WO2009114010A1 - Macrocyclic lactone compounds and methods for their use - Google Patents
Macrocyclic lactone compounds and methods for their use Download PDFInfo
- Publication number
- WO2009114010A1 WO2009114010A1 PCT/US2008/056501 US2008056501W WO2009114010A1 WO 2009114010 A1 WO2009114010 A1 WO 2009114010A1 US 2008056501 W US2008056501 W US 2008056501W WO 2009114010 A1 WO2009114010 A1 WO 2009114010A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- group
- independently
- poly
- member selected
- Prior art date
Links
- 0 CC1C(*)CCC(CC(*)C(CC(C(*)IC(C(*)C(*C(*)C(*)*)=O)O)=O)OC(C(C(*)C(*)C(*)*2)N2C(C(C2(O)OC(CC(*)I)(*3)C3*C2*)=O)=O)=O)C1 Chemical compound CC1C(*)CCC(CC(*)C(CC(C(*)IC(C(*)C(*C(*)C(*)*)=O)O)=O)OC(C(C(*)C(*)C(*)*2)N2C(C(C2(O)OC(CC(*)I)(*3)C3*C2*)=O)=O)=O)C1 0.000 description 5
- PLVABNXNTXQTQY-UHFFFAOYSA-N CC(CO)(CO)C(OC[NH+]1N=CN(C)[N-]1)=O Chemical compound CC(CO)(CO)C(OC[NH+]1N=CN(C)[N-]1)=O PLVABNXNTXQTQY-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/436—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having oxygen as a ring hetero atom, e.g. rapamycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/10—Ophthalmic agents for accommodation disorders, e.g. myopia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
Definitions
- the present invention relates to structures of demethyl, hydroxyl, demethylhydroxyl, epoxide, N-oxide, opened hemiketal ring and seco-macrocyclic lactones, their synthesis, pharmaceutical compositions and use for systemic and site specific therapeutic applications.
- FDA Food and Drug Administration
- Rapamycin resembles tacrolimus (binds to the same intracellular binding protein or immunophilin known as FKBP- 12) but differs in its mechanism of action. Whereas tacrolimus and cyclosporine inhibit T-cell activation by blocking lymphokine (e.g., 1L2) gene transcription, sirolimus inhibits T-cell activation and T lymphocyte proliferation by binding to mammalian target of rapamycin (mTOR). Rapamycin can act in synergy with cyclosporine or tacrolimus in suppressing the immune system.
- lymphokine e.g., 1L2
- mTOR mammalian target of rapamycin
- Rapamycin is also useful in preventing or treating systemic lupus erythematosus [U.S. Pat. No. 5,078,999], pulmonary inflammation [U.S. Pat. No. 5,080,899], insulin dependent diabetes mellitus [U.S. Pat. No. 5,321,009], skin disorders, such as psoriasis [U.S.
- rapamycin can be used to treat various disease conditions, the utility of the compound as a pharmaceutical drug has been limited by its very low and variable bioavailability and its high immunosuppressive potency and potential high toxicity. Also, rapamycin is only very slightly soluble in water. To overcome these problems, prodrugs and analogues of the compound have been synthesized. Water soluble prodrugs prepared by derivatizing rapamycin positions 31 and 42 (formerly positions 28 and 40) of the rapamycin structure to form glycinate, propionate, and pyrrolidino butyrate prodrugs have been described (U.S. Pat. No. 4,650,803).
- analogues of rapamycin described in the art include monoacyl and diacyl analogues (U.S. Pat. No. 4,316,885), acetal analogues (U.S. Pat. No. 5,151,413), silyl ethers (U.S. Pat. No. 5,120,842), hydroxyesters (U.S. Pat. No.
- Prodrugs and analogues of rapamycin are synthesized by chemical synthesis, where additional synthetic steps are required to protect and deprotect certain positions.
- Analogues can also be synthesized biologically, where the Streptomyces strain is genetically modified to produce these analogues of rapamycin.
- the analogues need to maintain necessary positions for protein binding or other cellular interactions and not generate steric hindrance in order to preserve its activity. The safety of these analogues requires extensively testing by series of preclinical and clinical experimentations.
- the present invention comprises novel macrocyclic lactones and novel uses for macrocyclic lactones, where the compositions can be synthesized chemically or biologically and which preserve at least some immunosuppressive, anti-proliferative, anti-fungal and anti-tumor properties for use in systemic and site specific applications.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a pharmaceutically acceptable excipient and a compound of the formula:
- R 1 , R 2 , R 3 , R 5 , R 6 , R 8 , M 1 , M 2 , M 3 , M 4 , M 5 , M 6 and M 7 are each independently a member selected from the group consisting of H, C 1-6 alkyl, OH and C 1-6 hydroxyalkyl; R 4 , R 7 and R 9 are each independently selected from the group consisting of C 1-6 alkoxy and OH;
- R 10 is a member selected from the group consisting of H, -OH, -OP(O)Me 2 , O
- each of L 1 and L 4 are independently selected from the group consisting of: wherein each M 8 is independently a member selected from the group consisting of C 1-6 alkyl, OH and C 1-6 hydroxyalkyl; each of L 2 and L 3 are independently selected from the group consisting of:
- the present invention provides a device for intracorporeal use, the device comprising an implant; and at least one source of a compound of the present invention.
- the present invention provides a method of inhibiting cell proliferation by administering to a subject in need thereof, a therapeutically effective amount of a compound of the present invention.
- the present invention provides macrocyclic lactone compounds of the following formula:
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , M 1 , M 2 , M 3 , M 4 , M 5 , M 6 , M 7 , L 1 , L 2 , L 3 and L 4 are as described above; with the proviso that when R 1 , R 6 , R 8 , M 6 and M 7 are Me, R 3 , R 5 , M 1 , M 2 , M 3 , M 4 and M 5 are H, R 4 , R 7 and R 9 are OMe, R 10 and M 8 are OH, L 2 and L 3 are -CH CH-, and L 1 and L 4 are Me , R 2 is other than OH; with the proviso that when R 1 , R 6 , R 8 , M 6 and M 7 are Me, R 2 , R 3 , R 5 , M 1 , M 2 , M 3 , M 4 and M 5
- the present invention provides a method of making a compound of the present invention, the method comprising contacting a macrocyclic lactone with an acid to replace an alkoxy group with a nucleophile, thereby making a compound of the present invention.
- the present invention provides a method of making a compound of the present invention, the method comprising contacting a macrocyclic lactone with an epoxidation agent to modify an alkene group to an epoxide, thereby making a compound of the present invention.
- the present invention provides a pharmaceutical composition including a pharmaceutically acceptable excipient and a compound of the formula:
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , M 1 , M 2 , M 3 , M 4 , M 5 , M 6 , M 7 , L 1 , L 2 , L 3 and L 4 are as described above; and salts, hydrates, isomers, metabolites, N-oxides and prodrugs thereof.
- the present invention provides a pharmaceutical composition including a pharmaceutically acceptable excipient and a compound of the formula:
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , M 1 , M 2 , M 3 , M 4 , M 5 , M 6 , M 7 , L 1 , L 2 , L 3 and L 4 are as described above; and salts, hydrates, isomers, metabolites, N-oxides and prodrugs thereof.
- the present invention provides a method of treating an ophthalmic condition or disease by administering to a subject in need thereof, a therapeutically effective amount of a compound of the present invention.
- Figure 2A-2FV shows compounds of the present invention.
- Figure 3 shows an example of stent configuration having an expandable structure.
- Figure 4 shows a preparative HPLC chromatogram of Compound AR.
- Figure 5 shows a Proton-NMR spectra of Compound AR.
- Figure 6 shows results of liquid chromatography and mass spectra of Compound AR.
- Figure 7(a) shows an analytical HPLC chromatogram of Compound AR.
- Figure 7(b) shows an analytical HPLC chromatogram of Compound AR with isomers.
- Figure 8 shows percentage proliferation of human smooth muscle cells after exposure to varying concentrations of rapamycin and Compound AR.
- Figure 9 shows quantitative coronary angiography (QCA) stenosis of the
- Figure 10 shows tissue concentration of Compound AR at different time points in a porcine coronary artery model.
- Figure 11 shows percentage of Compound AR released from the stent in a porcine coronary artery model.
- Figure 12(a) shows inhibition of IL-6, MMP-9 and MCP-I released by activated macrophages by exposure to macrocyclic lactone Compound AR and Sirolimus at 1OnM concentration.
- Figure 12(b) shows inhibition of IL-10 released by activated macrophages by exposure to macrocyclic lactone Compound AR and Sirolimus at 1 OnM concentration
- Figure 13 shows percentage proliferation of human smooth muscle cells after exposure to varying concentrations of 17,18-29,30-bis-epoxide macrocyclic lactone and Compound AR.
- Figure 14 shows the synthesis of 16-O-demethyl macrocyclic lactone of the present invention.
- Figure 15 shows the synthesis of 19, 20 bis-hydroxy macrocyclic lactone.
- Figure 16 shows the synthesis of 17,18-29,30-bis epoxide macrocyclic lactone.
- Figure 17 shows the synthesis of 31-hydroxyl, 44-hydroxyl and 47-hydroxyl macrocyclic lactones.
- Figure 18 shows the synthesis of 43-hydroxyl and 47-hydroxyl macrocyclic lactones.
- the term "acid” refers to any chemical compound that, when dissolved in water, gives a solution with a pH less than 7.0. Acids are generally described as a compound which donates a hydrogen ion (H+) (Bronsted-Lowry) or as an electron-pair acceptor (Lewis acid). Acids useful in the present invention include, but are not limited to, HCl, H 2 SO 4 , HNO 3 and acetic acid. One of skill in the art will appreciate that other acids are useful in the present invention.
- administering refers to systemic and local administration or a combination thereof such as oral administration, administration as a suppository, topical contact, parenteral, intravascular, intravenous, intraperitoneal, intramuscular, intralesional, intranasal, pulmonary, mucosal, transdermal, subcutaneous administration, intrathecal, intraocular, intravitreal administration, delivery through a temporary device such as catheter, porous balloon, delivery through implant such as polymeric implant, osmotic pump, prosthesis such as drug eluting stents or others to the subject.
- a temporary device such as catheter, porous balloon
- implant such as polymeric implant, osmotic pump, prosthesis such as drug eluting stents or others to the subject.
- alkoxy refers to alkyl with the inclusion of an oxygen atom, for example, methoxy, ethoxy, etc.
- Halo-substituted-alkoxy'- is as defined for alkoxy where some or all of the hydrogen atoms are substituted with halogen atoms.
- halo-substituted-alkoxy includes trifluoromethoxy, etc.
- alkoxy groups are useful in the present invention.
- alkyl refers to a straight or branched, saturated, aliphatic radical having the number of carbon atoms indicated.
- C 1 -C 6 alkyl includes, but is not limited to, methyl, ethyl, propyl, butyl, pentyl, hexyl, iso-propyl, iso-butyl, sec-butyl, tert-butyl, etc.
- alkyl groups are useful in the present invention.
- hydroxyalkyl refers to alkyl as defined above where at least one of the hydrogen atoms is substituted with a hydroxy group.
- hydroxyalkyl includes hydroxy-methyl, hydroxy-ethyl (1 - or 2-), hydroxy-propyl (1 -, 2- or 3-), hydroxy-butyl (1-, 2-, 3- or 4-), hydroxy-pentyl (1-, 2-, 3-, 4- or 5-), hydroxy-hexyl (1-, 2-, 3-, 4-, 5- or 6-), 1 ,2-dihydroxyethyl, and the like.
- hydroxyalkyl includes hydroxy-methyl, hydroxy-ethyl (1 - or 2-), hydroxy-propyl (1 -, 2- or 3-), hydroxy-butyl (1-, 2-, 3- or 4-), hydroxy-pentyl (1-, 2-, 3-, 4- or 5-), hydroxy-hexyl (1-, 2-, 3-, 4-, 5- or 6-), 1 ,2-dihydroxyethyl, and the like.
- the term "body lumen” refers to the lining or cavity of an artery, vein or an organ.
- the term “contacting” refers to the process of bringing into contact at least two distinct species such that they can react. It should be appreciated, however, that the resulting reaction product can be produced directly from a reaction between the added reagents or from an intermediate from one or more of the added reagents which can be produced in the reaction mixture.
- the term "hydrate” refers to a compound that is complexed to at least one water molecule. The compounds of the present invention can be complexed with from 1 to 100 water molecules.
- the term “implant” refers to a medical device inserted into a body in order to treat a condition. Implants include, but are not limited to, drug-eluting devices.
- the terms “inhibition”, “inhibits” and “inhibitor” refer to a compound that prohibits, reduces, diminishes or lessens, or to a method of prohibiting, reducing, diminishing or lessening a specific action or function.
- intracorporeal refers to an mammalian body.
- the term “isomer” refers to compounds of the present invention that possess asymmetric carbon atoms (optical centers) or double bonds, the racemates, diastereomers, enantiomers, geometric isomers, structural isomers and individual isomers are all intended to be encompassed within the scope of the present invention.
- organ refers to any organ of a mammal, such as, but not limited to, heart, lungs, brain, eye, stomach, spleen, bones, pancreas, kidneys, liver, intestines, uterus, colon, ovary, blood, skin, muscle, tissue, prostate, mammary and bladder.
- organs are useful in the present invention.
- Peracid refers to an acid in which an acidic -OH group has been replaced by an -OOH group.
- Peracids can be peroxy-carboxylic acids of the formula R-C(O)-OOH, where the R group can be groups such as H, alkyl, alkene or aryl.
- Peracids include, but are not limited to, peroxy-acetic acid and meta-chloro-peroxybenzoic acid (MCPBA).
- MCPBA meta-chloro-peroxybenzoic acid
- peroxide refers to a compound containing an oxygen-oxygen single bond.
- peroxides include, but are not limited to, hydrogen peroxide.
- hydrogen peroxide One of skill in the art will appreciate that other peroxides are useful in the present invention.
- the term "pharmaceutically acceptable excipient” refers to a substance that aids the administration of an active agent to and absorption by a subject.
- Pharmaceutical excipients useful in the present invention include, but are not limited to, polymers, solvents, antioxidants, binders, fillers, disintegrants, lubricants, coatings, sweeteners, flavors, stabilizers, colorants, metals, ceramics and semi-metals. See below for additional discussion of pharmaceutically acceptable excipients. One of skill in the art will recognize that other pharmaceutical excipients are useful in the present invention.
- polymer refers to a molecule composed of repeating structural units, or monomers, connected by covalent chemical bonds. Polymers useful in the present invention are described below. One of skill in the art will appreciate that other polymers are useful in the present invention.
- prodrug refers to compounds which are capable of releasing the active agent of the methods of the present invention, when the prodrug is administered to a mammalian subject. Release of the active ingredient occurs in vivo.
- Prodrugs can be prepared by techniques known to one skilled in the art. These techniques generally modify appropriate functional groups in a given compound. These modified functional groups however regenerate original functional groups by routine manipulation or in vivo.
- Prodrugs of the active agents of the present invention include active agents wherein a hydroxy, amidino, guanidino, amino, carboxylic or a similar group is modified.
- salt refers to acid or base salts of the compounds used in the methods of the present invention.
- pharmaceutically acceptable salts are mineral acid (hydrochloric acid, hydrobromic acid, phosphoric acid, and the like) salts, organic acid (acetic acid, propionic acid, glutamic acid, citric acid and the like) salts, quaternary ammonium (methyl iodide, ethyl iodide, and the like) salts. Additional information on suitable pharmaceutically acceptable salts can be found in Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, Pa., 1985, which is incorporated herein by reference.
- salts of the acidic compounds of the present invention are salts formed with bases, namely cationic salts such as alkali and alkaline earth metal salts, such as sodium, lithium, potassium, calcium, magnesium, as well as ammonium salts, such as ammonium, trimethyl-ammonium, diethylammonium, and tris-(hydroxymethy])-methyl-ammonium salts.
- bases namely cationic salts such as alkali and alkaline earth metal salts, such as sodium, lithium, potassium, calcium, magnesium, as well as ammonium salts, such as ammonium, trimethyl-ammonium, diethylammonium, and tris-(hydroxymethy])-methyl-ammonium salts.
- acid addition salts such as of mineral acids, organic carboxylic and organic sulfonic acids, e.g., hydrochloric acid, methanesulfonic acid, maleic acid, are also possible provided a basic group, such as pyridyl, constitutes part of the structure.
- the neutral forms of the compounds may be regenerated by contacting the salt with a base or acid and isolating the parent compound in the conventional manner.
- the parent form of the compound differs from the various salt forms in certain physical properties, such as solubility in polar solvents, but otherwise the salts are equivalent to the parent form of the compound for the purposes of the present invention.
- the term "source'- refers to a location on the device of the present invention providing a supply of the compound of the present invention or a supply of a therapeutic agent.
- the device of the present invention can have more than one source, such as a first and second. Each source can have a different compound and composition and be used to treat a different indication.
- the term "subject” refers to animals such as mammals, including, but not limited to, primates (e.g., humans), cows, sheep, goats, horses, pigs, dogs, cats, rabbits, rats, mice and the like. In certain embodiments, the subject is a human.
- therapeutic agent-' refers to any agent, compound or biological molecule that has a therapeutic effect on the patient to whom the therapeutic agent is administered.
- the terms “therapeutically effective amount or dose” or “therapeutically sufficient amount or dose” or “effective or sufficient amount or dose” refer to a dose that produces therapeutic effects for which it is administered.
- the exact dose will depend on the purpose of the treatment, and will be ascertainable by one skilled in the art using known techniques (see, e.g., Lieberman, Pharmaceutical Dosage Forms (vols. 1-3, 1992); Lloyd, The Art, Science and Technology of Pharmaceutical Compounding (1999); Pickar, Dosage Calculations (1999); and Remington: The Science and Practice of Pharmacy, 20th Edition, 2003, Gennaro, Ed., Lippincott, Williams & Wilkins). In sensitized cells, the therapeutically effective dose can often be lower than the conventional therapeutically effective dose for non-sensitized cells.
- vascular prosthesis refers to a prosthesis for the circulatory system of a mammal.
- Macrocyclic lactones their salts, prodrugs, tautomers and isomers will be referred to collectively as "macrocyclic lactones ' ' in this invention.
- the compounds of the present invention are macrocyclic lactone compounds of the following formula:
- R 1 , R 2 , R 3 , R 5 , R 6 , R 8 , M 1 , M 2 , M 3 , M 4 , M 5 , M 6 and M 7 are each independently a member selected from the group consisting of H, C ⁇ .(, alkyl, OH and C) -6 hydroxyalkyl; R 4 , R 7 and R 9 are each independently selected from the group consisting of Cj .(, alkoxy and OH;
- R 10 is a member selected from the group consisting of H, -OH, -OP(O)Me 2 , , , -O-(CH 2 ) n -OH and -O-(CH 2 ) m -O-(CH 2 ) 0 -CH 3 , wherein subscripts n and m are each independently from 2 to 8 and subscript o is from 1 to 6; each of L 1 and L are independently selected from the group consisting of: and wherein each M 8 is independently a member selected from the group consisting of C 1-6 alkyl, OH and C 1-6 hydroxyalkyl; each of L 2 and L 3 are independently selected from the group consisting of:
- R 10 is other than OH, -OP(O)Me 2 , , -O-(CH 2 ) n -OH and -O-(CH 2 ) m -O-(CH 2 ) o -CH3; and salts, hydrates, isomers, metabolites, N-oxides and prodrugs thereof.
- R 10 is
- R is In other embodiments, R 10 is -OP(O)Me 2 .
- R 10 is
- R is
- the compound is a compound of Figure 2.
- at least one of R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 and R 9 is OH.
- R 4 is OH.
- R 1 , R 6 and R 8 are each methyl
- R 2 , R 3 and R 5 are each H
- R 4 and R 10 are each OH
- R 7 and R 9 are each OMe, M 1 , M 2 , M 3 , M 4 and M 5
- the invention provides macrocyclic lactone compounds which are described in detail with Formula IA, IB, IC and ID.
- the composition contains macrocyclic lactones which include hydroxy, demethyl, hydroxydemethyl and epoxide macrocyclic lactones.
- N-oxidation position N-oxidation position
- dashed lines represent the position for ring-opening position
- R 1 is a
- R a is alky
- R a is alky
- R b is C 2-6 alkylene
- R c is C 1-5 alkyl, such as 40-O-(ethoxyethyl) rapamycin.
- the compounds of the present invention include certain demethyl macrocyclic lactones, such as 16-O-demethyl macrocyclic lactone, 39-O-demethyl macrocyclic lactone, 27-O-demethyl macrocyclic lactone, 16, 27-bis-O-demethyl macrocyclic lactone, 27, 39-bis-O-demethyl macrocyclic lactone, 16, 39-bis-O-demethyl macrocyclic lactone, individually or in combination with each other, as shown in Formula IA.
- demethyl macrocyclic lactones such as 16-O-demethyl macrocyclic lactone, 39-O-demethyl macrocyclic lactone, 27-O-demethyl macrocyclic lactone, 16, 27-bis-O-demethyl macrocyclic lactone, 27, 39-bis-O-demethyl macrocyclic lactone, 16, 39-bis-O-demethyl macrocyclic lactone, individually or in combination with each other, as shown in Formula IA.
- R 4 , R 7 and R 9 are selected from the group of consisting Of-OCH 3 and -OH.
- R 4 and R 7 are both, independently, selected from the group of consisting of -OH and -OCH 3 .
- R 7 and R 9 are both, independently, selected from the group of consisting of -OH and -OCH 3 .
- R 4 and R 9 are both, independently, selected from the group of consisting of -OH and -OCH 3 .
- R 4 , R 7 and R 9 are each, independently, selected from the group of consisting of -OH and
- the compounds of the present invention includes compositions of hydroxyl macrocyclic lactone, such as 1 1 -hydroxy] macrocyclic lactone, 12-hydroxyl macrocyclic lactone, 14-hydroxyl macrocyclic lactone, 24-hydroxyl macrocyclic lactone, 25-hydroxyl macrocyclic lactone, 25-methyl alcohol macrocyclic lactone, 31 -methyl alcohol macrocyclic lactone,35-methyl alcohol macrocyclic lactone, 13-mefhyl alcohol macrocyclic lactone, individually or in combination with each other, as shown in Formula IB.
- hydroxyl macrocyclic lactone such as 1 1 -hydroxy] macrocyclic lactone, 12-hydroxyl macrocyclic lactone, 14-hydroxyl macrocyclic lactone, 24-hydroxyl macrocyclic lactone, 25-hydroxyl macrocyclic lactone, 25-methyl alcohol macrocyclic lactone, 31 -methyl alcohol macrocyclic lactone,35-methyl alcohol macrocyclic lactone, 13-mefhyl alcohol macrocyclic lactone, individually or in combination with each other, as shown in Formula IB.
- R 1 , R 6 , R 8 , M 6 , M 7 , M 8 and M 8a are selected from the group of consisting Of-CH 3 , -CH 2 OH and -OH.
- R 2 , R 3 , R 5 , M 1 , M 2 , M 3 , M 4 and M 5 are selected from the group of consisting of -H and -OH.
- R 9 is selected from the group of consisting of -OH and -OCH 3 .
- the compounds of the present invention include compositions of epoxide macrocyclic lactone, such as 19, 20-21 , 22-29, 30 tris epoxide macrocyclic lactone, 17, 18-19, 20-21 , 22 tris epoxide macrocyclic lactone and 17, 18-29, 30 bis epoxide macrocyclic lactone, as shown in Formula IC.
- epoxide macrocyclic lactone such as 19, 20-21 , 22-29, 30 tris epoxide macrocyclic lactone, 17, 18-19, 20-21 , 22 tris epoxide macrocyclic lactone and 17, 18-29, 30 bis epoxide macrocyclic lactone, as shown in Formula IC.
- the compounds of the present invention include compositions of N-oxidized macrocyclic lactone, 1 1-hydroxyl with 10,14 ring opening macrocyclic lactone, seco-macrocyclic lactone ( with 1, 34 ring opened), as shown in Formula ID.
- each of R 1 , R 6 , R 8 , M 6 , M 7 , M 8 and M 8a are independently selected from the group of consisting of-CH 3 , -OH and C 1-6 hydroxyalkyl.
- Each of R 2 , R 3 , R 5 , M 1 , M 2 , M 3 , M 4 and M 5 are independently selected from the group of consisting of — H and -OH.
- R , R , R are independently selected from the group of consisting of -OH and -OCH 3 L 2 is selected from
- the compounds of the present invention are a combination of Formula IA and Formula IB, including composition of demethylhydroxy macrocyclic lactone such as 14-hydroxy-39-O-demethyl macrocyclic lactone, 16,39-bis-O-demethyl-24-hydroxy macrocyclic lactone, 16,27-bis-O-demethyl-24-hydroxy macrocyclic lactone, 27,39-bis-0-demethyl-24-hydroxy macrocyclic lactone individually or in combination with each other.
- demethylhydroxy macrocyclic lactone such as 14-hydroxy-39-O-demethyl macrocyclic lactone, 16,39-bis-O-demethyl-24-hydroxy macrocyclic lactone, 16,27-bis-O-demethyl-24-hydroxy macrocyclic lactone, 27,39-bis-0-demethyl-24-hydroxy macrocyclic lactone individually or in combination with each other.
- the compounds of the present invention are a combination of Formula IA and Formula IC, including composition of epoxide demethyl macrocyclic lactone such as 17,18-19,20-bis-epoxide-16-O-dem ethyl macrocyclic lactone,
- the compounds of the present invention are a combination of Formula IA, Formula IB and Formula IC, including composition of epoxide demethylhydroxyl macrocyclic lactone such as
- This invention also covers the compositions of salts, hydrates, isomers, tautomers, metabolites, N-oxides and prodrugs of compounds of the present invention of Formula IA, IB, IC and ID.
- the compound of the present invention has the following structure:
- This invention covers compounds in which the stereochemistry of the 16-position is racemic (R,S) as well as the individual R and S stereoisomers at the 16-position and all other isomers of the compound.
- the invention covers compounds with different polymorphic forms.
- different polymorphs form of 16-O-demethyl macrocyclic lactone are obtained from using dichloromethylene and from using a mixture of methanol and water
- the compounds of the present invention can be prepared by a variety of methods.
- the compounds of the present invention are synthesized biologically by genetically modifying the strains of organisms to produce the compounds of the present invention or by other means.
- the compounds of the present invention are prepared using chemical synthesis. Chemical synthesis of the compounds of the present invention can utilize the 17-18, 19-20, 21-22 triene structure in macrocyclic lactones, which facilitates the acid catalyzed nucleophilic substitution of the Cl 6 methoxy group and allows the introduction of a number of different substitutions and allows the selective manipulation of macrocyclic lactone effector domain.
- the Cl 6 methoxy group in macrocyclic lactones is manipulated towards acidic reagents to produce the compounds of the present invention.
- replacement of Cl 6 methoxy groups with different nucleophiles such as alcohols, thiols and electron rich aromatic groups can be accomplished. This method of synthesis can be performed without protection and deprotection steps.
- the present invention provides a method of making a compound of the present invention, the method comprising contacting a macrocyclic lactone with an acid to replace an alkoxy group with a nucleophile, thereby making a compound of the present invention.
- the macrocyclic lactone is rapamycin.
- the nucleophile is a member selected from the group consisting of -OH, -SH and electron rich aromatic groups.
- the present invention provides a method of making a compound of the present invention, the method comprising contacting a macrocyclic lactone with a suitable agent such as a peracid or peroxide, to modify an alkene group to an epoxide, thereby making a compound of the present invention.
- a suitable agent such as a peracid or peroxide
- Peracids useful in the methods of the present invention include, but are not limited to, peroxy-carboxylic acids of the formula R-C(O)-OOH, where the R group can be groups such as H, alkyl, alkene or aryl.
- the peracid can be peroxy-acetic acid or meta-chloro-peroxybenzoic acid (MCPBA).
- Peroxides useful in the methods of the present invention include, but are not limited to, hydrogen peroxide.
- One of skill in the art will appreciate that other epoxidation reagents are useful in the present invention.
- the compounds of the present invention are optionally deuterated.
- the present invention provides compounds of the present invention which have a similar potency to the corresponding parent macrocyclic lactone. In another embodiment, the present invention provides compounds of the present invention which are less potent than the corresponding parent macrocyclic lactone in order to improve the safety profile of the compounds.
- the compounds of the present invention can be administered in any appropriate manner.
- the compounds are administered orally, intramuscularly, intraperitoneally, subcutaneously, pulmonarily, mucosally, transdermally, intravascularly , intraocularly or intravitreally through the eye, and others.
- the compounds are administered site specifically through temporary or permanent drug delivery means such as an implant or a combination of systemic and site specific means. Examples include, but are not limited to catheter, stent, vascular wrap, pump, shunt or other temporary or permanent drug delivery means.
- the present invention provides a device for intracorporeal use, the device comprising a vascular prosthesis; and at least one source of a compound of the present invention.
- the present invention provides a device configured to release the compound to a body lumen or organ within an intracorporeal body to inhibit cell proliferation or cell migration.
- the device is configured to release the compound to a body lumen or organ within an intracorporeal body to inhibit smooth muscle cell proliferation or neo-vascularization.
- the drug delivery means is a device such as an implant including graft implants, vascular implants, non-vascular implants, implantable luminal prostheses, wound closure implants, drug delivery implants, sutures, biologic delivery implants, urinary tract implants, inter-uterine implants, organ implants, ophthalmic implants, bone implants including bone plates, bone screws, dental implants, spinal disks, or the like.
- the implant of the present invention can be implanted intraocularly or intravitreally by an intervention procedure.
- Such implants can be non-biodegradable, biodegradable, removable or permanent.
- implants can be placed in the duct, such as the tear duct.
- implants can be placed adjacent to the ocular body, or intraocularly, adjacent to the vitreal body or intravitreally.
- implants can be placed adjacent to the ocular body, or intraocularly, adjacent to the vitreal body or intravitreally.
- the implant typically allows for one or more of the following: support, contain, hold together, affix, plug, close, maintain, deliver drug, deliver biologies ⁇ to a body lumen, organ, vessel, conduit, muscle, tissue mass or bone for the prevention or treatment of disease conditions, such as for example hyper-proliferative diseases, restenosis, cardiovascular disease, inflammation, wound healing, cancer, aneurysm, diabetic disease, abdominal aortic aneurysm, hyper-calcemia, or others.
- disease conditions such as for example hyper-proliferative diseases, restenosis, cardiovascular disease, inflammation, wound healing, cancer, aneurysm, diabetic disease, abdominal aortic aneurysm, hyper-calcemia, or others.
- the implant of the present invention can be formed of metal, metal alloy, polymer, ceramic, semi-metal, nanocomposites or combination thereof.
- an implant can be made from metal such as tantalum, iron, magnesium, molybdenum or others; from a degradable or non degradable metal alloy such as 316L stainless steel, carbon steel, magnesium alloy, NI-Ti, Co-Cr such as L605, MP35 or other; from a polymer that is degradable or non-degradable such as poly lactic acid, poly glycolic acid, poly esters, polyamide, copolymers or others or blends of polymers; combination of metals and metals or metal alloys such as implant made from combination of layers of stainless steel and tantalum or others; nanocomposites such as nano carbon fibers or nano carbon tubules or others.
- the present invention provides a device wherein the implant is a vascular prosthesis.
- the vascular prosthesis comprises an expandable structure.
- the vascular prosthesis comprises a stent, graft, or a scaffold formed at least in part from an open lattice.
- the vascular prosthesis is a stent.
- the compounds of the present invention can be applied adjacent to the surface of the implant. For example the compounds of the present invention can be incorporated within the implant, contained within a coating, or carried on the implant.
- the present invention provides a device comprising a vascular prosthesis wherein the vascular prosthesis has a luminal and a tissue facing surface, and wherein the compound is associated with at least one of the luminal or tissue facing surfaces.
- the compounds of the present invention are applied on all implant surfaces. In another embodiment, the compounds of the present invention are applied only to the abluminal or luminal surface. In yet another embodiment, the compounds of the present invention are applied only to high stress or low stress areas.
- the compounds of the present invention are contained within an erodible or non-erodible filament or filaments that are adjacent to the implant.
- FIG. 3 An example of a stent configuration for carrying a compound of the present invention is illustrated in FIG. 3 in a contracted state.
- the stent body is formed of multiple rings 1 10.
- the rings are formed of crowns 120 and struts 130 in a generally expandable undulating configurations such as, zigzag, sawtooth, sinusoidal wave or other.
- the body is joined by links or connectors 140. It is understood that the connectors may be of any length or shape, or may not be needed if the crowns are directly attached to each other.
- the stent has a typical contracted state diameter of between 0.25 - 4mm, or more preferably between 0.7 to 1.5mm, and a length of between 5 and 100 mm.
- the stent diameter is typically at least twice and up to 10 times or more than that of the stent in its contracted state.
- a stent with a contracted diameter of between 0.7 to 1.5mm may expand radially to 2 to 10mm or more.
- Drug eluting stents with potent macrocyclic lactone compounds such as rapamycin (CypherTM) have resulted in late lumen loss in the range of approximately 0.01 mm to 0.2 mm at approximately 4 months to 12 months angiographic follow up.
- the late lumen loss with bare metal stents have ranged from approximately 0.70 mm to 1.2 mm for the same time period. Lower late lumen loss typically decreased the percent stenosis.
- the late lumen loss for stent carrying compounds of the present invention after approximately 4 to 12 months after implantation is greater than the late lumen loss for stent carrying the corresponding parent macrocyclic lactone by 0.05 mm to 0.6 mm, preferably by 0.1 mm to 0.4 mm, more preferably by 0.15 to 0.3 mm.
- late lumen loss after implantation ranges from 0.01 mm to 0.6 mm, preferably from 0.1 mm to 0.5 mm and most preferably from 0.2 mm to 0.4 mm.
- the present invention provides late lumen loss for stent carrying compounds of the present invention similar to a stent carrying the corresponding parent macrocyclic lactone.
- the present invention provides late lumen loss for stent carrying compounds of the present invention being higher than for a stent carrying the corresponding parent macrocyclic lactone. Higher late lumen can provide increased tissue coverage of the stent which can improve safety of the stent.
- the percent stenosis approximately 4 to 12 months after implantation for a stent carrying compounds of the present invention is greater by 1 to 30 percentage than the percent stenosis for a stent carrying the corresponding parent macrocyclic lactone, preferably from 3 to 20 percentage, more preferably from 5 to 15 percentage.
- the present invention provides percent stenosis for a stent carrying compounds of the present invention similar to a stent carrying the corresponding parent macrocyclic lactone.
- the present invention provides percent stenosis for a stent carrying compounds of the present invention higher than a stent carrying the corresponding parent macrocyclic lactone.
- the percent stenosis for a stent carrying compounds of the present invention is higher than a stent carrying the corresponding parent macrocyclic lactone but lower than the bare metal stent.
- Higher stenosis can provide increased tissue coverage of the stent which can improve safety of the stent.
- the present invention provides a device wherein the amount of compounds of the present invention on the implant is less than about 1 g/cm 2 .
- the amount of compounds on the implant can range from about 1 nanogram/cm 2 to about 1000 microgram/cm 2 , preferably from about 1 microgram/cm 2 to about 500 microgram/cm 2 , more preferably from about 10 microgram/cm 2 to about 400 microgram/cm 2 .
- the amount of compound on the implant is less than about 1 mg.
- the amount of compound on the implant is from about 1 ⁇ g to about 50 mg, preferably from about 100 ⁇ g to about 10 mg, more preferably from about 200 ⁇ g to about 500 ⁇ g.
- the present invention provides a device wherein the concentration of the compound of the present invention in the tissue adjacent to the implant is from about 0.001 ng/gm tissue to about 1000 ⁇ g/gm tissue, preferably from about 1 ng/gm tissue to about 500 ⁇ g/gm tissue, more preferably from about 100 ng/gm tissue to about 100 ⁇ g/gm tissue.
- the compounds of the present invention can be released from the implant over a period ranging from less than 5 minutes to 2 years, preferably from 3 days to 6 months, more preferably from 1 week to 3 months. In other embodiments, the compounds of the present invention can be released from the implant over a period greater than 1 day, preferably greater than 2 weeks, more preferably greater than 1 month. In another embodiment, the compounds of the present invention can require greater than 2 years to be fully released from the stent. In some embodiments, the amount of compound released over the given time period is at least 25%. In other embodiments, the amount of compound released is at least 50%. In still other embodiments, the amount of compound released is at least 75%. In yet other embodiments, the amount of compound released can be at least 80, 85, 90, 91 , 92, 93, 94, 95, 96, 97, 98 or 99%.
- the present invention provides a device wherein at least 75% of the compound is released from the device in a period from about 1 day to about 2 years. In another embodiment, at least 90% of the compound is released from the device in a period from about 3 day to about 6 months. In still another embodiment, at least 90% of the compound is released from the device in a period from about 1 week to about 3 months.
- the concentration of compound alone or within the polymer matrix, solvent or carrier can vary from 1 ug/ml to 5mg/ml, and preferably from 5ug/ml to 30ug/ml.
- the concentration of the compound of the present invention in the tissue adjacent to the site of administration can be from about 0.1 nM to 500 ⁇ M, preferably from about 1 nM to 100 ⁇ M, more preferably from about 10 nM to 1 O ⁇ M.
- concentrations of the compounds of the present invention are useful.
- the compounds of the present invention can be released from the implant via any means known in the art. In some embodiments, the implant releases the compound through active or passive means. In other embodiments, the implant releases the compound through osmotic pressure or diffusion. One of skill in the art will appreciate that other means of releasing the compound from the implant are useful in the present invention.
- the present invention provides a device that further includes a therapeutic agent, such as those described below.
- a therapeutic agent such as those described below.
- the therapeutic agent is released prior to, concurrent with, or subsequent to the release of the compound.
- the compound is released from a first source and the therapeutic agent is released from a second source.
- the compound and the therapeutic agent are released from a single source.
- the compounds of the present invention can be administered systemically on a daily, intermittent or one-time dose basis.
- the daily systemic dose can range from 0.1 mg to 20 mg preferably 0.5 mg to 10 mg, most preferably from 1 mg to 5 mg per day.
- One of skill in the art will appreciate that other doses are also useful in the present invention.
- the compounds of the present invention can be released from the implant at rates ranging from about 1 nanogram/cm 2 /day to about 1000 microgram /cm 2 /day, preferably from about 1 microgram/cm 2 /day to about 200 microgram/cm 2 /day, more preferably from about 5 microgram /cm 2 /day to about 100 microgram /cm 2 /day.
- the compounds of the present invention can be administered through the eye as an eye drop or an injection on a daily, intermittent or one time dose basis.
- the dose can range from 0.1 ⁇ g to 30mg, preferably from 10 ⁇ g to lOmg, most preferably from 100 ⁇ g to lmg per day.
- One of skill in the art will appreciate that other doses are also useful in the present invention.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a pharmaceutically acceptable excipient and a compound of the formula:
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , M 1 , M 2 , M 3 , M 4 , M 5 , M 6 , M 7 , L 1 , L 2 , L 3 and L 4 are as described above; and salts, hydrates, isomers, metabolites, N-oxides and prodrugs thereof.
- the present invention provides a pharmaceutical composition including a pharmaceutically acceptable excipient and a compound of the formula:
- the present invention provides a pharmaceutical composition including a pharmaceutically acceptable excipient and a compound of the formula:
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , M 1 , M 2 , M 3 , M 4 , M 5 , M 6 , M 7 , L 1 , L 2 , L 3 and L 4 are as described above; and salts, hydrates, isomers, metabolites, N-oxides and prodrugs thereof.
- the pharmaceutical composition includes a compound of Figure 2.
- R 1 , R 6 and R 8 are each methyl
- R 2 , R 3 and R 5 are each H
- I and R 10 are each OH
- R 7 and R 9 are each OMe
- M 1 , M 2 , M 3 , M 4 and M 5 are each H
- M 7 are each methyl, L 1 and L 4 are each ,and L and L 3 are each
- the present invention provides a pharmaceutical composition wherein the pharmaceutically acceptable excipient is a member selected from the group consisting of a polymer, a solvent, an antioxidant, a binder, a filler, a disintegrant, a lubricant, a coating, a sweetener, a flavor, a stabilizer, a colorant, a metal, a ceramic and a semi-metal.
- the pharmaceutically acceptable excipient is a polymer.
- the active ingredients of the present invention may be mixed with pharmaceutically acceptable carriers, diluents, adjuvants, excipients, or vehicles, such as preserving agents, fillers, polymers, disintegrating agents, glidants, wetting agents, emulsifying agents, suspending agents, sweetening agents, flavoring agents, perfuming agents, lubricating agents, acidifying agents, and dispensing agents, depending on the nature of the mode of administration and dosage forms.
- pharmaceutically acceptable carriers and excipients that may be used to formulate oral dosage forms, are described in the Handbook of Pharmaceutical Excipients, American Pharmaceutical Association (1986), incorporated herein by reference in its entirety.
- Examples of pharmaceutically acceptable carriers include water, ethanol, polyols, vegetable oils, fats, waxes polymers, including gel forming and non-gel forming polymers, and suitable mixtures thereof.
- excipients include starch, pregelatinized starch, Avicel, lactose, milk sugar, sodium citrate, calcium carbonate, dicalcium phosphate, and lake blend.
- disintegrating agents include starch, alginic acids, and certain complex silicates.
- Examples of lubricants include magnesium stearate, sodium lauryl sulphate, talc, as well as high molecular weight polyethylene glycols.
- Suitable nondegradable or slow degrading polymer coatings include, but are not limited to, polyacrylamide, poly-N-vinylpyrrollidone, polydimethyl acrylamide, polymers and copolymers of 2-acrylamido-2-methyl-propanesulfonic acid, acrylic acid and methacrylic acid, polyurethane, polyethylenes imine, ethylene vinyl alcohol copolymer, silicone, C-flex, nylons, polyamide, polyimide, polytetrafluoroethylene (PTFE), parylene, parylast, poly
- Suitable biodegradable polymer coatings include, but are not limited to, poly(lactic acid), polylactates, poly(glycolic acid), polyglycolates and copolymers, poly dioxanone, poly( ethyl glutamate), poly(hydroxybutyrate), polyhydroxyvalerate and copolymers, polycaprolactone, polyanhydride, poly(ortho esters); poly( ether esters), poly ethylene glycols, poly( ethylene oxide), poly (trimethyl carbonate), polyethylenecarbonate, copolymers of poly(ethylenecarbonate) and poly(trimethyl carbonate), poly(propylene carbonate), ⁇ oly(iminocarbonates), starch based polymers, cellulose acetate butyrate, polyester amides, polyester amines, polycyanoacrylates, polyphosphazenes, Poly N-vinyl-2-pyrrolidone, poly maleic anhydride, hyaluronic acid (hyaluronate), chondroit
- Suitable natural coatings include: fibrin, albumin, collagen, gelatin, glycosoaminoglycans, oligosaccharides and poly saccharides, chondroitin, chondroitin sulphates, hypoxyapatite, phospholipids, phosphorylcholine, glycolipids, fatty acids, proteins, cellulose, and mixtures, copolymers, or combinations thereof.
- Suitable non polymeric coatings include metallic coatings such as tungsten, magnesium, cobalt, zinc, iron, bismuth, tantalum, gold, platinum, stainless steel such as 316L, 304, titanium alloys; ceramics coatings such as silicon oxide; semi-metals such as carbon, nanoporous coatings; or combination thereof.
- the pharmaceutically acceptable excipient is a polymer is selected from the group consisting of polyurethane, polyethylene imine, ethylene vinyl alcohol copolymer, silicone, C-flex, nylons, polyamide, polyimide, polytetrafluoroethylene (PTFE), parylene, parylast, poly(methacrylate), poly(vinyl chloride), poly( dimethyl siloxane), poly( ethylene vinyl acetate), polycarbonate, polyacrylamide gels, poly (methyl methacrylate), poly(n-butyl methacrylate), poly (butyl methacrylate) copolymer or blended with poly( ethylene vinyl acetate), poly(methyl methacrylate), poly (2-hydroxy ethyl methacrylate), poly( ethylene glycol methacrylates), poly(ethylene carbonate), Poly L lactide-glycolide copolymer, poly L lactide-trimethylene carbonate copolymer and Poly L-lactide.
- polyurethane polyethylene imine
- the present invention provides a composition wherein the compound is present in an amount of at least 10% (w/w) in a mixture of the compound and the polymer.
- the compound is present in an amount of at least 20, 25, 30, 40, 50, 55, 60, 70, 75, 80 and 90% (w/w).
- the compound is present in an amount of at least 25% (w/w).
- the compound is present in an amount of at least 50% (w/w).
- the compound is present in an amount of at least 75% (w/w).
- One of skill in the art will appreciate that other compositions are useful in the present invention.
- the compounds of the present invention can be applied onto a stent without a coating.
- compounds of the present invention can be applied onto a stent in combination with a polymer coating such as a compounds of the present invention-polymer matrix.
- the compounds of the present invention can be fully or partially crystallized or in an amorphous form.
- the polymer can be non degradable, partially degradable or fully degradable.
- the coating can also be a non-polymeric such as metallic coating.
- the compounds of the present invention can be applied on a stent alone or contained in a coating with a polymer or non polymer topcoat.
- the stent includes an underlayer coating disposed between the stent surface and the compounds of the present invention or compounds of the present invention -polymer matrix.
- Suitable underlayer coatings can be polymeric such as paralyne C, parylene N, ethylene vinyl alcohol (EVOH), polycaprolactone, ethylvinyl hydroxylated acetate (EVA), or others or combination thereof or non polymeric such as metallic or ceramic or others.
- the coatings can be applied by any of the different methods which include but are not limited to spraying, ultrasonic deposition, dipping, inkjet dispension, plasma deposition, ion implantation, sputtering, evaporation, vapor deposition, pyrolysis, electroplating, glow discharge coating, or others or combination thereof.
- the coating thickness can range from 1 nanometer to 100 micrometers, preferably from 100 nanometers to 50 micrometers, more preferably from 1 micrometer to 20 micrometers.
- the compounds of the present invention can be combined with antioxidants or stabilizers to prevent degradation due to oxidation or other means.
- Antioxidants include but are not limited to butylated hydroxytoluene (BHT), ferrous sulfate, ethylenediamine-tetra-acetic acid (EDTA), or others.
- Stabilizers include, but are not limited to, amglene, hydroquinone, quinine, sodium metabisulfite or others.
- Antioxidants and stabilizers can be combined with the compounds directly or blended with the compound formulation such as compound-polymer matrix to reduce conformation change or degradation during manufacturing processes and increase shelf life or storage life of the compounds or compound containing implant.
- the amount of antioxidants such as BHT in the compounds can range from 0.01 % to 10%, preferable from 0.05% to 5% and most preferable from 0.1 % to 1%.
- the amount of stabilizers such as amylene in the compounds can range from 0.001% to 0.1%, preferably from 0.005% to 0.05%, most preferably from 0.01 % to 0.02%.
- antioxidants and stabilizers are useful in the present invention.
- the compounds of the present invention can be administered in combination with a therapeutic agent such as anti-platelet, anti-thrombotic, anti-inflammatory, anti-angiogenic, anti-proliferative, immunosuppressant, anti-cancer or other agents or combinations thereof.
- a therapeutic agent such as anti-platelet, anti-thrombotic, anti-inflammatory, anti-angiogenic, anti-proliferative, immunosuppressant, anti-cancer or other agents or combinations thereof.
- a therapeutic agent such as anti-platelet, anti-thrombotic, anti-inflammatory, anti-angiogenic, anti-proliferative, immunosuppressant, anti-cancer or other agents or combinations thereof.
- the therapeutic agents can be incorporated on the stent together with the compounds of the present invention and/or separately from compounds of the present invention. At least a portion of the therapeutic agent can be released from the stent prior to, concurrently or post release of the compounds of the present invention from the stent.
- the therapeutic agent can also be given separately through systemically or site specific administration prior to, during or post delivery of compounds of the present invention.
- compounds of the present invention are given with anti platelets or anti-thrombotics such as heparin, clopidogrel, Coumadin, aspirin, ticlid or others.
- compounds of the present invention are given with anti-inflammatory agents such as aspirin, diclofenac, indomethacin, sulindac, ketoprofen, flurbiprofen, ibuprofen, naproxen, piroxicam, tenoxicam, tolmetin, ketorolac, oxaprosin, mefenamic acid, fenoprofen, nambumetone (relafen), acetaminophen, and mixtures thereof; COX-2 inhibitors, such as nimesulide, NS-398, flosulid, L-745337, celecoxib, rofecoxib, SC-57666, DuP-697, parecoxib sodium, JTE-522, valdecoxib
- the compounds of the present invention are combined with at least one therapeutic agent for treatment of an ophthalmic condition or disorder.
- Any suitable therapeutic agent known to one of skill in the art can be combined with the compounds of the present invention for use in the treatment of ophthalmic conditions or diseases.
- Therapeutic agents that can be combined with the compounds of the present invention include, but are not limited to, lucentis, avastin, macugan, volociximab, olopatadine, mydriatcs, dexamethasone, pilocarpine, tropicamide, quinolone, galentamine, fluocinolone acetonide, triamcinolone acetonide, atropine, atropine sulfate, atropine hydrochloride, atropine methylbromide, atropine methylnitrate, atropine hyperduric, atropine N-oxide, phenylephrine, phenylephrine hydrochloride, hydroxyamphetamine, hydroxyamphetamine hydrobromide, hydroxyamphetamine hydrochloride, hydroxyamphetamine iodide, cyclopentolate, cyclopentolate hydrochloride, homatropine, homatropine hydrobromide
- Formulations of the compounds of the present invention for ophthalmic uses can include any polymer described above.
- the polymers useful in such formulations can be of any size.
- the polymers can have a molecular weight of between about 50 kilo Daltons (kD) and 8,000 kD.
- kD kilo Daltons
- One of skill in the art will appreciate that polymers of other sizes are useful in the present invention.
- the compounds of the present invention can be administrated alone or as part of compound-polymer formulation, compound-solvent formulation or compound-carrier formulation.
- All formulations of the present invention may include active and inactive ingredients.
- Active ingredients include, but are not limited to, anti-inflammatory agents, immunomodulating agent and anti-infective agents, antioxidants, antibody, antibiotics, anti-angiogenics, anti-vascular endotherlial growth factor agent, antihistamines and lubricant.
- Inactive ingredients include, but are not limited to, carrier, solvent, inorganic materials, pH-adjustor, radio-opaque, radioactive, fluorescent, NMR contrast or other
- solvent in the compound-solvent formulation examples include, but are not limited to, water, saline, alcohol, and dimethyl sulfoxide.
- carrier in the compound-carrier formulation examples include glycerin, paraffin, beeswax, ethylene glycol, propylene glycol, polyethylene glycol, and macrogels.
- inorganic materials include, but are not limited to, boric acid, calcium chloride, magnesium chloride, potassium chloride, sodium chloride, zinc chloride, sodium borate, povidone, and dibasic sodium phosphate.
- pH-adjustor examples include, but are not limited to, sodium hydroxide, hydrogen chloride, buffer, and other inorganic and organic acid/base.
- preservative examples include, but are not limited to, benzalkonium chloride, and a polyquaternium.
- examples of lubricant include, but are not limited to, carboxymethylcellulose sodium, polyethylene glycol, propylene glycol and ethylene glycol.
- the present invention provides a composition where less than about 5% of the compound is metabolized to rapamycin. In some other embodiments, less than about 1% of the compound is metabolized to rapamycin. In still other embodiments, less than about 0.1% of the compound is metabolized to rapamycin. [0147] In other embodiments, the present invention provides a composition in a dosage form, having a daily systemic dose of the compound of from about 0.1 mg to about 20 mg. In some other embodiments, the daily systemic dose of the compound is from about 0.5 mg to about 10 mg. In another embodiment, the daily systemic dose of the compound is from about 1 mg to about 5 mg.
- the compounds of the present invention can be used to treat conditions responsive to the class of compounds commonly known as macrocyclic trienes or macrocyclic lactones.
- the compounds of the present invention can be used to treat diseases in mammals alone or in combination with other agents, including conditions such as: a) Treatment and prevention of acute or chronic organ or tissue transplant rejection, e.g. for the treatment of recipients of heart, lung, combined heart-lung, liver, kidney, pancreatic, skin or corneal transplants. They can also be used for the prevention of graft- versus-host disease, such as following bone marrow transplantation. b) Treatment and prevention of transplant vasculopathies, e.g. atherosclerosis. c) Treatment and prevention of cell proliferation and migration leading to vessel intimal thickening, blood vessel obstruction, obstructive vascular atherosclerosis, restenosis.
- autoimmune disease and of inflammatory conditions such as inflammatory conditions with an etiology including an autoimmune component such as arthritis (for example rheumatoid arthritis, arthritis chronica progrediente and arthritis deformans) and rheumatic diseases.
- e Treatment and prevention of asthma.
- f Treatment of multi-drug resistance conditions such as multidrug resistant cancer or multidrug resistant AIDS.
- proliferative disorders e.g. tumors, cancer, hyperproliferative skin disorder and the like.
- Treatment of infections such as fungal, bacterial and viral, i) Treatment or prevention of cellular proliferation in vascular shunts.
- k Prevention of neo-vascularization.
- the present invention provides a method of inhibiting cell proliferation or migration by administering to a subject in need thereof, a therapeutically effective amount of a compound of the present invention.
- the present invention provides a method wherein the compound of the present invention is administered systemically, locally or via a combination thereof.
- the administration of the compound of the present invention is via oral administration, administration as a suppository, topical contact, parenteral, intravascular, intravenous, intraperitoneal, intramuscular, intralesional, intranasal, pulmonary, mucosal, transdermal, ophthalmic, subcutaneous administration or intrathecal administration.
- the administration of the compound of the present invention is via delivery through a temporary device or an implant.
- the temporary device is selected from the group consisting of a catheter and a porous balloon.
- the implant is a vascular prosthesis.
- the vascular prosthesis comprises an expandable structure.
- the vascular prosthesis comprises a stent, graft, or a scaffold formed at least in part from an open lattice.
- the inhibition concentration (IC 50 ) of compounds of the present invention is approximately equal to the IC 50 of its corresponding parent macrocyclic lactone (prior to the modifications in Figure 1).
- the IC 50 is higher than the IC 50 of its corresponding parent macrocyclic lactone.
- the IC 50 is lower than the IC 50 of its corresponding parent macrocyclic lactone.
- the IC 50 is two to thousand times lower than the IC 50 of its corresponding parent macrocyclic lactone.
- the IC 50 of a compound of the present invention is 1.5 to 1 ,000 times higher than its corresponding parent macrocyclic lactone, preferably 2 to 100 times higher than the corresponding parent macrocyclic lactone and more preferably 5 to 50 times higher than the corresponding parent macrocyclic lactone. In another embodiment the IC 50 of a compound of the present invention is from about 0.1 nM to about 1 ⁇ M, preferably from about 1 nM to about 0.5 ⁇ M, more preferably from about 5 nM to about 100 nM. [0157] Other means of measuring the effectiveness of the compounds of the present invention include measuring effective concentration (ECs 0 ).
- the EC 50 is approximately equal to the EC 50 of the corresponding parent macrocyclic lactone. In another embodiment, the EC 50 is higher than the EC 50 of the corresponding parent macrocyclic lactone. In yet another embodiment, the EC 50 is lower than the EC 50 of the corresponding parent macrocyclic lactone.
- the present invention provides a method wherein the effective dose of the compound is from about 0.1 mg to about 20 mg. In some other embodiments, the effective dose of the compound is from about 0.5 mg to about 10 mg. In still other embodiments, the effective dose of the compound is from about 1 mg to about 5 mg.
- cytokine inhibition Pro-inflammatory cytokine IL-6 is synthesized in response to diverse inflammatory stimuli and acts as a key regulatory protein in the inflammatory cascade. IL-6 plays a pivotal role in stimulating the acute-phase response after injury, including the release of fibrinogen and C-reactive protein.
- IL-6 may also be directly involved in restenosis, as it has been shown to stimulate leukocyte recruitment into the vessel wall, and vascular smooth muscle cell proliferation, factors that are essential to the pathogenesis of hyperproliferative diseases such as restenosis.
- MMP-9 Matrix metalloproteinases
- Plasma active MMP-9 levels may be a useful independent predictor of bare metal stent ISR. (Elevated Plasma Active Matrix Metalloproteinase-9 Level Is Associated With Coronary Artery In-Stent Restenosis, Arterioscler Thromb Vase Biol. 2006;26:el 21 -el 25.)
- Monocyte chemoattractant protein 1 (MCP-I) is a potent monocyte chemoattractant secreted by many cells in vitro, including vascular smooth muscle and endothelial cells. Eliminating MCP-I gene or blockade of MCP-I signals has been shown to decrease atherogenesis in hypercholesterolemic mice. MCP-I has been shown to play a role in pathogenesis of neointimal hyperplasia in monkeys. (Importance of Monocyte
- MCP-I is also strongly expressed in a small subset of cells in macrophage-rich regions of human and rabbit atherosclerotic lesions (Expression of Monocyte Chemoattractant Protein 1 in Macrophage-Rich Areas of Human and Rabbit Atherosclerotic Lesions, PNAS, VoI 88, 5252-5256). Inhibition of MCP-I can have therapeutic impact on treatment and prevention of inflammatory, proliferative and other disease conditions discussed above.
- Interleukin-10 is an anti-inflammatory cytokine with a powerful inhibitory effect on monocytes. IL-10 has been shown to reduce postinjury intimal hyperplasia (lnterleukin-10 Inhibits Intimal Hyperplasia After Angioplasty or Stent Implantation in
- the compounds, pharmaceutical compositions and devices of the present invention are useful for the treatment of ophthalmic conditions and diseases.
- the compounds, pharmaceutical compositions and devices of the present invention are useful in the treatment of any ophthalmic condition or disease.
- Ophthalmic conditions and diseases that can be treated by the compounds and devices of the present invention include, but are not limited to, disorders of the eyelid, disorders of the lacrimal system and orbit, tear duct blockage, disorders of conjunctiva, disorders of the sclera, cornea, iris and ciliary body, disorders of the lens, disorders of the choroid and retina, Age-related Macular Degeneration (AMD), Diabetic Macular Edema (DME), glaucoma, disorders of the vitreous body and globe, disorders of the optic nerve and visual pathways, disorders of the ocular muscles, binocular movement, accommodation and refraction, visual disturbances and blindness, etc.
- AMD Age-related Macular Degeneration
- DME Diabetic Macular Edema
- glaucoma disorders of
- Additional ophthalmic conditions and diseases that can be treated with the compounds and devices of the present invention include inhibition of cell proliferation, prevention of inflammation, prevention of neovascularization, protection of neurovascular system, and prevention of immune response after transplantation.
- ophthalmic conditions and diseases can be treated using the compounds and devices of the present invention.
- Surgical treatment methods include retinal implant, high speed laser eye surgery, endothelial keratoplasty, cataract surgery, glaucoma surgery, refractive surgery, corneal surgery, vitreo-retinal surgery, eye muscle surgery, oculoplastic surgery, uses of stem cells to create corneas or part of corneas that can be transplanted into the eyes.
- Ophthalmic conditions and diseases can be treated using compounds, pharmaceutical compositions and devices of the present invention, as described above.
- the compounds and pharmaceutical compositions of the present invention can be administered via any method known to one of skill in the art.
- the compounds of the present invention are administered via implant, injection or eye drop.
- the administration is through an intraocular or intravitreal body of the eye.
- the administration is via the implant.
- the compounds are administered via an implant where the compound is released via a metallic, ceramic or polymer coating.
- the compound can be released by any means known in the art.
- release of the compound from the implant can be via osmotic pressure of diffusion.
- the compounds of the present invention are combined with at least one other therapeutic agent for treatment of an ophthalmic condition or disorder.
- Any suitable therapeutic agent known to one of skill in the art can be combined with the compounds of the present invention for use in the treatment of ophthalmic conditions or diseases.
- the therapeutic agents include, but are not limited to, anti- inflammatory agents, immunomodulating agent and anti-infective agents, antioxidants, antibody, antibiotics, anti-angiogenics, anti-vascular endotherlial growth factor agent, antihistamines and lubricant.
- Therapeutic agents that can be combined with the compounds of the present invention include, but are not limited to, lucentis, avastin, macugan, volociximab, olopatadine, mydriatcs, dexamethasone, pilocarpine, tropicamide, quinolone, galentamine, fluocinolone acetonide, triamcinolone acetonide, atropine, atropine sulfate, atropine hydrochloride, atropine methylbromide, atropine metliylnitrate, atropine hyperduric, atropine N-oxide, phenylephrine, phenylephrine hydrochloride, hydroxyamphetamine, hydroxyamphetamine hydrobromide, hydroxyamphetamine hydrochloride, hydroxyamphetamine iodide, cyclopentolate, cyclopentolate hydrochloride, homatropine, homatropine hydro
- Compound AR was purified by preparative HPLC on an Ascentis Cl 8 (21.2 x 250mm, lO ⁇ m) column from SUPECO using Methanol: water (80:20) as mobile phase from 0-12 min then changed to 100% Methanol from 12.01-20min at a flow rate of 15 ml/min.
- the loading concentration was 350 mg/ml and the injection volume was 100 ul.
- the compound was monitored by UV absorbance at 254 nm. Under these conditions, Compound AR eluted between 9.0-1 1.5 min whereas the starting materials and by products eluted between 17.0-20.0 min.
- the preparative HPLC can also be conducted using a gradient of acetonitrile:water (starting from 70:30) as mobile phase and monitored by UV absorbance at 278 nm.
- the preparative HPLC chromatogram using this method of separation is shown in Figure 4.
- the total content of Compound AR was determined by a reverse phase HPLC using a Supelco Cl 8 (4.6 x 150mm, 5 ⁇ m) column from Sigma Aldrich using methanol: water (90: 10) as mobile phase and a flow rate of 1 ml/min.
- Compound AR was monitored by UV absorbance at 254nm.
- Compound AR had a retention time of 7.97 min.
- Figure 7 shows the analytical HPLC chromatogram of Compound AR with total content of >98%.
- the purity of Compound AR was determined by a reverse phase HPLC using a YMC ODS-AL Cl 8 (4.6x250mm, 5 ⁇ m) column from Waters Corporation using an acetonitrile:water gradient mobile phase and a flow rate of 1.0 ml/min.
- Figure 7b shows major isomers of Compound AR with a purity > 98% as monitored by UV absorbance at 278nm.
- BHT Butylated Hydroxytoluene
- the IC 50 of Compound AR and rapamycin was approximately 0.005 and 0.001 ⁇ M, respectively, after smooth muscle cells were exposure to Compound AR and rapamycin for 8 hours.
- the IC50 of Compound AR was approximately five times higher than that of rapamycin.
- PBMA poly(n-butyl methacrylate)
- a microprocessor-controlled ultrasonic sprayer was used to apply 450 ug of the drug containing PBMA solution to the entire surface of al 8 mm metal stent (available from Elixir Medical Corp, Sunnyvale, Calif.). After coating, the stent was placed in a vacuum chamber. The stent was then mounted on the balloon of a 3.0 x 20 mm PTCA delivery catheter. The catheter was then inserted in a coil and packaged in a Tyvek® pouch. The pouch was sterilized by ethylene oxide. The Tyvek® pouch was further packaged in a foil pouch with oxygen scavengers and nitrogen purge and vacuum sealed.
- Example 4 In vivo Testing of Stents Eluting Compound AR [0183] The efficacy of a Compound AR eluting stent system (as prepared above) from
- Example 3 was evaluated by comparing 28 ⁇ 2 day angiographic outcomes in porcine coronary arteries to the rapamycin eluting stent system, CypherTM Coronary Stent (Cordis Corporation) in the non-diseased porcine coronary artery model.
- the nonatherosclerotic swine model was chosen as this model has been used extensively for stent and angioplasty studies resulting in a large volume of data on the vascular response properties and its correlation to human vascular response (Schwartz et al, Circulation. 2002; 106: 1867-1873). The animals were housed and cared for in accordance the Guide for the Care and Use of Laboratory Animals as established by the National Research Council.
- Vessel angiography was performed under fluoroscopic guidance, a 7 Fr. guide catheter was inserted through the sheath and advanced to the appropriate location where intracoronary nitroglycerin was administered.
- a segment of coronary artery ranging from 2.25 to 4.0 mm mean lumen diameter was chosen and a 0.014" guidewire inserted.
- Quantitative Coronary Angiography was performed to document the reference vessel diameter.
- the appropriately sized stent was advanced to the deployment site.
- the balloon was inflated at a steady rate to a pressure sufficient to achieve a balloon to artery ratio of 1.30: 1.0. Pressure was maintained for approximately 10 seconds.
- Angiography was performed to document post-procedural vessel patency and diameter.
- MLD minimal lumen diameter
- RVD Diameter stenosis [1 - (MLD/RVD)] x 100%
- RVD is a calculation of the reference diameter at the position of the obstruction (measure obtained by a software-based iterative linear regression technique to generate an intrapolation of a projected vessel without the lesion) (final angiogram only)
- Balloon to artery ratio [balloon/pre-stent mean luminal diameter]
- the appropriately sized stent was advanced to the deployment site.
- the balloon was inflated at a steady rate to a pressure sufficient to achieve a balloon to artery ratio of 1 :1. Pressure was maintained for approximately 10 seconds.
- Angiography was performed to document post-procedural vessel patency and diameter. A total of 9 stents (3 per time point) were implanted.
- the IC 50 of 17, 18-29, 30-bis-epoxide macrocyclic lactone and Compound AR was approximately 0.1 and 0.005 ⁇ M, respectively, after smooth muscle cells were exposed to 17, 18-29, 30-bis-epoxide macrocyclic lactone and Compound AR for 8 hours ( Figure 13).
- the IC 50 of 17, 18-29, 30-bis-epoxide macrocyclic lactone was approximately 20 times higher than that of Compound AR.
- m-chloro-peroxybenzoic acid 0.93g ( 3.22 mmol) was added to a solution of 0.50g (0.5371mmol) rapamycin in 10 ml CHCl 3 at room temperature. The mixture was stirred at room temperature for 24 hours. IfTLC indicated some rapamycin was still unreacted, additional m-chloroperoxybenzoic acid 0.5Og and 5 mL CHCI 3 was added. Stirring was continued at room temperature until no Rapamycin was indicated by TLC. After completion of the reaction, the solution was diluted with dichlorom ethane, treated with aqueous sodium sulfite until the washings give a negative test with starch-iodide paper.
- MMP-9 levels in the 1st, 3rd and 7th day after stent implantation were positively correlated to the late loss index 6 months after stent implantation (Elevated matrix metalloproteinase expression after stent implantation is associated with restenosis. Int J Cardiol. 2006; 1 12(l):85-90).
- Compound AR and Sirolimus did not show and significant inhibition of release of IL-6.
- Compound AR significantly reduced the production of both the cytokines MMP-9 and MCP-I as compared to Sirolimus which increased the production of MMP-9 and did not impact the production of MCP-I .
- Compound AR and Sirolimus did not show any difference in inhibition of release of IL-6.
- Compound AR reduced the production of cytokine MMP-9 while Sirolimus increased the production of MMP-9.
- Compound AR reduced the production of cytokine MCP-I as compared to Sirolimus which did not impact the production of MCP-I .
- Compound AR and Sirolimus both inhibit the production of anti-inflammatory cytokine IL-10.
- Compounds claimed in the present invention can provide better therapeutic response with higher levels of anti -inflammatory effect (such as greater inhibition of pro-inflammatory cytokine MCP-I) and higher levels of anti-cell proliferative and anti-cell migratory effect (such as greater inhibition of pro-proliferative and migration cytokine MMP-9).
- Example 10 Testing of Compound AR eluting Stents in Human Clinical Trial: [0205] Clinical testing of the Compound AR coated stent was conducted on 15 human subjects. Safety of the Compound AR coated stent was evaluated clinically through the evaluation of major adverse cardiac events defined as: death, myocardial infarction (both Q-wave and non-Q-wave), and target lesion revascularization. Efficacy was evaluated through angiographic and intravascular ultrasound (IVUS) results at 4 months. The primary endpoint of the study was angiographic in-stent late lumen loss. Secondary endpoints were Major Adverse Cardiac Events (MACE) and additional angiographic and IVUS evaluation. The clinical study was approved by local Ethics Committee and all patients signed an Ethics approved informed consent before entry into the clinical study.
- MACE Adverse Cardiac Events
- Clopidogrel 75mg/day were continued through for at least six months.
- the left or right femoral artery was accessed using standard techniques and an arterial sheath was introduced and advanced into the artery.
- Index procedure vessel angiography was performed under fluoroscopic guidance, a 6 or 7 Fr. guide catheter was inserted through the sheath and advanced to the appropriate location; intracoronary nitroglycerin was administered. A segment of coronary artery ranging from 3.0mm to 3.5 mm mean lumen diameter was chosen and a 0.014" guidewire inserted. Quantitative Coronary Angiography (QCA) was performed to document the reference vessel diameter. Predilatation of the lesion was performed prior to stent implantation using standard technique. [0208] Following predilatation, the appropriately sized stent (3.0 x 18 mm or 3.5 x 18 mm was advanced to the deployment site.
- QCA Quantitative Coronary Angiography
- the balloon was inflated at a steady rate to a pressure to fully deploy the stent. Pressure was maintained for approximately 30 seconds. Post dilatation of the stent could be performed as needed to assure good stent apposition to the vessel wall. Angiographic and intravascular ultrasound imaging (IVUS) was performed and recorded.
- IVUS intravascular ultrasound imaging
- RVD Diameter stenosis [1 - (MLD/RVD)] x 100%]
- RVD is a calculation of the reference diameter at the position of the obstruction (measure obtained by a software-based iterative linear regression technique to generate an intrapolation of a projected vessel without the lesion) (final angiogram only).
Abstract
Description











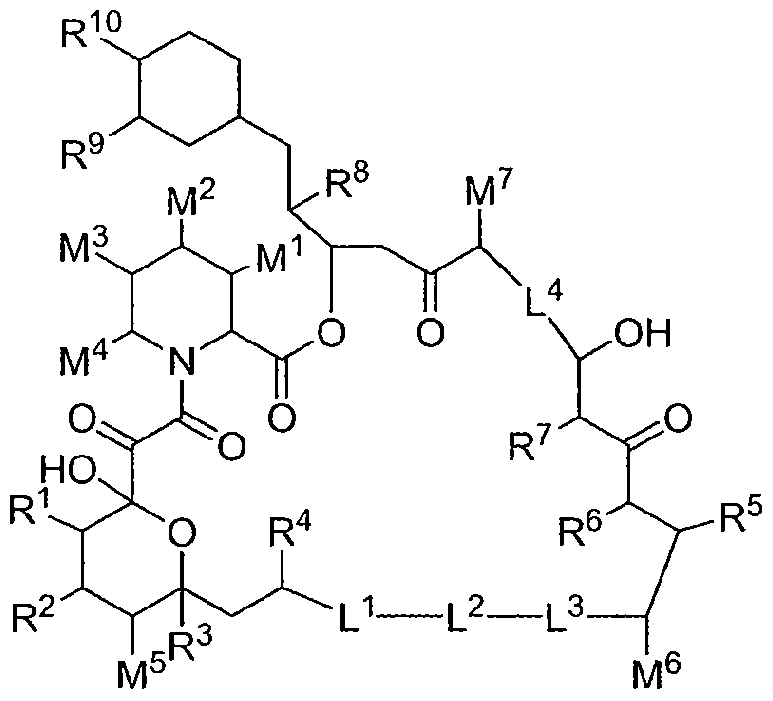



























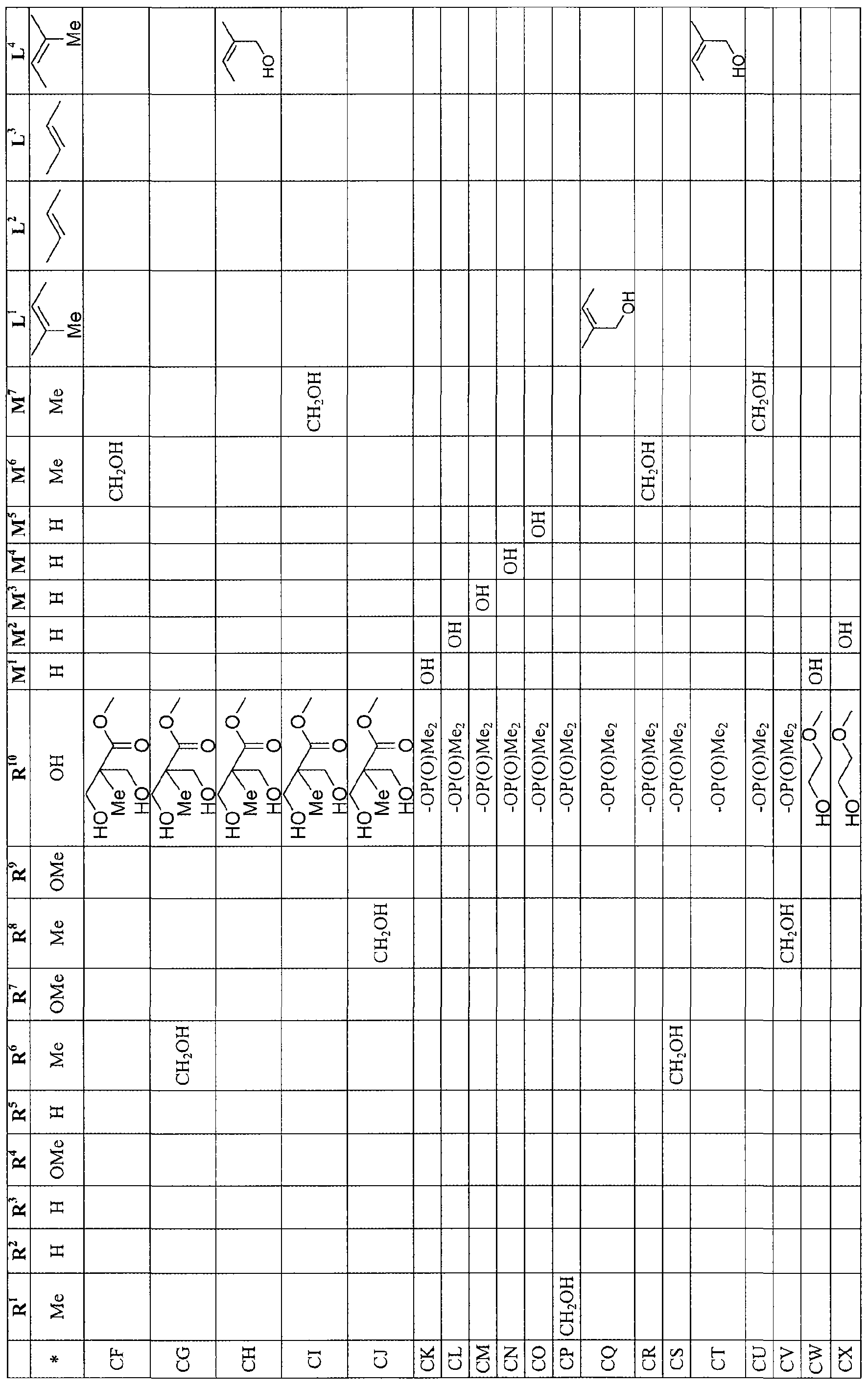













Claims




































Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP08731883A EP2254412A4 (en) | 2008-03-11 | 2008-03-11 | Macrocyclic lactone compounds and methods for their use |
| PCT/US2008/056501 WO2009114010A1 (en) | 2008-03-11 | 2008-03-11 | Macrocyclic lactone compounds and methods for their use |
| CN200880128027.1A CN102215682A (en) | 2008-03-11 | 2008-03-11 | Macrocyclic lactone compounds and methods for their use |
| JP2010550655A JP2011525890A (en) | 2008-03-11 | 2008-03-11 | Macrocyclic lactone compounds and methods for their use |
| BRPI0822405A BRPI0822405A8 (en) | 2008-03-11 | 2008-03-11 | METHOD FOR TREATMENT OF AN OPHTHALMIC CONDITION OR DISEASE, PHARMACEUTICAL COMPOSITION, DEVICE FOR INTRABODY USE, METHOD FOR INHIBITING CELL PROLIFERATION, COMPOUND, AND METHOD FOR MANUFACTURING A COMPOUND |
| IL208026A IL208026A0 (en) | 2008-03-11 | 2010-09-06 | Macrocyclic lactone compounds and methods for their use |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/US2008/056501 WO2009114010A1 (en) | 2008-03-11 | 2008-03-11 | Macrocyclic lactone compounds and methods for their use |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009114010A1 true WO2009114010A1 (en) | 2009-09-17 |
Family
ID=41065507
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2008/056501 WO2009114010A1 (en) | 2008-03-11 | 2008-03-11 | Macrocyclic lactone compounds and methods for their use |
Country Status (6)
| Country | Link |
|---|---|
| EP (1) | EP2254412A4 (en) |
| JP (1) | JP2011525890A (en) |
| CN (1) | CN102215682A (en) |
| BR (1) | BRPI0822405A8 (en) |
| IL (1) | IL208026A0 (en) |
| WO (1) | WO2009114010A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10123996B2 (en) | 2006-09-13 | 2018-11-13 | Elixir Medical Corporation | Macrocyclic lactone compounds and methods for their use |
| US10307292B2 (en) | 2011-07-18 | 2019-06-04 | Mor Research Applications Ltd | Device for adjusting the intraocular pressure |
| US10695327B2 (en) | 2006-09-13 | 2020-06-30 | Elixir Medical Corporation | Macrocyclic lactone compounds and methods for their use |
| US11654036B2 (en) | 2020-05-26 | 2023-05-23 | Elixir Medical Corporation | Anticoagulant compounds and methods and devices for their use |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103387572B (en) * | 2013-07-17 | 2015-09-30 | 成都雅途生物技术有限公司 | Everolimus defects inspecting reference marker and preparation method thereof |
| CN107759616B (en) * | 2016-08-23 | 2020-11-17 | 上海微创医疗器械(集团)有限公司 | Compound and preparation method and application thereof |
| CN113801104B (en) * | 2020-06-15 | 2023-12-08 | 鲁南制药集团股份有限公司 | Preparation method of hydrolysis impurity of ivermectin Mo Sida cyclic lactone |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5665772A (en) * | 1992-10-09 | 1997-09-09 | Sandoz Ltd. | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| US20060229711A1 (en) * | 2005-04-05 | 2006-10-12 | Elixir Medical Corporation | Degradable implantable medical devices |
| US20070015697A1 (en) * | 2005-07-18 | 2007-01-18 | Peyman Gholam A | Enhanced ocular neuroprotection and neurostimulation |
| US20070059336A1 (en) * | 2004-04-30 | 2007-03-15 | Allergan, Inc. | Anti-angiogenic sustained release intraocular implants and related methods |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ZA935112B (en) * | 1992-07-17 | 1994-02-08 | Smithkline Beecham Corp | Rapamycin derivatives |
| WO1994012655A1 (en) * | 1992-11-20 | 1994-06-09 | Smithkline Beecham Corporation | Process for producing rapamycin derivatives |
| EP1904056B1 (en) * | 2005-07-18 | 2009-04-29 | Minu, L.L.C. | Use of a macrolide to restore corneal sensation |
| BRPI0716950A2 (en) * | 2006-09-13 | 2013-10-29 | Elixir Medical Corp | PHARMACEUTICAL COMPOSITION, INTRACORPOSE DEVICE, METHOD FOR INHIBITING CELL, COMPOUND PROLIFERATION, AND METHOD FOR PREPARING A COMPOUND |
-
2008
- 2008-03-11 WO PCT/US2008/056501 patent/WO2009114010A1/en active Application Filing
- 2008-03-11 BR BRPI0822405A patent/BRPI0822405A8/en not_active IP Right Cessation
- 2008-03-11 EP EP08731883A patent/EP2254412A4/en not_active Withdrawn
- 2008-03-11 JP JP2010550655A patent/JP2011525890A/en active Pending
- 2008-03-11 CN CN200880128027.1A patent/CN102215682A/en active Pending
-
2010
- 2010-09-06 IL IL208026A patent/IL208026A0/en unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5665772A (en) * | 1992-10-09 | 1997-09-09 | Sandoz Ltd. | O-alkylated rapamycin derivatives and their use, particularly as immunosuppressants |
| US20070059336A1 (en) * | 2004-04-30 | 2007-03-15 | Allergan, Inc. | Anti-angiogenic sustained release intraocular implants and related methods |
| US20060229711A1 (en) * | 2005-04-05 | 2006-10-12 | Elixir Medical Corporation | Degradable implantable medical devices |
| US20070015697A1 (en) * | 2005-07-18 | 2007-01-18 | Peyman Gholam A | Enhanced ocular neuroprotection and neurostimulation |
Non-Patent Citations (2)
| Title |
|---|
| See also references of EP2254412A4 * |
| VIGNOT ET AL.: "mTOR-targeted therapy of cancer with rapamycin derivatives.", ANNALS OF ONCOLOGY ADVANCE ACCESS, 22 February 2005 (2005-02-22), pages 5,7,9, XP008145753 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10123996B2 (en) | 2006-09-13 | 2018-11-13 | Elixir Medical Corporation | Macrocyclic lactone compounds and methods for their use |
| US10695327B2 (en) | 2006-09-13 | 2020-06-30 | Elixir Medical Corporation | Macrocyclic lactone compounds and methods for their use |
| US10307292B2 (en) | 2011-07-18 | 2019-06-04 | Mor Research Applications Ltd | Device for adjusting the intraocular pressure |
| US11654036B2 (en) | 2020-05-26 | 2023-05-23 | Elixir Medical Corporation | Anticoagulant compounds and methods and devices for their use |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2011525890A (en) | 2011-09-29 |
| CN102215682A (en) | 2011-10-12 |
| BRPI0822405A8 (en) | 2018-06-05 |
| EP2254412A4 (en) | 2012-05-09 |
| EP2254412A1 (en) | 2010-12-01 |
| IL208026A0 (en) | 2010-12-30 |
| BRPI0822405A2 (en) | 2018-05-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10123996B2 (en) | Macrocyclic lactone compounds and methods for their use | |
| EP2431036B1 (en) | Macrocyclic lactone compounds and methods for their use | |
| JP5913541B2 (en) | Polycyclic lactone compound and method of using the same | |
| US20130230571A1 (en) | Macrocyclic lactone compounds and methods for their use | |
| EP2254412A1 (en) | Macrocyclic lactone compounds and methods for their use | |
| US20230165839A1 (en) | Macrocyclic lactone compounds and methods for their use | |
| CN105380947A (en) | Macrolide compound and use method thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880128027.1 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08731883 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2010550655 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2008731883 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008731883 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3791/KOLNP/2010 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: PI0822405 Country of ref document: BR Kind code of ref document: A2 Effective date: 20100910 |