WO2011024971A1 - ポリアクリル酸(塩)系吸水性樹脂およびその製造方法 - Google Patents
ポリアクリル酸(塩)系吸水性樹脂およびその製造方法 Download PDFInfo
- Publication number
- WO2011024971A1 WO2011024971A1 PCT/JP2010/064641 JP2010064641W WO2011024971A1 WO 2011024971 A1 WO2011024971 A1 WO 2011024971A1 JP 2010064641 W JP2010064641 W JP 2010064641W WO 2011024971 A1 WO2011024971 A1 WO 2011024971A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- water
- cooling
- absorbent resin
- cooling device
- stirring
- Prior art date
Links
Images



Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L15/00—Chemical aspects of, or use of materials for, bandages, dressings or absorbent pads
- A61L15/16—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons
- A61L15/22—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons containing macromolecular materials
- A61L15/24—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L15/00—Chemical aspects of, or use of materials for, bandages, dressings or absorbent pads
- A61L15/16—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons
- A61L15/42—Use of materials characterised by their function or physical properties
- A61L15/60—Liquid-swellable gel-forming materials, e.g. super-absorbents
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F120/00—Homopolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride, ester, amide, imide or nitrile thereof
- C08F120/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F120/04—Acids; Metal salts or ammonium salts thereof
- C08F120/06—Acrylic acid; Methacrylic acid; Metal salts or ammonium salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/04—Acids; Metal salts or ammonium salts thereof
- C08F220/06—Acrylic acid; Methacrylic acid; Metal salts or ammonium salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/24—Crosslinking, e.g. vulcanising, of macromolecules
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/24—Crosslinking, e.g. vulcanising, of macromolecules
- C08J3/245—Differential crosslinking of one polymer with one crosslinking type, e.g. surface crosslinking
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2300/00—Characterised by the use of unspecified polymers
- C08J2300/14—Water soluble or water swellable polymers, e.g. aqueous gels
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2333/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers
- C08J2333/02—Homopolymers or copolymers of acids; Metal or ammonium salts thereof
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Materials Engineering (AREA)
- Epidemiology (AREA)
- Hematology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Veterinary Medicine (AREA)
- Dispersion Chemistry (AREA)
- Processes Of Treating Macromolecular Substances (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Absorbent Articles And Supports Therefor (AREA)
- Solid-Sorbent Or Filter-Aiding Compositions (AREA)
Abstract
Description
上記冷却工程で用いられる冷却装置の内面積が、上記加熱表面架橋工程で用いられる加熱装置の内面積の0.25~0.95倍であるところに特徴を有している。
また、上記課題を解決するための本発明の吸水性樹脂粉末の連続製造方法(第2の方法)は、アクリル酸(塩)で単量体水溶液を調製する工程、該単量体水溶液の重合工程、重合時または重合後の含水ゲル状架橋重合体の細粒化工程、得られた粒子状の含水ゲル状架橋重合体の乾燥工程、乾燥物の粉砕および分級工程、分級後の加熱表面架橋工程、表面架橋後の冷却工程を含む吸水性樹脂粉末の連続製造方法であって、
上記冷却工程で用いられる冷却装置の内容積が、上記加熱表面架橋工程で用いられる加熱装置の内容積の0.25~0.95倍であるところに特徴を有している。
なお、好ましくは第1の方法と第2の方法は併用される。
(a)「吸水性樹脂」
「吸水性樹脂」とは、水膨潤性水不溶性の「高分子ゲル化剤(gelling agent)」を意味し、以下の物性を有するものをいう。すなわち、水膨潤性として無加圧下吸水倍率(CRC)が、5g/g以上のものである。CRCは好ましくは10~100g/g、さらに好ましくは20~80g/gである。また、水不溶性として水可溶分(Extractables)は、0~50質量%であることが必要である。水可溶分は、好ましくは0~30質量%、さらに好ましくは0~20質量%、特に好ましくは0~10質量%である。
「ポリアクリル酸(塩)」とは、任意にグラフト成分を含み、繰り返し単位として、アクリル酸(塩)を主成分とする重合体を意味する。具体的には、架橋剤を除く単量体として、アクリル酸(塩)を、必須に50~100モル%、好ましくは70~100モル%、さらに好ましくは90~100モル%、特に好ましくは実質100モル%含む重合体を意味する。重合体としての塩は、ポリアクリル酸塩を含み、好ましくは一価塩、より好ましくはアルカリ金属塩またはアンモニウム塩、さらに好ましくはアルカリ金属塩、特に好ましくはナトリウム塩を含む。なお、形状は特に問わないが、粒子または粉体が好ましい。
「EDANA」は、European Disposables and Nonwovens Associationsの略称であり、「ERT」は、欧州標準(ほぼ世界標準)の吸水性樹脂の測定方法(ERT/EDANA Recomended Test Method)の略称である。本明細書においては、特に断りのない限り、ERT原本(公知文献:2002年改定)に基づいて、吸水性樹脂の物性を測定する。
「CRC」とは、Centrifuge Retention Capacity(遠心分離機保持容量)の略称であり、無加圧下吸水倍率(単に「吸水倍率」とも称することもある)を意味する。具体的には、不織布袋中の吸水性樹脂0.200gを0.9質量%食塩水で30分、自由膨潤させた後、遠心分離機で250Gにて水切りした後の吸水倍率(単位;g/g)である。
「AAP」とは、Absorption Against Pressureの略称であり、加圧下吸水倍率を意味する。具体的には、吸水性樹脂0.900gを0.9質量%食塩水に1時間、1.9kPaでの荷重下で膨潤させた後の吸水倍率(単位;g/g)である。なお、本発明および実施例では4.8kPaで測定した。
「Extractables」とは、水可溶分量(可溶分)を意味する。具体的には、0.9質量%食塩水200mlに、吸水性樹脂1.000gを添加し、16時間攪拌した後、溶解したポリマー量をpH滴定で測定した値(単位;質量%)である。
「Residual Monomers」とは、吸水性樹脂中に残存しているモノマー量を意味する。具体的には、0.9質量%食塩水200cm3に吸水性樹脂1.000gを投入し2時間攪拌後、該水溶液に溶出したモノマー量を高速液体クロマトグラフィーで測定した値(単位;質量ppm)である。
「PSD」とは、Particle Size Distributionの略称であり、ふるい分級により測定される粒度分布を意味する。なお、質量平均粒子径および粒子径分布幅は欧州公告特許第0349240号明細書7頁25~43行や国際公開第2004/069915号に記載された「(1) Average Particle Diameter and Distribution of Particle Diameter」と同様の方法で測定する。
「pH」(ERT400.2-02):吸水性樹脂のpHを意味する。
荷重下または無荷重下における膨潤ゲルの粒子間を流れる液の流れを「通液性」という。この「通液性」の代表的な測定方法として、SFC(Saline Flow Conductivity)や、GBP(Gel Bed Permeability)がある。
「標準偏差」とは、データの散らばりの度合いを示す数値であり、n個からなるデータの値とその相加平均値との差、すなわち偏差の2乗を合計し、n-1で割った値の正の平方根をいう。変動に富む現象について、変動の度合いを知るために用いられる。なお、本明細書においては、目的とする所望の物性値に対する変動(振れ)を数値化するため、標準偏差を利用する。

本明細書において、範囲を示す「X~Y」は、「X以上Y以下」であることを意味する。また、質量の単位である「t(トン)」は、「Metric ton(メトリック トン)」であることを意味する。さらに、吸水性樹脂の物性の測定は、特に注釈のない限り、温度:20~25℃(単に「室温」、あるいは「常温」と称することもある)、相対湿度:40~50%の条件下で実施している。
(1)重合工程
(a)単量体(架橋剤を除く)
本発明の単量体は、上記のアクリル酸またはその塩を主成分としている。吸水特性や残存モノマーの低減の点から、重合体の酸基が中和されていることが好ましく、中和率は10~100モル%が好ましく、30~95モル%がより好ましく、50~90モル%がさらに好ましく、60~80モル%が特に好ましい。中和は重合後の重合体(含水ゲル)に行ってもよく、単量体に行ってもよいが、生産性やAAP向上の面などから、単量体を中和しておくことが好ましい。すなわち、本発明の単量体はアクリル酸部分中和塩を含む。
本発明では、吸水特性の観点から架橋剤(別称;内部架橋剤)を使用することが特に好ましい。架橋剤は物性面から、架橋剤を除く上記単量体に対して0.001~5モル%が好ましく、0.005~2モル%がより好ましく、0.01~1モル%がさらに好ましく、0.03~0.5モル%が特に好ましい。
アクリル酸の中和に用いられる塩基性物質としては、水酸化ナトリウム、水酸化カリウム、水酸化リチウムなどのアルカリ金属の水酸化物や炭酸(水素)ナトリウム、炭酸(水素)カリウムなどの炭酸(水素)塩等の一価塩基が好ましく、残存モノマー低減の点からアクリル酸アルカリ金属塩とすることが好ましく、水酸化ナトリウムでの中和塩が特に好ましい。なお、これらの中和処理での好ましい条件等は、国際公開2006/522181号に例示されており、該公報に記載の条件も本発明に適用され得る。中和温度は10~100℃が好ましく、30~90℃がより好ましい。この範囲内で適宜決定されるが、残存モノマー低減から後述の中和方法が好ましい。
これら単量体は、通常水溶液で重合され、その固形分濃度は通常10~90質量%であり、好ましくは20~80質量%、さらに好ましくは30~70質量%、特に好ましくは35~60質量%である。なお、重合は、飽和濃度を超えたスラリー(水分散液)で行ってもよいが、物性面から、好ましくは飽和濃度以下の水溶液で行う。
さらに、不飽和単量体水溶液は、単量体とともに、澱粉、ポリアクリル酸(塩)、ポリエチレンイミンなどの水溶性樹脂ないし吸水性樹脂を例えば0~50質量%、好ましくは0~20質量%、特に好ましくは0~10質量%、最も好ましくは0~3質量%有していてもよい。また、各種の発泡剤(炭酸塩、アゾ化合物、気泡など)、界面活性剤や後述の添加剤等を、例えば0~5質量%、好ましくは0~1質量%添加して、得られる吸水性樹脂や粒子状吸水剤の諸物性を改善してもよい。なお、その他成分を使用して得られたグラフト重合体(例;澱粉アクリル酸グラフト重合体)ないし吸水性樹脂組成物も、本発明ではポリアクリル酸(塩)系吸水性樹脂と総称する。
重合方法は、性能面や重合の制御の容易さから、噴霧重合または液滴重合でもよいが、好ましくは、通常、水溶液重合または逆相懸濁重合で行われる。従来、重合の制御や着色改善が困難であった水溶液重合が好ましく、連続水溶液重合が最も好ましい。特に1ラインで不飽和単量体水溶液を重合して吸水性樹脂を0.5t/hr以上、さらには1t/hr以上、よりさらには5t/hr以上、特に10t/hr以上の巨大スケールで製造するには、連続重合が好適に制御できる。よって好ましい連続重合として、連続ニーダー重合(例えば、米国特許第6987151号および同第670141号)、連続ベルト重合(例えば、米国特許第4893999号、同第6241928号および米国特許出願公開第2005/215734号)が挙げられる。
本発明で使用される重合開始剤としては、重合の形態によって適宜選択される。このような重合開始剤としては例えば、光分解型重合開始剤や熱分解型重合開始剤、レドックス系重合開始剤などのラジカル重合開始剤を例示できる。重合開始剤の使用量は上記単量体に対し、好ましくは0.0001~1モル%、より好ましくは0.001~0.5モル%の量使用される。
重合で得られた含水ゲル状架橋重合体(以下、「含水ゲル」と称することもある。)はそのまま乾燥を行っても良いが、重合時または重合後、必要により解砕機(ニーダー、ミートチョッパーなど)を用いてゲル解砕され粒子状(例えば、質量平均粒子径で0.1~5mm、さらには0.5~3mm)にされる。
本発明では残存モノマーの低減やゲル劣化防止(耐尿性)、黄変防止を達成するため、重合終了後にゲル細粒化工程を経て乾燥工程を行う。ゲル細粒化工程を経て乾燥を開始するまでの時間は、短いほど好ましい。すなわち、重合後の含水ゲル状架橋重合体は、重合機から排出後に、好ましくは1時間以内、より好ましくは0.5時間以内、さらに好ましくは0.1時間以内に乾燥を開始(乾燥機に投入)する。かかる時間とするためには、重合後にゲルの貯蔵工程を行うことなく、直接、細分化ないし乾燥することが好ましい。また、残存モノマーの低減や低着色を達成するため、重合後から乾燥開始までの含水ゲル状架橋重合体の温度は、好ましくは50~80℃、さらに好ましくは60~70℃に制御する。
上述の含水ゲル状架橋重合体を乾燥する工程後、必要により粒度を調整してもよい。後述の表面架橋での物性向上のため、好ましくは特定粒度にされる。粒度は重合工程(特に逆相懸濁重合の場合)、粉砕工程、分級工程、造粒工程、微粉回収工程などで適宜調整できる。以下、粒度は標準篩で規定(JIS Z8801-1(2000))される。
本発明の吸水性樹脂の製造方法では上記(1)~(4)を一例として得られた吸水性樹脂について、加熱表面架橋工程および特定の冷却工程を経ることを特徴とする。
この加湿混合工程は、上述の重合工程から分級工程を経て得られた吸水性樹脂粉体に、表面架橋剤を添加、混合する工程である。
本発明では乾燥後の表面架橋工程をさらに含む。本発明の製造方法は、高い加圧下吸水倍率(AAP)および通液性(SFC)の吸水性樹脂の製造方法や巨大スケール(特に1t/hr)での連続生産に適用され、特に吸水性樹脂の高温表面架橋に好適に適用できる。
本発明で用いることの出来る表面架橋剤としては、種々の有機または無機架橋剤を例示できるが、有機表面架橋剤が好ましく使用できる。表面架橋剤としては、得られる吸水性樹脂の物性面の点から、多価アルコール化合物、エポキシ化合物、多価アミン化合物またはそのハロエポキシ化合物との縮合物、オキサゾリン化合物、(モノ、ジ、またはポリ)オキサゾリジノン化合物、アルキレンカーボネート化合物が好ましい。特に高温での反応が必要な、多価アルコール化合物、アルキレンカーボネート化合物、オキサゾリジノン化合物からなる脱水反応性架橋剤が使用できる。脱水反応性架橋剤を使用しない場合、物性が低かったり、本発明の効果の差が現れにくかったりする場合もある。
また、上記有機表面架橋剤以外にイオン結合性の無機表面架橋剤(多価金属由来の架橋剤)を使用して、通液性などを向上させてもよい。使用できる無機表面架橋剤は、2価以上、好ましくは3価ないし4価の多価金属の塩(有機塩ないし無機塩)ないし水酸化物が挙げられる。使用できる多価金属としてはアルミニウム、ジルコニウムなどが挙げられ、乳酸アルミニムや硫酸アルミニムが使用可能である。これら無機表面架橋剤は有機表面架橋剤と同時または別途に使用される。多価金属による表面架橋は、国際公開第2007/121037号、同第2008/09843号、同第2008/09842号、米国特許第7157141号、同第6605673号、同第6620889号、米国特許出願公開第2005/0288182号、同第2005/0070671号、同第2007/0106013号、同第2006/0073969号に示されている。
表面架橋剤の使用量は、吸水性樹脂粒子100質量部に対して0.001~10質量部程度が好ましく、0.01~5質量部程度がより好ましい。表面架橋剤と共に、好ましくは水が使用され得る。使用される水の量は、吸水性樹脂粒子100質量部に対して0.5~20質量部が好ましく、より好ましくは0.5~10質量部の範囲である。無機表面架橋剤と有機表面架橋剤を併用する場合も、各々0.001~10質量部程度(より好ましくは0.01~5質量)使用することが好ましい。
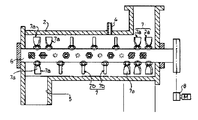
本発明では表面処理剤の混合に、連続高速回転攪拌型混合機を使用する。中でも横型の連続高速回転攪拌型混合機(例えば、図4、図5)が好適である。なお、表面処理剤とは上記表面架橋剤ないしその代替物(例えば、過硫酸塩などのラジカル重合開始剤、単量体)を指し、その溶液や分散液を含む概念である。攪拌速度は100~10000rpmが好ましく、300~2000rpmがより好ましい。滞留時間は180秒以内、さらには0.1~60秒、特に1~30秒程度が好ましい。
本発明においては、表面架橋工程や輸送管に供給される吸水性樹脂粒子(例えば、表面架橋剤を混合した後、表面架橋工程に導入されるまでの吸水性樹脂;粒子状吸水剤ともいう。)の温度は、好ましくは30℃以上、より好ましくは40℃以上、さらに好ましくは50℃以上である。また上限は100℃が好ましく、95℃がより好ましい。輸送管に供給される吸水性樹脂粒子(粒子状吸水剤)の温度を所定温度以上に保持することによって、粒子状吸水剤の物性の低下が抑制される。具体的には、生理食塩水流れ誘導性(SFC)などの物性維持に顕著な効果がある。
この加熱処理工程は、上記加湿混合工程で混合された、吸水性樹脂粉体と表面処理剤溶液との湿潤混合物を加熱し、表面架橋反応させる工程である。
(加熱装置)
本発明で用いられる加熱装置は、連続式が好ましく、溝型混合乾燥機、ロータリー乾燥機、ディスク乾燥機、流動床乾燥機、気流型乾燥機、赤外線乾燥機、パドル型乾燥機、振動流動乾燥機等が挙げられる。これらの中でも、物性向上および安定化の観点から、パドル型乾燥機が好ましい。
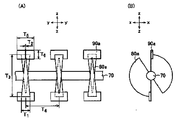
本発明の連続加熱装置としては、好ましくは、吸水性樹脂の投入口と排出口、および、複数の撹拌盤を備えた1本以上(好ましくは複数)の回転軸からなる撹拌手段と加熱手段とを有する横型連続撹拌装置(例えば、図1)が用いられる。
上記撹拌装置に表面処理剤を添加した後の吸水性樹脂には加熱処理を行う。必要な装置は上記横型連続撹拌装置である。攪拌動力指数の制御の面から、横型連続撹拌装置が0.1~10°の下向き傾斜角を有していることが好ましい。傾斜角は、0.5~5°がより好ましく、1~4°がさらに好ましい。傾斜角が上記範囲を満たさない場合、攪拌動力指数が過大または過小になり、吸水性樹脂の物性が低下することがある。
上記横型連続撹拌装置の縦横比(進行方向の装置の長さ/進行方向に断面の装置幅)は、1~20であることが好ましい。縦横比は、1.5~10がより好ましく、2~5がさらに好ましい。縦横比は装置内部の縦(進行方向)と横(進行方向に対して平面で直角)の長さの比で決定される。縦横比が上記範囲を満たさない場合、攪拌動力指数が過大または過小になり吸水性樹脂の物性が低下したり、あるいは装置内でのピストンフロー性が悪くなり、性能の安定性が悪くなることがある。
上記横型連続撹拌装置は、かき上げ羽根を有することが好ましい(かき上げ羽根は、例えば、図2の90a)。かき上げ羽根は上記特許文献31(特開2004-352941号公報)に記載されている。かき上げ羽根を用いれば攪拌動力指数が低く制御でき、その結果、吸水性樹脂の物性が向上する。
攪拌動力指数を上記好適範囲に制御する点からは、吸水性樹脂の平均滞留時間を0.05~2時間とすることが好ましい。平均滞留時間は0.1~1時間がより好ましく、0.2~0.8時間がさらに好ましい。
上記横型連続撹拌装置内部は平滑であることが好ましく、その表面粗さ(Rz)を800nm以下に制御する。この表面粗さ(Rz)は500nm以下が好ましく、300nm以下がより好ましく、200nm以下がさらに好ましく、185nm以下が特に好ましく、170nm以下が最も好ましい。横型連続攪拌装置内部の表面粗さ(Rz)が上記範囲を満たさない場合、吸水性樹脂粒子との摩擦抵抗が大きくなるため、攪拌動力指数が過大となり、物性が低下するおそれがある。
回転軸は1軸ないし複数、好ましくは2軸~10軸、特に2軸である。また、攪拌盤(例えば、図2)ないし攪拌羽根は、装置のサイズ(容量)によって適宜決定されるが、1軸あたり2~100枚、さらには5~50枚の範囲である。
表面架橋の物性安定や向上の面から、吸水性樹脂と表面処理剤溶液とが混合された後、撹拌装置へと導入される際に、横型の連続高速攪拌型混合機と横型撹拌装置内とを、周期的に遮蔽することが好ましい。周期的遮蔽の間隔は、0.001~5分が好ましく、0.005~1分がより好ましく、0.01~0.1分がさらに好ましく、0.01~0.05分が特に好ましい。周期的に遮蔽することにより、吸水性樹脂の、下流の連続装置への導入(混合機から加熱装置への導入、加熱装置から冷却装置への導入)を、周期的、すなわち間欠的(On-Off)に行うことができる。表面架橋工程で周期的遮蔽を行わない場合、得られる吸水性樹脂の物性が低下することがある。遮蔽率(吸水性樹脂が下流の連続装置へ遮蔽される時間の割合)としては、物性の安定化(標準偏差)の面から好ましくは1~80%、より好ましくは2~40%、さらに好ましくは5~30%、特に好ましくは5~20%、最も好ましくは5~10%の範囲である。周期的遮蔽を行っても、前記範囲(例えば1t/hr以上)の吸水性樹脂を次の装置へフィードできればよい。例えば、ロータリーバルブの場合、遮蔽間隔は回転数(rpm)の逆数(分)で定義され、遮蔽率は高速連続混合機から供給される混合物(湿潤粉体;吸水性樹脂と表面架橋剤溶液の混合物)を排出するのに必要なロータリーバルブの1分間当たりの理論回転数(rpm)(ロータリーバルブ1回転当たりの容積と排出される混合物の質量流量、嵩比重から求められる体積流量から得られる理論回転数)を実際のロータリーバルブの回転数(rpm)で割った値に100を乗じた値で定義される。なお、遮蔽率は、具体的には、混合機から単位時間当たりに排出される湿潤粉体(吸水性樹脂と表面架橋剤の混合物)を排出するのに必要なロータリーバルブの1分間当たりの回転数(rpm)を実際のロータリーバルブの回転数で割った値で規定できる。例えば、本実施例の場合で計算すると、1500×(1+3.5/100)/0.47/1000/0.02/60/25×100=11.0%となる。
表面処理剤が攪拌型混合機(例えば、図4、図5)に添加されて、吸水性樹脂と表面処理剤溶液とが混合された後は、加熱表面架橋処理を行う。加熱処理に必要な装置は好ましくは上記横型連続撹拌装置(例えば、図1)である。吸水性樹脂は加熱処理され、必要により第2の加熱処理を行った後、冷却処理される。加熱温度(ジャケットなどの伝熱面温度)は70~300℃、好ましくは120~250℃、より好ましくは150~250℃であり、加熱時間は、好ましくは1分~2時間の範囲である。加熱処理は、通常の乾燥機又は加熱炉で行うこともできる。本発明では、従来着色が激しかった高温加熱や空気(熱風)での乾燥でも、高度に白色の吸水性樹脂を提供することができる。
横型連続撹拌装置では、吸水性樹脂の充填率(容積比)は50~90%となるように連続供給されることが好ましい。充填率は、55~85%がより好ましく、60~80%がさらに好ましい。充填率が上記範囲を満たさない場合、攪拌動力指数が制御し難く、得られる吸水性樹脂の物性が低下することがある。充填率100%の位置は、先に述べたように、回転軸の攪拌盤の頂点部である。
(質量面積比)=(吸水性樹脂の単位時間あたりの質量流量)/(装置の伝熱面積)
本発明によれば、横型連続撹拌装置の撹拌速度を2~40rpmとすることで均一な加熱混合ができる。2rpmを下回ると、撹拌が不充分となり、一方、40rpmよりも速いと微粉が発生しやすくなる場合がある。より好ましい撹拌速度は5~30rpmである。また、装置内の滞留時間は、例えば10~180分、好ましくは20~120分である。10分未満では架橋反応が不充分となり易い。一方、180分を超えると吸水性能が低下することがある。
本発明では、横型連続撹拌装置内を微減圧とすることが好ましい。「減圧状態」とは、大気圧よりも気圧が低い状態を意味する。また「大気圧に対する減圧度」とは、大気圧との圧力差を意味し、気圧が大気圧よりも低い場合に正(プラス)の値として表現される。例えば、大気圧が標準大気圧(101.3kPa)である場合、「減圧度が10kPa」とは、気圧が91.3kPaであることを意味する。本願において、「大気圧に対する減圧度」は、単に「減圧度」とも称される。減圧にしない場合、混合機の吸気口から吸水性樹脂粉末がこぼれ出てしまうことがあり好ましくない。微減圧とすることで、吸水性樹脂からダスト(吸水性樹脂の超微粒子や必要により使用する無機微粒子)が除去でき、ダスト低減の観点からも好ましい。
上記横型連続撹拌装置内の雰囲気は空気でもよく、着色防止や燃焼防止ために、窒素などの不活性ガスでもよく、水蒸気が適宜追加されてもよい。また、温度や露点は適宜決定されるが、雰囲気温度(装置の上部空間のガス温度で規定)は30~200℃が好ましく、50~150℃がより好ましい。露点は0~100℃が好ましく、10~80℃がより好ましい。
(a)冷却温度
本発明においては、本発明の目的を達成するため、表面架橋後の加熱装置から取り出した吸水性樹脂を、好ましくは1分以内、さらに好ましくは30秒以内に冷却することが好ましい。従って、加熱装置と冷却装置は実質的に連結されていることが好ましい。また、物性面および生産性から、冷却開始時の吸水性樹脂の温度(材料温度で規定)は150~250℃であることが好ましく、冷却後の温度は40~100℃であることが好ましい。冷却後の温度は、50~90℃であることがより好ましく、50~80℃がさらに好ましい。
冷却に用いる装置としては、冷媒を用いて冷却装置内の温度、もしくは、吸水性樹脂粉末の温度をコントロールできる装置であれば特に限定はない。例えば、溝型混合冷却装置、ロータリー冷却装置、ディスク冷却装置、流動床冷却装置、パドル型冷却装置、振動流動冷却装置、気流併用パドル冷却装置等を挙げることができる。
すなわち、本発明では上記冷却装置および/または加熱装置が、吸水性樹脂の投入口と排出口、および、複数の撹拌盤を備えた1本以上の回転軸からなる撹拌手段と加熱手段とを有する横型連続撹拌装置であることが好ましく、その際、前記加熱装置に記載の好ましい攪拌動力指数、傾斜角(横型連続撹拌装置が0.1~10°、0.5~5°がより好ましく、1~4°がさらに好ましい)や縦横比(1.5~10がより好ましく、2~5がさらに好ましい)、かき上げ羽根、平均滞留時間、回転軸および攪拌盤などの好適条件は、上記内面積比ないし内容積比を維持する範囲において、そのまま冷却装置にも好ましく適用できる。
本発明では、冷却装置として、好ましくは、連続加熱装置と同様、吸水性樹脂の投入口と排出口、および、複数の撹拌盤を備えた1本以上の回転軸からなる撹拌手段と加熱手段とを有する横型連続撹拌装置が用いられる。なお、加熱手段には、高温の吸水性樹脂を冷却するための加熱手段が含まれる。冷却工程は、好ましくは連結された前記横型連続撹拌装置内(例えば、図3)で行う。
(攪拌動力指数)=((冷却時の装置の消費電力)-(空運転時の消費電力)×平均滞留時間)/(時間当たりの処理量×平均滞留時間)
特定の装置とその特定パラメーター(攪拌動力指数)によって、大スケール(特に1t/hr以上)へのスケールアップ時にも高物性の吸水性樹脂が連続的に安定的に得られる。攪拌動力指数は、冷却時の消費電力と空運転時の消費電力とから容易に求められる。攪拌動力指数が15W・hr/kgを超えると物性(特に通液性)が低下し、また、3W・hr/kgを下回っても物性(特に加圧下吸水倍率)が低下する。より好ましい攪拌動力指数は4~13W・hr/kgであり、さらに好ましくは5~11W・hr/kgであり、特に好ましくは5~10W・hr/kgであり、最も好ましくは5~9W・hr/kgの範囲である。
ここで前記した加熱装置(別称;加熱処理機、加熱機)の攪拌動力指数(4~13W・hr/kg、さらに好ましくは5~11W・hr/kg、特に好ましくは5~10W・hr/kg、最も好ましくは5~9W・hr/kg)と上記冷却装置の攪拌動力指数は同じでもよく異なってもよいが、物性面から好ましくは冷却装置(別称;冷却機)の攪拌動力指数の方が小さいことが好ましい。上記冷却装置の攪拌動力指数は、上記加熱装置の攪拌動力指数の0.99~0.25倍、さらには0.95~0.50倍、特に0.90~0.55倍の範囲であることが好ましい。
上記加熱装置と冷却装置において、本発明では冷却工程での冷却装置の内面積が表面架橋工程での加熱装置の内面積の0.25~0.95倍であることを特徴とする。すなわち、冷却装置の内面積を加熱装置の内面積よりも小さくする。ここで、内面積とは吸水性樹脂と接触する部分の面積で規定され、装置の内壁(側面)および底面からなる内面に加えて、攪拌羽根および攪拌軸を有する場合、それらとの接触面積を含めた合計面積である。攪拌羽根および攪拌軸を有する場合、装置側面(内面)は攪拌羽根および攪拌軸の最上部までの側面で規定される。なお、装置の内面の形状は、直方体に限定されず、円筒状やそれらの複合形状など各種形状が例示され、それらの側面および底面で内面積は規定できる。また内容積は有効容積(攪拌羽根までの容積)で規定できる。
本発明では、冷却装置の冷媒温度が冷却装置の雰囲気ガスの露点より10℃以上高く、冷却装置から排出される吸水性樹脂粒子の排出時温度より10℃以下低いことが好ましい。
冷却装置の空間に気流(気体の流れ)があることが好ましい。強制的な外部からの又は外部への通気がない場合、冷却後の吸水性樹脂に吸湿流動性(吸湿後の粉体流動性;Anti-Blocking性)が劣ってくるのみならず、さらに加圧下吸収倍率などの物性も安定しない。気流として、空気や不活性気体(窒素ガスなど)ないしそれらの混合物が用いられ、減圧、加圧、常圧のいずれの気流であってもよい。通常-50℃~100℃、好ましくは0~50℃、より好ましくは10~40℃の気流が冷却装置に通気されるように、冷却装置の外側に送風機構ないし減圧機構を有すればよい。尚、冷却装置によっては、回転攪拌軸(攪拌翼)を有するタイプのものもあるが、これらの回転は気流を生じせしめるものではない。本発明では、送風機構または減圧機構で、気流を通気する。気流を通気する方法としては、攪拌冷却する場合、冷却装置の一方の吸気口を開けて、別の口から吸引(減圧)する方法、あるいは、冷却装置の一方の排気口を開けて、別の口から送風(加圧)する方法、などが挙げられるが、特に限定されない。
(a)表面処理装置の数
攪拌動力指数や物性向上の面から、重合工程を連続ベルト重合または連続ニーダー重合で行い、かつ重合工程に対して、複数の表面処理工程が並列で行うことが好ましい。
本発明で表面架橋を2系列以上とするには分割工程を含み、好ましくは、粒子状含水ゲルまたはその乾燥物である粒子状吸水性樹脂の分割工程、より好ましくは、粒子状吸水性樹脂の分割工程を含む。
本発明で表面架橋物性の面から、表面架橋の前後に好ましくはホッパーが使用される。より好ましくは、逆角錐台形状や逆円錐台形状、ならびに逆角錐台の最大口径部分に同形状の角柱が付加された形状や逆円錐台の最大口径部分に同形状の円柱が付加されたホッパーが使用される。またその材質は特に限定されないが、ステンレス製が好ましく使用され、その表面粗さは好ましくは前記の範囲である。好適なホッパーやその形状はPCT/JP2009/54903号に例示され、かかるホッパーが推奨される。
表面架橋前後の吸水性樹脂の輸送方法は各種使用できるが、好ましくは、空気輸送が使用される。吸水性樹脂粒子および/または吸水性樹脂粉体の優れた物性が安定に保持されかつ閉塞現象が抑制されうるという観点から、一次空気及び必要により使用される二次空気(空気輸送中の追加空気)として、乾燥された空気を用いるのが好ましい。この空気の露点は通常-5℃以下であり、好ましくは-10℃以下であり、より好ましくは-12℃以下であり、特に好ましくは-15℃以下である。露点の範囲はコストパーフォマンスを考え、-100℃以上であり、-70℃以上であるのが好ましく、さらには-50℃程度で十分である。さらに、気体の温度は10~40℃、さらには15~35℃程度であることが好ましい。空気輸送時に用いる圧縮空気の露点を上記範囲内とすることで、特に、製品として包装する際、SFCの低下を抑えることができるため、好ましい。
上記以外に、必要により、蒸発した単量体のリサイクル工程、造粒工程、微粉除去工程、微粉リサイクル工程などを設けてもよい。さらには、経時色安定性効果やゲル劣化防止等のために、後述の添加剤を単量体ないしその重合物に使用してもよい。
(1)ポリアクリル酸(塩)系吸水性樹脂の物性
本発明のポリアクリル酸(塩)系吸水性樹脂を、衛生材料、特に紙おむつへの使用を目的とする場合、上記重合や表面架橋をもって、下記(a)~(e)の少なくとも1つ、さらにはAAPを含め2つ以上、特に3つ以上に制御されることが好ましい。下記を満たさない場合、後述の高濃度おむつでは十分な性能を発揮しないことがある。本発明の製造方法は下記の物性を達成する吸水性樹脂の製造に、より効果、特に物性の向上ないし安定化(小さい標準偏差)を発揮する。すなわち、下記目的物性の中でも、本発明の製造方法は好ましくは、吸水性樹脂の4.8kPa加圧下での0.9質量%の塩化ナトリウム水溶液に対する吸水倍率(AAP)が20g/g以上、0.69質量%生理食塩水流れ誘導性(SFC)が1(×10-7・cm3・s・g-1)以上、無加圧下吸水倍率(CRC)が20g/g以上である吸水性樹脂の製造方法、さらには下記範囲である製造方法に好適に適用され、物性が向上さらには安定化する。
おむつでのモレを防止するため、上記表面架橋とその後の冷却工程を達成手段の一例として、1.9kPaの加圧下さらには4.8kPaの加圧下での0.9質量%の塩化ナトリウム水溶液に対する吸水倍率(AAP)が、好ましくは20g/g以上、より好ましくは22g/g以上、さらに好ましくは24g/g以上に制御される。AAPは高いほど好ましいが、他の物性やコストとのバランスから、AAPの上限は1.9kPaなら40g/g、さらには4.8kPaなら30g/g程度でもよい。特に記載のない場合、AAPは4.8kPaでの値を示す。
おむつでのモレを防止するため、上記表面架橋とその後の冷却工程を達成手段の一例として、加圧下での液の通液特性である0.69質量%生理食塩水流れ誘導性SFC(米国特許5669894号で規定)は1(×10-7・cm3・s・g-1)以上、好ましくは25(×10-7・cm3・s・g-1)以上、より好ましくは50(×10-7・cm3・s・g-1)以上、さらに好ましくは70(×10-7・cm3・s・g-1)以上、特に好ましくは100(×10-7・cm3・s・g-1)以上に制御される。
無加圧下吸水倍率(CRC)は、好ましくは10g/g以上であり、より好ましくは20g/g以上、さらに好ましくは25g/g以上、特に好ましくは30g/g以上に制御される。CRCは、高いほど好ましく上限値は特に限定されないが、他の物性のバランスから、好ましくは50g/g以下、より好ましくは45g/g以下、さらに好ましくは40g/g以下である。
水可溶分量は好ましくは0~35質量%、より好ましくは25質量%以下であり、さらに好ましくは15質量%以下、特に好ましくは10質量%以下である。
上記重合を達成手段の一例として、残存モノマー(残存単量体)量は通常500質量ppm以下、好ましくは0~400質量ppm、より好ましくは0~300質量ppm、特に好ましくは0~200質量ppmを示す。
さらに、目的に応じて、酸化剤、酸化防止剤、水、多価金属化合物、シリカや金属石鹸等の水不溶性無機ないし有機粉末、消臭剤、抗菌剤、高分子ポリアミン、パルプや熱可塑性繊維などを吸水性樹脂中に0~3質量%、好ましくは0~1質量%添加してもよい。
本発明のポリアクリル酸(塩)系吸水性樹脂の用途は、特に限定されにないが、好ましくは、紙オムツ、生理ナプキン、失禁パット等の吸収性物品に使用され得る。特に、従来、原料由来の臭気、着色等が問題になっていた高濃度オムツ(1枚のオムツに多量の吸水性樹脂を使用したもの)に使用され、特に上記吸収性物品中の吸収体上層部に使用された場合に、特に優れた性能が発揮される。
以下、実施例によって本発明の効果が明らかにされるが、この実施例の記載に基づいて本発明が限定的に解釈されるべきではない。なお、以下におけるAAPやSFC等の測定方法は前述した通りである。また、下記物性(A)および(B)の測定方法については、下記に示す。なお、特に断りのない限り、粉砕工程、表面架橋工程および冷却工程は微減圧(0.28~0.31kPa)で行った。
冷却装置から排出された吸水性樹脂粉体を、850μmのふるいで分級し、850μm以上の凝集物の有無を、目視で確認、評価した。
連続運転中に冷却装置内の吸水性樹脂粒子および/またはその凝集物の付着状態を目視で評価した。
◎:全く付着物なし。
○:殆ど付着せず、付着した場合でもセルフクリーニングで付着が取れる。
ポリエチレングリコールジアクリレート(エチレングリコールの付加モル数:n=9)0.05モル%(対単量体)を内部架橋剤として含む、アクリル酸部分中和ナトリウム塩水溶液(中和率:71モル%、単量体濃度:38質量%)に過硫酸ナトリウムとL-アスコルビン酸とをラインミキシングにて連続混合した。このラインミキシングにおいて、過硫酸ナトリウムの混合比率は、単量体1モル当たり0.12gとし、L-アスコルビン酸の混合比率は、単量体1モル当たり0.005gとした。水平スチールベルト重合機に連続的に供給し、連続的に静置水溶液重合(ベルト滞留時間:約30分、厚み:約25mm)し、得られた含水ゲル状架橋重合体をミートチョッパーで粒子状に解砕し、これをバンド乾燥機の多孔板上に薄く広げて載せ、160~180℃で30分間連続熱風乾燥した。乾燥機出口でブロック状の乾燥重合体が得られた。得られた乾燥重合体を1.1t/hrで3段ロールグラニュレーターに連続供給することで粉砕した。得られた約60℃の吸水性樹脂粒子を、目開き850μmのふるい網を有する篩い分け装置で分級し、粒径が150~850μmの粒子含有量が90質量%以上である吸水性樹脂粒子(1)(質量平均粒子径:400μm)を得た。
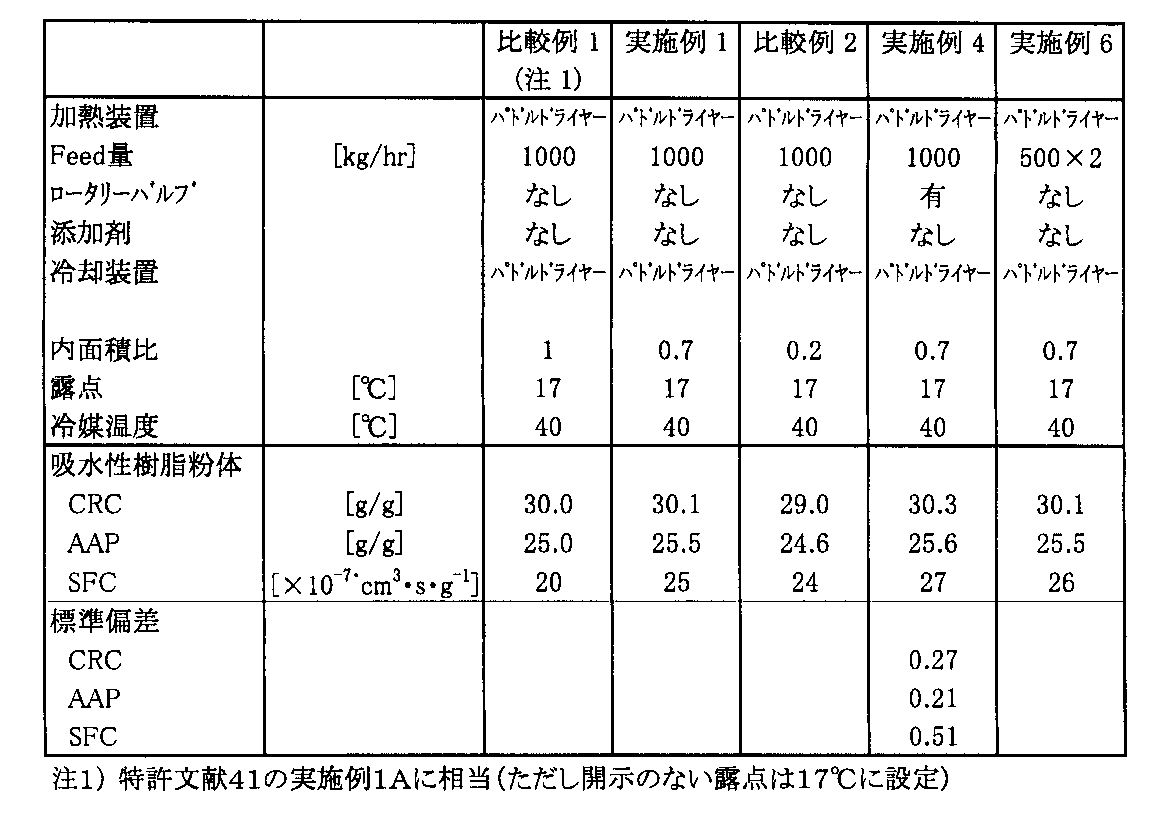
製造例1のパドルドライヤー(奈良機械(株)製)による加熱処理に引き続き、さらに直列に接続された同一形式のパドルドライヤーに40℃の冷媒(冷水)を流した。冷却装置の上部空間にわずかに吸引気流を通して内部を100mmH2Oの減圧としながら、冷却装置(ジャケットは40℃)により吸水剤粉末の冷却を行った。冷却後に850μmのふるいで分級した結果、得られた吸水性樹脂粉末の吸湿時の流動性は優れ、物性も時間ごとに安定し、さらに微粉(100μm以下)も実質なく、また、製造時の凝集も見られなかった。CRC,AAP,SFCの評価結果を表1に示す。なお、冷却時に気流によって吸水性樹脂微粒子の一部を除去した。
比較例1において、冷却装置として、加熱装置(パドルドライヤー)とほぼ相似形で、その内面積が加熱装置の0.7倍のパドルドライヤーを用いた以外は、同様の操作を行った。結果を表1に示す。
比較例1において、冷却装置として、加熱装置(パドルドライヤー)とほぼ相似形で、その内面積が加熱装置の0.2倍のパドルドライヤーを用いた以外は、結果を表1に示す。
上記製造例1で得られた吸水性樹脂粒子(1)を3.6kg/hrで連続的に高速混合機に供給し、吸水性樹脂粒子100質量部に対して、1,4-ブタンジオール/プロピレングリコール/水=0.32/0.5/2.73質量部からなる水性液(表面処理剤と称す)を高速混合機中で噴霧添加した。さらに表面処理剤が噴霧添加された湿潤混合物を、平均滞留時間が約40分になるように出口堰高さを調整した、総内容積4.6Lのパドル型低速攪拌型間接加熱乾燥機((株)栗本鐵工所製CDドライヤーCD-80型、以下、加熱装置と称す)に連続的に供給した。熱媒温度は212℃であった。
比較例3において、冷却装置(CDドライヤー)の内面積を、加熱装置(CDドライヤー)の0.7倍(ほぼ相似形に近い状態で縮小したもの)に変更した以外は、同様の操作を行った。結果を表2に示す。
比較例3において、冷却装置(CDドライヤー)の内面積を、加熱装置(CDドライヤー)の0.2倍(ほぼ相似形に近い状態で縮小したもの)に変更した以外は、同様の操作を行った。結果を表2に示す。
実施例1において、0.6g/分(対吸水性樹脂粒子(1)で1質量%)で噴霧添加する水を硫酸アルミニウム16水和物の50%水溶液に変更することで、共有結合性架橋剤およびイオン結合性架橋剤(硫酸アルミニウム)で架橋された吸水性樹脂を得た。ここで、表2にあるように、イオン結合性架橋剤は通液性の向上に寄与すると推定される。
比較例5において、冷却装置(パドルドライヤー)の内面積を、加熱装置(パドルドライヤー)の0.7倍(ほぼ相似形に近い状態で縮小したもの)に変更した以外は、同様の操作を行った。結果を表2に示す。
実施例1において、周期的遮蔽装置(ロータリーバルブ)を冷却装置(パドルドライヤー)の出入口に設置した。結果を表1に示す。なお、ロ-タリーバルブの1回転当たりの容積は0.02[m3/lev(1回転)]、回転数25rpmであった。遮蔽間隔は0.04分(回転数の逆数で定義、25rpmのため、1/25=0.04分)であった。
実施例2において、実施例4と同様の周期的遮蔽装置(ロータリーバルブ)を冷却装置の出入口に設置した。結果を表2に示す。
実施例1において、表面架橋を並列2系列(500kg/hr×2系列)とする以外は同様に行った。結果を表1に示す。

実施例1において、導入する空気の露点を40℃に調整した。吸水性樹脂の一部に凝集が見られ、内部の一部に付着が見られた。実施例1の露点(好ましくは冷媒温度と10℃以上の差)が好適であったことが分かる。
実施例1において、冷媒を10℃に調整した。吸水性樹脂の一部に凝集が見られ、内部の一部に付着が見られた。実施例1の冷媒温度(好ましくは30℃以上の温水)が好適であったことが分かる。
実施例2で得られた吸水性樹脂粉体を、内面の表面粗さ(Rz)200nmの配管内に圧縮空気(露点-15℃、温度35℃)を通すことで、該吸水性樹脂粉体を空気輸送して包装した。該空気輸送後のSFCは29.5であり、SFC低下率は1.7%であった。
露点20℃の圧縮空気を使用した以外は、実施例9と同様の空気輸送を行った。該空気輸送後のSFCは28.5であり、SFC低下率は5.0%であった。
表1、2にあるように、冷却装置の容量(内面積)を制御することで、加圧下吸水倍率や通液性、特に通液性が向上する。また、周期的遮蔽装置の併用や、表面架橋や冷却の並列(分割)、冷却装置へ導入する気体の露点の制御で、物性が向上し、かつ安定化する。
重合工程(ベルト上での静置重合)、ゲル細粒化工程(解砕工程)、乾燥工程、粉砕工程、分級工程及び各工程間の輸送工程の各装置が接続され、各工程を連続して行うことができるポリアクリル酸(塩)系吸水性樹脂の連続製造装置を用いた。この連続製造装置の生産能力は、1時間あたり約1500kgである。この連続製造装置を用いて、吸水性樹脂粒子を連続製造した。
先ず、単量体水溶液(2)として、75モル%が中和されたアクリル酸部分ナトリウム塩の水溶液を調製した。この単量体水溶液(2)は、内部架橋剤としてのポリエチレングリコールジアクリレート(平均n数9)を単量体の全モル数に対して0.055モル%含んでいた。上記単量体水溶液(2)において、上記単量体(上記アクリル酸部分ナトリウム塩)の濃度は、40質量%であった。得られた単量体水溶液(2)を定量ポンプでベルト上へ連続フィードした。フィードに用いた配管の途中で窒素ガスを連続的に配管内に吹き込み、単量体水溶液(2)における溶存酸素濃度を0.5mg/L以下にした。なお、上記「平均n数」とは、ポリエチレングリコール鎖中のメチレン鎖重合度の平均数を意味する。
次に、単量体水溶液(2)に、過硫酸ナトリウム0.10g/モルとL-アスコルビン酸0.005g/モルとをラインミキシングにて連続混合した。このラインミキシングにおいて、過硫酸ナトリウムの混合比率は、単量体1モル当たり0.12gとし、L-アスコルビン酸の混合比率は、単量体1モル当たり0.005gとした。このラインミキシングにより得られた連続混合物を、両端に堰を有する平面スチールベルトに厚み約30mmで供給して、連続的に30分間静置水溶液重合を行い、含水ゲル状架橋重合体(2)を得た。この含水ゲル状架橋重合体(2)を孔径7mmのミートチョッパーで約2mmに細粒化し、これを連続通風バンド乾燥機の移動する多孔板上に厚みが50mmとなるように広げて載せ、185℃で30分間乾燥し、乾燥重合体を得た。ここで重合機出口から乾燥機入口までの時間は1分以内であった。当該乾燥重合体の全量を3段ロールミルに連続供給することで粉砕した。この3段ロールミルのロールギャップは、上から順に、1.0mm/0.55mm/0.42mmであった。この粉砕の後、目開き850μmおよび150μmの金属篩網を有する篩い分け装置で分級して、150~850μmの粒子が約98質量%の吸水性樹脂粒子(2)を得た。この吸水性樹脂粒子(2)のCRCは35g/gであり、嵩比重は0.6g/cm3であった。
製造例2で使用した連続製造装置から引き続いて、表面処理工程(加湿混合工程、加熱工程および冷却工程)、整粒工程および各工程間を連結する輸送工程からなる連続製造装置を用いて、吸水性樹脂粉体(11)を製造した。すなわち、製造例2の分級工程と、表面処理工程は、輸送工程により連結されている。
吸水性樹脂粒子(2)を分級機から空気輸送(温度35℃、露点-15℃)で一時貯蔵ホッパーへと空気輸送し、定量フィーダーを経て高速連続混合機(タービュライザー;1000rpm;例えば、図4)に1.5t/hrで連続供給しつつ、表面処理剤溶液(11)をスプレーで噴霧し混合した(加湿混合工程)。この表面処理剤溶液(11)は、1,4-ブタンジオール、プロピレングリコール及び純水の混合液であった。この表面処理剤溶液(11)は、吸水性樹脂粒子(2)100質量部に対して、1,4-ブタンジオール0.3質量部、プロピレングリコール0.5質量部及び純水2.7質量部の割合で吸水性樹脂粒子(2)に混合され、湿潤粉体である混合物(11)とされた。
次いで、得られた混合物(11)を1°の下向き傾斜角を有し、縦横比2.2、パドル回転数13rpm、2本の回転軸と、かき上げ羽根を有する撹拌盤とを有し、内面の表面粗さ(Rz)が500nmの横型連続攪拌装置(11)により表面処理を行った(加熱処理工程)。このとき、装置(11)内を、バグフィルターを備えた吸引排気装置によって吸引し、装置内を1kPaの減圧とした。また、上記装置(11)の入口(混合機との連結部)および出口(冷却機との連結部)にはロータリーバルブ(周期的遮蔽装置)を設置した。事前のテストによって、平均滞留時間45分、平均充填率75%となる排出堰の位置を把握しておき、そのように、排出堰の位置を設定した。表面処理に用いた加熱源は、2.5MPaの加圧蒸気であり、上記横型連続攪拌装置(11)の排出部付近に設けられた温度計により装置内の混合物(11)温度を測定し、その温度が200℃になるように蒸気流量を制御して加熱を行った。攪拌盤と攪拌軸の総表面積は24.4m2であり、この総表面積(伝熱面積)と処理量とから計算される質量面積比は61.5kg/m2/hrであった。また、表面処理時の攪拌動力は27.8kW、空運転での攪拌動力は13.5kW、平均滞留時間45分であり、攪拌動力指数は9.5W・hr/kgであった。
次いで、加熱装置である横型連続攪拌装置(11)に対して相似形で内面積が0.80倍である小型の横型連続攪拌装置(冷却装置)を用いて40℃の温水流量を調整することで、吸水性樹脂を60℃まで強制冷却した(冷却工程)。攪拌動力指数は7.4W・hr/kgであった。
さらに、60℃で排出された吸水性樹脂を篩い分け装置で850μm通過物を分級し、850μm on品(850μm非通過物)は再度粉砕したのち、前記850μm通過物と混合することで、全量が850μm通過物である整粒された製品としての吸水性樹脂粉体(11)を得た。
得られた吸水性樹脂粉体(11)のCRCは30.7(g/g)、SFCは29.9(×10-7・cm3・s・g-1)、AAPは25.2(g/g)、嵩比重は0.68g/cm3であった。また、各物性値の標準偏差は、CRC:0.16、SFC:0.48、AAP:0.13であった。なお、これらの物性値は、運転開始後5時間を経過するまで、1時間ごとにサンプリング(5点)を行って求めた測定値の平均値である。
実施例11において、相似形で内面積が0.80倍の小型の横型連続攪拌装置に代えて、加熱装置である横型連続攪拌装置(11)と同じ大きさの横型連続攪拌装置(冷却装置)を用いる以外は実施例11と同様に行った。冷却装置の内面積が加熱表面架橋工程で用いられる加熱装置の内面積の1.0倍とすることで、SFCは28.1(×10-7・cm3・s・g-1)、AAPは24.7(g/g)に低下した。
実施例11において、相似形で内面積が0.80倍の小型の横型連続攪拌装置に代えて、ほぼ相似形で0.1倍の小型の横型連続攪拌装置(冷却装置)を用いる以外は実施例11と同様に行った。冷却装置の内面積が加熱表面架橋工程で用いられる加熱装置の内面積の0.1倍としたため、表面架橋反応の停止が十分ではなく、吸水性樹脂の一部に凝集が見られ、さらに物性も安定しなかった。
上記特許文献1~41に比べて、吸水性樹脂の物性も向上し、安定化(標準偏差の低下)が行える。冷却工程の改良技術として特許文献41など知られているが、かかる装置面の改良技術は本願を示唆せず、本願の比較例に相当する。
20 横型ドラム
30 原料供給口
40 熱媒入口
40’ 熱媒入口
45’ 熱媒出口
50 吸水性樹脂排出口
70 回転軸
80 攪拌盤
80a 攪拌盤
80b 攪拌盤
81 キャリアーガス導入口
85 排気口
90 かきあげ羽根
90a かきあげ羽根
90b かきあげ羽根
100 攪拌装置(攪拌手段)
Claims (16)
- アクリル酸(塩)で単量体水溶液を調製する工程、該単量体水溶液の重合工程、重合時または重合後の含水ゲル状架橋重合体の細粒化工程、得られた粒子状の含水ゲル状架橋重合体の乾燥工程、乾燥物の粉砕工程および分級する工程、分級後の加熱表面架橋工程、表面架橋後の冷却工程を含む、ポリアクリル酸(塩)系吸水性樹脂の製造方法であって、
冷却工程で用いられる冷却装置の内面積が、加熱表面架橋工程で用いられる加熱装置の内面積の0.25~0.95倍であることを特徴とする、ポリアクリル酸(塩)系吸水性樹脂の製造方法。 - アクリル酸(塩)で単量体水溶液を調製する工程、該単量体水溶液の重合工程、重合時または重合後の含水ゲル状架橋重合体の細粒化工程、得られた粒子状の含水ゲル状架橋重合体の乾燥工程、乾燥物の粉砕工程および分級する工程、分級後の加熱表面架橋工程、表面架橋後の冷却工程を含む、ポリアクリル酸(塩)系吸水性樹脂の製造方法であって、
冷却工程で用いられる冷却装置の内容積が、加熱表面架橋工程で用いられる加熱装置の内容積の0.25~0.95倍であることを特徴とする、ポリアクリル酸(塩)系吸水性樹脂の製造方法。 - 上記冷却装置および/または加熱装置が、吸水性樹脂の投入口と排出口、および、複数の撹拌盤を備えた1本以上の回転軸からなる撹拌手段と加熱手段とを有する横型連続撹拌装置である、請求項1または2に記載の製造方法。
- 上記冷却工程での冷却装置の冷媒が、冷却装置内の気体の露点以上の温度に加熱されてなる、請求項1~3のいずれか1項に記載の製造方法。
- 上記冷却装置の冷媒温度が、冷却装置内の気体の露点より10℃以上高く、かつ、冷却装置から排出される吸水性樹脂粒子の排出時温度より10℃以下低い、請求項1~4のいずれか1項に記載の製造方法。
- 上記冷却装置の冷媒が30℃以上の温水である、請求項1~5のいずれか1項に記載の製造方法。
- 上記冷却装置が上記横型連続撹拌装置であり、かつ、該装置の攪拌動力指数が3~11W・hr/kgである、請求項3~6のいずれか1項に記載の製造方法。
- 上記冷却装置および上記加熱装置が上記横型連続撹拌装置であり、冷却装置の攪拌動力指数が加熱装置に比べて0.25~0.95倍である、請求項2~7のいずれか1項に記載の製造方法。
- 上記加熱表面架橋工程および/または冷却工程の吸水性樹脂の温度を測定する工程、冷媒温度または流量を制御する工程を含む、請求項1~8のいずれか1項に記載の製造方法。
- 上記冷却装置および/または加熱装置の内面が、表面粗さ800nm以下のステンレス鋼である、請求項1~9のいずれか1項に記載の製造方法。
- 上記表面架橋工程前および/または冷却工程後の吸水性樹脂粒子が、露点-5℃~-100℃で空気輸送されてなる、請求項1~10のいずれか1項に記載の製造方法。
- 上記重合工程一系列に対して、表面架橋工程が二系列以上である、請求項1~11のいずれか1項に記載の製造方法。
- 上記分級工程、表面架橋工程および冷却工程が減圧である、請求項1~12のいずれか1項に記載の製造方法。
- 上記加熱表面架橋工程で、表面架橋剤として、共有結合性架橋剤およびイオン結合性架橋剤とを併用する、請求項1~13のいずれか1項に記載の製造方法。
- 上記冷却装置が、その入口が周期的遮蔽された連続冷却装置である、請求項1~14のいずれか1項に記載の製造方法。
- 上記表面架橋前および冷却後の吸水性樹脂が、いずれも40~100℃の範囲である、請求項1~15のいずれか1項に記載の製造方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP10812020.5A EP2471843B1 (en) | 2009-08-27 | 2010-08-27 | Method for producing polyacrylic acid (salt) water absorbent resin |
| JP2011528882A JP5619010B2 (ja) | 2009-08-27 | 2010-08-27 | ポリアクリル酸(塩)系吸水性樹脂およびその製造方法 |
| US13/392,256 US9138505B2 (en) | 2009-08-27 | 2010-08-27 | Polyacrylic acid (salt)-type water absorbent resin and method for producing of same |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009196967 | 2009-08-27 | ||
| JP2009-196967 | 2009-08-27 | ||
| JP2009-197091 | 2009-08-27 | ||
| JP2009197063 | 2009-08-27 | ||
| JP2009-197063 | 2009-08-27 | ||
| JP2009197091 | 2009-08-27 | ||
| JP2009-197022 | 2009-08-27 | ||
| JP2009197022 | 2009-08-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011024971A1 true WO2011024971A1 (ja) | 2011-03-03 |
Family
ID=43628069
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/064646 WO2011024975A1 (ja) | 2009-08-27 | 2010-08-27 | ポリアクリル酸(塩)系吸水性樹脂およびその製造方法 |
| PCT/JP2010/064645 WO2011024974A1 (ja) | 2009-08-27 | 2010-08-27 | ポリアクリル酸(塩)系吸水性樹脂およびその製造方法 |
| PCT/JP2010/064641 WO2011024971A1 (ja) | 2009-08-27 | 2010-08-27 | ポリアクリル酸(塩)系吸水性樹脂およびその製造方法 |
| PCT/JP2010/064643 WO2011024972A1 (ja) | 2009-08-27 | 2010-08-27 | ポリアクリル酸(塩)系吸水性樹脂およびその製造方法 |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/064646 WO2011024975A1 (ja) | 2009-08-27 | 2010-08-27 | ポリアクリル酸(塩)系吸水性樹脂およびその製造方法 |
| PCT/JP2010/064645 WO2011024974A1 (ja) | 2009-08-27 | 2010-08-27 | ポリアクリル酸(塩)系吸水性樹脂およびその製造方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/064643 WO2011024972A1 (ja) | 2009-08-27 | 2010-08-27 | ポリアクリル酸(塩)系吸水性樹脂およびその製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (4) | US8907021B2 (ja) |
| EP (4) | EP2471843B1 (ja) |
| JP (4) | JP5629688B2 (ja) |
| CN (2) | CN102482434B (ja) |
| WO (4) | WO2011024975A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015046604A1 (ja) | 2013-09-30 | 2015-04-02 | 株式会社日本触媒 | 粒子状吸水剤の充填方法および粒子状吸水剤充填物のサンプリング方法 |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5629688B2 (ja) * | 2009-08-27 | 2014-11-26 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂およびその製造方法 |
| WO2011099586A1 (ja) | 2010-02-10 | 2011-08-18 | 株式会社日本触媒 | 吸水性樹脂粉末の製造方法 |
| WO2011111857A1 (ja) * | 2010-03-12 | 2011-09-15 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| US9415529B2 (en) | 2012-05-08 | 2016-08-16 | Basf Se | Method for operating an apparatus with at least one rotating shaft |
| EP2890411B1 (en) | 2012-08-29 | 2021-10-06 | Basf Se | Process for producing water-absorbing polymer particles |
| CN107376866B (zh) * | 2012-09-11 | 2021-06-01 | 株式会社日本触媒 | 聚丙烯酸(盐)系吸水剂的制造方法及其吸水剂 |
| WO2014041968A1 (ja) * | 2012-09-11 | 2014-03-20 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水剤の製造方法及びその吸水剤 |
| CN110698696A (zh) | 2012-12-03 | 2020-01-17 | 株式会社日本触媒 | 聚丙烯酸(盐)系吸水性树脂及含有其的物品 |
| US10662300B2 (en) | 2013-05-10 | 2020-05-26 | Nippon Shokubai Co., Ltd. | Method for producing polyacrylic acid (salt)-based water-absorbent resin |
| CN105452303B (zh) * | 2013-08-28 | 2017-04-26 | 株式会社日本触媒 | 吸水性树脂的制造方法 |
| CN106255708B (zh) * | 2014-04-25 | 2019-12-27 | 株式会社日本触媒 | 聚丙烯酸(盐)系吸水性树脂的制造方法 |
| JP6425341B2 (ja) * | 2014-12-26 | 2018-11-21 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂の製造方法 |
| JP6722507B2 (ja) * | 2015-05-14 | 2020-07-15 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| US11465126B2 (en) | 2016-11-16 | 2022-10-11 | Nippon Shokubai Co., Ltd. | Method for producing water-absorbent resin powder and production apparatus therefor |
| CN106749803B (zh) * | 2016-12-28 | 2019-02-19 | 山东昊月新材料股份有限公司 | 一种提高sap制备效率的工艺方法 |
| CN107892169A (zh) * | 2017-12-06 | 2018-04-10 | 佛山市尊聚自动化设备有限公司 | 一种钢管分离送料机构 |
| JP7335051B2 (ja) * | 2019-10-07 | 2023-08-29 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
| CN110724224A (zh) * | 2019-10-22 | 2020-01-24 | 江苏厚生新能源科技有限公司 | 应用于萃取槽除水的吸水树脂及其制备工艺、萃取装置 |
| CN111138003A (zh) * | 2020-01-08 | 2020-05-12 | 柯侨宾 | 一种社区污水处理设备用预处理机构 |
| US20230390735A1 (en) * | 2021-06-18 | 2023-12-07 | Lg Chem, Ltd. | Preparation method of superabsorbent polymer |
Citations (80)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US670141A (en) | 1900-11-20 | 1901-03-19 | William Otis Shepard | Policeman's club. |
| JPS5410387A (en) * | 1977-06-24 | 1979-01-25 | Sanyo Chem Ind Ltd | Preparation of water-soluble polymer gel |
| US4734478A (en) | 1984-07-02 | 1988-03-29 | Nippon Shokubai Kagaku Kogyo Co., Ltd. | Water absorbing agent |
| US4755562A (en) | 1986-06-10 | 1988-07-05 | American Colloid Company | Surface treated absorbent polymers |
| US4783510A (en) | 1986-06-04 | 1988-11-08 | Taiyo Fishery Co., Ltd. | Process for improving a water absorbent polyacrylic acid polymer and an improved polymer produced by said process |
| EP0349240A2 (en) | 1988-06-28 | 1990-01-03 | Nippon Shokubai Co., Ltd. | Water-absorbent resin and production process |
| US4893999A (en) | 1985-12-18 | 1990-01-16 | Chemische Fabrik Stockhausen Gmbh | Apparatus for the continuous production of polymers and copolymers of water-soluble monomers |
| JPH0249002A (ja) * | 1988-05-23 | 1990-02-19 | Nippon Shokubai Kagaku Kogyo Co Ltd | 親水性重合体の製造方法 |
| JPH03168218A (ja) * | 1989-11-28 | 1991-07-22 | Sumitomo Chem Co Ltd | 加硫方法 |
| US5140076A (en) | 1990-04-02 | 1992-08-18 | Nippon Shokubai Kagaku Kogyo Co., Ltd. | Method of treating the surface of an absorbent resin |
| EP0534228A1 (de) | 1991-09-18 | 1993-03-31 | Hoechst Aktiengesellschaft | Verfahren zum Modifizieren hydrophiler Polymerisate |
| US5206205A (en) | 1991-08-15 | 1993-04-27 | Kimberly-Clark Corporation | Thermal treatment of superabsorbents to enhance their rate of absorbency under load |
| JPH05112654A (ja) * | 1991-04-10 | 1993-05-07 | Nippon Shokubai Co Ltd | 粒子状含水ゲル状重合体および吸水性樹脂の製造方法 |
| EP0603292A1 (en) | 1991-09-09 | 1994-06-29 | The Dow Chemical Company | Superabsorbent polymers and process for producing |
| US5409771A (en) | 1990-06-29 | 1995-04-25 | Chemische Fabrik Stockhausen Gmbh | Aqueous-liquid and blood-absorbing powdery reticulated polymers, process for producing the same and their use as absorbents in sanitary articles |
| US5422405A (en) | 1992-12-16 | 1995-06-06 | Nippon Shokubai Co., Ltd. | Method for production of absorbent resin |
| US5610208A (en) | 1994-02-17 | 1997-03-11 | Nippon Shokubai Co., Ltd. | Water-absorbent agent, method for production thereof, and water-absorbent composition |
| US5669894A (en) | 1994-03-29 | 1997-09-23 | The Procter & Gamble Company | Absorbent members for body fluids having good wet integrity and relatively high concentrations of hydrogel-forming absorbent polymer |
| US5672633A (en) | 1993-09-29 | 1997-09-30 | Chemische Fabrik Stockhausen Gmbh | Powdery polymers capable of absorbing aqueous liquids, a process for their production and their use as absorbents |
| US6071976A (en) | 1995-12-27 | 2000-06-06 | Nippon Shokubai Co., Ltd. | Water absorbing agent, manufacturing method thereof, and manufacturing machine thereof |
| US6228930B1 (en) | 1997-06-18 | 2001-05-08 | Nippon Shokubai Co., Ltd. | Water-absorbent resin granule-containing composition and production process for water-absorbent resin granule |
| US6239230B1 (en) | 1999-09-07 | 2001-05-29 | Bask Aktiengesellschaft | Surface-treated superabsorbent polymer particles |
| US6241928B1 (en) | 1998-04-28 | 2001-06-05 | Nippon Shokubai Co., Ltd. | Method for production of shaped hydrogel of absorbent resin |
| US6254990B1 (en) | 1998-02-18 | 2001-07-03 | Nippon Shokubai Co., Ltd. | Surface-crosslinking process for water-absorbent resin |
| US6265488B1 (en) | 1998-02-24 | 2001-07-24 | Nippon Shokubai Co., Ltd. | Production process for water-absorbing agent |
| US6297319B1 (en) | 1998-11-05 | 2001-10-02 | Nippon Shokubai Co., Ltd. | Water-absorbing agent and production process therefor |
| US6372852B2 (en) | 1998-03-31 | 2002-04-16 | Nippon Shokubai Co., Ltd | Water-absorbing composition and production process for water-absorbing agent |
| US6472478B1 (en) | 1998-02-21 | 2002-10-29 | Basf Aktiengesellschaft | Process for crosslinking hydrogels with bis- and poly-2- oxazolidinones |
| US6503979B1 (en) | 1998-02-26 | 2003-01-07 | Basf Aktiengesellschaft | Method for cross-linking hydrogels with bis- and poly-2-oxazolidinones |
| US6514615B1 (en) | 1999-06-29 | 2003-02-04 | Stockhausen Gmbh & Co. Kg | Superabsorbent polymers having delayed water absorption characteristics |
| US6559239B1 (en) | 1998-11-26 | 2003-05-06 | Basf Aktiengesellschaft | Method for the secondary cross-linking of hydrogels with N-acyl-2-oxazolidinones |
| US6605673B1 (en) | 1999-03-05 | 2003-08-12 | Stockhausen Gmbh & Co., Kg | Powdery, cross-linked polymers which absorb aqueous liquids and blood, method for the production thereof and their use |
| US6620899B1 (en) | 1998-10-15 | 2003-09-16 | E. I. Du Pont De Nemours And Company | Polymers, containing a fluorocyclobutyl ring and their preparation |
| US6620889B1 (en) | 1999-03-05 | 2003-09-16 | Stockhausen Gmbh & Co. Kg | Powdery, crosslinked absorbent polymers, method for the production thereof, and their use |
| US6657015B1 (en) | 1998-11-26 | 2003-12-02 | Basf Aktiengesellschaft | Method for the secondary cross-linking of hydrogels with 2-oxotetrahydro-1,3-oxazines |
| US6720389B2 (en) | 2000-09-20 | 2004-04-13 | Nippon Shokubai Co., Ltd. | Water-absorbent resin and production process therefor |
| WO2004069915A2 (en) | 2003-02-10 | 2004-08-19 | Nippon Shokubai Co., Ltd. | Particulate water-absorbing agent |
| US6809158B2 (en) | 2000-10-20 | 2004-10-26 | Nippon Shokubai Co., Ltd. | Water-absorbing agent and process for producing the same |
| JP2004300425A (ja) * | 2003-03-14 | 2004-10-28 | Nippon Shokubai Co Ltd | 吸水性樹脂粉末の表面架橋処理方法 |
| US20040240316A1 (en) | 2003-05-30 | 2004-12-02 | Kozo Nogi | Method for production of water-absorbent resin and plow-shaped mixing device |
| JP2004345804A (ja) * | 2003-05-22 | 2004-12-09 | Nippon Shokubai Co Ltd | 吸水性樹脂粉体の輸送方法 |
| JP2004352941A (ja) | 2003-05-30 | 2004-12-16 | Nippon Shokubai Co Ltd | 吸水性樹脂の製造法 |
| US20050029352A1 (en) | 2003-08-08 | 2005-02-10 | Spears Kurt E. | System and method for automatic correction of illumination noise caused by ambient light |
| WO2005016393A1 (en) | 2003-07-31 | 2005-02-24 | Kimberly-Clark Worldwide, Inc. | Absorbent materials and articles |
| US20050048221A1 (en) | 2003-08-27 | 2005-03-03 | Yoshio Irie | Process for production of surface-treated particulate water-absorbent resin |
| US20050070671A1 (en) | 2003-09-19 | 2005-03-31 | Kazushi Torii | Water-absorbent resin having treated surface and process for producing the same |
| US6906159B2 (en) | 2000-08-03 | 2005-06-14 | Nippon Shokubai Co., Ltd. | Water-absorbent resin, hydropolymer, process for producing them, and uses of them |
| US20050215734A1 (en) | 2004-03-24 | 2005-09-29 | Yorimichi Dairoku | Method for continuous production of water-absorbent resin |
| US20050288182A1 (en) | 2004-06-18 | 2005-12-29 | Kazushi Torii | Water absorbent resin composition and production method thereof |
| US6987151B2 (en) | 2001-09-12 | 2006-01-17 | Dow Global Technologies Inc. | Continuous polymerization process for the manufacture of superabsorbent polymers |
| JP2006503949A (ja) * | 2002-10-25 | 2006-02-02 | ストックハウゼン ゲーエムベーハー | 吸水性ポリマー及び吸収性ポリマーの製造方法、複合体及び複合体の製造方法、化学製品並びに吸水性ポリマー又は複合体の使用 |
| US20060057389A1 (en) | 2002-10-25 | 2006-03-16 | Armin Reimann | Two-step mixing process for producing an absorbent polymer |
| US20060073969A1 (en) | 2003-02-10 | 2006-04-06 | Kazushi Torii | Vater-absorbent resin composition and its production process |
| US20060082197A1 (en) | 2004-10-15 | 2006-04-20 | Brian Luce | Lounge caddy |
| US20060082188A1 (en) | 2004-08-06 | 2006-04-20 | Mitchell Stephen A G | Electromechanical strut |
| US20060082189A1 (en) | 2004-10-18 | 2006-04-20 | Faisal Sultan | Upper auxiliary seal with positive attachment configuration |
| US20060111404A1 (en) | 2004-11-22 | 2006-05-25 | Incyte Corporation | Salts of N-[2-({(3R)-1-[trans-4-hydroxy-4-(6-methoxypyridin-3-yl)-cyclohexyl]pyrrolidin-3-yl}amino)-2-oxoethyl]-3-(trifluoromethyl)benzamide |
| US20060111402A1 (en) | 2004-09-30 | 2006-05-25 | Raymond Ng | Novel benzimidazole derivatives useful as selective androgen receptor modulators (SARMS) |
| US20060111403A1 (en) | 2003-01-28 | 2006-05-25 | Hughes Kenneth A | Cyano anthranilamide insecticides |
| US7098284B2 (en) | 2001-01-26 | 2006-08-29 | Nippon Shokubal Co., Ltd | Water-absorbing agent and production process therefor, and water-absorbent structure |
| JP2006522181A (ja) | 2003-03-26 | 2006-09-28 | ビーエーエスエフ アクチェンゲゼルシャフト | 色安定性の高吸水性ポリマー組成物 |
| US7157141B2 (en) | 2000-03-31 | 2007-01-02 | Stockhausen Gmbh | Pulverulent polymers crosslinked on the surface |
| US20070078231A1 (en) | 2005-09-30 | 2007-04-05 | Nippon Shokubai Co., Ltd. | Method for manufacturing particulate water-absorbing agent and particulate water-absorbing agent |
| US20070106013A1 (en) | 2003-06-24 | 2007-05-10 | Yoshifumi Adachi | Water absorbent resin composition and production method thereof |
| US20070121037A1 (en) | 2005-11-29 | 2007-05-31 | Casio Computer Co., Ltd. | Homeotropic alignment type semi-transmissive reflective liquid crystal display device |
| WO2007065840A1 (de) | 2005-12-07 | 2007-06-14 | Basf Se | Verfahren zum kontinuierlichen mischen von polymerpartikeln |
| US20070149760A1 (en) | 2005-12-22 | 2007-06-28 | Kenji Kadonaga | Method for surface crosslinking water-absorbing resin and method for manufacturing water-absorbing resin |
| US20070161759A1 (en) | 2004-02-24 | 2007-07-12 | Basf Aktiengesellschaft | Postcrosslinking of water-absorbing polymers |
| EP1824910A2 (en) | 2004-12-10 | 2007-08-29 | Nippon Shokubai Co.,Ltd. | Method for production of modified water absorbent resin |
| US20070293632A1 (en) | 2004-11-30 | 2007-12-20 | Basf Aktiengesellschaft, A German Corporation | Postcrosslinking of Water Absorbing Polymeric Particles |
| US7312278B2 (en) | 2001-06-08 | 2007-12-25 | Nippon Shokubai Co., Ltd. | Water-absorbing agent and production process therefor, and sanitary material |
| US20080009843A1 (en) | 2006-06-14 | 2008-01-10 | De La Torre Ralph | Surgical ablation system with chest wall platform |
| US20080009842A1 (en) | 2000-12-28 | 2008-01-10 | The General Hospital Corporation | Method and apparatus for emr treatment |
| JP2008038128A (ja) * | 2005-12-22 | 2008-02-21 | Nippon Shokubai Co Ltd | 吸水性樹脂の表面架橋方法および吸水性樹脂の製造方法 |
| US7378453B2 (en) | 2003-03-14 | 2008-05-27 | Nippon Shokubai Co., Ltd. | Surface crosslinking treatment method of water-absorbing resin powder |
| WO2008108277A1 (ja) * | 2007-03-01 | 2008-09-12 | Nippon Shokubai Co., Ltd. | 吸水性樹脂を主成分とする粒子状吸水剤 |
| US20080227932A1 (en) | 2005-08-24 | 2008-09-18 | Basf Se | Method for Producing Water-Absorbing Polymer Particles |
| WO2009001954A1 (en) | 2007-06-27 | 2008-12-31 | Nippon Shokubai Co., Ltd. | Method of producing water-absorbent resin |
| WO2009028568A1 (ja) | 2007-08-28 | 2009-03-05 | Nippon Shokubai Co., Ltd. | 吸水性樹脂の製造方法 |
| WO2009113673A1 (ja) * | 2008-03-13 | 2009-09-17 | 株式会社日本触媒 | 吸水性樹脂を主成分とする粒子状吸水剤の製造方法 |
Family Cites Families (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5410387U (ja) | 1977-06-21 | 1979-01-23 | ||
| JPS5992452U (ja) | 1982-12-14 | 1984-06-22 | 三洋電機株式会社 | カセツトホルダ機構 |
| US5250640A (en) | 1991-04-10 | 1993-10-05 | Nippon Shokubai Co., Ltd. | Method for production of particulate hydrogel polymer and absorbent resin |
| JP2000143720A (ja) | 1998-11-11 | 2000-05-26 | Sanyo Chem Ind Ltd | 吸水性樹脂の製造法 |
| DE19955861A1 (de) | 1999-11-20 | 2001-05-23 | Basf Ag | Verfahren zur kontinuierlichen Herstellung von vernetzten feinteiligen gelförmigen Polymerisaten |
| EP1130045B2 (en) * | 2000-02-29 | 2015-10-28 | Nippon Shokubai Co., Ltd. | Process for producing a water-absorbent resin powder |
| JP4676625B2 (ja) | 2000-02-29 | 2011-04-27 | 株式会社日本触媒 | 吸水性樹脂粉末の製造方法 |
| US6727345B2 (en) | 2001-07-03 | 2004-04-27 | Nippon Shokubai Co., Ltd. | Continuous production process for water-absorbent resin powder and powder surface detector used therefor |
| DE60231834D1 (de) | 2001-12-19 | 2009-05-14 | Nippon Catalytic Chem Ind | Wasser absorbierendes harz und seine herstellungsverfahren |
| BE1015154A5 (fr) * | 2002-10-23 | 2004-10-05 | Alloys For Technical Applic S | Ensemble destine a la pulverisation cathodique magnetron. |
| TWI302541B (en) * | 2003-05-09 | 2008-11-01 | Nippon Catalytic Chem Ind | Water-absorbent resin and its production process |
| JP4749679B2 (ja) * | 2003-05-09 | 2011-08-17 | 株式会社日本触媒 | 吸水性樹脂およびその製造方法 |
| KR100819613B1 (ko) * | 2003-09-19 | 2008-04-07 | 가부시키가이샤 닛폰 쇼쿠바이 | 수분 흡수제와 그 제조방법 |
| KR100858387B1 (ko) | 2004-02-05 | 2008-09-11 | 가부시키가이샤 닛폰 쇼쿠바이 | 입자상 수분흡수제 및 그 제조방법과 수분흡수성 물품 |
| TW200700095A (en) | 2005-02-01 | 2007-01-01 | Basf Ag | Polyamine-coated superabsorbent polymers |
| TW200704689A (en) | 2005-02-01 | 2007-02-01 | Basf Ag | Polyamine-coated superabsorbent polymers |
| TW200639200A (en) | 2005-02-01 | 2006-11-16 | Basf Ag | Polyamine-coated superabsorbent polymers |
| TWI344469B (en) | 2005-04-07 | 2011-07-01 | Nippon Catalytic Chem Ind | Polyacrylic acid (salt) water-absorbent resin, production process thereof, and acrylic acid used in polymerization for production of water-absorbent resin |
| DE102005018924A1 (de) | 2005-04-22 | 2006-10-26 | Stockhausen Gmbh | Wasserabsorbierende Polymergebilde mit verbesserten Absorptionseigenschaften |
| EP1888132B1 (de) | 2005-04-22 | 2015-08-12 | Evonik Degussa GmbH | Oberflächennachvernetzte superabsorber behandelt mit wasserlöslichem aluminiumsalz und zinkoxid |
| DE102005018922A1 (de) | 2005-04-22 | 2006-10-26 | Stockhausen Gmbh | Mit Polykationen oberflächenbehandeltes wasserabsorbierendes Polymergebilde |
| US7680118B2 (en) | 2006-04-13 | 2010-03-16 | Motorola, Inc. | Method and apparatus for reordering fragments within a MAC layer service data unit within a downlink frame |
| DE102006019157A1 (de) | 2006-04-21 | 2007-10-25 | Stockhausen Gmbh | Herstellung von hochpermeablen, superabsorbierenden Polymergebilden |
| BRPI0711452B8 (pt) | 2006-04-21 | 2021-06-22 | Evonik Degussa Gmbh | processo para a preparação de uma estrutura polimérica absorvente de água, estrutura polimérica absorvente de água, compósito, processo para a preparação de um compósito, uso da estrutura polimérica absorvente de água ou uso do compósito e uso de um sal |
| FR2903988B1 (fr) | 2006-07-18 | 2008-09-05 | Arkema France | Procede de preparation de polymeres (meth)acryliques |
| FR2904059B1 (fr) | 2006-07-21 | 2010-06-18 | Peugeot Citroen Automobiles Sa | Fourchette de maintien d'un porte-injecteur de moteur thermique. |
| CN101821323B (zh) * | 2007-10-09 | 2013-03-27 | 株式会社日本触媒 | 吸水性树脂的表面处理方法 |
| EP2263957B1 (en) | 2008-03-28 | 2013-02-13 | Nippon Shokubai Co., Ltd. | Method of transporting absorbent resin powder |
| SG194348A1 (en) | 2008-09-16 | 2013-11-29 | Nippon Catalytic Chem Ind | Production method and method for enhancing liquid permeability of water-absorbing resin |
| JP5629688B2 (ja) * | 2009-08-27 | 2014-11-26 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂およびその製造方法 |
-
2010
- 2010-08-27 JP JP2011528883A patent/JP5629688B2/ja active Active
- 2010-08-27 WO PCT/JP2010/064646 patent/WO2011024975A1/ja active Application Filing
- 2010-08-27 JP JP2011528884A patent/JP5619011B2/ja active Active
- 2010-08-27 CN CN201080038386.5A patent/CN102482434B/zh active Active
- 2010-08-27 CN CN201080038364.9A patent/CN102482433B/zh active Active
- 2010-08-27 EP EP10812020.5A patent/EP2471843B1/en active Active
- 2010-08-27 JP JP2011528882A patent/JP5619010B2/ja active Active
- 2010-08-27 US US13/392,639 patent/US8907021B2/en active Active
- 2010-08-27 WO PCT/JP2010/064645 patent/WO2011024974A1/ja active Application Filing
- 2010-08-27 WO PCT/JP2010/064641 patent/WO2011024971A1/ja active Application Filing
- 2010-08-27 WO PCT/JP2010/064643 patent/WO2011024972A1/ja active Application Filing
- 2010-08-27 EP EP10812024.7A patent/EP2471846B1/en active Active
- 2010-08-27 JP JP2011528885A patent/JP5732396B2/ja active Active
- 2010-08-27 EP EP10812023.9A patent/EP2471845B1/en active Active
- 2010-08-27 US US13/392,214 patent/US8859685B2/en active Active
- 2010-08-27 US US13/392,256 patent/US9138505B2/en active Active
- 2010-08-27 EP EP10812021.3A patent/EP2471844B1/en active Active
- 2010-08-27 US US13/392,240 patent/US9023951B2/en active Active
Patent Citations (85)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US670141A (en) | 1900-11-20 | 1901-03-19 | William Otis Shepard | Policeman's club. |
| JPS5410387A (en) * | 1977-06-24 | 1979-01-25 | Sanyo Chem Ind Ltd | Preparation of water-soluble polymer gel |
| US4734478A (en) | 1984-07-02 | 1988-03-29 | Nippon Shokubai Kagaku Kogyo Co., Ltd. | Water absorbing agent |
| US4893999A (en) | 1985-12-18 | 1990-01-16 | Chemische Fabrik Stockhausen Gmbh | Apparatus for the continuous production of polymers and copolymers of water-soluble monomers |
| US4783510A (en) | 1986-06-04 | 1988-11-08 | Taiyo Fishery Co., Ltd. | Process for improving a water absorbent polyacrylic acid polymer and an improved polymer produced by said process |
| US4755562A (en) | 1986-06-10 | 1988-07-05 | American Colloid Company | Surface treated absorbent polymers |
| US4824901A (en) | 1986-06-10 | 1989-04-25 | American Colloid Company | Surface treated absorbent polymers |
| JPH0249002A (ja) * | 1988-05-23 | 1990-02-19 | Nippon Shokubai Kagaku Kogyo Co Ltd | 親水性重合体の製造方法 |
| EP0349240A2 (en) | 1988-06-28 | 1990-01-03 | Nippon Shokubai Co., Ltd. | Water-absorbent resin and production process |
| JPH03168218A (ja) * | 1989-11-28 | 1991-07-22 | Sumitomo Chem Co Ltd | 加硫方法 |
| US5140076A (en) | 1990-04-02 | 1992-08-18 | Nippon Shokubai Kagaku Kogyo Co., Ltd. | Method of treating the surface of an absorbent resin |
| US5409771A (en) | 1990-06-29 | 1995-04-25 | Chemische Fabrik Stockhausen Gmbh | Aqueous-liquid and blood-absorbing powdery reticulated polymers, process for producing the same and their use as absorbents in sanitary articles |
| JPH05112654A (ja) * | 1991-04-10 | 1993-05-07 | Nippon Shokubai Co Ltd | 粒子状含水ゲル状重合体および吸水性樹脂の製造方法 |
| US5206205A (en) | 1991-08-15 | 1993-04-27 | Kimberly-Clark Corporation | Thermal treatment of superabsorbents to enhance their rate of absorbency under load |
| EP0603292A1 (en) | 1991-09-09 | 1994-06-29 | The Dow Chemical Company | Superabsorbent polymers and process for producing |
| EP0534228A1 (de) | 1991-09-18 | 1993-03-31 | Hoechst Aktiengesellschaft | Verfahren zum Modifizieren hydrophiler Polymerisate |
| JPH05202199A (ja) * | 1991-09-18 | 1993-08-10 | Cassella Ag | 親水性ポリマーの変性法 |
| US5422405A (en) | 1992-12-16 | 1995-06-06 | Nippon Shokubai Co., Ltd. | Method for production of absorbent resin |
| US5672633A (en) | 1993-09-29 | 1997-09-30 | Chemische Fabrik Stockhausen Gmbh | Powdery polymers capable of absorbing aqueous liquids, a process for their production and their use as absorbents |
| US5610208A (en) | 1994-02-17 | 1997-03-11 | Nippon Shokubai Co., Ltd. | Water-absorbent agent, method for production thereof, and water-absorbent composition |
| US5669894A (en) | 1994-03-29 | 1997-09-23 | The Procter & Gamble Company | Absorbent members for body fluids having good wet integrity and relatively high concentrations of hydrogel-forming absorbent polymer |
| US6071976A (en) | 1995-12-27 | 2000-06-06 | Nippon Shokubai Co., Ltd. | Water absorbing agent, manufacturing method thereof, and manufacturing machine thereof |
| US6228930B1 (en) | 1997-06-18 | 2001-05-08 | Nippon Shokubai Co., Ltd. | Water-absorbent resin granule-containing composition and production process for water-absorbent resin granule |
| US6254990B1 (en) | 1998-02-18 | 2001-07-03 | Nippon Shokubai Co., Ltd. | Surface-crosslinking process for water-absorbent resin |
| US6472478B1 (en) | 1998-02-21 | 2002-10-29 | Basf Aktiengesellschaft | Process for crosslinking hydrogels with bis- and poly-2- oxazolidinones |
| US6265488B1 (en) | 1998-02-24 | 2001-07-24 | Nippon Shokubai Co., Ltd. | Production process for water-absorbing agent |
| US6503979B1 (en) | 1998-02-26 | 2003-01-07 | Basf Aktiengesellschaft | Method for cross-linking hydrogels with bis- and poly-2-oxazolidinones |
| US6372852B2 (en) | 1998-03-31 | 2002-04-16 | Nippon Shokubai Co., Ltd | Water-absorbing composition and production process for water-absorbing agent |
| US6241928B1 (en) | 1998-04-28 | 2001-06-05 | Nippon Shokubai Co., Ltd. | Method for production of shaped hydrogel of absorbent resin |
| US6620899B1 (en) | 1998-10-15 | 2003-09-16 | E. I. Du Pont De Nemours And Company | Polymers, containing a fluorocyclobutyl ring and their preparation |
| US6297319B1 (en) | 1998-11-05 | 2001-10-02 | Nippon Shokubai Co., Ltd. | Water-absorbing agent and production process therefor |
| US6657015B1 (en) | 1998-11-26 | 2003-12-02 | Basf Aktiengesellschaft | Method for the secondary cross-linking of hydrogels with 2-oxotetrahydro-1,3-oxazines |
| US6559239B1 (en) | 1998-11-26 | 2003-05-06 | Basf Aktiengesellschaft | Method for the secondary cross-linking of hydrogels with N-acyl-2-oxazolidinones |
| US6605673B1 (en) | 1999-03-05 | 2003-08-12 | Stockhausen Gmbh & Co., Kg | Powdery, cross-linked polymers which absorb aqueous liquids and blood, method for the production thereof and their use |
| US6620889B1 (en) | 1999-03-05 | 2003-09-16 | Stockhausen Gmbh & Co. Kg | Powdery, crosslinked absorbent polymers, method for the production thereof, and their use |
| US6514615B1 (en) | 1999-06-29 | 2003-02-04 | Stockhausen Gmbh & Co. Kg | Superabsorbent polymers having delayed water absorption characteristics |
| US6239230B1 (en) | 1999-09-07 | 2001-05-29 | Bask Aktiengesellschaft | Surface-treated superabsorbent polymer particles |
| US7157141B2 (en) | 2000-03-31 | 2007-01-02 | Stockhausen Gmbh | Pulverulent polymers crosslinked on the surface |
| US6906159B2 (en) | 2000-08-03 | 2005-06-14 | Nippon Shokubai Co., Ltd. | Water-absorbent resin, hydropolymer, process for producing them, and uses of them |
| US7091253B2 (en) | 2000-08-03 | 2006-08-15 | Nippon Shokubai Co., Ltd. | Water-absorbent resin, hydropolymer, process for producing them, and uses of them |
| US6720389B2 (en) | 2000-09-20 | 2004-04-13 | Nippon Shokubai Co., Ltd. | Water-absorbent resin and production process therefor |
| US7183456B2 (en) | 2000-09-20 | 2007-02-27 | Nippon Shokubai Co., Ltd. | Water-absorbent resin and production process therefor |
| US6809158B2 (en) | 2000-10-20 | 2004-10-26 | Nippon Shokubai Co., Ltd. | Water-absorbing agent and process for producing the same |
| US20080009842A1 (en) | 2000-12-28 | 2008-01-10 | The General Hospital Corporation | Method and apparatus for emr treatment |
| US7098284B2 (en) | 2001-01-26 | 2006-08-29 | Nippon Shokubal Co., Ltd | Water-absorbing agent and production process therefor, and water-absorbent structure |
| US7312278B2 (en) | 2001-06-08 | 2007-12-25 | Nippon Shokubai Co., Ltd. | Water-absorbing agent and production process therefor, and sanitary material |
| US6987151B2 (en) | 2001-09-12 | 2006-01-17 | Dow Global Technologies Inc. | Continuous polymerization process for the manufacture of superabsorbent polymers |
| US20060057389A1 (en) | 2002-10-25 | 2006-03-16 | Armin Reimann | Two-step mixing process for producing an absorbent polymer |
| JP2006503949A (ja) * | 2002-10-25 | 2006-02-02 | ストックハウゼン ゲーエムベーハー | 吸水性ポリマー及び吸収性ポリマーの製造方法、複合体及び複合体の製造方法、化学製品並びに吸水性ポリマー又は複合体の使用 |
| US20060111403A1 (en) | 2003-01-28 | 2006-05-25 | Hughes Kenneth A | Cyano anthranilamide insecticides |
| US20060073969A1 (en) | 2003-02-10 | 2006-04-06 | Kazushi Torii | Vater-absorbent resin composition and its production process |
| WO2004069915A2 (en) | 2003-02-10 | 2004-08-19 | Nippon Shokubai Co., Ltd. | Particulate water-absorbing agent |
| JP2004300425A (ja) * | 2003-03-14 | 2004-10-28 | Nippon Shokubai Co Ltd | 吸水性樹脂粉末の表面架橋処理方法 |
| US7378453B2 (en) | 2003-03-14 | 2008-05-27 | Nippon Shokubai Co., Ltd. | Surface crosslinking treatment method of water-absorbing resin powder |
| JP2006522181A (ja) | 2003-03-26 | 2006-09-28 | ビーエーエスエフ アクチェンゲゼルシャフト | 色安定性の高吸水性ポリマー組成物 |
| JP2004345804A (ja) * | 2003-05-22 | 2004-12-09 | Nippon Shokubai Co Ltd | 吸水性樹脂粉体の輸送方法 |
| US20040240316A1 (en) | 2003-05-30 | 2004-12-02 | Kozo Nogi | Method for production of water-absorbent resin and plow-shaped mixing device |
| JP2004352941A (ja) | 2003-05-30 | 2004-12-16 | Nippon Shokubai Co Ltd | 吸水性樹脂の製造法 |
| US20070106013A1 (en) | 2003-06-24 | 2007-05-10 | Yoshifumi Adachi | Water absorbent resin composition and production method thereof |
| WO2005016393A1 (en) | 2003-07-31 | 2005-02-24 | Kimberly-Clark Worldwide, Inc. | Absorbent materials and articles |
| US20050029352A1 (en) | 2003-08-08 | 2005-02-10 | Spears Kurt E. | System and method for automatic correction of illumination noise caused by ambient light |
| US20050048221A1 (en) | 2003-08-27 | 2005-03-03 | Yoshio Irie | Process for production of surface-treated particulate water-absorbent resin |
| US20050070671A1 (en) | 2003-09-19 | 2005-03-31 | Kazushi Torii | Water-absorbent resin having treated surface and process for producing the same |
| US20070161759A1 (en) | 2004-02-24 | 2007-07-12 | Basf Aktiengesellschaft | Postcrosslinking of water-absorbing polymers |
| US20050215734A1 (en) | 2004-03-24 | 2005-09-29 | Yorimichi Dairoku | Method for continuous production of water-absorbent resin |
| US20050288182A1 (en) | 2004-06-18 | 2005-12-29 | Kazushi Torii | Water absorbent resin composition and production method thereof |
| US20060082188A1 (en) | 2004-08-06 | 2006-04-20 | Mitchell Stephen A G | Electromechanical strut |
| US20060111402A1 (en) | 2004-09-30 | 2006-05-25 | Raymond Ng | Novel benzimidazole derivatives useful as selective androgen receptor modulators (SARMS) |
| US20060082197A1 (en) | 2004-10-15 | 2006-04-20 | Brian Luce | Lounge caddy |
| US20060082189A1 (en) | 2004-10-18 | 2006-04-20 | Faisal Sultan | Upper auxiliary seal with positive attachment configuration |
| US20060111404A1 (en) | 2004-11-22 | 2006-05-25 | Incyte Corporation | Salts of N-[2-({(3R)-1-[trans-4-hydroxy-4-(6-methoxypyridin-3-yl)-cyclohexyl]pyrrolidin-3-yl}amino)-2-oxoethyl]-3-(trifluoromethyl)benzamide |
| US20070293632A1 (en) | 2004-11-30 | 2007-12-20 | Basf Aktiengesellschaft, A German Corporation | Postcrosslinking of Water Absorbing Polymeric Particles |
| EP1824910A2 (en) | 2004-12-10 | 2007-08-29 | Nippon Shokubai Co.,Ltd. | Method for production of modified water absorbent resin |
| US20080227932A1 (en) | 2005-08-24 | 2008-09-18 | Basf Se | Method for Producing Water-Absorbing Polymer Particles |
| US20070078231A1 (en) | 2005-09-30 | 2007-04-05 | Nippon Shokubai Co., Ltd. | Method for manufacturing particulate water-absorbing agent and particulate water-absorbing agent |
| US20070121037A1 (en) | 2005-11-29 | 2007-05-31 | Casio Computer Co., Ltd. | Homeotropic alignment type semi-transmissive reflective liquid crystal display device |
| WO2007065840A1 (de) | 2005-12-07 | 2007-06-14 | Basf Se | Verfahren zum kontinuierlichen mischen von polymerpartikeln |
| JP2008038128A (ja) * | 2005-12-22 | 2008-02-21 | Nippon Shokubai Co Ltd | 吸水性樹脂の表面架橋方法および吸水性樹脂の製造方法 |
| US20070149760A1 (en) | 2005-12-22 | 2007-06-28 | Kenji Kadonaga | Method for surface crosslinking water-absorbing resin and method for manufacturing water-absorbing resin |
| US20080009843A1 (en) | 2006-06-14 | 2008-01-10 | De La Torre Ralph | Surgical ablation system with chest wall platform |
| WO2008108277A1 (ja) * | 2007-03-01 | 2008-09-12 | Nippon Shokubai Co., Ltd. | 吸水性樹脂を主成分とする粒子状吸水剤 |
| WO2009001954A1 (en) | 2007-06-27 | 2008-12-31 | Nippon Shokubai Co., Ltd. | Method of producing water-absorbent resin |
| WO2009028568A1 (ja) | 2007-08-28 | 2009-03-05 | Nippon Shokubai Co., Ltd. | 吸水性樹脂の製造方法 |
| WO2009113673A1 (ja) * | 2008-03-13 | 2009-09-17 | 株式会社日本触媒 | 吸水性樹脂を主成分とする粒子状吸水剤の製造方法 |
| WO2009113679A1 (ja) * | 2008-03-13 | 2009-09-17 | 株式会社日本触媒 | 吸水性樹脂を主成分とする粒子状吸水剤の製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2471843A4 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015046604A1 (ja) | 2013-09-30 | 2015-04-02 | 株式会社日本触媒 | 粒子状吸水剤の充填方法および粒子状吸水剤充填物のサンプリング方法 |
| EP4159307A1 (en) | 2013-09-30 | 2023-04-05 | Nippon Shokubai Co., Ltd. | Method for filling particulate water absorbing agent and method for sampling filled particulate water absorbing agent |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5619010B2 (ja) | ポリアクリル酸(塩)系吸水性樹脂およびその製造方法 | |
| JP5504334B2 (ja) | 吸水性樹脂の製造方法 | |
| JP5616347B2 (ja) | 吸水性樹脂の製造方法 | |
| JP5977839B2 (ja) | ポリアクリル酸(塩)系吸水性樹脂およびその製造方法 | |
| JP5605855B2 (ja) | 吸水性樹脂粉末の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10812020 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011528882 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13392256 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1201000824 Country of ref document: TH |
|
| REEP | Request for entry into the european phase |
Ref document number: 2010812020 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010812020 Country of ref document: EP |