-26 | Protiva Biotherapeutics, Inc. | Sirna silencing of apolipoprotein b |
| EP1774962A1 (en) * | 2005-10-14 | 2007-04-18 | Industrial Technology Research Institute | Lipid carrier and method of preparing the same |
| JP2008509205A (en) * | 2004-08-13 | 2008-03-27 | アイシー・ベック・リミテッド | Vector containing polymer-modified siRNA liposomes |
| WO2009082817A1 (en) | 2007-12-27 | 2009-07-09 | Protiva Biotherapeutics, Inc. | Silencing of polo-like kinase expression using interfering rna |
| WO2009088891A1 (en) * | 2008-01-02 | 2009-07-16 | Alnylam Pharmaceuticals, Inc. | Screening method for selected amino lipid-containing compositions |
| WO2009129385A1 (en) * | 2008-04-16 | 2009-10-22 | Abbott Laboratories | Cationic lipids and uses thereof |
| WO2009127060A1 (en) | 2008-04-15 | 2009-10-22 | Protiva Biotherapeutics, Inc. | Novel lipid formulations for nucleic acid delivery |
| WO2009129395A1 (en) * | 2008-04-16 | 2009-10-22 | Abbott Laboratories | Cationic lipids and uses thereof |
| WO2009129319A2 (en) | 2008-04-15 | 2009-10-22 | Protiva Biotherapeutics, Inc. | Silencing of csn5 gene expression using interfering rna |
| WO2010030739A1 (en) * | 2008-09-10 | 2010-03-18 | Abbott Laboratories | Polyethylene glycol lipid conjugates and uses thereof |
| WO2010030730A1 (en) * | 2008-09-10 | 2010-03-18 | Abbott Laboratories | Polyethylene glycol lipid conjugates and uses thereof |
| US7807815B2 (en) | 2004-07-02 | 2010-10-05 | Protiva Biotherapeutics, Inc. | Compositions comprising immunostimulatory siRNA molecules and DLinDMA or DLenDMA |
| US7838658B2 (en) | 2005-10-20 | 2010-11-23 | Ian Maclachlan | siRNA silencing of filovirus gene expression |
| WO2011000107A1 (en) | 2009-07-01 | 2011-01-06 | Protiva Biotherapeutics, Inc. | Novel lipid formulations for delivery of therapeutic agents to solid tumors |
| WO2011011447A1 (en) | 2009-07-20 | 2011-01-27 | Protiva Biotherapeutics, Inc. | Compositions and methods for silencing ebola virus gene expression |
| JP2011509258A (en) * | 2008-01-02 | 2011-03-24 | テクミラ ファーマシューティカルズ コーポレイション | Improved compositions and methods for delivery of nucleic acids |
| US7915399B2 (en) | 2006-06-09 | 2011-03-29 | Protiva Biotherapeutics, Inc. | Modified siRNA molecules and uses thereof |
| WO2011038160A2 (en) | 2009-09-23 | 2011-03-31 | Protiva Biotherapeutics, Inc. | Compositions and methods for silencing genes expressed in cancer |
| WO2011076807A2 (en) | 2009-12-23 | 2011-06-30 | Novartis Ag | Lipids, lipid compositions, and methods of using them |
| WO2011136369A1 (en) | 2010-04-28 | 2011-11-03 | 協和発酵キリン株式会社 | Cationic lipid |
| WO2011136368A1 (en) | 2010-04-28 | 2011-11-03 | 協和発酵キリン株式会社 | Cationic lipid |
| WO2011141705A1 (en) | 2010-05-12 | 2011-11-17 | Protiva Biotherapeutics, Inc. | Novel cationic lipids and methods of use thereof |
| WO2011141704A1 (en) | 2010-05-12 | 2011-11-17 | Protiva Biotherapeutics, Inc | Novel cyclic cationic lipids and methods of use |
| WO2012006372A1 (en) | 2010-07-06 | 2012-01-12 | Novartis Ag | Delivery of rna to different cell types |
| WO2012006377A2 (en) | 2010-07-06 | 2012-01-12 | Novartis Ag | Delivery of rna to trigger multiple immune pathways |
| WO2012006369A2 (en) | 2010-07-06 | 2012-01-12 | Novartis Ag | Immunisation of large mammals with low doses of rna |
| WO2012006378A1 (en) | 2010-07-06 | 2012-01-12 | Novartis Ag | Liposomes with lipids having an advantageous pka- value for rna delivery |
| WO2012006376A2 (en) | 2010-07-06 | 2012-01-12 | Novartis Ag | Virion-like delivery particles for self-replicating rna molecules |
| US8101741B2 (en) | 2005-11-02 | 2012-01-24 | Protiva Biotherapeutics, Inc. | Modified siRNA molecules and uses thereof |
| WO2012031046A2 (en) | 2010-08-31 | 2012-03-08 | Novartis Ag | Lipids suitable for liposomal delivery of protein-coding rna |
| WO2012031043A1 (en) | 2010-08-31 | 2012-03-08 | Novartis Ag | Pegylated liposomes for delivery of immunogen-encoding rna |
| WO2012170889A1 (en) | 2011-06-08 | 2012-12-13 | Shire Human Genetic Therapies, Inc. | Cleavable lipids |
| WO2012170930A1 (en) | 2011-06-08 | 2012-12-13 | Shire Human Genetic Therapies, Inc | Lipid nanoparticle compositions and methods for mrna delivery |
| WO2013006825A1 (en) | 2011-07-06 | 2013-01-10 | Novartis Ag | Liposomes having useful n:p ratio for delivery of rna molecules |
| WO2013033563A1 (en) | 2011-08-31 | 2013-03-07 | Novartis Ag | Pegylated liposomes for delivery of immunogen-encoding rna |
| WO2013065825A1 (en) | 2011-11-02 | 2013-05-10 | 協和発酵キリン株式会社 | Cationic lipid |
| WO2013089152A1 (en) | 2011-12-12 | 2013-06-20 | 協和発酵キリン株式会社 | Lipid nanoparticles containing combinations of cationic lipids |
| WO2013089151A1 (en) | 2011-12-12 | 2013-06-20 | 協和発酵キリン株式会社 | Lipid nanoparticles for drug delivery system containing cationic lipids |
| WO2013126803A1 (en) | 2012-02-24 | 2013-08-29 | Protiva Biotherapeutics Inc. | Trialkyl cationic lipids and methods of use thereof |
| WO2013149141A1 (en) | 2012-03-29 | 2013-10-03 | Shire Human Genetic Therapies, Inc. | Lipid-derived neutral nanoparticles |
| US8569256B2 (en) | 2009-07-01 | 2013-10-29 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods for the delivery of therapeutic agents |
| WO2013185069A1 (en) | 2012-06-08 | 2013-12-12 | Shire Human Genetic Therapies, Inc. | Pulmonary delivery of mrna to non-lung target cells |
| WO2014007398A1 (en) | 2012-07-06 | 2014-01-09 | 協和発酵キリン株式会社 | Cationic lipid |
| WO2014013995A1 (en) | 2012-07-16 | 2014-01-23 | 協和発酵キリン株式会社 | Rnai pharmaceutical composition capable of suppressing expression of kras gene |
| WO2014089486A1 (en) | 2012-12-07 | 2014-06-12 | Shire Human Genetic Therapies, Inc. | Lipidic nanoparticles for mrna delivering |
| WO2014108515A1 (en) | 2013-01-10 | 2014-07-17 | Novartis Ag | Influenza virus immunogenic compositions and uses thereof |
| WO2014144196A1 (en) | 2013-03-15 | 2014-09-18 | Shire Human Genetic Therapies, Inc. | Synergistic enhancement of the delivery of nucleic acids via blended formulations |
| WO2014152774A1 (en) | 2013-03-14 | 2014-09-25 | Shire Human Genetic Therapies, Inc. | Methods and compositions for delivering mrna coded antibodies |
| US8957199B2 (en) | 2008-11-26 | 2015-02-17 | Chugai Seiyaku Kabushiki Kaisha | Oligoribonucleotide or peptide nucleic acid capable of inhibiting activity of hepatitis C virus |
| US9018187B2 (en) | 2009-07-01 | 2015-04-28 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods for the delivery of therapeutic agents |
| WO2015061491A1 (en) | 2013-10-22 | 2015-04-30 | Shire Human Genetic Therapies, Inc. | Mrna therapy for phenylketonuria |
| WO2015061461A1 (en) | 2013-10-22 | 2015-04-30 | Shire Human Genetic Therapies, Inc. | Cns delivery of mrna and uses thereof |
| WO2015061500A1 (en) | 2013-10-22 | 2015-04-30 | Shire Human Genetic Therapies, Inc. | Mrna therapy for argininosuccinate synthetase deficiency |
| WO2015061467A1 (en) | 2013-10-22 | 2015-04-30 | Shire Human Genetic Therapies, Inc. | Lipid formulations for delivery of messenger rna |
| US9035039B2 (en) | 2011-12-22 | 2015-05-19 | Protiva Biotherapeutics, Inc. | Compositions and methods for silencing SMAD4 |
| US9126966B2 (en) | 2011-08-31 | 2015-09-08 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods of use thereof |
| US9139554B2 (en) | 2008-10-09 | 2015-09-22 | Tekmira Pharmaceuticals Corporation | Amino lipids and methods for the delivery of nucleic acids |
| US9181545B2 (en) | 2004-06-07 | 2015-11-10 | Protiva Biotherapeutics, Inc. | Lipid encapsulating interfering RNA |
| US9181321B2 (en) | 2013-03-14 | 2015-11-10 | Shire Human Genetic Therapies, Inc. | CFTR mRNA compositions and related methods and uses |
| US9193827B2 (en) | 2010-08-26 | 2015-11-24 | Massachusetts Institute Of Technology | Poly(beta-amino alcohols), their preparation, and uses thereof |
| WO2015200465A1 (en) | 2014-06-24 | 2015-12-30 | Shire Human Genetic Therapies, Inc. | Stereochemically enriched compositions for delivery of nucleic acids |
| US9227917B2 (en) | 2012-08-13 | 2016-01-05 | Massachusetts Institute Of Technology | Amine-containing lipidoids and uses thereof |
| WO2016004318A1 (en) | 2014-07-02 | 2016-01-07 | Shire Human Genetic Therapies, Inc. | Encapsulation of messenger rna |
| US9238716B2 (en) | 2011-03-28 | 2016-01-19 | Massachusetts Institute Of Technology | Conjugated lipomers and uses thereof |
| US9254265B2 (en) | 2010-08-31 | 2016-02-09 | Novartis Ag | Small liposomes for delivery of immunogen encoding RNA |
| WO2016054421A1 (en) | 2014-10-02 | 2016-04-07 | Protiva Biotherapeutics, Inc | Compositions and methods for silencing hepatitis b virus gene expression |
| US9315472B2 (en) | 2013-05-01 | 2016-04-19 | Massachusetts Institute Of Technology | 1,3,5-triazinane-2,4,6-trione derivatives and uses thereof |
| WO2016090262A1 (en) | 2014-12-05 | 2016-06-09 | Shire Human Genetic Therapies, Inc. | Messenger rna therapy for treatment of articular disease |
| WO2016149508A1 (en) | 2015-03-19 | 2016-09-22 | Shire Human Genetic Therapies, Inc. | Mrna therapy for pompe disease |
| US9512073B2 (en) | 2011-10-27 | 2016-12-06 | Massachusetts Institute Of Technology | Amino acid-, peptide-and polypeptide-lipids, isomers, compositions, and uses thereof |
| WO2016197132A1 (en) | 2015-06-04 | 2016-12-08 | Protiva Biotherapeutics Inc. | Treating hepatitis b virus infection using crispr |
| US9556110B2 (en) | 2008-11-07 | 2017-01-31 | Massachusetts Institute Of Technology | Aminoalcohol lipidoids and uses thereof |
| KR20170012366A (en) | 2014-06-04 | 2017-02-02 | 교와 핫꼬 기린 가부시키가이샤 | Ckap5-gene-silencing rnai pharmaceutical composition |
| WO2017019891A2 (en) | 2015-07-29 | 2017-02-02 | Protiva Biotherapeutics, Inc. | Compositions and methods for silencing hepatitis b virus gene expression |
| WO2017111172A1 (en) | 2015-12-25 | 2017-06-29 | 協和発酵キリン株式会社 | Compounds as cationic lipids |
| EP3192788A1 (en) * | 2006-10-03 | 2017-07-19 | Arbutus Biopharma Corporation | Lipid containing formulations |
| WO2017177169A1 (en) | 2016-04-08 | 2017-10-12 | Rana Therapeutics, Inc. | Multimeric coding nucleic acid and uses thereof |
| US9840479B2 (en) | 2014-07-02 | 2017-12-12 | Massachusetts Institute Of Technology | Polyamine-fatty acid derived lipidoids and uses thereof |
| WO2017218524A1 (en) | 2016-06-13 | 2017-12-21 | Rana Therapeutics, Inc. | Messenger rna therapy for the treatment of ornithine transcarbamylase deficiency |
| US9850269B2 (en) | 2014-04-25 | 2017-12-26 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| WO2018006052A1 (en) | 2016-06-30 | 2018-01-04 | Protiva Biotherapeutics, Inc. | Compositions and methods for delivering messenger rna |
| US9957499B2 (en) | 2013-03-14 | 2018-05-01 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US9956271B2 (en) | 2010-11-30 | 2018-05-01 | Translate Bio, Inc. | mRNA for use in treatment of human genetic diseases |
| EP3318248A1 (en) | 2009-12-01 | 2018-05-09 | Translate Bio, Inc. | Delivery of mrna for the augmentation of proteins and enzymes in human genetic diseases |
| WO2018089846A1 (en) | 2016-11-10 | 2018-05-17 | Translate Bio, Inc. | Subcutaneous delivery of messenger rna |
| WO2018089801A1 (en) | 2016-11-10 | 2018-05-17 | Translate Bio, Inc. | Improved process of preparing mrna-loaded lipid nanoparticles |
| WO2018129544A1 (en) | 2017-01-09 | 2018-07-12 | Whitehead Institute For Biomedical Research | Methods of altering gene expression by perturbing transcription factor multimers that structure regulatory loops |
| US10022455B2 (en) | 2014-05-30 | 2018-07-17 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| WO2018157154A2 (en) | 2017-02-27 | 2018-08-30 | Translate Bio, Inc. | Novel codon-optimized cftr mrna |
| WO2018165257A1 (en) | 2017-03-07 | 2018-09-13 | Translate Bio, Inc. | Polyanionic delivery of nucleic acids |
| WO2018213476A1 (en) | 2017-05-16 | 2018-11-22 | Translate Bio, Inc. | Treatment of cystic fibrosis by delivery of codon-optimized mrna encoding cftr |
| US10144942B2 (en) | 2015-10-14 | 2018-12-04 | Translate Bio, Inc. | Modification of RNA-related enzymes for enhanced production |
| WO2018236849A1 (en) | 2017-06-19 | 2018-12-27 | Translate Bio, Inc. | Messenger rna therapy for the treatment of friedreich's ataxia |
| US10201618B2 (en) | 2015-06-19 | 2019-02-12 | Massachusetts Institute Of Technology | Alkenyl substituted 2,5-piperazinediones, compositions, and uses thereof |
| EP3450553A1 (en) | 2014-03-24 | 2019-03-06 | Translate Bio, Inc. | Mrna therapy for treatment of ocular diseases |
| WO2019118806A1 (en) | 2017-12-14 | 2019-06-20 | Solid Biosciences Inc. | Non-viral production and delivery of genes |
| WO2019126593A1 (en) | 2017-12-20 | 2019-06-27 | Translate Bio, Inc. | Improved composition and methods for treatment of ornithine transcarbamylase deficiency |
| US10342761B2 (en) | 2014-07-16 | 2019-07-09 | Novartis Ag | Method of encapsulating a nucleic acid in a lipid nanoparticle host |
| WO2019152802A1 (en) | 2018-02-02 | 2019-08-08 | Translate Bio, Inc. | Cationic polymers |
| EP3536787A1 (en) | 2012-06-08 | 2019-09-11 | Translate Bio, Inc. | Nuclease resistant polynucleotides and uses thereof |
| EP3542825A1 (en) | 2014-11-10 | 2019-09-25 | Ethris GmbH | Induction of osteogenesis by delivering bmp encoding rna |
| US10463751B2 (en) | 2012-04-02 | 2019-11-05 | Modernatx, Inc. | Modified polynucleotides for the production of cytoplasmic and cytoskeletal proteins |
| WO2019222424A1 (en) | 2018-05-16 | 2019-11-21 | Translate Bio, Inc. | Ribose cationic lipids |
| WO2019222277A1 (en) | 2018-05-15 | 2019-11-21 | Translate Bio, Inc. | Subcutaneous delivery of messenger rna |
| WO2019226925A1 (en) | 2018-05-24 | 2019-11-28 | Translate Bio, Inc. | Thioester cationic lipids |
| WO2019232103A1 (en) | 2018-05-30 | 2019-12-05 | Translate Bio, Inc. | Messenger rna vaccines and uses thereof |
| WO2019232095A1 (en) | 2018-05-30 | 2019-12-05 | Translate Bio, Inc. | Vitamin cationic lipids |
| WO2019232208A1 (en) | 2018-05-30 | 2019-12-05 | Translate Bio, Inc. | Cationic lipids comprising a steroidal moiety |
| WO2019232097A1 (en) | 2018-05-30 | 2019-12-05 | Translate Bio, Inc. | Phosphoester cationic lipids |
| US10501513B2 (en) | 2012-04-02 | 2019-12-10 | Modernatx, Inc. | Modified polynucleotides for the production of oncology-related proteins and peptides |
| US10501512B2 (en) | 2012-04-02 | 2019-12-10 | Modernatx, Inc. | Modified polynucleotides |
| WO2020023533A1 (en) | 2018-07-23 | 2020-01-30 | Translate Bio, Inc. | Dry power formulations for messenger rna |
| WO2020047061A1 (en) | 2018-08-29 | 2020-03-05 | Translate Bio, Inc. | Improved process of preparing mrna-loaded lipid nanoparticles |
| EP3620447A1 (en) | 2012-03-29 | 2020-03-11 | Translate Bio MA, Inc. | Ionizable cationic lipids |
| WO2020056294A1 (en) | 2018-09-14 | 2020-03-19 | Translate Bio, Inc. | Composition and methods for treatment of methylmalonic acidemia |
| WO2020081933A1 (en) | 2018-10-19 | 2020-04-23 | Translate Bio, Inc. | Pumpless encapsulation of messenger rna |
| WO2020097376A1 (en) | 2018-11-09 | 2020-05-14 | Translate Bio, Inc. | Multi-peg lipid compounds |
| WO2020097379A2 (en) | 2018-11-09 | 2020-05-14 | Translate Bio, Inc. | Peg lipidoid compounds |
| WO2020097511A2 (en) | 2018-11-09 | 2020-05-14 | Translate Bio, Inc. | Messenger rna therapy for treatment of ocular diseases |
| WO2020097384A1 (en) | 2018-11-09 | 2020-05-14 | Translate Bio, Inc. | 2,5-dioxopiperazine lipids with intercalated ester, thioester, disulfide and anhydride moieities |
| WO2020102172A2 (en) | 2018-11-12 | 2020-05-22 | Translate Bio, Inc. | Methods for inducing immune tolerance |
| WO2020106946A1 (en) | 2018-11-21 | 2020-05-28 | Translate Bio, Inc. | TREATMENT OF CYSTIC FIBROSIS BY DELIVERY OF NEBULIZED mRNA ENCODING CFTR |
| WO2020106903A1 (en) | 2018-11-21 | 2020-05-28 | Translate Bio, Inc. | Cationic lipid compounds and compositions thereof for use in the delivery of messenger rna |
| US10703789B2 (en) | 2012-04-02 | 2020-07-07 | Modernatx, Inc. | Modified polynucleotides for the production of secreted proteins |
| EP3677567A1 (en) | 2013-07-23 | 2020-07-08 | Arbutus Biopharma Corporation | Compositions and methods for delivering messenger rna |
| WO2020146344A1 (en) | 2019-01-07 | 2020-07-16 | Translate Bio, Inc. | Composition and methods for treatment of primary ciliary dyskinesia |
| WO2020165352A1 (en) | 2019-02-14 | 2020-08-20 | Ethris Gmbh | Treatment of ciliopathies |
| WO2020214946A1 (en) | 2019-04-18 | 2020-10-22 | Translate Bio, Inc. | Cystine cationic lipids |
| US10815530B2 (en) | 2014-08-14 | 2020-10-27 | Technion Research & Development Foundation Limited | Compositions and methods for therapeutics prescreening |
| WO2020219427A1 (en) | 2019-04-22 | 2020-10-29 | Translate Bio, Inc. | Thioester cationic lipids |
| WO2020227085A1 (en) | 2019-05-03 | 2020-11-12 | Translate Bio, Inc. | Di-thioester cationic lipids |
| WO2020232276A1 (en) | 2019-05-14 | 2020-11-19 | Translate Bio, Inc. | Improved process of preparing mrna-loaded lipid nanoparticles |
| WO2020237227A1 (en) | 2019-05-22 | 2020-11-26 | Massachusetts Institute Of Technology | Circular rna compositions and methods |
| WO2020243540A1 (en) | 2019-05-31 | 2020-12-03 | Translate Bio, Inc. | Macrocyclic lipids |
| WO2020257716A1 (en) | 2019-06-21 | 2020-12-24 | Translate Bio, Inc. | Tricine and citric acid lipids |
| WO2020257611A1 (en) | 2019-06-21 | 2020-12-24 | Translate Bio, Inc. | Cationic lipids comprising an hydroxy moiety |
| WO2021007278A1 (en) | 2019-07-08 | 2021-01-14 | Translate Bio, Inc. | Improved mrna-loaded lipid nanoparticles and processes of making the same |
| WO2021016430A1 (en) | 2019-07-23 | 2021-01-28 | Translate Bio, Inc. | Stable compositions of mrna-loaded lipid nanoparticles and processes of making |
| WO2021021988A1 (en) | 2019-07-30 | 2021-02-04 | Translate Bio, Inc. | Treatment of cystic fibrosis by delivery of nebulized mrna encoding cftr |
| WO2021055609A1 (en) | 2019-09-20 | 2021-03-25 | Translate Bio, Inc. | Mrna encoding engineered cftr |
| WO2021072172A1 (en) | 2019-10-09 | 2021-04-15 | Translate Bio, Inc. | Compositions, methods and uses of messenger rna |
| WO2021081058A1 (en) | 2019-10-21 | 2021-04-29 | Translate Bio, Inc. | Compositions, methods and uses of messenger rna |
| WO2021113777A2 (en) | 2019-12-04 | 2021-06-10 | Orna Therapeutics, Inc. | Circular rna compositions and methods |
| WO2021127641A1 (en) | 2019-12-20 | 2021-06-24 | Translate Bio, Inc. | Improved process of preparing mrna-loaded lipid nanoparticles |
| WO2021127394A2 (en) | 2019-12-20 | 2021-06-24 | Translate Bio, Inc. | Rectal delivery of messenger rna |
| WO2021142245A1 (en) | 2020-01-10 | 2021-07-15 | Translate Bio, Inc. | Compounds, pharmaceutical compositions and methods for modulating expression of muc5b in lung cells and tissues |
| WO2021173840A1 (en) | 2020-02-25 | 2021-09-02 | Translate Bio, Inc. | Improved processes of preparing mrna-loaded lipid nanoparticles |
| WO2021195218A1 (en) | 2020-03-24 | 2021-09-30 | Generation Bio Co. | Non-viral dna vectors and uses thereof for expressing gaucher therapeutics |
| WO2021195214A1 (en) | 2020-03-24 | 2021-09-30 | Generation Bio Co. | Non-viral dna vectors and uses thereof for expressing factor ix therapeutics |
| US11166996B2 (en) | 2018-12-12 | 2021-11-09 | Flagship Pioneering Innovations V, Inc. | Anellovirus compositions and methods of use |
| WO2021226468A1 (en) | 2020-05-07 | 2021-11-11 | Translate Bio, Inc. | Improved compositions for cftr mrna therapy |
| WO2021226436A1 (en) | 2020-05-07 | 2021-11-11 | Translate Bio, Inc. | Optimized nucleotide sequences encoding sars-cov-2 antigens |
| WO2021226463A1 (en) | 2020-05-07 | 2021-11-11 | Translate Bio, Inc. | Composition and methods for treatment of primary ciliary dyskinesia |
| US11174500B2 (en) | 2018-08-24 | 2021-11-16 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| WO2021231697A1 (en) | 2020-05-14 | 2021-11-18 | Translate Bio, Inc. | Peg lipidoid compounds |
| WO2021231901A1 (en) | 2020-05-15 | 2021-11-18 | Translate Bio, Inc. | Lipid nanoparticle formulations for mrna delivery |
| WO2021236855A1 (en) | 2020-05-19 | 2021-11-25 | Orna Therapeutics, Inc. | Circular rna compositions and methods |
| WO2022006527A1 (en) | 2020-07-02 | 2022-01-06 | Maritime Therapeutics, Inc. | Compositions and methods for reverse gene therapy |
| WO2022023284A1 (en) | 2020-07-27 | 2022-02-03 | Anjarium Biosciences Ag | Compositions of dna molecules, methods of making therefor, and methods of use thereof |
| WO2022076562A1 (en) | 2020-10-06 | 2022-04-14 | Translate Bio, Inc. | Improved process and formulation of lipid nanoparticles |
| WO2022081544A1 (en) | 2020-10-12 | 2022-04-21 | Translate Bio, Inc. | Improved process of preparing mrna-loaded lipid nanoparticles |
| WO2022081548A1 (en) | 2020-10-12 | 2022-04-21 | Translate Bio, Inc. | Improved process of preparing ice-based lipid nanoparticles |
| WO2022099194A1 (en) | 2020-11-09 | 2022-05-12 | Translate Bio, Inc. | Improved compositions for delivery of codon-optimized mrna |
| WO2022115547A1 (en) | 2020-11-25 | 2022-06-02 | Translate Bio, Inc. | Stable liquid lipid nanoparticle formulations |
| WO2022155404A1 (en) | 2021-01-14 | 2022-07-21 | Translate Bio, Inc. | Methods and compositions for delivering mrna coded antibodies |
| WO2022204549A1 (en) | 2021-03-25 | 2022-09-29 | Translate Bio, Inc. | Optimized nucleotide sequences encoding the extracellular domain of human ace2 protein or a portion thereof |
| WO2022225918A1 (en) | 2021-04-19 | 2022-10-27 | Translate Bio, Inc. | Improved compositions for delivery of mrna |
| WO2022223556A1 (en) | 2021-04-20 | 2022-10-27 | Anjarium Biosciences Ag | Compositions of dna molecules encoding amylo-alpha-1, 6-glucosidase, 4-alpha-glucanotransferase, methods of making thereof, and methods of use thereof |
| WO2022232289A1 (en) | 2021-04-27 | 2022-11-03 | Generation Bio Co. | Non-viral dna vectors expressing therapeutic antibodies and uses thereof |
| WO2022232286A1 (en) | 2021-04-27 | 2022-11-03 | Generation Bio Co. | Non-viral dna vectors expressing anti-coronavirus antibodies and uses thereof |
| WO2023278754A1 (en) | 2021-07-01 | 2023-01-05 | Translate Bio, Inc. | Compositions for delivery of mrna |
| US11564893B2 (en) | 2015-08-17 | 2023-01-31 | Modernatx, Inc. | Methods for preparing particles and related compositions |
| US11576872B2 (en) | 2017-05-08 | 2023-02-14 | Flagship Pioneering Innovations V, Inc. | Compositions for facilitating membrane fusion and uses thereof |
| WO2023021421A1 (en) | 2021-08-16 | 2023-02-23 | Glaxosmithkline Biologicals Sa | Low-dose lyophilized rna vaccines and methods for preparing and using the same |
| WO2023021427A1 (en) | 2021-08-16 | 2023-02-23 | Glaxosmithkline Biologicals Sa | Freeze-drying of lipid nanoparticles (lnps) encapsulating rna and formulations thereof |
| US11639370B2 (en) | 2010-10-11 | 2023-05-02 | Glaxosmithkline Biologicals Sa | Antigen delivery platforms |
| WO2023081526A1 (en) | 2021-11-08 | 2023-05-11 | Orna Therapeutics, Inc. | Lipid nanoparticle compositions for delivering circular polynucleotides |
| WO2023086893A1 (en) | 2021-11-10 | 2023-05-19 | Translate Bio, Inc. | Composition and methods for treatment of primary ciliary dyskinesia |
| WO2023135273A2 (en) | 2022-01-14 | 2023-07-20 | Anjarium Biosciences Ag | Compositions of dna molecules encoding factor viii, methods of making thereof, and methods of use thereof |
| WO2023144798A1 (en) | 2022-01-31 | 2023-08-03 | Genevant Sciences Gmbh | Ionizable cationic lipids for lipid nanoparticles |
| US11744801B2 (en) | 2017-08-31 | 2023-09-05 | Modernatx, Inc. | Methods of making lipid nanoparticles |
| WO2023177655A1 (en) | 2022-03-14 | 2023-09-21 | Generation Bio Co. | Heterologous prime boost vaccine compositions and methods of use |
| US11786607B2 (en) | 2017-06-15 | 2023-10-17 | Modernatx, Inc. | RNA formulations |
| WO2023214405A1 (en) | 2022-05-01 | 2023-11-09 | Yeda Research And Development Co. Ltd. | Reexpression of hnf4a to alleviate cancer-associated cachexia |
| WO2023215481A1 (en) | 2022-05-05 | 2023-11-09 | The Board Of Trustees Of The Leland Stanford Junior University | INTERFERING RNA THERAPY FOR PLN-R14del CARDIOMYOPATHY |
| WO2023239756A1 (en) | 2022-06-07 | 2023-12-14 | Generation Bio Co. | Lipid nanoparticle compositions and uses thereof |
| US11896636B2 (en) | 2011-07-06 | 2024-02-13 | Glaxosmithkline Biologicals Sa | Immunogenic combination compositions and uses thereof |
| US11905525B2 (en) | 2017-04-05 | 2024-02-20 | Modernatx, Inc. | Reduction of elimination of immune responses to non-intravenous, e.g., subcutaneously administered therapeutic proteins |
Families Citing this family (245)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2354607T3 (en) | 2002-06-28 | 2011-03-16 | Protiva Biotherapeutics Inc. | PROCEDURE AND APPLIANCE TO PRODUCE LIPOSOMES. |
| US9228186B2 (en) | 2002-11-14 | 2016-01-05 | Thermo Fisher Scientific Inc. | Methods and compositions for selecting siRNA of improved functionality |
| CN101291653B (en) * | 2003-07-16 | 2012-06-27 | 普洛体维生物治疗公司 | Lipid encapsulated interfering rna |
| CA2551022C (en) * | 2003-09-15 | 2013-06-04 | Protiva Biotherapeutics, Inc. | Polyethyleneglycol-modified lipid compounds and uses thereof |
| EP1735009A4 (en) | 2004-03-12 | 2011-03-30 | Alnylam Pharmaceuticals Inc | iRNA AGENTS TARGETING VEGF |
| US9315862B2 (en) * | 2004-10-05 | 2016-04-19 | California Institute Of Technology | Aptamer regulated nucleic acids and uses thereof |
| US9393315B2 (en) | 2011-06-08 | 2016-07-19 | Nitto Denko Corporation | Compounds for targeting drug delivery and enhancing siRNA activity |
| HUE043492T2 (en) | 2005-08-23 | 2019-08-28 | Univ Pennsylvania | Rna containing modified nucleosides and methods of use thereof |
| US20070054873A1 (en) * | 2005-08-26 | 2007-03-08 | Protiva Biotherapeutics, Inc. | Glucocorticoid modulation of nucleic acid-mediated immune stimulation |
| US20070218122A1 (en) * | 2005-11-18 | 2007-09-20 | Protiva Biotherapeutics, Inc. | siRNA silencing of influenza virus gene expression |
| ES2682284T3 (en) | 2006-02-14 | 2018-09-19 | Bp Corporation North America Inc. | Xylanases, nucleic acids that encode them and methods to make and use them |
| EP2316962B1 (en) | 2006-03-07 | 2014-07-09 | Cargill, Incorporated | Aldolases, nucleic acids encoding them and methods for making and using them |
| CA2645225A1 (en) | 2006-03-07 | 2007-09-13 | Verenium Corporation | Aldolases, nucleic acids encoding them and methods for making and using them |
| EP3872179A1 (en) | 2006-05-11 | 2021-09-01 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for inhibiting expression of the pcsk9 gene |
| US8598333B2 (en) * | 2006-05-26 | 2013-12-03 | Alnylam Pharmaceuticals, Inc. | SiRNA silencing of genes expressed in cancer |
| US8138160B2 (en) | 2006-08-03 | 2012-03-20 | Warsaw Orthopedic, Inc. | Reagents, methods and systems to suppress pro-inflammatory cytokines |
| EP2056880A4 (en) * | 2006-08-16 | 2010-10-13 | Protiva Biotherapeutics Inc | Nucleic acid modulation of toll-like receptor-mediated immune stimulation |
| EP2057179A4 (en) * | 2006-08-24 | 2010-11-10 | British Columbia Cancer Agency | Compositions and methods for treating myelosuppression |
| KR100817024B1 (en) * | 2006-11-09 | 2008-03-26 | 재단법인 목암생명공학연구소 | Composite for specifically transporting a nucleic acid or a drug to liver and pharmaceutical composition comprising the same |
| US8158595B2 (en) | 2006-11-09 | 2012-04-17 | California Institute Of Technology | Modular aptamer-regulated ribozymes |
| KR100825519B1 (en) * | 2007-01-05 | 2008-04-25 | 주식회사 바이오폴리메드 | A chitosan based polymer conjugate and a method for producing the same |
| CN103555735B (en) | 2007-04-27 | 2016-03-09 | 加利福尼亚大学董事会 | Plant CO 2sensor, encode their nucleic acid and manufacture and their method of use |
| WO2009002719A1 (en) * | 2007-06-22 | 2008-12-31 | The Board Of Regents Of The University Of Texas System | Liposomal inhibitory nucleic acid against stat proteins |
| WO2009011855A2 (en) * | 2007-07-16 | 2009-01-22 | California Institute Of Technology | Selection of nucleic acid-based sensor domains within nucleic acid switch platform |
| US8716255B2 (en) | 2007-08-10 | 2014-05-06 | British Columbia Cancer Agency Branch | Microrna compositions and methods for the treatment of myelogenous leukemia |
| US8367815B2 (en) * | 2007-08-28 | 2013-02-05 | California Institute Of Technology | Modular polynucleotides for ligand-controlled regulatory systems |
| US20120165387A1 (en) | 2007-08-28 | 2012-06-28 | Smolke Christina D | General composition framework for ligand-controlled RNA regulatory systems |
| US8865667B2 (en) | 2007-09-12 | 2014-10-21 | California Institute Of Technology | Higher-order cellular information processing devices |
| CN110577945A (en) | 2007-10-03 | 2019-12-17 | 维莱尼姆公司 | xylanases, nucleic acids encoding them, and methods for making and using them |
| CA2702494A1 (en) | 2007-10-19 | 2009-04-23 | The Regents Of The University Of California | Compositions and methods for ameliorating cns inflammation, psychosis, delirium, ptsd or ptss |
| EP2219587A4 (en) * | 2007-11-14 | 2012-11-21 | Univ California | Sterol-modified amphiphilic lipids |
| US9029524B2 (en) * | 2007-12-10 | 2015-05-12 | California Institute Of Technology | Signal activated RNA interference |
| CN104651381A (en) | 2008-01-03 | 2015-05-27 | 巴斯夫酶有限责任公司 | Transferases and oxidoreductases, nucleic acids encoding them and methods for making and using them |
| EP2238261B1 (en) | 2008-01-03 | 2013-12-04 | Verenium Corporation | Isomerases, nucleic acids encoding them and methods for making and using them |
| AU2009241591A1 (en) * | 2008-01-31 | 2009-11-05 | Alnylam Pharmaceuticals, Inc. | Optimized methods for delivery of DSRNA targeting the PCSK9 gene |
| EA019531B1 (en) * | 2008-03-05 | 2014-04-30 | Элнилэм Фармасьютикалз, Инк. | COMPOSITIONS AND METHODS FOR INHIBITING EXPRESSION OF Eg5 AND VEGF GENES |
| EP2264167B1 (en) | 2008-03-31 | 2016-10-12 | National Institute of Advanced Industrial Science and Technology | Double-stranded lipid-modified rna having high rna interference effect |
| US8815818B2 (en) | 2008-07-18 | 2014-08-26 | Rxi Pharmaceuticals Corporation | Phagocytic cell delivery of RNAI |
| US20110224447A1 (en) * | 2008-08-18 | 2011-09-15 | Bowman Keith A | Novel Lipid Nanoparticles and Novel Components for Delivery of Nucleic Acids |
| CN108165548B (en) | 2008-09-22 | 2022-10-14 | 菲奥医药公司 | Reduced size self-delivering RNAi compounds |
| EP2334793B1 (en) | 2008-09-25 | 2016-04-06 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for inhibiting expression of serum amyloid a gene |
| IL302142B1 (en) | 2008-10-20 | 2024-03-01 | Alnylam Pharmaceuticals Inc | Compositions and Methods for Inhabiting Expression of TRANSTHYRETIN |
| EP3207944B1 (en) | 2008-11-10 | 2020-01-15 | Arbutus Biopharma Corporation | Novel lipids and compositions for the delivery of therapeutics |
| CN102292069B (en) * | 2008-11-26 | 2014-07-30 | 中外制药株式会社 | Vesicle preparation |
| AU2009324534B2 (en) | 2008-12-10 | 2015-07-30 | Alnylam Pharmaceuticals, Inc. | GNAQ targeted dsRNA compositions and methods for inhibiting expression |
| WO2010078536A1 (en) | 2009-01-05 | 2010-07-08 | Rxi Pharmaceuticals Corporation | Inhibition of pcsk9 through rnai |
| WO2010090762A1 (en) | 2009-02-04 | 2010-08-12 | Rxi Pharmaceuticals Corporation | Rna duplexes with single stranded phosphorothioate nucleotide regions for additional functionality |
| US8329882B2 (en) | 2009-02-18 | 2012-12-11 | California Institute Of Technology | Genetic control of mammalian cells with synthetic RNA regulatory systems |
| US20100267806A1 (en) * | 2009-03-12 | 2010-10-21 | David Bumcrot | LIPID FORMULATED COMPOSITIONS AND METHODS FOR INHIBITING EXPRESSION OF Eg5 AND VEGF GENES |
| US9145555B2 (en) | 2009-04-02 | 2015-09-29 | California Institute Of Technology | Integrated—ligand-responsive microRNAs |
| AU2010245933B2 (en) * | 2009-05-05 | 2016-06-16 | Arbutus Biopharma Corporation | Methods of delivering oligonucleotides to immune cells |
| WO2010147992A1 (en) | 2009-06-15 | 2010-12-23 | Alnylam Pharmaceuticals, Inc. | Methods for increasing efficacy of lipid formulated sirna |
| EA201270019A1 (en) * | 2009-06-15 | 2012-06-29 | Элнилэм Фармасьютикалз, Инк. | BENTROVAL RNA INCLUDED IN LIPID COMPOSITION AND WHICH IS THE PCSK9 GENE |
| CA2767129C (en) * | 2009-07-01 | 2015-01-06 | Protiva Biotherapeutics, Inc. | Compositions and methods for silencing apolipoprotein b |
| EP2464336A4 (en) | 2009-08-14 | 2013-11-20 | Alnylam Pharmaceuticals Inc | Lipid formulated compositions and methods for inhibiting expression of a gene from the ebola virus |
| WO2011038031A1 (en) | 2009-09-22 | 2011-03-31 | Alnylam Pharmaceuticals, Inc. | Dual targeting sirna agents |
| WO2011056883A1 (en) | 2009-11-03 | 2011-05-12 | Alnylam Pharmaceuticals, Inc. | Lipid formulated compositions and methods for inhibiting expression of transthyretin (ttr) |
| AU2014250713B2 (en) * | 2009-12-01 | 2016-07-28 | Translate Bio, Inc. | Delivery Of mRNA For The Augmentation Of Proteins And Enzymes In Human Genetic Diseases |
| AU2016250459B2 (en) * | 2009-12-01 | 2018-06-28 | Translate Bio, Inc. | Delivery Of mRNA For The Augmentation Of Proteins And Enzymes In Human Genetic Diseases |
| EP2526113B1 (en) | 2010-01-22 | 2016-08-10 | Sirna Therapeutics, Inc. | Post-synthetic chemical modification of rna at the 2'-position of the ribose ring via "click" chemistry |
| KR20180044433A (en) | 2010-03-24 | 2018-05-02 | 알엑스아이 파마슈티칼스 코포레이션 | Rna interference in dermal and fibrotic indications |
| EP2550001B1 (en) | 2010-03-24 | 2019-05-22 | Phio Pharmaceuticals Corp. | Rna interference in ocular indications |
| WO2011120053A1 (en) | 2010-03-26 | 2011-09-29 | Mersana Therapeutics, Inc. | Modified polymers for delivery of polynucleotides, method of manufacture, and methods of use thereof |
| US9006417B2 (en) | 2010-06-30 | 2015-04-14 | Protiva Biotherapeutics, Inc. | Non-liposomal systems for nucleic acid delivery |
| US20130210663A1 (en) | 2010-08-04 | 2013-08-15 | Cizzle Biotechnology Limited | Methods and compounds for the diagnosis and treatment of cancer |
| CA2807552A1 (en) | 2010-08-06 | 2012-02-09 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| US8466122B2 (en) | 2010-09-17 | 2013-06-18 | Protiva Biotherapeutics, Inc. | Trialkyl cationic lipids and methods of use thereof |
| EP4108671A1 (en) | 2010-10-01 | 2022-12-28 | ModernaTX, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| AU2012236099A1 (en) | 2011-03-31 | 2013-10-03 | Moderna Therapeutics, Inc. | Delivery and formulation of engineered nucleic acids |
| US10196637B2 (en) | 2011-06-08 | 2019-02-05 | Nitto Denko Corporation | Retinoid-lipid drug carrier |
| US9011903B2 (en) | 2011-06-08 | 2015-04-21 | Nitto Denko Corporation | Cationic lipids for therapeutic agent delivery formulations |
| US9464124B2 (en) | 2011-09-12 | 2016-10-11 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| CN103974724B (en) | 2011-10-03 | 2019-08-30 | 现代泰克斯公司 | Nucleosides, nucleotide and nucleic acid of modification and application thereof |
| HUE048622T2 (en) | 2011-11-18 | 2020-08-28 | Alnylam Pharmaceuticals Inc | Rnai agents, compositions and methods of use thereof for treating transthyretin (ttr) associated diseases |
| US9061063B2 (en) | 2011-12-07 | 2015-06-23 | Alnylam Pharmaceuticals, Inc. | Biodegradable lipids for the delivery of active agents |
| CA2859387A1 (en) | 2011-12-16 | 2013-06-20 | Moderna Therapeutics, Inc. | Modified nucleoside, nucleotide, and nucleic acid compositions |
| US9572897B2 (en) | 2012-04-02 | 2017-02-21 | Modernatx, Inc. | Modified polynucleotides for the production of cytoplasmic and cytoskeletal proteins |
| EP2864485A4 (en) | 2012-06-22 | 2016-03-02 | Univ California | Compositions and methods for mediating plant stomatal development in response to carbon dioxide and applications for engineering drought tolerance in plants |
| JP6108197B2 (en) | 2012-07-02 | 2017-04-05 | 日油株式会社 | Method for producing tertiary amino group-containing lipid |
| JP6144355B2 (en) | 2012-11-26 | 2017-06-07 | モデルナティエックス インコーポレイテッドModernaTX,Inc. | Chemically modified mRNA |
| SG10201707569YA (en) | 2012-12-12 | 2017-10-30 | Broad Inst Inc | Delivery, Engineering and Optimization of Systems, Methods and Compositions for Sequence Manipulation and Therapeutic Applications |
| WO2014160243A1 (en) | 2013-03-14 | 2014-10-02 | The Trustees Of The University Of Pennsylvania | Purification and purity assessment of rna molecules synthesized with modified nucleosides |
| US8980864B2 (en) | 2013-03-15 | 2015-03-17 | Moderna Therapeutics, Inc. | Compositions and methods of altering cholesterol levels |
| WO2014140856A2 (en) | 2013-03-15 | 2014-09-18 | Graham Lord | Mir-142 and antagonists thereof for treating disease |
| EP3597755A1 (en) | 2013-06-17 | 2020-01-22 | The Broad Institute, Inc. | Delivery, use and therapeutic applications of the crispr-cas systems and compositions for targeting disorders and diseases using viral components |
| MX2015017312A (en) | 2013-06-17 | 2017-04-10 | Broad Inst Inc | Delivery and use of the crispr-cas systems, vectors and compositions for hepatic targeting and therapy. |
| KR20160019553A (en) | 2013-06-17 | 2016-02-19 | 더 브로드 인스티튜트, 인코퍼레이티드 | Delivery, engineering and optimization of systems, methods and compositions for targeting and modeling diseases and disorders of post mitotic cells |
| WO2015048744A2 (en) | 2013-09-30 | 2015-04-02 | Moderna Therapeutics, Inc. | Polynucleotides encoding immune modulating polypeptides |
| EA201690675A1 (en) | 2013-10-03 | 2016-08-31 | Модерна Терапьютикс, Инк. | POLYNUCLEOTES ENCODING THE RECEPTOR OF LOW DENSITY LIPOPROTEINS |
| US10202601B2 (en) | 2013-11-22 | 2019-02-12 | Mina Therapeutics Limited | C/EBPα short activating RNA compositions and methods of use |
| JP6883987B2 (en) | 2013-12-04 | 2021-06-09 | フィオ ファーマシューティカルズ コーポレーションPhio Pharmaceuticals Corp. | Methods for wound healing procedures utilizing chemically modified oligonucleotides |
| EP3080271B1 (en) | 2013-12-12 | 2020-02-12 | The Broad Institute, Inc. | Systems, methods and compositions for sequence manipulation with optimized functional crispr-cas systems |
| RU2016128077A (en) | 2013-12-12 | 2018-12-06 | Те Брод Инститьют Инк. | DELIVERY, APPLICATION AND APPLICATIONS IN THE THERAPY OF CRISPR-CAS SYSTEMS AND COMPOSITIONS FOR TREATMENT OF CONDITIONED HBV AND VIRAL DISEASES AND DISORDERS |
| WO2015089419A2 (en) | 2013-12-12 | 2015-06-18 | The Broad Institute Inc. | Delivery, use and therapeutic applications of the crispr-cas systems and compositions for targeting disorders and diseases using particle delivery components |
| CA2932436A1 (en) | 2013-12-12 | 2015-06-18 | The Broad Institute, Inc. | Compositions and methods of use of crispr-cas systems in nucleotide repeat disorders |
| MX2016007328A (en) | 2013-12-12 | 2017-07-19 | Broad Inst Inc | Delivery, use and therapeutic applications of the crispr-cas systems and compositions for genome editing. |
| EP3080259B1 (en) | 2013-12-12 | 2023-02-01 | The Broad Institute, Inc. | Engineering of systems, methods and optimized guide compositions with new architectures for sequence manipulation |
| EP3137119B1 (en) | 2014-04-28 | 2020-07-01 | Phio Pharmaceuticals Corp. | Methods for treating cancer using a nucleic acid targeting mdm2 |
| WO2015168605A1 (en) | 2014-05-01 | 2015-11-05 | Rxi Pharmaceuticals Corporation | Methods for treatment of disorders in the front of the eye utilizing nucleic acid molecules |
| EP3154694A1 (en) | 2014-06-13 | 2017-04-19 | Children's Medical Center Corporation | Products and methods to isolate mitochondria |
| EP3766916B1 (en) | 2014-06-25 | 2022-09-28 | Acuitas Therapeutics Inc. | Novel lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| ES2780904T3 (en) | 2014-08-17 | 2020-08-27 | Broad Inst Inc | Genomic editing using Cas9 nickases |
| ES2928500T3 (en) | 2014-08-29 | 2022-11-18 | Alnylam Pharmaceuticals Inc | Patisiran for use in the treatment of transthyretin-mediated amyloidosis |
| EP3188799B1 (en) | 2014-09-05 | 2022-07-06 | Phio Pharmaceuticals Corp. | Methods for treating aging and skin disorders using nucleic acids targeting tyr or mmp1 |
| WO2016049163A2 (en) | 2014-09-24 | 2016-03-31 | The Broad Institute Inc. | Use and production of chd8+/- transgenic animals with behavioral phenotypes characteristic of autism spectrum disorder |
| WO2016049258A2 (en) | 2014-09-25 | 2016-03-31 | The Broad Institute Inc. | Functional screening with optimized functional crispr-cas systems |
| EP3230452A1 (en) | 2014-12-12 | 2017-10-18 | The Broad Institute Inc. | Dead guides for crispr transcription factors |
| EP3230451B1 (en) | 2014-12-12 | 2021-04-07 | The Broad Institute, Inc. | Protected guide rnas (pgrnas) |
| WO2016094874A1 (en) | 2014-12-12 | 2016-06-16 | The Broad Institute Inc. | Escorted and functionalized guides for crispr-cas systems |
| WO2016094880A1 (en) | 2014-12-12 | 2016-06-16 | The Broad Institute Inc. | Delivery, use and therapeutic applications of crispr systems and compositions for genome editing as to hematopoietic stem cells (hscs) |
| EP3234192B1 (en) | 2014-12-19 | 2021-07-14 | The Broad Institute, Inc. | Unbiased identification of double-strand breaks and genomic rearrangement by genome-wide insert capture sequencing |
| WO2016106236A1 (en) | 2014-12-23 | 2016-06-30 | The Broad Institute Inc. | Rna-targeting system |
| AU2015369725A1 (en) | 2014-12-24 | 2017-06-29 | Massachusetts Institute Of Technology | CRISPR having or associated with destabilization domains |
| WO2016205745A2 (en) | 2015-06-18 | 2016-12-22 | The Broad Institute Inc. | Cell sorting |
| CA3012631A1 (en) | 2015-06-18 | 2016-12-22 | The Broad Institute Inc. | Novel crispr enzymes and systems |
| CA3012607A1 (en) | 2015-06-18 | 2016-12-22 | The Broad Institute Inc. | Crispr enzymes and systems |
| US9790490B2 (en) | 2015-06-18 | 2017-10-17 | The Broad Institute Inc. | CRISPR enzymes and systems |
| SG10201912329YA (en) | 2015-06-18 | 2020-02-27 | Broad Inst Inc | Crispr Enzyme Mutations Reducing Off-Target Effects |
| CN107922364B (en) | 2015-06-29 | 2021-12-31 | 爱康泰生治疗公司 | Lipid and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2017007813A1 (en) | 2015-07-06 | 2017-01-12 | Rxi Pharmaceuticals Corporation | Nucleic acid molecules targeting superoxide dismutase 1 (sod1) |
| WO2017007825A1 (en) | 2015-07-06 | 2017-01-12 | Rxi Pharmaceuticals Corporation | Methods for treating neurological disorders using a synergistic small molecule and nucleic acids therapeutic approach |
| ES2842300T3 (en) | 2015-07-31 | 2021-07-13 | Alnylam Pharmaceuticals Inc | Transthyretin (TTR) RNAi Compositions and Methods for Their Use for the Treatment or Prevention of TTR-Associated Diseases |
| US9856481B2 (en) | 2015-08-13 | 2018-01-02 | Ann & Robert H. Lurie Children's Hospital | MicroRNA treatment of fibrosis |
| WO2017031370A1 (en) | 2015-08-18 | 2017-02-23 | The Broad Institute, Inc. | Methods and compositions for altering function and structure of chromatin loops and/or domains |
| CN108366966A (en) | 2015-08-24 | 2018-08-03 | 光环生物干扰疗法公司 | Polynucleotides nano particle and application thereof for adjusting gene expression |
| LT3350157T (en) | 2015-09-17 | 2022-02-25 | Modernatx, Inc. | Compounds and compositions for intracellular delivery of therapeutic agents |
| WO2017069958A2 (en) | 2015-10-09 | 2017-04-27 | The Brigham And Women's Hospital, Inc. | Modulation of novel immune checkpoint targets |
| WO2017070151A1 (en) | 2015-10-19 | 2017-04-27 | Rxi Pharmaceuticals Corporation | Reduced size self-delivering nucleic acid compounds targeting long non-coding rna |
| CA3024543A1 (en) | 2015-10-22 | 2017-04-27 | The Broad Institute, Inc. | Type vi-b crispr enzymes and systems |
| EP3368687B1 (en) | 2015-10-27 | 2021-09-29 | The Broad Institute, Inc. | Compositions and methods for targeting cancer-specific sequence variations |
| CN113636947A (en) | 2015-10-28 | 2021-11-12 | 爱康泰生治疗公司 | Novel lipid and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2018081480A1 (en) | 2016-10-26 | 2018-05-03 | Acuitas Therapeutics, Inc. | Lipid nanoparticle formulations |
| WO2017075478A2 (en) | 2015-10-28 | 2017-05-04 | The Broad Institute Inc. | Compositions and methods for evaluating and modulating immune responses by use of immune cell gene signatures |
| WO2017075465A1 (en) | 2015-10-28 | 2017-05-04 | The Broad Institute Inc. | Compositions and methods for evaluating and modulating immune responses by detecting and targeting gata3 |
| WO2017075451A1 (en) | 2015-10-28 | 2017-05-04 | The Broad Institute Inc. | Compositions and methods for evaluating and modulating immune responses by detecting and targeting pou2af1 |
| EP3397261A4 (en) | 2015-11-30 | 2019-07-03 | Flagship Pioneering Innovations V, Inc. | Methods and compositions relating to chondrisomes |
| WO2017096326A1 (en) | 2015-12-02 | 2017-06-08 | Massachusetts Institute Of Technology | Method for efficient generation of neurons from non-neuronal cells |
| ES2924407T3 (en) | 2015-12-10 | 2022-10-06 | Modernatx Inc | Compositions and methods for the delivery of therapeutic agents |
| US20190233814A1 (en) | 2015-12-18 | 2019-08-01 | The Broad Institute, Inc. | Novel crispr enzymes and systems |
| JP7114465B2 (en) | 2015-12-22 | 2022-08-08 | モデルナティエックス インコーポレイテッド | Compounds and compositions for intracellular delivery of drugs |
| CA3026112A1 (en) | 2016-04-19 | 2017-10-26 | The Broad Institute, Inc. | Cpf1 complexes with reduced indel activity |
| WO2017184768A1 (en) | 2016-04-19 | 2017-10-26 | The Broad Institute Inc. | Novel crispr enzymes and systems |
| US20200263190A1 (en) | 2016-04-19 | 2020-08-20 | The Broad Institute, Inc. | Novel crispr enzymes and systems |
| CN109642231A (en) | 2016-06-17 | 2019-04-16 | 博德研究所 | VI type CRISPR ortholog and system |
| US20210222164A1 (en) | 2016-06-29 | 2021-07-22 | The Broad Institute, Inc. | Crispr-cas systems having destabilization domain |
| US20200283743A1 (en) | 2016-08-17 | 2020-09-10 | The Broad Institute, Inc. | Novel crispr enzymes and systems |
| CN110114461A (en) | 2016-08-17 | 2019-08-09 | 博德研究所 | Novel C RISPR enzyme and system |
| US20190262399A1 (en) | 2016-09-07 | 2019-08-29 | The Broad Institute, Inc. | Compositions and methods for evaluating and modulating immune responses |
| WO2018067991A1 (en) | 2016-10-07 | 2018-04-12 | The Brigham And Women's Hospital, Inc. | Modulation of novel immune checkpoint targets |
| US11583504B2 (en) | 2016-11-08 | 2023-02-21 | Modernatx, Inc. | Stabilized formulations of lipid nanoparticles |
| US11739308B2 (en) | 2017-03-15 | 2023-08-29 | The Broad Institute, Inc. | Cas13b orthologues CRISPR enzymes and systems |
| JP7220154B2 (en) | 2017-03-15 | 2023-02-09 | モデルナティエックス インコーポレイテッド | Crystalline forms of amino lipids |
| RS63953B1 (en) | 2017-03-15 | 2023-02-28 | Modernatx Inc | Compound and compositions for intracellular delivery of therapeutic agents |
| AU2018251801A1 (en) | 2017-04-12 | 2019-11-07 | Massachusetts Institute Of Technology | Novel type VI crispr orthologs and systems |
| WO2018191657A1 (en) | 2017-04-13 | 2018-10-18 | Acuitas Therapeutics, Inc. | Lipids for delivery of active agents |
| WO2018191719A1 (en) | 2017-04-13 | 2018-10-18 | Acuitas Therapeutics, Inc. | Lipid delivery of therapeutic agents to adipose tissue |
| WO2018191750A2 (en) | 2017-04-14 | 2018-10-18 | The Broad Institute Inc. | Novel delivery of large payloads |
| WO2018200943A1 (en) | 2017-04-28 | 2018-11-01 | Acuitas Therapeutics, Inc. | Novel carbonyl lipids and lipid nanoparticle formulations for delivery of nucleic acids |
| WO2018204777A2 (en) | 2017-05-05 | 2018-11-08 | The Broad Institute, Inc. | Methods for identification and modification of lncrna associated with target genotypes and phenotypes |
| EP3638797A1 (en) | 2017-06-13 | 2020-04-22 | Flagship Pioneering Innovations V, Inc. | Compositions comprising curons and uses thereof |
| US11639329B2 (en) | 2017-08-16 | 2023-05-02 | Acuitas Therapeutics, Inc. | Lipids for use in lipid nanoparticle formulations |
| US11542225B2 (en) | 2017-08-17 | 2023-01-03 | Acuitas Therapeutics, Inc. | Lipids for use in lipid nanoparticle formulations |
| US11524932B2 (en) | 2017-08-17 | 2022-12-13 | Acuitas Therapeutics, Inc. | Lipids for use in lipid nanoparticle formulations |
| WO2019048631A1 (en) | 2017-09-08 | 2019-03-14 | Mina Therapeutics Limited | Hnf4a sarna compositions and methods of use |
| WO2019048645A1 (en) | 2017-09-08 | 2019-03-14 | Mina Therapeutics Limited | Stabilized cebpa sarna compositions and methods of use |
| KR20200089656A (en) | 2017-09-19 | 2020-07-27 | 알닐람 파마슈티칼스 인코포레이티드 | Compositions and methods for treating transthyretin (TTR) mediated amyloidosis |
| EP3684397A4 (en) | 2017-09-21 | 2021-08-18 | The Broad Institute, Inc. | Systems, methods, and compositions for targeted nucleic acid editing |
| US11547614B2 (en) | 2017-10-31 | 2023-01-10 | The Broad Institute, Inc. | Methods and compositions for studying cell evolution |
| WO2019089828A1 (en) | 2017-10-31 | 2019-05-09 | Acuitas Therapeutics, Inc. | Lamellar lipid nanoparticles |
| EP3710039A4 (en) | 2017-11-13 | 2021-08-04 | The Broad Institute, Inc. | Methods and compositions for treating cancer by targeting the clec2d-klrb1 pathway |
| EP4242307A3 (en) | 2018-04-12 | 2023-12-27 | MiNA Therapeutics Limited | Sirt1-sarna compositions and methods of use |
| CA3106035A1 (en) | 2018-08-07 | 2020-02-13 | The Broad Institute, Inc. | Cas12b enzymes and systems |
| WO2020041380A1 (en) | 2018-08-20 | 2020-02-27 | The Broad Institute, Inc. | Methods and compositions for optochemical control of crispr-cas9 |
| JP2021535226A (en) | 2018-09-04 | 2021-12-16 | ザ ボード オブ リージェンツ オブ ザ ユニバーシティー オブ テキサス システム | Compositions and Methods for Organ-Specific Delivery of Nucleic Acids |
| GB2606038B (en) | 2018-09-04 | 2023-05-03 | Univ Texas | Compositions and methods for organ specific delivery of nucleic acids |
| EP3852911A2 (en) | 2018-09-21 | 2021-07-28 | Acuitas Therapeutics, Inc. | Systems and methods for manufacturing lipid nanoparticles and liposomes |
| WO2020077007A1 (en) | 2018-10-09 | 2020-04-16 | The University Of British Columbia | Compositions and systems comprising transfection-competent vesicles free of organic-solvents and detergents and methods related thereto |
| US20220072154A1 (en) * | 2018-11-09 | 2022-03-10 | Arbutus Biopharma Corporation | Negatively charged peg-lipid conjugates |
| WO2020146805A1 (en) | 2019-01-11 | 2020-07-16 | Acuitas Therapeutics, Inc. | Lipids for lipid nanoparticle delivery of active agents |
| US20220177863A1 (en) | 2019-03-18 | 2022-06-09 | The Broad Institute, Inc. | Type vii crispr proteins and systems |
| EP3947646A1 (en) | 2019-04-05 | 2022-02-09 | Precision BioSciences, Inc. | Methods of preparing populations of genetically-modified immune cells |
| EP3953473A1 (en) | 2019-04-12 | 2022-02-16 | MiNA Therapeutics Limited | Sirt1-sarna compositions and methods of use |
| WO2020236972A2 (en) | 2019-05-20 | 2020-11-26 | The Broad Institute, Inc. | Non-class i multi-component nucleic acid targeting systems |
| MA56520A (en) | 2019-06-18 | 2022-04-27 | Janssen Sciences Ireland Unlimited Co | CONSTRUCTION OF RECOMBINANT INTERLEUKIN-12 AND ITS USES |
| CN114630675A (en) | 2019-06-18 | 2022-06-14 | 爱尔兰詹森科学公司 | Combination of Hepatitis B Virus (HBV) vaccine and anti-PD-1 or anti-PD-L1 antibody |
| WO2020255010A1 (en) | 2019-06-18 | 2020-12-24 | Janssen Sciences Ireland Unlimited Company | Combination of recombinant interleukin 12 construct and hepatitis b virus (hbv) vaccines |
| JP2022536945A (en) | 2019-06-18 | 2022-08-22 | ヤンセン・サイエンシズ・アイルランド・アンリミテッド・カンパニー | Combination of hepatitis B virus (HBV) vaccine and RNAi targeting HBV |
| EP3986460A2 (en) | 2019-06-18 | 2022-04-27 | Janssen Sciences Ireland Unlimited Company | Combination of hepatitis b virus (hbv) vaccines and anti-pd-1 antibody |
| WO2020255062A1 (en) | 2019-06-20 | 2020-12-24 | Janssen Sciences Ireland Unlimited Company | Lipid nanoparticle or liposome delivery of hepatitis b virus (hbv) vaccines |
| MX2022001720A (en) | 2019-08-14 | 2022-03-11 | Acuitas Therapeutics Inc | Improved lipid nanoparticles for delivery of nucleic acids. |
| BR112022004759A2 (en) | 2019-09-19 | 2022-06-21 | Modernatx Inc | Branched tail lipid compositions and compounds for intracellular delivery of therapeutic agents |
| EP4055167A2 (en) | 2019-11-08 | 2022-09-14 | Phio Pharmaceuticals Corp. | Chemically modified oligonucleotides targeting bromodomain containing protein 4 (brd4) for immunotherapy |
| WO2021138537A1 (en) | 2019-12-31 | 2021-07-08 | Phio Pharmaceuticals Corp. | Chemically modified oligonucleotides with improved systemic delivery |
| IL296781A (en) | 2020-03-30 | 2022-11-01 | BioNTech SE | Rna compositions targeting claudin-18.2 |
| EP4164668A1 (en) | 2020-06-15 | 2023-04-19 | Research Institute at Nationwide Children's Hospital | Adeno-associated virus vector delivery for muscular dystrophies |
| AU2021303478A1 (en) | 2020-07-08 | 2023-02-16 | Janssen Sciences Ireland Unlimited Company | RNA replicon vaccines against HBV |
| WO2022076547A1 (en) | 2020-10-07 | 2022-04-14 | Precision Biosciences, Inc. | Lipid nanoparticle compositions |
| EP4251170A1 (en) | 2020-11-25 | 2023-10-04 | Akagera Medicines, Inc. | Lipid nanoparticles for delivery of nucleic acids, and related methods of use |
| GB2603454A (en) | 2020-12-09 | 2022-08-10 | Ucl Business Ltd | Novel therapeutics for the treatment of neurodegenerative disorders |
| TW202245809A (en) | 2020-12-18 | 2022-12-01 | 美商詹森藥物公司 | Combination therapy for treating hepatitis b virus infection |
| EP4267740A1 (en) | 2020-12-28 | 2023-11-01 | Arcturus Therapeutics, Inc. | Transcription activator-like effector nucleases (talens) targeting hbv |
| US11524023B2 (en) | 2021-02-19 | 2022-12-13 | Modernatx, Inc. | Lipid nanoparticle compositions and methods of formulating the same |
| AU2022246144A1 (en) | 2021-03-26 | 2023-09-21 | Mina Therapeutics Limited | Tmem173 sarna compositions and methods of use |
| EP4319803A1 (en) | 2021-04-08 | 2024-02-14 | Vaxthera SAS | Coronavirus vaccine comprising a mosaic protein |
| CA3173953A1 (en) | 2021-06-11 | 2023-12-10 | Tyson D. BOWEN | Rna polymerase iii promoters and methods of use |
| AU2022301302A1 (en) | 2021-07-01 | 2024-01-25 | Indapta Therapeutics, Inc. | Engineered natural killer (nk) cells and related methods |
| AU2022318664A1 (en) | 2021-07-30 | 2024-02-29 | Tune Therapeutics, Inc. | Compositions and methods for modulating expression of methyl-cpg binding protein 2 (mecp2) |
| CA3227103A1 (en) | 2021-07-30 | 2023-02-02 | Matthew P. GEMBERLING | Compositions and methods for modulating expression of frataxin (fxn) |
| WO2023015265A2 (en) | 2021-08-04 | 2023-02-09 | Phio Pharmaceuticals Corp. | Chemically modified oligonucleotides |
| WO2023015264A1 (en) | 2021-08-04 | 2023-02-09 | Phio Pharmaceuticals Corp. | Immunotherapy of cancer utilizing natural killer cells treated with chemically modified oligonucleotides |
| WO2023023055A1 (en) | 2021-08-16 | 2023-02-23 | Renagade Therapeutics Management Inc. | Compositions and methods for optimizing tropism of delivery systems for rna |
| CA3231523A1 (en) | 2021-09-14 | 2023-03-23 | Renagade Therapeutics Management Inc. | Acyclic lipids and methods of use thereof |
| WO2023044333A1 (en) | 2021-09-14 | 2023-03-23 | Renagade Therapeutics Management Inc. | Cyclic lipids and methods of use thereof |
| WO2023081756A1 (en) | 2021-11-03 | 2023-05-11 | The J. David Gladstone Institutes, A Testamentary Trust Established Under The Will Of J. David Gladstone | Precise genome editing using retrons |
| WO2023091490A1 (en) | 2021-11-16 | 2023-05-25 | Senda Biosciences, Inc. | Novel ionizable lipids and lipid nanoparticles and methods of using the same |
| WO2023091787A1 (en) | 2021-11-22 | 2023-05-25 | Senda Biosciences, Inc. | Novel ionizable lipids and lipid nanoparticles and methods of using the same |
| WO2023099884A1 (en) | 2021-12-01 | 2023-06-08 | Mina Therapeutics Limited | Pax6 sarna compositions and methods of use |
| GB202117758D0 (en) | 2021-12-09 | 2022-01-26 | Ucl Business Ltd | Therapeutics for the treatment of neurodegenerative disorders |
| WO2023122752A1 (en) | 2021-12-23 | 2023-06-29 | Renagade Therapeutics Management Inc. | Constrained lipids and methods of use thereof |
| WO2023133595A2 (en) | 2022-01-10 | 2023-07-13 | Sana Biotechnology, Inc. | Methods of ex vivo dosing and administration of lipid particles or viral vectors and related systems and uses |
| WO2023141602A2 (en) | 2022-01-21 | 2023-07-27 | Renagade Therapeutics Management Inc. | Engineered retrons and methods of use |
| WO2023147090A1 (en) | 2022-01-27 | 2023-08-03 | BioNTech SE | Pharmaceutical compositions for delivery of herpes simplex virus antigens and related methods |
| WO2023150647A1 (en) | 2022-02-02 | 2023-08-10 | Sana Biotechnology, Inc. | Methods of repeat dosing and administration of lipid particles or viral vectors and related systems and uses |
| WO2023170435A1 (en) | 2022-03-07 | 2023-09-14 | Mina Therapeutics Limited | Il10 sarna compositions and methods of use |
| WO2023183616A1 (en) | 2022-03-25 | 2023-09-28 | Senda Biosciences, Inc. | Novel ionizable lipids and lipid nanoparticles and methods of using the same |
| WO2023196818A1 (en) | 2022-04-04 | 2023-10-12 | The Regents Of The University Of California | Genetic complementation compositions and methods |
| WO2023196931A1 (en) | 2022-04-07 | 2023-10-12 | Renagade Therapeutics Management Inc. | Cyclic lipids and lipid nanoparticles (lnp) for the delivery of nucleic acids or peptides for use in vaccinating against infectious agents |
| WO2023218420A1 (en) | 2022-05-13 | 2023-11-16 | Janssen Pharmaceuticals, Inc. | Mrna compositions for inducing latent hiv-1 reversal |
| WO2023218431A1 (en) | 2022-05-13 | 2023-11-16 | BioNTech SE | Rna compositions targeting hiv |
| WO2023230295A1 (en) | 2022-05-25 | 2023-11-30 | BioNTech SE | Rna compositions for delivery of monkeypox antigens and related methods |
| WO2023230587A2 (en) * | 2022-05-25 | 2023-11-30 | Akagera Medicines, Inc. | Lipid nanoparticles for delivery of nucleic acids and methods of use thereof |
| WO2023232747A1 (en) | 2022-05-30 | 2023-12-07 | BioNTech SE | Complexes for delivery of nucleic acids |
| WO2023233290A1 (en) | 2022-05-31 | 2023-12-07 | Janssen Sciences Ireland Unlimited Company | Rnai agents targeting pd-l1 |
| WO2023250511A2 (en) | 2022-06-24 | 2023-12-28 | Tune Therapeutics, Inc. | Compositions, systems, and methods for reducing low-density lipoprotein through targeted gene repression |
| WO2024007020A1 (en) | 2022-06-30 | 2024-01-04 | Indapta Therapeutics, Inc. | Combination of engineered natural killer (nk) cells and antibody therapy and related methods |
| WO2024015881A2 (en) | 2022-07-12 | 2024-01-18 | Tune Therapeutics, Inc. | Compositions, systems, and methods for targeted transcriptional activation |
| WO2024033901A1 (en) | 2022-08-12 | 2024-02-15 | LifeEDIT Therapeutics, Inc. | Rna-guided nucleases and active fragments and variants thereof and methods of use |
| US20240067968A1 (en) | 2022-08-19 | 2024-02-29 | Tune Therapeutics, Inc. | Compositions, systems, and methods for regulation of hepatitis b virus through targeted gene repression |
| WO2024040222A1 (en) | 2022-08-19 | 2024-02-22 | Generation Bio Co. | Cleavable closed-ended dna (cedna) and methods of use thereof |
| WO2024049979A2 (en) | 2022-08-31 | 2024-03-07 | Senda Biosciences, Inc. | Novel ionizable lipids and lipid nanoparticles and methods of using the same |
| WO2024064642A2 (en) | 2022-09-19 | 2024-03-28 | Tune Therapeutics, Inc. | Compositions, systems, and methods for modulating t cell function |
| WO2024063789A1 (en) | 2022-09-23 | 2024-03-28 | BioNTech SE | Compositions for delivery of malaria antigens and related methods |
| WO2024064931A1 (en) | 2022-09-23 | 2024-03-28 | BioNTech SE | Compositions for delivery of liver stage antigens and related methods |
| WO2024064934A1 (en) | 2022-09-23 | 2024-03-28 | BioNTech SE | Compositions for delivery of plasmodium csp antigens and related methods |
| WO2024063788A1 (en) | 2022-09-23 | 2024-03-28 | BioNTech SE | Compositions for delivery of malaria antigens and related methods |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5208036A (en) | 1985-01-07 | 1993-05-04 | Syntex (U.S.A.) Inc. | N-(ω, (ω-1)-dialkyloxy)- and N-(ω, (ω-1)-dialkenyloxy)-alk-1-yl-N,N,N-tetrasubstituted ammonium lipids and uses therefor |
| US5264618A (en) | 1990-04-19 | 1993-11-23 | Vical, Inc. | Cationic lipids for intracellular delivery of biologically active molecules |
| US5279833A (en) | 1990-04-04 | 1994-01-18 | Yale University | Liposomal transfection of nucleic acids into animal cells |
| US5283185A (en) | 1991-08-28 | 1994-02-01 | University Of Tennessee Research Corporation | Method for delivering nucleic acids into cells |
| US5753613A (en) | 1994-09-30 | 1998-05-19 | Inex Pharmaceuticals Corporation | Compositions for the introduction of polyanionic materials into cells |
| US5785992A (en) | 1994-09-30 | 1998-07-28 | Inex Pharmaceuticals Corp. | Compositions for the introduction of polyanionic materials into cells |
| US5885613A (en) | 1994-09-30 | 1999-03-23 | The University Of British Columbia | Bilayer stabilizing components and their use in forming programmable fusogenic liposomes |
| WO2002087541A1 (en) * | 2001-04-30 | 2002-11-07 | Protiva Biotherapeutics Inc. | Lipid-based formulations for gene transfer |
| US20050008689A1 (en) | 1997-05-14 | 2005-01-13 | Inex Pharmaceuticals Corporation | High efficiency encapsulation of charged therapeutic agents in lipid vesicles |
| WO2005026372A1 (en) | 2003-09-15 | 2005-03-24 | Protiva Biotherapeutics, Inc. | Polyethyleneglycol-modified lipid compounds and uses thereof |
Family Cites Families (88)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4394448A (en) * | 1978-02-24 | 1983-07-19 | Szoka Jr Francis C | Method of inserting DNA into living cells |
| EP0032578B1 (en) * | 1980-01-16 | 1984-07-25 | Hans Georg Prof. Dr. Weder | Process and dialysis-installation for the preparation of bilayer-vesicles and their use |
| US4598051A (en) * | 1980-03-12 | 1986-07-01 | The Regents Of The University Of California | Liposome conjugates and diagnostic methods therewith |
| US4515736A (en) * | 1983-05-12 | 1985-05-07 | The Regents Of The University Of California | Method for encapsulating materials into liposomes |
| US4897355A (en) * | 1985-01-07 | 1990-01-30 | Syntex (U.S.A.) Inc. | N[ω,(ω-1)-dialkyloxy]- and N-[ω,(ω-1)-dialkenyloxy]-alk-1-yl-N,N,N-tetrasubstituted ammonium lipids and uses therefor |
| US5550289A (en) * | 1985-01-07 | 1996-08-27 | Syntex (U.S.A.) Inc. | N-(1,(1-1)-dialkyloxy)-and N-(1,(1-1)-dialkenyloxy alk-1-yl-N-N,N-tetrasubstituted ammonium lipids and uses therefor |
| US5320906A (en) * | 1986-12-15 | 1994-06-14 | Vestar, Inc. | Delivery vehicles with amphiphile-associated active ingredient |
| US5703055A (en) * | 1989-03-21 | 1997-12-30 | Wisconsin Alumni Research Foundation | Generation of antibodies through lipid mediated DNA delivery |
| FR2645866B1 (en) * | 1989-04-17 | 1991-07-05 | Centre Nat Rech Scient | NEW LIPOPOLYAMINES, THEIR PREPARATION AND THEIR USE |
| JPH03126211A (en) | 1989-10-12 | 1991-05-29 | Nippon Chemicon Corp | Electrolyte for electrolytic capacitor |
| US5013556A (en) * | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5225212A (en) * | 1989-10-20 | 1993-07-06 | Liposome Technology, Inc. | Microreservoir liposome composition and method |
| US6465188B1 (en) * | 1990-06-11 | 2002-10-15 | Gilead Sciences, Inc. | Nucleic acid ligand complexes |
| JP2774417B2 (en) | 1991-08-07 | 1998-07-09 | 株式会社ディ・ディ・エス研究所 | Branched-chain saccharide complex having peptide skeleton and fine particle carrier |
| AU3467193A (en) | 1991-12-17 | 1993-07-19 | Regents Of The University Of California, The | Gene therapy for cystic fibrosis transmembrane conductance regulator activity (CFTR) |
| US5858784A (en) | 1991-12-17 | 1999-01-12 | The Regents Of The University Of California | Expression of cloned genes in the lung by aerosol- and liposome-based delivery |
| CA2130264A1 (en) * | 1992-02-19 | 1993-09-02 | Harris Busch | Oligonucleotide modulation of cell growth |
| CA2134773A1 (en) | 1992-06-04 | 1993-12-09 | Robert J. Debs | Methods and compositions for in vivo gene therapy |
| US5334761A (en) | 1992-08-28 | 1994-08-02 | Life Technologies, Inc. | Cationic lipids |
| JP2854203B2 (en) | 1992-09-03 | 1999-02-03 | 株式会社ディ・ディ・エス研究所 | Method for producing liposomes |
| US5578475A (en) | 1993-07-12 | 1996-11-26 | Life Technologies, Inc. | Composition and methods for transfecting eukaryotic cells |
| US5674908A (en) | 1993-12-20 | 1997-10-07 | Life Technologies, Inc. | Highly packed polycationic ammonium, sulfonium and phosphonium lipids |
| FR2714830B1 (en) | 1994-01-10 | 1996-03-22 | Rhone Poulenc Rorer Sa | Composition containing nucleic acids, preparation and uses. |
| US6075012A (en) | 1994-02-11 | 2000-06-13 | Life Technologies, Inc. | Reagents for intracellular delivery of macromolecules |
| US6989434B1 (en) | 1994-02-11 | 2006-01-24 | Invitrogen Corporation | Reagents for intracellular delivery of macromolecules |
| WO1995035301A1 (en) | 1994-06-22 | 1995-12-28 | Megabios Corporation | Cationic amphiphiles |
| FR2722506B1 (en) | 1994-07-13 | 1996-08-14 | Rhone Poulenc Rorer Sa | COMPOSITION CONTAINING NUCLEIC ACIDS, PREPARATION AND USES |
| US5820873A (en) * | 1994-09-30 | 1998-10-13 | The University Of British Columbia | Polyethylene glycol modified ceramide lipids and liposome uses thereof |
| US5627159A (en) | 1994-10-27 | 1997-05-06 | Life Technologies, Inc. | Enhancement of lipid cationic transfections in the presence of serum |
| AU701106B2 (en) | 1995-06-07 | 1999-01-21 | Promega Biosciences, Inc. | Novel carbamate-based cationic lipids |
| CA2223179A1 (en) | 1995-06-07 | 1996-12-19 | Bob Dale Brown | Phosphonic acid-based cationic lipids |
| AU723163B2 (en) * | 1995-06-07 | 2000-08-17 | Tekmira Pharmaceuticals Corporation | Lipid-nucleic acid particles prepared via a hydrophobic lipid-nucleic acid complex intermediate and use for gene transfer |
| US5705385A (en) * | 1995-06-07 | 1998-01-06 | Inex Pharmaceuticals Corporation | Lipid-nucleic acid particles prepared via a hydrophobic lipid-nucleic acid complex intermediate and use for gene transfer |
| US5981501A (en) * | 1995-06-07 | 1999-11-09 | Inex Pharmaceuticals Corp. | Methods for encapsulating plasmids in lipid bilayers |
| US5811406A (en) | 1995-06-07 | 1998-09-22 | Regents Of The University Of California | Dry powder formulations of polynucleotide complexes |
| US20030069173A1 (en) | 1998-03-16 | 2003-04-10 | Life Technologies, Inc. | Peptide-enhanced transfections |
| WO1996040961A1 (en) | 1995-06-07 | 1996-12-19 | Life Technologies, Inc. | Peptide-enhanced cationic lipid transfections |
| US6051429A (en) | 1995-06-07 | 2000-04-18 | Life Technologies, Inc. | Peptide-enhanced cationic lipid transfections |
| US7422902B1 (en) * | 1995-06-07 | 2008-09-09 | The University Of British Columbia | Lipid-nucleic acid particles prepared via a hydrophobic lipid-nucleic acid complex intermediate and use for gene transfer |
| EP0869937A4 (en) | 1995-07-21 | 2004-07-21 | Promega Biosciences Inc | Novel amide-based cationic lipids |
| US6339173B1 (en) | 1996-07-22 | 2002-01-15 | Promega Biosciences, Inc. | Amide-based cationic lipids |
| US6330349B1 (en) | 1995-11-30 | 2001-12-11 | Chromavision Medical Systems, Inc. | Automated method for image analysis of residual protein |
| WO1997019675A2 (en) | 1995-11-30 | 1997-06-05 | Vical Incorporated | Complex cationic lipids |
| US5817856A (en) | 1995-12-11 | 1998-10-06 | Yissum Research Development Company Of The Hebrew University Of Jerusalem | Radiation-protective phospholipid and method |
| US6284267B1 (en) | 1996-08-14 | 2001-09-04 | Nutrimed Biotech | Amphiphilic materials and liposome formulations thereof |
| US6210707B1 (en) | 1996-11-12 | 2001-04-03 | The Regents Of The University Of California | Methods of forming protein-linked lipidic microparticles, and compositions thereof |
| US6034135A (en) | 1997-03-06 | 2000-03-07 | Promega Biosciences, Inc. | Dimeric cationic lipids |
| US5877220A (en) | 1997-03-06 | 1999-03-02 | Genta, Incorporated | Amide-based oligomeric cationic lipids |
| WO1998051285A2 (en) | 1997-05-15 | 1998-11-19 | Genzyme Corporation | Cationic amphiphile formulations |
| EP1023048B1 (en) | 1997-10-10 | 2007-03-07 | Inex Pharmaceuticals Corp. | Methods for encapsulating nucleic acids in lipid bilayers |
| US6410328B1 (en) | 1998-02-03 | 2002-06-25 | Protiva Biotherapeutics Inc. | Sensitizing cells to compounds using lipid-mediated gene and compound delivery |
| CA2271582A1 (en) | 1998-05-14 | 1999-11-14 | Sean C. Semple | Method for administration of therapeutic agents, including antisense, with repeat dosing |
| CA2335393C (en) | 1998-07-20 | 2008-09-23 | Inex Pharmaceuticals Corporation | Liposomal encapsulated nucleic acid-complexes |
| US6900049B2 (en) | 1998-09-10 | 2005-05-31 | Cell Genesys, Inc. | Adenovirus vectors containing cell status-specific response elements and methods of use thereof |
| EP1129064B1 (en) | 1998-11-12 | 2008-01-09 | Invitrogen Corporation | Transfection reagents |
| US6649780B1 (en) * | 1998-12-22 | 2003-11-18 | Valentis, Inc. | Cationic lipids |
| EP1173600A2 (en) | 1999-04-20 | 2002-01-23 | The University Of British Columbia | Cationic peg-lipids and methods of use |
| US6696424B1 (en) | 1999-05-28 | 2004-02-24 | Vical Incorporated | Cytofectin dimers and methods of use thereof |
| US20010048940A1 (en) | 1999-06-18 | 2001-12-06 | Jennifer D. Tousignant | Cationic amphiphile micellar complexes |
| CN1193059C (en) | 1999-07-14 | 2005-03-16 | 阿尔萨公司 | Neutral lipopolymer and liposomal compositions contg. same |
| JP2003504390A (en) * | 1999-07-15 | 2003-02-04 | イネックス ファーマスーティカルズ コーポレイション | Method and apparatus for the production of lipid vesicles |
| JP4782966B2 (en) | 2000-01-10 | 2011-09-28 | イッサム・リサーチ・ディベロップメント・カンパニー・オブ・ザ・ヘブルー・ユニバーシティ・オブ・エルサレム・リミテッド | Use of lipid conjugates in the treatment of disease |
| US20070026394A1 (en) * | 2000-02-11 | 2007-02-01 | Lawrence Blatt | Modulation of gene expression associated with inflammation proliferation and neurite outgrowth using nucleic acid based technologies |
| WO2002034236A2 (en) | 2000-10-25 | 2002-05-02 | The University Of British Columbia | Lipid formulations for target delivery |
| AU2002232387A1 (en) | 2000-10-27 | 2002-05-06 | Invitrogen Corporation | Method for introducing antisense oligonucleotides into eucaryotic cells |
| US20040259247A1 (en) * | 2000-12-01 | 2004-12-23 | Thomas Tuschl | Rna interference mediating small rna molecules |
| US20040142892A1 (en) * | 2001-04-30 | 2004-07-22 | The University Of British Columbia | Autogene nucleic acids encoding a secretable RNA polymerase |
| US20040063654A1 (en) * | 2001-11-02 | 2004-04-01 | Davis Mark E. | Methods and compositions for therapeutic use of RNA interference |
| EP1509203B1 (en) | 2002-05-15 | 2016-04-13 | California Pacific Medical Center | Delivery of nucleic acid-like compounds |
| US7148342B2 (en) * | 2002-07-24 | 2006-12-12 | The Trustees Of The University Of Pennyslvania | Compositions and methods for sirna inhibition of angiogenesis |
| MXPA05007651A (en) | 2003-01-16 | 2005-10-26 | Univ Pennsylvania | COMPOSITIONS AND METHODS FOR siRNA INHIBITION OF ICAM-1. |
| US7803781B2 (en) * | 2003-02-28 | 2010-09-28 | Isis Pharmaceuticals, Inc. | Modulation of growth hormone receptor expression and insulin-like growth factor expression |
| US7771711B2 (en) | 2003-06-18 | 2010-08-10 | Yissum Research Development Company Of The Hebrew University Of Jerusalem | Sphingolipids' polyalkylamines conjugates |
| CN101291653B (en) | 2003-07-16 | 2012-06-27 | 普洛体维生物治疗公司 | Lipid encapsulated interfering rna |
| WO2005121348A1 (en) | 2004-06-07 | 2005-12-22 | Protiva Biotherapeutics, Inc. | Lipid encapsulated interfering rna |
| CA2569645C (en) | 2004-06-07 | 2014-10-28 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods of use |
| CA2572439A1 (en) | 2004-07-02 | 2006-01-12 | Protiva Biotherapeutics, Inc. | Immunostimulatory sirna molecules and uses therefor |
| CA2580707C (en) * | 2004-09-24 | 2014-07-08 | Alnylam Pharmaceuticals, Inc. | Rnai modulation of apob and uses thereof |
| AU2005306533B2 (en) | 2004-11-17 | 2012-05-31 | Arbutus Biopharma Corporation | siRNA silencing of apolipoprotein B |
| DE102004060863A1 (en) * | 2004-12-17 | 2006-06-22 | Dr. Johannes Heidenhain Gmbh | Angle measuring device |
| WO2007086883A2 (en) | 2005-02-14 | 2007-08-02 | Sirna Therapeutics, Inc. | Cationic lipids and formulated molecular compositions containing them |
| US20060228406A1 (en) | 2005-03-17 | 2006-10-12 | Invitrogen Corporation | Transfection reagent for non-adherent suspension cells |
| CA2721380A1 (en) | 2008-04-15 | 2009-10-22 | Protiva Biotherapeutics, Inc. | Silencing of csn5 gene expression using interfering rna |
| WO2009129387A2 (en) | 2008-04-16 | 2009-10-22 | Abbott Laboratories | Cationic lipids and uses thereof |
| WO2009132131A1 (en) | 2008-04-22 | 2009-10-29 | Alnylam Pharmaceuticals, Inc. | Amino lipid based improved lipid formulation |
| EP3207944B1 (en) | 2008-11-10 | 2020-01-15 | Arbutus Biopharma Corporation | Novel lipids and compositions for the delivery of therapeutics |
| WO2011000106A1 (en) | 2009-07-01 | 2011-01-06 | Protiva Biotherapeutics, Inc. | Improved cationic lipids and methods for the delivery of therapeutic agents |
| US20120136073A1 (en) | 2010-11-15 | 2012-05-31 | Life Technologies Corporation | Amine-Containing Transfection Reagents and methods for making and using same |
-
2005
- 2005-06-07 WO PCT/CA2005/000886 patent/WO2005121348A1/en active Application Filing
- 2005-06-07 JP JP2007526139A patent/JP4796062B2/en not_active Expired - Fee Related
- 2005-06-07 US US11/148,152 patent/US7799565B2/en not_active Expired - Fee Related
- 2005-06-07 AU AU2005252273A patent/AU2005252273B2/en not_active Ceased
- 2005-06-07 AT AT05757651T patent/ATE536418T1/en active
- 2005-06-07 CA CA2569664A patent/CA2569664C/en not_active Expired - Fee Related
- 2005-06-07 EP EP05757651A patent/EP1766035B1/en active Active
-
2010
- 2010-08-06 US US12/852,379 patent/US9181545B2/en not_active Expired - Fee Related
-
2015
- 2015-11-09 US US14/936,169 patent/US9926560B2/en not_active Expired - Fee Related
-
2018
- 2018-03-26 US US15/936,284 patent/US20190071669A1/en not_active Abandoned
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5208036A (en) | 1985-01-07 | 1993-05-04 | Syntex (U.S.A.) Inc. | N-(ω, (ω-1)-dialkyloxy)- and N-(ω, (ω-1)-dialkenyloxy)-alk-1-yl-N,N,N-tetrasubstituted ammonium lipids and uses therefor |
| US5279833A (en) | 1990-04-04 | 1994-01-18 | Yale University | Liposomal transfection of nucleic acids into animal cells |
| US5264618A (en) | 1990-04-19 | 1993-11-23 | Vical, Inc. | Cationic lipids for intracellular delivery of biologically active molecules |
| US5283185A (en) | 1991-08-28 | 1994-02-01 | University Of Tennessee Research Corporation | Method for delivering nucleic acids into cells |
| US5753613A (en) | 1994-09-30 | 1998-05-19 | Inex Pharmaceuticals Corporation | Compositions for the introduction of polyanionic materials into cells |
| US5785992A (en) | 1994-09-30 | 1998-07-28 | Inex Pharmaceuticals Corp. | Compositions for the introduction of polyanionic materials into cells |
| US5885613A (en) | 1994-09-30 | 1999-03-23 | The University Of British Columbia | Bilayer stabilizing components and their use in forming programmable fusogenic liposomes |
| US20050008689A1 (en) | 1997-05-14 | 2005-01-13 | Inex Pharmaceuticals Corporation | High efficiency encapsulation of charged therapeutic agents in lipid vesicles |
| WO2002087541A1 (en) * | 2001-04-30 | 2002-11-07 | Protiva Biotherapeutics Inc. | Lipid-based formulations for gene transfer |
| US20030077829A1 (en) | 2001-04-30 | 2003-04-24 | Protiva Biotherapeutics Inc.. | Lipid-based formulations |
| WO2005026372A1 (en) | 2003-09-15 | 2005-03-24 | Protiva Biotherapeutics, Inc. | Polyethyleneglycol-modified lipid compounds and uses thereof |
Non-Patent Citations (4)
| Title |
|---|
| ARPICCO S ET AL: "Preparation and characterization of novel cationic lipids developed for gene transfection.", PROCEED INT SYMP CONTROL REL BIOACT MATER., vol. 26, 1999, pages 759 - 760, XP008103434 * |
| ARPICCO S ET AL: "Synthesis, characterization and transfection activity of new saturated and unsaturated cationic lipids.", II FARMACO., vol. 59, no. 11, November 2004 (2004-11-01), pages 869 - 878, XP004641713 * |
| CEVC G.: "How membrane chain-melting phase-transition temperature is affected by the lipid chain asymmetry and degree of unsaturation: an effective chain-length model.", BIOCHEMISTRY., vol. 30, no. 29, July 1991 (1991-07-01), pages 7186 - 7193, XP008115986 * |
| KEOUGH K.M.W.: "Influence of chain unsaturation and chain position on thermotropism and intermolecular interactions in membranes.", BIOCHEM SOC TRANSACTIONS., vol. 18, no. 5, 1990, pages 835 - 837, XP008103661 * |
Cited By (385)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9926560B2 (en) | 2004-06-07 | 2018-03-27 | Protiva Biotherapeutics, Inc. | Lipid encapsulating interfering RNA |
| US9181545B2 (en) | 2004-06-07 | 2015-11-10 | Protiva Biotherapeutics, Inc. | Lipid encapsulating interfering RNA |
| US7807815B2 (en) | 2004-07-02 | 2010-10-05 | Protiva Biotherapeutics, Inc. | Compositions comprising immunostimulatory siRNA molecules and DLinDMA or DLenDMA |
| WO2006016097A2 (en) * | 2004-08-13 | 2006-02-16 | Ic Vec Limited | Vector comprising polymer modified sirna liposomes |
| WO2006016097A3 (en) * | 2004-08-13 | 2006-11-23 | Ic Vec Ltd | Vector comprising polymer modified sirna liposomes |
| JP2008509205A (en) * | 2004-08-13 | 2008-03-27 | アイシー・ベック・リミテッド | Vector containing polymer-modified siRNA liposomes |
| WO2006053430A1 (en) * | 2004-11-17 | 2006-05-26 | Protiva Biotherapeutics, Inc. | Sirna silencing of apolipoprotein b |
| EP1774962A1 (en) * | 2005-10-14 | 2007-04-18 | Industrial Technology Research Institute | Lipid carrier and method of preparing the same |
| US7838658B2 (en) | 2005-10-20 | 2010-11-23 | Ian Maclachlan | siRNA silencing of filovirus gene expression |
| US8513403B2 (en) | 2005-11-02 | 2013-08-20 | Protiva Biotherapeutics, Inc. | Modified siRNA molecules and uses thereof |
| US8188263B2 (en) | 2005-11-02 | 2012-05-29 | Protiva Biotherapeutics, Inc. | Modified siRNA molecules and uses thereof |
| US9074208B2 (en) | 2005-11-02 | 2015-07-07 | Protiva Biotherapeutics, Inc. | Modified siRNA molecules and uses thereof |
| US8101741B2 (en) | 2005-11-02 | 2012-01-24 | Protiva Biotherapeutics, Inc. | Modified siRNA molecules and uses thereof |
| US7915399B2 (en) | 2006-06-09 | 2011-03-29 | Protiva Biotherapeutics, Inc. | Modified siRNA molecules and uses thereof |
| EP3192788A1 (en) * | 2006-10-03 | 2017-07-19 | Arbutus Biopharma Corporation | Lipid containing formulations |
| US11420931B2 (en) | 2006-10-03 | 2022-08-23 | Arbutus Biopharma Corporation | Lipid containing formulations |
| WO2009082817A1 (en) | 2007-12-27 | 2009-07-09 | Protiva Biotherapeutics, Inc. | Silencing of polo-like kinase expression using interfering rna |
| JP2011509258A (en) * | 2008-01-02 | 2011-03-24 | テクミラ ファーマシューティカルズ コーポレイション | Improved compositions and methods for delivery of nucleic acids |
| WO2009088891A1 (en) * | 2008-01-02 | 2009-07-16 | Alnylam Pharmaceuticals, Inc. | Screening method for selected amino lipid-containing compositions |
| WO2009127060A1 (en) | 2008-04-15 | 2009-10-22 | Protiva Biotherapeutics, Inc. | Novel lipid formulations for nucleic acid delivery |
| WO2009129319A2 (en) | 2008-04-15 | 2009-10-22 | Protiva Biotherapeutics, Inc. | Silencing of csn5 gene expression using interfering rna |
| EP2770057A1 (en) | 2008-04-15 | 2014-08-27 | Protiva Biotherapeutics Inc. | Silencing of CSN5 gene expression using interfering RNA |
| WO2009129395A1 (en) * | 2008-04-16 | 2009-10-22 | Abbott Laboratories | Cationic lipids and uses thereof |
| WO2009129385A1 (en) * | 2008-04-16 | 2009-10-22 | Abbott Laboratories | Cationic lipids and uses thereof |
| WO2010030730A1 (en) * | 2008-09-10 | 2010-03-18 | Abbott Laboratories | Polyethylene glycol lipid conjugates and uses thereof |
| WO2010030739A1 (en) * | 2008-09-10 | 2010-03-18 | Abbott Laboratories | Polyethylene glycol lipid conjugates and uses thereof |
| US10653780B2 (en) | 2008-10-09 | 2020-05-19 | The University Of British Columbia | Amino lipids and methods for the delivery of nucleic acids |
| US9139554B2 (en) | 2008-10-09 | 2015-09-22 | Tekmira Pharmaceuticals Corporation | Amino lipids and methods for the delivery of nucleic acids |
| US11414393B2 (en) | 2008-11-07 | 2022-08-16 | Massachusetts Institute Of Technology | Aminoalcohol lipidoids and uses thereof |
| US10844028B2 (en) | 2008-11-07 | 2020-11-24 | Massachusetts Institute Of Technology | Aminoalcohol lipidoids and uses thereof |
| US10189802B2 (en) | 2008-11-07 | 2019-01-29 | Massachusetts Institute Of Technology | Aminoalcohol lipidoids and uses thereof |
| US9556110B2 (en) | 2008-11-07 | 2017-01-31 | Massachusetts Institute Of Technology | Aminoalcohol lipidoids and uses thereof |
| US8957199B2 (en) | 2008-11-26 | 2015-02-17 | Chugai Seiyaku Kabushiki Kaisha | Oligoribonucleotide or peptide nucleic acid capable of inhibiting activity of hepatitis C virus |
| US8569256B2 (en) | 2009-07-01 | 2013-10-29 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods for the delivery of therapeutic agents |
| US9018187B2 (en) | 2009-07-01 | 2015-04-28 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods for the delivery of therapeutic agents |
| WO2011000107A1 (en) | 2009-07-01 | 2011-01-06 | Protiva Biotherapeutics, Inc. | Novel lipid formulations for delivery of therapeutic agents to solid tumors |
| US9187748B2 (en) | 2009-07-20 | 2015-11-17 | Protiva Biotherapeutics, Inc. | Compositions and methods for silencing ebola virus gene expression |
| US8716464B2 (en) | 2009-07-20 | 2014-05-06 | Thomas W. Geisbert | Compositions and methods for silencing Ebola virus gene expression |
| WO2011011447A1 (en) | 2009-07-20 | 2011-01-27 | Protiva Biotherapeutics, Inc. | Compositions and methods for silencing ebola virus gene expression |
| WO2011038160A2 (en) | 2009-09-23 | 2011-03-31 | Protiva Biotherapeutics, Inc. | Compositions and methods for silencing genes expressed in cancer |
| EP3403647A1 (en) | 2009-12-01 | 2018-11-21 | Translate Bio, Inc. | Delivery of mrna for the augmentation of proteins and enzymes in human genetic diseases |
| US10576166B2 (en) | 2009-12-01 | 2020-03-03 | Translate Bio, Inc. | Liver specific delivery of messenger RNA |
| EP3338765A1 (en) | 2009-12-01 | 2018-06-27 | Translate Bio, Inc. | Steroid derivative for the delivery of mrna in human genetic diseases |
| EP3318248A1 (en) | 2009-12-01 | 2018-05-09 | Translate Bio, Inc. | Delivery of mrna for the augmentation of proteins and enzymes in human genetic diseases |
| EP4115874A1 (en) | 2009-12-01 | 2023-01-11 | Translate Bio, Inc. | Delivery of mrna for the augmentation of proteins and enzymes in human genetic diseases |
| WO2011076807A2 (en) | 2009-12-23 | 2011-06-30 | Novartis Ag | Lipids, lipid compositions, and methods of using them |
| EP3721943A1 (en) | 2009-12-23 | 2020-10-14 | Novartis AG | Lipids, lipid compositions and methods of using them |
| US9301923B2 (en) | 2009-12-23 | 2016-04-05 | Novartis Ag | Lipids, lipid compositions, and methods of using them |
| WO2011136368A1 (en) | 2010-04-28 | 2011-11-03 | 協和発酵キリン株式会社 | Cationic lipid |
| US9920028B2 (en) | 2010-04-28 | 2018-03-20 | Kyowa Hakko Kirin Co., Ltd. | Cationic lipid |
| WO2011136369A1 (en) | 2010-04-28 | 2011-11-03 | 協和発酵キリン株式会社 | Cationic lipid |
| US9408914B2 (en) | 2010-04-28 | 2016-08-09 | Kyowa Hakko Kirin Co., Ltd. | Cationic lipid |
| US9845306B2 (en) | 2010-04-28 | 2017-12-19 | Kyowa Hakko Kirin Co., Ltd. | Cationic lipid |
| WO2011141705A1 (en) | 2010-05-12 | 2011-11-17 | Protiva Biotherapeutics, Inc. | Novel cationic lipids and methods of use thereof |
| WO2011141704A1 (en) | 2010-05-12 | 2011-11-17 | Protiva Biotherapeutics, Inc | Novel cyclic cationic lipids and methods of use |
| US11839686B2 (en) | 2010-07-06 | 2023-12-12 | Glaxosmithkline Biologicals Sa | Lipid formulations with viral immunogens |
| US11291682B2 (en) | 2010-07-06 | 2022-04-05 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11851660B2 (en) | 2010-07-06 | 2023-12-26 | Glaxosmithkline Biologicals Sa | Immunisation of large mammals with low doses of RNA |
| US11845925B2 (en) | 2010-07-06 | 2023-12-19 | Glaxosmithkline Biologicals Sa | Immunisation of large mammals with low doses of RNA |
| US11857562B2 (en) | 2010-07-06 | 2024-01-02 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11786467B2 (en) | 2010-07-06 | 2023-10-17 | Glaxosmithkline Biologicals Sa | Lipid formulations with immunogens |
| US11857681B2 (en) | 2010-07-06 | 2024-01-02 | Glaxosmithkline Biologicals Sa | Lipid formulations with RNA encoding immunogens |
| US11291635B2 (en) | 2010-07-06 | 2022-04-05 | Glaxosmithkline Biological Sa | Virion-like delivery particles for self-replicating RNA molecules |
| US11865080B2 (en) | 2010-07-06 | 2024-01-09 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11883534B2 (en) | 2010-07-06 | 2024-01-30 | Glaxosmithkline Biologicals Sa | Immunisation with lipid formulations with RNA encoding immunogens |
| US11773395B1 (en) | 2010-07-06 | 2023-10-03 | Glaxosmithkline Biologicals Sa | Immunization of large mammals with low doses of RNA |
| US11891608B2 (en) | 2010-07-06 | 2024-02-06 | Glaxosmithkline Biologicals Sa | Immunization of large mammals with low doses of RNA |
| US11638693B2 (en) | 2010-07-06 | 2023-05-02 | Glaxosmithkline Biologicals Sa | Vaccine for eliciting immune response comprising RNA encoding an immunogen and lipid formulations comprising mole percentage of lipids |
| US11766401B2 (en) | 2010-07-06 | 2023-09-26 | Glaxosmithkline Biologicals Sa | Methods of administering lipid formulations with immunogens |
| US11759475B2 (en) | 2010-07-06 | 2023-09-19 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| WO2012006372A1 (en) | 2010-07-06 | 2012-01-12 | Novartis Ag | Delivery of rna to different cell types |
| WO2012006377A2 (en) | 2010-07-06 | 2012-01-12 | Novartis Ag | Delivery of rna to trigger multiple immune pathways |
| WO2012006369A2 (en) | 2010-07-06 | 2012-01-12 | Novartis Ag | Immunisation of large mammals with low doses of rna |
| US11850305B2 (en) | 2010-07-06 | 2023-12-26 | Glaxosmithkline Biologicals Sa | Method of making lipid formulations with RNA encoding immunogens |
| US11739334B2 (en) | 2010-07-06 | 2023-08-29 | Glaxosmithkline Biologicals Sa | Immunisation of large mammals with low doses of RNA |
| US11905514B2 (en) | 2010-07-06 | 2024-02-20 | Glaxosmithkline Biological Sa | Immunisation of large mammals with low doses of RNA |
| US11638694B2 (en) | 2010-07-06 | 2023-05-02 | Glaxosmithkline Biologicals Sa | Vaccine for eliciting immune response comprising lipid formulations and RNA encoding multiple immunogens |
| US11730754B2 (en) | 2010-07-06 | 2023-08-22 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11913001B2 (en) | 2010-07-06 | 2024-02-27 | Glaxosmithkline Biologicals Sa | Immunisation of large mammals with low doses of RNA |
| US11717529B2 (en) | 2010-07-06 | 2023-08-08 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11707482B2 (en) | 2010-07-06 | 2023-07-25 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US20220125723A1 (en) | 2010-07-06 | 2022-04-28 | Glaxosmithkline Biologicals Sa | Lipid formulations with viral immunogens |
| US11324770B2 (en) | 2010-07-06 | 2022-05-10 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11696923B2 (en) | 2010-07-06 | 2023-07-11 | Glaxosmithkline Biologicals, Sa | Delivery of RNA to trigger multiple immune pathways |
| EP3115061A1 (en) | 2010-07-06 | 2017-01-11 | GlaxoSmithKline Biologicals SA | Virion-like delivery particles for self-replicating rna molecules |
| WO2012006378A1 (en) | 2010-07-06 | 2012-01-12 | Novartis Ag | Liposomes with lipids having an advantageous pka- value for rna delivery |
| US11690865B2 (en) | 2010-07-06 | 2023-07-04 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11690862B1 (en) | 2010-07-06 | 2023-07-04 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| WO2012006376A2 (en) | 2010-07-06 | 2012-01-12 | Novartis Ag | Virion-like delivery particles for self-replicating rna molecules |
| US11690863B2 (en) | 2010-07-06 | 2023-07-04 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11690864B2 (en) | 2010-07-06 | 2023-07-04 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11690861B2 (en) | 2010-07-06 | 2023-07-04 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US11026964B2 (en) | 2010-07-06 | 2021-06-08 | Glaxosmithkline Biologicals Sa | Delivery of RNA to different cell types |
| US11666534B2 (en) | 2010-07-06 | 2023-06-06 | Glaxosmithkline Biologicals Sa | Methods of administering lipid formulations with viral immunogens |
| EP4015632A1 (en) | 2010-07-06 | 2022-06-22 | GlaxoSmithKline Biologicals SA | Delivery of rna to different cell types |
| US11655475B2 (en) | 2010-07-06 | 2023-05-23 | Glaxosmithkline Biologicals Sa | Immunisation of large mammals with low doses of RNA |
| EP3243526A1 (en) | 2010-07-06 | 2017-11-15 | GlaxoSmithKline Biologicals S.A. | Delivery of rna to trigger multiple immune pathways |
| US11596645B2 (en) | 2010-07-06 | 2023-03-07 | Glaxosmithkline Biologicals Sa | Delivery of RNA to trigger multiple immune pathways |
| US9193827B2 (en) | 2010-08-26 | 2015-11-24 | Massachusetts Institute Of Technology | Poly(beta-amino alcohols), their preparation, and uses thereof |
| EP2611461B1 (en) | 2010-08-31 | 2022-03-09 | GlaxoSmithKline Biologicals SA | Pegylated liposomes for delivery of immunogen-encoding rna |
| WO2012031046A2 (en) | 2010-08-31 | 2012-03-08 | Novartis Ag | Lipids suitable for liposomal delivery of protein-coding rna |
| EP2611467B1 (en) | 2010-08-31 | 2022-07-20 | GlaxoSmithKline Biologicals SA | Small liposomes for delivery of immunogen-encoding rna |
| US11759422B2 (en) | 2010-08-31 | 2023-09-19 | Glaxosmithkline Biologicals Sa | Pegylated liposomes for delivery of immunogen-encoding RNA |
| EP4066819B1 (en) | 2010-08-31 | 2023-03-01 | GlaxoSmithKline Biologicals SA | Small liposomes for delivery of immunogen-encoding rna |
| EP4043040B1 (en) | 2010-08-31 | 2023-01-11 | GlaxoSmithKline Biologicals SA | Small liposomes for delivery of immunogen-encoding rna |
| WO2012031043A1 (en) | 2010-08-31 | 2012-03-08 | Novartis Ag | Pegylated liposomes for delivery of immunogen-encoding rna |
| EP4066857B1 (en) | 2010-08-31 | 2022-12-21 | GlaxoSmithKline Biologicals SA | Pegylated liposomes for delivery of immunogen-encoding rna |
| EP3970742B1 (en) | 2010-08-31 | 2022-05-25 | GlaxoSmithKline Biologicals S.A. | Pegylated liposomes for delivery of immunogen-encoding rna |
| EP3981427B1 (en) | 2010-08-31 | 2022-05-25 | GlaxoSmithKline Biologicals S.A. | Pegylated liposomes for delivery of immunogen-encoding rna |
| EP4066856B1 (en) | 2010-08-31 | 2022-12-07 | GlaxoSmithKline Biologicals SA | Pegylated liposomes for delivery of immunogen-encoding rna |
| EP3542789A2 (en) | 2010-08-31 | 2019-09-25 | GlaxoSmithKline Biologicals SA | Lipids suitable for liposomal delivery of protein-coding rna |
| US9254265B2 (en) | 2010-08-31 | 2016-02-09 | Novartis Ag | Small liposomes for delivery of immunogen encoding RNA |
| EP4066855B1 (en) | 2010-08-31 | 2022-12-28 | GlaxoSmithKline Biologicals SA | Pegylated liposomes for delivery of immunogen-encoding rna |
| EP4008357B1 (en) | 2010-08-31 | 2022-12-28 | GlaxoSmithKline Biologicals SA | Small liposomes for delivery of immunogen-encoding rna |
| US11639370B2 (en) | 2010-10-11 | 2023-05-02 | Glaxosmithkline Biologicals Sa | Antigen delivery platforms |
| US9956271B2 (en) | 2010-11-30 | 2018-05-01 | Translate Bio, Inc. | mRNA for use in treatment of human genetic diseases |
| US11135274B2 (en) | 2010-11-30 | 2021-10-05 | Translate Bio, Inc. | MRNA for use in treatment of human genetic diseases |
| US9238716B2 (en) | 2011-03-28 | 2016-01-19 | Massachusetts Institute Of Technology | Conjugated lipomers and uses thereof |
| US10117934B2 (en) | 2011-03-28 | 2018-11-06 | Massachusetts Institute Of Technology | Conjugated lipomers and uses thereof |
| US10933139B2 (en) | 2011-03-28 | 2021-03-02 | Massachusetts Institute Of Technology | Conjugated lipomers and uses thereof |
| US11951179B2 (en) | 2011-06-08 | 2024-04-09 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for MRNA delivery |
| US10238754B2 (en) | 2011-06-08 | 2019-03-26 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for MRNA delivery |
| US10507183B2 (en) | 2011-06-08 | 2019-12-17 | Translate Bio, Inc. | Cleavable lipids |
| EP4043025A1 (en) | 2011-06-08 | 2022-08-17 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mrna delivery |
| EP4074693A1 (en) | 2011-06-08 | 2022-10-19 | Translate Bio, Inc. | Cleavable lipids |
| EP3354644A1 (en) | 2011-06-08 | 2018-08-01 | Translate Bio, Inc. | Cleavable lipids |
| EP3336082A1 (en) | 2011-06-08 | 2018-06-20 | Translate Bio, Inc. | Cleavable lipids |
| US11547764B2 (en) | 2011-06-08 | 2023-01-10 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for MRNA delivery |
| EP3586861A1 (en) | 2011-06-08 | 2020-01-01 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mrna delivery |
| US9717690B2 (en) | 2011-06-08 | 2017-08-01 | Rana Therapeutics, Inc. | Cleavable lipids |
| US9597413B2 (en) | 2011-06-08 | 2017-03-21 | Shire Human Genetic Therapies, Inc. | Pulmonary delivery of mRNA |
| US11338044B2 (en) | 2011-06-08 | 2022-05-24 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| EP3998064A1 (en) | 2011-06-08 | 2022-05-18 | Translate Bio, Inc. | Cleavable lipids |
| EP4212514A1 (en) | 2011-06-08 | 2023-07-19 | Translate Bio, Inc. | Cleavable lipids |
| WO2012170889A1 (en) | 2011-06-08 | 2012-12-13 | Shire Human Genetic Therapies, Inc. | Cleavable lipids |
| US11730825B2 (en) | 2011-06-08 | 2023-08-22 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US9308281B2 (en) | 2011-06-08 | 2016-04-12 | Shire Human Genetic Therapies, Inc. | MRNA therapy for Fabry disease |
| US11291734B2 (en) | 2011-06-08 | 2022-04-05 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| WO2012170930A1 (en) | 2011-06-08 | 2012-12-13 | Shire Human Genetic Therapies, Inc | Lipid nanoparticle compositions and methods for mrna delivery |
| US10413618B2 (en) | 2011-06-08 | 2019-09-17 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US11234936B2 (en) | 2011-06-08 | 2022-02-01 | Translate Bio, Inc. | Cleavable lipids |
| EP3674292A1 (en) | 2011-06-08 | 2020-07-01 | Translate Bio, Inc. | Cleavable lipids |
| US11185595B2 (en) | 2011-06-08 | 2021-11-30 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US11951180B2 (en) | 2011-06-08 | 2024-04-09 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for MRNA delivery |
| US11052159B2 (en) | 2011-06-08 | 2021-07-06 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US11951181B2 (en) | 2011-06-08 | 2024-04-09 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US10702478B2 (en) | 2011-06-08 | 2020-07-07 | Translate Bio, Inc. | Cleavable lipids |
| US10507249B2 (en) | 2011-06-08 | 2019-12-17 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US10888626B2 (en) | 2011-06-08 | 2021-01-12 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US10350303B1 (en) | 2011-06-08 | 2019-07-16 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| EP3821879A1 (en) | 2011-07-06 | 2021-05-19 | GlaxoSmithKline Biologicals S.A. | Liposomes having useful n:p ratio for delivery of rna molecules |
| US11896636B2 (en) | 2011-07-06 | 2024-02-13 | Glaxosmithkline Biologicals Sa | Immunogenic combination compositions and uses thereof |
| WO2013006825A1 (en) | 2011-07-06 | 2013-01-10 | Novartis Ag | Liposomes having useful n:p ratio for delivery of rna molecules |
| WO2013033563A1 (en) | 2011-08-31 | 2013-03-07 | Novartis Ag | Pegylated liposomes for delivery of immunogen-encoding rna |
| EP3508220A1 (en) | 2011-08-31 | 2019-07-10 | GlaxoSmithKline Biologicals S.A. | Pegylated liposomes for delivery of immunogen-encoding rna |
| US9126966B2 (en) | 2011-08-31 | 2015-09-08 | Protiva Biotherapeutics, Inc. | Cationic lipids and methods of use thereof |
| US11458158B2 (en) | 2011-10-27 | 2022-10-04 | Massachusetts Institute Of Technology | Amino acid-, peptide- and polypeptide-lipids, isomers, compositions, and uses thereof |
| US10682374B2 (en) | 2011-10-27 | 2020-06-16 | Massachusetts Intstitute Of Technology | Amino acid-, peptide- and polypeptide-lipids, isomers, compositions, and uses thereof |
| US9512073B2 (en) | 2011-10-27 | 2016-12-06 | Massachusetts Institute Of Technology | Amino acid-, peptide-and polypeptide-lipids, isomers, compositions, and uses thereof |
| US10086013B2 (en) | 2011-10-27 | 2018-10-02 | Massachusetts Institute Of Technology | Amino acid-, peptide- and polypeptide-lipids, isomers, compositions, and uses thereof |
| WO2013065825A1 (en) | 2011-11-02 | 2013-05-10 | 協和発酵キリン株式会社 | Cationic lipid |
| EP3301086A1 (en) | 2011-11-02 | 2018-04-04 | Kyowa Hakko Kirin Co., Ltd. | Production of a composition containing a complex between a membrane composed of a lipid monolayer and a nucleic acid, and, a lipid membrane for encapsulating the complex therein |
| US9839616B2 (en) | 2011-12-12 | 2017-12-12 | Kyowa Hakko Kirin Co., Ltd. | Lipid nano particles comprising cationic lipid for drug delivery system |
| WO2013089152A1 (en) | 2011-12-12 | 2013-06-20 | 協和発酵キリン株式会社 | Lipid nanoparticles containing combinations of cationic lipids |
| WO2013089151A1 (en) | 2011-12-12 | 2013-06-20 | 協和発酵キリン株式会社 | Lipid nanoparticles for drug delivery system containing cationic lipids |
| US9035039B2 (en) | 2011-12-22 | 2015-05-19 | Protiva Biotherapeutics, Inc. | Compositions and methods for silencing SMAD4 |
| EP3988104A1 (en) | 2012-02-24 | 2022-04-27 | Arbutus Biopharma Corporation | Trialkyl cationic lipids and methods of use thereof |
| WO2013126803A1 (en) | 2012-02-24 | 2013-08-29 | Protiva Biotherapeutics Inc. | Trialkyl cationic lipids and methods of use thereof |
| US10786455B2 (en) | 2012-03-29 | 2020-09-29 | Translate Bio, Inc. | Lipid-derived neutral nanoparticles |
| US9877919B2 (en) | 2012-03-29 | 2018-01-30 | Translate Bio, Inc. | Lipid-derived neutral nanoparticles |
| EP3865123A1 (en) | 2012-03-29 | 2021-08-18 | Translate Bio, Inc. | Lipid-derived neutral nanoparticles |
| US11497716B2 (en) | 2012-03-29 | 2022-11-15 | Translate Bio, Inc. | Lipid-derived neutral nanoparticles |
| US10137086B2 (en) | 2012-03-29 | 2018-11-27 | Translate Bio, Inc. | Lipid-derived neutral nanoparticles |
| EP3620447A1 (en) | 2012-03-29 | 2020-03-11 | Translate Bio MA, Inc. | Ionizable cationic lipids |
| US10137087B2 (en) | 2012-03-29 | 2018-11-27 | Translate Bio, Inc. | Lipid-derived neutral nanoparticles |
| WO2013149141A1 (en) | 2012-03-29 | 2013-10-03 | Shire Human Genetic Therapies, Inc. | Lipid-derived neutral nanoparticles |
| US11564998B2 (en) | 2012-04-02 | 2023-01-31 | Modernatx, Inc. | Modified polynucleotides for the production of cytoplasmic and cytoskeletal proteins |
| US10501513B2 (en) | 2012-04-02 | 2019-12-10 | Modernatx, Inc. | Modified polynucleotides for the production of oncology-related proteins and peptides |
| US10463751B2 (en) | 2012-04-02 | 2019-11-05 | Modernatx, Inc. | Modified polynucleotides for the production of cytoplasmic and cytoskeletal proteins |
| US10493167B2 (en) | 2012-04-02 | 2019-12-03 | Modernatx, Inc. | In vivo production of proteins |
| US10772975B2 (en) | 2012-04-02 | 2020-09-15 | Modernatx, Inc. | Modified Polynucleotides for the production of biologics and proteins associated with human disease |
| US10703789B2 (en) | 2012-04-02 | 2020-07-07 | Modernatx, Inc. | Modified polynucleotides for the production of secreted proteins |
| US10583203B2 (en) | 2012-04-02 | 2020-03-10 | Modernatx, Inc. | In vivo production of proteins |
| US10501512B2 (en) | 2012-04-02 | 2019-12-10 | Modernatx, Inc. | Modified polynucleotides |
| EP3884949A1 (en) | 2012-06-08 | 2021-09-29 | Translate Bio, Inc. | Pulmonary delivery of mrna to non-lung target cells |
| WO2013185069A1 (en) | 2012-06-08 | 2013-12-12 | Shire Human Genetic Therapies, Inc. | Pulmonary delivery of mrna to non-lung target cells |
| EP3536787A1 (en) | 2012-06-08 | 2019-09-11 | Translate Bio, Inc. | Nuclease resistant polynucleotides and uses thereof |
| US11254936B2 (en) | 2012-06-08 | 2022-02-22 | Translate Bio, Inc. | Nuclease resistant polynucleotides and uses thereof |
| US10245229B2 (en) | 2012-06-08 | 2019-04-02 | Translate Bio, Inc. | Pulmonary delivery of mRNA to non-lung target cells |
| US11090264B2 (en) | 2012-06-08 | 2021-08-17 | Translate Bio, Inc. | Pulmonary delivery of mRNA to non-lung target cells |
| WO2014007398A1 (en) | 2012-07-06 | 2014-01-09 | 協和発酵キリン株式会社 | Cationic lipid |
| WO2014013995A1 (en) | 2012-07-16 | 2014-01-23 | 協和発酵キリン株式会社 | Rnai pharmaceutical composition capable of suppressing expression of kras gene |
| US9913907B2 (en) | 2012-07-16 | 2018-03-13 | Kyowa Hakko Kirin Co., Ltd. | RNAi pharmaceutical composition for suppressing expression of KRAS gene |
| US9227917B2 (en) | 2012-08-13 | 2016-01-05 | Massachusetts Institute Of Technology | Amine-containing lipidoids and uses thereof |
| US9439968B2 (en) | 2012-08-13 | 2016-09-13 | Massachusetts Institute Of Technology | Amine-containing lipidoids and uses thereof |
| EP4331620A2 (en) | 2012-12-07 | 2024-03-06 | Translate Bio, Inc. | Lipidic nanoparticles for mrna delivery |
| WO2014089486A1 (en) | 2012-12-07 | 2014-06-12 | Shire Human Genetic Therapies, Inc. | Lipidic nanoparticles for mrna delivering |
| EP3628335A1 (en) | 2012-12-07 | 2020-04-01 | Translate Bio, Inc. | Lipidic nanoparticles for mrna delivery in the lungs |
| WO2014108515A1 (en) | 2013-01-10 | 2014-07-17 | Novartis Ag | Influenza virus immunogenic compositions and uses thereof |
| US11820977B2 (en) | 2013-03-14 | 2023-11-21 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| WO2014152774A1 (en) | 2013-03-14 | 2014-09-25 | Shire Human Genetic Therapies, Inc. | Methods and compositions for delivering mrna coded antibodies |
| US10899830B2 (en) | 2013-03-14 | 2021-01-26 | Translate Bio, Inc. | Methods and compositions for delivering MRNA coded antibodies |
| US9957499B2 (en) | 2013-03-14 | 2018-05-01 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| EP3932947A1 (en) | 2013-03-14 | 2022-01-05 | Translate Bio MA, Inc. | Methods and compositions for delivering mrna coded antibodies |
| US10420791B2 (en) | 2013-03-14 | 2019-09-24 | Translate Bio, Inc. | CFTR MRNA compositions and related methods and uses |
| EP3446712A1 (en) | 2013-03-14 | 2019-02-27 | Translate Bio Ma, Inc. | Cftr mrna compositions and related methods and uses |
| US10876104B2 (en) | 2013-03-14 | 2020-12-29 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US10087247B2 (en) | 2013-03-14 | 2018-10-02 | Translate Bio, Inc. | Methods and compositions for delivering mRNA coded antibodies |
| US11692189B2 (en) | 2013-03-14 | 2023-07-04 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US9713626B2 (en) | 2013-03-14 | 2017-07-25 | Rana Therapeutics, Inc. | CFTR mRNA compositions and related methods and uses |
| US10584165B2 (en) | 2013-03-14 | 2020-03-10 | Translate Bio, Inc. | Methods and compositions for delivering mRNA coded antibodies |
| US11510937B2 (en) | 2013-03-14 | 2022-11-29 | Translate Bio, Inc. | CFTR MRNA compositions and related methods and uses |
| US9181321B2 (en) | 2013-03-14 | 2015-11-10 | Shire Human Genetic Therapies, Inc. | CFTR mRNA compositions and related methods and uses |
| US10130649B2 (en) | 2013-03-15 | 2018-11-20 | Translate Bio, Inc. | Synergistic enhancement of the delivery of nucleic acids via blended formulations |
| EP3388834A1 (en) | 2013-03-15 | 2018-10-17 | Translate Bio, Inc. | Synergistic enhancement of the delivery of nucleic acids via blended formulations |
| EP4332576A2 (en) | 2013-03-15 | 2024-03-06 | Translate Bio, Inc. | Synergistic enhancement of the delivery of nucleic acids via blended formulations |
| WO2014144196A1 (en) | 2013-03-15 | 2014-09-18 | Shire Human Genetic Therapies, Inc. | Synergistic enhancement of the delivery of nucleic acids via blended formulations |
| US10646504B2 (en) | 2013-03-15 | 2020-05-12 | Translate Bio, Inc. | Synergistic enhancement of the delivery of nucleic acids via blended formulations |
| EP3757570A1 (en) | 2013-03-15 | 2020-12-30 | Translate Bio, Inc. | Synergistic enhancement of the delivery of nucleic acids via blended formulations |
| US9315472B2 (en) | 2013-05-01 | 2016-04-19 | Massachusetts Institute Of Technology | 1,3,5-triazinane-2,4,6-trione derivatives and uses thereof |
| EP3677567A1 (en) | 2013-07-23 | 2020-07-08 | Arbutus Biopharma Corporation | Compositions and methods for delivering messenger rna |
| WO2015061461A1 (en) | 2013-10-22 | 2015-04-30 | Shire Human Genetic Therapies, Inc. | Cns delivery of mrna and uses thereof |
| US10052284B2 (en) | 2013-10-22 | 2018-08-21 | Translate Bio, Inc. | Lipid formulations for delivery of messenger RNA |
| EP3501605A1 (en) | 2013-10-22 | 2019-06-26 | Translate Bio, Inc. | Mrna therapy for argininosuccinate synthetase deficiency |
| WO2015061500A1 (en) | 2013-10-22 | 2015-04-30 | Shire Human Genetic Therapies, Inc. | Mrna therapy for argininosuccinate synthetase deficiency |
| WO2015061467A1 (en) | 2013-10-22 | 2015-04-30 | Shire Human Genetic Therapies, Inc. | Lipid formulations for delivery of messenger rna |
| US10208295B2 (en) | 2013-10-22 | 2019-02-19 | Translate Bio, Inc. | MRNA therapy for phenylketonuria |
| WO2015061491A1 (en) | 2013-10-22 | 2015-04-30 | Shire Human Genetic Therapies, Inc. | Mrna therapy for phenylketonuria |
| EP3871696A1 (en) | 2013-10-22 | 2021-09-01 | Translate Bio MA, Inc. | Lipid formulations for delivery of messenger rna |
| US11377642B2 (en) | 2013-10-22 | 2022-07-05 | Translate Bio, Inc. | mRNA therapy for phenylketonuria |
| US10493031B2 (en) | 2013-10-22 | 2019-12-03 | Translate Bio, Inc. | Lipid formulations for delivery of messenger RNA |
| US10959953B2 (en) | 2013-10-22 | 2021-03-30 | Translate Bio, Inc. | Lipid formulations for delivery of messenger RNA |
| US9522176B2 (en) | 2013-10-22 | 2016-12-20 | Shire Human Genetic Therapies, Inc. | MRNA therapy for phenylketonuria |
| US11224642B2 (en) | 2013-10-22 | 2022-01-18 | Translate Bio, Inc. | MRNA therapy for argininosuccinate synthetase deficiency |
| EP3574923A1 (en) | 2013-10-22 | 2019-12-04 | Translate Bio, Inc. | Mrna therapy for phenylketonuria |
| US11890377B2 (en) | 2013-10-22 | 2024-02-06 | Translate Bio, Inc. | Lipid formulations for delivery of messenger RNA |
| EP4276176A2 (en) | 2013-10-22 | 2023-11-15 | Translate Bio, Inc. | Mrna therapy for argininosuccinate synthetase deficiency |
| EP4036241A1 (en) | 2013-10-22 | 2022-08-03 | Translate Bio, Inc. | Cns delivery of mrna and uses thereof |
| US9629804B2 (en) | 2013-10-22 | 2017-04-25 | Shire Human Genetic Therapies, Inc. | Lipid formulations for delivery of messenger RNA |
| US10780052B2 (en) | 2013-10-22 | 2020-09-22 | Translate Bio, Inc. | CNS delivery of MRNA and uses thereof |
| EP3450553A1 (en) | 2014-03-24 | 2019-03-06 | Translate Bio, Inc. | Mrna therapy for treatment of ocular diseases |
| EP3699274A1 (en) | 2014-03-24 | 2020-08-26 | Translate Bio, Inc. | Mrna therapy for the treatment of ocular diseases |
| US11884692B2 (en) | 2014-04-25 | 2024-01-30 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US11059841B2 (en) | 2014-04-25 | 2021-07-13 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US9850269B2 (en) | 2014-04-25 | 2017-12-26 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US10155785B2 (en) | 2014-04-25 | 2018-12-18 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| EP3587409A1 (en) | 2014-05-30 | 2020-01-01 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US10293057B2 (en) | 2014-05-30 | 2019-05-21 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US10286083B2 (en) | 2014-05-30 | 2019-05-14 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US10912844B2 (en) | 2014-05-30 | 2021-02-09 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US10286082B2 (en) | 2014-05-30 | 2019-05-14 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US10493166B2 (en) | 2014-05-30 | 2019-12-03 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US11433144B2 (en) | 2014-05-30 | 2022-09-06 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US10022455B2 (en) | 2014-05-30 | 2018-07-17 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| KR20170012366A (en) | 2014-06-04 | 2017-02-02 | 교와 핫꼬 기린 가부시키가이샤 | Ckap5-gene-silencing rnai pharmaceutical composition |
| US11104652B2 (en) | 2014-06-24 | 2021-08-31 | Translate Bio, Inc. | Stereochemically enriched compositions for delivery of nucleic acids |
| WO2015200465A1 (en) | 2014-06-24 | 2015-12-30 | Shire Human Genetic Therapies, Inc. | Stereochemically enriched compositions for delivery of nucleic acids |
| US10138213B2 (en) | 2014-06-24 | 2018-11-27 | Translate Bio, Inc. | Stereochemically enriched compositions for delivery of nucleic acids |
| US9840479B2 (en) | 2014-07-02 | 2017-12-12 | Massachusetts Institute Of Technology | Polyamine-fatty acid derived lipidoids and uses thereof |
| US9668980B2 (en) | 2014-07-02 | 2017-06-06 | Rana Therapeutics, Inc. | Encapsulation of messenger RNA |
| WO2016004318A1 (en) | 2014-07-02 | 2016-01-07 | Shire Human Genetic Therapies, Inc. | Encapsulation of messenger rna |
| US10342761B2 (en) | 2014-07-16 | 2019-07-09 | Novartis Ag | Method of encapsulating a nucleic acid in a lipid nanoparticle host |
| US10815530B2 (en) | 2014-08-14 | 2020-10-27 | Technion Research & Development Foundation Limited | Compositions and methods for therapeutics prescreening |
| WO2016054421A1 (en) | 2014-10-02 | 2016-04-07 | Protiva Biotherapeutics, Inc | Compositions and methods for silencing hepatitis b virus gene expression |
| EP3542825A1 (en) | 2014-11-10 | 2019-09-25 | Ethris GmbH | Induction of osteogenesis by delivering bmp encoding rna |
| EP3884964A1 (en) | 2014-12-05 | 2021-09-29 | Translate Bio, Inc. | Messenger rna therapy for treatment of articular disease |
| US9943595B2 (en) | 2014-12-05 | 2018-04-17 | Translate Bio, Inc. | Messenger RNA therapy for treatment of articular disease |
| US10864267B2 (en) | 2014-12-05 | 2020-12-15 | Translate Bio, Inc. | Messenger RNA therapy for treatment of articular disease |
| WO2016090262A1 (en) | 2014-12-05 | 2016-06-09 | Shire Human Genetic Therapies, Inc. | Messenger rna therapy for treatment of articular disease |
| EP3900702A1 (en) | 2015-03-19 | 2021-10-27 | Translate Bio, Inc. | Mrna therapy for pompe disease |
| US11712463B2 (en) | 2015-03-19 | 2023-08-01 | Translate Bio, Inc. | MRNA therapy for pompe disease |
| WO2016149508A1 (en) | 2015-03-19 | 2016-09-22 | Shire Human Genetic Therapies, Inc. | Mrna therapy for pompe disease |
| US10172924B2 (en) | 2015-03-19 | 2019-01-08 | Translate Bio, Inc. | MRNA therapy for pompe disease |
| US11090368B2 (en) | 2015-03-19 | 2021-08-17 | Translate Bio, Inc. | MRNA therapy for Pompe disease |
| WO2016197132A1 (en) | 2015-06-04 | 2016-12-08 | Protiva Biotherapeutics Inc. | Treating hepatitis b virus infection using crispr |
| US10695444B2 (en) | 2015-06-19 | 2020-06-30 | Massachusetts Institute Of Technology | Alkenyl substituted 2,5-piperazinediones, compositions, and uses thereof |
| US10201618B2 (en) | 2015-06-19 | 2019-02-12 | Massachusetts Institute Of Technology | Alkenyl substituted 2,5-piperazinediones, compositions, and uses thereof |
| WO2017019891A2 (en) | 2015-07-29 | 2017-02-02 | Protiva Biotherapeutics, Inc. | Compositions and methods for silencing hepatitis b virus gene expression |
| US11564893B2 (en) | 2015-08-17 | 2023-01-31 | Modernatx, Inc. | Methods for preparing particles and related compositions |
| US10995354B2 (en) | 2015-10-14 | 2021-05-04 | Translate Bio, Inc. | Modification of RNA-related enzymes for enhanced production |
| US10144942B2 (en) | 2015-10-14 | 2018-12-04 | Translate Bio, Inc. | Modification of RNA-related enzymes for enhanced production |
| WO2017111172A1 (en) | 2015-12-25 | 2017-06-29 | 協和発酵キリン株式会社 | Compounds as cationic lipids |
| US10428349B2 (en) | 2016-04-08 | 2019-10-01 | Translate Bio, Inc. | Multimeric coding nucleic acid and uses thereof |
| US11124804B2 (en) | 2016-04-08 | 2021-09-21 | Translate Bio, Inc. | Multimeric coding nucleic acid and uses thereof |
| WO2017177169A1 (en) | 2016-04-08 | 2017-10-12 | Rana Therapeutics, Inc. | Multimeric coding nucleic acid and uses thereof |
| US10266843B2 (en) | 2016-04-08 | 2019-04-23 | Translate Bio, Inc. | Multimeric coding nucleic acid and uses thereof |
| EP3825400A1 (en) | 2016-04-08 | 2021-05-26 | Translate Bio Ma, Inc. | Multimeric coding nucleic acid and uses thereof |
| US10835583B2 (en) | 2016-06-13 | 2020-11-17 | Translate Bio, Inc. | Messenger RNA therapy for the treatment of ornithine transcarbamylase deficiency |
| EP3842530A1 (en) | 2016-06-13 | 2021-06-30 | Translate Bio, Inc. | Messenger rna therapy for the treatment of ornithine transcarbamylase deficiency |
| WO2017218524A1 (en) | 2016-06-13 | 2017-12-21 | Rana Therapeutics, Inc. | Messenger rna therapy for the treatment of ornithine transcarbamylase deficiency |
| WO2018006052A1 (en) | 2016-06-30 | 2018-01-04 | Protiva Biotherapeutics, Inc. | Compositions and methods for delivering messenger rna |
| WO2018089846A1 (en) | 2016-11-10 | 2018-05-17 | Translate Bio, Inc. | Subcutaneous delivery of messenger rna |
| WO2018089801A1 (en) | 2016-11-10 | 2018-05-17 | Translate Bio, Inc. | Improved process of preparing mrna-loaded lipid nanoparticles |
| EP4249501A2 (en) | 2017-01-09 | 2023-09-27 | Whitehead Institute for Biomedical Research | Methods of altering gene expression by perturbing transcription factor multimers that structure regulatory loops |
| WO2018129544A1 (en) | 2017-01-09 | 2018-07-12 | Whitehead Institute For Biomedical Research | Methods of altering gene expression by perturbing transcription factor multimers that structure regulatory loops |
| US11253605B2 (en) | 2017-02-27 | 2022-02-22 | Translate Bio, Inc. | Codon-optimized CFTR MRNA |
| WO2018157154A2 (en) | 2017-02-27 | 2018-08-30 | Translate Bio, Inc. | Novel codon-optimized cftr mrna |
| WO2018165257A1 (en) | 2017-03-07 | 2018-09-13 | Translate Bio, Inc. | Polyanionic delivery of nucleic acids |
| US11905525B2 (en) | 2017-04-05 | 2024-02-20 | Modernatx, Inc. | Reduction of elimination of immune responses to non-intravenous, e.g., subcutaneously administered therapeutic proteins |
| US11576872B2 (en) | 2017-05-08 | 2023-02-14 | Flagship Pioneering Innovations V, Inc. | Compositions for facilitating membrane fusion and uses thereof |
| WO2018213476A1 (en) | 2017-05-16 | 2018-11-22 | Translate Bio, Inc. | Treatment of cystic fibrosis by delivery of codon-optimized mrna encoding cftr |
| US11173190B2 (en) | 2017-05-16 | 2021-11-16 | Translate Bio, Inc. | Treatment of cystic fibrosis by delivery of codon-optimized mRNA encoding CFTR |
| US11786607B2 (en) | 2017-06-15 | 2023-10-17 | Modernatx, Inc. | RNA formulations |
| WO2018236849A1 (en) | 2017-06-19 | 2018-12-27 | Translate Bio, Inc. | Messenger rna therapy for the treatment of friedreich's ataxia |
| US11744801B2 (en) | 2017-08-31 | 2023-09-05 | Modernatx, Inc. | Methods of making lipid nanoparticles |
| WO2019118806A1 (en) | 2017-12-14 | 2019-06-20 | Solid Biosciences Inc. | Non-viral production and delivery of genes |
| WO2019126593A1 (en) | 2017-12-20 | 2019-06-27 | Translate Bio, Inc. | Improved composition and methods for treatment of ornithine transcarbamylase deficiency |
| US11167043B2 (en) | 2017-12-20 | 2021-11-09 | Translate Bio, Inc. | Composition and methods for treatment of ornithine transcarbamylase deficiency |
| WO2019152802A1 (en) | 2018-02-02 | 2019-08-08 | Translate Bio, Inc. | Cationic polymers |
| WO2019222277A1 (en) | 2018-05-15 | 2019-11-21 | Translate Bio, Inc. | Subcutaneous delivery of messenger rna |
| WO2019222424A1 (en) | 2018-05-16 | 2019-11-21 | Translate Bio, Inc. | Ribose cationic lipids |
| WO2019226925A1 (en) | 2018-05-24 | 2019-11-28 | Translate Bio, Inc. | Thioester cationic lipids |
| WO2019232097A1 (en) | 2018-05-30 | 2019-12-05 | Translate Bio, Inc. | Phosphoester cationic lipids |
| WO2019232208A1 (en) | 2018-05-30 | 2019-12-05 | Translate Bio, Inc. | Cationic lipids comprising a steroidal moiety |
| WO2019232095A1 (en) | 2018-05-30 | 2019-12-05 | Translate Bio, Inc. | Vitamin cationic lipids |
| WO2019232103A1 (en) | 2018-05-30 | 2019-12-05 | Translate Bio, Inc. | Messenger rna vaccines and uses thereof |
| WO2020023533A1 (en) | 2018-07-23 | 2020-01-30 | Translate Bio, Inc. | Dry power formulations for messenger rna |
| US11174500B2 (en) | 2018-08-24 | 2021-11-16 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| WO2020047061A1 (en) | 2018-08-29 | 2020-03-05 | Translate Bio, Inc. | Improved process of preparing mrna-loaded lipid nanoparticles |
| WO2020056294A1 (en) | 2018-09-14 | 2020-03-19 | Translate Bio, Inc. | Composition and methods for treatment of methylmalonic acidemia |
| WO2020081933A1 (en) | 2018-10-19 | 2020-04-23 | Translate Bio, Inc. | Pumpless encapsulation of messenger rna |
| WO2020097376A1 (en) | 2018-11-09 | 2020-05-14 | Translate Bio, Inc. | Multi-peg lipid compounds |
| WO2020097384A1 (en) | 2018-11-09 | 2020-05-14 | Translate Bio, Inc. | 2,5-dioxopiperazine lipids with intercalated ester, thioester, disulfide and anhydride moieities |
| WO2020097511A2 (en) | 2018-11-09 | 2020-05-14 | Translate Bio, Inc. | Messenger rna therapy for treatment of ocular diseases |
| WO2020097379A2 (en) | 2018-11-09 | 2020-05-14 | Translate Bio, Inc. | Peg lipidoid compounds |
| WO2020102172A2 (en) | 2018-11-12 | 2020-05-22 | Translate Bio, Inc. | Methods for inducing immune tolerance |
| WO2020106946A1 (en) | 2018-11-21 | 2020-05-28 | Translate Bio, Inc. | TREATMENT OF CYSTIC FIBROSIS BY DELIVERY OF NEBULIZED mRNA ENCODING CFTR |
| WO2020106903A1 (en) | 2018-11-21 | 2020-05-28 | Translate Bio, Inc. | Cationic lipid compounds and compositions thereof for use in the delivery of messenger rna |
| US11166996B2 (en) | 2018-12-12 | 2021-11-09 | Flagship Pioneering Innovations V, Inc. | Anellovirus compositions and methods of use |
| US11446344B1 (en) | 2018-12-12 | 2022-09-20 | Flagship Pioneering Innovations V, Inc. | Anellovirus compositions and methods of use |
| WO2020146344A1 (en) | 2019-01-07 | 2020-07-16 | Translate Bio, Inc. | Composition and methods for treatment of primary ciliary dyskinesia |
| WO2020165352A1 (en) | 2019-02-14 | 2020-08-20 | Ethris Gmbh | Treatment of ciliopathies |
| EP4223306A2 (en) | 2019-02-14 | 2023-08-09 | Ethris GmbH | Treatment of ciliopathies |
| WO2020214946A1 (en) | 2019-04-18 | 2020-10-22 | Translate Bio, Inc. | Cystine cationic lipids |
| WO2020219427A1 (en) | 2019-04-22 | 2020-10-29 | Translate Bio, Inc. | Thioester cationic lipids |
| WO2020227085A1 (en) | 2019-05-03 | 2020-11-12 | Translate Bio, Inc. | Di-thioester cationic lipids |
| WO2020232276A1 (en) | 2019-05-14 | 2020-11-19 | Translate Bio, Inc. | Improved process of preparing mrna-loaded lipid nanoparticles |
| WO2020237227A1 (en) | 2019-05-22 | 2020-11-26 | Massachusetts Institute Of Technology | Circular rna compositions and methods |
| WO2020243540A1 (en) | 2019-05-31 | 2020-12-03 | Translate Bio, Inc. | Macrocyclic lipids |
| WO2020257716A1 (en) | 2019-06-21 | 2020-12-24 | Translate Bio, Inc. | Tricine and citric acid lipids |
| WO2020257611A1 (en) | 2019-06-21 | 2020-12-24 | Translate Bio, Inc. | Cationic lipids comprising an hydroxy moiety |
| WO2021007278A1 (en) | 2019-07-08 | 2021-01-14 | Translate Bio, Inc. | Improved mrna-loaded lipid nanoparticles and processes of making the same |
| WO2021016430A1 (en) | 2019-07-23 | 2021-01-28 | Translate Bio, Inc. | Stable compositions of mrna-loaded lipid nanoparticles and processes of making |
| WO2021021988A1 (en) | 2019-07-30 | 2021-02-04 | Translate Bio, Inc. | Treatment of cystic fibrosis by delivery of nebulized mrna encoding cftr |
| WO2021055609A1 (en) | 2019-09-20 | 2021-03-25 | Translate Bio, Inc. | Mrna encoding engineered cftr |
| WO2021072172A1 (en) | 2019-10-09 | 2021-04-15 | Translate Bio, Inc. | Compositions, methods and uses of messenger rna |
| WO2021081058A1 (en) | 2019-10-21 | 2021-04-29 | Translate Bio, Inc. | Compositions, methods and uses of messenger rna |
| EP4289951A2 (en) | 2019-12-04 | 2023-12-13 | Orna Therapeutics, Inc. | Circular rna compositions and methods |
| WO2021113777A2 (en) | 2019-12-04 | 2021-06-10 | Orna Therapeutics, Inc. | Circular rna compositions and methods |
| WO2021127394A2 (en) | 2019-12-20 | 2021-06-24 | Translate Bio, Inc. | Rectal delivery of messenger rna |
| WO2021127641A1 (en) | 2019-12-20 | 2021-06-24 | Translate Bio, Inc. | Improved process of preparing mrna-loaded lipid nanoparticles |
| WO2021142245A1 (en) | 2020-01-10 | 2021-07-15 | Translate Bio, Inc. | Compounds, pharmaceutical compositions and methods for modulating expression of muc5b in lung cells and tissues |
| WO2021173840A1 (en) | 2020-02-25 | 2021-09-02 | Translate Bio, Inc. | Improved processes of preparing mrna-loaded lipid nanoparticles |
| WO2021195218A1 (en) | 2020-03-24 | 2021-09-30 | Generation Bio Co. | Non-viral dna vectors and uses thereof for expressing gaucher therapeutics |
| WO2021195214A1 (en) | 2020-03-24 | 2021-09-30 | Generation Bio Co. | Non-viral dna vectors and uses thereof for expressing factor ix therapeutics |
| WO2021226463A1 (en) | 2020-05-07 | 2021-11-11 | Translate Bio, Inc. | Composition and methods for treatment of primary ciliary dyskinesia |
| WO2021226468A1 (en) | 2020-05-07 | 2021-11-11 | Translate Bio, Inc. | Improved compositions for cftr mrna therapy |
| WO2021226436A1 (en) | 2020-05-07 | 2021-11-11 | Translate Bio, Inc. | Optimized nucleotide sequences encoding sars-cov-2 antigens |
| WO2021231697A1 (en) | 2020-05-14 | 2021-11-18 | Translate Bio, Inc. | Peg lipidoid compounds |
| WO2021231901A1 (en) | 2020-05-15 | 2021-11-18 | Translate Bio, Inc. | Lipid nanoparticle formulations for mrna delivery |
| WO2021236855A1 (en) | 2020-05-19 | 2021-11-25 | Orna Therapeutics, Inc. | Circular rna compositions and methods |
| WO2022006527A1 (en) | 2020-07-02 | 2022-01-06 | Maritime Therapeutics, Inc. | Compositions and methods for reverse gene therapy |
| WO2022023284A1 (en) | 2020-07-27 | 2022-02-03 | Anjarium Biosciences Ag | Compositions of dna molecules, methods of making therefor, and methods of use thereof |
| WO2022076562A1 (en) | 2020-10-06 | 2022-04-14 | Translate Bio, Inc. | Improved process and formulation of lipid nanoparticles |
| WO2022081548A1 (en) | 2020-10-12 | 2022-04-21 | Translate Bio, Inc. | Improved process of preparing ice-based lipid nanoparticles |
| WO2022081544A1 (en) | 2020-10-12 | 2022-04-21 | Translate Bio, Inc. | Improved process of preparing mrna-loaded lipid nanoparticles |
| WO2022099194A1 (en) | 2020-11-09 | 2022-05-12 | Translate Bio, Inc. | Improved compositions for delivery of codon-optimized mrna |
| WO2022115547A1 (en) | 2020-11-25 | 2022-06-02 | Translate Bio, Inc. | Stable liquid lipid nanoparticle formulations |
| WO2022155404A1 (en) | 2021-01-14 | 2022-07-21 | Translate Bio, Inc. | Methods and compositions for delivering mrna coded antibodies |
| WO2022204549A1 (en) | 2021-03-25 | 2022-09-29 | Translate Bio, Inc. | Optimized nucleotide sequences encoding the extracellular domain of human ace2 protein or a portion thereof |
| WO2022225918A1 (en) | 2021-04-19 | 2022-10-27 | Translate Bio, Inc. | Improved compositions for delivery of mrna |
| WO2022223556A1 (en) | 2021-04-20 | 2022-10-27 | Anjarium Biosciences Ag | Compositions of dna molecules encoding amylo-alpha-1, 6-glucosidase, 4-alpha-glucanotransferase, methods of making thereof, and methods of use thereof |
| WO2022232286A1 (en) | 2021-04-27 | 2022-11-03 | Generation Bio Co. | Non-viral dna vectors expressing anti-coronavirus antibodies and uses thereof |
| WO2022232289A1 (en) | 2021-04-27 | 2022-11-03 | Generation Bio Co. | Non-viral dna vectors expressing therapeutic antibodies and uses thereof |
| WO2023278754A1 (en) | 2021-07-01 | 2023-01-05 | Translate Bio, Inc. | Compositions for delivery of mrna |
| WO2023021427A1 (en) | 2021-08-16 | 2023-02-23 | Glaxosmithkline Biologicals Sa | Freeze-drying of lipid nanoparticles (lnps) encapsulating rna and formulations thereof |
| WO2023021421A1 (en) | 2021-08-16 | 2023-02-23 | Glaxosmithkline Biologicals Sa | Low-dose lyophilized rna vaccines and methods for preparing and using the same |
| WO2023081526A1 (en) | 2021-11-08 | 2023-05-11 | Orna Therapeutics, Inc. | Lipid nanoparticle compositions for delivering circular polynucleotides |
| WO2023086893A1 (en) | 2021-11-10 | 2023-05-19 | Translate Bio, Inc. | Composition and methods for treatment of primary ciliary dyskinesia |
| WO2023135273A2 (en) | 2022-01-14 | 2023-07-20 | Anjarium Biosciences Ag | Compositions of dna molecules encoding factor viii, methods of making thereof, and methods of use thereof |
| WO2023144798A1 (en) | 2022-01-31 | 2023-08-03 | Genevant Sciences Gmbh | Ionizable cationic lipids for lipid nanoparticles |
| WO2023177655A1 (en) | 2022-03-14 | 2023-09-21 | Generation Bio Co. | Heterologous prime boost vaccine compositions and methods of use |
| WO2023214405A1 (en) | 2022-05-01 | 2023-11-09 | Yeda Research And Development Co. Ltd. | Reexpression of hnf4a to alleviate cancer-associated cachexia |
| WO2023215481A1 (en) | 2022-05-05 | 2023-11-09 | The Board Of Trustees Of The Leland Stanford Junior University | INTERFERING RNA THERAPY FOR PLN-R14del CARDIOMYOPATHY |
| WO2023239756A1 (en) | 2022-06-07 | 2023-12-14 | Generation Bio Co. | Lipid nanoparticle compositions and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2569664A1 (en) | 2005-12-22 |
| ATE536418T1 (en) | 2011-12-15 |
| CA2569664C (en) | 2013-07-16 |
| EP1766035A4 (en) | 2009-04-22 |
| US20110060032A1 (en) | 2011-03-10 |
| JP2008501730A (en) | 2008-01-24 |
| AU2005252273B2 (en) | 2011-04-28 |
| US9926560B2 (en) | 2018-03-27 |
| US7799565B2 (en) | 2010-09-21 |
| EP1766035A1 (en) | 2007-03-28 |
| EP1766035B1 (en) | 2011-12-07 |
| US20060008910A1 (en) | 2006-01-12 |
| US9181545B2 (en) | 2015-11-10 |
| US20160115477A1 (en) | 2016-04-28 |
| US20190071669A1 (en) | 2019-03-07 |
| AU2005252273A1 (en) | 2005-12-22 |
| JP4796062B2 (en) | 2011-10-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9926560B2 (en) | Lipid encapsulating interfering RNA | |
| AU2005251403B2 (en) | Cationic lipids and methods of use | |
| EP1648519B1 (en) | Lipid encapsulated interfering rna | |
| US20060051405A1 (en) | Compositions for the delivery of therapeutic agents and uses thereof | |
| EP1828219A1 (en) | Sirna silencing of apolipoprotein b | |
| AU2011253734A1 (en) | Cationic lipids and methods of use | |
| NZ583623A (en) | Lipid encapsulated interfering RNA |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2569664 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007526139 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005252273 Country of ref document: AU |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005757651 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200580022582.2 Country of ref document: CN |
|
| ENP | Entry into the national phase |
Ref document number: 2005252273 Country of ref document: AU Date of ref document: 20050607 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005252273 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005757651 Country of ref document: EP |