, 3 -BENZOXAK IN DERIVATIVES AS NON-STEROIDAL GLUCOCRTICOID RECEPTOR MODULATORS
The present invention relates to compounds which are non-steroidal glucocorticoid receptor modulators, pharmaceutical compositions comprising the compounds, the use of the compounds for the manufacture of medicaments particularly for the treatment of inflammatory and/or allergic conditions, processes for the preparation of the compounds, and chemical intermediates in the processes for the manufacture of the compounds.
Nuclear receptors are a class of structurally related proteins involved in the regulation of gene expression. The steroid hormone receptors are a subset of this family whose natural ligands typically comprise endogenous steroids such as estradiol (estrogen receptor), progesterone (progesterone receptor) and Cortisol (glucocorticoid receptor). Man-made ligands to these receptors play an important role in human health, in particular the use of glucocorticoid agonists to treat a wide range of inflammatory conditions.
Glucocorticoids exert their actions at the glucocorticoid receptor (GR) through at least two intracellular mechanisms, transactivation and transrepression (see: Schacke, H, Docke, W-D. & Asadullah, K (2002) Pharmacol and Therapeutics 96: 23-43; Ray, A., Siegel, M.D., Prefontaine, K.E. & Ray, P. (1995) Chest 107: 139S; and Konig, H., Ponta, H., Rahmsdorf, HJ. & Herrlich, P. (1992) EMBO J 11: 2241-2246). Transactivation involves direct binding of the glucocorticoid receptor to distinct deoxyribonucleic acid (DNA) response elements (GREs) within gene promoters, usually but not always increasing the transcription of the downstream gene product. Recently, it has been shown that the GR can also regulate gene expression through an additional pathway (transrepression) in which the GR does not bind directly to DNA. This mechanism involves interaction of the GR with other transcription factors, in particular NFkB and AP1 , leading to inhibition of their pro-transcriptional activity (Schacke, H, Docke, W-D. & Asadullah, K (2002) Pharmacol and Therapeutics 96: 23-43; Ray, A., Siegel, M. D., Prefontaine, K.E. & Ray, P. (1995) Chest 107: 139S). Many of the genes involved in the inflammatory response are transcriptionally activated through the NFkB and AP1 pathways and therefore inhibition of this pathway by glucocorticoids may explain their anti-inflammatory effect (see: Barnes, PJ. & Adcock, I. (1993) Trend Pharmacol Sci 14: 436-441; Cato, A.C. & Wade, E. (1996) Bioessays 18: 371-378).
Despite the effectiveness of glucocorticoids in treating a wide range of conditions, a number of side-effects are associated with pathological increases in endogenous Cortisol or the use of exogenous, and particularly systemically administered, glucocorticoids. These include reduction in bone mineral density (Wong, C.A., Walsh, L.J., Smith, CJ. et al. (2000) Lancet 355: 1399-1403), slowing of growth (Allen, D. B. (2000) Allergy 55: suppl 62, 15-18), skin bruising (Pauwels, R.A., Lofdahl, CG. , Latinen, L.A. et al. (1999) N Engl J Med 340: 1948-1953), development of cataracts (Gumming, R.G., Mitchell, P. & Leeder, S. R. (1997) N Engl J Med 337: 8-14) and dysregulation of lipid and glucose metabolism (Faul, J.L., Tormey, W., Tormey, V. & Burke, C (1998) BMJ 317: 1491 ; Andrews, R.C. & Walker, B. R. (1999) CHn Sci 96: 513-523). The side-effects are serious enough often to limit the dose of glucocorticoid that can be used to treat the underlying pathology leading to reduced efficacy of treatment.
It has been suggested that excessive activation of the transactivation-GRE pathway may mediate some of these side-effects (see Schacke, H, Docke, W-D. & Asadullah, K (2002) Pharmacol and Therapeutics 96: 23-43). Development of glucocorticoids that selectively modulate the transrepression pathway compared with the transactivation pathway may therefore have a superior anti-inflammatory to side-effect therapeutic index, allowing more effective and safer treatment of the patient. This new class of glucocorticoids could be used to treat more effectively and more safely the whole spectrum of disease currently treated by current glucocorticoids.
Current known glucocorticoids have proved useful in the treatment of inflammation, tissue rejection, auto-immunity, various malignancies, such as leukemias and lymphomas, Cushing's syndrome, rheumatic fever, polyarteritis nodosa, granulomatous polyarteritis, inhibition of myeloid cell lines, immune proliferation/apoptosis, HPA axis suppression and regulation, hypercortisolemia, modulation of the Th1/Th2 cytokine balance, chronic kidney disease, stroke and spinal cord injury, hypercalcemia, hypergylcemia, acute adrenal insufficiency, chronic primary adrenal insufficiency, secondary adrenal insufficiency, congenital adrenal hyperplasia, cerebral edema, thrombocytopenia and Little's syndrome.
Glucocorticoids are especially useful in disease states involving systemic inflammation such as inflammatory bowel disease, systemic lupus erythematosus, polyarteritis nodosa, Wegener's granulomatosis, giant cell arteritis, rheumatoid arthritis, osteoarthritis, seasonal rhinitis, allergic rhinitis, urticaria, angioneurotic edema, chronic obstructive pulmonary
disease, asthma, tendonitis, bursitis, Crohn's disease, ulcerative colitis, autoimmune chronic active hepatitis, organ transplantation, hepatitis and cirrhosis. Glucocorticoids have also been used as immunostimulants and repressors and as wound healing and tissue repair agents.
Glucocorticoids have also found use in the treatment of diseases such as inflammatory scalp alopecia, panniculitis, psoriasis, discoid lupus erythemnatosus, inflamed cysts, atopic dermatitis, pyoderma gangrenosum, pemphigus vulgaris, bullous pemphigoid, systemic lupus erythematosus, dermatomyositis, herpes gestationis, eosinophilic fasciitis, relapsing polychondritis, inflammatory vasculitis, sarcoidosis, Sweet's disease, type 1 reactive leprosy, capillary hemangiomas, contact dermatitis, atopic dermatitis, lichen planus, exfoliative dermatitus, erythema nodosum, acne, hirsutism, toxic epidermal necrolysis, erythema multiform, cutaneous T-cell lymphoma.
WO00/32584, WO02/10143, WO03/082827, WO/03082280 and DE10261874 disclose certain non-steroidal glucocorticoid receptor modulators.
The present invention provides compounds of formula (I):
wherein R
1 represents 1-ethylpropyl, 1 -methyiethyl or 2-methylpropyl
and physiologically functional derivatives thereof (hereinafter "the compounds of the invention").
In one embodiment of the invention R1 represents 1-ethylpropyl. In a second embodiment of the invention R1 represents 1 -methyiethyl. In another embodiment of the invention R1 represents 2-methylpropyl.
The compounds of formula (I) each contain two chiral centres and there are four possible stereoisomers of each compound of formula (I). Further, at least one of the possible stereoisomers of each compound of formula (I) modulates the glucocorticoid receptor.
The terms D1 and D2 are used herein to refer to the diastereomers of a compound of formula (I), based on the order of their elution using the chromatography methodology described herein (LCMS System A). D1 refers to the first diastereomer to elute, and D2 refers to the second diastereomer to elute.
The terms D1E1, D1E2, D2E1 and D2E2 are used herein to refer to the isomers of a compound of formula (I). D1E1 refers to the first enantiomer to elute, and D1E2 refers to the second enantiomer to elute, during chiral separation of diastereomer D1 according to the methodology described herein. D2E1 refers to the first enantiomer to elute, and D2E2 refers to the second enantiomer to elute, during chiral separation of diastereomer D2 according to the methodology described herein.
It will be appreciated by those skilled in the art that although the absolute retention time on chromatography can be variable, the order of elution remains the same when a similar column and conditions are employed. However, the use of a different chromatography column and conditions may alter the order of elution.
A mixture of isomers, such as a racemic mixture, may be preferred, for example, a mixture of all four isomers, or a racemic mixture of two isomers may be preferred, for example diastereomer D2. Thus, in one embodiment of the invention the diastereomer D2 is preferred.
Alternatively, a single isomer may be preferred, for example the isomer D2E2. Therefore, in one embodiment of the invention the isomer D2E2 is preferred.
For example, when the group R1 represents 1-ethylpropyl, preferably the compound is isomer D2E2. Isomer D2E2 of the compound of formula (I) wherein the group R1 represents 1-ethylpropyl is characterised by having a circular dichroism as described below:
Circular Dichroism (Jasco Spectrophotometer Model J-720) acetonitrile, room temperature, 0.00038M, v = 350 - 200 nm, cell length = 0.1 cm, 212.0 nm (de = -8.54; E = 17206), 242.6 nm (de = 13.45; E = 28860), 277.0 nm (de = 6.27; E = 13074).
Additionally or alternatively, isomer D2E2 of the compound of formula (I) wherein the group R1 represents 1-ethylpropyl is characterised by having a retention time of about 6.54 min when eluted on an analytical chiral HPLC on a 25 x 0.46 cm Chiralpak AD column using a mobile phase of 10% ethanol in heptane at 1 mL/min. Isomer D2E2 is the later running enantiomer of the racemic mixture of isomers D2E1 and D2E2.
Compounds of the invention which are of particular interest include: 2-{[1-(1-ethylpropyl)-1 ,2,3,4-tetrahydro-1-naphthalenyl]methyl}-3,3,3-trifluoro-2-hydroxy-Λ/- (4-methyl-1-oxo-1H~2,3-benzoxazin-6-yl)propanamide isomer D2E2; 3,3,3-trifluoro-2-hydroxy-2-{[1-(1-methylethyl)-1 ,2,3,4-tetrahydro-1-naphthalenyl]methyl}- Λ/-(4-methyl-1-oxo-1/-/-2,3-benzoxazin-6-yl)propanamide isomer D2E2; and physiologically functional derivatives thereof.
The compounds of the invention may provide agonism of the glucocorticoid receptor.
It has been found that at least one of the possible stereoisomers of each of the compounds of formula (I) binds to the glucocorticoid receptor. Further, it appears that at least one of the possible stereoisomers of each of the compounds of formula (I) has glucocorticoid receptor agonist activity. Additionally, it appears that at least one of the possible stereoisomers of each of the compounds of formula (I) possesses advantageous selectivity in respect of maintaining transrepression activity whilst reducing the transactivation activity. These observations are believed to be indicative that the compounds of the invention may provide anti-inflammatory properties with fewer or less severe related side effects.
It will be appreciated by those skilled in the art that at least one isomer (e.g. an enantiomer in a diastereomer) has the described activity. The other isomers may have similar activity, less activity, no activity or may have some antagonist activity in a functional assay. For example, in the case of the compound of formula (I) wherein the group R1 represents 1-ethylpropyl, both the racemic mixture of isomers D2E1 and D2E2
(ie. the diastereomer D2), and the single isomer D2E2 alone have been shown to demonstrate glucocorticoid receptor agonist activity.
The invention includes physiologically functional derivatives of the compounds of formula (I). By the term "physiologically functional derivative" is meant a chemical derivative of a compound of formula (I) having the same physiological function as the free compound of formula (I), for example, by being convertible in the body thereto and includes any pharmaceutically acceptable esters, carbonates and carbamates, solvates of compounds of formula (I) and solvates of any pharmaceutically acceptable esters, carbonates and carbamates or salts of compounds of formula (I), which, upon administration to the recipient, are capable of providing (directly or indirectly) compounds of formula (I) or active metabolite or residue thereof. Thus one particular embodiment of the invention embraces compounds of formula (I) and salts and solvates thereof. A further embodiment of the invention embraces compounds of formula (I) and salts thereof. A further embodiment embraces compounds of formula (I).
Solvates of the compounds of formula (I) and physiologically functional derivatives thereof which are suitable for use in medicine are those wherein the associated solvent is pharmaceutically acceptable. However, solvates having non-pharmaceutically acceptable counter-ions or associated solvents are within the scope of the present invention, for example, for use as intermediates in the preparation of other compounds of formula (I) and their pharmaceutically acceptable salts, solvates, and physiologically functional derivatives.
The compounds of the invention are expected to have potentially beneficial anti¬ inflammatory or anti-allergic effects, particularly upon topical administration, demonstrated by, for example, their ability to bind to the glucocorticoid receptor and to illicit a response via that receptor. Hence, the compounds of the invention may be useful in the treatment of inflammatory and/or allergic disorders.
Examples of disease states in which the compounds of the invention are expected to have utility include skin diseases such as eczema, psoriasis, allergic dermatitis, neurodermatitis, pruritis and hypersensitivity reactions; inflammatory conditions of the nose, throat or lungs such as asthma (including allergen-induced asthmatic reactions), rhinitis (including hayfever), nasal polyps, chronic obstructive pulmonary disease (COPD),
interstitial lung disease, and fibrosis; inflammatory bowel conditions such as ulcerative colitis and Crohn's disease; and auto-immune diseases such as rheumatoid arthritis.
It will be appreciated by those skilled in the art that reference herein to treatment extends to prophylaxis as well as the treatment of established conditions.
As mentioned above, compounds of the invention are expected to be of use in human or veterinary medicine, in particular as anti-inflammatory and anti-allergic agents.
There is thus provided as a further aspect of the invention a compound of the invention for use in human or veterinary medicine, particularly in the treatment of patients with inflammatory and/or allergic conditions, such as rheumatoid arthritis, asthma, COPD, allergy or rhinitis.
A further aspect of the present invention provides a compound of formula (I), for use in the treatment of patients with skin disease such as eczema, psoriasis, allergic dermatitis, neurodermatitis, pruritis and/or hypersensitivity reactions.
According to another aspect of the invention, there is provided the use of a compound of the invention for the manufacture of a medicament for the treatment of patients with inflammatory and/or allergic conditions, such as rheumatoid arthritis, asthma, COPD, allergy or rhinitis.
According to yet to another aspect of the invention, there is provided the use of a compound of the invention for the manufacture of a medicament for the treatment of patients with skin disease such as eczema, psoriasis, allergic dermatitis, neurodermatitis, pruritis and hypersensitivity reactions.
In a further or alternative aspect, there is provided a method for the treatment of a human or animal subject with an inflammatory and/or allergic condition, which method comprises administering to said human or animal subject an effective amount of a compound of the invention.
In yet a further or alternative aspect, there is provided a method for the treatment of a human or animal subject with for skin disease such as eczema, psoriasis, allergic
dermatitis, neurodermatitis, pruritis and hypersensitivity reactions, which method comprises administering to said human or animal subject an effective amount of a compound of the invention.
The compounds according to the invention may be formulated for administration in any convenient way, and the invention therefore also includes within its scope pharmaceutical compositions comprising a compound of the invention together, if desirable, in admixture with one or more physiologically acceptable diluents or carriers.
Further, there is provided a process for the preparation of such pharmaceutical compositions which comprises mixing the ingredients.
The compounds of the invention may, for example, be formulated for oral, buccal, sublingual, parenteral, local rectal administration or other local administration.
Local administration as used herein, includes administration by insufflation and inhalation. Examples of various types of preparation for local administration include ointments, lotions, creams, gels, foams, preparations for delivery by transdermal patches, powders, sprays, aerosols, capsules or cartridges for use in an inhaler or insufflator or drops (e.g. eye or nose drops), solutions/suspensions for nebulisation, suppositories, pessaries, retention enemas and chewable or suckable tablets or pellets (e.g. for the treatment of aphthous ulcers) or liposome or microencapsulation preparations.
Ointments, creams and gels, may, for example, be formulated with an aqueous or oily base with the addition of suitable thickening and/or gelling agent and/or solvents. Such bases may thus, for example, include water and/or an oil such as liquid paraffin or a vegetable oil such as arachis oil or castor oil, or a solvent such as polyethylene glycol. Thickening agents and gelling agents which may be used according to the nature of the base include soft paraffin, aluminium stearate, cetostearyl alcohol, polyethylene glycols, woolfat, beeswax, carboxypolymethylene and cellulose derivatives, and/or glyceryl monostearate and/or non-ionic emulsifying agents.
Lotions may be formulated with an aqueous or oily base and will in general also contain one or more emulsifying agents, stabilising agents, dispersing agents, suspending agents or thickening agents.
Powders for external application may be formed with the aid of any suitable powder base, for example, talc, lactose or starch. Drops may be formulated with an aqueous or non¬ aqueous base also comprising one or more dispersing agents, solubilising agents, suspending agents or preservatives.
Spray compositions may for example be formulated as aqueous solutions or suspensions or as aerosols delivered from pressurised packs, such as a metered dose inhaler, with the use of a suitable liquefied propellant. Aerosol compositions suitable for inhalation can be either a suspension or a solution and generally contain a compound of formula (I) and a suitable propellant such as a fluorocarbon or hydrogen-containing chlorofluorocarbon or mixtures thereof, particularly hydrofluoroalkanes, especially 1 ,1 ,1 ,2-tetrafluoroethane, 1 ,1 ,1 ,2,3,3,3-heptafluoro-n-propane or a mixture thereof. The aerosol composition may optionally contain additional formulation excipients well known in the art such as surfactants e.g. oleic acid or lecithin and cosolvents e.g. ethanol.
Advantageously, the formulations of the invention may be buffered by the addition of suitable buffering agents.
Capsules and cartridges for use in an inhaler or insufflator, of for example gelatine, may be formulated containing a powder mix for inhalation of a compound of the invention and a suitable powder base such as lactose or starch. Each capsule or cartridge may generally contain between 20μg-10mg of the compound of formula (I). Alternatively, the compound of the invention may be presented without excipients such as lactose.
The proportion of the active compound of formula (I) in the local compositions according to the invention depends on the precise type of formulation to be prepared but will generally be within the range of from 0.001 to 10% by weight. Generally, however for most types of preparations advantageously the proportion used will be within the range of from 0.005 to 1% and preferably from 0.01 to 0.5%. However, in powders for inhalation or insufflation the proportion used will be within the range of from 0.1 to 5%.
Aerosol formulations are preferably arranged so that each metered dose or "puff' of aerosol contains from 20μg to 10mg preferably from 20μg-2000μg, more preferably from 20μg-500μg of a compound of formula (I). Administration may be once daily or several
times daily, for example 2, 3, 4 or 8 times, giving for example 1, 2 or 3 doses each time. The overall daily dose with an aerosol will be within the range from 100μg to 10mg preferably from 200μg to 2000μg. The overall daily dose and the metered dose delivered by capsules and cartridges in an inhaler or insufflator will generally be double that delivered with aerosol formulations.
In the case of suspension aerosol formulations, the particle size of the particular (e.g., micronised) drug should be such as to permit inhalation of substantially all the drug into the lungs upon administration of the aerosol formulation and will thus be less than 100 microns, desirably less than 20 microns, and, in particular, in the range of from 1 to 10 microns, such as from 1 to 5 microns, more preferably from 2 to 3 microns.
The formulations of the invention may be prepared by dispersal or dissolution of the medicament and a compound of the invention in the selected propellant in an appropriate container, for example, with the aid of sonication or a high-shear mixer. The process is desirably carried out under controlled humidity conditions.
The chemical and physical stability and the pharmaceutical acceptability of the aerosol formulations according to the invention may be determined by techniques well known to those skilled in the art. Thus, for example, the chemical stability of the components may be determined by HPLC assay, for example, after prolonged storage of the product. Physical stability data may be gained from other conventional analytical techniques such as, for example, by leak testing, by valve delivery assay (average shot weights per actuation), by dose reproducibility assay (active ingredient per actuation) and spray distribution analysis.
The stability of the suspension aerosol formulations according to the invention may be measured by conventional techniques, for example, by measuring flocculation size distribution using a back light scattering instrument or by measuring particle size distribution by cascade impaction or by the "twin impinger" analytical process. As used herein reference to the "twin impinger" assay means "Determination of the deposition of the emitted dose in pressurised inhalations using apparatus A" as defined in British Pharmacopaeia 1988, pages A204-207, Appendix XVII C. Such techniques enable the "respirable fraction" of the aerosol formulations to be calculated. One method used to calculate the "respirable fraction" is by reference to "fine particle fraction" which is the
amount of active ingredient collected in the lower impingement chamber per actuation expressed as a percentage of the total amount of active ingredient delivered per actuation using the twin impinger method described above.
MDI canisters generally comprise a container capable of withstanding the vapour pressure of the propellant used such as a plastic or plastic-coated glass bottle or preferably a metal can, for example, aluminium or an alloy thereof which may optionally be anodised, lacquer-coated and/or plastic-coated (for example incorporated herein by reference WO96/32099 wherein part or all of the internal surfaces are coated with one or more fluorocarbon polymers optionally in combination with one or more non-fluorocarbon polymers), which container is closed with a metering valve. The cap may be secured onto the can via ultrasonic welding, screw fitting or crimping. MDIs taught herein may be prepared by methods of the art (e.g., see Byron, above and WO/96/32099). Preferably the canister is fitted with a cap assembly, wherein a drug-metering valve is situated in the cap, and said cap is crimped in place.
The metering valves are designed to deliver a metered amount of the formulation per actuation and incorporate a gasket to prevent leakage of propellant through the valve. The gasket may comprise any suitable elastomeric material such as, for example, low density polyethylene, chlorobutyl, black and white butadiene-acrylonitrile rubbers, butyl rubber and neoprene. Suitable valves are commercially available from manufacturers well known in the aerosol industry, for example, from Valois, France (e.g. DF10, DF30, DF60), Bespak pic, UK (e.g. BK300, BK357) and 3M-Neotechnic Ltd, UK (e.g. Spraymiser™).
Conventional bulk manufacturing methods and machinery well known to those skilled in the art of pharmaceutical aerosol manufacture may be employed for the preparation of large-scale batches for the commercial production of filled canisters. Thus, for example, in one bulk manufacturing method a metering valve is crimped onto an aluminium can to form an empty canister. The particulate medicament is added to a charge vessel and liquefied propellant is pressure filled through the charge vessel into a manufacturing vessel, together with liquefied propellant containing the surfactant. The drug suspension is mixed before recirculation to a filling machine and an aliquot of the drug suspension is then filled through the metering valve into the canister.
In an alternative process, an aliquot of the liquefied formulation is added to an open canister under conditions which are sufficiently cold to ensure formulation does not vaporise, and then a metering valve crimped onto the canister.
Typically, in batches prepared for pharmaceutical use, each filled canister is check- weighed, coded with a batch number and packed into a tray for storage before release testing.
In one aspect the present invention provides a pharmaceutical aerosol formulation comprising a compound of formula (I), or a physiologically functional derivative thereof, and a fluorocarbon or hydrogen containing chlorofluorocarbon as propellant, optionally in combination with a surfactant and/or cosolvent.
Topical preparations may be administered by one or more applications per day to the affected area; over skin areas occlusive dressings may advantageously be used. Continuous or prolonged delivery may be achieved by an adhesive reservoir system.
For internal administration the compounds according to the invention may, for example, be formulated in conventional manner for oral, parenteral or rectal administration. Formulations for oral administration include syrups, elixirs, powders, granules, tablets and capsules which typically contain conventional excipients such as binding agents, fillers, lubricants, disintegrants, wetting agents, suspending agents, emulsifying agents, preservatives, buffer salts, flavouring, colouring and/or sweetening agents as appropriate. Dosage unit forms are, however, preferred as described below.
The compound according to the invention may in general be given by internal administration in cases where systemic adreno-cortical therapy is indicated.
Slow release or enteric coated formulations may be advantageous, particularly for the treatment of inflammatory bowel disorders.
In some embodiments, the compound of formula (I) will be formulated for oral administration. In other embodiments the compounds of formula (I) will be formulated for inhaled administration.
The compound and pharmaceutical formulations according to the invention may be used in combination with or include one or more other therapeutic agents, for example selected from anti-inflammatory agents, anticholinergic agents (particularly an M1ZM2ZM3 receptor antagonist), β2-adrenoreceptor agonists, antiinfective agents (e.g. antibiotics, antivirals), or antihistamines. The invention thus provides, in a further aspect, a combination comprising a compound of formula (I) or a pharmaceutically acceptable salt, solvate or physiologically functional derivative thereof together with one or more other therapeutically active agents, for example selected from an anti-inflammatory agent (for example another corticosteroid or an NSAID), . an anticholinergic agent, a β2- adrenoreceptor agonist, an antiinfective agent (e.g. an antibiotic or an antiviral), or an antihistamine. One embodiment of the invention encompasses combinations comprising a compound of formula (I) or a pharmaceutically acceptable salt, solvate or physiologically functional derivative thereof together with a β2-adrenoreceptor agonist, andZor an anticholinergic, andZor a PDE-4 inhibitor. Suitable combinations are those comprising one or two other therapeutic agents.
It will be clear to a person skilled in the art that, where appropriate, the other therapeutic ingredient(s) may be used in the form of salts, (e.g. as alkali metal or amine salts or as acid addition salts), or prodrugs, or as esters (e.g. lower alkyl esters), or as solvates (e.g. hydrates) to optimise the activity andZor stability andZor physical characteristics (e.g. solubility) of the therapeutic ingredient. It will be clear also that where appropriate, the therapeutic ingredients may be used in optically pure form.
Suitable combinations include a combination comprising of a compound of the invention together with a β2-adrenoreceptor agonist.
Examples of β2-adrenoreceptor agonists include salmeterol (e.g. as racemate or a single enantiomer such as the R-enantiomer), salbutamol, formoterol, salmefamol, fenoterol or terbutaline and salts thereof, for example the xinafoate salt of salmeterol, the sulphate salt or free base of salbutamol or the fumarate salt of formoterol. In one embodiment the β2-adrenoreceptor agonists are long-acting β2-adrenoreceptor agonists, for example those having a therapeutic effect over a 24 hour period such as salmeterol or formoterol.
Suitable long acting β2-adrenoreceptor agonists may include those described in WO 02Z066422, WO 02Z070490, WO 02/076933, WO 03/024439, WO 03/072539, WO
03/091204, WO 04/016578, WO 2004/022547, WO 2004/037807, WO 2004/037773, WO 2004/037768, WO 2004/039762, WO 2004/039766, WO01/42193 and WO03/042160.
Examples of long-acting β2-adrenoreceptor agonists may include compounds of formula (XX):
or a salt or solvate thereof, wherein: m is an integer of from 2 to 8; n is an integer of from 3 to 11 , with the proviso that m + n is 5 to 19, R
21 is -XSO
2NR
26R
27 wherein X is -(CH
2)
P- or C
2-6 alkenylene; R
26 and R
27 are independently selected from hydrogen, C
1-6alkyl, C
3-7cycloalkyl, C(O)NR
28R
29, phenyl, and phenyl (d
-4alkyl)-, or R
26 and R
27, together with the nitrogen to which they are bonded, form a 5-, 6-, or 7- membered nitrogen containing ring, and R
26 and R
27 are each optionally substituted by one or two groups selected from halo, C
1-6alkyl, C
1-6haloalkyl, C
1-6alkoxy, hydroxy- substituted C
1^aIkOXy
1 -CO
2R
28, -SO
2NR
28R
29, -CONR
28R
29, -NR
28C(O)R
29, or a 5-, 6- or 7-membered heterocylic ring; R
28 and R
29 are independently selected from hydrogen, C^alkyl, C
3.
6cycloalkyl, phenyl, and phenyl (C
1-4alkyl)-; and p is an integer of from 0 to 6, preferably from 0 to 4; R
22 and R
23 are independently selected from hydrogen, C
1-6alkyl, Ci
-6alkoxy, halo, phenyl, and Ci
-6haloalkyl; and R
24 and R
25 are independently selected from hydrogen and C
1-4alkyl with the proviso that the total number of carbon atoms in R
24 and R
25 is not more than 4.
Other examples of long-acting β2-adrenoreceptor agonists may include: 3-(4-{[6-({(2R)-2-hydroxy-2-[4-hydroxy-3-(hydroxymethyl)phenyl]ethyl}amino) hexyl]oxy}butyl)benzenesulfonamide;
3-(3-{[7-({(2R)-2~hydroxy-2-[4-hydroxy-3-hydroxymethyl)phenyl]ethyl}- amino)heptyl]oxy}propyl)benzenesulfonamide; 4-{(1R)-2-[(6-{2-t(2,6-dichlorobenzyl)oxy]ethoxy}hexyl)amino]-1-hydroxyethyl}-2- (hydroxymethyl)phenol; 4-{(1 R)-2-[(6-{4-t3-(cyclopentylsulfonyl)phenyl]butoxy}hexyl)amino]-1 -hydroxyethyl}-2- (hydroxymethyl)phenol; N-[2-hydroxyl-5-[(1 R)-1 -hydroxy-2-[[2-4-[[(2R)-2-hydroxy-2- phenylethyl]amino]phenyl]ethyl]amino]ethyl]phenyl]foramide, and N-2{2-[4-(3-phenyl-4-methoxyphenyl)aminophenyl]ethyl}-2-hydroxy-2-(8-hydroxy-2(1H)- quinolinon-5-yl)ethylamine.
Suitable anti-inflammatory agents include corticosteroids. Suitable corticosteroids which may be used in combination with the compounds of the invention are those oral and inhaled corticosteroids and their pro-drugs which have anti-inflammatory activity. Examples include methyl prednisolone, prednisolone, dexamethasone, fluticasone propionate, 6α,9α-difluoro-11 β-hydroxy-16α-methyl-17α-[(4-methyl-1 ,3-thiazole-5- carbonyl)oxy]-3-oxo-androsta-1,4-diene-17β-carbothioic acid S-fluoromethyl ester, 6α,9α- difluoro-17α-[(2-furanylcarbonyl)oxy]-11 β-hydroxy-16α-methyl-3-oxo-androsta-1 ,4-diene- 17β-carbothioic acid S-fluoromethyl ester, 6α,9α-difluoro-11β-hydroxy-16α-methyl-3-oxo- 17α-propionyloxy- androsta-1 ,4-diene-17β-carbothioic acid S-(2-oxo-tetrahydro-furan-3S- yl) ester, beclomethasone esters (e.g. the 17-propionate ester or the 17,21-dipropionate ester), budesonide, flunisolide, mometasone esters (e.g. the furoate ester), triamcinolone acetonide, rofleponide, ciclesonide (16α,17-[[(R)-cyclohexylmethylene]bis(oxy)]-11β,21- dihydroxy-pregna-1,4-diene-3,20-dione), butixocort propionate, RPR-106541 , and ST- 126. Preferred corticosteroids include fluticasone propionate, 6α,9α-difluoro-11β-hydroxy- 16α-methyl-17α-[(4-methyl-1 ,3-thiazole-5-carbonyl)oxy]-3-oxo-androsta-1 ,4-diene-17β- carbothioic acid S-fluoromethyl ester and 6α,9α~difluoro~17α-[(2-furanylcarbonyl)oxy]-11β- hydroxy-16α-methyl-3-oxo-androsta-1 ,4-diene-17β-carbothioic acid S-fluoromethyl ester, more preferably 6α,9α-difluoro-17α-[(2-furanylcarbonyl)oxy]-11 β-hydroxy-16α-methyl-3- oxo-androsta-1 ,4-diene-17β-carbothioic acid S-fluoromethyl ester.
Non-steroidal compounds having glucocorticoid agonism that may possess selectivity for transrepression over transactivation and that may be useful in combination therapy include those covered in the following patents: WO03/082827, WO01/10143,
WO98/54159, WO04/005229, WO04/009016, WO04/009017, WO04/018429, WO03/104195, WO03/082787, WO03/082280, WO03/059899, WO03/101932, WO02/02565, WO01/16128, WO00/66590, WO03/086294, WO04/026248, WO03/061651 , WO03/08277.
Suitable anti-inflammatory agents include non-steroidal anti-inflammatory drugs (NSAI D's).
Suitable NSAID's include sodium cromoglycate, nedocromil sodium, phosphodiesterase (PDE) inhibitors (e.g. theophylline, PDE4 inhibitors or mixed PDE3/PDE4 inhibitors), leukotriene antagonists, inhibitors of leukotriene synthesis (e.g. montelukast), iNOS inhibitors, tryptase and elastase inhibitors, beta-2 integrin antagonists and adenosine receptor agonists or antagonists (e.g. adenosine 2a agonists), cytokine antagonists (e.g. chemokine antagonists, such as a CCR3 antagonist) or inhibitors of cytokine synthesis, or 5-lipoxygenase inhibitors. Suitable other β2-adrenoreceptor agonists include salmeterol (e.g. as the xinafoate), salbutamol (e.g. as the sulphate or the free base), formoterol (e.g. as the fumarate), fenoterol or terbutaline and salts thereof. An iNOS (inducible nitric oxide synthase inhibitor) is preferably for oral administration. Suitable iNOS inhibitors include those disclosed in WO93/13055, WO98/30537, WO02/50021, WO95/34534 and WO99/62875. Suitable CCR3 inhibitors include those disclosed in WO02/26722.
Of particular interest is use of the compounds of formula (I) in combination with a phosphodiesterase 4 (PDE4) inhibitor, especially in the case of a formulation adapted for inhalation. The PDE4-specific inhibitor useful in this aspect of the invention may be any compound that is known to inhibit the PDE4 enzyme or which is discovered to act as a PDE4 inhibitor, and which are only PDE4 inhibitors, not compounds which inhibit other members of the PDE family, such as PDE3 and PDE5, as well as PDEΞ4.
Compounds of interest include c/s-4-cyano-4-(3-cyclopentyloxy-4- methoxyphenyl)cyclohexan-1-carboxylic acid, 2-carbomethoxy-4-cyano-4-(3- cyclopropylmethoxy-4-difluoromethoxyphenyl)cyclohexan-1 -one and c/s-[4-cyano-4-(3- cyclopropylmethoxy-4-difluoromethoxyphenyl)cyclohexan-1 -ol]. Also, c/s-4-cyano-4-[3- (cyclopentyloxy)-4-methoxyphenyl]cyclohexane-1-carboxylic acid (also known as cilomilast) and its salts, esters, pro-drugs or physical forms, which is described in U.S.
patent 5,552,438 issued 03 September, 1996; this patent and the compounds it discloses are incorporated herein in full by reference.
Other compounds of interest include AWD-12-281 from Elbion (Hofgen, N. et al. 15th EFMC lnt Symp Med Chem (Sept 6-10, Edinburgh) 1998, Abst P.98; CAS reference No. 247584020-9); a 9-benzyladenine derivative nominated NCS-613 (INSERM); D-4418 from Chiroscience and Schering-Plough; a benzodiazepine PDE4 inhibitor identified as Cl- 1018 (PD-168787) and attributed to Pfizer; a benzodioxole derivative disclosed by Kyowa Hakko in WO99/16766; K-34 from Kyowa Hakko; V-11294A from Napp (Landells, L.J. et al. Eur Resp J [Annu Cong Eur Resp Soc (Sept 19-23, Geneva) 1998] 1998, 12 (Suppl. 28): Abst P2393); roflumilast (CAS reference No 162401-32-3) and a pthalazinone (WO99/47505, the disclosure of which is hereby incorporated by reference) from Byk- Gulden; Pumafentrine, (-)-p-[(4aR*,10bS*)-9-ethoxy-1 ,2,3,4,4a,10b-hexahydro-8- methoxy-2-methylbenzo[c][1 ,6]naphthyridin-6-yl]-N,N-diisopropylbenzamide which is a mixed PDE3/PDE4 inhibitor which has been prepared and published on by Byk-Gulden, now Altana; arofylline under development by Almirall-Prodesfarma; VM554/UM565 from Vernalis; or T-440 (Tanabe Seiyaku; Fuji, K. et al. J Pharmacol Exp Ther,1998, 284(1): 162), and T2585.
Further compounds of interest are disclosed in the published international patent application WO04/024728 (Glaxo Group Ltd), PCT/EP2003/014867 (Glaxo Group Ltd) and PCT/EP2004/005494 (Glaxo Group Ltd).
Suitable anticholinergic agents are those compounds that act as antagonists at the muscarinic receptors, in particular those compounds which are antagonists of the M1 or M3 receptors, dual antagonists of the M1/M3 or M2/M3, receptors or pan-antagonists of the Mi/M2/M3 receptors. Exemplary compounds for administration via inhalation include ipratropium (e.g. as the bromide, CAS 22254-24-6, sold under the name Atrovent), oxitropium (e.g. as the bromide, CAS 30286-75-0) and tiotropium (e.g. as the bromide, CAS 136310-93-5, sold under the name Spiriva). Also of interest are revatropate (e.g. as the hydrobromide, CAS 262586-79-8) and LAS-34273 which is disclosed in WO01/04118. Exemplary compounds for oral administration include pirenzepine (CAS 28797-61-7), darifenacin (CAS 133099-04-4, or CAS 133099-07-7 for the hydrobromide sold under the name Enablex), oxybutynin (CAS 5633-20-5, sold under the name Ditropan), terodiline (CAS 15793-40-5), tolterodine (CAS 124937-51-5, or CAS 124937-52-6 for the tartrate,
sold under the name Detrol), otilonium (e.g. as the bromide, CAS 26095-59-0, sold under the name Spasmomen), trospium chloride (CAS 10405-02-4) and solifenacin (CAS 242478-37-1 , or CAS 242478-38-2 for the succinate also known as YM-905 and sold under the name Vesicare).
Other suitable anticholinergic agents include compounds of formula (XXI), which are disclosed in US patent application 60/487981 :
in which the preferred orientation of the alkyl chain attached to the tropane ring is endo; R31 and R32 are, independently, selected from the group consisting of straight or branched chain lower alkyl groups having preferably from 1 to 6 carbon atoms, cycloalkyl groups having from 5 to 6 carbon atoms, cycloalkyl-alkyl having 6 to 10 carbon atoms, 2-thienyl, 2-pyridyl, phenyl, phenyl substituted with an alkyl group having not in excess of 4 carbon atoms and phenyl substituted with an alkoxy group having not in excess of 4 carbon atoms; X" represents an anion associated with the positive charge of the N atom. X" may be but is not limited to chloride, bromide, iodide, sulfate, benzene sulfonate, and toluene sulfonate, including, for example: (3-encyo)-3-(2,2-di-2-thienylethenyl)-8,8-dimethyl-8-azoniabicyclo[3.2.1]octane bromide; (3-encfo)-3-(2,2-diphenylethenyl)-8,8-dimethyl-8-azoniabicyclo[3.2.1 ]octane bromide; (3-encfo)-3-(2,2-diphenylethenyl)-8,8-dimethyl-8-azoniabicyclo[3.2.1]octane 4- methylbenzenesulfonate; (3-enc/o)-8,8-dimethyl-3-[2-phenyl-2-(2-thienyl)ethenyl]-8-azoniabicyclo[3.2.1]octane bromide; and/or (3-e/7cfo)-8,8-dimethyl-3-[2-phenyl-2-(2-pyridinyl)ethenyl]-8-azoniabicyclo[3.2.1]octane bromide.
Further suitable anticholinergic agents include compounds of formula (XXII) or (XXIII), which are disclosed in US patent application 60/511009:
(XXII) (XXlII)
wherein: the H atom indicated is in the exo position; R41 represents an anion associated with the positive charge of the N atom. R41 may be but is not limited to chloride, bromide, iodide, sulfate, benzene sulfonate and toluene sulfonate; R42 and R43 are independently selected from the group consisting of straight or branched chain lower alkyl groups (having preferably from 1 to 6 carbon atoms), cycloalkyl groups (having from 5 to 6 carbon atoms), cycloalkyl-alkyl (having 6 to 10 carbon atoms), heterocycloalkyl (having 5 to 6 carbon atoms) and N or O as the heteroatom, heterocycloalkyl-alkyl (having 6 to10 carbon atoms) and N or O as the heteroatom, aryl, optionally substituted aryl, heteroaryl, and optionally substituted heteroaryl; R44 is selected from the group consisting of (CrC6)alkyl, (C3-C12)cycloalkyl, (C3- C7)heterocycloalkyl, (CrC6)alkyl(C3-C12)cycloalkyl, (C1-C6)alkyl(C3-C7)heterocycloalkyl, aryl, heteroaryl, (CrC6)alkyl-aryl, (C1-C6)alkyl-heteroaryl, -OR45, -CH2OR45, -CH2OH, -CN, -CF3, -CH2O(CO)R46, -CO2R47, -CH2NH2, -CH2N(R47)SO2R45, -SO2N(R47)(R48), - CON(R47XR48), -CH2N(R48)CO(R46), -CH2N(R48)SO2(R46), -CH2N(R48)CO2(R45), - CH2N(R48)CONH(R47); R45 is selected from the group consisting of (CrC6)alkyl, (CrC6)alkyl(C3-C12)cycloalkyl, (C1-C6)alkyl(C3-C7)heterocycloalkyl, (CrC6)alkyl-aryl, (CrC6)alkyl-heteroaryl; R46 is selected from the group consisting of (Ci-C6)alkyl, (C3-C12)cycloalkyl, (C3- C7)heterocycloalkyl, (CrC6)alkyl(C3-C12)cycloalkyl, (CrC6)alkyl(C3-C7)heterocycloalkyl, aryl, heteroaryl, (CrC6)alkyl-aryl, (Ci-C6)alkyl-heteroaryl; R47 and R48 are, independently, selected from the group consisting of H, (CrC6)alkyl, (C3- C12)cycloalkyl, (C3-C7)heterocycloalkyl, (CrC6)alkyl(C3-Ci2)cycloalkyl, (CrC6)alkyl(C3- C7)heterocycloalkyl, (CrC6)alkyl-aryl, and (C1-C6)alkyl-heteroaryl, including, for example:
(Endo)-3-(2-methoxy-2,2-di-thiophen-2-yl-ethyl)-8,8-clinnethyl-8-azonia- bicyclo[3.2.1]octane iodide; 3-((Endo)-8-methyl-8-aza-bicyclo[3.2.1]oct-3-yl)-2,2-diphenyl-propionitrile; (Endo)-8-methyl-3-(2,2,2-triphenyl-ethyl)-8-aza-bicyclo[3.2.1]octane; 3-((Endo)-8-methyl-8-aza-bicyclo[3.2.1]oct-3-yl)-2,2-diphenyl-propionamide; 3-((Endo)-8-methyl-8-aza-bicyclo[3.2.1]oct-3-yl)-2,2-diphenyl-propionic acid; (Endo)-3-(2-cyano-2,2-diphenyl-ethyl)-8,8-dimethyl-8-azonia-bicyclo[3.2.1]octane iodide; (Endo)-3-(2-cyano-2,2-diphenyl-ethyl)-8,8-dimethyl-8-azonia-bicyclo[3.2.1]octane bromide; 3-((Endo)-8-methyl-8-aza-bicyclo[3.2.1]oct-3-yl)-2,2-diphenyl-propan-1-ol; Λ/-Benzyl-3-((endo)-8-methyl-8-aza-bicyclo[3.2.1]oct-3-yl)-2,2-diphenyl--propionamide; (Endo)-3-(2-carbamoyl-2,2-diphenyl-ethyl)-8,8-dimethyl-8-azonia-bicyclo[3.2.1]octane iodide; 1-Benzyl-3-[3-((endo)-8-methyl-8-aza-bicyclo[3.2.1]oct-3-yl)-2,2-diphenyl-propyl]-urea; 1-Ethyl-3-[3-((endo)-8-methyl-8-aza-bicyclo[3.2.1]oct-3-yl)-2,2-diphenyl-propyl]-urea; Λ/-[3-((Endo)-8-methyl-8-aza-bicyclo[3.2.1]oct-3-yl)-2,2-diphenyl-propyl]-acetamide; /V-[3-((Endo)-8-methyl-8-aza-bicyclo[3.2.1]oct-3-yl)-2,2-diphenyl-propyl]-benzamide; 3-((Endo)-8-methyl-8-aza-bicyclo[3.2.1]oct-3-yl)-2,2-di-thiophen-2-yl-propionitrile; (Endo)-3-(2-cyano-2,2-di-thiophen-2-yl-ethyl)-8,8-dimethyl-8-azonia-bicyclo[3.2.1]octane iodide; Λ/-[3-((Endo)-8-methyl-8-aza-bicyclo[3.2.1]oct-3-yl)-2,2-diphenyl-propyl]- benzenesulfonamide; [3-((Endo)-8-methyl-8-aza-bicyclo[3.2.1]oct-3-yl)-2,2-diphenyl-propyl]-urea; ^^-((EndoJ-δ-methyl-δ-aza-bicyclop^.iloct-S-yO^^-diphenyl-propyl]- methanesulfonamide; and/or (Endo)-3-{2,2-diphenyl-3-[(1-phenyl-methanoyl)-arrιino]-propyl}-8,8-dimethyl-8-azonia- bicyclo[3.2.1]octane bromide. For example compounds useful in the present invention include: (Endo)-3-(2-methoxy-2,2-di-thiophen-2-yl-ethyl)-8,8-dimethyl-8-azonia- bicyclo[3.2.1]octane iodide; (Endo)-3-(2-cyano-2,2-diphenyl-ethyl)-8,8-dimethyl-8-azonia-bicyclo[3.2.1]octane iodide; (Endo)-3-(2-cyano-2,2-diphenyl-ethyl)-8,8-dimethyl-8-azonia-bicyclo[3.2.1]octane bromide; (Endo)-3-(2-carbamoyl-2,2-diphenyl-ethyl)-8,8-dimethyl-8-azonia-bicyclo[3.2.1]octane iodide;
(Endo)-3-(2-cyano-2,2-di-thiophen-2-yi-ethyl)-8,8-dimethyl-8-azonia-bicyclo[3.2.1]octane iodide; and/or (Endo)-3-{2,2-diphenyl-3-[(1-phenyl-methanoyl)-amino]-propyl}-8,8-dimethyl-8-azonia- bicyclo[3.2.1]octane bromide.
Suitable antihistamines (also referred to as H1 -receptor antagonists) include any one or more of the numerous antagonists known which inhibit H 1 -receptors, and are safe for human use. First generation antagonists, include derivatives of ethanolamines, ethylenediamines, and alkylamines, e.g diphenylhydramine, pyrilamine, clemastine, chloropheniramine. Second generation antagonists, which are non-sedating, include loratidine, desloratidine, terfenadine, astemizole, acrivastine, azelastine, levocetirizine fexofenadine and cetirizine.
Examples of suitable antihistamines include loratidine, desloratidine, fexofenadine and cetirizine.
The invention provides in one aspect, a pharmaceutical composition comprising a compound of formula (I), or a physiologically functional derivative thereof, in admixture with one or more physiologically acceptable diluents or carriers which further comprises another therapeutically active agent.
A further aspect of the invention provides a pharmaceutical composition comprising a compound of formula (I), or a physiologically functional derivative thereof, in admixture with one or more physiologically acceptable diluents or carriers which further comprises a β2-adrenoreceptor agonist.
In another aspect of the invention a combination is provided comprising a compound of formula (I), or a physiologically functional derivative thereof, together with a PDE4 inhibitor.
The invention thus provides, in a further aspect, a combination comprising a compound of formula (I) a pharmaceutically acceptable salt, solvate or physiologically functional derivative thereof together with a PDE4 inhibitor.
The invention thus provides, in a further aspect, a combination comprising a compound of formula (I) a pharmaceutically acceptable salt, solvate or physiologically functional derivative thereof together with a β2-adrenoreceptor agonist.
The invention thus provides, in a further aspect, a combination comprising a compound of formula (I) a pharmaceutically acceptable salt, solvate or physiologically functional derivative thereof together with an anticholinergic.
The invention thus provides, in a further aspect, a combination comprising a compound of formula (I) a pharmaceutically acceptable salt, solvate or physiologically functional derivative thereof together with an antihistamine.
The invention thus provides, in a further aspect, a combination comprising a compound of formula (I) a pharmaceutically acceptable salt, solvate or physiologically functional derivative thereof together with a PDE4 inhibitor and a β2-adrenoreceptor agonist.
The invention thus provides, in a further aspect, a combination comprising a compound of formula (I) a pharmaceutically acceptable salt, solvate or physiologically functional derivative thereof together with an anticholinergic and a PDE-4 inhibitor.
The combinations referred to above may conveniently be presented for use in the form of a pharmaceutical formulation and thus pharmaceutical formulations comprising a combination as defined above together with a pharmaceutically acceptable diluent or carrier represent a further aspect of the invention.
The individual compounds of such combinations may be administered either sequentially or simultaneously in separate or combined pharmaceutical formulations. In one embodiment the individual compounds will be administered simultaneously in a combined pharmaceutical formulation. Appropriate doses of known therapeutic agents will be readily appreciated by those skilled in the art.
There are four possible isomers of compounds of formula (I). These are called isomers D1 E1 , D1 E2, D2E1 and D2E2 herein.
Isomer D2E1 of the compound of formula (I) wherein R1 represents 1-ethylpropyl is characterised in having a retention time in analytical chiral HPLC on a 25 x 0.46 cm Chiralpak AD column using a mobile phase of 10% ethanol in heptane eluting at 1 mL/min of about 5.59 min. Isomer D2E2 the compound of formula (I) wherein R1 represents 1- ethylpropyl has a retention time of about 6.54 min under the same conditions. Isomers D1E1 and D1E2 are characterised in having retention times of about 16.05 min and 18.61 min respectively by analytical chiral HPLC on a 25 x 0.46 cm Chiralpak AD column using a mobile phase of 15% ethanol in heptane eluting at 1 mL/min.
It will be appreciated by those skilled in the art that although the absolute retention time on chiral chromatography can be variable, the order of elution of the enantiomers remains the same when the same chiral column and conditions are employed.
Preferred isomers of the compounds of formula (I) may be prepared by chromatographic separation of the isomer from a mixture of enantiomeric isomers (e.g. a racemic mixture, such as a diastereomer D2).
There are also provided methods for the preparative separation of isomer D2E2 of a compound of formula (I) from a mixture of isomers D2E1 and D2E2 (e.g. a racemic mixture, such as diastereomer D2) by chromatography.
According to another aspect of the invention there is provided a mixture of isomer D2E2 of a compound of formula (I) with one or more other isomers e.g. a racemic mixture of isomers D2E1 and D2E2 (diastereomer D2).
A mixture (e.g. racemic mixture) of enantiomeric isomers D2E1 and D2E2 may be prepared by chromatographic separation from a mixture of isomers D1E1 , D1 E2, D2E1 and D2E2.
The racemic mixture of isomers D2E1 and D2E2 of the compound of formula (I) wherein R1 represents 1-ethylpropyl (diastereomer D2) has an LCMS: tret of about 3.98 min, while the racemic mixture of isomers D1 E1 and D1 E2 of the compound of formula (I) wherein R1 represents 1-ethylpropyl (diastereomer D1) has an LCMS: tret of about 3.86 min under the conditions described for LCMS System A.
The invention also provides a mixture (e.g. a racemic mixture) of isomers D1 E1 , D1 E2, D2E1 and D2E2.
There are also provided methods for the separation of a mixture of isomers D2E1 and D2E2 (diastereomer D2) from a mixture of isomers D1 E1 , D1 E2, D2E1 and D2E2 (e.g. a mixture of D1 and D2) by chromatography.
A process according to the invention for the preparation of compounds of formula (I) comprises treatment of a compound of formula (II):
wherein R
1 represents 1-ethylpropyl, 1-methylethyl or 2-methylpropyl
with a trifluoromethylating agent of formula (III)
CF3R10 (III)
wherein R10 represents an activating group, for example, trimethylsilyl (TMS).
The reaction will generally be performed in the presence of an inert solvent, such as dimethylformamide (DMF) and a base, such as caesium fluoride (CsF) at a non- extreme temperature, for example, 0-1200C, and more suitably at room temperature.
Alternative conditions with PhSCF3, Et3GeNa in HMPA (Yokoyama, Y. and Mochida, K. Syn. Lett. 1997, 907-8) may also be suitable. Further alternative reagents include CF3H, CF3I, PhSOCF3 and PhSO2CF3. Alternative methods featuring trifluoromethylating reagents are described in: Prakash, G.K.S., Hu, J. and Olah, G.A. Org. Lett. 2003, 5, 3253-6; Caron, S., Do, N. M., Arpin, P. and Larivee, A. Synthesis, 2003, 1693-98; Langlois, B.R. et al Synthesis 2003, 185-194; and further methods cited in these papers. Further conditions which may be suitable are described in Langlois, B.R. et al Angew. Chem. Int. Ed. 2003, 42, 3133-3136.
Alternatively, the reaction of compound of formula (II) and a compound of formula (III) wherein R10 represents TMS may be effected in a solvent such as DMF in the presence of excess (5 equivalents) lithium fluoride (LiF) or caesium carbonate (Cs2CO3) at a non- extreme temperature, for example, between room temperature and 1000C, preferably at room temperature, wherein the compound of formula (III) is present in a large excess (10 equivalents). Where R10 represents a trialkylsilyl group the final step may include deprotection using a suitable reagent, for example, when R10 is TMS it may be removed by treatment with TBAF (tetrabutylammonium fluoride) in tetrahydrofuran.
Compounds of formula (II) may be prepared from compounds of formula (IV):
wherein R
1 represents 1-ethylpropyl, 1-methylethyl or 2-methylpropyl by treatment with an activating agent and 6-amino-4-methyl-1 H-2,3-benzoxazin-1-one.
A suitable method for preparing 6-amino-4-methyl-1H-2,3-benzoxazin-1-one is described in WO 98/54159. An example of a suitable activating agent is thionyl chloride.
The reaction is generally effected in the presence of a suitable solvent, for example, dimethylacetamide or dimethylformamide in the presence of thionyl chloride at a non- extreme temperature. For example, the reaction may be carried out in dimethylacetamide in the presence of thionyl chloride at a temperature of -15 to 250C, such as -50C.
The compounds of formula (IV) have proved difficult to synthesise. We have now provided a process which enables the compound to be made.
In this process, compounds of formula (IV) may be prepared by reaction of compounds of formula (V):
wherein R1 represents 1-ethylpropyl, 1-methylethyl or 2-methylpropyl and A is a group selected from -CO-2-furanyl, -CO-C(=CH2)OC1-6alkyl, -ethynyl or -C(=CH2)CO2C1-6alkyl by reaction with a suitable oxidising agent.
When A is -CO-2-furanyl, -CO-C(=CH2)OC1-6alkyl or -C(=CH2)CO2C1-6alkyl, suitable oxidising agents include ozone, potassium permanganate or sodium periodate with a catalytic ruthenium salt. When A is an ethynyl group, suitable oxidising conditions include bromination followed by treatment with permanganate (see Y.-L.Wu et ai, Tetrahedron Lett., 2002, 43, 2427-2430). Other suitable oxidising conditions include osmium tetroxide/te/t-butylhydroperoxide, osmium tetroxide/sodium periodate and MoOPH (oxodiperoxymolybdenum-pyridine-hexamethylphosphoric triamide) (as set out in, for example in Oxidations in Organic Chemistry, ACS Monograph 186, M. Hudlicky, 1990).
Ozonolysis may be carried out in any suitable solvent, for example methanol or dichloromethane or mixtures of those solvents in a ratio between 100:0 and 0:100. The ozonolysis may take place over a temperature range from -78 0C to 0 0C. When A is -CO- 2-furanyl the ozonolysis is optimally carried out in methanol at -78 0C. A variety of work- up conditions may be used to decompose ozonide intermediates; those work-up conditions include treatment with Me2S, Ph3P or H2O2. In this case, dimethylsulphide is preferred.
Compounds of formula (V) may be prepared by reaction of compounds of formula (Vl)
wherein R
1 represents 1-ethylpropyl, 1-methylethyl or 2-methylpropyl and X' is selected from Br, I and OTf
by reaction with trialkylSnC(=CH
2)CO
2Ci
-6alkyl or with carbon monoxide and a stannane such as trialkylfuran-2-ylstannane, trialkylethynylstannane, or a (1- alkoxyvinyl)trialkylstannane or carbon monoxide and a boronic acid such as furan-2- ylboronic acid in the presence of a source of Pd(O). The source of Pd(O) may for example be Pd(OAc)
2, PdCI
2(MeCN)
2, Pd(PPh
3) or Pd
2(dba)
3. Preferably, the reaction is carried out in the presence of a phosphine ligand, for example Ph
3P, (2-furyl)
3P or (o-tolyl)
3P. Preferably, the reaction is carried out at a temperature in the range of 25
0C to 140
0C. The reaction solvent is preferably an aprotic solvent and it may, for example, be selected from toluene, xylene, benzene or DMF. Most preferably, the reaction is carried out on a starting material with X'=l, and the reaction is with (2-furyl)tributylstannane and carbon monoxide in the presence of palladium acetate and triphenylphosphine in toluene at 110
0C. General conditions for reactions of this type are described in further detail in: J. K. Stille et a/., J. Org. Chem., 1990, 55, 3114-3118 and R. Grigg et a/., Tetrahedron, 2001 , 57, 1347-1359.
OTf represents OSO2CF3, known as triflate.
Compounds of formula (Vl) may be prepared from a compound of formula (VII):
wherein R
1 represents 1-ethylpropyl, 1 -methylethyl or 2-methylpropyl and X' is as defined above for compounds of formula (Vl)
by reaction with a suitable olefinating reagent. Suitable olefinating reagents include Wittig reagents, for example methyltriphenylphosphonium salts. Peterson, Tebbe, Petasis and Lombardo reagents are also suitable. Reactions of this type are described in further detail in: R. C. Hartley et ai, J. Chem. Soc, Perkin Trans. 7, 2002, 2763-2793 and Tetrahedron Lett., 1985, 26, 5579-5580. A Wittig reaction on compound (VII) may suitably be carried out in a polar solvent, for example a solvent selected from diethylether, tetrahydrofuran, ethylene glycol, dimethylether, diglyme or dioxane, in the presence of a strong base, for example n-BuLi, sec-BuU, f-BuLi, LDA, LiHMDS, NaHMDS, KHMDS, NaH or KO1Bu, at a temperature in the range of -78 0C to +70 0C. Preferably, a Wittig reaction is carried out
using methyltriphenyphosphonium bromide in Et2O as the solvent with π-BuLi or KO1Bu as the base at a temperature of 0 0C warming to room temperature.
Compounds of formula (VII) may be prepared by reaction of compounds of formula (VIII):
wherein X' is as defined above for compounds of formula (Vl) and M' is MgQ or ZnQ, where Q is Cl, Br or I
with compounds of formula (IX):
wherein R
1 represents 1-ethylpropyl, 1 -methylethyl or 2-methylpropyl.
The compound of formula (IX) can be prepared in situ by reaction of R1zinc halide with acryloyl chloride in the presence of a palladium(O) source and can then be further reacted with compounds of formula (VIII) as defined above to afford compound of formula (VII).
Preferably the reaction is carried out in a polar solvent, for example a solvent selected from tetrahydrofuran and diethylether at a temperature in the range of from -78 0C to +25 0C. If M' is a magnesium halide, the reaction is carried out in the presence of a copper(l) salt. In one embodiment, the reaction is carried out with a magnesium bromide reagent in diethylether at -78 0C in the presence of a CuBr.Me2S complex. The reaction is suitable for use with compounds of formula (VIII) in which X' is bromine atom.
If M' is a zinc halide, the reaction is carried out in the presence of a complex of LiCI and CuCN. In one embodiment, the reaction is carried out using a compound of formula (VIII) in which M1 is ZnQ where Q represents Br in the presence of a 2:1 LiChCuCN complex as well as one equivalent of TMSCI in THF at -78 0C. The reaction is particularly suitable for use with compounds of formula (VIII) in which X' is a bromine or an iodine atom.
Compounds of formula (VIII) where X' represents bromine or iodine are commercially available.
There are numerous approaches to the synthesis of the vinyl ketone (IX).
In one approach, the zinc reagent R1ZnQ is coupled with acryloyl chloride in the presence of a palladium(O) source such as Pd(OAc)2, PdCI2(MeCN)2, Pd(PPh3)4 or Pd2(dba)3 with a phosphine ligand such as Ph3P, (2-furyl)3P or (o-tolyl)3P over a temperature range of from -25 0C to +75 0C in an ether solvent such as THF, dioxane or ether. Optimal conditions are in THF at 0 0C. By this method, the vinyl ketone (IX) may be isolated or used in situ as described above.
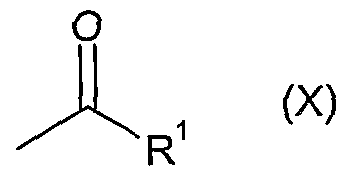
Another approach to access the vinyl ketone (IX) involves a Mannich reaction with the appropriate ketone of formula (X):
wherein R
1 represents 1-ethylpropyl, 1 -methylethyl or 2-methylpropyl
using a formaldehyde source such as trioxymethylene, paraformaldehyde or formaldehyde itself along with a secondary amine salt such as dimethylamine hydrochloride or methylanilinium trifluoroacetate. Suitable solvents include THF and dioxane with temperatures in the range O0C to 1000C. Optimally methylanilinium trifluoroacetate and paraformaldehyde are used in THF with a temperature range 250C to 7O0C. The vinyl ketone (IX) generated by this method is isolated before reaction with (VIII) as outlined above.
Certain compounds of formula (II), (IV), (V), (Vl) and (VII) are new and form an aspect of the invention.
In addition processes for preparing formulations including one or more compounds of formula (I) form an aspect of this invention.
Compositions comprising a compound of the invention also constitute an aspect of the invention.
Solvates of compounds of formula (I), or physiologically functional derivatives thereof or solvates thereof, which are not physiologically acceptable may be useful intermediates in the preparation of other compounds of formula (I), or physiologically functional derivatives thereof or salts thereof.
Processes for the preparation of the compounds of the invention and processes for the preparation of the novel intermediates also from an aspect of the invention.
Compounds of the invention may be expected to demonstrate good anti-inflammatory properties. They also may be expected to have an attractive side-effect profile, demonstrated, for example, by increased selectivity for glucocorticoid receptor mediated transrepression over transactivation and are expected to be compatible with a convenient regime of treatment in human patients.
The invention will now be illustrated by way of the following non-limiting examples.
EXAMPLES
SYNTHETIC EXPERIMENTAL
Abbreviations
THF Tetrahydrofuran EtOAc Ethyl acetate SPE Solid phase extraction n-BuLi n-butyl lithium HCI Hydrochloric acid CDCI3 Deuterochloroform MeCN Acetonitrile DCM Dichloromethane EtOH Ethanol DMF N, Λ/-dimethylformamide TMSCF3 Trimethyl(trifluoromethyl)silane Na2SO4 Sodium sulfate DMA Λ/,Λ/'-dimethylacetamide KF Potassium fluoride KOH Potassium hydroxide MgSO4 Magnesium sulphate RT Room Temperature
General Experimental Conditions
LC/MS System A Column: 3.3cm x 4.6mm ID, 3um ABZ+PLUS; Flow Rate: 3ml/min; Injection Volume: 5μl; Temp. RT; UV Detection Range: 215 to 330nm
Solvents: A: 0.1% Formic Acid + IOmMolar Ammonium Acetate. B: 95% Acetonitrile + 0.05% Formic Acid
Gradient: Time A% B% 0.00 100 0 0.70 100 0 4.20 0 100 5.30 0 100 5.50 100 0
Mass Spectra The mass spectra were recorded on Waters Micromass ZQ spectrometer using electrospray positive and negative ionisation modes (ES+ve and ES-ve).
Chiral Separations Chiral separations were carried out on Chiralpak AD columns made by Daicel Industries Ltd. The Techocel OA-4900 analytical column (equivalent to Sumichiral OA-4900) is available from HPLC Technology Company Ltd, Welwyn Garden City, UK.
Circular Dichroism Circular dichroism was carried out on a Jasco Spectrophotometer Model J-720 at room temperature in MeCN as a solvent in the range 350 - 200 nm
NMR 1H NMR spectra were obtained in CDCI3 on a Bruker DPX 400 spectrometer working at 400.13 MHz and 9.4 Tesla using as internal standard the signal from the residual protonated solvent at 7.25 ppm.
Experimental
Method A Method A is the process for the conversion of 2-iodophenylalkyl ketone such as Intermediate 1 into a keto-amide such as Intermediate 5 via a four stage process. The four stages are firstly a methylenation, then a "one pot" palladium based cyclisation, carbonylation and furylation, followed by ozonolysis with a basic work-up and finally amide formation.
Intermediate 1 5-ethyl-1-(2-iodophenyl)-4-heptanone Tetrakis(triphenylphosphine)palladium (0.29g, 0.25 mmol) was cooled to O0C and a solution of 1-ethylpropyl zinc bromide (0.5M in THF, 50 ml_, 25 mmol) was added. Acryloyl chloride (2.2 ml_, 27.1 mmol) was added slowly keeping the temperature below 50C and the mixture was then stirred at O0C for 1.5 hours. Dried lithium chloride (2.12g, 50 mmol) and copper (I) cyanide (2.24g, 25 mmol) in dry THF was stirred for 10 minutes then cooled to -750C. A solution of benzyl zinc bromide (0.5M in THF, 50 mL, 25 mmol) was added slowly maintaining the temperature below -670C. The resulting mixture was allowed to warm to O0C, stirred for 20 minutes then again cooled to -750C before the slow addition of chlorotrimethylsilane (6.4ml_, 50.4 mmol) keeping the temperature below - 7O0C. The solution of the vinyl ketone prepared above was added slowly below -680C. After the addition the mixture was stirred at -750C for 2.5 hours then allowed to come to room temperature, stirred at this temperature for 2 hours and then partitioned between diethyl ether and water. The aqueous phase was extracted with ether and the combined organic solutions were washed with brine twice, dried (Na2SO4) and evaporated in vacuo to an oil. Purification was by silica chromatography using hexane and hexane:ether 10:1 to give the title compound (3.12 g, 36%). LCMS: tret 3.87 min; 345 (MH)+; 362 (MNH4)+ 1H-NMR: δ H (CDCI3, 400 MHz) 7.84 (d, 1H), 7.25 (m, 2H), 6.90 (t, 1H), 2.72 (t, 2H), 2.50 (t, 2H), 2.34 (m, 1 H), 1.88 (m, 2H), 1.61 (m, 2H), 1.48 (m, 2H), 0.86 (t, 6H)
Intermediate 2 1 -\A-{ 1 -ethylpropyD-4-penten- 1 -yll-2-iodobenzene A stirred suspension of methyltriphenylphosphonium bromide (75.6 g, 0.21 mol) in dry THF (600 mL) was stirred at O0C whilst a 1.6M solution of n-BuLi in hexane (120 mL, 0.19 mol)
was added slowly below 5°C. The mixture was stirred at O0C for 30 minutes and then a solution of 5-ethyl-1-(2-iodophenyl)-4-heptanone (Intermediate 1) (30.1 g, 87.5 mmol) in THF (75 ml_) was added slowly below O0C. After stirring for 15 minutes the mixture was stirred at room temperature for 1 hour, refluxed overnight and then cooled to 1O0C. Saturated ammonium chloride solution (250 ml.) was added maintaining the temperature at ca 1O0C and the mixture was stirred for 15 minutes and then diluted with water (250 mL). After stirring for a further 15 minutes the mixture was extracted with diethyl ether. The combined ether extracts were washed with 1:1 saturated ammonium chloride solutionibrine (500 mL) then dried (Na2SO4) and evaporated in vacuo. The residue was triturated with hexane and filtered. The filtrate was applied to a column of silica and eluted with hexane to give the title compound (22.5 g, 76%). 1H-NMR: δH (CDCI3, 400 MHz) 7.78 (d, 1 H), 7.50 (m, 2H), 6.90 (t, 1 H), 4.88 (s, 1 H), 4.77 (s, 1 H), 2.73 (t, 2H), 2.03 (t, 2H), 1.80 (m, 3H), 1.40 (m, 4H), 0.83 (t, 6H)
Intermediate s 2-[1-(1-ethylpropyl)-1 ,2,3,4-tetrahydro-1-naphthalenvn-1-(2-furanyl)ethanone To palladium acetate (68 mg, 2.92 mmol) and triphenyl phosphine (1.53 g, 5.84 mmol) was added toluene (225 mL) and (2-furyl)tributylstannane (6.78 g, 19 mmol). A solution of 1-[4-(1-ethylpropyl)-4-penten-1-yl]-2-iodobenzene (Intermediate 2) (5 g, 14.6 mmol) in toluene (75 mL) was then added and the apparatus was purged three times with carbon monoxide. The mixture was then placed in a pre-heated 1100C oil bath and stirred for three hours under carbon monoxide, diluted with ether (300 mL), washed with 2M HCI (300 mL) and with water (300 mL). The organic layer was separated, poured into a solution of KF (40 g) in water (600 mL), stirred for 0.5 hour then filtered. The organic layer was separated, washed with water (300 mL), dried and evaporated in vacuo. The preparation was repeated using a further 7.5 g (21.9 mmol) of 1-[4-(1-ethylpropyl)-4- penten-1-yl]-2-iodobenzene (Intermediate 2) and the crude products were combined. Purification was by Biotage chromatography. The solvent gradients used were cyclohexane:DCM 1 :0 to 0:1 followed by DCM:EtOAc 3:1 to 0:1 which gave the title compound (2.19 g, 20%). LCMS: tret 3.95 min; 311 (MH)+, no MH" ion observed 1H-NMR: δH (CDCI3, 400 MHz) 7.25 (d, 1H), 6.96 (m, 3H), 6.77 (m, 1 H), 6.33 (m,1 H), 3.28 (d, 1H), 3.04 (d, 1H), 2.69 (m, 2H), 1.97 (m, 1H), 1.78 (m, 5H), 1.28 (m, 1H), 1.09 (m, 5H), 0.78 (t, 3H)
Intermediate 4 S-fi-d-ethylpropyπ-I .Σ.SΛ-tetrahydro-i-naphthalenvn^-oxoDropanoic acid Ozone was bubbled through a solution of 2-[1-(1-ethylpropyl)-1 ,2,3,4-tetrahydro-1- naphthalenyl]-1-(2-furanyl)ethanone (Intermediate 3) (2.19 g, 7.1 mmol) in anhydrous methanol (200 mL) at -780C for 1 hour followed by oxygen for 10 minutes and nitrogen for 10 minutes. Dimethyl sulfide (15 mL, 205 mmol) was added and the mixture was stirred for 1 hour at room temperature then evaporated in vacuo. The residue was dissolved in methanol (200 mL) and KOH pellets (3 g) were added then stirred for 1.5 hours. Volatiles were removed in vacuo and the residue was partitioned between 2M KOH solution (300 mL) and a 1:1 mixture of cyclohexane and diethyl ether (300 mL). The aqueous phase was acidified and extracted with DCM (300 mL). The extract was dried (MgSO4) and evaporated to give the title compound (2.01 g, 99%) which was used without further purification. 1H-NMR: δH (CDCI3, 400 MHz) 7.22 (d, 1 H), 7.05 (m, 3H), 3.38 (d, 1H), 3.30 (d, 1 H), 2.70 (m, 2H), 2.0-1.61 (m, 6H), 1.31 (m, 1H), 1.15-0.98 (m, 5H), 0.72 (t, 3H)
Intermediate 5 3-f1-(1-ethylpropyl)-1 ,2,3,4-tetrahvdro-1-naphthalenvn-Λ/-(4-methyl-1-oxo-1H-2,3- benzoxazin-6-yl)-2-oxopropanamide 3-[1 -(1 -ethylpropyl)-1 ,2,3,4-tetrahydro-1 -naphthalenyl]-2-oxopropanoic acid (Intermediate) 4 (333 mg,1.15 mmol) was dried by azeotroping from toluene (3 x 5 mL) and the dried material was dissolved in DMA (2.9 mL) then cooled to -1O0C. Thionyl chloride (243 mg, 2.08 mmol) was added and stirring at -1O0C was continued for 1 hour. A solution of 6-amino-4-methyl-1 H-2,3-benzoxazin-1-one (407 mg, 2.31 mmol) in DMA (9.5 mL) was added at -60C and the mixture was allowed to come to room temperature. After stirring at this temperature for 3 hours, the reaction mixture was poured into a 1 :1 DCM:2M HCI mixture. The aqueous layer was extracted with DCM and the combined extracts were dried (MgSO4), evaporated in vacuo and the residue was triturated with DCM. Filtration and evaporation of the filtrate gave a brown oil. Purification was by silica SPE. The solvent gradient used was cyclohexane: EtOAc 20:1 to 0:1 to give the title compound (69 mg, 33%) as yellow crystals. LCMS: trβt 3.94 min; 464 (M+18)+ ;445 (MH)" 1H-NMR: δH (CDCI3, 400 MHz) 8.85 (br, 1 H), 8.31 (d, 1 H), 8.12 (d, 1H), 7.70 (d, 1H), 7.26 (d, 1 H), 7.05 (m, 3H), 3.52 (d, 1H), 3.35 (d, 1H), 2.82 (m, 2H), 2.56 (s, 3H), 2.0-1.68 (m, 6H), 1.32 (m, 1 H), 1.12 (m, 5H), 0.78 (t, 3H)
Intermediate 6 6-(2-iodophenyl)-2-methyl-3-hexanone A solution of zinc chloride (0.5M in THF, 80 ml_, 40 mmol) was cooled to O0C and to this was added a solution of isopropyl magnesium bromide (1M in THF, 40 ml_, 40 mmol). The mixture was warmed to room temperature and stirred for 1 hour then added carefully to tetrakis(triphenylphosphine)palladium(0) (693 mg, 0.6 mmol). Acryloyl chloride (3.62g, 40 mmol) was then added dropwise, the resulting exothermic reaction being controlled with an ice bath. When addition was complete the ice bath was removed and the mixture was stirred for 1 hour to give a solution of vinyl isopropyl ketone. Lithium chloride (3.39 g, 80 mmol) was dried under vacuum at 12O0C for 5 hours then cooled to room temperature. Copper(l) cyanide (3.58g, 40 mmol) was added followed by THF (20 ml_) and the whole was stirred at -780C for 15 minutes. A solution of 2-iodobenzylzinc bromide (0.5M in THF, 80 mL, 40 mmol) was added and the reaction mixture was warmed to -2O0C for 15 minutes then re-cooled to -780C for 15 minutes. Chlorotrimethylsilane (8.1 g, 75 mmol) was added followed by the vinyl isopropyl ketone solution prepared above. The mixture was allowed to come to room temperature and stirred overnight then poured onto water (200 mL) and extracted with diethyl ether (200 mL). The organic phase was washed with water (200 mL) and the washings were extracted with ether (2 x 200 mL). The combined organic solutions were dried over magnesium sulfate and evaporated under vacuum. The residue was triturated with cyclohexane:EtOAc 5:1 and filtered to remove inorganic salts. Purification (Biotage, solvent gradient of cyclohexane: EtOAc 1 :0 to 25:2) gave the title compound (5.98g, 47%). 1H-NMR:δH (CDCI3, 400 MHz) 7.82 (d, 1 H), 7.31-7.15 (m, 2H), 6.90 (m, 1H), 2.72 (t, 2H), 2.61 (m, 1 H), 2.52 (t, 2H), 1.90 (m, 2H), 1.11 (d, 6H)
Intermediate 7 3-π -f 1 -methylethyl)-1 ,2,3,4-tetrahvdro-1 -naphthalenvH-/\/-(4-methyl-1 -oxo-1 H-2,3- benzoxazin-6-yl)-2-oxopropanamide Prepared from 6-(2-iodophenyl)-2-methyl-3-hexanone (Intermediate 6) according to Method A. LCMS: tret 3.71 min; 419 (MH)+; 417 (MH)-
Intermediate 8
1-(2-iodophenyl)-6-methyl-4-heptanone To dried lithium chloride (1.06g, 25 mmol) and copper(l) cyanide (1.12 g, 12.5 mmol) was added THF (12.5 ml_). After stirring this solution for 10 minutes it was cooled to -780C and a solution of 2-iodobenzylzinc bromide (0.5M in THF, 25 ml_, 12.5 mmol). When addition was complete, the mixture was warmed to O0C for 20 minutes then cooled back to -780C. Tetrakis(triphenylphosphine)palladium(0) (145 mg, 0.125 mmol) was cooled to O0C under nitrogen. A solution of isobutylzinc bromide (0.5M in THF, 25 mL, 12.5 mmol) was added and after 5 minutes acryloyl chloride (1.1 mL,13.5 mmol) was added and stirring was continued at O0C for 1 hour. To the Cu/Zn complex solution was added chlorotrimethylsilane (3.2 mL, 25.2 mmol) at -780C. The vinyl ketone solution prepared above was then added, stirred at -780C for 3 hours and warmed to room temperature with stirring for a further 1 hour. The crude reaction mixture was poured onto water (100 mL) and diethyl ether (100 mL). The aqueous layer was extracted with ether (2 x 100 mL) and the combined ether solutions were washed with brine (2 x 100 mL) then dried over magnesium sulfate. Evaporation of the solvent under vacuum gave a crude product. Purification (5Og silica SPE, cyclohexane: EtOAc 95:5) gave the title compound (3.66 g, 89%). 1H-NMR:δH (CDCI3, 400 MHz) 7.8 (d, 1 H), 7.3-7.18 (m, 2H), 6.90 (m, 1H), 2.72 (t, 2H), 2.47 (t, 2H), 2.31 (d, 2H), 2.16 (m, 1H), 1.88 (m, 2H), 0.93 (d, 6H)
Intermediate 9 Λ/-(4-methyl-1-oxo-1H-2.3-benzoxazin-6-vπ-3-π-(2-methylpropyπ-1.2,3.4-tetrahvdro-1- naphthalenyll-2-oxopropanamide Prepared from 1-(2-iodophenyl)-6-methyl-4-heptanone (Intermediate 8) according to method A. 1H-NMR:δH (CDCI3, 400 MHz) 9.18 (s, 1H), 8.34 (d, 1 H), 8.23 (s, 1H), 7.78 (dd, 1 H). 7.26 (m, 2H), 7.08 (m, 2H), 3.5 (d, 1H), 3.36 (d, 1 H), 2.81 (m, 2H), 2.59 (s, 3H), 2.08-1.79 (m, 6H), 1.4 (m, 1 H), 0.97 (d, 3H), 0.76 (d, 3H)
Example 1 2-{f1-(1-ethylpropyn-1 ,2,3,4-tetrahvdro-1-naphthalenyllmethyl)-3,3,3-trifluoro-2-hvdroχy-Λ/- (4-methyl-1 -oxo-1 H-2,3-benzoxazin-6-yl)propanamide Cesium fluoride (130 mg, 856 μmol) was dried at 1200C for 2 hours under vacuum. To this was added a solution of 3-[1-(1-ethylpropyl)-1 ,2,3,4-tetrahydro-1-naphthalenyl]-Λ/-(4- methyl-1 -oxo-1 H-2,3-benzoxazin-6-yl)-2-oxopropanamide
(Intermediate 5) (77 mg, 172 μmol) in DMF (2mL) followed by trimethyl(trifluoromethyl)silane (274μL, 1.85 mmol). The mixture was stirred at room temperature overnight then diluted with DCM (20 ml_) and washed with 2M HCI (10 mL). The aqueous phase was extracted with DCM and the combined organic solutions were dried (MgSO4) and evaporated in vacuo. Purification (2Og silica SPE, solvent gradient of cyclohexane:EtOAc 9:1 to 0:1) gave (in order of elution) diastereomer 2 and diasteromer 1. The recovery was Example 1-D1 (diastereomer 1 , 34.3 mg) and Example 1-D2 (diastereomer 2, 35.1 mg)
Example 1-D1 (racemic diastereomer 1) LCMS: tret 3.86 min; 534 (M +18)+; 515 (MH)" 1H-NMR: δH (CDCI3, 400 MHz) 8.23 (d, 1H), 8.02 (d, 1 H), 7.33 (m, 1 H), 7.22 (d, 1 H), 6.95 (d, 1H), 6.81-6.70 (m, 2H), 2.91 (d, 1 H) ,2.72 (m, 2H), 2.58 (s, 3H), 2.48 (d, 1H), 2.07-1.7 (m, 5H), 1.54 (m, 1 H), 1.32 (m, 1 H), 1.10 (t, 3H), 0.96 (m, 2H), 0.69 (t, 3H)
Example 1-D2 (racemic diastereomer 2) LCMS: tret 3.98 min; 534 (M +18)+; 515 (MH)" 1H-NMR: δH (CDCI3, 400 MHz) 8.33 (d, 1H), 8.28 (d, 1H), 7.69 (m, 1H), 7.45 (d, 1H), 7.30 (m, 1 H), 7.19 (m, 2H), 3.15 (d, 1 H), 2.70 (m, 2H), 2.61 (s, 3H), 2.57 (d, 1 H), 1.88-1.52 (m, 5H), 1.28 (m, 2H), 0.99 (m, 5H), 0.72 (t, 3H)
Example 1-D1 was separated into its enantiomers using a 2 x 25 cm Chiralpak AD column eluting with 2% EtOH in heptane with a flow rate of 15 mL/min. Enantiomer 1 eluted at around 36 min and enantiomer 2 eluted at around 44 min.
Example 1-D1E1 (enantiomer 1 of diastereomer 1) Analytical chiral HPLC (25 x 0.46 cm Techocel OA-4900 column, 15% EtOH in heptane eluting at 1 mL/min) retention time 17.85 min.
Example 1-D1E2 (enantiomer 2 of diastereomer 1) Analytical chiral HPLC (25 x 0.46 cm Techocel OA-4900 column, 15% EtOH in heptane eluting at 1 mL/min) retention time 15.38 min.
Example 1-D2 was separated into its enantiomers using a 2 x 25 Chiralpak AD column eluting with 10% EtOH in heptane with a flow rate of 15 mL/min. Example 1-D2E1
(enantiomer 1) eluted around 7.16 minutes (1.1 mg) and Example 1-D2E2 (enantiomer 2) eluted around 8.41 minutes (1.1 mg).
Example 1-D2E1 (enantiomer 1 of diastereomer 2) Analytical chiral HPLC (25 x 0.46cm Chiralpak AD column, 10% EtOH in heptane eluting at 1mL/min) retention time 5.59 min LCMS: tret 3.98 min; 534 (MNH4)+; 515 (MH)" Circular Dichroism (0.000329M, cell length = 0.1 cm) 210.0 nm (de = 8.51; E = 16886) 243.0 nm (de = -12.78; E = 28070) 277.0 nm (de = -6.09; E = 12828)
Example 1-D2E2 (enantiomer 2 of diastereomer 2) Analytical chiral HPLC (25 x 0.46cm Chiralpak AD column, 10% EtOH in heptane eluting at 1mL/min) retention time 6.54 min LCMS: tret= 3.98 min; (MNH4)* = 534, MH" = 515 Circular Dichroism (0.00038M, cell length = 0.1 cm) 212.0 nm (de = -8.54; E = 17206) 242.6 nm (de = 13.45; E = 28860) 277.0 nm (de = 6.27; E = 13074)
Example 2 3.3,3-trifluoro-2-hvdroxy-2-{ri-(1-methylethvπ-1.2,3,4-tetrahvdro-1-naphthalenvπmethyl)- Λ/-(4-methyl-1 -oxo-1 H-2,3-benzoxazin-6-yl)propanamide Cesium fluoride (96 mg, 633 μmol) was dried at 12O0C for 2 hours under vacuum. The flask was filled with nitrogen and cooled to room temperature. A solution of 3-[1-(1- methylethyl)-1 ,2,3,4-tetrahydro-1 -naphthalenyl]-Λ/-(4-methyl-1 -oxo-1 H-2,3-benzoxazin-6- yl)-2-oxopropanamide (Intermediate 7) (53 mg, 127 μmol) in DMF (2 mL) was then added followed by TMSCF3 (187 μL, 1.27 mmol) and the mixture was stirred at room temperature for 3 hours. The reaction mixture was concentrated under vacuum, diluted with DCM (20 mL) and washed with 2M HCI solution (10 mL). The aqueous layer was extracted with DCM (20 mL) and the combined organic layers were dried over magnesium sulfate then evaporated under vacuum to give a crude product. Purification (2Og silica SPE, gradient of cyclohexane: EtOAc 9:1 to 8:2) gave (in order of elution) diastereomer 2
and diastereomer 1. The recovery was Example 2-D1 (diastereomer 1 , 22.6 mg) and Example 2-D2 (diastereomer 2, 20.4 mg).
Example 2-D1 (racemic diastereomer 1) LCMS: tret 3.74 min; 489 (MH)+; 487 (MH)- 1H-NMR: δH (CDCI3, 400 MHz) 8.25 (d, 1H), 8.20 (s, 1H), 8.05 (s, 1H), 7.38 (dd, 1H), 7.23 (d, 1 H), 6.95 (d, 1H), 6.75 (m, 2H), 3.57 (s, 1 H), 2.90 (d, 1 H), 2.75 (m, 2H), 2.59 (s, 3H), 2.39 (d, 1H), 2.21 (m, 1H), 2.01 (m, 1 H), 1.98-1.72 (m, 3H), 1.1 (d, 3H), 0.6 (d,3H)
Example 2-D2 (racemic diastereomer 2) LCMS: tret 3.86 min; 506 (MH)+; 487 (MH)" 1H-NMR: δH (CDCI3, 400 MHz) 9.0 (s, 1H), 8.35 (d, 1H), 8.28 (s, 1H), 7.70 (dd, 1H), 7.39 (d, 1H), 7.30 (m, 1 H), 7.22 (m, 2H), 3.05 (d, 1H), 2.85 (s, 1 H), 2.74 (m, 2H), 2.6 (m, 4H), 1.85 (m, 2H), 1.6 (m, 3H), 1.0 (d, 3H), 0.7 (d, 3H)
Example 2-D2 (6 mg) was separated into its enantiomers using a 2 x 25 cm Chiralpak AD column eluting with 5% EtOH in heptane with a flow rate of 15 mL/min. Example 2-D2E1 (enantiomer 1) eluted around 17.1 min (1.4 mg) and Example 2-D2E2 (enantiomer 2) eluted around 20.8 min (1.3 mg).
Example 2-D2E1 (enantiomer 1 of diastereomer 2) Analytical chiral HPLC (25 x 0.46 cm Chiralpak AD column, 5% EtOH in heptane eluting at 1 mL/min) retention time 13.71 min LCMS: tret 3.86 min; 489 (MH)+
Example 2-D2E2 (enantiomer 2 of diasteromer 2) Analytical chiral HPLC (25 x 0.46 cm Chiralpak AD column, 5% EtOH in heptane eluting at 1 mL/min) retention time 16.71 min LCMS: tret: 3.86 min; 489 (MH)+
Example 3 3.3.3-trifluoro-2-hvdroxy-Λ/-(4-methyl-1 -oxo-1 H-2.3-benzoxazin-6-yl)-2-(f 1 -(2- methylpropyO-1 ,2Λ4-tetrahvdro-1-naphthalenyl1methyl)propanamide Cesium fluoride (21.1 mg, 0.139 mmol) was dried at 12O0C under vacuum for 1 hour then cooled to room temperature. A solution of intermediate 9 (30 mg, 0.0694 mmol) in dry
DMF (1 ml_) and TMSCF3 (50 μl_, 4.9 equivalents) were added under nitrogen and the mixture was stirred at room temperature overnight. The reaction mixture was poured into 1M HCI and extracted with DCM (2 x 50 ml_). The combined extracts were dried over sodium sulfate and evaporated to give a crude product. Purification (1Og silica SPE, cyclohexane:EtOAc 85:15 followed by cyclohexane: EtOAc 80:20) gave (in order of elution) diastereomer 2 pure and diastereomer 1 containing ca 7% diastereomer 2. The recovery was Example 3-D1 (diastereomer 1 , 3.6 mg) and Example 3-D2 (diastereomer 2, 3.8 mg).
Example 3-D1 (racemic diastereomer 1) LCMS: tre{ 3.78 min; 520 (MNH4)+; 501 (MH)-
Example 3-D2 (racemic diastereomer 2) LCMS: tret 3.85 min; 520 (MNH4)+; 503 (MH)+; 501 (MH)- 1H-NMR: δH (CDCI3, 400 MHz) 9.0 (s, 1H), 8.35 (m, 2H), 7.68 (d, 1 H), 7.42 (d, 1 H), 7.27 (m, 1 H), 7.20 (m, 2H), 3.10 (d, 1 H), 2.83-2.55 (m, 8H), 1.88-1.60 (m, 6H), 0.95 (d, 3H), 0.83 (d, 3H)
Example 3-D2 was separated into its enantiomers using a 2 x 25 cm Chiralpak AD column eluting with 5% EtOH in heptane with a flow rate of 15 mL/min. Example 3-D2E1 (enantiomer 1) eluted around 18.5 min (0.6 mg) and Example 3-D2E2 (enantiomer 2) eluted around 21.4 min (0.6 mg).
Example 3-D2E1 (enantiomer 1 of diastereomer 2) Analytical chiral HPLC (25 x 0.46 cm Chiralpak AD column, 5% EtOH in heptane eluting at 1 mL/min) retention time 11.55 min LCMS: tret 3.85 min; 520 (MNH4)4"; 503 (MH)+; 501 (MH)-
Example 3-D2E2 (enantiomer 2 of diastereomer 2) Analytical chiral HPLC (25 x 0.46 cm Chiralpak AD column, 5% EtOH in heptane eluting at 1 mL/min) retention time 14.05 min LCMS: tret 3.85 min; 520 (MNH4)+; 503 (MH)+; 501 (MH)-
BIOLOGICAL EXPERIMENTAL
Glucocorticoid receptor binding assay The ability of compounds to bind to the glucocorticoid receptor was determined by assessing their ability to compete with fluorescent-labelled glucocortioid using a kit supplied by Pan Vera (Madison, Wl, USA). Compounds were solvated and diluted in DMSO, and transferred directly into assay plates. Fluorescent glucocortioid and partially purified glucocorticoid receptor were added to the plates and incubated at 4°C for 16 hours in the dark. Binding of the compound was assessed by analysing the displacement of fluorescent ligand by measuring the decrease in fluorescence polarisation signal from the mixture.
The PlC50 values for compounds of Examples 1-D1 , 1-D2, 1-D2E2, 2-D1 , 2-D2, 2-D2E2, 3-D1 , 3-D2, 3-D1 E2, 3-D2E1 and 3-D2E2 are > 7 and for 1-D2E2 >8 for the glucocorticoid receptor binding assay.
Glucocorticoid mediated Transrepression of NFkB activity.
Human A549 lung epithelial cells were engineered to contain a secreted placental alkaline phosphatase gene under the control of the distal region of the NFkB dependent ELAM promoter as previously described in Ray, K.P., Farrow, S., Daly, M., Talabot, F. and Searle, N. "Induction of the E-selectin promoter by interleukin 1 and tumour necrosis factor alpha, and inhibition by glucocorticoids" Biochemical Journal. 1997 328 707-15.
Compounds were solvated and diluted in DMSO, and transferred directly into assay plates such that the final concentration of DMSO was 0.7%. Following the addition of cells (4OK per well), plates were incubated for 1 hr prior to the addition of 3ng/ml human recombinant TNFα. Following continued incubation for 16 hours, alkaline phosphatase activity was determined by measuring the change in optical density at 405nM with time following the addition of 0.7 volumes of assay buffer (1mg/ml p-nitrophenylphosphate dissolved in 1 M diethanolamine, 0.28M NaCI, 0.5mM MgCI2).
The plC50 values for the diastereomer Example 3-D2 is >7.5 and for the diastereomer Examples 1-D2, 2-D2 and single enantiomers Examples 1-D2E2 and 2-D2E2 are > 8 for the NFkB assay.
Glucocorticoid mediated Transactivation of MMTV driven gene expression Human A549 lung epithelial cells or human MG63 osteosarcoma were engineered to contain a renialla luciferase gene under the control of the distal region of the LTR from the mouse mammary tumour virus as previously described (Austin, R.H., Maschera, B., Walker, A., Fairbairn, L., Meldrum, E., Farrow, S. and Uings, I.J. Mometasone furoate is a less specific glucocorticoid than fluticasone propionate. European Respiratory Journal 2002 20 1386-1392).
Compounds were solvated and diluted in DMSO, and transferred directly into assay plates such that the final concentration of DMSO was 0.7%. Following the addition of cells (4OK per well), plates were incubated for 6hr. Luciferase activity was determined using the Firelight kit (Packard, Pangbourne, UK).
The diastereomers Examples 1-D2, 2-D2 and 3-D2, and the single enantiomers Examples 1-D2E2, 2-D2E2 and 3-D2E2 all have reduced efficacy in the MMTV transactivation assay compared with the efficacies in the NFkB assay.
Example 1-D2, Example 1-D2E1 and Example 1-D2E2 have reduced efficacy in both A549 and MG63 cell lines
Throughout the specification and the claims which follow, unless the context requires otherwise, the word 'comprise', and variations such as 'comprises' and 'comprising', will be understood to imply the inclusion of a stated integer or step or group of integers but not to the exclusion of any other integer or step or group of integers or steps.
The application of which this description and claims forms part may be used as a basis for priority in respect of any subsequent application. The claims of such subsequent application may be directed to any feature or combination of features described herein. They may take the form of product, composition, process, or use claims and may include, by way of example and without limitation, the following claims.
The patents and patent applications described in this application are herein incorporated by reference.