WO2011095174A1 - Human herpes virus 6 and 7 u20 polypeptide and polynucleotides for use as a medicament or diagnosticum - Google Patents
Human herpes virus 6 and 7 u20 polypeptide and polynucleotides for use as a medicament or diagnosticum Download PDFInfo
- Publication number
- WO2011095174A1 WO2011095174A1 PCT/DK2011/050032 DK2011050032W WO2011095174A1 WO 2011095174 A1 WO2011095174 A1 WO 2011095174A1 DK 2011050032 W DK2011050032 W DK 2011050032W WO 2011095174 A1 WO2011095174 A1 WO 2011095174A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- less
- sequence identity
- sequence
- seq
- polynucleotide
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/005—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from viruses
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N7/00—Viruses; Bacteriophages; Compositions thereof; Preparation or purification thereof
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/569—Immunoassay; Biospecific binding assay; Materials therefor for microorganisms, e.g. protozoa, bacteria, viruses
- G01N33/56983—Viruses
- G01N33/56994—Herpetoviridae, e.g. cytomegalovirus, Epstein-Barr virus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/25—Tumour necrosing factors [TNF]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/40—Regulators of development
- C12N2501/48—Regulators of apoptosis
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/16011—Herpesviridae
- C12N2710/16511—Roseolovirus, e.g. human herpesvirus 6, 7
- C12N2710/16522—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/16011—Herpesviridae
- C12N2710/16511—Roseolovirus, e.g. human herpesvirus 6, 7
- C12N2710/16533—Use of viral protein as therapeutic agent other than vaccine, e.g. apoptosis inducing or anti-inflammatory
Definitions
- the present invention relates to human herpes virus U20 polypeptides, biologically active variants or fragments thereof, as well as to polynucleotides encoding
- the present invention relates to U20 polypeptides, biologically active variants or fragments thereof for use as a medicament for the treatment of diseases related to TNF receptor activity such as in autoimmune, inflammatory, degenerative or cancerous diseases.
- TNFa cytokine tumor necrosis factor-alpha
- TNFa cytokine tumor necrosis factor-alpha
- TNFa exerts its activity by forming trimeric structures that bind to trimeric receptors.
- the TNF receptors include TNFR1 (also known as TNF receptor superfamily 1 A or TNFRSF1 A) and TNFR2 (known as TNFRSF1 B).
- TNFR1 also known as TNF receptor superfamily 1 A or TNFRSF1 A
- TNFR2 TNFRSF1 B
- TNFR1 TNFR1 -associated death domain protein
- TRADD TNFR1 -associated death domain protein
- TRAF2 TNFR-associated factor 2
- the protein RIP1 is recruited and is subsequently modified by ubiquitination to serve as a docking site for further recruited proteins in the NF-kB signalling pathway.
- hTNFa human TNFa
- therapeutic strategies have been designed to inhibit or counteract hTNFa activity.
- antibodies that bind to, and neutralize, hTNFa have been suggested as a means to inhibit hTNFa activity.
- Examples of patents and patent application disclosing hTNFa binding antibodies and treatments targeting hTNFa are EP 1 309 691 B1 , US 6,379,666 B1 , US 6,509,015 B1 and WO 2006/056779 A2.
- Alternative means for inhibiting human TNFa is the use of soluble TNFa receptors.
- WO 2006/038027 A2 discloses antibodies that bind Tumor Necrosis Factor Receptor 1 , and which can be used for treating inflammatory diseases.
- a general problem in relation to antibody-based therapies is the development of an immune response in the patient towards the applied antibodies, which can be both harmful for the patient and inhibit the effect of the treatment.
- HHV-6A and -6B Human herpesvirus 6 (HHV-6) was isolated in 1986 and later divided into two variants, HHV-6A and -6B, based on differences in restriction enzyme cleavage patterns and reactivity with monoclonal antibodies.
- HHV-6A and -6B belong to the betaherpesvirus subfamily and belong to the Roseolovirus genus together with the closely related HHV- 7.
- Infection by HHV-6B may give rise to a febrile illness during the first years of life, known as exanthem subitum, after which the virus remains in a latent state in the infected individual for life. It is possible that primary infection with HHV-6A or HHV-7 may cause a similar disease with rash and fever.
- HHV-6B is considered an
- opportunistic pathogen and its reactivation from the latent state may cause severe disease in immunocompromised individuals.
- HHV-6A and -6B have unique characteristics making them atypical among the human herpesvirus family. Compared with other herpesviruses, the heparin-like molecules seem to play a minor role in HHV-6 surface interactions required for infection.
- the intracellular maturation pathway of HHV-6 is different from that of other herpesviruses, and few viral glycoproteins are detected on the host cell plasma membrane of HHV-6 infected cells.
- HHV-6B The genome of HHV-6B is 162 kbp long with a unique segment (U) of about 144 kbp flanked in both ends by direct repeats (DR) of about 9 kbp each (Dominguez et al.,
- HHV-6B is predicted to have 97 unique genes, of which 88 have counterparts in HHV-6A and 82 in HHV-7 (Dominguez et al., 1999, J Virol 73:8040-8052). Most of the genes from HHV-6A, -6B, and -7 can be divided into gene blocks.
- U2 to U19 belongs to the betaherpesvirus genes found only in betaherpesvirinae (Dominguez et al., 1999).
- the expression from the U18-U20 has been studied in HHV-6A and HHV-6B, indicating possible differences in their expression pattern. Inspection of the amino acid sequence of the predicted U20 polypeptide from HHV-6B indicates that it may be a glycoprotein of 434 amino acids with 95.6% nucleotide identity to its homologue in HHV-6A.
- the function of U20 is entirely unknown. Although most glycoproteins are positioned in the cell membrane (or secreted), it has not yet been defined whether or not U20 is actually expressed on the cell surface or whether it is secreted from infected cells.
- This invention pertains to isolated human herpes virus U20 polypeptides, biologically active variants and fragments thereof that bind to a human tumor necrosis factor (TNF) receptor, which also serves as a receptor for tumor necrosis factor -alpha (TNF-alpha).
- TNF tumor necrosis factor
- Various aspects of the invention relate human herpes virus U20 polypeptides, biologically active variants and fragments thereof for use as a medicament or diagnosticum, pharmaceutical compositions, as well as polynucleotides, recombinant expression vectors and host cells for making such human herpes virus U20
- polypeptides are polypeptides, biologically active variants and fragments thereof.
- an object of the present invention relates to providing a new medicament in treatments requiring decreasing TNF receptor activity, such as decreasing human TNF- alpha inhibition of the TNF receptor.
- one aspect of the present invention relates to an isolated polypeptide for use as medicament or diagnosticum comprising
- variant has at least 70% sequence identity to said SEQ ID NO. 1 , 3 or 5; or
- c) a biologically active fragment of at least 50 contiguous amino acids of any of a) through b), wherein said fragment is a fragment of SEQ ID NO 1 , 3 or 5.
- a second aspect of the present invention relates to An isolated polynucleotide for use as a medicament comprising a nucleic acid or its complementary sequence, said polynucleotide being selected from the group consisting of:
- polynucleotides may be expressed in a vector system.
- another aspect of the present invention relates to an expression vector comprising the polynucleotides as described herein and above.
- the vectors according to the invention may be amplified and stored in a host cell. Similarly, the polynucleotides of the invention may be expressed in such a host cell. Thus, in yet another aspect of the present invention relates to an isolated host cell transfected or transduced with the vector as described herein and above.
- a further aspect of the present invention pertains to a pharmaceutical composition
- a pharmaceutical composition comprising
- kits of parts comprising a therapeutic agent of the invention and the isolated polypeptide of the invention, or the isolated polynucleotide of the invention, or the vector of the invention, or the host cell of the invention as a combination for the simultaneous, separate or successive administration in TNFR1 related disease therapy.
- Another aspect of the present invention relates to a pharmaceutical composition for treating, ameliorating and/or preventing TNF receptor related diseases comprising a) the isolated polypeptide according to the present invention or b) the isolated nucleic acid sequence according to the present
- Yet another aspect of the present invention relates to a kit comprising
- a further aspect of the present invention relates to a method for treating, ameliorating and/or preventing TNF receptor related diseases comprising administration of
- kit of the invention in a therapeutically effective amount to an individual in need thereof.
- Another aspect of the present invention relates to an isolated polypeptide of as described herein and above
- kits of the invention for the treatment of TNF receptor related diseases.
- Yet another aspect of the present invention relates to the use of a) the isolated polypeptide according to the present invention or b) the isolated nucleic acid sequence according to the present
- a further aspect of the present invention relates to a method for decreasing TNF receptor activity comprising
- Figure 1 shows that HHV-6B rescues cells from TNFoc-induced apoptosis.
- PARP cleavage is a marker for apoptosis.
- GAPDH is included as loading control.
- camptothecin-induced apoptosis camptothecin-induced apoptosis.
- FIG. 2 shows that HHV-6B blocks TNF induction by interfering with TNFR1 or a protein in close proximity to TNFR1 .
- HHV-6B completely blocks induction of both the pathway originating from FADD signalling (caspase pathway) and the pathway originating from TRAF2 signalling ( ⁇ - ⁇ ). This indicates a block directly of TNFR1 or TRADD (see fig. 8).
- FIG. 3 shows that U20 blocks TNFoc-induced TNFR1 signalling.
- Figure 4 shows that U20 blocks TNFoc induction at a site in close proximity to TNFR1 .
- TNFoc/CHX and stained with an antibody against active caspase-3 (arrow) and DAPI DNA stain. Staining of active caspase-3 was only observed in wt cells.
- Figure 5 shows that U20 is predicted to be a transmembrane protein.
- Bioinformatics analysis reveals a highly likely N- terminal signal peptide for sorting to the membrane, a possible Immunoglobulin-like domain, a highly likely transmembrane a-helix, and several N-glycosylation sites on the N-terminal side of the a-helix. Together this indicates a protein destined for the outer membrane with the large N-terminal domain outside the cell.
- FIG. 6 shows that U20 blocks TRADD translocation to the cytoplasm.
- Wt or U20 stably transfected (U20-S) HCT1 16 cells treated with TNFa/CHX and analysed by confocal microscopy at different time points after treatment.
- TRADD rapidly translocate to be dispersed troughout the cytoplasm. This translocation is completely blocked in U20-S cells.
- Figure 7 shows that U20 blocks binding of the HTR-19 TNFa antibody to TNFR1 .
- A) Flowcytometry analysis of wt HCT1 16 cells treated with isotype control, HTR-19 anti- TNFa antibody, or HTR-19 after pre-treatment with TNFa. TNFa blocks binding of the HTR-19 antibody to TNFR1 .
- B) Flowcytometry analysis of U20 stably transfected (U20- S) HCT1 16 cells treated with isotype control (dashed line), HTR-19 anti-TNFa antibody (grey), or HTR-19 antibody after pre-treatment with TNFa (white). U20 completely abrogates binding of the HTR-19 antibody to TNFR1 . These data indicate that U20 interacts directly with TNFR1 in the membrane.
- Figure 8 shows part of the TNFR1 signaling pathway.
- TNFa binds TNFR1 and forms a trimer complex.
- the TNFR1 trimer recruits adaptor proteins TRADD, TRAF2 and FADD. This can lead to two pathways, which inhibit one another.
- FADD recruits pro-caspase-8 and induces cleavage of it into active caspase-8.
- Active caspase-8 cleaves pro-caspase-3 into active caspase-3, which is an effector caspase involved in onset of apoptosis.
- TRAF2 induces phosphorylation of IKB, which is then no longer able to block NFKB translocation to the nucleus.
- Cycloheximide inhibits cell survival by preventing de novo protein synthesis.
- Figure 9 shows an alignment between the U20 protein from HHV-6A, HHV-6B and HHV-7. In addition a consensus sequence and a degree of similarity are shown.
- Figure 10 shows a structural model prediction of U20 from HHV-6B. The amino acid numbering for the predicted different domains and the predicted glycolysation sites are also indicated.
- Figure 1 1 shows an alignment between the U20 nucleic acid sequences from HHV-6A, HHV-6B and HHV-7. In addition a consensus sequence and a degree of similarity are shown.
- HCT1 16 cells were treated with CHX (10 ⁇ ), TNFa (25 ng/ml) or a CHX/TNFa in combination for 4 hours and analysed for PARP cleavage and GAPDH (loading control).
- B) Mock infected or HHV-6B infected HCT1 16 cells (48 hpi) were treated with a combination of CHX/TNFa (10 ⁇ /25 ng/ml) for 4 hours and analysed for PARP cleavage.
- C) Mock infected or HHV-6B infected HCT1 16 cells (48 hpi) were treated with Camptothesine and analysed for PARP cleavage.
- HCT1 16 cells were either mock infected or infected with HHV-6B for 48 hpi and treated with CHX/TNFa (10 ⁇ /25 ng/ml) for 4 hours and analysed for PARP cleavage, caspase-8 cleavage, caspase-3 cleavage, ⁇ phosphorylation, total ⁇ , 7C7 (infection marker) and RCC1 (loading control).
- Mock or HHV-6B infected cells 48 hpi
- HCT1 16 cells transfected with U20-V5 and analysed by lysate fractionation into cytoplasm, membranes and nuclear fractions. WB were stained with anti-V5 antibody, and antibodies against GAPDH, COX IV and RCC1 as fraction purity controls.
- F & G HCT1 16 cells transfected with a U20 expression plasmid for 48 hours, treated with CHX/TNF (10 ⁇ /25 ng/ml) for 4 hours and analysed for PARP cleavage and 7-AAD incorporation by flowcytometry (average of four independent experiments).
- H PCR with U20 end-specific primers on cDNA from a stable U20 expressing clone (U20-S).
- CHX/TNFalpha (10 ⁇ /25 ng/ml) for 4 hours and analysed for the cleavage of caspase-8 and -3 and the phosphorylation of ⁇ .
- C Wt and U20-S cells treated with CHX/TNF (10 ⁇ /25 ng/ml) for 4 hours and analysed with antibodies against PARP, RIP, TRAF2, TRADD, FADD and GAPDH.
- U20 inhibits TNFalpha internalization.
- A) Flowcytometry analysis of wt and U20-S cells treated with TNF -biotin for 60 min at 4°C and stained with avidin-FITC for 30 min at 4 ⁇ C.
- isolated as used herein in reference to nucleic acids or proteins, is intended to refer to a polynucleotide in which the nucleotide sequences encoding the protein or polypeptide is free of other nucleotide sequences which may naturally flank the nucleic acid in genomic DNA.
- sequence identity indicates a quantitative measure of the degree of homology between two amino acid sequences or between two nucleic acid sequences of equal length. If the two sequences to be compared are not of equal length, they must be aligned to give the best possible fit, allowing the insertion of gaps or, alternatively, truncation at the ends of the polypeptide sequences or nucleotide sequences.
- quence identity can be calculated as Nre ⁇ , wherein Ndif is the total number of non-identical residues in the two sequences when aligned and wherein Nref is the number of residues in one of the sequences, preferably sequence identity is calculated over the full length reference as provided herein.
- the percentage of sequence identity between one or more sequences may also be based on alignments using the clustalW software (http:/www.ebi.ac.uk/clustalW/index.html) with default settings. For nucleotide sequence alignments these settings are:
- Sequence identity is determined in one embodiment by utilising fragments of SEQ ID NO:3 peptides comprising at least 25 contiguous amino acids and having an amino acid sequence which is at least 80%, such as 85%, for example 90%, such as 95%, for example 99% identical to the amino acid sequence of SEQ ID NO: 1 , wherein the percent identity is determined with the algorithm GAP, BESTFIT, or FASTA in the Wisconsin Genetics Software Package Release 7.0, using default gap weights.
- predetermined sequence is a defined sequence used as a basis for a sequence comparison; a predetermined sequence may be a subset of a larger sequence, for example, as a segment of a full-length DNA or gene sequence given in a sequence listing, such as a polynucleotide sequence of SEQ ID NO:2, or may comprise a complete DNA or gene sequence. Generally, a predetermined sequence is at least 20 nucleotides in length, frequently at least 25 nucleotides in length, and often at least 50 nucleotides in length. Likewise, the predetermined seequence is that of the
- polypeptides of the invention are polypeptides of the invention.
- two polynucleotides may each (1 ) comprise a sequence (i.e., a portion of the complete polynucleotide sequence) that is similar between the two polynucleotides, and (2) may further comprise a sequence that is divergent between the two polynucleotides
- sequence comparisons between two (or more) polynucleotides are typically performed by comparing sequences of the two polynucleotides over a
- a “comparison window” to identify and compare local regions of sequence similarity.
- a “comparison window”, as used herein, refers to a conceptual segment of at least 20 contiguous nucleotide positions wherein a polynucleotide sequence may be compared to a predetermined sequence of at least 20 contiguous nucleotides and wherein the portion of the polynucleotide sequence in the comparison window may comprise additions or deletions (i.e., gaps) of 20 percent or less as compared to the
- predetermined sequence which does not comprise additions or deletions
- Optimal alignment of sequences for aligning a comparison window may be conducted by the local homology algorithm of Smith and Waterman (1981 ) Adv. Appl. Math. 2: 482, by the homology alignment algorithm of Needleman and Wunsch (1970) J. Mol. Biol. 48: 443, by the search for similarity method of Pearson and Lipman (1988) Proc. Natl. Acad. Sci.
- BLAST Basic Local Alignment Search Tool
- Homologs of the disclosed polypeptides are typically characterised by possession of at least 94% sequence identity counted over the full length alignment with the disclosed amino acid sequence using the NCBI Basic Blast 2.0, gapped blastp with databases such as the nr or swissprot database. Alternatively, one may manually align the sequences and count the number of identical amino acids. This number divided by the total number of amino acids in your sequence multiplied by 100 results in the percent identity.
- expression vector refers to a DNA molecule used as a vehicle to transfer recombinant genetic material into a host cell.
- the four major types of vectors are plasmids, bacteriophages and other viruses, cosmids, and artifical chromosomes.
- the vector itself is generally a DNA sequence that consists of an insert (a heterologous nucleic acid sequence, transgene) and a larger sequence that serves as the
- backbone of the vector.
- the purpose of a vector which transfers genetic information to the host is typically to isolate, multiply, or express the insert in the target cell.
- Vectors called expression vectors are specifically adapted for the expression of the heterologous sequences in the target cell, and generally have a promoter sequence that drives expression of the heterologous sequences.
- Simpler vectors called transcription vectors are only capable of being transcribed but not translated: they can be replicated in a target cell but not expressed, unlike expression vectors. Transcription vectors are used to amplify the inserted heterologous
- transcripts may subsequently be isolated and used in as templates suitable in vitro translations systems.
- recombinant when used with reference to a cell, or nucleic acid, peptide or vector, indicates that the cell, or nucleic acid, peptide or vector, has been modified by the introduction of a heterologous nucleic acid or the alteration of a native nucleic acid, or that the cell is derived from a cell so modified.
- recombinant cells express genes that are not found within the native (non-recombinant) form of the cell or express native genes that are otherwise abnormally expressed, under expressed or not expressed at all.
- the expression “recombinant” also relates to a cell, wherein further regulatory elements have been included in order to initiate or enhance expression of an otherwise silent endogenous gene, or wherein a manipulation of the regulatory elements have been performed for the same purpose.
- hybridization under stringent conditions means that after washing for 1 h with 1 times SSC and 0.1 % SDS at 50 degree C, preferably at 55 degree C, more preferably at 62degree C and most preferably at 68 degree C, particularly for 1 h in 0.2 times SSC and 0.1 % SDS at 50 degree C, preferably at 55 degree C, more preferably at 62 degree C and most preferably at 68 degree C, a positive hybridization signal is observed.
- a nucleotide sequence which hybridizes under the above washing conditions with the nucleotide sequence of SEQ ID NO:2 (HHV 6B U20 gene) and/or SEQ ID NO:7 (HHV 6B U20 mRNA) or a nucleotide sequence corresponding thereto in the scope of the degeneracy of the genetic code is encompassed by the present invention.
- nucleic acid or “polynucleotide” refers to polynucleotides, such as deoxyribonucleic acid (DNA) or ribonucleic acid (RNA), oligonucleotides, fragments generated by the polymerase chain reaction (PCR), and fragments generated by any of ligation, scission, endonuclease action, and exonuclease action.
- DNA deoxyribonucleic acid
- RNA ribonucleic acid
- PCR polymerase chain reaction
- Polynucleotides can be composed of monomers that are naturally-occurring nucleotides (such as DNA and RNA), or analogs of naturally-occurring nucleotides (e.g., (alpha-enantiomeric forms of naturally-occurring nucleotides), or a combination of both.
- Modified nucleotides can have alterations in sugar moieties and/or in pyrimidine or purine base moieties.
- Sugar modifications include, for example, replacement of one or more hydroxyl groups with halogens, alkyl groups, amines, and azido groups, or sugars can be functionalized as ethers or esters.
- the entire sugar moiety can be replaced with sterically and electronically similar structures, such as aza-sugars and carbocyclic sugar analogs.
- nucleic acid monomers can be linked by phosphodiester bonds or analogs of such linkages. Analogs of phosphodiester linkages include phosphorothioate, phosphorodithioate, phosphoroselenoate, phosphorodiselenoate, phosphoroanilothioate, phosphoranilidate, phosphoramidate, and the like.
- polynucleotide also includes so-called "peptide nucleic acids,” which comprise naturally-occurring or modified nucleic acid bases attached to a polyamide backbone. Nucleic acids can be either single stranded or double stranded.
- natural nucleotide refers to any of the four deoxyribonucleotides, dA, dG, dT, and dC (constituents of DNA), and the four ribonucleotides, A, G, U, and C
- RNA Ribonuents of RNA
- Each natural nucleotide comprises or essentially consists of a sugar moiety (ribose or deoxyribose), a phosphate moiety, and a natural/standard base moiety.
- Natural nucleotides bind to complementary nucleotides according to well-known rules of base pairing (Watson and Crick), where adenine (A) pairs with thymine (T) or uracil (U); and where guanine (G) pairs with cytosine (C), wherein corresponding base-pairs are part of complementary, anti-parallel nucleotide strands.
- the base pairing results in a specific hybridization between predetermined and complementary nucleotides.
- the base pairing is the basis by which enzymes are able to catalyze the synthesis of an oligonucleotide complementary to the template oligonucleotide.
- building blocks normally the triphosphates of ribo or deoxyribo derivatives of A, T, U, C, or G
- A, T, U, C, or G building blocks
- the recognition of an oligonucleotide sequence by its complementary sequence is mediated by corresponding and interacting bases forming base pairs.
- base pairing In nature, the specific interactions leading to base pairing are governed by the size of the bases and the pattern of hydrogen bond donors and acceptors of the bases.
- a six membered ring a pyrimidine in natural oligonucleotides
- a five membered ring a purine in natural
- oligonucleotides with a middle hydrogen bond linking two ring atoms, and hydrogen bonds on either side joining functional groups appended to each of the rings, with donor groups paired with acceptor groups.
- complement of a polynucleotide refers to a polynucleotide having a complementary nucleotide sequence and reverse orientation as compared to a reference nucleotide sequence.
- sequence 5' ATGCACGGG 3' is complementary to 5' CCCGTGCAT 3'.
- degenerate nucleotide sequence denotes a sequence of nucleotides that includes one or more degenerate codons as compared to a reference polynucleotide that encodes a polypeptide.
- Degenerate codons contain different triplets of nucleotides, but encode the same amino acid residue (i.e., GAU and GAC triplets each encode Asp).
- structural gene refers to a polynucleotide that is transcribed into messenger RNA (mRNA), which is then translated into a sequence of amino acids characteristic of a specific polypeptide.
- a "polynucleotide construct” is a polynucleotide, either single- or double-stranded, that has been modified through human intervention to contain segments of nucleic acid combined and juxtaposed in an arrangement not existing in nature.
- Codon DNA is a single-stranded DNA molecule that is formed from an mRNA template by the enzyme reverse transcriptase. Typically, a primer complementary to portions of mRNA is employed for the initiation of reverse
- cDNA refers to a double- stranded DNA molecule consisting of such a single-stranded DNA molecule and its complementary DNA strand.
- cDNA also refers to a clone of a cDNA molecule synthesized from an RNA template.
- a nucleotide is herein defined as a monomer of RNA or DNA.
- a nucleotide is a ribose or a deoxyribose ring attached to both a base and a phosphate group. Both mono-, di-, and tri-phosphate nucleosides are referred to as nucleotides.
- nucleotides' refers to both natural nucleotides and non- natural nucleotides capable of being incorporated - in a template-directed manner - into an oligonucleotide, preferably by means of an enzyme comprising DNA or RNA dependent DNA or RNA polymerase activity, including variants and functional equivalents of natural or recombinant DNA or RNA polymerases.
- Corresponding binding partners in the form of coding elements and complementing elements comprising a nucleotide part are capable of interacting with each other by means of hydrogen bonds. The interaction is generally termed "base-pairing".
- Nucleotides may differ from natural nucleotides by having a different phosphate moiety, sugar moiety and/or base moiety. Nucleotides may accordingly be bound to their respective neighbour(s) in a template or a complementing template by a natural bond in the form of a phosphodiester bond, or in the form of a non-natural bond, such as e.g. a peptide bond as in the case of PNA (peptide nucleic acids). Nucleotides according to the invention includes ribonucleotides comprising a nucleobase selected from the group consisting of adenine (A), uracil (U), guanine (G), and cytosine (C), and
- deoxyribonucleotide comprising a nucleobase selected from the group consisting of adenine (A), thymine (T), guanine (G), and cytosine (C).
- Nucleobases are capable of associating specifically with one or more other nucleobases via hydrogen bonds. Thus it is an important feature of a nucleobase that it can only form stable hydrogen bonds with one or a few other nucleobases, but that it can not form stable hydrogen bonds with most other nucleobases usually including itself.
- base-pairing The specific interaction of one nucleobase with another nucleobase is generally termed "base-pairing". The base pairing results in a specific hybridisation between predetermined and complementary nucleotides.
- nucleotides that comprise nucleobases that are capable of base-pairing.
- nucleobases that are capable of base-pairing.
- A adenine
- T thymine
- U uracil
- G guanine
- C cytosine
- a nucleotide comprising A is complementary to a nucleotide comprising either T or U
- a nucleotide comprising G is complementary to a nucleotide comprising C.
- oligonucleotide refers to a single stranded or double stranded oligomer or polymer of ribonucleic acid (RNA) or deoxyribonucleic acid (DNA) or mimetics thereof.
- RNA ribonucleic acid
- DNA deoxyribonucleic acid
- oligonucleotides composed of naturally-occurring bases, sugars and covalent internucleoside linkages (e.g., backbone) as well as oligonucleotides having non-naturally-occurring portions which function similarly to respective naturally-occurring portions (see disclosed in U.S. Pat. Nos. 4,469,863; 4,476,301 ; 5,023,243; 5,177,196; 5,188,897; 5,264,423; 5,276,019; 5,278,302;
- oligonucleotide thus also refers to any
- oligonucleotides of natural and non-natural nucleotides may be linked by natural phosphodiester bonds or by non-natural bonds.
- Preferred oligonucleotides comprise only natural nucleotides linked by phosphodiester bonds.
- Oligonucleotide is used interchangeably with polynucleotide.
- the oligomer or polymer sequences of the present invention are formed from the chemical or enzymatic addition of monomer subunits.
- oligonucleotide as used herein includes linear oligomers of natural or modified monomers or linkages, including deoxyribonucleotides, ribonucleotides, anomeric forms thereof, peptide nucleic acid monomers (PNAs), locked nucleotide acid monomers (LNA), and the like, capable of specifically binding to a single stranded polynucleotide tag by way of a regular pattern of monomer-to-monomer interactions, such as Watson-Crick type of base pairing, base stacking, Hoogsteen or reverse Hoogsteen types of base pairing, or the like.
- PNAs peptide nucleic acid monomers
- LNA locked nucleotide acid monomers
- oligonucleotides ranging in size from a few monomeric units, e.g. 3-4, to several tens of monomeric units, e.g. 40-60.
- ATGCCTG an oligonucleotide is represented by a sequence of letters, such as "ATGCCTG,” it will be understood that the nucleotides are in 5' ⁇ 3' order from left to right and the "A” denotes deoxyadenosine, "C” denotes deoxycytidine, “G” denotes deoxyguanosine, and "T” denotes thymidine, unless otherwise noted.
- oligonucleotides of the invention comprise the four natural nucleotides; however, they may also comprise methylated or non-natural nucleotide analogs.
- Suitable oligonucleotides may be prepared by the phosphoramidite method described by Beaucage and Carruthers (Tetrahedron Lett., 22, 1859-1862, 1981 ), or by the triester method according to Matteucci, et al. (J. Am. Chem. Soc, 103, 3185, 1981 ), both incorporated herein by reference, or by other chemical methods using either a commercial automated oligonucleotide synthesizer or VLSIPS.TM. technology.
- double-stranded When oligonucleotides are referred to as “double-stranded,” it is understood by those of skill in the art that a pair of oligonucleotides exist in a hydrogen-bonded, helical configuration typically associated with, for example, DNA.
- double-stranded As used herein is also meant to refer to those forms which include such structural features as bulges and loops. For example as described in US 5.770.722 for a unimolecular double-stranded DNA. It is clear to those skilled in the art when oligonucleotides having natural or non-natural nucleotides may be employed, e.g.
- oligonucleotides consisting of natural nucleotides are required.
- nucleotides are conjugated together in a string using synthetic procedures, they are always referred to as oligonucleotides.
- a plurality of individual nucleotides linked together in a single molecule may form a polynucleotide.
- Polynucleotide covers any derivatized nucleotides such as DNA, RNA, PNA, LNA etc. Any oligonucleotide is also a polynucleotide, but every polynucleotide is not an oligonucleotide.
- polypeptide peptide
- protein protein
- amino acid polymers in which one or more amino acid residue is an artificial chemical analogue of a corresponding naturally occurring amino acid, as well as to naturally occurring amino acid polymers.
- Amino acid Entity comprising an amino terminal part (NH 2 ) and a carboxy terminal part (COOH) separated by a central part comprising a carbon atom, or a chain of carbon atoms, comprising at least one side chain or functional group.
- NH 2 refers to the amino group present at the amino terminal end of an amino acid or peptide
- COOH refers to the carboxy group present at the carboxy terminal end of an amino acid or peptide.
- the generic term amino acid comprises both natural and non-natural amino acids. Natural amino acids of standard nomenclature as listed in J. Biol.
- amino acid residue is meant to encompass amino acids, either standard amino acids, non-standard amino acids or pseudo-amino acids, which have been reacted with at least one other species, such as 2, for example 3, such as more than 3 other species.
- amino acid residues may comprise an acyl bond in place of a free carboxyl group and/or an amine-bond and/or amide bond in place of a free amine group.
- reacted amino acids residues may comprise an ester or thioester bond in place of an amide bond
- isolated polypeptide is a polypeptide that is essentially free from contaminating cellular components, such as carbohydrate, lipid, or other proteinaceous impurities associated with the polypeptide in nature.
- a preparation of isolated polypeptide contains the polypeptide in a highly purified form, i.e., at least about 80% pure, at least about 90% pure, at least about 95% pure, greater than 95% pure, or greater than 99% pure.
- SDS sodium dodecyl sulfate
- amino-terminal and “carboxyl-terminal” are used herein to denote positions within polypeptides. Where the context allows, these terms are used with reference to a particular sequence or portion of a polypeptide to denote proximity or relative position. For example, a certain sequence positioned carboxyl-terminal to a reference sequence within a polypeptide is located proximal to the carboxyl terminus of the reference sequence, but is not necessarily at the carboxyl terminus of the complete polypeptide.
- variants of U20 gene variants of fragments thereof When being polypeptides, variants are determined on the basis of their degree of identity or their homology with a predetermined amino acid sequence, said predetermined amino acid sequence being of SEQ ID NO: 1 , 3 or 5, when the variant is a fragment, a fragment of any of the aforementioned amino acid sequences, respectively.
- a "fusion protein” is a hybrid protein expressed by a polynucleotide comprising nucleotide sequences of at least two genes.
- a fusion protein can comprise at least part of a polypeptide according to the present invention fused with a polypeptide that binds an affinity matrix.
- Such a fusion protein provides a means to isolate large quantities of a polypeptide according to the present invention using affinity chromatography.
- complement/anti-complement pair denotes non-identical moieties that form a non-covalently associated, stable pair under appropriate conditions.
- biotin and avidin are prototypical members of a complement/anti- complement pair.
- Other exemplary complement/anti-complement pairs include receptor/ligand pairs, antibody/antigen (or hapten or epitope) pairs, sense/antisense polynucleotide pairs, and the like.
- the complement/anti-complement pair preferably has a binding affinity of less than 10 9 M " .
- affinity tag is used herein to denote a polypeptide segment that can be attached to a second polypeptide to provide for purification or detection of the second polypeptide or provide sites for attachment of the second polypeptide to a substrate.
- affinity tag any peptide or protein for which an antibody or other specific binding agent is available can be used as an affinity tag.
- Affinity tags include a poly-histidine tract, protein A (Nilsson et al., EMBO J. 4:1075 (1985); Nilsson et al., Methods Enzymol.
- recombinant host cell (or simply “host cell”), as used herein, is intended to refer to a cell into which a vector has been introduced. It should be understood that such terms are intended to refer not only to the particular subject cell but to the progeny of such a cell. Because certain modifications may occur in succeeding generations due to either mutation or environmental influences, such progeny may not, in fact, be identical to the parent cell, but are still included within the scope of the term "host cell” as used herein. Preferably host cells are used for the expression of the proteins of the invention.
- operatively linked is intended to mean that the two DNA fragments are joined such that the amino acid sequences encoded by the two DNA fragments remain in-frame. Similarly, two linked polypeptides maintain in frame when they are operably linked.
- regulatory sequence is intended to include promoters, enhancers and other expression control elements (e.g., polyadenylation signals) that control the transcription or translation of the antibody chain genes. It will be appreciated by those skilled in the art that the design of the expression vector, including the selection of regulatory sequences may depend on such factors as the choice of the host cell to be
- Preferred regulatory sequences for mammalian host cell expression include viral elements that direct high levels of protein expression in mammalian cells, such as promoters and/or enhancers derived from cytomegalovirus (CMV) (such as the CMV promoter/enhancer), Simian Virus 40 (SV40) (such as the SV40 promoter/enhancer), adenovirus, (e.g., the adenovirus major late promoter (AdMLP)) and polyoma.
- CMV cytomegalovirus
- SV40 Simian Virus 40
- AdMLP adenovirus major late promoter
- human TNFa (abbreviated herein as hTNFa, hTNF-alpha, hTNF- a, or simply hTNF), as used herein, is intended to refer to a human cytokine that exists as a 17 kD secreted form and a 26 kD membrane associated form, the biologically active form of which is composed of a trimer of noncovalently bound 17 kD molecules.
- human TNFa is intended to include recombinant human TNFa (rhTNFa). rhTNFa can be prepared by standard recombinant expression methods or purchased commercially. TNFa receptor
- TNFa receptor refers to two known forms of TNF receptors, also known as TNFR1 (or p55 or TNFRSF1 A) and TNFR2 (or p75 or TNFRSF1 B).
- linker refers to an amino acid or nucleic acid sequences linking in frame two amino acid sequences or two nucleic acid sequences respectively.
- Linkers may comprise one or more sequences enabling purification, increased solubility, detection or separation of domains. Examples of such sequences are epitopes for one or more antibodies, restriction enzymes, purification tags or sequences predicted to form alpha helices or disorganised domains.
- the length of a linker may be 1 -500 amino acids, 1 -200 amino acids, such as 1 -150, such as 1 -100, such as 1 -80, such as 1 -60, such as 1 -40, such as 5-40.
- diagnostic refers in the present context to a compound or composition used in diagnosis of a disease or medical state.
- the diagnosticum is the U20 protein or active derivative thereof for use in the diagnosis of a disease or condition.
- biologically active refers to variants and fragments of polypeptides and polynucleotides of the present invention. Such variants and fragments of polypeptides and polynucleotides are also suitable for use according to the invention, insofar as these polypeptides display substantially the same biological activity.
- substantially same biologically activity can be defined as having an activity being at least 20% of the full length U20 polypeptide or polynucleotides, such as at least 30%, such as at least 40%, such as at least 50%, such as at least 60%, such as at least 70%, such as at least 80%, or such as at least 90%.
- active derivatives may also have a higher activity than the full length U20 polypeptide or polynucleotides such as at the most 20% more activity, such as at the most 40% more activity, such as at the most 60% more activity, such as at the most 80% more activity, such as at the most 100% more activity, such as at the most 150% more activity, or such as at the most 200%, or such as at the most 500% more activity than the full length U20 polypeptide or polynucleotides.
- the biological activity may be measured as the ability of the polypeptides or polynucleotides to influence TNF receptor activity. The biological activity can thus be measured as the level of apoptosis in response to signalling throug the TNF receptor.
- example 1 and figure 2 providing an assay for measuring the level of apoptosis, where cells have been induced to apoptosis in the presence of hTNF-alpha.
- the biologic activity of the polypeptides or polynucleotides to influence TNF receptor activity can also be measured by examining the activation state of proteins involved in the TNF receptor signal transduction.
- FIG 4A This invention pertains to isolated U20 proteins or active derivatives thereof that bind to human TNFa-receptor (TNFR1 ).
- U20 proteins or derivatives thereof for use as a medicament or diagnosticum, pharmaceutical compositions, as well as nucleic acids, recombinant expression vectors and host cells for making such U20 proteins and derivatives.
- the invention furthermore relates to the U20 proteins or active derivatives thereof as part of pharmaceutical and diagnostic kits. Methods of using the U20 protein according to the invention to detect human TNFa receptor or to inhibit the interaction between TNFa and the receptor, either in vitro or in vivo, are also encompassed by the invention.
- HHV U20 In addition to full-length HHV U20, substantially full-length HHV U20, to pro- HHV U20, to C-terminal peptides, to N-terminal peptides and to truncated forms of HHV U20, the present invention provides for biologically active variants of the polypeptides.
- An HHV U20 polypeptide or fragment is biologically active if it exhibits a biological activity of naturally occurring HHV U20. It is to be understood that the invention relates to substantially purified HHV U20as herein defined.
- One biological activity is the ability to compete with naturally occurring HHV U20 in a receptor-binding assay.
- Biologically active variants may also be defined with reference to one or more of the other in vitro and/or in vivo biological assays described in the examples.
- a preferred biological activity is the ability to elicit substantially the same response as in the apoptosis assay described in the Examples and Figures 2, and 6.
- the polypeptides according to the present invention can be used as a medicament where the medicament is active by modulating, or interfering with the activity of TNF receptors.
- the invention relates to an isolated polypeptide for use as medicament or diagnosticum comprising
- SEQ ID NO: 1 is the amino acid sequence of the U20 protein from HHV-6B (NCBI accession number NP 050200.1 )
- SEQ ID NO: 3 is the amino acid sequence of the U20 protein from HHV-6A (NCBI accession number NP_042913.1 )
- SEQ ID NO: 5 is the amino acid sequence of the U20 protein from HHV-7 (NCBI accession number YP_073760.1 ).
- An alignment of U20 from HHV-6B, HHV-6A and HHV-7 is shown in figure 9.
- polypeptides of the present invention are in one embodiment modified so as to preferably improve the resistance to proteolytic degradation and stability or to optimize solubility properties or to render the polypeptide more suitable as a therapeutic agent.
- the polypeptide may comprise amino acid residues other than naturally occurring L-amino acid residues.
- the polypeptides may comprise D-amino acid residues.
- the polypeptides may also comprise non-naturally occurring, synthetic amino acids.
- the polypeptides may further comprising one or more blocking groups, preferably in the form of chemical substituents suitable to protect and/or stabilize the N- and C- termini of the polypeptide from undesirable degradation.
- One or more blocking groups include protecting groups which do not adversely affect in vivo activities of the polypeptide.
- the one or more blocking groups are for example introduced by alkylation or acylation of the N-terminus.
- Such N-terminal blocking groups are selected from N- terminal blocking groups comprising Ci to C 5 branched or non-branched alkyl groups and acyl groups, such as formyl and acetyl groups, as well as substituted forms thereof, such as the acetamidomethyl (Acm) group.
- one or more blocking groups are selected from N-terminal blocking groups comprising desamino analogs of amino acids, which are either coupled to the N-terminus of the peptide or used in place of the N-terminal amino acid residue.
- the polypeptides may also comprise C-terminal blocking groups, wherein the carboxyl group of the C-terminus is either incorporated or not, such as esters, ketones, and amides, as well as descarboxylated amino acid analogues.
- the one or more blocking groups are selected from C-terminal blocking groups comprising ester or ketone- forming alkyl groups, such as lower (Ci to C 6 ) alkyl groups, for example methyl, ethyl and propyl, and amide-forming amino groups, such as primary amines (-NH 2 ), and mono- and di-alkylamino groups, such as methylamino, ethylamino, dimethylamino, diethylamino, methylethylamino, and the like.
- C-terminal blocking groups comprising ester or ketone- forming alkyl groups, such as lower (Ci to C 6 ) alkyl groups, for example methyl, ethyl and propyl, and amide-forming amino groups, such as primary amines (-NH 2 ), and mono- and di-alkylamino groups, such as methylamino, ethylamino, dimethylamino, diethylamino,
- Free amino group(s) at the N-terminal end and free carboxyl group(s) at the termini can be removed altogether from the polypeptide to yield desamino and descarboxylated forms thereof without significantly affecting the biological activity of the polypeptide.
- Functional assays ie. assays for the biological activity
- U20 function is conserved.
- variant refers to polypeptides or proteins which are homologous to the basic protein, which is suitably U20 (such as SEQ ID NO.: 1 , 3 or 5), but which differs from the base sequence from which they are derived in that one or more amino acids within the sequence are substituted for other amino acids.
- Amino acid substitutions may be regarded as "conservative” where an amino acid is replaced with a different amino acid with broadly similar properties. Non-conservative substitutions are where amino acids are replaced with amino acids of a different type. Broadly speaking, fewer non-conservative substitutions will be possible without altering the biological activity of the polypeptide.
- amino acids may be grouped according to shared characteristics.
- a conservative amino acid substitution is a substitution of one amino acid within a predetermined group of amino acids for another amino acid within the same group, wherein the amino acids within a predetermined groups exhibit similar or substantially similar characteristics.
- Conservative amino acid substitutions refer to the interchangeability of residues having similar side chains.
- a group of amino acids having aliphatic side chains is glycine, alanine, valine, leucine, and isoleucine; a group of amino acids having aliphatic-hydroxyl side chains is serine and threonine, a group of amino acids having amide-containing side chains is asparagine and glutamine; a group of amino acids having aromatic side chains is phenylalanine, tyrosine, and tryptophan; a group of amino acids having basic side chains is lysine, arginine, and histidine; and a group of amino acids having sulfur-containing side chains is cysteine and methionine.
- Preferred conservative amino acids substitution groups are: valine-leucine-isoleucine, phenylalanine-tyrosine, lysine-arginine, alanine-valine, and asparagine-glutamine.
- Amino acids having polar side chains (Asp, Glu, Lys, Arg, His, Asn, Gin, Ser, Thr, Tyr, and Cys,)
- Amino acids having non-polar side chains (Gly, Ala, Val, Leu, lie, Phe, Trp, Pro, and Met)
- Hydrophobic amino acids Al, Cys, Gly, lie, Leu, Met, Phe, Pro, Trp, Tyr, Val
- Hydrophilic amino acids Arg, Ser, Thr, Asn, Asp, Gin, Glu, His, Lys
- Preferred conservative amino acids substitution groups are: valine-leucine-isoleucine, phenylalanine-tyrosine, lysine-arginine, alanine-valine, and asparagine-glutamine.
- the alignment in Figure 9 can be used to predict which amino acid residues can be substituted without substantially affecting the biological acitivity of the protein.
- a variant or a fragment thereof according to the invention may comprise, within the same variant of the sequence or fragments thereof, or among different variants of the sequence or fragments thereof, at least one substitution, such as a plurality of substitutions introduced independently of one another.
- nonstandard amino acids include the sulfur-containing taurine and the neurotransmitters GABA and dopamine. Other examples are lanthionine, 2-Aminoisobutyric acid, and dehydroalanine. Further non standard amino are ornithine and citrulline.
- Non-standard amino acids are usually formed through modifications to standard amino acids. For example, taurine can be formed by the decarboxylation of cysteine, while dopamine is synthesized from tyrosine and hydroxyproline is made by a
- proline common in collagen
- non-natural amino acids are those listed e.g. in 37 C.F.R. section 1 .822(b)(4), all of which are incorporated herein by reference.
- a functional equivalent according to the invention may comprise any amino acid including non-standard amino acids. In preferred embodiments a functional equivalent comprises only standard amino acids.
- the standard and/or non-standard amino acids may be linked by peptide bonds or by non-peptide bonds.
- the term peptide also embraces post-translational modifications introduced by chemical or enzyme-catalyzed reactions, as are known in the art. Such post-translational modifications can be introduced prior to partitioning, if desired.
- Amino acids as specified herein will preferentially be in the L-stereoisomeric form.
- Amino acid analogs can be employed instead of the 20 naturally-occurring amino acids. Several such analogs are known, including fluorophenylalanine, norleucine, azetidine-2- carboxylic acid, S-aminoethyl cysteine, 4-methyl tryptophan and the like.
- variants are variant(s) of SEQ ID NO:1 , 3 or 5 having at least 80% sequence identity, such as preferably at least 81 % sequence identity, more preferably e.g. at least 82% sequence identity, such as more preferably at least 83% sequence identity, e.g. more preferably at least 84% sequence identity, more preferably such as at least 85% sequence identity, more preferably e.g. at least 86% sequence identity, more preferably such as at least 87% sequence identity, more preferably e.g. at least 88% sequence identity, more preferably such as at least 89% sequence identity, more preferably e.g. at least 90% sequence identity, more preferably such as at least 91 % sequence identity, more preferably e.g.
- sequence identity such as at least 93% sequence identity, more preferably e.g. at least 94% sequence identity, more preferably such as at least 95% sequence identity, more preferably e.g. at least 96% sequence identity, more preferably such as at least 97% sequence identity, more preferably e.g. at least 98% sequence identity, more preferably such as at least 99% sequence identity, more preferably e.g. at least 99.5% sequence identity identity with the predetermined sequence of U20 from HHV (SEQ ID No: 1 , 3 and 5).
- variants are variant(s) of SEQ ID NO:1 , 3 or 5 having at least 80% sequence identity, such as preferably at least 81 % sequence identity, more preferably e.g. at least 82% sequence identity, such as more preferably at least 83% sequence identity, e.g. more preferably at least 84% sequence identity, more preferably such as at least 85% sequence identity, more preferably e.g. at least 86% sequence identity, more preferably such as at least 87% sequence identity, more preferably e.g. at least 88% sequence identity, more preferably such as at least 89% sequence identity, more preferably e.g. at least 90% sequence identity, more preferably such as at least 91 % sequence identity, more preferably e.g.
- sequence identity such as at least 93% sequence identity, more preferably e.g. at least 94% sequence identity, more preferably such as at least 95% sequence identity, more preferably e.g. at least 96% sequence identity, more preferably such as at least 97% sequence identity, more preferably e.g. at least 98% sequence identity, more preferably such as at least 99% sequence identity, more preferably e.g. at least 99.5% sequence identity identity with the predetermined sequence of U20 from HHV 6B (SEQ ID No: 1 ).
- variants are variant(s) of SEQ ID NO:1 , 3 or 5 having at least 80% sequence identity, such as preferably at least 81 % sequence identity, more preferably e.g. at least 82% sequence identity, such as more preferably at least 83% sequence identity, e.g. more preferably at least 84% sequence identity, more preferably such as at least 85% sequence identity, more preferably e.g. at least 86% sequence identity, more preferably such as at least 87% sequence identity, more preferably e.g. at least 88% sequence identity, more preferably such as at least 89% sequence identity, more preferably e.g. at least 90% sequence identity, more preferably such as at least 91 % sequence identity, more preferably e.g.
- sequence identity such as at least 93% sequence identity, more preferably e.g. at least 94% sequence identity, more preferably such as at least 95% sequence identity, more preferably e.g. at least 96% sequence identity, more preferably such as at least 97% sequence identity, more preferably e.g. at least 98% sequence identity, more preferably such as at least 99% sequence identity, more preferably e.g. at least 99.5% sequence identity identity with the predetermined sequence of U20 from HHV6A (SEQ ID No: 3).
- variants are variant(s) of SEQ ID NO:1 , 3 or 5 having at least 80% sequence identity, such as preferably at least 81 % sequence identity, more preferably e.g. at least 82% sequence identity, such as more preferably at least 83% sequence identity, e.g. more preferably at least 84% sequence identity, more preferably such as at least 85% sequence identity, more preferably e.g. at least 86% sequence identity, more preferably such as at least 87% sequence identity, more preferably e.g. at least 88% sequence identity, more preferably such as at least 89% sequence identity, more preferably e.g. at least 90% sequence identity, more preferably such as at least 91 % sequence identity, more preferably e.g.
- sequence identity such as at least 93% sequence identity, more preferably e.g. at least 94% sequence identity, more preferably such as at least 95% sequence identity, more preferably e.g. at least 96% sequence identity, more preferably such as at least 97% sequence identity, more preferably e.g. at least 98% sequence identity, more preferably such as at least 99% sequence identity, more preferably e.g. at least 99.5% sequence identity identity with the predetermined sequence of U20 from HHV 7 (SEQ ID No: 5).
- the polypeptide fragment according to the invention wherein the fragment has a stretch of at least 50 contiguous amino acids contains less than 410 consecutive amino acid residues of SEQ ID NO: 1 , 3 or 5, such as less than 400 consecutive amino acid residues, such as less than 395 consecutive amino acid residues, e.g. less than 390 consecutive amino acid residues, such as less than 385 consecutive amino acid residues, e.g. less than 380 consecutive amino acid residues, such as less than 370 consecutive amino acid residues, e.g. less than 360 consecutive amino acid residues, such as less than 350 consecutive amino acid residues, e.g. less than 345 consecutive amino acid residues, such as less than 340 consecutive amino acid residues, e.g.
- less than 335 consecutive amino acid residues such as less than 330 consecutive amino acid residues, e.g. less than 325 consecutive amino acid residues, such as less than 300 consecutive amino acid residues, e.g. less than 295 consecutive amino acid residues, such as less than 290 consecutive amino acid residues, e.g. less than 285 consecutive amino acid residues, such as less than 280 consecutive amino acid residues, e.g. less than 275 consecutive amino acid residues, such as less than 270 consecutive amino acid residues, e.g. less than 265 consecutive amino acid residues, such as less than 260 consecutive amino acid residues, such as less than 255 consecutive amino acid residues, e.g.
- less than 250 consecutive amino acid residues such as less than 245 consecutive amino acid residues, e.g. less than 240 consecutive amino acid residues, such as less than 235 consecutive amino acid residues, e.g. less than 230 consecutive amino acid residues, such as less than 225 consecutive amino acid residues, such as less than 220 consecutive amino acid residues, such as less than 215 consecutive amino acid residues, e.g. less than 210 consecutive amino acid residues, such as less than 205 consecutive amino acid residues, e.g. less than 200 consecutive amino acid residues, such as less than 195 consecutive amino acid residues, e.g. less than 190 consecutive amino acid residues, such as less than 185 consecutive amino acid residues, e.g.
- less than 180 consecutive amino acid residues such as less than 175 consecutive amino acid residues, e.g. less than 170 consecutive amino acid residues, such as less than 165 consecutive amino acid residues, e.g. less than 160 consecutive amino acid residues, such as less than 155 consecutive amino acid residues, e.g. less than 150 consecutive amino acid residues, such as less than 145 consecutive amino acid residues, e.g. less than 140 consecutive amino acid residues, such as less than 135 consecutive amino acid residues, e.g. less than 130 consecutive amino acid residues, such as less than 125 consecutive amino acid residues, e.g. less than 120 consecutive amino acid residues, such as less than 1 15 consecutive amino acid residues, e.g.
- less than 1 10 consecutive amino acid residues such as less than 105 consecutive amino acid residues, e.g. less than 100 consecutive amino acid residues, such as less than 95 consecutive amino acid residues, e.g. less than 90 consecutive amino acid residues, such as less than 85 consecutive amino acid residues, e.g. less than 80 consecutive amino acid residues, such as less than 75, e.g. less than 60 consecutive amino acid residues of SEQ ID NO: 1 , 3 or 5.
- polypeptide variant according to the present invention, wherein the polypeptide variant is a variant of SEQ ID NO:1 , 3 or 5 having at least 80% sequence identity, such as preferably at least 81 % sequence identity, more preferably e.g. at least 82% sequence identity, such as more preferably at least 83% sequence identity, e.g. more preferably at least 84% sequence identity, more preferably such as at least 85% sequence identity, more preferably e.g. at least 86% sequence identity, more preferably such as at least 87% sequence identity, more preferably e.g. at least 88% sequence identity, more preferably such as at least 89% sequence identity, more preferably e.g.
- sequence identity more preferably such as at least 91 % sequence identity, more preferably e.g. at least 92% sequence identity, such as at least 93% sequence identity, more preferably e.g. at least 94% sequence identity, more preferably such as at least 95% sequence identity, more preferably e.g. at least 96% sequence identity, more preferably such as at least 97% sequence identity, more preferably e.g. at least 98% sequence identity, more preferably such as at least 99% sequence identity, more preferably e.g. at least 99.5% sequence identity to said SEQ ID NO. 1 , 3 or 5, or a fragement of SEQ ID NO: 1 , 3 or 5.
- polypeptide according to the present invention wherein the polypeptide variant fragment contains less than 99.5%, such as less than 98%, e.g. less than 97%, such as less than 96%, e.g. less than 95%, such as less than 94%, e.g. less than 93%, such as less than 92%, e.g. less than 91 %, such as less than 90%, e.g. less than 88%, such as less than 86%, e.g. less than 84%, e.g. less than 82%, such as less than 80%, e.g. less than 75%, such as less than 70%, e.g. less than 65%, such as less than 60%, e.g.
- less than 55% such as less than 50%, e.g. less than 45%, such as less than 40%, e.g. less than 35%, such as less than 30%, e.g. less than 25%, such as less than 20%, such as less than 15%, e.g. less than 10% of the amino acid residues of SEQ ID NO:1 .
- polypeptide according to the present invention wherein the polypeptide variant fragment contains less than 99.5%, such as less than 98%, e.g. less than 97%, such as less than 96%, e.g. less than 95%, such as less than 94%, e.g. less than 93%, such as less than 92%, e.g. less than 91 %, such as less than 90%, e.g. less than 88%, such as less than 86%, e.g. less than 84%, e.g. less than 82%, such as less than 80%, e.g. less than 75%, such as less than 70%, e.g. less than 65%, such as less than 60%, e.g.
- polypeptide according to the present invention wherein the polypeptide variant fragment contains less than 99.5%, such as less than 98%, e.g. less than 97%, such as less than 96%, e.g. less than 95%, such as less than 94%, e.g. less than 93%, such as less than 92%, e.g. less than 91 %, such as less than 90%, e.g.
- less than 88% such as less than 86%, e.g. less than 84%, e.g. less than 82%, such as less than 80%, e.g. less than 75%, such as less than 70%, e.g. less than 65%, such as less than 60%, e.g. less than 55%, such as less than 50%, e.g. less than 45%, such as less than 40%, e.g. less than 35%, such as less than 30%, e.g. less than 25%, such as less than 20%, such as less than 15%, e.g. less than 10% of the amino acid residues of SEQ ID NO:5.
- polypeptide variant fragment contains than 410 consecutive amino acid residues of SEQ ID NO: 1 , such as less than 400 consecutive amino acid residues, such as less than 395 consecutive amino acid residues, e.g. less than 390 consecutive amino acid residues, such as less than 385 consecutive amino acid residues, e.g. less than 380 consecutive amino acid residues, such as less than 370 consecutive amino acid residues, e.g. less than 360 consecutive amino acid residues, such as less than 350 consecutive amino acid residues, e.g. less than 345 consecutive amino acid residues, such as less than 340 consecutive amino acid residues, e.g.
- less than 335 consecutive amino acid residues such as less than 330 consecutive amino acid residues, e.g. less than 325 consecutive amino acid residues, such as less than 300 consecutive amino acid residues, e.g. less than 295 consecutive amino acid residues, such as less than 290 consecutive amino acid residues, e.g. less than 285 consecutive amino acid residues, such as less than 280 consecutive amino acid residues, e.g. less than 275 consecutive amino acid residues, such as less than 270 consecutive amino acid residues, e.g. less than 265 consecutive amino acid residues, such as less than 260 consecutive amino acid residues, such as less than 255 consecutive amino acid residues, e.g.
- less than 250 consecutive amino acid residues such as less than 245 consecutive amino acid residues, e.g. less than 240 consecutive amino acid residues, such as less than 235 consecutive amino acid residues, e.g. less than 230 consecutive amino acid residues, such as less than 225 consecutive amino acid residues, such as less than 220 consecutive amino acid residues, such as less than 215 consecutive amino acid residues, e.g. less than 210 consecutive amino acid residues, such as less than 205 consecutive amino acid residues, e.g. less than 200 consecutive amino acid residues, such as less than 195 consecutive amino acid residues, e.g. less than 190 consecutive amino acid residues, such as less than 185 consecutive amino acid residues, e.g.
- less than 180 consecutive amino acid residues such as less than 175 consecutive amino acid residues, e.g. less than 170 consecutive amino acid residues, such as less than 165 consecutive amino acid residues, e.g. less than 160 consecutive amino acid residues, such as less than 155 consecutive amino acid residues, e.g. less than 150 consecutive amino acid residues, such as less than 145 consecutive amino acid residues, e.g. less than 140 consecutive amino acid residues, such as less than 135 consecutive amino acid residues, e.g. less than 130 consecutive amino acid residues, such as less than 125 consecutive amino acid residues, e.g. less than 120 consecutive amino acid residues, such as less than 1 15 consecutive amino acid residues, e.g.
- less than 1 10 consecutive amino acid residues such as less than 105 consecutive amino acid residues, e.g. less than 100 consecutive amino acid residues, such as less than 95 consecutive amino acid residues, e.g. less than 90 consecutive amino acid residues, such as less than 85 consecutive amino acid residues, e.g. less than 80 consecutive amino acid residues, such as less than 75, e.g. less than 60 consecutive amino acid residues of SEQ ID NO: 1 .
- amino acids including the terminal amino acids, may be modified in a given polypeptide, either by natural processes such as glycosylation and other post- translational modifications, or by chemical modification techniques which are well known in the art. Among the known modifications which may be present in
- polypeptides of the present invention are, to name an illustrative few, acetylation, acylation, ADP-ribosylation, amidation, covalent attachment of flavin, covalent attachment of a heme moiety, covalent attachment of a polynucleotide or
- polynucleotide derivative covalent attachment of a lipid or lipid derivative, covalent attachment of phosphotidylinositol, cross-linking, cyclization, disulfide bond formation, demethylation, formation of covalent cross-links, formation of cystine, formation of pyroglutamate, formylation, gamma-carboxylation, glycation, glycosylation, GPI anchor formation, hydroxylation, iodination, methylation, myristoylation, oxidation, proteolytic processing, phosphorylation, prenylation, racemization, selenoylation, sulfation, transfer-RNA mediated addition of amino acids to proteins such as arginylation, and ubiquitination.
- glycosylation, lipid attachment, sulfation, gamma-carboxylation of glutamic acid residues, hydroxylation and ADP-ribosylation, for instance, are described in most basic texts, such as, for instance, I. E. Creighton, Proteins-Structure and Molecular
- Functional equivalents may further comprise chemical modifications such as ubiquitination, labeling (e.g., with radionuclides, various enzymes, etc.), pegylation (derivatization with polyethylene glycol), or by insertion (or substitution by chemical synthesis) of amino acids (amino acids) such as ornithine, which do not normally occur in human proteins.
- chemical modifications such as ubiquitination, labeling (e.g., with radionuclides, various enzymes, etc.), pegylation (derivatization with polyethylene glycol), or by insertion (or substitution by chemical synthesis) of amino acids (amino acids) such as ornithine, which do not normally occur in human proteins.
- One or more amino acid residues may be modified, where the modification(s) preferably being selected from the group consisting of in vivo or in vitro chemical derivatization, such as acetylation or carboxylation, glycosylation, such as glycosylation resulting from exposing the polypeptide to enzymes which affect glycosylation, for example mammalian glycosylating or deglycosylating enzymes, phosphorylation, such as modification of amino acid residues which results in phosphorylated amino acid residues, for example phosphotyrosine, phosphoserine and phosphothreonine.
- modification(s) preferably being selected from the group consisting of in vivo or in vitro chemical derivatization, such as acetylation or carboxylation, glycosylation, such as glycosylation resulting from exposing the polypeptide to enzymes which affect glycosylation, for example mammalian glycosylating or deglycosylating enzymes, phosphorylation, such as modification of amino
- sterically similar compounds may be formulated to mimic the key portions of the peptide structure and that such compounds may also be used in the same manner as the peptides of the invention. This may be achieved by techniques of modelling and chemical designing known to those of skill in the art. For example, esterification and other alkylations may be employed to modify the amino terminus of, e.g., a di-arginine peptide backbone, to mimic a tetra peptide structure. It will be understood that all such sterically similar constructs fall within the scope of the present invention.
- the protein may comprise a protein tag to allow subsequent purification and optionally removal of the tag using an endopeptidase.
- the tag may also comprise a protease cleavage site to facilitate subsequent removal of the tag.
- affinity tags include a polyhis tag, a GST tag, a HA tag, a Flag tag, a C- myc tag, a HSV tag, a V5 tag, a maltose binding protein tag, a cellulose binding domain tag.
- the tag is a polyhistag.
- the tag is in the C-terminal portion of the protein.
- the isolated polypeptide(s) may be attached to a carrier.
- a carrier comprises an avidin moiety, such as streptavidin, which is optionally biotinylated.
- the carrier may be attached as covalently bound, to a solid support or a semi-solid support.
- the isolated polypeptide(s) may operably be fused to an affinity tag, such as a His-tag.
- the isolated polypeptide is part of a fusion polypeptide operably fused to an N-terminal flanking sequence.
- the isolated polypeptides are operably fused to an C-terminal flanking sequence.
- Peptides with N-terminal alkylations and C-terminal esterifications are also encompassed within the present invention.
- Functional equivalents also comprise glycosylated and covalent or aggregative conjugates formed with the same molecules, including dimers or unrelated chemical moieties. Such functional equivalents are prepared by linkage of functionalities to groups which are found in fragment including at any one or both of the N- and C-termini, by means known in the art.
- fragment thereof may refer to any portion of the given amino acid sequence. Fragments may comprise more than one portion from within the full-length protein, joined together. Suitable fragments may be deletion or addition mutants.
- the addition of at least one amino acid may be an addition of from preferably 2 to 250 amino acids, such as from 10 to 20 amino acids, for example from 20 to 30 amino acids, such as from 40 to 50 amino acids. Fragments may include small regions from the protein or combinations of these. Suitable fragments may be deletion or addition mutants.
- the addition or deletion of at least one amino acid may be an addition or deletion of from preferably 2 to 250 amino acids, such as from 10 to 20 amino acids, for example from 20 to 30 amino acids, such as from 40 to 50 amino acids.
- the deletion and/or the addition may - independently of one another - be a deletion and/or an addition within a sequence and/or at the end of a sequence.
- a functional homologue may be a deletion mutant of HHV U20 polypeptides as identified by SEQ ID NO: 1 , 3 or 5, sharing at least 70% and accordingly, a functional homologue preferably have at least 75% sequence identity, for example at least 80% sequence identity, such as at least 85 % sequence identity, for example at least 90 % sequence identi ity, such as at least 91 % sequence identity, for example at least 91 % sequence identi ity, such as at least 92 % sequence identity, for example at least 93 % sequence identi ity, such as at least 94 % sequence identity, for example at least 95 % sequence identi ity, such as at least 96 % sequence identity, for example at least 97% sequence identi ity, such as at least 98 % sequence identity, for example 99% sequence identity.
- Deletion mutants suitably comprise at least 20 or 40 consecutive amino acid and more preferably at least 80 or 100 consecutive amino acids in length. Accordingly such a fragment may be a shorter sequence of the sequence as identified by SEQ ID NO: 1 , 3 or 5, comprising at least 20 consecutive amino acids, for example at least 30 consecutive amino acids, such as at least 40 consecutive amino acids, for example at least 50 consecutive amino acids, such as at least 60 consecutive amino acids, for example at least 70 consecutive amino acids, such as at least 80 consecutive amino acids, for example at least 90 consecutive amino acids, such as at least 95 consecutive amino acids, such as at least 100 consecutive amino acids, such as at least 105 amino acids, for example at least 1 10 consecutive amino acids, such as at least 1 15 consecutive amino acids, for example at least 120 consecutive amino acids, wherein said deletion mutants preferably share at least 75% sequence identity, for example at least 80% sequence identity, such as at least 85 % sequence identity, for example at least 90 % sequence identity, such as at least 91 % sequence identity, for example at least 91 % sequence identity
- functional homologues of HHV U20 comprises at the most 450, more preferably at the most 400, even more preferably at the most 300, yet more preferably at the most 200, such as at the most 175, for example at the most 160, such as at the most 150 amino acids, for example at the most 140 amino acids.
- fragment thereof may refer to any portion of the given amino acid sequence. Fragments may comprise more than one portion from within the full-length protein, joined together. Portions will suitably comprise at least 5 and preferably at least 10 consecutive amino acids from the basic sequence. They may include small regions from the protein or combinations of these.
- polypeptides are not always entirely linear.
- polypeptides may be branched as a result of ubiquitination, and they may be circular, with or without branching, generally as a result of posttranslational events, including natural processing events and events brought about by human manipulation which do not occur naturally.
- Circular, branched and branched circular polypeptides may be synthesized by non-translational natural processes and by entirely synthetic methods, as well and are all within the scope of the present invention.
- Modifications can occur anywhere in a polypeptide, including the peptide backbone, the amino acid side-chains and the amino or carboxyl termini.
- blockage of the amino or carboxyl group in a polypeptide, or both, by a covalent modification is common in naturally occurring and synthetic polypeptides and such modifications may be present in polypeptides of the present invention, as well.
- the amino terminal residue of polypeptides made in E. coli, prior to proteolytic processing almost invariably will be N-formylmethionine.
- the modifications that occur in a polypeptide often will be a function of how it is made.
- polypeptides made by expressing a cloned gene in a host for instance, the nature and extent of the modifications in large part will be determined by the host cell's posttranslational modification capacity and the modification signals present in the polypeptide amino acid sequence. For instance, glycosylation often does not occur in bacterial hosts such as E. coli. Accordingly, when glycosylation is desired, a polypeptide should be expressed in a glycosylating host, generally a eukaryotic cell. Insect cells often carry out the same posttranslational glycosylations as mammalian cells and, for this reason, insect cell expression systems have been developed to efficiently express mammalian proteins having native patterns of glycosylation, inter alia. Similar considerations apply to other modifications.
- the active derivative of U20 is selected from the group consisting of the following positions in SEQ ID NO:1 (U20 polypeptide sequence of HHV-6B): position 16-434, 16-319, 1 -319, 16- 52, 80-100, 152-182, 100-152, 78-182, 182-300, 182-320, 16-50 + linker + 180- 320, 79-320, 16-382, 16-343, 1 -343, 16-52 + linker + 319 - 343, 80-100 + linker + 319 - 343, 152-182 + linker + 319 - 343, 100-152 + linker + 319-343, 78-182 + linker + 319-343, 182 - 343, 16-50 + linker + 180-343, 79-343, 16-382 + linker + 319-343, 319 ⁇ 134, or 183 ⁇ 134.
- SEQ ID NO:1 U20 polypeptide sequence of HHV-6B
- Immunoglobulin domain including a linker to the
- polypeptides of the invention has an amino acid sequence as identified in SEQ ID NO.: 10 corresponding to the extracellular domain of HHV 6B U20 polypeptide without the signal peptide. It is appreciated that biologically active variants and fragments of the polypeptide of said domain are also within the scope of the invention.
- HHV-6A Due to the high similarity between HHV-6B and 6A domains in HHV-6A may have the same effect as the domains from HHV-6B listed in Table 2. Thus the domain sequences or combination of domains from HHV-6A, as listed in table 3, may be particularly relevant.
- the active derivative of U20 is selected from the group consisting of the following positions in SEQ ID NO:3 (U20 polypeptide sequence of HHV-6A): position 16-422, 16-319, 1 -319, 16- 52, 80-100, 152-182, 100-152, 78-182, 182-300, 182-320, 16-50 + linker + 180- 320, 79-320, 16-382, 16-343, 1-343, 16-52 + linker + 319-343, 80-100 + linker + 319-343, 152-182 + linker + 319-343, 100-152 + linker + 319-343, 78-182 + linker + 319-343, 182-343, 16-50 + linker + 180-343, 79-343, 16-382 + linker + 319-343, 319 ⁇ 122, and 183-422.
- SEQ ID NO:3 U20 polypeptide sequence of HHV-6A
- Immunoglobulin domain including a linker to the
- Extracellular domain including signal peptid and
- linker + N-terminal domain including predicted a-helix (2T), long (319-343) "linker” and transmembrane helix
- polypeptides of the invention has an amino acid sequence as identified in SEQ ID NO.: 1 1 corresponding to the extracellular domain of HHV 6A U20 polypeptide without the signal peptide. It is appreciated that biologically active variants and fragments of the polypeptide of said domain are also within the scope of the invention.
- Glycolysation is the enzymatic process that links saccharides to produce glycans, attached to proteins, lipids, or other organic molecules. This enzymatic process produces one of the fundamental biopolymers found in cells (along with DNA, RNA, and proteins). Glycosylation is a form of co-translational and post-translational modification. Glycans serve a variety of structural and functional roles in membrane and secreted proteins. The majority of proteins synthesized in the rough ER undergo glycosylation. It is an enzyme-directed site-specific process, as opposed to the non- enzymatic chemical reaction of glycation. Glycosylation is also present in the cytoplasm and nucleus as the O-GlcNAc modification.
- glycans Five classes of glycans are produced: relinked glycans attached to a nitrogen of asparagine or arginine side chains, O-linked glycans attached to the hydroxy oxygen of serine, threonine, tyrosine, hydroxylysine, or hydroxyproline side chains, or to oxygens on lipids such as ceramide; phospho-glycans linked through the phosphate of a phospho-serine; C-linked glycans, a rare form of glycosylation where a sugar is added to a carbon on a tryptophan side chain, and glypiation which is the addition of a GPI anchor which links proteins to lipids through glycan linkages.
- a glycolysation may both be chemically and enzymatically attached the the protein or peptide.
- the U20 protein from HHV-6B comprises several glycosylation sites such as position 58, 78, 107, 133, 145, 154, 161 , and 227.
- the U20 protein or derivative thereof comprises at least one glycosylated amino acid.
- the at least one glycosylation site is selected from the group consisting of position 58, 78, 107, 133, 145, 154, 161 , and 227 in SEQ ID NO: 1 .
- the U20 protein or active derivative thereof comprises at least one glycosylated amino acid, such as at least two, such as at least three, such as at least four, such as at least five, such as at least six, such as at least seven, such as at least eight glycosylated amino acids.
- the U20 protein or active derivative thereof comprises 1 -20 glycosylated amino acid, such as 1 -10 glycosylated amino acid, such as 1 -5
- glycosylated amino acid such as 3-8 glycosylated amino acid, such as 1 glycosylated amino acid, such as 2 glycosylated amino acid, or such as 3 glycosylated amino acid.
- glycosylation sites are position 58, 78, 107, 133, 154, 161 227, 266 of SEQ ID NO: 3.
- the at least one glycosylation site is selected from the group consisting of position 58, 78, 107, 133, 154, 161 , 227, 266 of SEQ ID NO: 3.
- glycosylation sites are position 50, 55, 98, 129, 198
- the at least one glycosylation site is selected from the group consisting of position 50, 55, 98, 129, 198 265 of SEQ ID NO: 5.
- the glycosylation positions in HHV-6B, HHV-6A and HHV-7 are related as follows See also figure 9 showing an alignment between HHV-6B, HHV-6A and HHV-7):
- the invention provides medical use of genomic DNA and cDNA coding for HHV U20, including for example the nucleotide sequence (SEQ ID No. 2, 4 or 6), the sequences coding for HHV U20 without signal peptide.
- the invention also provides the cDNA sequence coding for HHV U 20.
- One aspect of the present invention pertains to an isolated polynucleotide for use as a medicament comprising a nucleic acid or its complementary sequence, said
- polynucleotide being selected from the group consisting of:
- a polynucleotide comprising a nucleic acid having 70% sequence identity to SEQ ID NO.: 2, 4, 6, 7, 8 or 9 or
- the polynucleotide is SEQ ID NO.: 2, 4, 6, 7, 8 or 9.
- polynucleotide is a polynucleotide comprising a nucleic acid having 70% sequence identity to SEQ ID NO.: 2, 4, 6, 7, 8 or 9.
- the polynucleotide is capable of hybridising to a polynucleotide having the sequence of SEQ ID NO.: 2, 4, 6, 7, 8 or 9.
- polynucleotide is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N
- a polynucleotide encoding a biologically active sequence variant of the amino acid sequence wherein the variant has at least 70% sequence identity to said SEQ ID NO. 1 , 3 or 5 and iii) a polynucleotide encoding a biologically active fragment of at least 50 contiguous amino acids of any of a) through b), wherein said fragment is a fragment of SEQ ID NO 1 , 3 or 5.
- polynucleotide of the present invention is complementary to SEQ ID NO.: 2, 4, 6, 7, 8 or 9.
- polynucleotide of the invention may in another embodiment may be any polynucleotide of the invention.
- the polynucleotide of the invention may in another embodiment may be any polynucleotide of the invention.
- nucleic acid having 70% sequence identity to SEQ ID NO.: 2, 4, 6, 7, 8 or 9.
- the polynucleotide may comprise the nucleotide sequence of a naturally occurring allelic nucleic acid variant.
- the polynucleotide of the invention may encode a variant polypeptide, wherein the variant polypeptide has the polypeptide sequence of a naturally occurring polypeptide variant.
- the nucleic acid sequence of the polynucleotide may differ by a single nucleotide from a nucleic acid sequence selected from the group consisting of SEQ ID NO: 2, 4, 6, 7, 8 and 9. However, the polynucleotide may also differ from a nucleic acid sequence selected from the group consisting of SEQ ID NO: 2, 4, 6, 7, 8 and 9 by 2, 3, 4, 5, 6, 7, 8, 9, 10, or more nucleotides.
- the nucleic acid sequence of the polynucleotide has at least 80% sequence identity, such as preferably at least 81 % sequence identity, more preferably e.g. at least 82% sequence identity, such as more preferably at least 83% sequence identity, e.g. more preferably at least 84% sequence identity, more preferably such as at least 85% sequence identity, more preferably e.g. at least 86% sequence identity, more preferably such as at least 87% sequence identity, more preferably e.g. at least 88% sequence identity, more preferably such as at least 89% sequence identity, more preferably e.g. at least 90% sequence identity, more preferably such as at least 91 % sequence identity, more preferably e.g.
- sequence identity such as at least 93% sequence identity, more preferably e.g. at least 94% sequence identity, more preferably such as at least 95% sequence identity, more preferably e.g. at least 96% sequence identity, more preferably such as at least 97% sequence identity, more preferably e.g. at least 98% sequence identity, more preferably such as at least 99% sequence identity, more preferably e.g. at least 99.5% sequence identity to a nucleotide sequence selected from the group consisting of SEQ ID No. 2, 4, 6, 7, 8, and 9;
- the nucleic acid sequence of the polynucleotide may contain less than 99.5%, such as less than 98%, e.g. less than 97%, such as less than 96%, e.g. less than 95%, such as less than 94%, e.g. less than 93%, such as less than 92%, e.g. less than 91 %, such as less than 90%, e.g. less than 88%, such as less than 86%, e.g. less than 84%, e.g. less than 82%, such as less than 80%, e.g. less than 75%, such as less than 70%, e.g. less than 65%, such as less than 60%, e.g.
- nucleotide sequence selected from the group consisting of SEQ ID No. 2, 4, 6, 7, 8, and 9.
- the encoded polypeptide has at least 70% sequence identity to SEQ ID No. 3, more preferably at least 75%, more preferably at least 80%, more preferably at least 95%, more preferably at least 98%, more preferably wherein said polypeptide has the sequence of SEQ ID No. 3.
- the encoded polypeptide has at least 70% sequence identity to SEQ ID No. 5, more preferably at least 75%, more preferably at least 80%, more preferably at least 95%, more preferably at least 98%, more preferably wherein said polypeptide has the sequence of SEQ ID No. 5.
- the encoded polypeptide has at least 70% sequence identity to SEQ ID No. 7, more preferably at least 75%, more preferably at least 80%, more preferably at least 95%, more preferably at least 98%, more preferably wherein said polypeptide has the sequence of SEQ ID No. 7.
- the isolated polynucleotide of the invention comprises a nucelic acid sequence having at least 70%, preferably at least 75%, more preferably at least 80%, preferably at least 85%, more preferred at least 90%, more preferred at least 95%, more preferred at least 98% sequence identity to the polynucleotide sequence presented as SEQ ID NO: 2.
- the isolated polynucleotide of the invention comprises a nucelic acid sequence having at least 70%, preferably at least 75%, more preferably at least 80%, preferably at least 85%, more preferred at least 90%, more preferred at least 95%, more preferred at least 98% sequence identity to the polynucleotide sequence presented as SEQ ID NO: 4.
- the isolated polynucleotide of the invention comprises a nucelic acid sequence having at least 70%, preferably at least 75%, more preferably at least 80%, preferably at least 85%, more preferred at least 90%, more preferred at least 95%, more preferred at least 98% sequence identity to the polynucleotide sequence presented as SEQ ID NO: 6.
- the polynucleotide is capable of hybridizing to the nucleic acid selected from the group consisting of SEQ ID NO: SEQ ID NO: 2, 4, 6, 7, 8 and 9, or a fragment hereof, under stringent conditions as described below.
- a portion of the polynucleotide may hybridize under stringent conditions to a nucleotide probe corresponding to at least 10 consecutive nucleotides of a nucleotide sequence selected from the group consisting of SEQ ID NO: SEQ ID NO: 2, 4, 6, 7, 8 and 9, or a fragment hereof.
- the invention relates to the use of the nucleic acids and proteins of the present invention to design probes to isolate other genes, which encode proteins with structural or functional properties of the HHV U20 proteins of the invention.
- the probes can be a variety of base pairs in length.
- a nucleic acid probe can be between about 10 base pairs in length to about 150 base pairs in length.
- the nucleic acid probe can be greater than about 150 base pairs in length.
- oligonucleotide also referred to herein as nucleic acid
- T m 80 ' ⁇ (assuming 2° for each A or T and 4°C for each G or C).
- the oligonucleotide should preferably be labeled to facilitate detection of hybridisation. Labelling may be with ⁇ - 32 ⁇ ATP (specific activity 6000 Ci/mmole) and T4
- oligonucleotides Other labeling techniques can also be used. Unincorporated label should preferably be removed by gel filtration chromatography or other established methods. The amount of radioactivity incorporated into the probe should be quantitated by measurement in a scintillation counter. Preferably, specific activity of the resulting probe should be approximately 4 x 10 6 dpm/pmole.
- the bacterial culture containing the pool of full-length clones should preferably be thawed and 100 ⁇ _ of the stock used to inoculate a sterile culture flask containing 25 ml of sterile L-broth containing ampicillin at 100 pg/ml.
- the invention relates to nucleic acid sequences (e.
- nucleic acids of HHV U20 g., DNA, RNA
- the invention relates to a complement of nucleic acid of HHV U20.
- it relates to complements of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 6, SEQ ID NO:7, SEQ ID No. 8, or SEQ ID No. 9
- the invention relates to an RNA counterpart of the DNA nucleic acid of HHV U20.
- it relates to RNA counterparts of SEQ ID NO: 2, SEQ ID NO: 4 and , SEQ ID NO: 6, exemplified by SEQ ID No 7, SEQ ID No 8, and SEQ ID No 9.
- polynucleotide of the present invention is DNA, RNA, LNA or PNA.
- the invention relates to an isolated polynucleotide comprising a polynucleotide encoding the U20 protein according to the invention.
- the invention relates to an isolated polynucleotide comprising a
- polynucleotide encoding the U20 protein or an active derivative thereof according to the invention. Such a polynucleotide may be used for expression purposes, cloning purposes, storage purposes.
- SEQ ID NO:2 discloses the nucleic acid sequence for U20 from HHV-6B
- SEQ ID NO:4 discloses the nucleic acid sequence for U20 from HHV-6A
- SEQ ID NO:6 discloses the nucleic acid sequence for U20 from HHV-7.
- the person skilled in the art would know how to construct polynucleotides encoding the peptide sequences disclosed on Table 1 and Table 2.
- the human herpes virus U20 protein of the present invention may be produced by any suitable method.
- the U20 protein composition may according to the present invention be produced by any suitable methods.
- Natural sources of U20 protein may be cells infected by HHV- 6A, HHV-6B or HHV-7 either in vitro or in vivo during natural infections.
- U20 protein may be produced recombinantly (see more details herein below in the section "recombinant production").
- Purification of proteins in general involves one or more steps of removal of or separation from contaminating nucleic acids, phages and/or viruses, other proteins and/or other biological macromolecules.
- the obtaining of U20 polypeptides from a composition comprising U20, such as milk or a culture medium or an extract of host cells may comprise one or more protein isolation steps. Any suitable protein isolation step may be used with the present invention. The skilled person will in general readily be able to identify useful protein isolation steps for U20 polypeptides if such are required.
- the protein isolation steps useful with the present invention may be commonly used methods for protein purification including for example chromatographic methods such as for example gas chromatography, liquid chromatography, ion exchange
- Purification of U20 may comprise one or more of the aforementioned methods in any combination.
- purification of U20 may for example comprise one or more centrifugation steps.
- Said centrifugation may be employed for example for defattening purposes and/or to remove cells/cellular debris or the like and/or to separate supernatant from precipitate
- purification of U20 may for example comprise one or more precipitation steps, for example precipitation using ammonium sulphate, for example at a concentration in 10 to 75%, preferably in the range of 30 to 60%, such as in the range of 40-45%.
- U20 will generally be present in the supernatant.
- Purification of U20 may comprise one or more steps of filtration, for example filtration through a filter paper and/or filtration using another filter with a pore size of the range of 0.1 ⁇ to 100 ⁇ , for example in the range of 0.5 to 50 ⁇ , such as in the range of 0.5 to 20 ⁇ , such as in the range of 0.5-1 ⁇ .
- Purification of U20 may comprise one or more chromatographic steps, for example any of the chromatographic methods mentioned above.
- the method comprises a hydrophobic interaction chromatography.
- Functional equivalents of U20 are preferably produced recombinantly.
- Wild type U20 may in one preferred embodiment also be recombinantly produced.
- Useful recombinant production methods includes conventional methods known in the art, such as by expression of heterologuos U20 of functional homologues thereof in suitable host cells such as E. coli, S. cerevisiae or S. pombe or insect or mammalian cells suitable for production of recombinant proteins (see below).
- suitable host cells such as E. coli, S. cerevisiae or S. pombe or insect or mammalian cells suitable for production of recombinant proteins (see below).
- suitable host cells such as E. coli, S. cerevisiae or S. pombe or insect or mammalian cells suitable for production of recombinant proteins (see below).
- suitable host cells such as E. coli, S. cerevisiae or S. pombe or insect or mammalian cells suitable for production of re
- U20 is produced in a transgenic plant or animal.
- a transgenic plant or animal in this context is meant a plant or animal which has been genetically modified to contain and express a nucleic acid encoding human U20 or functional homolgues hereof.
- U20 or a functional homolgue thereof is produced recombinantly by host cells.
- U20 is produced by host cells comprising a first nucleic acid sequence encoding U20 or a functional homologue thereof operably associated with a second nucleic acid capable of directing expression in said host cells.
- the second nucleic acid sequence may thus comprise or even consist of a promoter that will direct the expression of protein of interest in said cells.
- a skilled person will be readily capable of identifying useful second nucleic acid sequence for use in a given host cell.
- the process of producing recombinant U20 or a functional homologue thereof in general comprises the steps of:
- composition comprising U20 may thus be an extract of said host cells or a composition purified from an extract of said host cells and/or from the culture medium.
- the recombinant U20 thus produced may be isolated by any conventional method for example by any of the protein purification methods described herein above.
- the skilled person will be able to identify a suitable protein isolation steps for purifying any protein of interest.
- the recombinantly produced U20 or the functional homologue thereof is excreted by the host cells.
- the process of producing a recombinant protein of interest may comprise the following steps
- composition comprising U20 or a functional homologue thereof may thus in this embodiment of the invention be the culture medium or a composition prepared from the culture medium.
- said composition is an extract prepared from animals, parts thereof or cells or an isolated fraction of such an extract.
- U20 is recombinantly produced in vitro in host cells and is isolated from cell lysate, cell extract or from tissue culture supernatant.
- U20 is produced by host cells that are modified in such a way that they express the protein of interest.
- said host cells are transformed to produce and excrete U20.
- the polynucleotides encoding U20 may be derived from the human U20 gene(s) or derived from the human U20 mRNA transcript(s), (see elsewhere herein).
- the gene expression construct is suitable for expression in mammalian cell lines or transgenic plants or animals.
- the host cell culture is cultured in a transgene animal.
- a transgenic plant or animal in this context is meant a plant or animal which has been genetically modified to contain and express a nucleic acid encoding human U20 or a functional homologue thereof as defined herein above
- the gene expression construct of the present invention comprises a viral based vector, such as a DNA viral based vector, a RNA viral based vector, or a chimeric viral based vector.
- a viral based vector such as a DNA viral based vector, a RNA viral based vector, or a chimeric viral based vector.
- DNA viruses are cytomegalo virus, Herpex Simplex, Epstein-Barr virus, Simian virus 40, Bovine papillomavirus, Adeno-associated virus, Adenovirus, Vaccinia virus, and Baculo virus.
- the gene expression construct may for example only comprise a plasmid based vector.
- the invention provides an expression construct encoding human U20 or functional homologues thereof, featured by comprising one or more intron sequences from the human U20 gene including functional derivatives hereof. Additionally, it may contain a promoter region derived from a viral gene or a eukaryotic gene, including mammalian and insect genes. The promoter region is preferably selected to be different from the native human U20 promoter, and preferably in order to optimize the yield of human U20, the promoter region is selected to function most optimally with the vector and host cells in question.
- the promoter region is selected from a group comprising Rous sarcoma virus long terminal repeat promoter, and cytomegalovirus immediate- early promoter, and elongation factor-1 alpha promoter.
- the promoter region is derived from a gene of a microorganism, such as other viruses, yeasts and bacteria.
- the promoter region may comprise enhancer elements, such as the QBI SP163 element of the 5' end untranslated region of the mouse vascular endothelian growth factor gene
- the host cell culture is may be eukaryotic, and for example a mammalian cell culture or a yeast cell culture.
- Useful mammalian cells may for example be human embryonal kidney cells (HEK cells), such as the cell lines deposited at the American Type Culture Collection with the numbers CRL-1573 and CRL-10852, chick embryo fibroblast, hamster ovary cells, baby hamster kidney cells, human cervical carcinoma cells, human melanoma cells, human kidney cells, human umbilical vascular endothelium cells, human brain endothelium cells, human oral cavity tumor cells, monkey kidney cells, mouse fibroblast, mouse kidney cells, mouse connective tissue cells, mouse oligodendritic cells, mouse macrophage, mouse fibroblast, mouse neuroblastoma cells, mouse pre-B cell, mouse B lymphoma cells, mouse plasmacytoma cells, mouse teratocacinoma cells, rat astrocytoma cells
- HEK cells human embryonal kidney cells
- recombinantly produced U20 When recombinantly produced U20 is used with the present invention it is preferred that said recombinantly produced U20 has a size distribution profile that is similar to naturally occurring U20.
- the aforementioned methods are well known to the skilled person and may for example be performed as described in the Current Protocols in Molecular Biology, 2001 , by John Wiley and Sons, Inc. edited by Frederick M. Ausubel et al.
- One aspect of the invention relates to the use of an isolated U20 polypeptides, biologically active variants or fragments thereof, polynucleotides of the invention, vectors of the invention, pharmaceutical composition, or kits of the invention for the manufacture of a medicament for the treatment of diseases related to TNF receptor activity such as autoimmune, inflammatory, degenerative or cancerous disease.
- one aspect concerns the isolated polypeptide or an isolated nucleic acid sequence as defined herein or a vector of the invention ora host cell of the invention or a pharmaceutical composition of the present invention or a kit of parts of the invention or a kit of the invention for the treatment of TNF receptor related diseases.
- the TNF receptor is the TNFR1 or the TNFR2, in one embodiment the TNF receptor is TNFR1 .
- the methods and pharmaceutical compositions of the present invention thus influence the TNF receptor activity by decreasing the TNF receptor activity in the signal transduction pathway of TNF receptors. In one embodiment of the present invention the TNF receptor activity is reduced with respect to induction of apoptosis.
- Apoptosis through the TNF receptor pathway is in one embodiment influenced by the action of TNF-alpha, or for example lymphotoxin (also referred to as TNF-beta) on the TNF receptor, for example TNFR1 in one particular embodiment, where the complex formation in response to binding af the TNF-alpha and/or TNF-beta to the receptor induces apoptosis.
- TNF-alpha or for example lymphotoxin (also referred to as TNF-beta) on the TNF receptor, for example TNFR1 in one particular embodiment, where the complex formation in response to binding af the TNF-alpha and/or TNF-beta to the receptor induces apoptosis.
- TNF-alpha or for example lymphotoxin (also referred to as TNF-beta) on the TNF receptor, for example TNFR1 in one particular embodiment, where the complex formation in response to binding af the TNF-alpha and/or TNF-
- the pharmaceutical composition of the present invention b) obtaining a decrease in TNF receptor activity compared to a standard level of TNF receptor activity in an individual.
- the TNF receptor activity is decreased by at least 10%, such as at least 15%, such as at least 25%, such as at least 30%, such as at least 35%, such as at least 40%, such as at least 45%, such as at least 50%, such as at least 55%, such as at least 60%, such as at least 70%, such as at least 75%, such as at least 80%, such as at least 85%, such as at least 90%, such as at least 95%, such as at least 96%, such as at least 97%, such as at least 98%, such as at least 99%, such as 100% of a standard TNF receptor activity in an individual.
- the present invention relates in one aspect to a pharmaceutical composition for treating, ameliorating and/or preventing TNF receptor related diseases comprising a) the isolated polypeptide of the present invention or b) the isolated nucleic acid sequence of the present invention or c) the vector of the present invention or d) the host cell of the present invention; and in another aspect to a method for treating, ameliorating and/or preventing TNF receptor related diseases comprising
- the isolated polypeptide of the invention or b) the isolated nucleic acid sequence of the invention or c) the vector of any of the invention d) the host cell of the invention e) the pharmaceutical composition of the invention or f) the kit of parts of the invention or g) the kit of the invention, in a therapeutically effective amount to an individual in need thereof.
- the present invention in one aspect also pertains to use an isolated polypeptide of the present invention or an isolated nucleic acid sequence present invention or a vector of present invention or a host cell present invention or a pharmaceutical composition or a kit of parts or a kit of the invention for the treatment of TNF receptor related diseases.
- the methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity, for exampel in response to TNF-alpha activity on the receptor.
- the present invention can be used to treat diseases resulting from TNF-alpha activity, in particular the hTNFa.
- the isolated polypeptide of the present invention, the isolated nucleic acid sequence of the present invention, the vector of any of the present invention, the host cell of the present invention, the pharmaceutical composition may be used in the prevention, amelioration and/or treatment of TNF-alpha related disease, in a cell, tissue, organ, animal, or patient including, but not limited to, at least one of an immune related disease, a cardiovascular disease, an infectious disease, a malignant disease or a neurologic disease.
- the methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used for modulating or treating at least one immune related disease, in a cell, tissue, organ, animal, or patient including, but not limited to, at least one of rheumatoid arthritis, juvenile rheumatoid arthritis, systemic onset juvenile rheumatoid arthritis, psoriatic arthritis, ankylosing spondilitis, gastric ulcer, seronegative arthropathies, osteoarthritis, inflammatory bowel disease, ulcerative colitis, systemic lupus
- polyneuropathy polyneuropathy, or-ganomegaly, endocrinopathy, monoclonal gammopathy, skin changes syndrome, antiphospholipid syndrome, pemphigus, scleroderma, mixed connective tissue disease, idiopathic Addison's disease, diabetes mellitus, chronic active hepatitis, primary billiary cirrhosis, vitiligo, vasculitis, post-MI cardiotomy syndrome, type IV hypersensitivity, contact dermatitis, hypersensitivity pneumonitis, allograft rejection, granulomas due to intracellular organisms, drug sensitivity, metabolic/idiopathic, Wilson's disease, hemachromatosis, alpha-1 -antitrypsin deficiency, diabetic retinopathy, hashimoto's thyroiditis, osteoporosis, hypothalamic- pituitary-adrenal axis evaluation, primary biliary cirrhosis, thyroiditis, encephalomyelitis,
- the methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used for modulating or treating at least onejmmune related disease, in a cell, tissue, organ, animal, or patient including, such as, at least one of rheumatoid arthritis, juvenile rheumatoid arthritis,systemic onset juvenile rheumatoid arthritis, psoriatic arthritits, Crohn's disease and other inflammatory bowel disease.
- the methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used for modulating or treating at least onejmmune related disease, in a cell, tissue, organ, animal, or patient including, such as rheumatoid arthritis.
- the methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used for modulating or treating at least onejmmune related disease, in a cell, tissue, organ, animal, or patient including, such as juvenile rheumatoid arthritis.
- the methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used for modulating or treating at least onejmmune related disease, in a cell, tissue, organ, animal, or patient including, such as systemic onset juvenile rheumatoid arthritis.
- compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used for modulating or treating at least one immune related disease, in a cell, tissue, organ, animal, or patient including, such as psoriatic arthritits.
- the methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used for modulating or treating at least onejmmune related disease, in a cell, tissue, organ, animal, or patient including, such as Crohn's disease.
- the methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used in the prevention, amelioration and/or treatment of at least one cardiovascular disease in a cell, tissue, organ, animal, or patient, including, but not limited to, at least one of cardiac stun syndrome, myocardial infarction, congestive heart failure, stroke, ischemic stroke, hemorrhage, arteriosclerosis, atherosclerosis, restenosis, diabetic ateriosclerotic disease, hypertension, arterial hypertension, renovascular hypertension, syncope, shock, syphilis of the cardiovascular system, heart failure, cor pulmonale, primary pulmonary hypertension, cardiacarrhythmias, atrial ectopic beats, atrial flutter, atrial fibrillation (sustained or paroxysmal), post perfusion syndrome, cardiopulmonary bypass inflammation response, chaotic or multifocal atrial tachycardia, regular narrow QRS tachycardia, specific arrythmi
- lymphederma lipedema, unstable angina, reperfusion injury, post pump syndrome, ischemia-reperfusion injury, and the like.
- the methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used in the prevention, amelioration and/or treatment of at least one infectious disease in a cell, tissue, organ, animal or patient, including, but not limited to, at least one of: acute or chronic bacterial infection, acute and chronic parasitic or infectious processes, including bacterial, viral and fungal infections, HIV infection/HIV neuropathy, meningitis, hepatitis (A,B or C, or the like), septic arthritis, peritonitis, pneumonia, epiglottitis, e.
- coli 0157:h7 hemolytic uremic syndrome/thrombolytic thrombocytopenic purpura, malaria, dengue hemorrhagic fever, leishmaniasis, leprosy, toxic shock syndrome, streptococcal myositis, gas gangrene, mycobacterium tuberculosis, mycobacterium avium
- the methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used in the prevention, amelioration and/or treatment of at least one malignant disease in a cell, tissue, organ, animal or patient, including, but not limited to, at least one of: leukemia, acute leukemia, acute lymphoblastic leukemia (ALL), B-cell, T-cell or FAB ALL, acute myeloid leukemia (AML), chromic myelocytic leukemia (CML), chronic lymphocytic leukemia (CLL), hairy cell leukemia, myelodyplastic syndrome (MDS), a lymphoma
- ALL acute leukemia
- ALL acute lymphoblastic leukemia
- B-cell B-cell
- T-cell or FAB ALL acute myeloid leukemia
- AML acute myeloid leukemia
- CML chromic myelocytic leukemia
- CLL chronic lymphocytic leukemia
- the methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used in the prevention, amelioration and/or treatment of at least one neurologic disease in a cell, tissue, organ, animal or patient, including, but not limited to, at least one of: neurodegenerative diseases, multiple sclerosis, migraine headache, AIDS dementia complex, demyelinating diseases, such as multiple sclerosis and acute transverse myelitis; extrapyramidal and cerebellar disorders' such as lesions of the corticospinal system; disorders of the basal ganglia or cerebellar disorders; hyperkinetic movement disorders such as Huntington's Chorea and senile chorea; drug-induced movement disorders, such as those induced by drugs which block CNS dopamine receptors; hypokinetic movement disorders, such as Parkinson's disease
- supranucleo Palsy structural lesions of the cerebellum; spinocerebellar degenerations, such as spinal ataxia, Friedreich's ataxia, cerebellar cortical degenerations, multiple systems degenerations (Mencel, Dejerine-Thomas, Shi-Drager, and Machado- Joseph); systemic disorders (Refsum's disease, abetalipoprotemia, ataxia, telangiectasia, and mitochondrial multi system disorder); demyelinating core disorders, such as multiple sclerosis, acute transverse myelitis; and disorders of the motor unit' such as
- neurogenic muscular atrophies anterior horn cell degeneration, such as amyotrophic lateral sclerosis, infantile spinal muscularatrophy and juvenile spinal muscular atrophy
- Alzheimer's disease Down's Syndrome in middle age
- Diffuse Lewy body disease Senile Dementia of Lewy body type
- Wernicke-Korsakoff syndrome chronic alcoholism
- Creutzfeldt-Jakob disease Subacute sclerosing panencephalitis
- polypeptides, nuclaic acid molecules, host cell, vectors of the present invention offering a novel method for decreasing the TNF receptor activity can be used in one aspect relating to a pharmaceutical composition
- a pharmaceutical composition comprising
- compositions containing the polypeptides, polynucleotides, vectors, host cell, kit, or kit of parts of the present invention may be prepared by conventional techniques, e.g. as described in Remington: The Science and Practice of Pharmacy 1995, edited by E. W. Martin, Mack Publishing Company, 19th edition, Easton, Pa.
- the compositions may appear in conventional forms, for example capsules, tablets, aerosols, solutions, suspensions or topical applications.
- the present invention provides pharmaceutical compositions for treating, ameliorating and/or preventing TNF receptor related diseases comprising a) the isolated polypeptide of the invention or b) the isolated polynucleotide of the invention or c) the vector of the present invention or d) the host cell of the invention or e) the kit of the invention or f) the kit of parts of the invention.
- the present invention relates to a pharmaceutical composition.
- the pharmaceutical composition may be formulated in a number of different manners, depending on the purpose for the particular pharmaceutical composition.
- the pharmaceutical composition may be formulated in a manner so it is useful for a particular administration form. Preferred administration forms are described herein below.
- the pharmaceutical composition is formulated so it is a liquid.
- the composition may be a protein solution or the composition may be a protein suspension.
- Said liquid may be suitable for parenteral administration, for example for injection or infusion.
- the liquid may be any useful liquid, however it is frequently preferred that the liquid is an aqueous liquid.
- Sterility may be conferred by any conventional method, for example filtration, irradiation or heating.
- it is preferred that the liquid has been subjected to a virus reduction step, in particular if the liquid is formulated for parenteral administration.
- Virus reduction may for example be performed by nanofiltration or virus filtering over a suitable filter, such as a Planova filter consisting of several layers.
- the Planova filter may be any suitable size for example 75N, 35N, 20N or 15N or filters of different size may be used, for example Planova 20N.
- Virus reduction may also comprise a step of prefiltering with another filter, for example using a filter with a pore size of the the range of 0.01 to 1 ⁇ , such as in the range of 0.05 to 0.5 ⁇ , for example around 0.1 ⁇ .
- Virus reductions may also include an acidic treatment step.
- the pharmaceutical composition may be packaged in single dosage units, which may be more convenient for the user.
- compositions for bolus injections may be packages in dosage units of for example at the most 10 ml, preferably at the most 8 ml, more preferably at the most 6 ml, such as at the most 5 ml, for example at the most 4 ml, such as at the most 3 ml, for example around 2 ml.
- the pharmaceutical composition may be packaged in any suitable container.
- a single dosage of the pharmaceutical composition may be packaged in injection syringes or in a container useful for infusion.
- the pharmaceutical composition is a dry composition.
- the dry composition may be used as such, but for most purposes the composition is a dry composition for storage only. Prior to use the dry composition may be dissolved or suspended in a suitable liquid composition, for example sterile water.
- the pharmaceutical composition according to the present invention may also comprise a first nucleic acid sequence encoding HHV 6B (SEQ ID NO.:2) or alternatively encoding HHV 6A or HHV 7, SEQ ID NO.: 4 and 6, respectively, or SEQ ID NO: 7, 8, or 9, or fragments, functional equivalents, variants, or complementary sequences thereof.
- Said first nucleic acid sequence is preferably operably associated with a second nucleic acid sequence directing expression of the first nucleic acid in the individual to be treated with the pharmaceutical composition, more preferably in the cells of said individual, which are diseased.
- the second nucleic acid sequence is capable of directing expression of the first nucleic acid sequence in a human being.
- the second nucleic acid sequence is capable of directing expression of the first nucleic acid sequence in cancer cells, such as malignant cells. It is furthermore preferred that the first and the second nucleic acid sequences are included in a suitable vector.
- the pharmaceutical composition may be applied topically to the site of the site, for example in the form of a lotion, a creme, an ointment, a spray, such as an aerosol spray or a nasal spray, rectal or vaginal suppositories, drops, such as eye drops or nasal drops, a patch, an occlusive dressing or the like.
- compositions may be prepared by any conventional technique, e.g. as described in Remington: The Science and Practice of Pharmacy 1995, edited by E. W. Martin, Mack Publishing Company, 19th edition, Easton, Pa.
- the pharmaceutically acceptable additives may be any conventionally used pharmaceutically acceptable additive, which should be selected according to the specific formulation, intended administration route etc.
- the pharmaceutically acceptable additives may be any of the additives mentioned in Nema et al, 1997.
- the pharmaceutically acceptable additive may be any accepted additive from FDA ' s "inactive ingredients list", which for example is available on the internet address http://www.fda.gov/cder/drug/iig/default.htm.
- the pharmaceutical composition comprises an isotonic agent.
- the composition may comprise at least one pharmaceutically acceptable additive which is an isotonic agent.
- the pharmaceutical composition may be isotonic, hypotonic or hypertonic. However it is often preferred that a pharmaceutical composition for infusion or injection is essentially isotonic, when it is administrated. Hence, for storage the pharmaceutical composition may preferably be isotonic or hypertonic. If the pharmaceutical composition is hypertonic for storage, it may be diluted to become an isotonic solution prior to administration.
- the isotonic agent may be an ionic isotonic agent such as a salt or a non-ionic isotonic agent such as a carbohydrate.
- ionic isotonic agents include but are not limited to NaCI, CaCI 2 , KG and MgCI 2.
- non-ionic isotonic agents include but are not limited to mannitol and glycerol.
- the pharmaceutical composition may comprise no buffer at all or only micromolar amounts of buffer.
- the buffer is TRIS.
- TRIS buffer is known under various other names for example tromethamine including tromethamine USP, THAM, Trizma, Trisamine, Tris amino and trometamol. The designation TRIS covers all the
- the buffer may furthermore for example be selected from USP compatible buffers for parenteral use, in particular, when the pharmaceutical formulation is for parenteral use.
- the buffer may be selected from the group consisting of monobasic acids such as acetic, benzoic, gluconic, glyceric and lactic, dibasic acids such as aconitic, adipic, ascorbic, carbonic, glutamic, malic, succinic and tartaric, polybasic acids such as citric and phosphoric and bases such as ammonia, diethanolamine, glycine, triethanolamine, and TRIS.
- compositions may comprise at least one pharmaceutically acceptable additive which is a stabiliser.
- the stabiliser may be selected from the group consisting of poloxamers, Tween-20, Tween-40, Tween-60, Tween-80, Brij, metal ions, amino acids, polyethylene glycol, Triton, EDTA, ascorbic acid, Triton X-100, NP40 or CHAPS.
- the pharmaceutical composition according to the invention may also comprise one or more cryoprotectant agents.
- the composition comprises freeze-dried protein or the composition should be stored frozen, it may be desirable to add a cryoprotecting agent to the pharmaceutical composition.
- the cryoprotectant agent may be any useful cryoprotectant agent, for example the cryoprotectant agent may be selected from the group consisting of dextran, glycerin, polyethylenglycol, sucrose, trehalose and mannitol. Accordingly, the pharmaceutically acceptable additives may comprise one or more selected from the group consisting of isotonic salt, hypertonic salt, buffer and stabilisers. Furthermore, the pharmaceutically acceptable additives may comprise one or more selected from the group consisting of isotonic agents, buffer, stabilisers and cryoprotectant agents. For example, the pharmaceutically acceptable additives comprise glucosemonohydrate, glycine, NaCI and polyethyleneglycol 3350.
- composition of the present invention Whilst it is possible for the composition of the present invention to be administered as the raw composition, it is preferred to present it in the form of a pharmaceutical formulation. Accordingly, the present invention further provides a pharmaceutical formulation, for medicinal application, which comprises a composition of the present invention or a pharmaceutically acceptable salt thereof, as herein defined, and a pharmaceutically acceptable carrier therefore.
- a pharmaceutical formulation for medicinal application, which comprises a composition of the present invention or a pharmaceutically acceptable salt thereof, as herein defined, and a pharmaceutically acceptable carrier therefore.
- compositions of the present invention may be formulated in a wide variety of oral administration dosage forms.
- the pharmaceutical compositions and dosage forms may comprise the compositions of the invention or its pharmaceutically acceptable salt or a crystal form thereof as the active component.
- the pharmaceutically acceptable carriers can be either solid or liquid. Solid form preparations include powders, tablets, pills, capsules, cachets, suppositories, and dispersible granules.
- a solid carrier can be one or more substances which may also act as diluents, flavoring agents, solubilizers, lubricants, suspending agents, binders, preservatives, wetting agents, tablet disintegrating agents, or an encapsulating material.
- the composition will be about 0.5% to 75% by weight of a composition or compositions of the invention, with the remainder consisting of suitable pharmaceutical excipients.
- suitable pharmaceutical excipients include pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, sodium saccharine, talcum, cellulose, glucose, gelatin, sucrose, magnesium carbonate, and the like.
- the carrier is a finely divided solid which is a mixture with the finely divided active component.
- the active component is mixed with the carrier having the necessary binding capacity in suitable proportions and compacted in the shape and size desired.
- the powders and tablets preferably contain from 1 to about 70 %t of the active composition.
- Suitable carriers are magnesium carbonate, magnesium stearate, talc, sugar, lactose, pectin, dextrin, starch, gelatin, tragacanth, methylcellulose, sodium carboxymethylcellulose, a low melting wax, cocoa butter, and the like.
- the term "preparation" is intended to include the formulation of the active composition with encapsulating material as carrier providing a capsule in which the active component, with or without carriers, is surrounded by a carrier, which is in association with it.
- Tablets, powders, capsules, pills, cachets, and lozenges can be as solid forms suitable for oral administration.
- Multiple- unit-dosage granules can be prepared as well. Tablets and granules of the above cores can be coated with concentrated solutions of sugar, etc.
- the cores can also be coated with polymers which change the dissolution rate in the gastrointestinal tract, such as anionic polymers having a pka of above 5.5.
- anionic polymers having a pka of above 5.5.
- Such polymers are hydroxypropylmethyl cellulose phthalate, cellulose acetate phthalate, and polymers sold under the trade mark Eudragit S100 andU OO.
- In preparation of gelatine capsules these can be soft or hard. In the former case the active compound is mixed with oil, and in the latter case the multiple-unit-dosage granules are filled therein.
- Drops according to the present invention may comprise sterile or non-sterile aqueous or oil solutions or suspensions, and may be prepared by dissolving the active ingredient in a suitable aqueous solution, optionally including a bactericidal and/or fungicidal agent and/or any other suitable preservative, and optionally including a surface active agent.
- a suitable aqueous solution optionally including a bactericidal and/or fungicidal agent and/or any other suitable preservative, and optionally including a surface active agent.
- the resulting solution may then be clarified by filtration, transferred to a suitable container which is then sealed and sterilized by autoclaving or maintaining at 98-100 degree C for half an hour.
- the solution may be sterilized by filtration and transferred to the container aseptically.
- bactericidal and fungicidal agents suitable for inclusion in the drops are phenylmercuric nitrate or acetate (0.002%), benzalkonium chloride (0.01 %) and chlorhexidine acetate (0.01 %).
- Suitable solvents for the preparation of an oily solution include glycerol, diluted alcohol and propylene glycol.
- solid form preparations which are intended to be converted, shortly before use, to liquid form preparations for oral administration.
- liquid forms include solutions, suspensions, and emulsions.
- These preparations may contain, in addition to the active component, colorants, flavours, stabilizers, buffers, artificial and natural sweeteners, dispersants, thickeners, solubilising agents, and the like.
- liquid form preparations including emulsions, syrups, elixirs, aqueous solutions, aqueous suspensions, toothpaste, gel dentrifrice, chewing gum, or solid form preparations which are intended to be converted shortly before use to liquid form preparations.
- Emulsions may be prepared in solutions in aqueous propylene glycol solutions or may contain emulsifying agents such as lecithin, sorbitan monooleate, or acacia.
- Aqueous solutions can be prepared by dissolving the active component in water and adding suitable colorants, flavours, stabilizing and thickening agents.
- Aqueous suspensions can be prepared by dispersing the finely divided active component in water with viscous material, such as natural or synthetic gums, resins, methylcellulose, sodium carboxymethylcellulose, and other well known suspending agents.
- Solid form preparations include solutions, suspensions, and emulsions, and may contain, in addition to the active component, colorants, flavours, stabilizers, buffers, artificial and natural sweeteners, dispersants, thickeners, solubilising agents, and the like.
- compositions of the present invention may be formulated for parenteral administration (e.g., by injection, for example bolus injection or continuous infusion) and may be presented in unit dose form in ampoules, pre-filled syringes, small volume infusion or in multi-dose containers with an added preservative.
- the compositions may take such forms as suspensions, solutions, or emulsions in oily or aqueous vehicles, for example solutions in aqueous polyethylene glycol.
- oily or nonaqueous carriers, diluents, solvents or vehicles examples include propylene glycol, polyethylene glycol, vegetable oils (e.g., olive oil), and injectable organic esters (e.g., ethyl oleate), and may contain formulatory agents such as preserving, wetting, emulsifying or suspending, stabilizing and/or dispersing agents.
- the active ingredient may be in powder form, obtained by aseptic isolation of sterile solid or by lyophilisation from solution for constitution before use with a suitable vehicle, e.g., sterile, pyrogen-free water.
- Oils useful in parenteral formulations include petroleum, animal, vegetable, or synthetic oils. Specific examples of oils useful in such formulations include peanut, soybean, sesame, cottonseed, corn, olive, petrolatum, and mineral. Suitable fatty acids for use in parenteral formulations include oleic acid, stearic acid, and isostearic acid. Ethyl oleate and isopropyl myristate are examples of suitable fatty acid esters.
- Suitable soaps for use in parenteral formulations include fatty alkali metal, ammonium, and triethanolamine salts
- suitable detergents include (a) cationic detergents such as, for example, dimethyl dialkyl ammonium halides, and alkyl pyridinium halides; (b) anionic detergents such as, for example, alkyl, aryl, and olefin sulfonates, alkyl, olefin, ether, and monoglyceride sulfates, and sulfosuccinates, (c) nonionic detergents such as, for example, fatty amine oxides, fatty acid alkanolamides, and
- amphoteric detergents such as, for example, alkyl-.beta.-aminopropionates, and 2-alkyl-imidazoline quaternary ammonium salts, and (e) mixtures thereof.
- the parenteral formulations typically will contain from about 0.5 to about 25% by weight of the active ingredient in solution. Preservatives and buffers may be used. In order to minimize or eliminate irritation at the site of injection, such compositions may contain one or more nonionic surfactants having a hydrophile-lipophile balance (HLB) of from about 12 to about 17. The quantity of surfactant in such formulations will typically range from about 5to about 15% by weight. Suitable surfactants include polyethylene sorbitan fatty acid esters, such as sorbitan monooleate and the high molecular weight adducts of ethylene oxide with a hydrophobic base, formed by the condensation of propylene oxide with propylene glycol.
- HLB hydrophile-lipophile balance
- parenteral formulations can be presented in unit-dose or multi-dose sealed containers, such as ampules and vials, and can be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid excipient, for example, water, for injections, immediately prior to use.
- sterile liquid excipient for example, water
- Extemporaneous injection solutions and suspensions can be prepared from sterile powders, granules, and tablets of the kind previously described.
- compositions of the invention can also be delivered topically.
- Regions for topical administration include the skin surface and also mucous membrane tissues of the vagina, rectum, nose, mouth, and throat.
- Compositions for topical administration via the skin and mucous membranes should not give rise to signs of irritation, such as swelling or redness.
- the topical composition may include a pharmaceutically acceptable carrier adapted for topical administration.
- the composition may take the form of a suspension, solution, ointment, lotion, sexual lubricant, cream, foam, aerosol, spray, suppository, implant, inhalant, tablet, capsule, dry powder, syrup, balm or lozenge, for example. Methods for preparing such compositions are well known in the pharmaceutical industry.
- compositions of the present invention may be formulated for topical administration to the epidermis as ointments, creams or lotions, or as a transdermal patch.
- Ointments and creams may, for example, be formulated with an aqueous or oily base with the addition of suitable thickening and/or gelling agents.
- Lotions may be formulated with an aqueous or oily base and will in general also containing one or more emulsifying agents, stabilizing agents, dispersing agents, suspending agents, thickening agents, or coloring agents.
- Formulations suitable for topical administration in the mouth include lozenges comprising active agents in a flavored base, usually sucrose and acacia or tragacanth; pastilles comprising the active ingredient in an inert base such as gelatin and glycerin or sucrose and acacia; and mouthwashes comprising the active ingredient in a suitable liquid carrier.
- Creams, ointments or pastes according to the present invention are semi-solid formulations of the active ingredient for external application. They may be made by mixing the active ingredient in finely-divided or powdered form, alone or in solution or suspension in an aqueous or non-aqueous fluid, with the aid of suitable machinery, with a greasy or non-greasy base.
- the base may comprise hydrocarbons such as hard, soft or liquid paraffin, glycerol, beeswax, a metallic soap; a mucilage; an oil of natural origin such as almond, corn, arachis, castor or olive oil; wool fat or its derivatives or a fatty acid such as steric or oleic acid together with an alcohol such as propylene glycol or a macrogel.
- the formulation may incorporate any suitable surface active agent such as an anionic, cationic or non-ionic surfactant such as a sorbitan ester or a
- Suspending agents such as natural gums, cellulose derivatives or inorganic materials such as silicaceous silicas, and other ingredients such as lanolin, may also be included.
- Lotions according to the present invention include those suitable for application to the skin or eye.
- An eye lotion may comprise a sterile aqueous solution optionally containing a bactericide and may be prepared by methods similar to those for the preparation of drops.
- Lotions or liniments for application to the skin may also include an agent to hasten drying and to cool the skin, such as an alcohol or acetone, and/or a moisturizer such as glycerol or an oil such as castor oil or arachis oil.
- Transdermal administration typically involves the delivery of a pharmaceutical agent for percutaneous passage of the drug into the systemic circulation of the patient.
- the skin sites include anatomic regions for transdermally administering the drug and include the forearm, abdomen, chest, back, buttock, mastoidal area, and the like.
- Transdermal delivery is accomplished by exposing a source of the complex to a patient's skin for an extended period of time.
- Transdermal patches have the added advantage of providing controlled delivery of a pharmaceutical agent-chemical modifier complex to the body. See Transdermal Drug Delivery: Developmental Issues and Research Initiatives, Hadgraft and Guy (eds.), Marcel Dekker, Inc., (1989); Controlled Drug Delivery: Fundamentals and Applications, Robinson and Lee (eds.), Marcel Dekker Inc., (1987); and Transdermal Delivery of Drugs, Vols. 1 -3, Kydonieus and Berner (eds.), CRC Press, (1987).
- Such dosage forms can be made by dissolving, dispersing, or otherwise incorporating the pharmaceutical agent-chemical modifier complex in a proper medium, such as an elastomeric matrix material.
- Absorption enhancers can also be used to increase the flux of the compound across the skin. The rate of such flux can be controlled by either providing a rate-controlling membrane or dispersing the compound in a polymer matrix or gel.
- a simple adhesive patch can be prepared from a backing material and an acrylate adhesive.
- the pharmaceutical agent-chemical modifier complex and any enhancer are formulated into the adhesive casting solution and allowed to mix thoroughly.
- the solution is cast directly onto the backing material and the casting solvent is evaporated in an oven, leaving an adhesive film.
- the release liner can be attached to complete the system.
- a polyurethane matrix patch can be employed to deliver the
- the layers of this patch comprise a backing, a polyurethane drug/enhancer matrix, a membrane, an adhesive, and a release liner.
- the polyurethane matrix is prepared using a room temperature curing polyurethane prepolymer. Addition of water, alcohol, and complex to the prepolymer results in the formation of a tacky firm elastomer that can be directly cast only the backing material.
- a further embodiment of this invention will utilize a hydrogel matrix patch.
- the hydrogel matrix will comprise alcohol, water, drug, and several hydrophilic polymers. This hydrogel matrix can be incorporated into a transdermal patch between the backing and the adhesive layer.
- the liquid reservoir patch will also find use in the methods described herein.
- This patch comprises an impermeable or semipermeable, heat sealable backing material, a heat sealable membrane, an acrylate based pressure sensitive skin adhesive, and a siliconized release liner.
- the backing is heat sealed to the membrane to form a reservoir which can then be filled with a solution of the complex, enhancers, gelling agent, and other excipients.
- Foam matrix patches are similar in design and components to the liquid reservoir system, except that the gelled pharmaceutical agent-chemical modifier solution is constrained in a thin foam layer, typically a polyurethane. This foam layer is situated between the backing and the membrane which have been heat sealed at the periphery of the patch.
- the rate of release is typically controlled by a membrane placed between the reservoir and the skin, by diffusion from a monolithic device, or by the skin itself serving as a rate-controlling barrier in the delivery system. See U.S. Pat. Nos. 4,816,258; 4,927,408; 4,904,475; 4,588,580, 4,788,062; and the like.
- the rate of drug delivery will be dependent, in part, upon the nature of the membrane. For example, the rate of drug delivery across membranes within the body is generally higher than across dermal barriers.
- the rate at which the complex is delivered from the device to the membrane is most advantageously controlled by the use of rate-limiting membranes which are placed between the reservoir and the skin. Assuming that the skin is sufficiently permeable to the complex (i.e., absorption through the skin is greater than the rate of passage through the membrane), the membrane will serve to control the dosage rate experienced by the patient.
- Suitable permeable membrane materials may be selected based on the desired degree of permeability, the nature of the complex, and the mechanical considerations related to constructing the device.
- Exemplary permeable membrane materials include a wide variety of natural and synthetic polymers, such as polydimethylsiloxanes (silicone rubbers), ethylenevinylacetate copolymer (EVA), polyurethanes, polyurethane- polyether copolymers, polyethylenes, polyamides, polyvinylchlorides (PVC), polypropylenes, polycarbonates, polytetrafluoroethylenes (PTFE), cellulosic materials, e.g., cellulose triacetate and cellulose nitrate/acetate, and hydrogels, e.g., 2- hydroxyethylmethacrylate (HEMA).
- siloxanes silicone rubbers
- EVA ethylenevinylacetate copolymer
- PVC polyurethanes
- polyurethane- polyether copolymers poly
- compositions according to this invention may also include one or more
- preservatives or bacteriostatic agents e.g., methyl hydroxybenzoate, propyl hydroxybenzoate, chlorocresol, benzalkonium chlorides, and the like.
- bacteriostatic agents e.g., methyl hydroxybenzoate, propyl hydroxybenzoate, chlorocresol, benzalkonium chlorides, and the like.
- active ingredients such as antimicrobial agents, particularly antibiotics, anesthetics, analgesics, and antipruritic agents.
- compositions of the present invention may be formulated for administration as suppositories.
- a low melting wax such as a mixture of fatty acid glycerides or cocoa butter is first melted and the active component is dispersed homogeneously, for example, by stirring. The molten homogeneous mixture is then poured into convenient sized molds, allowed to cool, and to solidify.
- the active composition may be formulated into a suppository comprising, for example, about 0.5% to about 50% of a composition of the invention, disposed in a polyethylene glycol (PEG) carrier (e.g., PEG 1000 [96%] and PEG 4000 [4%].
- PEG polyethylene glycol
- compositions of the present invention may be formulated for vaginal
- compositions of the present invention may be formulated for nasal administration.
- the solutions or suspensions are applied directly to the nasal cavity by conventional means, for example with a dropper, pipette or spray.
- the formulations may be provided in a single or multidose form. In the latter case of a dropper or pipette this may be achieved by the patient administering an appropriate, predetermined volume of the solution or suspension. In the case of a spray this may be achieved for example by means of a metering atomizing spray pump.
- compositions of the present invention may be formulated for aerosol
- the composition will generally have a small particle size for example of the order of 5 microns or less. Such a particle size may be obtained by means known in the art, for example by micronization.
- the active ingredient is provided in a pressurized pack with a suitable propellant such as a chlorofluorocarbon (CFC) for example dichlorodifluoromethane, trichlorofluoromethane, or dichlorotetrafluoroethane, carbon dioxide or other suitable gas.
- CFC chlorofluorocarbon
- the aerosol may conveniently also contain a surfactant such as lecithin.
- the dose of drug may be controlled by a metered valve.
- the active ingredients may be provided in a form of a dry powder, for example a powder mix of the composition in a suitable powder base such as lactose, starch, starch derivatives such as hydroxypropylmethyl cellulose and
- the powder carrier will form a gel in the nasal cavity.
- the powder composition may be presented in unit dose form for example in capsules or cartridges of e.g., gelatin or blister packs from which the powder may be administered by means of an inhaler.
- formulations can be prepared with enteric coatings adapted for sustained or controlled release administration of the active ingredient.
- the pharmaceutical preparations are preferably in unit dosage forms.
- the preparation is subdivided into unit doses containing appropriate quantities of the active component.
- the unit dosage form can be a packaged preparation, the package containing discrete quantities of preparation, such as packeted tablets, capsules, and powders in vials or ampoules.
- the unit dosage form can be a capsule, tablet, cachet, or lozenge itself, or it can be the appropriate number of any of these in packaged form.
- salts of the instant compounds where they can be prepared, are also intended to be covered by this invention. These salts will be ones which are acceptable in their application to a pharmaceutical use. By that it is meant that the salt will retain the biological activity of the parent compound and the salt will not have untoward or deleterious effects in its application and use in treating diseases.
- compositions are prepared in a standard manner. If the parent compound is a base it is treated with an excess of an organic or inorganic acid in a suitable solvent. If the parent compound is an acid, it is treated with an inorganic or organic base in a suitable solvent.
- the compounds of the invention may be administered in the form of an alkali metal or earth alkali metal salt thereof, concurrently, simultaneously, or together with a pharmaceutically acceptable carrier or diluent, especially and preferably in the form of a pharmaceutical composition thereof, whether by oral, rectal, or parenteral (including subcutaneous) route, in an effective amount.
- Examples of pharmaceutically acceptable acid addition salts for use in the present inventive pharmaceutical composition include those derived from mineral acids, such as hydrochloric, hydrobromic, phosphoric, metaphosphoric, nitric and sulfuric acids, and organic acids, such as tartaric, acetic, citric, malic, lactic, fumaric, benzoic, glycolic, gluconic, succinic, p-toluenesulphonic acids, and arylsulphonic, for example.
- mineral acids such as hydrochloric, hydrobromic, phosphoric, metaphosphoric, nitric and sulfuric acids
- organic acids such as tartaric, acetic, citric, malic, lactic, fumaric, benzoic, glycolic, gluconic, succinic, p-toluenesulphonic acids, and arylsulphonic, for example.
- organic acids such as tartaric, acetic, citric, malic, lactic, fumaric, benzoic, glycolic, glu
- the pharmaceutical composition may be prepared so it is suitable for one or more particular administration methods.
- the method of treatment described herein may involve different administration methods.
- any administration method wherein at least one isolated U20 encoding polynucleotide, transcriptional product and/or polypeptide thereof, functional equivalent thereof, variants or fragments thereof may be administered to an individual in a manner so that active an isolated U20 encoding polynucleotide, transcriptional product and/or polypeptide thereof, functional equivalent thereof, variants or fragments thereof may reach the site of disease may be employed with the present invention.
- the main routes of drug delivery, in the treatment method are intravenous, oral, and topical, as will be described below.
- Other drug-administration methods such as subcutaneous injection or via inhalation, which are effective to deliver the drug to a target site or to introduce the drug into the bloodstream, are also contemplated.
- the mucosal membrane to which the pharmaceutical preparation of the invention is administered may be any mucosal membrane of the mammal to which the biologically active substance is to be given, e.g. in the nose, vagina, eye, mouth, genital tract, lungs, gastrointestinal tract, or rectum, preferably the mucosa of the nose, mouth or vagina.
- compositions of the invention may be administered parenterally, that is by intravenous, intramuscular, subcutaneous intranasal, intrarectal, intravaginal or intraperitoneal administration.
- the subcutaneous and intramuscular forms of parenteral administration are generally preferred.
- Appropriate dosage forms for such administration may be prepared by conventional techniques.
- the compositions may also be administered by inhalation, that is by intranasal and oral inhalation administration.
- Appropriate dosage forms for such administration such as an aerosol formulation or a metered dose inhaler, may be prepared by conventional techniques.
- compositions according to the invention may be administered with at least one other compound.
- the compounds may be administered simultaneously, either as separate formulations or combined in a unit dosage form, or administered sequentially.
- the invention relates to a pharmaceutical composition according to the invention, further comprising at least one additional therapeutic agent.
- at least one additional therapeutic agent may be used in the methods for treating TNF receptor related diseases.
- the isolated polypeptide of the present invention, the isolated nucleic acid sequence of the present invention, the vector of the present invention, the host cell of the present invention may be used in a pharmaceutical composition or in a method comprising administering an effective amount to a cell, tissue, organ, animal or patient in need.
- Such a pharmaceutical composition or method can optionally further comprise coadministration or combination therapy for treating such immune diseases, wherein the administering of said composition further comprises administering, before concurrently, and/or after, at least one selected from at least one TNF antagonist (e.g., but not limited to a TNF antibody or fragment, a soluble TNF receptor or fragment, fusion proteins thereof, or a small molecule TNF antagonist), an antirheumatic (e.g., methotrexate, auranofin, aurothioglucose, azathioprine, etanercept, gold sodium thiomalate, hydroxychloroquine sulfate, leflunomide, sulfasalzine), a muscle relaxant, a narcotic, a non-steroid anti-inflammatory drug (NSAID), an analgesic, an anesthetic, a sedative, a local anethetic, a neuromuscular blocker, an antimicrobial (e.g., a
- aminoglycoside an antifungal, an antiparasitic, an antiviral, a carbapenem,
- cephalosporin a flurorquinolone, a macrolide, a penicillin, a sulfonamide, a
- an antipsoriatic e.g. a corticosteriod, an anabolic steroid, a diabetes related agent, a mineral, a nutritional, a thyroid agent, a vitamin, a calcium related hormone, an antidiarrheal, an antitussive, an antiemetic, an antiulcer, a laxative, an anticoagulant, an erythropieitin (e.g. epoetin alpha), a filgrastim (e.g. G- CSF, Neupogen), a sargramostim (GM-CSF, Leukine), an immunization, an antipsoriatic, a corticosteriod, an anabolic steroid, a diabetes related agent, a mineral, a nutritional, a thyroid agent, a vitamin, a calcium related hormone, an antidiarrheal, an antitussive, an antiemetic, an antiulcer, a laxative, an anticoagulant, an erythro
- an immunosuppressive e.g., basiliximab, cyclosporine, daclizumab
- a growth hormone e.g., a hormone replacement drug, an estrogen receptor modulator, a mydriatic, a cycloplegic, an alkylating agent, an antimetabolite, a mitoticinhibitor, a radiopharmaceutical, an antidepressant, antimanic agent, an antipsychotic, an anxiolytic, a hypnotic, a sympathomimetic, a stimulant, donepezil, tacrine, an asthma medication, a beta agonist, an inhaled steroid, a leukotriene inhibitor, a methylxanthine, a cromolyn, an epinephrine or analog, dornase alpha (Pulmozyme), a cytokine or a cytokine antagonist.
- an immunosuppressive e.g., basiliximab
- Suitable dosages are well known in the art. See, e.g., Wells et al., eds., Pharmacotherapy Handbook, 2nd Edition, Appleton and Lange, Stamford, CT (2000); PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe Edition, Tarascon Publishing, Loma Linda, CA (2000).
- TNF antagonists suitable for compositions, combination therapy, co-administration, devices and/or methods of the present invention include, but are not limited to, anti- TNF antibodies, antigen-binding fragments thereof, and receptor molecules which bind specifically to TNF; compounds which prevent and/or inhibit TNF synthesis, TNF release or its action on target cells, such as thalidomide, tenidap, phosphodiesterase inhibitors (e.g, pentoxifylline and rolipram), A2b adenosine receptor agonists and A2b adenosine receptor enhancers; compounds which prevent and/or inhibit TNF receptor signalling, such as mitogen activated protein (MAP) kinase inhibitors; compounds which block and/or inhibit membrane TNF cleavage, such as metalloproteinase inhibitors; compounds which block and/or inhibit TNF activity, such as angiotensin converting enzyme (ACE) inhibitors (e.g., captopril); and compounds which block and/or inhibit TNF
- the additional therapeutic agent is selected from the group consisting of anti-TNF-alpha monoclonal antibodies: infliximab (Remicade), adalimumab (Humira), certolizumab pegol (Cimzia), golimumab (Simponi), TNF-alpha receptor fusion protein: etanercept (Enbrel), or TNF-alpha inhibitory molecules:
- the invention relates to a pharmaceutical composition, wherein the additional therapeutic is selected from the group consisting of anti-TNF-alpha monoclonal antibodies: infliximab (Remicade), adalimumab (Humira), certolizumab pegol (Cimzia), golimumab (Simponi), TNF-alpha receptor fusion protein: etanercept (Enbrel), or TNF-alpha inhibitory molecules: pentoxifylline or bupropion.
- the additional therapeutic is selected from the group consisting of anti-TNF-alpha monoclonal antibodies: infliximab (Remicade), adalimumab (Humira), certolizumab pegol (Cimzia), golimumab (Simponi), TNF-alpha receptor fusion protein: etanercept (Enbrel), or TNF-alpha inhibitory molecules: pentoxifylline or bupropion.
- one aspect of the invention relates to a kit of parts comprising a therapeutic agent as described above and the isolated polypeptide of the invention, or the isolated polynucleotide of the invention, or the vector of the invention, or the host cell of the invention as a combination for the simultaneous, separate or successive administration in TNFR1 related disease therapy.
- the dosage requirements of monomeric alpha-lactalbuminin complex, preferably LAC to be administered will vary with the particular drug composition employed, the route of administration and the particular subject being treated. Ideally, a patient to be treated by the present method will receive a pharmaceutically effective amount of the compound in the maximum tolerated dose, generally no higher than that required before drug resistance develops.
- the daily oral dosage regimen will preferably be from about 0.01 to about 80 mg/kg of total body weight.
- the daily parenteral dosage regimen may be about 0.001 to about 80 mg/kg of total body weight.
- the daily topical dosage regimen will preferably be from 0.1 mg to 150 mg, administered one to four, preferably two or three times daily.
- the daily inhalation dosage regimen will preferably be from about 0.01 mg/kg to about 1 mg/kg per day. It will also be recognized by one of skill in the art that the optimal quantity and spacing of individual dosages of a compound or a pharmaceutically acceptable salt thereof will be determined by the nature and extent of the condition being treated, the form, route and site of administration, and the particular patient being treated, and that such optimums can be determined by conventional techniques. It will also be appreciated by one of skill in the art that the optimal course of treatment, i.e., the number of doses of a compound or a pharmaceutically acceptable salt thereof given per day for a defined number of days, can be ascertained by those skilled in the art using conventional course of treatment determination tests.
- the daily dose of the active compound varies and is dependant on the type of administrative route, but as a general rule it is 1 to 100 mg/dose of active compound at personal administration, and 2 to 200 mg/dose in topical administration.
- the number of applications per 24 hours depend of the administration route, but may vary, e. g. in the case of a topical application in the no. se from 3 to 8 times per 24 hours, i. e. , depending on the flow of phlegm produced by the body treated in therapeutic use.
- the compound according to the present invention is given in an effective amount to an individual in need there of.
- the amount of compound according to the present invention in one preferred embodiment is in the range of from about 0.01 milligram per kg body weight per dose to about 20 milligram per kg body weight per dose, such as from about 0.02 milligram per kg body weight per dose to about 18 milligram per kg body weight per dose, for example from about 0.04 milligram per kg body weight per dose to about 16 milligram per kg body weight per dose, such as from about 0.06 milligram per kg body weight per dose to about 14 milligram per kg body weight per dose, for example from about 0.08 milligram per kg body weight per dose to about 12 milligram per kg body weight per dose, such as from about 0.1 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 0.2 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 0.3 milligram per kg body weight per dose to about 10 mill
- unit dosage form refers to physically discrete units suitable as unitary dosages for human and animal subjects, each unit containing a
- a compound predetermined quantity of a compound, alone or in combination with other agents, calculated in an amount sufficient to produce the desired effect in association with a pharmaceutically acceptable diluent, carrier, or vehicle.
- the specifications for the unit dosage forms of the present invention depend on the particular compound or compounds employed and the effect to be achieved, as well as the pharmacodynamics associated with each compound in the host.
- the dose administered should be an "effective amount” or an amount necessary to achieve an "effective level" in the individual patient.
- the effective level is used as the preferred endpoint for dosing, the actual dose and schedule can vary, depending on individual differences in pharmacokinetics, drug distribution, and metabolism.
- the "effective level” can be defined, for example, as the blood or tissue level desired in the patient that corresponds to a concentration of one or more compounds according to the invention.
- HHV U20 polypeptides for use in the invention may be accomplished using conventional techniques which do not require detailed explanation to one of ordinary skill in the art. For review, however, those of ordinary skill may wish to consult Maniatis et al., in Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, (NY 1982). Expression vectors may be used for generating producer cells for recombinant production of NsG33 polypeptides for medical use, and for generating therapeutic cells secreting NsG33 polypeptides for naked or encapsulated therapy.
- construction of recombinant expression vectors employs standard ligation techniques.
- the genes are sequenced using, for example, the method of Messing, et al., (Nucleic Acids Res., 9: 309-, 1981 ), the method of Maxam, et al., (Methods in Enzymology, 65: 499, 1980), or other suitable methods which will be known to those skilled in the art.
- these should contain regulatory sequences necessary for expression of the encoded gene in the correct reading frame.
- Expression of a gene is controlled at the transcription, translation or post-translation levels. Transcription initiation is an early and critical event in gene expression. This depends on the promoter and enhancer sequences and is influenced by specific cellular factors that interact with these sequences.
- the transcriptional unit of many genes consists of the promoter and in some cases enhancer or regulator elements (Banerji et al., Cell 27: 299 (1981 ); Corden et al., Science 209: 1406 (1980); and Breathnach and Chambon, Ann. Rev. Biochem. 50: 349 (1981 )).
- LTR long terminal repeat
- CMV cytomegalovirus
- the invention relates to a recombinant vector comprising the nucleic acid sequence according to the invention.
- the invention relates to a vector according to the invention, wherein the vector is selected from the group consisting of plasmids, cosmids, phages, bacterial artificial chromosomes (BAC), Phagemids and P1 -derived artificial chromosomes.
- the vector may comprise one or more regulatory sequences e.g. controlling the expression of the U20 protein or active derivative thereof.
- the regulatory sequence may either form part of the final protein or peptid or it may not form part of the final protein or peptid.
- the at least one regulatory sequence may be a promoter operably linked to the polynucleotide.
- promoters are promoters selected from CMV (cytomegalovirus), SV40 (simian adenovirus), AdMLP, elongation factor-1 alpha, Rous sarcoma virus long terminal repeat promoter, T7 promoter.
- the choice of vector may depend on the type of host cell which can be used to amplify the vector and express the polypeptides of the present invention.
- the vector For cloning and expression purposes it may be useful to have the vector cloned in a host cell. In this way the vector can be maintained, cloned and purified and thus provide a source for almost unlimited numbers of the vectors.
- stable producer cell lines expressing U20 protein or active derivative thereof according to the invention may also be advantageous.
- In order to maintain a producer cell selection marker may be incorporated in the expression vector.
- the invention relates to an isolated host cell transformed or transduced with the recombinant vector according to the invention. Different types of host cells may be used.
- the invention relates to host cells selected from the group consisting of bacteria, fungi and molds.
- bacteria are selected from the group consisting of Bacillus licheniformis, Bacillus subtilis and E.coli and the fungi and molds are selected from the group consisting of Aspergillus niger and Aspergillus oryzae.
- the host cell may thus be selected from E.coli, Yeast, Saccharomyces cerevisiae, S. pombe or insect cells.
- the host cells may in one embodiment be a mammalian cell, such as human, feline, porcine, simian, canine, murine, rat, mouse or rabbit. In one particular embodiment the host cell is mammalian.
- the host cells may be a human embryonal kidney cells (HEK cells), such as the cell lines deposited at the American Type Culture Collection with the numbers CRL-1573 and CRL-10852, chick embryo fibroblast, hamster ovary cells, baby hamster kidney cells, human cervical carcinoma cells, human melanoma cells, human kidney cells, human umbilical vascular endothelium cells, human brain endothelium cells, human oral cavity tumor cells, monkey kidney cells, mouse fibroblast, mouse kidney cells, mouse connective tissue cells, mouse oligodendritic cells, mouse macrophage, mouse fibroblast, mouse neuroblastoma cells, mouse pre-B cell, mouse B lymphoma cells, mouse plasmacytoma cells, mouse teratocacinoma cells, rat astrocytoma cells, rat mammary epithelium cells, COS, CHO, BHK, 293, VERO, HeLa, MDCK, WI38, HTC1 16 cell or NIH 3
- HEK cells human embryon
- kits comprising a) at least one component selected from
- the instruction manual provides instructions to how the components should be used, dosing regimes and diseases for which the kit is applicable.
- kits comprising the U20 protein or an active derivative thereof according to the invention may be used in the clinic when U20 or an active derivative thereof is going to be provided to a patient suffereing from disease which may require treatment with U20 or an active derivative thereof.
- a kit comprising the pharmaceutical composition may be used for similar purposes.
- a kit comprising the polynucleotide, the vector or the host cell may be used for production of the U20 protein or an active derivative thereof.
- the instruction manual provides the user with instruction on how to use the different components.
- the polypeptides, methods, compositions and kits of the invention may also be used as a diagnosticum, Thus, in an aspect the invention relates to an isolated U20 protein or an active derivative thereof according to the invention for use as a diagnosticum. Since the polypeptides of the invention interacts with the TNF receptor, for example TNFR1 , the polypeptides, methods, compositions or kit may be used for measuring the amount of receptors expressed in a sample in a similar way as a standard antibody. The expression can be detected using a standard sandwich assay known to the person skilled in the art. If no secondary antibody able to bind to the polypeptides of the present invention (after binding to the receptor) a tag may be positioned, in frame, on the polypeptides, thereby allowing binding and detection of the secondary antibody.
- the invention relates to the polypeptides for use in determining the expression level of a TNF receptor in a sample.
- the amount of receptor may be used for determining the amount of a pharmaceutical composition, or polypeptide of the present invention is needed in a specific treatment of an individual.
- the polypeptide may also be used as a diagnosticum to provide precise measurements of TNFa in a sample. Since the polypeptides binds to the TNF-alpha receptor, the polypeptide may be used to prevent the binding of TNFa to its receptor in a cellular assay, such as a biological assay where cells secrete or are induced to secrete TNFa to the supernatant.
- a cellular assay such as a biological assay where cells secrete or are induced to secrete TNFa to the supernatant.
- U20 polypeptide or amino acids may prevent the consumption of TNFa from the sample and thus provide precise measurements of amount of secreted TNFa.
- HHV-6B rescues cells from TNF -induced apoptosis
- HHV-6B In order to determine whether or not HHV-6B is able to rescue cells from extrinsic induced apoptosis, we first established criteria for rapid induction of apoptosis through TNF receptorl (TNFR1 ). Treatment of HCT1 16 cells with TNFa and cycloheximide (CHX) and subsequent Western blot (WB) analysis of poly (ADP-ribose) polymerase (PARP) cleavage. Activation of caspase 3 leads to cleavage in PARP within its nuclear localization domain to yield a lower molecular weight (MW) fragment, which can be identified by WB.
- CHX TNFa and cycloheximide
- PARP Western blot
- caspase 3 Activation of caspase 3 leads to cleavage in PARP within its nuclear localization domain to yield a lower molecular weight (MW) fragment, which can be identified by WB.
- MW molecular weight
- CHX treatment blocks phosphorylation of ⁇ , which directs the TNFR1 response toward formation of complex II, and thereby directing it against apoptosis.
- Treatment with Camptothecin which induces activation of the intrinsic apoptotic pathway, caused apoptosis in both wild type (wt) cells and HHV-6B-infected cells.
- TNFoc When TNFoc binds to TNFR1 it can induce several different downstream pathways, depending on intracellular protein complexes that subsequently are formed.
- the first pathway we analyzed is mediated through FADD and TRADD, which mediates cleavage of pro-caspase-3 into active caspase-3. Caspase-3 is essential for cleavage of a large number of proteins destined for degradation during apoptosis.
- the first caspase to be activated in this pathway is caspase-8, which upon activation cleaves pro-caspase-3.
- HHV-6B infection blocked the cleavage of pro- caspase-8 to active caspase-8 ( Figure 2A).
- TNFR1 -induced pathway leads to IKBOC phosphorylation, through TRAF2, which results in NFKB translocation to the nucleus, and subsequent transcription of either pro-apoptotic or anti-apoptotic genes, depending on cell type and cellular gene expression profile.
- WB analysis of IKBOC phosphorylation showed that HHV-6B infection could block this pathway ( Figure 2A).
- HCT1 16 cells transfected with U20 expression plasmid showed a reduced resistance to TNFoc-induced apoptosis when analyzed by WB for PARP cleavage ( Figure 3D).
- the lower levels of rescue during transient expression could be explained by a transfection efficiency that only approached 50%.
- HCT1 16 cells, transiently expressing U20 by flow cytometry for the apoptosis marker 7-aminoactinomycin D (7-AAD). This analysis also indicated a reduced resistance to TNFoc-induced apoptosis in U20-expressing cells ( Figure 3E-G).
- U20 appears to block TNFoc induced apoptosis, and could potentially be the protein solely responsible for this block during infection with HHV-6B.
- confocal microscopy was used to visualize active caspase-3.
- U20 is indeed a gene and it is transcribed during both productive and non-productive infections.
- Theoretical analyses identified U20 as a membrane protein with a single membrane spanning oc-helix (aa 319 - 343) and an N-terminal signal peptide (aa 1 - 15) for ER sorting to the plasma membrane (Figure 5C).
- the orientation of U20 in the membrane is predicted to be with the N-terminal end at the outside of the cell and the C-terminal end in the cytoplasm.
- Theoretic analysis further predicted an immunoglobulin-like fold spanning aa 182 - 300. Furthermore, analysis indicates a highly organized N-terminal part and a highly disordered C-terminal part.
- a structural model of U20 from HHV-6B is shown in Figure 10. Taken together, these data strongly support that U20 is expressed on the surface of the cells, and oriented with a large N-terminal extracellular part and a small C-terminal intracellular part.
- TNFR1 When TNFR1 binds TNFa it rapidly forms a signaling complex, which includes the adaptor protein TRADD.
- TRADD the adaptor protein
- Complex I is a complex that is formed immediately following TNFa stimulation and contains TNFR1 with the recruited proteins TRADD, TRAF2 and RIP, but not FADD or caspase 8.
Abstract
Isolated human herpes virus U20 polypeptides are provided, as well as biologically active variants and fragments thereof, which bind to a human tumor necrosis factor (TNF) receptor, which also serves as a receptor for tumor necrosis factor -alpha (TNF- alpha). The polypeptides are also provided for use as medicaments, and as components in pharmaceutical compositions. The use of the polypeptides is also provided in methods for decreasing TNF receptor activity and for treatment of TNF receptor associated disorders.
Description
HUMAN HERPES VIRUS 6 AND 7 U20 POLYPEPTIDE AND POLYNUCLEOTIDES FOR USE AS A MEDICAMENT OR DIAGNOSTICUM
All patent and non-patent references cited in the application, or in the present application, are also hereby incorporated by reference in their entirety.
Field of invention
The present invention relates to human herpes virus U20 polypeptides, biologically active variants or fragments thereof, as well as to polynucleotides encoding
polypeptides identical to the human herpes virus U20 polypeptides, variants or fragments thereof. In particular the present invention relates to U20 polypeptides, biologically active variants or fragments thereof for use as a medicament for the treatment of diseases related to TNF receptor activity such as in autoimmune, inflammatory, degenerative or cancerous diseases. Background of invention
Infections may initiate an inflammatory reaction. A key regulator during this process is the cytokine tumor necrosis factor-alpha (TNFa), which regulates the expression of other pro-inflammatory cytokines in the signal transduction pathway of TNF receptors. TNFa exerts its activity by forming trimeric structures that bind to trimeric receptors. The TNF receptors include TNFR1 (also known as TNF receptor superfamily 1 A or TNFRSF1 A) and TNFR2 (known as TNFRSF1 B). However, there are 19 ligands and 30 receptors within the TNF superfamily, and they are all thought to be important in controlling immune functions during health and disease (Croft, 2009, Nature Rev Immunol 9:271 -285). The success of anti-TNFa antibodies in minimizing
proinflammatory reactions and pathogenic responses during autoimmune and inflammatory diseases indicates the significance of TNFa-TNFR1/2 interactions for the function of the immune system.
Signalling through the TNFR1 is complex and involves the recruitment and modification of many proteins leading to a cascade activation that may result in cell death or cell survival (Karin & Gallagher, 2009, Immunol Rev 228:225-240). The specific details in determining the exact outcome are still debated. In general, upon binding of TNFa, TNFR1 recruits the adaptor protein TNFR1 -associated death domain protein (TRADD) and TNFR-associated factor 2 (TRAF2). In addition, the protein RIP1 is recruited and is subsequently modified by ubiquitination to serve as a docking site for further recruited
proteins in the NF-kB signalling pathway. This arm of the pathway is thought to enhance cell survival, whereas another arm of the signalling cascade may lead to cell death following TRADD recruitment of Fas-associated death domain (FADD). This complex may then recruit caspase 8 to activate a signalling cascade that leads to apoptosis of the cell. For an overview of part of the signalling pathway through TNFR1 see Figure 8.
Because of the harmful role of human TNFa (hTNFa) in a variety of human disorders, therapeutic strategies have been designed to inhibit or counteract hTNFa activity. In particular, antibodies that bind to, and neutralize, hTNFa have been suggested as a means to inhibit hTNFa activity. Examples of patents and patent application disclosing hTNFa binding antibodies and treatments targeting hTNFa are EP 1 309 691 B1 , US 6,379,666 B1 , US 6,509,015 B1 and WO 2006/056779 A2. Alternative means for inhibiting human TNFa is the use of soluble TNFa receptors. WO 2006/038027 A2 discloses antibodies that bind Tumor Necrosis Factor Receptor 1 , and which can be used for treating inflammatory diseases.
A general problem in relation to antibody-based therapies is the development of an immune response in the patient towards the applied antibodies, which can be both harmful for the patient and inhibit the effect of the treatment.
Human herpesvirus 6 (HHV-6) was isolated in 1986 and later divided into two variants, HHV-6A and -6B, based on differences in restriction enzyme cleavage patterns and reactivity with monoclonal antibodies. HHV-6A and -6B belong to the betaherpesvirus subfamily and belong to the Roseolovirus genus together with the closely related HHV- 7. Infection by HHV-6B may give rise to a febrile illness during the first years of life, known as exanthem subitum, after which the virus remains in a latent state in the infected individual for life. It is possible that primary infection with HHV-6A or HHV-7 may cause a similar disease with rash and fever. HHV-6B is considered an
opportunistic pathogen and its reactivation from the latent state may cause severe disease in immunocompromised individuals.
HHV-6A and -6B have unique characteristics making them atypical among the human herpesvirus family. Compared with other herpesviruses, the heparin-like molecules seem to play a minor role in HHV-6 surface interactions required for infection.
Furthermore, the intracellular maturation pathway of HHV-6 is different from that of
other herpesviruses, and few viral glycoproteins are detected on the host cell plasma membrane of HHV-6 infected cells.
The genome of HHV-6B is 162 kbp long with a unique segment (U) of about 144 kbp flanked in both ends by direct repeats (DR) of about 9 kbp each (Dominguez et al.,
1999). The left end of the unique segment is the best-conserved region between HHV- 6A and HHV-6B, whereas the DRs and the region from the open reading frames U86 to U100 are more divergent. HHV-6B is predicted to have 97 unique genes, of which 88 have counterparts in HHV-6A and 82 in HHV-7 (Dominguez et al., 1999, J Virol 73:8040-8052). Most of the genes from HHV-6A, -6B, and -7 can be divided into gene blocks. Seven of these are the conserved herpesvirus core gene blocks, spanning U27 to U82 in HHV-6B, whereas U2 to U19 belongs to the betaherpesvirus genes found only in betaherpesvirinae (Dominguez et al., 1999). The expression from the U18-U20 has been studied in HHV-6A and HHV-6B, indicating possible differences in their expression pattern. Inspection of the amino acid sequence of the predicted U20 polypeptide from HHV-6B indicates that it may be a glycoprotein of 434 amino acids with 95.6% nucleotide identity to its homologue in HHV-6A. The function of U20 is entirely unknown. Although most glycoproteins are positioned in the cell membrane (or secreted), it has not yet been defined whether or not U20 is actually expressed on the cell surface or whether it is secreted from infected cells.
Though many alternative therapies for inhibiting the harmful role of human TNFa (hTNFa) in a variety of human disorders are available a need remains for alternative or complementary treatments. Hence, an improved treatment of diseases involving human TNFa (hTNFa) would be advantageous, and in particular a more efficient and/or reliable therapy targeting alternative parts of the hTNFa pathway would be
advantageous. Summary of invention
This invention pertains to isolated human herpes virus U20 polypeptides, biologically active variants and fragments thereof that bind to a human tumor necrosis factor (TNF) receptor, which also serves as a receptor for tumor necrosis factor -alpha (TNF-alpha). Various aspects of the invention relate human herpes virus U20 polypeptides, biologically active variants and fragments thereof for use as a medicament or
diagnosticum, pharmaceutical compositions, as well as polynucleotides, recombinant expression vectors and host cells for making such human herpes virus U20
polypeptides, biologically active variants and fragments thereof.
Thus, an object of the present invention relates to providing a new medicament in treatments requiring decreasing TNF receptor activity, such as decreasing human TNF- alpha inhibition of the TNF receptor.
In particular, it is an object of the present invention to provide a new medicament in treatment requiring decreasing TNF receptor activity, such as decreasing human TNF- alpha inhibition of the TNF receptor that solves the above mentioned problems of the prior art with patients acquiring for example an immune response against TNFalpha antibodies.
Thus, one aspect of the present invention relates to an isolated polypeptide for use as medicament or diagnosticum comprising
a) an amino acid sequence consisting of SEQ ID No. 1 , 3 or 5; b) a biologically active sequence variant of the amino acid sequence,
wherein the variant has at least 70% sequence identity to said SEQ ID NO. 1 , 3 or 5; or
c) a biologically active fragment of at least 50 contiguous amino acids of any of a) through b), wherein said fragment is a fragment of SEQ ID NO 1 , 3 or 5.
A second aspect of the present invention relates to An isolated polynucleotide for use as a medicament comprising a nucleic acid or its complementary sequence, said polynucleotide being selected from the group consisting of:
a) i) a polynucleotide encoding an amino acid sequence consisting of SEQ ID No. 1 , 3 or 5;or
ii) a biologically active sequence variant of the amino acid sequence, wherein the variant has at least 70% sequence identity to said SEQ ID
NO. 1 , 3 or 5; and
iii) a biologically active fragment of at least 50 contiguous amino acids of any of a) through b), wherein said fragment is a fragment of SEQ ID NO 1 , 3 or 5 or
b) SEQ ID NO.: 2, 4, 6, 7, 8 or 9 or
a) a polynucleotide comprising a nucleic acid having 70% sequence identity to SEQ ID NO.: 2, 4, 6, 7, 8 or 9 or
b) a polynucleotide hybridising to SEQ ID NO.: 2, 4, 6, 7, 8 or 9 and c) a polynucleotide complementary to any of a) to d).
The previously described polynucleotides may be expressed in a vector system. Thus, another aspect of the present invention relates to an expression vector comprising the polynucleotides as described herein and above.
The vectors according to the invention may be amplified and stored in a host cell. Similarly, the polynucleotides of the invention may be expressed in such a host cell. Thus, in yet another aspect of the present invention relates to an isolated host cell transfected or transduced with the vector as described herein and above.
A further aspect of the present invention pertains to a pharmaceutical composition comprising
the isolated polypeptide according to the present invention or the isolated nucleic acid sequence according to the present invention or
the vector of according to the present invention or the host cell according to the present invention
and a pharmaceutical acceptable carrier and/or diluent.
Another aspect pertains to a kit of parts comprising a therapeutic agent of the invention and the isolated polypeptide of the invention, or the isolated polynucleotide of the invention, or the vector of the invention, or the host cell of the invention as a combination for the simultaneous, separate or successive administration in TNFR1 related disease therapy.
Another aspect of the present invention relates to a pharmaceutical composition for treating, ameliorating and/or preventing TNF receptor related diseases comprising a) the isolated polypeptide according to the present invention or b) the isolated nucleic acid sequence according to the present
invention or
c) the vector of according to the present invention or
d) the host cell according to the present invention e) the kit of parts of the invention.
Yet another aspect of the present invention relates to a kit comprising
a) at least one component selected from
a. the isolated polypeptide according to the present invention or b. the isolated nucleic acid sequence according to the present invention or
c. the vector according to the present invention or
d. the host cell according to the present invention or e. the pharmaceutical composition according to the present invention
and
b) an instruction manual.
A further aspect of the present invention relates to a method for treating, ameliorating and/or preventing TNF receptor related diseases comprising administration of
a) the isolated polypeptide of the invention or
b) the isolated nucleic acid sequence of the invention or
c) the vector of the invention or
d) the host cell of the invention or
e) the pharmaceutical composition of the invention
f) the kit of parts of the invention
g) the kit of the invention in a therapeutically effective amount to an individual in need thereof.
Another aspect of the present invention relates to an isolated polypeptide of as described herein and above
an isolated nucleic acid sequence according to the present invention or a vector of the invention or
a host cell of the invention or
a pharmaceutical composition of the invention or
a kit of part of the invention or
a kit of the invention for the treatment of TNF receptor related diseases.
Yet another aspect of the present invention relates to the use of a) the isolated polypeptide according to the present invention or b) the isolated nucleic acid sequence according to the present
invention or
c) the vector according to the present invention
d) the host cell according to the present invention
e) the pharmaceutical composition according to the present invention f) the kit of parts of the invention or
g) the kit of the invention
for the manufacture of a medicament for treatment of TNF receptor related diseases.
A further aspect of the present invention relates to a method for decreasing TNF receptor activity comprising
a) contacting a TNF receptor with
i) the isolated polypeptide according to the present invention or ii) the isolated nucleic acid sequence according to the present invention or
iii) the vector of according to the invention or
iv) the host cell according to the invention or
v) the pharmaceutical composition of the present invention vi) the kit of parts of the invention or
vii) the kit of the invention
b) obtaining a decrease in TNF receptor activity compared to a standard level of TNF receptor activity in an individual.
Description of Drawings
Figure 1
Figure 1 shows that HHV-6B rescues cells from TNFoc-induced apoptosis.
A) Western blot (WB) analysis of PARP cleavage in HCT1 16 cells treated with TNFa, Cycloheximide (CHX) or a combination of them. PARP cleavage is a marker for apoptosis. GAPDH is included as loading control. B) WB analysis of PARP cleavage in HCT1 16 cells treated with or without TNFa/CHX and with or without HHV-6B infection. C) WB analysis of HCT1 16 cells treated with or without camptothecin (Campt.) and
with or without HHV-6B infection. HHV-6B is unable to rescue the cells from
camptothecin-induced apoptosis.
Figure 2
Figure 2 shows that HHV-6B blocks TNF induction by interfering with TNFR1 or a protein in close proximity to TNFR1 . A) WB analysis of HCT1 16 cells treated with or without TNFoc/CHX and infected with or without HHV-6B. Analysis is performed with antibodies against active caspase-8, active caspase-3 and phosphorylated ΙκΒ (ρ-ΙκΒ). Antibodies against RCC1 are included as a loading control. HHV-6B completely blocks induction of both the pathway originating from FADD signalling (caspase pathway) and the pathway originating from TRAF2 signalling (ρ-ΙκΒ). This indicates a block directly of TNFR1 or TRADD (see fig. 8).
B) Flowcytometry analysis of TNFR1 on the surface of HCT1 16 cells infected with or without HHV-6B. The analysis shows similar levels of TNFR1 on HHV-6B-infected- and non-infected cells.
Figure 3
Figure 3 shows that U20 blocks TNFoc-induced TNFR1 signalling. A) WB analysis of PARP cleavage in wt HCT1 16 cells or HCT1 16 cells stably expressing U20 (U20-S). U20 completely rescues HCT1 16 cells from TNFoc/CHX-induced apoptosis. Probing with antibodies against GAPDH is included as loading control. B) WB analysis of wt or U20-S HCT1 16 cells treated with or without camptothecin. U20 is unable to rescue cells from camptothecin-induced apoptosis. C) PCR with U20-specific primers on cDNA from wt HCT1 16 cells or U20-S HCT1 16 cell clone. The U20-S clone does express an mRNA with the size expected for U20. D) WB analysis of PARP cleavage in HCT1 16 cells transfected transiently with either mock or U20 plasmid (U20-T). U20 expression is able to rescue the cells partially from TNFoc/CHX-induced apoptosis. E - F)
Flowcytometry analysis of 7-aminoactinomycin D (7-AAD) incorporation in HCT1 16 cells transfected transiently with either mock or U20 plasmid. 7-AAD stains apoptotic cells. U20 expression is able to rescue the cells partially from TNFoc/CHX-induced apoptosis. A representative figure from four independent experiments is shown. G) Average of four experiments as shown in E & F. Error bars represent SD.
Figure 4
Figure 4 shows that U20 blocks TNFoc induction at a site in close proximity to TNFR1 . A) WB analysis of wt or U20 stably transfected HCT1 16 cells (U20-S) treated with or without TNFoc/CHX. The membrane was stained with antibodies against active caspase-3, active caspase-8 or phosphorylated ΙκΒ (ρ-ΙκΒ). U20 completely blocks the FADD pathway leading to caspase cleavage, and the TRAF2 pathway leading to ΙκΒ phosphorylation. Probing with antibodies against RCC1 is included as a loading control. B) Confocal microscopy of wt and U20-S cells treated with or without
TNFoc/CHX and stained with an antibody against active caspase-3 (arrow) and DAPI DNA stain. Staining of active caspase-3 was only observed in wt cells. C) WB analysis of wt or U20 stable HCT1 16 cells treated with or without TNFoc/CHX. The membrane was probed with antibodies against PARP, TRAF2 and TRADD. Antibodies against GAPDH are included as a loading control. U20 and wt cells show similar levels of both TRADD and TRAF2, indicating lack of signalling is not due to downregulation of the adaptor proteins. D) Flowcytometry analysis of TNFR1 expression on the surface of wt HCT1 16 cells and the U20-S cells. U20 and wt cells show similar levels of TNFR1 on their surface. Therefore, lack of signalling from TNFR1 is not due to downregulation of TNFR1 .
Figure 5
Figure 5 shows that U20 is predicted to be a transmembrane protein. A) PCR conducted with U20 specific primers on cDNA from HHV-6B-infected HCT1 16 cells. U20 mRNA is expressed at detectable levels at 8 hours post infection (hpi). B) Realtime PCR conducted with U20 specific primers on cDNA from HHV-6B-infected HCT1 16 cells. U20 mRNA was quantified relative to β-Actin. U20 mRNA could be detected as early as 2 hpi, but with a marked increase around 8 hpi. D) Schematic representation of U20 protein motifs. Bioinformatics analysis reveals a highly likely N- terminal signal peptide for sorting to the membrane, a possible Immunoglobulin-like domain, a highly likely transmembrane a-helix, and several N-glycosylation sites on the N-terminal side of the a-helix. Together this indicates a protein destined for the outer membrane with the large N-terminal domain outside the cell.
Figure 6
Figure 6 shows that U20 blocks TRADD translocation to the cytoplasm. Wt or U20 stably transfected (U20-S) HCT1 16 cells treated with TNFa/CHX and analysed by confocal microscopy at different time points after treatment. In wt cells TRADD rapidly translocate to be dispersed troughout the cytoplasm. This translocation is completely
blocked in U20-S cells. These data indicate that the TNFR1 signalling complexes I & II are not formed in U20-S cells.
Figure 7
Figure 7 shows that U20 blocks binding of the HTR-19 TNFa antibody to TNFR1 . A) Flowcytometry analysis of wt HCT1 16 cells treated with isotype control, HTR-19 anti- TNFa antibody, or HTR-19 after pre-treatment with TNFa. TNFa blocks binding of the HTR-19 antibody to TNFR1 . B) Flowcytometry analysis of U20 stably transfected (U20- S) HCT1 16 cells treated with isotype control (dashed line), HTR-19 anti-TNFa antibody (grey), or HTR-19 antibody after pre-treatment with TNFa (white). U20 completely abrogates binding of the HTR-19 antibody to TNFR1 . These data indicate that U20 interacts directly with TNFR1 in the membrane.
Figure 8
Figure 8 shows part of the TNFR1 signaling pathway. TNFa binds TNFR1 and forms a trimer complex. The TNFR1 trimer recruits adaptor proteins TRADD, TRAF2 and FADD. This can lead to two pathways, which inhibit one another. In one pathway FADD recruits pro-caspase-8 and induces cleavage of it into active caspase-8. Active caspase-8 cleaves pro-caspase-3 into active caspase-3, which is an effector caspase involved in onset of apoptosis. In the other pathway TRAF2 induces phosphorylation of IKB, which is then no longer able to block NFKB translocation to the nucleus.
Cycloheximide (CHX) inhibits cell survival by preventing de novo protein synthesis. Figure 9
Figure 9 shows an alignment between the U20 protein from HHV-6A, HHV-6B and HHV-7. In addition a consensus sequence and a degree of similarity are shown.
Figure 10
Figure 10 shows a structural model prediction of U20 from HHV-6B. The amino acid numbering for the predicted different domains and the predicted glycolysation sites are also indicated.
Figure 1 1
Figure 1 1 shows an alignment between the U20 nucleic acid sequences from HHV-6A, HHV-6B and HHV-7. In addition a consensus sequence and a degree of similarity are shown.
Figure 12
HHV-6 infection blocks TNFR1 signalling. A) HCT1 16 cells were treated with CHX (10 μΜ), TNFa (25 ng/ml) or a CHX/TNFa in combination for 4 hours and analysed for
PARP cleavage and GAPDH (loading control). B) Mock infected or HHV-6B infected HCT1 16 cells (48 hpi) were treated with a combination of CHX/TNFa (10 μΜ/25 ng/ml) for 4 hours and analysed for PARP cleavage. C) Mock infected or HHV-6B infected HCT1 16 cells (48 hpi) were treated with Camptothesine and analysed for PARP cleavage. D) HCT1 16 cells were either mock infected or infected with HHV-6B for 48 hpi and treated with CHX/TNFa (10 μΜ/25 ng/ml) for 4 hours and analysed for PARP cleavage, caspase-8 cleavage, caspase-3 cleavage, ΙκΒα phosphorylation, total ΙκΒα, 7C7 (infection marker) and RCC1 (loading control). Mock or HHV-6B infected cells (48 hpi) were stained with TNFR1 antibody and analysed by flowcytometry with an Alexa 488 conjugated secondary antibody.
Figure 13
Early protein U20 is sufficient for blocking TNFR1 signalling. A) Schematic
representation of U20 protein based on bioinformatics analysis. C) cDNA from MOLT3 cells infected with HHV-6B for up to 48 hpi were analysed by PCR with U20 end- specific primers. C) cDNA from HCT1 16 cells infected with HHV-6B for up to 24 hpi were analysed by relative qPCR and quantified against Actin. D) HCT1 16 cells transfected with a plasmid with V5 tagged U20 and analysed by confocal microscopy with a V5 specific antibody and an Alexa 546 conjugated secondare antibody. Arrows points to intracellular U20 and arrowhead points to outermembrane localized U20. E) HCT1 16 cells transfected with U20-V5 and analysed by lysate fractionation into cytoplasm, membranes and nuclear fractions. WB were stained with anti-V5 antibody, and antibodies against GAPDH, COX IV and RCC1 as fraction purity controls. F & G) HCT1 16 cells transfected with a U20 expression plasmid for 48 hours, treated with CHX/TNF (10 μΜ/25 ng/ml) for 4 hours and analysed for PARP cleavage and 7-AAD incorporation by flowcytometry (average of four independent experiments). H) PCR with U20 end-specific primers on cDNA from a stable U20 expressing clone (U20-S). I) PARP cleavage analysis of wt and U20-S cells treated with CHX/TNF (10 μΜ/25 ng/ml) for 4 hours. J) PARP cleavage analysis of wt and U20-S cells treated with Camptothesine.
Figure 14
U20 inhibit death receptor signalling. A) Wt and U20-S cells treated with
CHX/TNFalpha (10 μΜ/25 ng/ml) for 4 hours and analysed for the cleavage of caspase-8 and -3 and the phosphorylation of ΙκΒ. B) Confocal images of wt and U20-S cells treated with CHX/TNF (10 μΜ/25 ng/ml) for 4 hours and stained with an
antibody specific for cleaved caspase-3 and an Alexa 546 conjugated secondary antibody. Arrows indicate cleaved caspase-3 C) Wt and U20-S cells treated with CHX/TNF (10 μΜ/25 ng/ml) for 4 hours and analysed with antibodies against PARP, RIP, TRAF2, TRADD, FADD and GAPDH. D) Flowcytometry analysis of TNFR1 expression on the surface of wt and U20-S cells. E) PARP cleavage analysis of wt and U20-S cells treated with CHX/TNF (10 μΜ/25 ng/ml) or activating antibodies against the FAS/CD95 receptor (2R2 and DX2). F) PARP cleavage analysis of wt and U20-S cells treated with CHX/TNF (10 μΜ/25 ng/ml) or CHX/TRAIL ligand (10 μΜ/20 μΜ). Figure 15
U20 inhibits TNFalpha internalization. A) Flowcytometry analysis of wt and U20-S cells treated with TNF -biotin for 60 min at 4°C and stained with avidin-FITC for 30 min at 4<C. B) Confocal microscopy images of wt and U20-S cells treated with TNFalpha- biotin for 60 min at 4<C, avidin-FITC for 30 min at 4°C, fixed and stained with avidin- FITC. FITC staining is shown in the first row, and a DIC image of the cells in the second row. C) Confocal microscopy images of wt and U20-S cells treated with TNF - biotin for 60 min at 4<C and avidin-FITC for 30 min at 4<C, shifted to 37°C and fixed after 0 or 60 minutes and stained with DAPI. Arrows indicate internalized TNF . D) Live confocal microscopy images of wt and U20-S cells treated with TNF -biotin for 60 min at 4<C, followed by avidin-FITC staining for 30 min at 4<C, and then shifted to 37<C and microscopied after 5, 15, 30, 45 and 60 minutes.
Figure 16
U20 sensitize cells to Etoposide killing through programmed necrosis. A & B) xCELLigence analysis of wt or U20-S cells either mock treated or treated with etoposide or etoposide and zVAD. Data is normalised to the point of compound addition (arrow) and followed for 48 hours after addition. C) xCELLigence analysis of wt cells either mock treated or treated with CHX/TRAIL-L or CHX/TRAIL-L and zVAD. Data is normalised to the point of compound addition (arrow) and followed for 48 hours after addition. D) xCELLigence analysis of wt cells infected with HHV-6B (24 hpi) and either mock treated or treated with etoposide or etoposide and zVAD. Data is normalised to the point of compound addition (arrow) and followed for 48 hours after addition. The present invention will now be described in more detail in the following.
Detailed description of the invention
Definitions
Prior to discussing the present invention in further details, the following terms and conventions are defined:
Isolated
The term "isolated", as used herein in reference to nucleic acids or proteins, is intended to refer to a polynucleotide in which the nucleotide sequences encoding the protein or polypeptide is free of other nucleotide sequences which may naturally flank the nucleic acid in genomic DNA.
Sequence identity
The term "sequence identity" indicates a quantitative measure of the degree of homology between two amino acid sequences or between two nucleic acid sequences of equal length. If the two sequences to be compared are not of equal length, they must be aligned to give the best possible fit, allowing the insertion of gaps or, alternatively, truncation at the ends of the polypeptide sequences or nucleotide sequences. The se-
(Nref-Ndlf )l00
quence identity can be calculated as Nre{ , wherein Ndif is the total number of non-identical residues in the two sequences when aligned and wherein Nref is the number of residues in one of the sequences, preferably sequence identity is calculated over the full length reference as provided herein. Hence, the DNA sequence
AGTCAGTC will have a sequence identity of 75% with the sequence AATCAATC (Ndif=2 and Nref=8). A gap is counted as non-identity of the specific residue(s), i.e. the DNA sequence AGTGTC will have a sequence identity of 75% with the DNA sequence AGTCAGTC (Ndif=2 and Nref=8).
With respect to all embodiments of the invention relating to nucleotide sequences, the percentage of sequence identity between one or more sequences may also be based on alignments using the clustalW software (http:/www.ebi.ac.uk/clustalW/index.html) with default settings. For nucleotide sequence alignments these settings are:
Alignment=3Dfull, Gap Open 10.00, Gap Ext. 0.20, Gap separation Dist. 4, DNA weight matrix: identity (IUB).
Sequence identity is determined in one embodiment by utilising fragments of SEQ ID NO:3 peptides comprising at least 25 contiguous amino acids and having an amino
acid sequence which is at least 80%, such as 85%, for example 90%, such as 95%, for example 99% identical to the amino acid sequence of SEQ ID NO: 1 , wherein the percent identity is determined with the algorithm GAP, BESTFIT, or FASTA in the Wisconsin Genetics Software Package Release 7.0, using default gap weights.
The following terms are used to describe the sequence relationships between two or more polynucleotides: "predetermined sequence", "comparison window", "sequence identity", "percentage of sequence identity", and "substantial identity". A "predetermined sequence" is a defined sequence used as a basis for a sequence comparison; a predetermined sequence may be a subset of a larger sequence, for example, as a segment of a full-length DNA or gene sequence given in a sequence listing, such as a polynucleotide sequence of SEQ ID NO:2, or may comprise a complete DNA or gene sequence. Generally, a predetermined sequence is at least 20 nucleotides in length, frequently at least 25 nucleotides in length, and often at least 50 nucleotides in length. Likewise, the predetermined seequence is that of the
polypeptides of the invention.
Since two polynucleotides may each (1 ) comprise a sequence (i.e., a portion of the complete polynucleotide sequence) that is similar between the two polynucleotides, and (2) may further comprise a sequence that is divergent between the two
polynucleotides, sequence comparisons between two (or more) polynucleotides are typically performed by comparing sequences of the two polynucleotides over a
"comparison window" to identify and compare local regions of sequence similarity. A "comparison window", as used herein, refers to a conceptual segment of at least 20 contiguous nucleotide positions wherein a polynucleotide sequence may be compared to a predetermined sequence of at least 20 contiguous nucleotides and wherein the portion of the polynucleotide sequence in the comparison window may comprise additions or deletions (i.e., gaps) of 20 percent or less as compared to the
predetermined sequence (which does not comprise additions or deletions) for optimal alignment of the two sequences.
Optimal alignment of sequences for aligning a comparison window may be conducted by the local homology algorithm of Smith and Waterman (1981 ) Adv. Appl. Math. 2: 482, by the homology alignment algorithm of Needleman and Wunsch (1970) J. Mol.
Biol. 48: 443, by the search for similarity method of Pearson and Lipman (1988) Proc. Natl. Acad. Sci. (U.S.A.) 85: 2444, by computerized implementations of these algorithms (GAP, BESTFIT, FASTA, and T FAST A in the Wisconsin Genetics Software Package Release 7.0, Genetics Computer Group, 575 Science Dr., Madison, Wis.), or by inspection, and the best alignment (i.e., resulting in the highest percentage of homology or identity over the comparison window) generated by the various methods is selected.
The NCBI Basic Local Alignment Search Tool (BLAST) is available from several sources, including the National Center for Biotechnology Information (NBCI, Bethesda, Md.) and on the Internet, for use in connection with the sequence analysis programs blastp, blastn, blastx, tblastn and tblastx. It can be accessed at
http://www.ncbi.nlm.nih.gov/BLAST/. A description of how to determine sequence identity using this program is available at
http://www.ncbi.nlm.nih.gov/BLAST/blast_help.html.
Homologs of the disclosed polypeptides are typically characterised by possession of at least 94% sequence identity counted over the full length alignment with the disclosed amino acid sequence using the NCBI Basic Blast 2.0, gapped blastp with databases such as the nr or swissprot database. Alternatively, one may manually align the sequences and count the number of identical amino acids. This number divided by the total number of amino acids in your sequence multiplied by 100 results in the percent identity.
Expression vector
The term "expression vector" refers to a DNA molecule used as a vehicle to transfer recombinant genetic material into a host cell. The four major types of vectors are plasmids, bacteriophages and other viruses, cosmids, and artifical chromosomes. The vector itself is generally a DNA sequence that consists of an insert (a heterologous nucleic acid sequence, transgene) and a larger sequence that serves as the
"backbone" of the vector. The purpose of a vector which transfers genetic information to the host is typically to isolate, multiply, or express the insert in the target cell. Vectors called expression vectors (expression constructs) are specifically adapted for the expression of the heterologous sequences in the target cell, and generally have a promoter sequence that drives expression of the heterologous sequences. Simpler vectors called transcription vectors are only capable of being transcribed but not
translated: they can be replicated in a target cell but not expressed, unlike expression vectors. Transcription vectors are used to amplify the inserted heterologous
sequences. The transcripts may subsequently be isolated and used in as templates suitable in vitro translations systems.
Recombinant
The term "recombinant" when used with reference to a cell, or nucleic acid, peptide or vector, indicates that the cell, or nucleic acid, peptide or vector, has been modified by the introduction of a heterologous nucleic acid or the alteration of a native nucleic acid, or that the cell is derived from a cell so modified. Thus, for example, recombinant cells express genes that are not found within the native (non-recombinant) form of the cell or express native genes that are otherwise abnormally expressed, under expressed or not expressed at all. Further, the expression "recombinant" also relates to a cell, wherein further regulatory elements have been included in order to initiate or enhance expression of an otherwise silent endogenous gene, or wherein a manipulation of the regulatory elements have been performed for the same purpose.
The term "hybridization under stringent conditions" is defined according to Sambrook et al., Molecular Cloning, A Laboratory Manual, Cold Spring Harbor, Laboratory Press (1989), 1 .101 -1 .104. Preferably, hybridization under stringent conditions means that after washing for 1 h with 1 times SSC and 0.1 % SDS at 50 degree C, preferably at 55 degree C, more preferably at 62degree C and most preferably at 68 degree C, particularly for 1 h in 0.2 times SSC and 0.1 % SDS at 50 degree C, preferably at 55 degree C, more preferably at 62 degree C and most preferably at 68 degree C, a positive hybridization signal is observed. A nucleotide sequence which hybridizes under the above washing conditions with the nucleotide sequence of SEQ ID NO:2 (HHV 6B U20 gene) and/or SEQ ID NO:7 (HHV 6B U20 mRNA) or a nucleotide sequence corresponding thereto in the scope of the degeneracy of the genetic code is encompassed by the present invention.
Polynucleotide
As used herein, "nucleic acid" or "polynucleotide" refers to polynucleotides, such as deoxyribonucleic acid (DNA) or ribonucleic acid (RNA), oligonucleotides, fragments
generated by the polymerase chain reaction (PCR), and fragments generated by any of ligation, scission, endonuclease action, and exonuclease action. Polynucleotides can be composed of monomers that are naturally-occurring nucleotides (such as DNA and RNA), or analogs of naturally-occurring nucleotides (e.g., (alpha-enantiomeric forms of naturally-occurring nucleotides), or a combination of both. Modified nucleotides can have alterations in sugar moieties and/or in pyrimidine or purine base moieties. Sugar modifications include, for example, replacement of one or more hydroxyl groups with halogens, alkyl groups, amines, and azido groups, or sugars can be functionalized as ethers or esters. Moreover, the entire sugar moiety can be replaced with sterically and electronically similar structures, such as aza-sugars and carbocyclic sugar analogs.
Examples of modifications in a base moiety include alkylated purines and pyrimidines, acylated purines or pyrimidines, or other well-known heterocyclic substitutes. Nucleic acid monomers can be linked by phosphodiester bonds or analogs of such linkages. Analogs of phosphodiester linkages include phosphorothioate, phosphorodithioate, phosphoroselenoate, phosphorodiselenoate, phosphoroanilothioate, phosphoranilidate, phosphoramidate, and the like. The term "polynucleotide" also includes so-called "peptide nucleic acids," which comprise naturally-occurring or modified nucleic acid bases attached to a polyamide backbone. Nucleic acids can be either single stranded or double stranded.
The term "natural nucleotide" refers to any of the four deoxyribonucleotides, dA, dG, dT, and dC (constituents of DNA), and the four ribonucleotides, A, G, U, and C
(constituents of RNA) are the natural nucleotides. Each natural nucleotide comprises or essentially consists of a sugar moiety (ribose or deoxyribose), a phosphate moiety, and a natural/standard base moiety. Natural nucleotides bind to complementary nucleotides according to well-known rules of base pairing (Watson and Crick), where adenine (A) pairs with thymine (T) or uracil (U); and where guanine (G) pairs with cytosine (C), wherein corresponding base-pairs are part of complementary, anti-parallel nucleotide strands. The base pairing results in a specific hybridization between predetermined and complementary nucleotides. The base pairing is the basis by which enzymes are able to catalyze the synthesis of an oligonucleotide complementary to the template oligonucleotide. In this synthesis, building blocks (normally the triphosphates of ribo or deoxyribo derivatives of A, T, U, C, or G) are directed by a template oligonucleotide to form a complementary oligonucleotide with the correct, complementary sequence. The recognition of an oligonucleotide sequence by its complementary sequence is
mediated by corresponding and interacting bases forming base pairs. In nature, the specific interactions leading to base pairing are governed by the size of the bases and the pattern of hydrogen bond donors and acceptors of the bases. A large purine base (A or G) pairs with a small pyrimidine base (T, U or C). Additionally, base pair recognition between bases is influenced by hydrogen bonds formed between the bases. In the geometry of the Watson-Crick base pair, a six membered ring (a pyrimidine in natural oligonucleotides) is juxtaposed to a ring system composed of a fused, six membered ring and a five membered ring (a purine in natural
oligonucleotides), with a middle hydrogen bond linking two ring atoms, and hydrogen bonds on either side joining functional groups appended to each of the rings, with donor groups paired with acceptor groups.
The term "complement of a polynucleotide" refers to a polynucleotide having a complementary nucleotide sequence and reverse orientation as compared to a reference nucleotide sequence. For example, the sequence 5' ATGCACGGG 3' is complementary to 5' CCCGTGCAT 3'.
The term "degenerate nucleotide sequence" denotes a sequence of nucleotides that includes one or more degenerate codons as compared to a reference polynucleotide that encodes a polypeptide. Degenerate codons contain different triplets of nucleotides, but encode the same amino acid residue (i.e., GAU and GAC triplets each encode Asp).
The term "structural gene" refers to a polynucleotide that is transcribed into messenger RNA (mRNA), which is then translated into a sequence of amino acids characteristic of a specific polypeptide.
A "polynucleotide construct" is a polynucleotide, either single- or double-stranded, that has been modified through human intervention to contain segments of nucleic acid combined and juxtaposed in an arrangement not existing in nature.
"Complementary DNA (cDNA)" is a single-stranded DNA molecule that is formed from an mRNA template by the enzyme reverse transcriptase. Typically, a primer complementary to portions of mRNA is employed for the initiation of reverse
transcription. Those skilled in the art also use the term "cDNA" to refer to a double-
stranded DNA molecule consisting of such a single-stranded DNA molecule and its complementary DNA strand. The term "cDNA" also refers to a clone of a cDNA molecule synthesized from an RNA template. A nucleotide is herein defined as a monomer of RNA or DNA. A nucleotide is a ribose or a deoxyribose ring attached to both a base and a phosphate group. Both mono-, di-, and tri-phosphate nucleosides are referred to as nucleotides.
The term 'nucleotides' as used herein refers to both natural nucleotides and non- natural nucleotides capable of being incorporated - in a template-directed manner - into an oligonucleotide, preferably by means of an enzyme comprising DNA or RNA dependent DNA or RNA polymerase activity, including variants and functional equivalents of natural or recombinant DNA or RNA polymerases. Corresponding binding partners in the form of coding elements and complementing elements comprising a nucleotide part are capable of interacting with each other by means of hydrogen bonds. The interaction is generally termed "base-pairing". Nucleotides may differ from natural nucleotides by having a different phosphate moiety, sugar moiety and/or base moiety. Nucleotides may accordingly be bound to their respective neighbour(s) in a template or a complementing template by a natural bond in the form of a phosphodiester bond, or in the form of a non-natural bond, such as e.g. a peptide bond as in the case of PNA (peptide nucleic acids). Nucleotides according to the invention includes ribonucleotides comprising a nucleobase selected from the group consisting of adenine (A), uracil (U), guanine (G), and cytosine (C), and
deoxyribonucleotide comprising a nucleobase selected from the group consisting of adenine (A), thymine (T), guanine (G), and cytosine (C). Nucleobases are capable of associating specifically with one or more other nucleobases via hydrogen bonds. Thus it is an important feature of a nucleobase that it can only form stable hydrogen bonds with one or a few other nucleobases, but that it can not form stable hydrogen bonds with most other nucleobases usually including itself. The specific interaction of one nucleobase with another nucleobase is generally termed "base-pairing". The base pairing results in a specific hybridisation between predetermined and complementary nucleotides. Complementary nucleotides according to the present invention are nucleotides that comprise nucleobases that are capable of base-pairing. Of the naturally occurring nucleobases adenine (A) pairs with thymine (T) or uracil (U); and guanine (G) pairs with cytosine (C). Accordingly, e.g. a nucleotide comprising A is
complementary to a nucleotide comprising either T or U, and a nucleotide comprising G is complementary to a nucleotide comprising C.
The term 'oligonucleotide' is used herein interchangebly with polynucleotide. As used herein the term "oligonucleotide" refers to a single stranded or double stranded oligomer or polymer of ribonucleic acid (RNA) or deoxyribonucleic acid (DNA) or mimetics thereof. This term includes oligonucleotides composed of naturally-occurring bases, sugars and covalent internucleoside linkages (e.g., backbone) as well as oligonucleotides having non-naturally-occurring portions which function similarly to respective naturally-occurring portions (see disclosed in U.S. Pat. Nos. 4,469,863; 4,476,301 ; 5,023,243; 5,177,196; 5,188,897; 5,264,423; 5,276,019; 5,278,302;
5,286,717; 5,321 ,131 ; 5,399,676; 5,405,939; 5,453,496; 5,455,233; 5,466,677;
5,476,925; 5,519,126; 5,536,821 ; 5,541 ,306; 5,550,1 1 1 ; 5,563,253; 5,571 ,799;
5,587,361 ; and 5,625,050). The term oligonucleotide thus also refers to any
combination of oligonucleotides of natural and non-natural nucleotides. The natural and/or non-natural nucleotides may be linked by natural phosphodiester bonds or by non-natural bonds. Preferred oligonucleotides comprise only natural nucleotides linked by phosphodiester bonds. Oligonucleotide is used interchangeably with polynucleotide. The oligomer or polymer sequences of the present invention are formed from the chemical or enzymatic addition of monomer subunits. The term "oligonucleotide" as used herein includes linear oligomers of natural or modified monomers or linkages, including deoxyribonucleotides, ribonucleotides, anomeric forms thereof, peptide nucleic acid monomers (PNAs), locked nucleotide acid monomers (LNA), and the like, capable of specifically binding to a single stranded polynucleotide tag by way of a regular pattern of monomer-to-monomer interactions, such as Watson-Crick type of base pairing, base stacking, Hoogsteen or reverse Hoogsteen types of base pairing, or the like. Usually monomers are linked by phosphodiester bonds or analogs thereof to form oligonucleotides ranging in size from a few monomeric units, e.g. 3-4, to several tens of monomeric units, e.g. 40-60. Whenever an oligonucleotide is represented by a sequence of letters, such as "ATGCCTG," it will be understood that the nucleotides are in 5'→ 3' order from left to right and the "A" denotes deoxyadenosine, "C" denotes deoxycytidine, "G" denotes deoxyguanosine, and "T" denotes thymidine, unless otherwise noted. Usually oligonucleotides of the invention comprise the four natural nucleotides; however, they may also comprise methylated or non-natural nucleotide analogs. Suitable oligonucleotides may be prepared by the phosphoramidite method
described by Beaucage and Carruthers (Tetrahedron Lett., 22, 1859-1862, 1981 ), or by the triester method according to Matteucci, et al. (J. Am. Chem. Soc, 103, 3185, 1981 ), both incorporated herein by reference, or by other chemical methods using either a commercial automated oligonucleotide synthesizer or VLSIPS.TM. technology. When oligonucleotides are referred to as "double-stranded," it is understood by those of skill in the art that a pair of oligonucleotides exist in a hydrogen-bonded, helical configuration typically associated with, for example, DNA. In addition to the 100% complementary form of double-stranded oligonucleotides, the term "double-stranded" as used herein is also meant to refer to those forms which include such structural features as bulges and loops. For example as described in US 5.770.722 for a unimolecular double-stranded DNA. It is clear to those skilled in the art when oligonucleotides having natural or non-natural nucleotides may be employed, e.g. where processing by enzymes is called for, usually oligonucleotides consisting of natural nucleotides are required. When nucleotides are conjugated together in a string using synthetic procedures, they are always referred to as oligonucleotides.
A plurality of individual nucleotides linked together in a single molecule may form a polynucleotide. Polynucleotide covers any derivatized nucleotides such as DNA, RNA, PNA, LNA etc. Any oligonucleotide is also a polynucleotide, but every polynucleotide is not an oligonucleotide.
Polypeptide
The terms "polypeptide", "peptide" and "protein" are used interchangeably herein to refer to a polymer of amino acid residues. The terms apply to amino acid polymers in which one or more amino acid residue is an artificial chemical analogue of a corresponding naturally occurring amino acid, as well as to naturally occurring amino acid polymers.
Amino acid: Entity comprising an amino terminal part (NH2) and a carboxy terminal part (COOH) separated by a central part comprising a carbon atom, or a chain of carbon atoms, comprising at least one side chain or functional group. NH2 refers to the amino group present at the amino terminal end of an amino acid or peptide, and COOH refers to the carboxy group present at the carboxy terminal end of an amino acid or peptide. The generic term amino acid comprises both natural and non-natural amino acids. Natural amino acids of standard nomenclature as listed in J. Biol. Chem., 243:3552-59
(1969) and adopted in 37 C.F.R., section 1 .822(b)(2) belong to the group of amino acids listed in Table 2 herein below. Non-natural amino acids are those not listed in Table 2. Examples of non-natural amino acids are those listed e.g. in 37 C.F.R. section 1 .822(b)(4), all of which are incorporated herein by reference. Further examples of non- natural amino acids are listed herein below. Amino acid residues described herein can be in the "D" or or "L" isomeric form.
Symbols Amino acid
1 -Letter 3-Letter
Y Tyr tyrosine
G Gly glycine
F Phe phenylalanine
M Met methionine
A Ala alanine
S Ser serine
I lie isoleucine
L Leu leucine
T Thr threonine
V Val valine
P Pro proline
K Lys lysine
H His histidine
Q Gin glutamine
E Glu glutamic acid
W Trp tryptophan
R Arg arginine
D Asp aspartic acid
N Asn asparagine
C Cys cysteine
Table 2. Natural amino acids and their respective codes.
Amino acid residue: the term "amino acid residue" is meant to encompass amino acids, either standard amino acids, non-standard amino acids or pseudo-amino acids, which have been reacted with at least one other species, such as 2, for example 3, such as more than 3 other species. In particular amino acid residues may comprise an acyl bond in place of a free carboxyl group and/or an amine-bond and/or amide bond in place of a free amine group. Furthermore, reacted amino acids residues may comprise an ester or thioester bond in place of an amide bond
An "isolated polypeptide" is a polypeptide that is essentially free from contaminating cellular components, such as carbohydrate, lipid, or other proteinaceous impurities associated with the polypeptide in nature. Typically, a preparation of isolated polypeptide contains the polypeptide in a highly purified form, i.e., at least about 80% pure, at least about 90% pure, at least about 95% pure, greater than 95% pure, or greater than 99% pure. One way to show that a particular protein preparation contains an isolated polypeptide is by the appearance of a single band following sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis of the protein preparation and
Coomassie Brilliant Blue staining of the gel. However, the term "isolated" does not exclude the presence of the same polypeptide in alternative physical forms, such as dimers or alternatively glycosylated or derivatized forms. The terms "amino-terminal" and "carboxyl-terminal" are used herein to denote positions within polypeptides. Where the context allows, these terms are used with reference to a particular sequence or portion of a polypeptide to denote proximity or relative position. For example, a certain sequence positioned carboxyl-terminal to a reference sequence within a polypeptide is located proximal to the carboxyl terminus of the reference sequence, but is not necessarily at the carboxyl terminus of the complete polypeptide.
Functional equivalents and variants are used interchangeably herein. In one preferred embodiment of the invention there is also provided variants of U20 gene variants of fragments thereof. When being polypeptides, variants are determined on the basis of their degree of identity or their homology with a predetermined amino acid sequence, said predetermined amino acid sequence being of SEQ ID NO: 1 , 3 or 5, when the variant is a fragment, a fragment of any of the aforementioned amino acid sequences, respectively.
A "fusion protein" is a hybrid protein expressed by a polynucleotide comprising nucleotide sequences of at least two genes. For example, a fusion protein can comprise at least part of a polypeptide according to the present invention fused with a polypeptide that binds an affinity matrix. Such a fusion protein provides a means to isolate large quantities of a polypeptide according to the present invention using affinity chromatography.
The term "complement/anti-complement pair" denotes non-identical moieties that form a non-covalently associated, stable pair under appropriate conditions. For instance, biotin and avidin (or streptavidin) are prototypical members of a complement/anti- complement pair. Other exemplary complement/anti-complement pairs include receptor/ligand pairs, antibody/antigen (or hapten or epitope) pairs, sense/antisense polynucleotide pairs, and the like. Where subsequent dissociation of the
complement/anti-complement pair is desirable, the complement/anti-complement pair preferably has a binding affinity of less than 109 M" .
The term "affinity tag" is used herein to denote a polypeptide segment that can be attached to a second polypeptide to provide for purification or detection of the second polypeptide or provide sites for attachment of the second polypeptide to a substrate. In principal, any peptide or protein for which an antibody or other specific binding agent is available can be used as an affinity tag. Affinity tags include a poly-histidine tract, protein A (Nilsson et al., EMBO J. 4:1075 (1985); Nilsson et al., Methods Enzymol. 198:3 (1991 )), glutathione S transferase (Smith and Johnson, Gene 67:31 (1988)), Glu- Glu affinity tag (Grussenmeyer et al., Proc. Natl. Acad. Sci. USA 82:7952 (1985)), substance P, FLAG peptide (Hopp et al., Biotechnology 6:1204 (1988)), streptavidin binding peptide, or other antigenic epitope or binding domain. See, in general, Ford et al., Protein Expression and Purification 2:95 (1991 ). DNAs encoding affinity tags are available from commercial suppliers (e.g., Pharmacia Biotech, Piscataway, N.J.). Host cell
The term "recombinant host cell" (or simply "host cell"), as used herein, is intended to refer to a cell into which a vector has been introduced. It should be understood that such terms are intended to refer not only to the particular subject cell but to the progeny of such a cell. Because certain modifications may occur in
succeeding generations due to either mutation or environmental influences, such progeny may not, in fact, be identical to the parent cell, but are still included within the scope of the term "host cell" as used herein. Preferably host cells are used for the expression of the proteins of the invention.
Operatively linked
The term "operatively linked", "operably linked" or "linked in frame" as used in this context, is intended to mean that the two DNA fragments are joined such that the amino acid sequences encoded by the two DNA fragments remain in-frame. Similarly, two linked polypeptides maintain in frame when they are operably linked.
Regulatory sequence
The term "regulatory sequence" is intended to include promoters, enhancers and other expression control elements (e.g., polyadenylation signals) that control the transcription or translation of the antibody chain genes. It will be appreciated by those skilled in the art that the design of the expression vector, including the selection of regulatory sequences may depend on such factors as the choice of the host cell to be
transformed, the level of expression of protein desired, etc. Preferred regulatory sequences for mammalian host cell expression include viral elements that direct high levels of protein expression in mammalian cells, such as promoters and/or enhancers derived from cytomegalovirus (CMV) (such as the CMV promoter/enhancer), Simian Virus 40 (SV40) (such as the SV40 promoter/enhancer), adenovirus, (e.g., the adenovirus major late promoter (AdMLP)) and polyoma. Human TNFa
The term "human TNFa" (abbreviated herein as hTNFa, hTNF-alpha, hTNF- a, or simply hTNF), as used herein, is intended to refer to a human cytokine that exists as a 17 kD secreted form and a 26 kD membrane associated form, the biologically active form of which is composed of a trimer of noncovalently bound 17 kD molecules. The term human TNFa is intended to include recombinant human TNFa (rhTNFa). rhTNFa can be prepared by standard recombinant expression methods or purchased commercially.
TNFa receptor
The term "TNFa receptor" as used herein refers to two known forms of TNF receptors, also known as TNFR1 (or p55 or TNFRSF1 A) and TNFR2 (or p75 or TNFRSF1 B).
Linker
In the present context the term "linker" refers to an amino acid or nucleic acid sequences linking in frame two amino acid sequences or two nucleic acid sequences respectively. Linkers may comprise one or more sequences enabling purification, increased solubility, detection or separation of domains. Examples of such sequences are epitopes for one or more antibodies, restriction enzymes, purification tags or sequences predicted to form alpha helices or disorganised domains. The length of a linker may be 1 -500 amino acids, 1 -200 amino acids, such as 1 -150, such as 1 -100, such as 1 -80, such as 1 -60, such as 1 -40, such as 5-40.
Diagnosticum
The term "diagnosticum" refers in the present context to a compound or composition used in diagnosis of a disease or medical state. In the present text the diagnosticum is the U20 protein or active derivative thereof for use in the diagnosis of a disease or condition.
Biologically active
In the present context the term 'biologically active' refers to variants and fragments of polypeptides and polynucleotides of the present invention. Such variants and fragments of polypeptides and polynucleotides are also suitable for use according to the invention, insofar as these polypeptides display substantially the same biological activity.
The term "substantially same biologically activity" can be defined as having an activity being at least 20% of the full length U20 polypeptide or polynucleotides, such as at least 30%, such as at least 40%, such as at least 50%, such as at least 60%, such as at least 70%, such as at least 80%, or such as at least 90%. It is to be understood that active derivatives may also have a higher activity than the full length U20 polypeptide or polynucleotides such as at the most 20% more activity, such as at the most 40% more activity, such as at the most 60% more activity, such as at the most 80% more
activity, such as at the most 100% more activity, such as at the most 150% more activity, or such as at the most 200%, or such as at the most 500% more activity than the full length U20 polypeptide or polynucleotides. In the present invention the biological activity may be measured as the ability of the polypeptides or polynucleotides to influence TNF receptor activity. The biological activity can thus be measured as the level of apoptosis in response to signalling throug the TNF receptor. This is shown in example 1 and figure 2, providing an assay for measuring the level of apoptosis, where cells have been induced to apoptosis in the presence of hTNF-alpha. The biologic activity of the polypeptides or polynucleotides to influence TNF receptor activity can also be measured by examining the activation state of proteins involved in the TNF receptor signal transduction. This is shown in figure 4A. This invention pertains to isolated U20 proteins or active derivatives thereof that bind to human TNFa-receptor (TNFR1 ). Various aspects of the invention relate to U20 proteins or derivatives thereof for use as a medicament or diagnosticum, pharmaceutical compositions, as well as nucleic acids, recombinant expression vectors and host cells for making such U20 proteins and derivatives. The invention furthermore relates to the U20 proteins or active derivatives thereof as part of pharmaceutical and diagnostic kits. Methods of using the U20 protein according to the invention to detect human TNFa receptor or to inhibit the interaction between TNFa and the receptor, either in vitro or in vivo, are also encompassed by the invention.
Polypeptides
In addition to full-length HHV U20, substantially full-length HHV U20, to pro- HHV U20, to C-terminal peptides, to N-terminal peptides and to truncated forms of HHV U20, the present invention provides for biologically active variants of the polypeptides. An HHV U20 polypeptide or fragment is biologically active if it exhibits a biological activity of naturally occurring HHV U20. It is to be understood that the invention relates to substantially purified HHV U20as herein defined.
One biological activity is the ability to compete with naturally occurring HHV U20 in a receptor-binding assay.
Biologically active variants may also be defined with reference to one or more of the other in vitro and/or in vivo biological assays described in the examples.
A preferred biological activity is the ability to elicit substantially the same response as in the apoptosis assay described in the Examples and Figures 2, and 6. The polypeptides according to the present invention can be used as a medicament where the medicament is active by modulating, or interfering with the activity of TNF receptors. Thus in one aspect the invention relates to an isolated polypeptide for use as medicament or diagnosticum comprising
a) an amino acid sequence consisting of SEQ ID No. 1 , 3 or 5;b) a biologically active sequence variant of the amino acid sequence, wherein the variant has at least 70% sequence identity to said SEQ ID NO. 1 , 3 or 5; or c)a biologically active fragment of at least 50 contiguous amino acids of any of a) through b), wherein said fragment is a fragment of SEQ ID NO 1 , 3 or 5. SEQ ID NO: 1 is the amino acid sequence of the U20 protein from HHV-6B (NCBI accession number NP 050200.1 ) SEQ ID NO: 3 is the amino acid sequence of the U20 protein from HHV-6A (NCBI accession number NP_042913.1 ) and SEQ ID NO: 5 is the amino acid sequence of the U20 protein from HHV-7 (NCBI accession number YP_073760.1 ). An alignment of U20 from HHV-6B, HHV-6A and HHV-7 is shown in figure 9.
One or more amino acid residues of the polypeptides of the present invention are in one embodiment modified so as to preferably improve the resistance to proteolytic degradation and stability or to optimize solubility properties or to render the polypeptide more suitable as a therapeutic agent. Thus, the polypeptide may comprise amino acid residues other than naturally occurring L-amino acid residues. For example, the polypeptides may comprise D-amino acid residues. However, the polypeptides may also comprise non-naturally occurring, synthetic amino acids. The polypeptides may further comprising one or more blocking groups, preferably in the form of chemical substituents suitable to protect and/or stabilize the N- and C- termini of the polypeptide from undesirable degradation. One or more blocking groups include protecting groups which do not adversely affect in vivo activities of the polypeptide. The one or more blocking groups are for example introduced by alkylation or acylation of the N-terminus. Such N-terminal blocking groups are selected from N-
terminal blocking groups comprising Ci to C5 branched or non-branched alkyl groups and acyl groups, such as formyl and acetyl groups, as well as substituted forms thereof, such as the acetamidomethyl (Acm) group. In another embodiment, one or more blocking groups are selected from N-terminal blocking groups comprising desamino analogs of amino acids, which are either coupled to the N-terminus of the peptide or used in place of the N-terminal amino acid residue.
The polypeptides may also comprise C-terminal blocking groups, wherein the carboxyl group of the C-terminus is either incorporated or not, such as esters, ketones, and amides, as well as descarboxylated amino acid analogues. The one or more blocking groups are selected from C-terminal blocking groups comprising ester or ketone- forming alkyl groups, such as lower (Ci to C6) alkyl groups, for example methyl, ethyl and propyl, and amide-forming amino groups, such as primary amines (-NH2), and mono- and di-alkylamino groups, such as methylamino, ethylamino, dimethylamino, diethylamino, methylethylamino, and the like.
Free amino group(s) at the N-terminal end and free carboxyl group(s) at the termini can be removed altogether from the polypeptide to yield desamino and descarboxylated forms thereof without significantly affecting the biological activity of the polypeptide. Functional assays (ie. assays for the biological activity) can for example be used in order to determine if U20 function is conserved.
As used herein "variant" refers to polypeptides or proteins which are homologous to the basic protein, which is suitably U20 (such as SEQ ID NO.: 1 , 3 or 5), but which differs from the base sequence from which they are derived in that one or more amino acids within the sequence are substituted for other amino acids. Amino acid substitutions may be regarded as "conservative" where an amino acid is replaced with a different amino acid with broadly similar properties. Non-conservative substitutions are where amino acids are replaced with amino acids of a different type. Broadly speaking, fewer non-conservative substitutions will be possible without altering the biological activity of the polypeptide.
A person skilled in the art will know how to make and assess 'conservative' amino acid substitutions, by which one amino acid is substituted for another with one or more shared chemical and/or physical characteristics. Conservative amino acid substitutions are less likely to affect the functionality of the protein. Amino acids may be grouped
according to shared characteristics. A conservative amino acid substitution is a substitution of one amino acid within a predetermined group of amino acids for another amino acid within the same group, wherein the amino acids within a predetermined groups exhibit similar or substantially similar characteristics.
Conservative amino acid substitutions refer to the interchangeability of residues having similar side chains. For example, a group of amino acids having aliphatic side chains is glycine, alanine, valine, leucine, and isoleucine; a group of amino acids having aliphatic-hydroxyl side chains is serine and threonine, a group of amino acids having amide-containing side chains is asparagine and glutamine; a group of amino acids having aromatic side chains is phenylalanine, tyrosine, and tryptophan; a group of amino acids having basic side chains is lysine, arginine, and histidine; and a group of amino acids having sulfur-containing side chains is cysteine and methionine. Preferred conservative amino acids substitution groups are: valine-leucine-isoleucine, phenylalanine-tyrosine, lysine-arginine, alanine-valine, and asparagine-glutamine.
Within the meaning of the term "conservative amino acid substitution" as applied herein, one amino acid may be substituted for another within the groups of amino acids indicated herein below:
Lower levels of similarity:
Polarity:
i) Amino acids having polar side chains (Asp, Glu, Lys, Arg, His, Asn, Gin, Ser, Thr, Tyr, and Cys,) ii) Amino acids having non-polar side chains (Gly, Ala, Val, Leu, lie, Phe, Trp, Pro, and Met)
Hydrophilic or hydrophobic:
iii) Hydrophobic amino acids (Ala, Cys, Gly, lie, Leu, Met, Phe, Pro, Trp, Tyr, Val) iv) Hydrophilic amino acids (Arg, Ser, Thr, Asn, Asp, Gin, Glu, His, Lys)
Charges:
v) Neutral amino acids ( Ala, Asn, Cys, Gin, Gly, lie, Leu, Met, Phe, Pro, Ser, Thr, Trp, Tyr, Val)
vi) Basic amino acids (Arg, His, Lys)
vii) Acidic amino acids ((asp, Glu)
High level of similarity:
viii) Acidic amino acids and their amides (Gin, Asn, Glu, Asp)
ix) Amino acids having aliphatic side chains (Gly, Ala Val, Leu, lie)
x) Amino acids having aromatic side chains (Phe, Tyr, Trp)
xi) Amino acids having basic side chains (Lys, Arg, His)
xii) Amino acids having hydroxy side chains (Ser, Thr)
xiii) Amino acids having sulphor-containing side chains (Cys, Met).
Preferred conservative amino acids substitution groups are: valine-leucine-isoleucine, phenylalanine-tyrosine, lysine-arginine, alanine-valine, and asparagine-glutamine. The alignment in Figure 9 can be used to predict which amino acid residues can be substituted without substantially affecting the biological acitivity of the protein.
Substutions within the following groups ('strong' conservation group) are to be regarded as conservative substitutions within the meaning of the present invention
-STA, NEQK, NHQK, NDEQ, QHRK, MILV, MILF, HY, FYW. Substutions within the following groups ('weak' conservation group) are to be regarded as semi-conservative substitutions within the meaning of the present invention
-CSA, ATV, SAG, STNK, STPA, SGND, SNDEQK, NDEQHK, NEQHRK, VLIM, HFY.
Accordingly, a variant or a fragment thereof according to the invention may comprise, within the same variant of the sequence or fragments thereof, or among different variants of the sequence or fragments thereof, at least one substitution, such as a plurality of substitutions introduced independently of one another.
It is clear from the above outline that the same variant or fragment thereof may comprise more than one conservative amino acid substitution from more than one group of conservative amino acids as defined herein above. Aside from the twenty standard amino acids and two special amino acids, seleno- cysteine and pyrrolysine, there are a vast number of "nonstandard amino acids" which are not incorporated into protein in vivo. Examples of nonstandard amino acids include the sulfur-containing taurine and the neurotransmitters GABA and dopamine. Other examples are lanthionine, 2-Aminoisobutyric acid, and dehydroalanine. Further non standard amino are ornithine and citrulline.
Non-standard amino acids are usually formed through modifications to standard amino acids. For example, taurine can be formed by the decarboxylation of cysteine, while dopamine is synthesized from tyrosine and hydroxyproline is made by a
posttranslational modification of proline (common in collagen). Examples of non-natural amino acids are those listed e.g. in 37 C.F.R. section 1 .822(b)(4), all of which are incorporated herein by reference.
Both standard and non standard amino acid residues described herein can be in the "D" or or "L" isomeric form.
It is contemplated that a functional equivalent according to the invention may comprise any amino acid including non-standard amino acids. In preferred embodiments a functional equivalent comprises only standard amino acids.
The standard and/or non-standard amino acids may be linked by peptide bonds or by non-peptide bonds. The term peptide also embraces post-translational modifications introduced by chemical or enzyme-catalyzed reactions, as are known in the art. Such post-translational modifications can be introduced prior to partitioning, if desired. Amino acids as specified herein will preferentially be in the L-stereoisomeric form. Amino acid analogs can be employed instead of the 20 naturally-occurring amino acids. Several such analogs are known, including fluorophenylalanine, norleucine, azetidine-2- carboxylic acid, S-aminoethyl cysteine, 4-methyl tryptophan and the like. Suitably variants are variant(s) of SEQ ID NO:1 , 3 or 5 having at least 80% sequence identity, such as preferably at least 81 % sequence identity, more preferably e.g. at least 82% sequence identity, such as more preferably at least 83% sequence identity, e.g. more preferably at least 84% sequence identity, more preferably such as at least 85% sequence identity, more preferably e.g. at least 86% sequence identity, more preferably such as at least 87% sequence identity, more preferably e.g. at least 88% sequence identity, more preferably such as at least 89% sequence identity, more preferably e.g. at least 90% sequence identity, more preferably such as at least 91 % sequence identity, more preferably e.g. at least 92% sequence identity, such as at least 93% sequence identity, more preferably e.g. at least 94% sequence identity, more preferably such as at least 95% sequence identity, more preferably e.g. at least 96%
sequence identity, more preferably such as at least 97% sequence identity, more preferably e.g. at least 98% sequence identity, more preferably such as at least 99% sequence identity, more preferably e.g. at least 99.5% sequence identity identity with the predetermined sequence of U20 from HHV (SEQ ID No: 1 , 3 and 5).
Suitably variants are variant(s) of SEQ ID NO:1 , 3 or 5 having at least 80% sequence identity, such as preferably at least 81 % sequence identity, more preferably e.g. at least 82% sequence identity, such as more preferably at least 83% sequence identity, e.g. more preferably at least 84% sequence identity, more preferably such as at least 85% sequence identity, more preferably e.g. at least 86% sequence identity, more preferably such as at least 87% sequence identity, more preferably e.g. at least 88% sequence identity, more preferably such as at least 89% sequence identity, more preferably e.g. at least 90% sequence identity, more preferably such as at least 91 % sequence identity, more preferably e.g. at least 92% sequence identity, such as at least 93% sequence identity, more preferably e.g. at least 94% sequence identity, more preferably such as at least 95% sequence identity, more preferably e.g. at least 96% sequence identity, more preferably such as at least 97% sequence identity, more preferably e.g. at least 98% sequence identity, more preferably such as at least 99% sequence identity, more preferably e.g. at least 99.5% sequence identity identity with the predetermined sequence of U20 from HHV 6B (SEQ ID No: 1 ).
Suitably variants are variant(s) of SEQ ID NO:1 , 3 or 5 having at least 80% sequence identity, such as preferably at least 81 % sequence identity, more preferably e.g. at least 82% sequence identity, such as more preferably at least 83% sequence identity, e.g. more preferably at least 84% sequence identity, more preferably such as at least 85% sequence identity, more preferably e.g. at least 86% sequence identity, more preferably such as at least 87% sequence identity, more preferably e.g. at least 88% sequence identity, more preferably such as at least 89% sequence identity, more preferably e.g. at least 90% sequence identity, more preferably such as at least 91 % sequence identity, more preferably e.g. at least 92% sequence identity, such as at least 93% sequence identity, more preferably e.g. at least 94% sequence identity, more preferably such as at least 95% sequence identity, more preferably e.g. at least 96% sequence identity, more preferably such as at least 97% sequence identity, more preferably e.g. at least 98% sequence identity, more preferably such as at least 99%
sequence identity, more preferably e.g. at least 99.5% sequence identity identity with the predetermined sequence of U20 from HHV6A (SEQ ID No: 3).
Suitably variants are variant(s) of SEQ ID NO:1 , 3 or 5 having at least 80% sequence identity, such as preferably at least 81 % sequence identity, more preferably e.g. at least 82% sequence identity, such as more preferably at least 83% sequence identity, e.g. more preferably at least 84% sequence identity, more preferably such as at least 85% sequence identity, more preferably e.g. at least 86% sequence identity, more preferably such as at least 87% sequence identity, more preferably e.g. at least 88% sequence identity, more preferably such as at least 89% sequence identity, more preferably e.g. at least 90% sequence identity, more preferably such as at least 91 % sequence identity, more preferably e.g. at least 92% sequence identity, such as at least 93% sequence identity, more preferably e.g. at least 94% sequence identity, more preferably such as at least 95% sequence identity, more preferably e.g. at least 96% sequence identity, more preferably such as at least 97% sequence identity, more preferably e.g. at least 98% sequence identity, more preferably such as at least 99% sequence identity, more preferably e.g. at least 99.5% sequence identity identity with the predetermined sequence of U20 from HHV 7 (SEQ ID No: 5). The polypeptide fragment according to the invention, wherein the fragment has a stretch of at least 50 contiguous amino acids contains less than 410 consecutive amino acid residues of SEQ ID NO: 1 , 3 or 5, such as less than 400 consecutive amino acid residues, such as less than 395 consecutive amino acid residues, e.g. less than 390 consecutive amino acid residues, such as less than 385 consecutive amino acid residues, e.g. less than 380 consecutive amino acid residues, such as less than 370 consecutive amino acid residues, e.g. less than 360 consecutive amino acid residues, such as less than 350 consecutive amino acid residues, e.g. less than 345 consecutive amino acid residues, such as less than 340 consecutive amino acid residues, e.g. less than 335 consecutive amino acid residues, such as less than 330 consecutive amino acid residues, e.g. less than 325 consecutive amino acid residues, such as less than 300 consecutive amino acid residues, e.g. less than 295 consecutive amino acid residues, such as less than 290 consecutive amino acid residues, e.g. less than 285 consecutive amino acid residues, such as less than 280 consecutive amino acid residues, e.g. less than 275 consecutive amino acid residues, such as less than 270 consecutive amino acid residues, e.g. less than 265 consecutive amino acid residues,
such as less than 260 consecutive amino acid residues, such as less than 255 consecutive amino acid residues, e.g. less than 250 consecutive amino acid residues, such as less than 245 consecutive amino acid residues, e.g. less than 240 consecutive amino acid residues, such as less than 235 consecutive amino acid residues, e.g. less than 230 consecutive amino acid residues, such as less than 225 consecutive amino acid residues,such as less than 220 consecutive amino acid residues, such as less than 215 consecutive amino acid residues, e.g. less than 210 consecutive amino acid residues, such as less than 205 consecutive amino acid residues, e.g. less than 200 consecutive amino acid residues, such as less than 195 consecutive amino acid residues, e.g. less than 190 consecutive amino acid residues, such as less than 185 consecutive amino acid residues, e.g. less than 180 consecutive amino acid residues, such as less than 175 consecutive amino acid residues, e.g. less than 170 consecutive amino acid residues, such as less than 165 consecutive amino acid residues, e.g. less than 160 consecutive amino acid residues, such as less than 155 consecutive amino acid residues, e.g. less than 150 consecutive amino acid residues, such as less than 145 consecutive amino acid residues, e.g. less than 140 consecutive amino acid residues, such as less than 135 consecutive amino acid residues, e.g. less than 130 consecutive amino acid residues, such as less than 125 consecutive amino acid residues, e.g. less than 120 consecutive amino acid residues, such as less than 1 15 consecutive amino acid residues, e.g. less than 1 10 consecutive amino acid residues, such as less than 105 consecutive amino acid residues, e.g. less than 100 consecutive amino acid residues, such as less than 95 consecutive amino acid residues, e.g. less than 90 consecutive amino acid residues, such as less than 85 consecutive amino acid residues, e.g. less than 80 consecutive amino acid residues, such as less than 75, e.g. less than 60 consecutive amino acid residues of SEQ ID NO: 1 , 3 or 5.
The polypeptide variant according to the present invention, wherein the polypeptide variant is a variant of SEQ ID NO:1 , 3 or 5 having at least 80% sequence identity, such as preferably at least 81 % sequence identity, more preferably e.g. at least 82% sequence identity, such as more preferably at least 83% sequence identity, e.g. more preferably at least 84% sequence identity, more preferably such as at least 85% sequence identity, more preferably e.g. at least 86% sequence identity, more preferably such as at least 87% sequence identity, more preferably e.g. at least 88% sequence identity, more preferably such as at least 89% sequence identity, more preferably e.g. at least 90% sequence identity, more preferably such as at least 91 %
sequence identity, more preferably e.g. at least 92% sequence identity, such as at least 93% sequence identity, more preferably e.g. at least 94% sequence identity, more preferably such as at least 95% sequence identity, more preferably e.g. at least 96% sequence identity, more preferably such as at least 97% sequence identity, more preferably e.g. at least 98% sequence identity, more preferably such as at least 99% sequence identity, more preferably e.g. at least 99.5% sequence identity to said SEQ ID NO. 1 , 3 or 5, or a fragement of SEQ ID NO: 1 , 3 or 5.
The polypeptide according to the present invention, wherein the polypeptide variant fragment contains less than 99.5%, such as less than 98%, e.g. less than 97%, such as less than 96%, e.g. less than 95%, such as less than 94%, e.g. less than 93%, such as less than 92%, e.g. less than 91 %, such as less than 90%, e.g. less than 88%, such as less than 86%, e.g. less than 84%, e.g. less than 82%, such as less than 80%, e.g. less than 75%, such as less than 70%, e.g. less than 65%, such as less than 60%, e.g. less than 55%, such as less than 50%, e.g. less than 45%, such as less than 40%, e.g. less than 35%, such as less than 30%, e.g. less than 25%, such as less than 20%, such as less than 15%, e.g. less than 10% of the amino acid residues of SEQ ID NO:1 .
The polypeptide according to the present invention, wherein the polypeptide variant fragment contains less than 99.5%, such as less than 98%, e.g. less than 97%, such as less than 96%, e.g. less than 95%, such as less than 94%, e.g. less than 93%, such as less than 92%, e.g. less than 91 %, such as less than 90%, e.g. less than 88%, such as less than 86%, e.g. less than 84%, e.g. less than 82%, such as less than 80%, e.g. less than 75%, such as less than 70%, e.g. less than 65%, such as less than 60%, e.g. less than 55%, such as less than 50%, e.g. less than 45%, such as less than 40%, e.g. less than 35%, such as less than 30%, e.g. less than 25%, such as less than 20%, such as less than 15%, e.g. less than 10% of the amino acid residues of SEQ ID NO:3. The polypeptide according to the present invention, wherein the polypeptide variant fragment contains less than 99.5%, such as less than 98%, e.g. less than 97%, such as less than 96%, e.g. less than 95%, such as less than 94%, e.g. less than 93%, such as less than 92%, e.g. less than 91 %, such as less than 90%, e.g. less than 88%, such as less than 86%, e.g. less than 84%, e.g. less than 82%, such as less than 80%, e.g. less than 75%, such as less than 70%, e.g. less than 65%, such as less than 60%, e.g. less than 55%, such as less than 50%, e.g. less than 45%, such as less than 40%, e.g.
less than 35%, such as less than 30%, e.g. less than 25%, such as less than 20%, such as less than 15%, e.g. less than 10% of the amino acid residues of SEQ ID NO:5.
The polypeptide according to the present invention, wherein the polypeptide variant fragment contains than 410 consecutive amino acid residues of SEQ ID NO: 1 , such as less than 400 consecutive amino acid residues, such as less than 395 consecutive amino acid residues, e.g. less than 390 consecutive amino acid residues, such as less than 385 consecutive amino acid residues, e.g. less than 380 consecutive amino acid residues, such as less than 370 consecutive amino acid residues, e.g. less than 360 consecutive amino acid residues, such as less than 350 consecutive amino acid residues, e.g. less than 345 consecutive amino acid residues, such as less than 340 consecutive amino acid residues, e.g. less than 335 consecutive amino acid residues, such as less than 330 consecutive amino acid residues, e.g. less than 325 consecutive amino acid residues, such as less than 300 consecutive amino acid residues, e.g. less than 295 consecutive amino acid residues, such as less than 290 consecutive amino acid residues, e.g. less than 285 consecutive amino acid residues, such as less than 280 consecutive amino acid residues, e.g. less than 275 consecutive amino acid residues, such as less than 270 consecutive amino acid residues, e.g. less than 265 consecutive amino acid residues, such as less than 260 consecutive amino acid residues, such as less than 255 consecutive amino acid residues, e.g. less than 250 consecutive amino acid residues, such as less than 245 consecutive amino acid residues, e.g. less than 240 consecutive amino acid residues, such as less than 235 consecutive amino acid residues, e.g. less than 230 consecutive amino acid residues, such as less than 225 consecutive amino acid residues, such as less than 220 consecutive amino acid residues, such as less than 215 consecutive amino acid residues, e.g. less than 210 consecutive amino acid residues, such as less than 205 consecutive amino acid residues, e.g. less than 200 consecutive amino acid residues, such as less than 195 consecutive amino acid residues, e.g. less than 190 consecutive amino acid residues, such as less than 185 consecutive amino acid residues, e.g. less than 180 consecutive amino acid residues, such as less than 175 consecutive amino acid residues, e.g. less than 170 consecutive amino acid residues, such as less than 165 consecutive amino acid residues, e.g. less than 160 consecutive amino acid residues, such as less than 155 consecutive amino acid residues, e.g. less than 150 consecutive amino acid residues, such as less than 145 consecutive amino acid residues, e.g. less than 140 consecutive amino acid residues, such as less than 135
consecutive amino acid residues, e.g. less than 130 consecutive amino acid residues, such as less than 125 consecutive amino acid residues, e.g. less than 120 consecutive amino acid residues, such as less than 1 15 consecutive amino acid residues, e.g. less than 1 10 consecutive amino acid residues, such as less than 105 consecutive amino acid residues, e.g. less than 100 consecutive amino acid residues, such as less than 95 consecutive amino acid residues, e.g. less than 90 consecutive amino acid residues, such as less than 85 consecutive amino acid residues, e.g. less than 80 consecutive amino acid residues, such as less than 75, e.g. less than 60 consecutive amino acid residues of SEQ ID NO: 1 .
Many amino acids, including the terminal amino acids, may be modified in a given polypeptide, either by natural processes such as glycosylation and other post- translational modifications, or by chemical modification techniques which are well known in the art. Among the known modifications which may be present in
polypeptides of the present invention are, to name an illustrative few, acetylation, acylation, ADP-ribosylation, amidation, covalent attachment of flavin, covalent attachment of a heme moiety, covalent attachment of a polynucleotide or
polynucleotide derivative, covalent attachment of a lipid or lipid derivative, covalent attachment of phosphotidylinositol, cross-linking, cyclization, disulfide bond formation, demethylation, formation of covalent cross-links, formation of cystine, formation of pyroglutamate, formylation, gamma-carboxylation, glycation, glycosylation, GPI anchor formation, hydroxylation, iodination, methylation, myristoylation, oxidation, proteolytic processing, phosphorylation, prenylation, racemization, selenoylation, sulfation, transfer-RNA mediated addition of amino acids to proteins such as arginylation, and ubiquitination.
Such modifications are well known to those of skill and have been described in great detail in the scientific literature. Several particularly common modifications,
glycosylation, lipid attachment, sulfation, gamma-carboxylation of glutamic acid residues, hydroxylation and ADP-ribosylation, for instance, are described in most basic texts, such as, for instance, I. E. Creighton, Proteins-Structure and Molecular
Properties, 2nd Ed., W. H. Freeman and Company, New York, 1993. Many detailed reviews are available on this subject, such as, for example, those provided by Wold, F., in Posttranslational Covalent Modification of Proteins, B. C. Johnson, Ed., Academic Press, New York, pp 1 -12, 1983; Seifter et al., Meth. Enzymol. 182: 626-646, 1990 and
Rattan et al., Protein Synthesis: Posttranslational Modifications and Aging, Ann. N.Y. Acad. Sci. 663: 48-62, 1992.
Functional equivalents may further comprise chemical modifications such as ubiquitination, labeling (e.g., with radionuclides, various enzymes, etc.), pegylation (derivatization with polyethylene glycol), or by insertion (or substitution by chemical synthesis) of amino acids (amino acids) such as ornithine, which do not normally occur in human proteins. One or more amino acid residues may be modified, where the modification(s) preferably being selected from the group consisting of in vivo or in vitro chemical derivatization, such as acetylation or carboxylation, glycosylation, such as glycosylation resulting from exposing the polypeptide to enzymes which affect glycosylation, for example mammalian glycosylating or deglycosylating enzymes, phosphorylation, such as modification of amino acid residues which results in phosphorylated amino acid residues, for example phosphotyrosine, phosphoserine and phosphothreonine.
In addition to the peptidyl compounds described herein, sterically similar compounds may be formulated to mimic the key portions of the peptide structure and that such compounds may also be used in the same manner as the peptides of the invention. This may be achieved by techniques of modelling and chemical designing known to those of skill in the art. For example, esterification and other alkylations may be employed to modify the amino terminus of, e.g., a di-arginine peptide backbone, to mimic a tetra peptide structure. It will be understood that all such sterically similar constructs fall within the scope of the present invention.
In addition, the protein may comprise a protein tag to allow subsequent purification and optionally removal of the tag using an endopeptidase. The tag may also comprise a protease cleavage site to facilitate subsequent removal of the tag. Non-limiting examples of affinity tags include a polyhis tag, a GST tag, a HA tag, a Flag tag, a C- myc tag, a HSV tag, a V5 tag, a maltose binding protein tag, a cellulose binding domain tag. Preferably for production and purification, the tag is a polyhistag. Preferably, the tag is in the C-terminal portion of the protein.
The isolated polypeptide(s) may be attached to a carrier. An example of such as carrier comprises an avidin moiety, such as streptavidin, which is optionally biotinylated.
The carrier may be attached as covalently bound, to a solid support or a semi-solid support.
The isolated polypeptide(s) may operably be fused to an affinity tag, such as a His-tag.
In one embodiment, the isolated polypeptide is part of a fusion polypeptide operably fused to an N-terminal flanking sequence. In addition, or alternatively, the isolated polypeptides are operably fused to an C-terminal flanking sequence. Peptides with N-terminal alkylations and C-terminal esterifications are also encompassed within the present invention. Functional equivalents also comprise glycosylated and covalent or aggregative conjugates formed with the same molecules, including dimers or unrelated chemical moieties. Such functional equivalents are prepared by linkage of functionalities to groups which are found in fragment including at any one or both of the N- and C-termini, by means known in the art.
The term "fragment thereof" may refer to any portion of the given amino acid sequence. Fragments may comprise more than one portion from within the full-length protein, joined together. Suitable fragments may be deletion or addition mutants. The addition of at least one amino acid may be an addition of from preferably 2 to 250 amino acids, such as from 10 to 20 amino acids, for example from 20 to 30 amino acids, such as from 40 to 50 amino acids. Fragments may include small regions from the protein or combinations of these. Suitable fragments may be deletion or addition mutants. The addition or deletion of at least one amino acid may be an addition or deletion of from preferably 2 to 250 amino acids, such as from 10 to 20 amino acids, for example from 20 to 30 amino acids, such as from 40 to 50 amino acids. The deletion and/or the addition may - independently of one another - be a deletion and/or an addition within a sequence and/or at the end of a sequence.
A functional homologue may be a deletion mutant of HHV U20 polypeptides as identified by SEQ ID NO: 1 , 3 or 5, sharing at least 70% and accordingly, a functional homologue preferably have at least 75% sequence identity, for example at least 80% sequence identity, such as at least 85 % sequence identity, for example at least 90 %
sequence identi ity, such as at least 91 % sequence identity, for example at least 91 % sequence identi ity, such as at least 92 % sequence identity, for example at least 93 % sequence identi ity, such as at least 94 % sequence identity, for example at least 95 % sequence identi ity, such as at least 96 % sequence identity, for example at least 97% sequence identi ity, such as at least 98 % sequence identity, for example 99% sequence identity.
Deletion mutants suitably comprise at least 20 or 40 consecutive amino acid and more preferably at least 80 or 100 consecutive amino acids in length. Accordingly such a fragment may be a shorter sequence of the sequence as identified by SEQ ID NO: 1 , 3 or 5, comprising at least 20 consecutive amino acids, for example at least 30 consecutive amino acids, such as at least 40 consecutive amino acids, for example at least 50 consecutive amino acids, such as at least 60 consecutive amino acids, for example at least 70 consecutive amino acids, such as at least 80 consecutive amino acids, for example at least 90 consecutive amino acids, such as at least 95 consecutive amino acids, such as at least 100 consecutive amino acids, such as at least 105 amino acids, for example at least 1 10 consecutive amino acids, such as at least 1 15 consecutive amino acids, for example at least 120 consecutive amino acids, wherein said deletion mutants preferably share at least 75% sequence identity, for example at least 80% sequence identity, such as at least 85 % sequence identity, for example at least 90 % sequence identity, such as at least 91 % sequence identity, for example at least 91 % sequence identity, such as at least 92 % sequence identity, for example at least 93 % sequence identity, such as at least 94 % sequence identity, for example at least 95 % sequence identity, such as at least 96 % sequence identity, for example at least 97% sequence identity, such as at least 98 % sequence identity, for example 99% sequence identity with full length SEQ ID NO: 1 , 3 or 5.
It is preferred that functional homologues of HHV U20 comprises at the most 450, more preferably at the most 400, even more preferably at the most 300, yet more preferably at the most 200, such as at the most 175, for example at the most 160, such as at the most 150 amino acids, for example at the most 140 amino acids.
The term "fragment thereof" may refer to any portion of the given amino acid sequence. Fragments may comprise more than one portion from within the full-length protein, joined together. Portions will suitably comprise at least 5 and preferably at least 10
consecutive amino acids from the basic sequence. They may include small regions from the protein or combinations of these.
It will be appreciated, as is well known and as noted above, that polypeptides are not always entirely linear. For instance, polypeptides may be branched as a result of ubiquitination, and they may be circular, with or without branching, generally as a result of posttranslational events, including natural processing events and events brought about by human manipulation which do not occur naturally. Circular, branched and branched circular polypeptides may be synthesized by non-translational natural processes and by entirely synthetic methods, as well and are all within the scope of the present invention.
Modifications can occur anywhere in a polypeptide, including the peptide backbone, the amino acid side-chains and the amino or carboxyl termini. In fact, blockage of the amino or carboxyl group in a polypeptide, or both, by a covalent modification, is common in naturally occurring and synthetic polypeptides and such modifications may be present in polypeptides of the present invention, as well. For instance, the amino terminal residue of polypeptides made in E. coli, prior to proteolytic processing, almost invariably will be N-formylmethionine. The modifications that occur in a polypeptide often will be a function of how it is made. For polypeptides made by expressing a cloned gene in a host, for instance, the nature and extent of the modifications in large part will be determined by the host cell's posttranslational modification capacity and the modification signals present in the polypeptide amino acid sequence. For instance, glycosylation often does not occur in bacterial hosts such as E. coli. Accordingly, when glycosylation is desired, a polypeptide should be expressed in a glycosylating host, generally a eukaryotic cell. Insect cells often carry out the same posttranslational glycosylations as mammalian cells and, for this reason, insect cell expression systems have been developed to efficiently express mammalian proteins having native patterns of glycosylation, inter alia. Similar considerations apply to other modifications.
It will be appreciated that the same type of modification may be present to the same or varying degree at several sites in a given polypeptide. Also, a given polypeptide may contain many types of modifications.
Specific domains of U20 may also be specifically relevant for the decreasing of TNF receptor activity, preferably for the inhibition of TNF-alpha - TNF-alpha receptor interaction. The domain sequences or combination of domains listed in table 1 may be particularly relevant. Thus in an embodiment according to the invention the active derivative of U20 is selected from the group consisting of the following positions in SEQ ID NO:1 (U20 polypeptide sequence of HHV-6B): position 16-434, 16-319, 1 -319, 16- 52, 80-100, 152-182, 100-152, 78-182, 182-300, 182-320, 16-50 + linker + 180- 320, 79-320, 16-382, 16-343, 1 -343, 16-52 + linker + 319 - 343, 80-100 + linker + 319 - 343, 152-182 + linker + 319 - 343, 100-152 + linker + 319-343, 78-182 + linker + 319-343, 182 - 343, 16-50 + linker + 180-343, 79-343, 16-382 + linker + 319-343, 319^134, or 183^134.
Explanations of the different domain combinations are described in Table 2.
TABLE 2
AA in U20 from HHV- 6B (SEQ ID NO: 1) Description
16 - 434 U20 except the predicted signal peptid
16 - 319 Extracellular domain except predicted signal peptid
1 - 319 Extracellular domain including predicted signal peptid
16 - 52 N-terminal domain including predicted a-helix (2T)
80 - 100 Short predicted a-helix
152 - 182 Long predicted a-helix
100 - 152 β-Sheet 2
78 - 182 Core domain
182 - 300 Immunoglobulin domain
Immunoglobulin domain including a linker to the
182 - 320 membrane helix
(16 - 50) + linker + Immunoglobulin and N-terminal domain separated by a (180 - 320) linker sequence
79 - 320 Core domain and Immunoglobulin domain
16 - 382 Core domain plus N-terminal domain
Extracellular domain except signal peptid and
16 - 343 transmembrane helix
1 - 343 Extracellular domain including signal peptid and
transmembran helix
(16 - 52) + linker + N-terminal domain including predicted a-helix (2T), long (319 - 343) "linker" and transmembrane helix
(80 - 100) + linker + Short a-helix and linker sequence and transmembrane (319 - 343) helix
(152 - 182) + linker + Long a-helix and linker sequence and transmembrane (319 - 343) helix
(100 - 152) + linker +
(319 - 343) β-Sheet 2 and linker sequence and transmembrane helix
(78 - 182) + linker + Core domain and linker sequence and transmembrane (319 - 343) helix
182 - 343 Immunoglobulin domain and transmembrane helix
(16 - 50) + linker + Immunoglobulin and N-terminal domain separated by a (180 - 343) linker sequence (incl. transmembran helix)
Core domain plus immunoglobulin domain plus
79 - 343 transmembrane helix
(16 - 382) + linker + Core domain and N-terminal domain and transmembrane (319 - 343) helix
319 - 434 Transmembrane helix and C-terminal domain
Immunoglobulin domain and transmembrane helix and C-
183 - 434 terminal domain
In one embodiment the polypeptides of the invention has an amino acid sequence as identified in SEQ ID NO.: 10 corresponding to the extracellular domain of HHV 6B U20 polypeptide without the signal peptide. It is appreciated that biologically active variants and fragments of the polypeptide of said domain are also within the scope of the invention.
Due to the high similarity between HHV-6B and 6A domains in HHV-6A may have the same effect as the domains from HHV-6B listed in Table 2. Thus the domain sequences or combination of domains from HHV-6A, as listed in table 3, may be particularly relevant. Thus, in an embodiment according to the invention the active derivative of U20 is selected from the group consisting of the following positions in SEQ ID NO:3 (U20 polypeptide sequence of HHV-6A): position 16-422, 16-319, 1 -319, 16- 52, 80-100, 152-182, 100-152, 78-182, 182-300, 182-320, 16-50 + linker + 180-
320, 79-320, 16-382, 16-343, 1-343, 16-52 + linker + 319-343, 80-100 + linker + 319-343, 152-182 + linker + 319-343, 100-152 + linker + 319-343, 78-182 + linker + 319-343, 182-343, 16-50 + linker + 180-343, 79-343, 16-382 + linker + 319-343, 319^122, and 183-422.
TABLE 3
AA in U20 from HHV- 6A (SEQIDNO:3) Description
16-422 U20 except the predicted signal peptid
16-319 Extracellular domain except predicted signal peptid
1 -319 Extracellular domain including predicted signal peptid
16-52 N-terminal domain including predicted a-helix (2T)
80-100 Short predicted a-helix
152-182 Long predicted a-helix
100-152 β-Sheet 2
78-182 Core domain
182-300 Immunoglobulin domain
Immunoglobulin domain including a linker to the
182-320 membrane helix
(16-50) + linker + Immunoglobulin and N-terminal domain separated by a (180-320) linker sequence
79 - 320 Core domain and Immunoglobulin domain
16-382 Core domain plus N-terminal domain
Extracellular domain except signal peptid and
16-343 transmembrane helix
Extracellular domain including signal peptid and
1 -343 transmembran helix
(16-52) + linker + N-terminal domain including predicted a-helix (2T), long (319-343) "linker" and transmembrane helix
(80 - 100) + linker + Short a-helix and linker sequence and transmembrane (319-343) helix
(152-182) + linker + Long a-helix and linker sequence and transmembrane (319-343) helix
(100- 152) + linker + β-Sheet 2 and linker sequence and transmembrane helix
(319 - 343)
(78 - 182) + linker + Core domain and linker sequence and transmembrane (319 - 343) helix
182 - 343 Immunoglobulin domain and transmembrane helix
(16 - 50) + linker + Immunoglobulin and N-terminal domain separated by a (180 - 343) linker sequence (incl. transmembran helix)
Core domain plus immunoglobulin domain plus
79 - 343 transmembrane helix
(16 - 382) + linker + Core domain and N-terminal domain and transmembrane (319 - 343) helix
319 - 422 Transmembrane helix and C-terminal domain
Immunoglobulin domain and transmembrane helix and C-
183 - 422 terminal domain
In another embodiment the polypeptides of the invention has an amino acid sequence as identified in SEQ ID NO.: 1 1 corresponding to the extracellular domain of HHV 6A U20 polypeptide without the signal peptide. It is appreciated that biologically active variants and fragments of the polypeptide of said domain are also within the scope of the invention.
The person skilled in the art would know how to arrive at similar domain or domain combinations in the U20 amino acid sequence from HHV-7 (SEQ ID NO:5), e.g. by using the sequence alignment disclosed in figure 9. Similar, the person skilled in the art would know how to put in a linker at other positions in the sequences according to the invention.
Glycosylation
Glycolysation is the enzymatic process that links saccharides to produce glycans, attached to proteins, lipids, or other organic molecules. This enzymatic process produces one of the fundamental biopolymers found in cells (along with DNA, RNA, and proteins). Glycosylation is a form of co-translational and post-translational modification. Glycans serve a variety of structural and functional roles in membrane and secreted proteins. The majority of proteins synthesized in the rough ER undergo glycosylation. It is an enzyme-directed site-specific process, as opposed to the non- enzymatic chemical reaction of glycation. Glycosylation is also present in the cytoplasm
and nucleus as the O-GlcNAc modification. Five classes of glycans are produced: relinked glycans attached to a nitrogen of asparagine or arginine side chains, O-linked glycans attached to the hydroxy oxygen of serine, threonine, tyrosine, hydroxylysine, or hydroxyproline side chains, or to oxygens on lipids such as ceramide; phospho-glycans linked through the phosphate of a phospho-serine; C-linked glycans, a rare form of glycosylation where a sugar is added to a carbon on a tryptophan side chain, and glypiation which is the addition of a GPI anchor which links proteins to lipids through glycan linkages. In the present context a glycolysation may both be chemically and enzymatically attached the the protein or peptide.
The U20 protein from HHV-6B comprises several glycosylation sites such as position 58, 78, 107, 133, 145, 154, 161 , and 227. Thus, in an embodiment the U20 protein or derivative thereof comprises at least one glycosylated amino acid. In a further embodiment the at least one glycosylation site is selected from the group consisting of position 58, 78, 107, 133, 145, 154, 161 , and 227 in SEQ ID NO: 1 .
In an embodiment the U20 protein or active derivative thereof comprises at least one glycosylated amino acid, such as at least two, such as at least three, such as at least four, such as at least five, such as at least six, such as at least seven, such as at least eight glycosylated amino acids.
In another embodiment the U20 protein or active derivative thereof comprises 1 -20 glycosylated amino acid, such as 1 -10 glycosylated amino acid, such as 1 -5
glycosylated amino acid, such as 3-8 glycosylated amino acid, such as 1 glycosylated amino acid, such as 2 glycosylated amino acid, or such as 3 glycosylated amino acid.
In HHV-6A examples of glycosylation sites are position 58, 78, 107, 133, 154, 161 227, 266 of SEQ ID NO: 3. Thus, In a further embodiment the at least one glycosylation site is selected from the group consisting of position 58, 78, 107, 133, 154, 161 , 227, 266 of SEQ ID NO: 3.
In HHV-7 examples of glycosylation sites are position 50, 55, 98, 129, 198
265 of SEQ ID NO: 5. Thus, In an additional embodiment the at least one glycosylation site is selected from the group consisting of position 50, 55, 98, 129, 198 265 of SEQ ID NO: 5.
The glycosylation positions in HHV-6B, HHV-6A and HHV-7 are related as follows See also figure 9 showing an alignment between HHV-6B, HHV-6A and HHV-7):
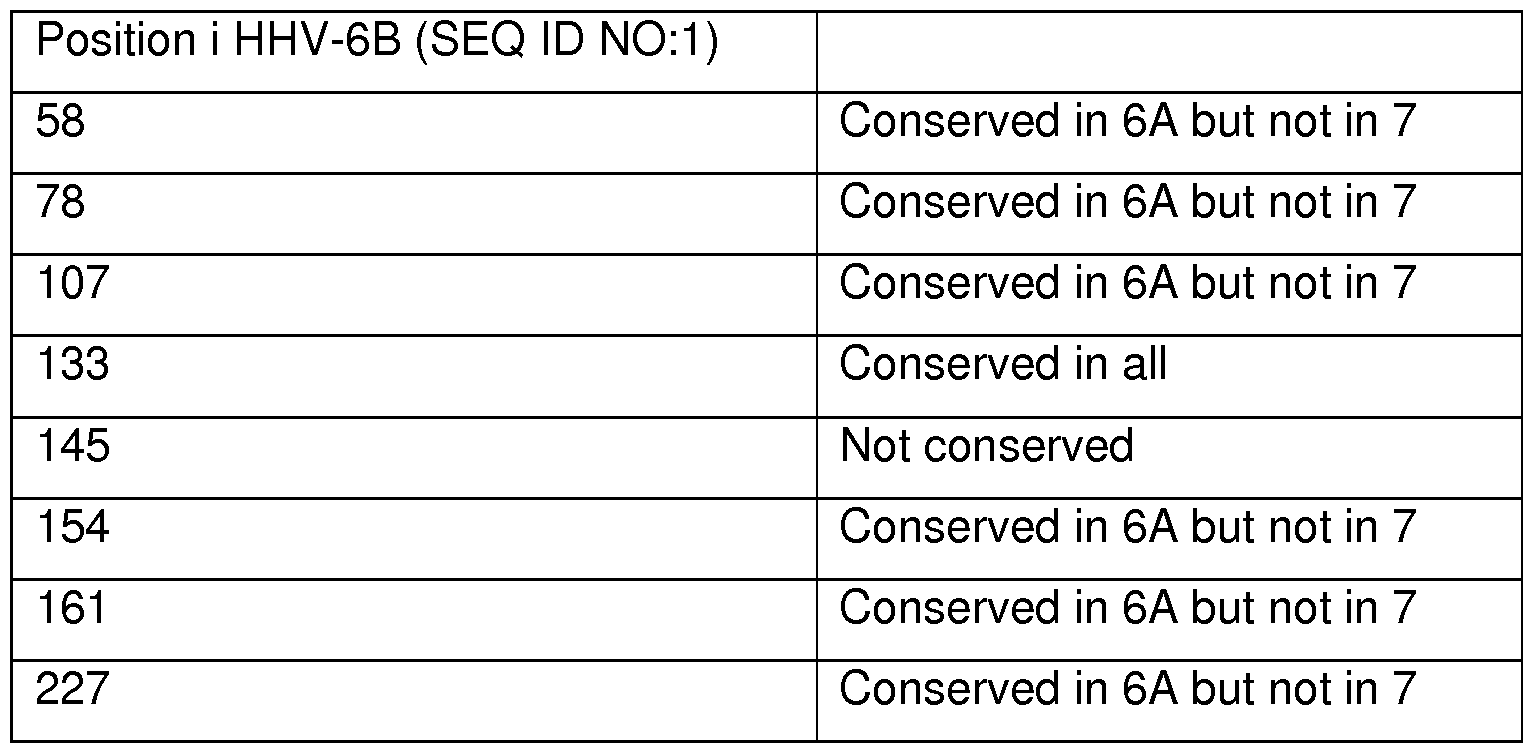
The person skilled in the art would know how to identify other or corresponding glycosylation sites in HHV-6B, HHV-6A and HHV-7.
Polynucleotide
The invention provides medical use of genomic DNA and cDNA coding for HHV U20, including for example the nucleotide sequence (SEQ ID No. 2, 4 or 6), the sequences coding for HHV U20 without signal peptide. The invention also provides the cDNA sequence coding for HHV U 20.
Variants of these sequences are also included within the scope of the present invention.
One aspect of the present invention pertains to an isolated polynucleotide for use as a medicament comprising a nucleic acid or its complementary sequence, said
polynucleotide being selected from the group consisting of:
a) i) a polynucleotide encoding an amino acid sequence consisting of SEQ ID No. 1 , 3 or 5;or
ii) a biologically active sequence variant of the amino acid sequence, wherein the variant has at least 70% sequence identity to said SEQ ID NO. 1 , 3 or 5; and
iii) a biologically active fragment of at least 50 contiguous amino acids of any of a) through b), wherein said fragment is a fragment of SEQ ID NO 1 , 3 or 5 or
b) SEQ ID NO.: 2, 4, 6, 7, 8 or 9 or
d) a polynucleotide comprising a nucleic acid having 70% sequence identity to SEQ ID NO.: 2, 4, 6, 7, 8 or 9 or
e) a polynucleotide hybridising to SEQ ID NO.: 2, 4, 6, 7, 8 or 9 and f) a polynucleotide complementary to any of a) to d).
In one preferred embodiment of the polynucleotide, the polynucleotide is
i) a polynucleotide encoding an amino acid sequence consisting of SEQ ID No. 1 , 3 or 5; or
ii) a biologically active sequence variant of the amino acid
sequence, wherein the variant has at least 70% sequence identity to said SEQ ID NO. 1 , 3 or 5 and
iii) iii) a biologically active fragment of at least 50 contiguous amino acids of any of a) through b), wherein said fragment is a fragment of SEQ ID NO 1 , 3 or 5.
In another preferred embodiment of the invention, the polynucleotide is SEQ ID NO.: 2, 4, 6, 7, 8 or 9.
In another preferred embodiment of the invention the polynucleotide is a polynucleotide comprising a nucleic acid having 70% sequence identity to SEQ ID NO.: 2, 4, 6, 7, 8 or 9.
In yet another preferred embodiment of the invention the polynucleotide is capable of hybridising to a polynucleotide having the sequence of SEQ ID NO.: 2, 4, 6, 7, 8 or 9.
In a further, preferred embodiment of the invention the polynucleotide is
complementary to
i) a polynucleotide encoding an amino acid sequence consisting of SEQ ID No. 1 , 3 or 5; or
ii) a polynucleotide encoding a biologically active sequence variant of the amino acid sequence, wherein the variant has at least 70% sequence identity to said SEQ ID NO. 1 , 3 or 5 and iii) a polynucleotide encoding a biologically active fragment of at least 50 contiguous amino acids of any of a) through b), wherein said fragment is a fragment of SEQ ID NO 1 , 3 or 5.
Thus, in one embodiment the polynucleotide of the present invention is complementary to SEQ ID NO.: 2, 4, 6, 7, 8 or 9.
However, the polynucleotide of the invention may in another embodiment may be
complementary to a polynucleotide comprising a nucleic acid having 70% sequence identity to SEQ ID NO.: 2, 4, 6, 7, 8 or 9.
The polynucleotide may comprise the nucleotide sequence of a naturally occurring allelic nucleic acid variant.
The polynucleotide of the invention may encode a variant polypeptide, wherein the variant polypeptide has the polypeptide sequence of a naturally occurring polypeptide variant.
The nucleic acid sequence of the polynucleotide may differ by a single nucleotide from a nucleic acid sequence selected from the group consisting of SEQ ID NO: 2, 4, 6, 7, 8 and 9. However, the polynucleotide may also differ from a nucleic acid sequence selected from the group consisting of SEQ ID NO: 2, 4, 6, 7, 8 and 9 by 2, 3, 4, 5, 6, 7, 8, 9, 10, or more nucleotides.
In one embodiment, the nucleic acid sequence of the polynucleotide has at least 80% sequence identity, such as preferably at least 81 % sequence identity, more preferably e.g. at least 82% sequence identity, such as more preferably at least 83% sequence identity, e.g. more preferably at least 84% sequence identity, more preferably such as at least 85% sequence identity, more preferably e.g. at least 86% sequence identity, more preferably such as at least 87% sequence identity, more preferably e.g. at least 88% sequence identity, more preferably such as at least 89% sequence identity, more
preferably e.g. at least 90% sequence identity, more preferably such as at least 91 % sequence identity, more preferably e.g. at least 92% sequence identity, such as at least 93% sequence identity, more preferably e.g. at least 94% sequence identity, more preferably such as at least 95% sequence identity, more preferably e.g. at least 96% sequence identity, more preferably such as at least 97% sequence identity, more preferably e.g. at least 98% sequence identity, more preferably such as at least 99% sequence identity, more preferably e.g. at least 99.5% sequence identity to a nucleotide sequence selected from the group consisting of SEQ ID No. 2, 4, 6, 7, 8, and 9;
The nucleic acid sequence of the polynucleotide may contain less than 99.5%, such as less than 98%, e.g. less than 97%, such as less than 96%, e.g. less than 95%, such as less than 94%, e.g. less than 93%, such as less than 92%, e.g. less than 91 %, such as less than 90%, e.g. less than 88%, such as less than 86%, e.g. less than 84%, e.g. less than 82%, such as less than 80%, e.g. less than 75%, such as less than 70%, e.g. less than 65%, such as less than 60%, e.g. less than 55%, such as less than 50%, e.g. less than 45%, such as less than 40%, e.g. less than 35%, such as less than 30%, e.g. less than 25%, such as less than 20%, such as less than 15%, e.g. less than 10% of nucleotide sequence selected from the group consisting of SEQ ID No. 2, 4, 6, 7, 8, and 9.
In a preferred embodiment the encoded polypeptide has at least 70% sequence identity to SEQ ID No. 3, more preferably at least 75%, more preferably at least 80%, more preferably at least 95%, more preferably at least 98%, more preferably wherein said polypeptide has the sequence of SEQ ID No. 3.
In a preferred embodiment the encoded polypeptide has at least 70% sequence identity to SEQ ID No. 5, more preferably at least 75%, more preferably at least 80%, more preferably at least 95%, more preferably at least 98%, more preferably wherein said polypeptide has the sequence of SEQ ID No. 5.
In a preferred embodiment the encoded polypeptide has at least 70% sequence identity to SEQ ID No. 7, more preferably at least 75%, more preferably at least 80%, more preferably at least 95%, more preferably at least 98%, more preferably wherein said polypeptide has the sequence of SEQ ID No. 7.
In one embodiment, the isolated polynucleotide of the invention comprises a nucelic acid sequence having at least 70%, preferably at least 75%, more preferably at least 80%, preferably at least 85%, more preferred at least 90%, more preferred at least 95%, more preferred at least 98% sequence identity to the polynucleotide sequence presented as SEQ ID NO: 2.
In one embodiment, the isolated polynucleotide of the invention comprises a nucelic acid sequence having at least 70%, preferably at least 75%, more preferably at least 80%, preferably at least 85%, more preferred at least 90%, more preferred at least 95%, more preferred at least 98% sequence identity to the polynucleotide sequence presented as SEQ ID NO: 4.
In one embodiment, the isolated polynucleotide of the invention comprises a nucelic acid sequence having at least 70%, preferably at least 75%, more preferably at least 80%, preferably at least 85%, more preferred at least 90%, more preferred at least 95%, more preferred at least 98% sequence identity to the polynucleotide sequence presented as SEQ ID NO: 6. In yet another embodiment the polynucleotide is capable of hybridizing to the nucleic acid selected from the group consisting of SEQ ID NO: SEQ ID NO: 2, 4, 6, 7, 8 and 9, or a fragment hereof, under stringent conditions as described below.
A portion of the polynucleotide may hybridize under stringent conditions to a nucleotide probe corresponding to at least 10 consecutive nucleotides of a nucleotide sequence selected from the group consisting of SEQ ID NO: SEQ ID NO: 2, 4, 6, 7, 8 and 9, or a fragment hereof.
In another embodiment, the invention relates to the use of the nucleic acids and proteins of the present invention to design probes to isolate other genes, which encode proteins with structural or functional properties of the HHV U20 proteins of the invention. The probes can be a variety of base pairs in length. For example, a nucleic acid probe can be between about 10 base pairs in length to about 150 base pairs in length.
Alternatively, the nucleic acid probe can be greater than about 150 base pairs in length. Experimental methods are provided in Ausubel et al., "Current Protocols in Molecular Biology", J. Wiley (ed.) (1999), the entire teachings of which are herein incorporated by reference in their entirety.
The design of the oligonucleotide (also referred to herein as nucleic acid) probe should preferably follow these parameters:
i) it should be designed to an area of the sequence which has the fewest
ambiguous bases, if any and
ii) it should be designed to have a calculated Tm of about 80 'Ό (assuming 2° for each A or T and 4°C for each G or C).
The oligonucleotide should preferably be labeled to facilitate detection of hybridisation. Labelling may be with γ-32Ρ ATP (specific activity 6000 Ci/mmole) and T4
polynucleotide kinase using commonly employed techniques for labeling
oligonucleotides. Other labeling techniques can also be used. Unincorporated label should preferably be removed by gel filtration chromatography or other established methods. The amount of radioactivity incorporated into the probe should be quantitated by measurement in a scintillation counter. Preferably, specific activity of the resulting probe should be approximately 4 x 106 dpm/pmole. The bacterial culture containing the pool of full-length clones should preferably be thawed and 100 μΙ_ of the stock used to inoculate a sterile culture flask containing 25 ml of sterile L-broth containing ampicillin at 100 pg/ml. In yet another embodiment, the invention relates to nucleic acid sequences (e. g., DNA, RNA) that hybridise to nucleic acids of HHV U20. In particular, nucleic acids which hybridise to SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 6, SEQ ID NO:7, SEQ ID No. 8, or SEQ ID No. 9 under high, moderate or reduced stringency conditions as described above.
In still another embodiment, the invention relates to a complement of nucleic acid of HHV U20. In particular, it relates to complements of SEQ ID NO: 2, SEQ ID NO: 4, SEQ ID NO: 6, SEQ ID NO:7, SEQ ID No. 8, or SEQ ID No. 9
In another embodiment, the invention relates to an RNA counterpart of the DNA nucleic acid of HHV U20. In particular, it relates to RNA counterparts of SEQ ID NO: 2, SEQ ID
NO: 4 and , SEQ ID NO: 6, exemplified by SEQ ID No 7, SEQ ID No 8, and SEQ ID No 9.
It is appreciated that the polynucleotide of the present invention is DNA, RNA, LNA or PNA.
In many instances it would advantageously also to have an isolated polynucleotide comprising a polynucleotide encoding the U20 protein according to the invention. Thus, in aspect the invention relates to an isolated polynucleotide comprising a
polynucleotide encoding the U20 protein or an active derivative thereof according to the invention. Such a polynucleotide may be used for expression purposes, cloning purposes, storage purposes. SEQ ID NO:2 discloses the nucleic acid sequence for U20 from HHV-6B, SEQ ID NO:4 discloses the nucleic acid sequence for U20 from HHV-6A, SEQ ID NO:6 discloses the nucleic acid sequence for U20 from HHV-7. The person skilled in the art would know how to construct polynucleotides encoding the peptide sequences disclosed on Table 1 and Table 2. Similarly, the person skilled in the art would know how to construct polynucleotides encoding the corresponding peptides from HHV-7 listed in table 1 and Table 2, e.g. by using the alignment disclosed in Figure 9.
Production of U20
The human herpes virus U20 protein of the present invention may be produced by any suitable method. The U20 protein composition may according to the present invention be produced by any suitable methods. Natural sources of U20 protein may be cells infected by HHV- 6A, HHV-6B or HHV-7 either in vitro or in vivo during natural infections. In addition, supernatants from HHV-6A, HHV-6B or HHV-7 in vitro infected cells or serum or plasma from individuals infected with HHV-6A, HHV-6B or HHV-7 Alternatively U20 protein may be produced recombinantly (see more details herein below in the section "recombinant production").
Purification
Purification of proteins in general involves one or more steps of removal of or separation from contaminating nucleic acids, phages and/or viruses, other proteins
and/or other biological macromolecules. The obtaining of U20 polypeptides from a composition comprising U20, such as milk or a culture medium or an extract of host cells (see herein below in the section "Recombinant production") may comprise one or more protein isolation steps. Any suitable protein isolation step may be used with the present invention. The skilled person will in general readily be able to identify useful protein isolation steps for U20 polypeptides if such are required.
The protein isolation steps useful with the present invention may be commonly used methods for protein purification including for example chromatographic methods such as for example gas chromatography, liquid chromatography, ion exchange
chromatography and/or affinity chromatography; filtration methods such as for example gel filtration and ultrafiltration; precipitation, such as ammonium sulphate precipitation and/or gradient separation such as sucrose gradient separation. Purification of U20 may comprise one or more of the aforementioned methods in any combination.
The aforementioned methods are well known to the skilled person and may for example be performed as described in the "Protein Separation Handbook Collection" including the titles "Antibody Purification", "The Recombinant Protein Handbook", "Protein Purification", "Ion Exchange Chromatography", "Affinity Chromatography", "Hydrophobic Interaction Chromatography", "Gel Filtration", "Reversed Phase
Chromatography", "Expanded Bed Adsorption" and "Chromatofocusing" prepared by Amersham Biosciences and available from GE.
In particular, purification of U20 may for example comprise one or more centrifugation steps. Said centrifugation may be employed for example for defattening purposes and/or to remove cells/cellular debris or the like and/or to separate supernatant from precipitate
In particular, purification of U20 may for example comprise one or more precipitation steps, for example precipitation using ammonium sulphate, for example at a concentration in 10 to 75%, preferably in the range of 30 to 60%, such as in the range of 40-45%. U20 will generally be present in the supernatant.
Purification of U20 may comprise one or more steps of filtration, for example filtration through a filter paper and/or filtration using another filter with a pore size of the range of
0.1 μηι to 100 μηι, for example in the range of 0.5 to 50 μηι, such as in the range of 0.5 to 20 μηι, such as in the range of 0.5-1 μηι.
Purification of U20 may comprise one or more chromatographic steps, for example any of the chromatographic methods mentioned above. In one preferred embodiment the method comprises a hydrophobic interaction chromatography.
Recombinant production
Functional equivalents of U20 are preferably produced recombinantly. Wild type U20 may in one preferred embodiment also be recombinantly produced. Useful recombinant production methods includes conventional methods known in the art, such as by expression of heterologuos U20 of functional homologues thereof in suitable host cells such as E. coli, S. cerevisiae or S. pombe or insect or mammalian cells suitable for production of recombinant proteins (see below). The skilled person will appreciate that different types of signal peptides are used in different producer cells, and that signal peptides are switched accordingly. The skilled person will also know that codon utilisation can be optimised in respect of different host cells for production.The skilled person will in general readily be able to identify useful recombinant techniques for the production of recombinant proteins in general and U20 specifically. In one embodiment U20 is produced in a transgenic plant or animal. By a transgenic plant or animal in this context is meant a plant or animal which has been genetically modified to contain and express a nucleic acid encoding human U20 or functional homolgues hereof. In a preferred embodiment of the invention, U20 or a functional homolgue thereof is produced recombinantly by host cells.
Thus, in one aspect of the present invention, U20 is produced by host cells comprising a first nucleic acid sequence encoding U20 or a functional homologue thereof operably associated with a second nucleic acid capable of directing expression in said host cells. The second nucleic acid sequence may thus comprise or even consist of a promoter that will direct the expression of protein of interest in said cells. A skilled person will be readily capable of identifying useful second nucleic acid sequence for use in a given host cell.
The process of producing recombinant U20 or a functional homologue thereof in general comprises the steps of:
-providing a host cell
-preparing a gene expression construct comprising a first nucleic acid encoding U20 or a functional homologue thereof operably linked to a second nucleic acid capble of directing expression of said protein of interest in the host cell
-transfecting/transducing the host cell with the construct,
-cultivating the host cell, thereby obtaining expression of U20 or the functional homologue thereof.
The composition comprising U20 may thus be an extract of said host cells or a composition purified from an extract of said host cells and/or from the culture medium.
The recombinant U20 thus produced may be isolated by any conventional method for example by any of the protein purification methods described herein above. The skilled person will be able to identify a suitable protein isolation steps for purifying any protein of interest.
In one embodiment of the invention, the recombinantly produced U20 or the functional homologue thereof is excreted by the host cells.
When the U20 or the functional homologue thereof is excreted the process of producing a recombinant protein of interest may comprise the following steps
-providing a host cell
-preparing a gene expression construct comprising a first nucleic acid encoding U20 or a functional homologue thereof operably linked to a second nucleic acid capable of directing expression of said U20 or functional homologue thereof in said host cell
- transfecting/transducing said host cell with the construct, -cultivating the host cell in a culture medium, thereby obtaining expression of U20 or the functional homologue thereof and secretion of the protein into the culture medium,
-thereby obtaining culture medium comprising U20 or a functional homologue thereof.
The composition comprising U20 or a functional homologue thereof may thus in this embodiment of the invention be the culture medium or a composition prepared from the culture medium.
In another embodiment of the invention said composition is an extract prepared from animals, parts thereof or cells or an isolated fraction of such an extract.
In a preferred embodiment of the invention, U20 is recombinantly produced in vitro in host cells and is isolated from cell lysate, cell extract or from tissue culture supernatant. In a more preferred embodiment U20 is produced by host cells that are modified in such a way that they express the protein of interest. In an even more preferred embodiment of the invention said host cells are transformed to produce and excrete U20.
According to the invention, the polynucleotides encoding U20 may be derived from the human U20 gene(s) or derived from the human U20 mRNA transcript(s), (see elsewhere herein). In a preferred embodiment the gene expression construct is suitable for expression in mammalian cell lines or transgenic plants or animals. In one embodiment the host cell culture is cultured in a transgene animal. By a transgenic plant or animal in this context is meant a plant or animal which has been genetically modified to contain and express a nucleic acid encoding human U20 or a functional homologue thereof as defined herein above
In a preferred embodiment the gene expression construct of the present invention comprises a viral based vector, such as a DNA viral based vector, a RNA viral based vector, or a chimeric viral based vector. Examples of DNA viruses are cytomegalo virus, Herpex Simplex, Epstein-Barr virus, Simian virus 40, Bovine papillomavirus, Adeno-associated virus, Adenovirus, Vaccinia virus, and Baculo virus. However, the gene expression construct may for example only comprise a plasmid based vector.
In one aspect the invention provides an expression construct encoding human U20 or functional homologues thereof, featured by comprising one or more intron sequences
from the human U20 gene including functional derivatives hereof. Additionally, it may contain a promoter region derived from a viral gene or a eukaryotic gene, including mammalian and insect genes. The promoter region is preferably selected to be different from the native human U20 promoter, and preferably in order to optimize the yield of human U20, the promoter region is selected to function most optimally with the vector and host cells in question.
In a preferred embodiment the promoter region is selected from a group comprising Rous sarcoma virus long terminal repeat promoter, and cytomegalovirus immediate- early promoter, and elongation factor-1 alpha promoter.
In another embodiment the promoter region is derived from a gene of a microorganism, such as other viruses, yeasts and bacteria.
In order to obtain a greater yield of recombinant U20 or functional homologue thereof, the promoter region may comprise enhancer elements, such as the QBI SP163 element of the 5' end untranslated region of the mouse vascular endothelian growth factor gene
One process for producing recombinant U20 according to the invention is characterised in that the host cell culture is may be eukaryotic, and for example a mammalian cell culture or a yeast cell culture. Useful mammalian cells may for example be human embryonal kidney cells (HEK cells), such as the cell lines deposited at the American Type Culture Collection with the numbers CRL-1573 and CRL-10852, chick embryo fibroblast, hamster ovary cells, baby hamster kidney cells, human cervical carcinoma cells, human melanoma cells, human kidney cells, human umbilical vascular endothelium cells, human brain endothelium cells, human oral cavity tumor cells, monkey kidney cells, mouse fibroblast, mouse kidney cells, mouse connective tissue cells, mouse oligodendritic cells, mouse macrophage, mouse fibroblast, mouse neuroblastoma cells, mouse pre-B cell, mouse B lymphoma cells, mouse plasmacytoma cells, mouse teratocacinoma cells, rat astrocytoma cells, rat mammary epithelium cells, COS, CHO, BHK, 293, VERO, HeLa, MDCK, WI38, HTC1 16 cells and NIH 3T3 cells.
However, the host cells may in one embodiments be either prokaryotic cells or yeast cells. Prokaryotic cells may for example be E. coli. Yeast cells may for example be Saccharomyces, Pichia or Hansenula.
When recombinantly produced U20 is used with the present invention it is preferred that said recombinantly produced U20 has a size distribution profile that is similar to naturally occurring U20. The aforementioned methods are well known to the skilled person and may for example be performed as described in the Current Protocols in Molecular Biology, 2001 , by John Wiley and Sons, Inc. edited by Frederick M. Ausubel et al.
Treatment
One aspect of the invention relates to the use of an isolated U20 polypeptides, biologically active variants or fragments thereof, polynucleotides of the invention, vectors of the invention, pharmaceutical composition, or kits of the invention for the manufacture of a medicament for the treatment of diseases related to TNF receptor activity such as autoimmune, inflammatory, degenerative or cancerous disease.
Thus, one aspect concerns the isolated polypeptide or an isolated nucleic acid sequence as defined herein or a vector of the invention ora host cell of the invention or a pharmaceutical composition of the present invention or a kit of parts of the invention or a kit of the invention for the treatment of TNF receptor related diseases.
The TNF receptor is the TNFR1 or the TNFR2, in one embodiment the TNF receptor is TNFR1 . The methods and pharmaceutical compositions of the present invention thus influence the TNF receptor activity by decreasing the TNF receptor activity in the signal transduction pathway of TNF receptors. In one embodiment of the present invention the TNF receptor activity is reduced with respect to induction of apoptosis. Apoptosis through the TNF receptor pathway is in one embodiment influenced by the action of TNF-alpha, or for example lymphotoxin (also referred to as TNF-beta) on the TNF receptor, for example TNFR1 in one particular embodiment, where the complex formation in response to binding af the TNF-alpha and/or TNF-beta to the receptor induces apoptosis.
Thus in one aspect the present invention relates to a a method for decreasing TNF receptor activity comprising
a) contacting a TNF receptor with
i) the isolated polypeptide of the present invention or ii) the isolated nucleic acid sequence of the present invention or iii) the vector of any of the present invention or
iv) the host cell of the present invention or
v) the pharmaceutical composition of the present invention b) obtaining a decrease in TNF receptor activity compared to a standard level of TNF receptor activity in an individual.
According to the methods and compositions of the present invention the TNF receptor activity is decreased by at least 10%, such as at least 15%, such as at least 25%, such as at least 30%, such as at least 35%, such as at least 40%, such as at least 45%, such as at least 50%, such as at least 55%, such as at least 60%, such as at least 70%, such as at least 75%, such as at least 80%, such as at least 85%, such as at least 90%, such as at least 95%, such as at least 96%, such as at least 97%, such as at least 98%, such as at least 99%, such as 100% of a standard TNF receptor activity in an individual.
Thus, in analogy, the present invention relates in one aspect to a pharmaceutical composition for treating, ameliorating and/or preventing TNF receptor related diseases comprising a) the isolated polypeptide of the present invention or b) the isolated nucleic acid sequence of the present invention or c) the vector of the present invention or d) the host cell of the present invention; and in another aspect to a method for treating, ameliorating and/or preventing TNF receptor related diseases comprising
administration of a) the isolated polypeptide of the invention or b) the isolated nucleic acid sequence of the invention or c) the vector of any of the invention d) the host cell of the invention e) the pharmaceutical composition of the invention or f) the kit of parts of the invention or g) the kit of the invention, in a therapeutically effective amount to an individual in need thereof.
Accordingly, the present invention in one aspect also pertains to use an isolated polypeptide of the present invention or an isolated nucleic acid sequence present invention or a vector of present invention or a host cell present invention or a
pharmaceutical composition or a kit of parts or a kit of the invention for the treatment of TNF receptor related diseases.
Thus, the methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity, for exampel in response to TNF-alpha activity on the receptor. A person skilled in the art will appreciate that the present invention can be used to treat diseases resulting from TNF-alpha activity, in particular the hTNFa.
The isolated polypeptide of the present invention, the isolated nucleic acid sequence of the present invention, the vector of any of the present invention, the host cell of the present invention, the pharmaceutical composition may be used in the prevention, amelioration and/or treatment of TNF-alpha related disease, in a cell, tissue, organ, animal, or patient including, but not limited to, at least one of an immune related disease, a cardiovascular disease, an infectious disease, a malignant disease or a neurologic disease.
The methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used for modulating or treating at least one immune related disease, in a cell, tissue, organ, animal, or patient including, but not limited to, at least one of rheumatoid arthritis, juvenile rheumatoid arthritis, systemic onset juvenile rheumatoid arthritis, psoriatic arthritis, ankylosing spondilitis, gastric ulcer, seronegative arthropathies, osteoarthritis, inflammatory bowel disease, ulcerative colitis, systemic lupus
erythematosis, antiphospholipid syndrome, iridocyclitis/uveitis/optic neuritis, idiopathic pulmonary fibrosis, systemic vasculitis/wegener's ranulomatosis, sarcoidosis, orchitis/vasectomy reversal procedures, allergic/atopic diseases, asthma, allergic rhinitis, eczema, allergic contact dermatitis, allergic conjunctivitis, hypersensitivity pneumonitis, transplants, organ transplant rejection, graft-versus-host disease, systemic inflammatory response syndrome, sepsis syndrome, gram positive sepsis, gram negative sepsis, culture negative sepsis, fungal sepsis, neutropenic fever, urosepsis, meningococcemia, trauma/hemorrhage, bums, ionizing radiation exposure, acute pancreatitis, adult respiratory distress syndrome, rheumatoid arthritis, alcohol- induced hepatitis, chronic inflammatory pathologies, sarcoidosis, Crohn's pathology, sickle cell anemia, diabetes, nephrosis, atopic diseases, hypersensitity reactions, allergic rhinitis, hay fever, perennial rhinitis, conjunctivitis, endometriosis, asthma,
urticaria, systemic anaphalaxis, dermatitis, pernicious anemia, hemolytic disesease, thrombocytopenia, graft rejection of any organ or tissue, kidney transplant rejection, heart transplant rejection, liver transplant rejection, pancreas transplant rejection, lung transplant rejection, bone marrow transplant (BMT) rejection, skin allograft rejection, cartilage transplant rejection, bone graft rejection, small bowel transplant rejection, fetal thymus implant rejection, par-athyroid transplant rejection, xenograft rejection of any organ or tissue, allograft rejection, anti-receptor hypersensitivity reactions, Graves disease, Raynoud's disease, type B insulin-resistant diabetes, asthma, myasthenia gravis, antibody-meditated cytotoxicity, type III hypersensitivity reactions, systemic lupus erythematosus, POEMS syndrome (polyneuropathy, organomegaly,
endocrinopathy, monoclonal gammopathy, and skin changes syndrome),
polyneuropathy, or-ganomegaly, endocrinopathy, monoclonal gammopathy, skin changes syndrome, antiphospholipid syndrome, pemphigus, scleroderma, mixed connective tissue disease, idiopathic Addison's disease, diabetes mellitus, chronic active hepatitis, primary billiary cirrhosis, vitiligo, vasculitis, post-MI cardiotomy syndrome, type IV hypersensitivity, contact dermatitis, hypersensitivity pneumonitis, allograft rejection, granulomas due to intracellular organisms, drug sensitivity, metabolic/idiopathic, Wilson's disease, hemachromatosis, alpha-1 -antitrypsin deficiency, diabetic retinopathy, hashimoto's thyroiditis, osteoporosis, hypothalamic- pituitary-adrenal axis evaluation, primary biliary cirrhosis, thyroiditis, encephalomyelitis, cachexia, cystic fibrosis, neonatal chronic lung disease, chronic obstructive pulmonary disease (COPD), familial hematophagocytic lymphohistiocytosis, dermatologic conditions, psoriasis, alopecia, nephrotic syndrome, nephritis, glomerular nephritis, acute renal failure, hemodialysis, uremia, toxicity, preeclampsia, okt3 therapy, anti-cd3 therapy, cytokine therapy, chemotherapy, radiation therapy (e.g., including but not limited toasthenia, anemia, cachexia, and the like), chronic salicylate intoxication, and the like. See, e.g., the Merck Manual, 12th-17th Editions, Merck & Company, Rahway, NJ (1972, 1977, 1982, 1987, 1992, 1999), Pharmacotherapy Handbook, Wells et al., eds., Second Edition, Appleton and Lange, Stamford, Conn. (1998, 2000).
In one embodiment the methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used for modulating or treating at least onejmmune related disease, in a cell, tissue, organ, animal, or patient including, such as, at least one of
rheumatoid arthritis, juvenile rheumatoid arthritis,systemic onset juvenile rheumatoid arthritis, psoriatic arthritits, Crohn's disease and other inflammatory bowel disease.
In one embodiment the methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used for modulating or treating at least onejmmune related disease, in a cell, tissue, organ, animal, or patient including, such as rheumatoid arthritis.
In another embodiment the methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used for modulating or treating at least onejmmune related disease, in a cell, tissue, organ, animal, or patient including, such as juvenile rheumatoid arthritis. In another embodiment the methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used for modulating or treating at least onejmmune related disease, in a cell, tissue, organ, animal, or patient including, such as systemic onset juvenile rheumatoid arthritis.
In yet another embodiment the methods, medicaments, composition and
pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used for modulating or treating at least one immune related disease, in a cell, tissue, organ, animal, or patient including, such as psoriatic arthritits.
In a further embodiment the methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used for modulating or treating at least onejmmune related disease, in a cell, tissue, organ, animal, or patient including, such as Crohn's disease.
The methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used in the prevention, amelioration and/or treatment of at least one cardiovascular disease in a cell, tissue, organ, animal, or patient, including, but not limited to, at least
one of cardiac stun syndrome, myocardial infarction, congestive heart failure, stroke, ischemic stroke, hemorrhage, arteriosclerosis, atherosclerosis, restenosis, diabetic ateriosclerotic disease, hypertension, arterial hypertension, renovascular hypertension, syncope, shock, syphilis of the cardiovascular system, heart failure, cor pulmonale, primary pulmonary hypertension, cardiacarrhythmias, atrial ectopic beats, atrial flutter, atrial fibrillation (sustained or paroxysmal), post perfusion syndrome, cardiopulmonary bypass inflammation response, chaotic or multifocal atrial tachycardia, regular narrow QRS tachycardia, specific arrythmias, ventricular fibrillation, His bundle arrythmias, atrioventricular block, bundle branch block, myocardial ischemic disorders, coronary artery disease, angina pectoris, myocardial infarction, cardiomyopathy, dilated congestive cardiomyopathy, restrictive cardiomyopathy, valvular heart diseases, endocarditis, pericardial disease, cardiac tumors, aordic and peripheral aneuryisms, aortic dissection, inflammation of the aorta, occulsion of the abdominal aorta and its branches, peripheral vascular disorders, occulsive arterial disorders, peripheral atherosclerotic disease, thromboangitis obliterans, functional peripheral arterial disorders, Raynaud's phenomenon and disease, acrocyanosis, erythromelalgia, venous diseases, venous thrombosis, varicose veins, arteriovenous fistula,
lymphederma, lipedema, unstable angina, reperfusion injury, post pump syndrome, ischemia-reperfusion injury, and the like.
The methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used in the prevention, amelioration and/or treatment of at least one infectious disease in a cell, tissue, organ, animal or patient, including, but not limited to, at least one of: acute or chronic bacterial infection, acute and chronic parasitic or infectious processes, including bacterial, viral and fungal infections, HIV infection/HIV neuropathy, meningitis, hepatitis (A,B or C, or the like), septic arthritis, peritonitis, pneumonia, epiglottitis, e. coli 0157:h7, hemolytic uremic syndrome/thrombolytic thrombocytopenic purpura, malaria, dengue hemorrhagic fever, leishmaniasis, leprosy, toxic shock syndrome, streptococcal myositis, gas gangrene, mycobacterium tuberculosis, mycobacterium avium
intracellular, Pneumocystis carinii pneumonia, pelvic inflammatory disease, orchitis/epidydimitis, legionella, lyme disease, influenza a, epstein-barr virus, vital- associated hemaphagocytic syndrome, vital encephalitis/aseptic meningitis, and the like;
The methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used in the prevention, amelioration and/or treatment of at least one malignant disease in a cell, tissue, organ, animal or patient, including, but not limited to, at least one of: leukemia, acute leukemia, acute lymphoblastic leukemia (ALL), B-cell, T-cell or FAB ALL, acute myeloid leukemia (AML), chromic myelocytic leukemia (CML), chronic lymphocytic leukemia (CLL), hairy cell leukemia, myelodyplastic syndrome (MDS), a lymphoma, Hodgkin's disease, a malignant lymphoma, non-hodgkin's lymphoma, Burkitt's lymphoma, multiple myeloma, Kaposi's sarcoma, colorectal carcinoma, pancreatic carcinoma, nasopharyngeal carcinoma, malignant histiocytosis,
paraneoplastic syndrome/hypercalcemia of malignancy, solid tumors,
adenocarcinomas, sarcomas, malignant melanoma, hemangioma, metastatic disease, cancer related bone resorption, cancer related bone pain, and the like. The methods, medicaments, composition and pharmaceutical compositions of the present invention offer a novel method for decreasing the TNF receptor activity can be used in the prevention, amelioration and/or treatment of at least one neurologic disease in a cell, tissue, organ, animal or patient, including, but not limited to, at least one of: neurodegenerative diseases, multiple sclerosis, migraine headache, AIDS dementia complex, demyelinating diseases, such as multiple sclerosis and acute transverse myelitis; extrapyramidal and cerebellar disorders' such as lesions of the corticospinal system; disorders of the basal ganglia or cerebellar disorders; hyperkinetic movement disorders such as Huntington's Chorea and senile chorea; drug-induced movement disorders, such as those induced by drugs which block CNS dopamine receptors; hypokinetic movement disorders, such as Parkinson's disease; progressive
supranucleo Palsy; structural lesions of the cerebellum; spinocerebellar degenerations, such as spinal ataxia, Friedreich's ataxia, cerebellar cortical degenerations, multiple systems degenerations (Mencel, Dejerine-Thomas, Shi-Drager, and Machado- Joseph); systemic disorders (Refsum's disease, abetalipoprotemia, ataxia, telangiectasia, and mitochondrial multi system disorder); demyelinating core disorders, such as multiple sclerosis, acute transverse myelitis; and disorders of the motor unit' such as
neurogenic muscular atrophies (anterior horn cell degeneration, such as amyotrophic lateral sclerosis, infantile spinal muscularatrophy and juvenile spinal muscular atrophy); Alzheimer's disease; Down's Syndrome in middle age; Diffuse Lewy body disease; Senile Dementia of Lewy body type; Wernicke-Korsakoff syndrome; chronic
alcoholism; Creutzfeldt-Jakob disease; Subacute sclerosing panencephalitis,
Hallerrorden-Spatz disease; and Dementia pugilistica, and the like. (Merck Manual, 16th Edition, Merck & Company, Rahway, NJ (1992)
Pharmaceutical composition
The polypeptides, nuclaic acid molecules, host cell, vectors of the present invention offering a novel method for decreasing the TNF receptor activity can be used in one aspect relating to a pharmaceutical composition comprising
a) the isolated polypeptide of the invention or
b) the isolated nucleic acid sequence of the invention or
c) the vector of the invention or
d) the host cell of the invention
and a pharmaceutical acceptable carrier and/or diluent.
Pharmaceutical formulation
Pharmaceutical compositions containing the polypeptides, polynucleotides, vectors, host cell, kit, or kit of parts of the present invention may be prepared by conventional techniques, e.g. as described in Remington: The Science and Practice of Pharmacy 1995, edited by E. W. Martin, Mack Publishing Company, 19th edition, Easton, Pa. The compositions may appear in conventional forms, for example capsules, tablets, aerosols, solutions, suspensions or topical applications.
The terms "medicament" and "pharmaceutical compostions" are used interchangeably herein.
The present invention provides pharmaceutical compositions for treating, ameliorating and/or preventing TNF receptor related diseases comprising a) the isolated polypeptide of the invention or b) the isolated polynucleotide of the invention or c) the vector of the present invention or d) the host cell of the invention or e) the kit of the invention or f) the kit of parts of the invention.
In one aspect the present invention relates to a pharmaceutical composition. The pharmaceutical composition may be formulated in a number of different manners, depending on the purpose for the particular pharmaceutical composition.
For example the pharmaceutical composition may be formulated in a manner so it is useful for a particular administration form. Preferred administration forms are described herein below. In one embodiment the pharmaceutical composition is formulated so it is a liquid. For example the composition may be a protein solution or the composition may be a protein suspension. Said liquid may be suitable for parenteral administration, for example for injection or infusion. The liquid may be any useful liquid, however it is frequently preferred that the liquid is an aqueous liquid. For many purposes, in particular when the liquid should be used for parenteral administration, it is furthermore preferred that the liquid is sterile. Sterility may be conferred by any conventional method, for example filtration, irradiation or heating. Furthermore, it is preferred that the liquid has been subjected to a virus reduction step, in particular if the liquid is formulated for parenteral administration.
Virus reduction may for example be performed by nanofiltration or virus filtering over a suitable filter, such as a Planova filter consisting of several layers. The Planova filter may be any suitable size for example 75N, 35N, 20N or 15N or filters of different size may be used, for example Planova 20N. Virus reduction may also comprise a step of prefiltering with another filter, for example using a filter with a pore size of the the range of 0.01 to 1 μηι, such as in the range of 0.05 to 0.5 μηι, for example around 0.1 μηι. Virus reductions may also include an acidic treatment step. The pharmaceutical composition may be packaged in single dosage units, which may be more convenient for the user. Hence, pharmaceutical compositions for bolus injections may be packages in dosage units of for example at the most 10 ml, preferably at the most 8 ml, more preferably at the most 6 ml, such as at the most 5 ml, for example at the most 4 ml, such as at the most 3 ml, for example around 2 ml.
The pharmaceutical composition may be packaged in any suitable container. In one example a single dosage of the pharmaceutical composition may be packaged in injection syringes or in a container useful for infusion.
In another embodiment of the present invention the pharmaceutical composition is a dry composition. The dry composition may be used as such, but for most purposes the composition is a dry composition for storage only. Prior to use the dry composition may be dissolved or suspended in a suitable liquid composition, for example sterile water.
The pharmaceutical composition according to the present invention may also comprise a first nucleic acid sequence encoding HHV 6B (SEQ ID NO.:2) or alternatively encoding HHV 6A or HHV 7, SEQ ID NO.: 4 and 6, respectively, or SEQ ID NO: 7, 8, or 9, or fragments, functional equivalents, variants, or complementary sequences thereof. Said first nucleic acid sequence is preferably operably associated with a second nucleic acid sequence directing expression of the first nucleic acid in the individual to be treated with the pharmaceutical composition, more preferably in the cells of said individual, which are diseased. Thus it is preferred that the second nucleic acid sequence is capable of directing expression of the first nucleic acid sequence in a human being. In embodiments of the invention wherein the clinical condition is cancer, it is preferred that the second nucleic acid sequence is capable of directing expression of the first nucleic acid sequence in cancer cells, such as malignant cells. It is furthermore preferred that the first and the second nucleic acid sequences are included in a suitable vector.
It is also comprised within the invention that the pharmaceutical composition may be applied topically to the site of the site, for example in the form of a lotion, a creme, an ointment, a spray, such as an aerosol spray or a nasal spray, rectal or vaginal suppositories, drops, such as eye drops or nasal drops, a patch, an occlusive dressing or the like.
Pharmaceutically acceptable additives
The pharmaceutical compositions may be prepared by any conventional technique, e.g. as described in Remington: The Science and Practice of Pharmacy 1995, edited by E. W. Martin, Mack Publishing Company, 19th edition, Easton, Pa.
The pharmaceutically acceptable additives may be any conventionally used pharmaceutically acceptable additive, which should be selected according to the specific formulation, intended administration route etc. For example the pharmaceutically acceptable additives may be any of the additives mentioned in Nema et al, 1997.
Furthermore, the pharmaceutically acceptable additive may be any accepted additive from FDA's "inactive ingredients list", which for example is available on the internet address http://www.fda.gov/cder/drug/iig/default.htm. In some embodiments of the present invention it is desirable that the pharmaceutical composition comprises an isotonic agent. In particular when the pharmaceutical composition is prepared for administration by injection or infusion it is often desirable that an isotonic agent is added. Accordingly, the composition may comprise at least one pharmaceutically acceptable additive which is an isotonic agent.
The pharmaceutical composition may be isotonic, hypotonic or hypertonic. However it is often preferred that a pharmaceutical composition for infusion or injection is essentially isotonic, when it is administrated. Hence, for storage the pharmaceutical composition may preferably be isotonic or hypertonic. If the pharmaceutical composition is hypertonic for storage, it may be diluted to become an isotonic solution prior to administration. The isotonic agent may be an ionic isotonic agent such as a salt or a non-ionic isotonic agent such as a carbohydrate.
Examples of ionic isotonic agents include but are not limited to NaCI, CaCI2, KG and MgCI2. Examples of non-ionic isotonic agents include but are not limited to mannitol and glycerol.
However, in other embodiments of the invention the pharmaceutical composition may comprise no buffer at all or only micromolar amounts of buffer. In a preferred embodiment the buffer is TRIS. TRIS buffer is known under various other names for example tromethamine including tromethamine USP, THAM, Trizma, Trisamine, Tris amino and trometamol. The designation TRIS covers all the
aforementioned designations.
The buffer may furthermore for example be selected from USP compatible buffers for parenteral use, in particular, when the pharmaceutical formulation is for parenteral use. For example the buffer may be selected from the group consisting of monobasic acids such as acetic, benzoic, gluconic, glyceric and lactic, dibasic acids such as aconitic, adipic, ascorbic, carbonic, glutamic, malic, succinic and tartaric, polybasic acids such as citric and phosphoric and bases such as ammonia, diethanolamine, glycine, triethanolamine, and TRIS.
The pharmaceutical compositions may comprise at least one pharmaceutically acceptable additive which is a stabiliser.
For example the stabiliser may be selected from the group consisting of poloxamers, Tween-20, Tween-40, Tween-60, Tween-80, Brij, metal ions, amino acids, polyethylene glycol, Triton, EDTA, ascorbic acid, Triton X-100, NP40 or CHAPS.
The pharmaceutical composition according to the invention may also comprise one or more cryoprotectant agents. In particular, when the composition comprises freeze-dried protein or the composition should be stored frozen, it may be desirable to add a cryoprotecting agent to the pharmaceutical composition.
The cryoprotectant agent may be any useful cryoprotectant agent, for example the cryoprotectant agent may be selected from the group consisting of dextran, glycerin, polyethylenglycol, sucrose, trehalose and mannitol. Accordingly, the pharmaceutically acceptable additives may comprise one or more selected from the group consisting of isotonic salt, hypertonic salt, buffer and stabilisers. Furthermore, the pharmaceutically acceptable additives may comprise one or more selected from the group consisting of isotonic agents, buffer, stabilisers and cryoprotectant agents. For example, the pharmaceutically acceptable additives comprise glucosemonohydrate, glycine, NaCI and polyethyleneglycol 3350.
Formulations
Whilst it is possible for the composition of the present invention to be administered as the raw composition, it is preferred to present it in the form of a pharmaceutical formulation. Accordingly, the present invention further provides a pharmaceutical
formulation, for medicinal application, which comprises a composition of the present invention or a pharmaceutically acceptable salt thereof, as herein defined, and a pharmaceutically acceptable carrier therefore. Oral administration
The compositions of the present invention may be formulated in a wide variety of oral administration dosage forms. The pharmaceutical compositions and dosage forms may comprise the compositions of the invention or its pharmaceutically acceptable salt or a crystal form thereof as the active component. The pharmaceutically acceptable carriers can be either solid or liquid. Solid form preparations include powders, tablets, pills, capsules, cachets, suppositories, and dispersible granules. A solid carrier can be one or more substances which may also act as diluents, flavoring agents, solubilizers, lubricants, suspending agents, binders, preservatives, wetting agents, tablet disintegrating agents, or an encapsulating material.
Preferably, the composition will be about 0.5% to 75% by weight of a composition or compositions of the invention, with the remainder consisting of suitable pharmaceutical excipients. For oral administration, such excipients include pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, sodium saccharine, talcum, cellulose, glucose, gelatin, sucrose, magnesium carbonate, and the like.
In powders, the carrier is a finely divided solid which is a mixture with the finely divided active component. In tablets, the active component is mixed with the carrier having the necessary binding capacity in suitable proportions and compacted in the shape and size desired. The powders and tablets preferably contain from 1 to about 70 %t of the active composition. Suitable carriers are magnesium carbonate, magnesium stearate, talc, sugar, lactose, pectin, dextrin, starch, gelatin, tragacanth, methylcellulose, sodium carboxymethylcellulose, a low melting wax, cocoa butter, and the like. The term "preparation" is intended to include the formulation of the active composition with encapsulating material as carrier providing a capsule in which the active component, with or without carriers, is surrounded by a carrier, which is in association with it.
Similarly, cachets and lozenges are included. Tablets, powders, capsules, pills, cachets, and lozenges can be as solid forms suitable for oral administration. . Multiple- unit-dosage granules can be prepared as well. Tablets and granules of the above cores can be coated with concentrated solutions of sugar, etc. The cores can also be coated
with polymers which change the dissolution rate in the gastrointestinal tract, such as anionic polymers having a pka of above 5.5. Such polymers are hydroxypropylmethyl cellulose phthalate, cellulose acetate phthalate, and polymers sold under the trade mark Eudragit S100 andU OO. In preparation of gelatine capsules these can be soft or hard. In the former case the active compound is mixed with oil, and in the latter case the multiple-unit-dosage granules are filled therein.
Drops according to the present invention may comprise sterile or non-sterile aqueous or oil solutions or suspensions, and may be prepared by dissolving the active ingredient in a suitable aqueous solution, optionally including a bactericidal and/or fungicidal agent and/or any other suitable preservative, and optionally including a surface active agent. The resulting solution may then be clarified by filtration, transferred to a suitable container which is then sealed and sterilized by autoclaving or maintaining at 98-100 degree C for half an hour. Alternatively, the solution may be sterilized by filtration and transferred to the container aseptically. Examples of bactericidal and fungicidal agents suitable for inclusion in the drops are phenylmercuric nitrate or acetate (0.002%), benzalkonium chloride (0.01 %) and chlorhexidine acetate (0.01 %). Suitable solvents for the preparation of an oily solution include glycerol, diluted alcohol and propylene glycol.
Also included are solid form preparations which are intended to be converted, shortly before use, to liquid form preparations for oral administration. Such liquid forms include solutions, suspensions, and emulsions. These preparations may contain, in addition to the active component, colorants, flavours, stabilizers, buffers, artificial and natural sweeteners, dispersants, thickeners, solubilising agents, and the like.
Other forms suitable for oral administration include liquid form preparations including emulsions, syrups, elixirs, aqueous solutions, aqueous suspensions, toothpaste, gel dentrifrice, chewing gum, or solid form preparations which are intended to be converted shortly before use to liquid form preparations. Emulsions may be prepared in solutions in aqueous propylene glycol solutions or may contain emulsifying agents such as lecithin, sorbitan monooleate, or acacia. Aqueous solutions can be prepared by dissolving the active component in water and adding suitable colorants, flavours, stabilizing and thickening agents. Aqueous suspensions can be prepared by dispersing the finely divided active component in water with viscous material, such as natural or
synthetic gums, resins, methylcellulose, sodium carboxymethylcellulose, and other well known suspending agents. Solid form preparations include solutions, suspensions, and emulsions, and may contain, in addition to the active component, colorants, flavours, stabilizers, buffers, artificial and natural sweeteners, dispersants, thickeners, solubilising agents, and the like.
Parenteral administration
The compositions of the present invention may be formulated for parenteral administration (e.g., by injection, for example bolus injection or continuous infusion) and may be presented in unit dose form in ampoules, pre-filled syringes, small volume infusion or in multi-dose containers with an added preservative. The compositions may take such forms as suspensions, solutions, or emulsions in oily or aqueous vehicles, for example solutions in aqueous polyethylene glycol. Examples of oily or nonaqueous carriers, diluents, solvents or vehicles include propylene glycol, polyethylene glycol, vegetable oils (e.g., olive oil), and injectable organic esters (e.g., ethyl oleate), and may contain formulatory agents such as preserving, wetting, emulsifying or suspending, stabilizing and/or dispersing agents. Alternatively, the active ingredient may be in powder form, obtained by aseptic isolation of sterile solid or by lyophilisation from solution for constitution before use with a suitable vehicle, e.g., sterile, pyrogen-free water.
Oils useful in parenteral formulations include petroleum, animal, vegetable, or synthetic oils. Specific examples of oils useful in such formulations include peanut, soybean, sesame, cottonseed, corn, olive, petrolatum, and mineral. Suitable fatty acids for use in parenteral formulations include oleic acid, stearic acid, and isostearic acid. Ethyl oleate and isopropyl myristate are examples of suitable fatty acid esters.
Suitable soaps for use in parenteral formulations include fatty alkali metal, ammonium, and triethanolamine salts, and suitable detergents include (a) cationic detergents such as, for example, dimethyl dialkyl ammonium halides, and alkyl pyridinium halides; (b) anionic detergents such as, for example, alkyl, aryl, and olefin sulfonates, alkyl, olefin, ether, and monoglyceride sulfates, and sulfosuccinates, (c) nonionic detergents such as, for example, fatty amine oxides, fatty acid alkanolamides, and
polyoxyethylenepolypropylene copolymers, (d) amphoteric detergents such as, for
example, alkyl-.beta.-aminopropionates, and 2-alkyl-imidazoline quaternary ammonium salts, and (e) mixtures thereof.
The parenteral formulations typically will contain from about 0.5 to about 25% by weight of the active ingredient in solution. Preservatives and buffers may be used. In order to minimize or eliminate irritation at the site of injection, such compositions may contain one or more nonionic surfactants having a hydrophile-lipophile balance (HLB) of from about 12 to about 17. The quantity of surfactant in such formulations will typically range from about 5to about 15% by weight. Suitable surfactants include polyethylene sorbitan fatty acid esters, such as sorbitan monooleate and the high molecular weight adducts of ethylene oxide with a hydrophobic base, formed by the condensation of propylene oxide with propylene glycol. The parenteral formulations can be presented in unit-dose or multi-dose sealed containers, such as ampules and vials, and can be stored in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid excipient, for example, water, for injections, immediately prior to use. Extemporaneous injection solutions and suspensions can be prepared from sterile powders, granules, and tablets of the kind previously described.
Topical administration
The compositions of the invention can also be delivered topically. Regions for topical administration include the skin surface and also mucous membrane tissues of the vagina, rectum, nose, mouth, and throat. Compositions for topical administration via the skin and mucous membranes should not give rise to signs of irritation, such as swelling or redness.
The topical composition may include a pharmaceutically acceptable carrier adapted for topical administration. Thus, the composition may take the form of a suspension, solution, ointment, lotion, sexual lubricant, cream, foam, aerosol, spray, suppository, implant, inhalant, tablet, capsule, dry powder, syrup, balm or lozenge, for example. Methods for preparing such compositions are well known in the pharmaceutical industry.
The compositions of the present invention may be formulated for topical administration to the epidermis as ointments, creams or lotions, or as a transdermal patch. Ointments and creams may, for example, be formulated with an aqueous or oily base with the
addition of suitable thickening and/or gelling agents. Lotions may be formulated with an aqueous or oily base and will in general also containing one or more emulsifying agents, stabilizing agents, dispersing agents, suspending agents, thickening agents, or coloring agents. Formulations suitable for topical administration in the mouth include lozenges comprising active agents in a flavored base, usually sucrose and acacia or tragacanth; pastilles comprising the active ingredient in an inert base such as gelatin and glycerin or sucrose and acacia; and mouthwashes comprising the active ingredient in a suitable liquid carrier. Creams, ointments or pastes according to the present invention are semi-solid formulations of the active ingredient for external application. They may be made by mixing the active ingredient in finely-divided or powdered form, alone or in solution or suspension in an aqueous or non-aqueous fluid, with the aid of suitable machinery, with a greasy or non-greasy base. The base may comprise hydrocarbons such as hard, soft or liquid paraffin, glycerol, beeswax, a metallic soap; a mucilage; an oil of natural origin such as almond, corn, arachis, castor or olive oil; wool fat or its derivatives or a fatty acid such as steric or oleic acid together with an alcohol such as propylene glycol or a macrogel. The formulation may incorporate any suitable surface active agent such as an anionic, cationic or non-ionic surfactant such as a sorbitan ester or a
polyoxyethylene derivative thereof. Suspending agents such as natural gums, cellulose derivatives or inorganic materials such as silicaceous silicas, and other ingredients such as lanolin, may also be included.
Lotions according to the present invention include those suitable for application to the skin or eye. An eye lotion may comprise a sterile aqueous solution optionally containing a bactericide and may be prepared by methods similar to those for the preparation of drops. Lotions or liniments for application to the skin may also include an agent to hasten drying and to cool the skin, such as an alcohol or acetone, and/or a moisturizer such as glycerol or an oil such as castor oil or arachis oil.
Transdermal Delivery
The pharmaceutical agent-chemical modifier complexes described herein can be administered transdermally. Transdermal administration typically involves the delivery of a pharmaceutical agent for percutaneous passage of the drug into the systemic circulation of the patient. The skin sites include anatomic regions for transdermally
administering the drug and include the forearm, abdomen, chest, back, buttock, mastoidal area, and the like.
Transdermal delivery is accomplished by exposing a source of the complex to a patient's skin for an extended period of time. Transdermal patches have the added advantage of providing controlled delivery of a pharmaceutical agent-chemical modifier complex to the body. See Transdermal Drug Delivery: Developmental Issues and Research Initiatives, Hadgraft and Guy (eds.), Marcel Dekker, Inc., (1989); Controlled Drug Delivery: Fundamentals and Applications, Robinson and Lee (eds.), Marcel Dekker Inc., (1987); and Transdermal Delivery of Drugs, Vols. 1 -3, Kydonieus and Berner (eds.), CRC Press, (1987). Such dosage forms can be made by dissolving, dispersing, or otherwise incorporating the pharmaceutical agent-chemical modifier complex in a proper medium, such as an elastomeric matrix material. Absorption enhancers can also be used to increase the flux of the compound across the skin. The rate of such flux can be controlled by either providing a rate-controlling membrane or dispersing the compound in a polymer matrix or gel.
Passive Transdermal Drug Delivery
A variety of types of transdermal patches will find use in the methods described herein. For example, a simple adhesive patch can be prepared from a backing material and an acrylate adhesive. The pharmaceutical agent-chemical modifier complex and any enhancer are formulated into the adhesive casting solution and allowed to mix thoroughly. The solution is cast directly onto the backing material and the casting solvent is evaporated in an oven, leaving an adhesive film. The release liner can be attached to complete the system.
Alternatively, a polyurethane matrix patch can be employed to deliver the
pharmaceutical agent-chemical modifier complex. The layers of this patch comprise a backing, a polyurethane drug/enhancer matrix, a membrane, an adhesive, and a release liner. The polyurethane matrix is prepared using a room temperature curing polyurethane prepolymer. Addition of water, alcohol, and complex to the prepolymer results in the formation of a tacky firm elastomer that can be directly cast only the backing material.
A further embodiment of this invention will utilize a hydrogel matrix patch. Typically, the hydrogel matrix will comprise alcohol, water, drug, and several hydrophilic polymers. This hydrogel matrix can be incorporated into a transdermal patch between the backing and the adhesive layer.
The liquid reservoir patch will also find use in the methods described herein. This patch comprises an impermeable or semipermeable, heat sealable backing material, a heat sealable membrane, an acrylate based pressure sensitive skin adhesive, and a siliconized release liner. The backing is heat sealed to the membrane to form a reservoir which can then be filled with a solution of the complex, enhancers, gelling agent, and other excipients.
Foam matrix patches are similar in design and components to the liquid reservoir system, except that the gelled pharmaceutical agent-chemical modifier solution is constrained in a thin foam layer, typically a polyurethane. This foam layer is situated between the backing and the membrane which have been heat sealed at the periphery of the patch.
For passive delivery systems, the rate of release is typically controlled by a membrane placed between the reservoir and the skin, by diffusion from a monolithic device, or by the skin itself serving as a rate-controlling barrier in the delivery system. See U.S. Pat. Nos. 4,816,258; 4,927,408; 4,904,475; 4,588,580, 4,788,062; and the like. The rate of drug delivery will be dependent, in part, upon the nature of the membrane. For example, the rate of drug delivery across membranes within the body is generally higher than across dermal barriers. The rate at which the complex is delivered from the device to the membrane is most advantageously controlled by the use of rate-limiting membranes which are placed between the reservoir and the skin. Assuming that the skin is sufficiently permeable to the complex (i.e., absorption through the skin is greater than the rate of passage through the membrane), the membrane will serve to control the dosage rate experienced by the patient.
Suitable permeable membrane materials may be selected based on the desired degree of permeability, the nature of the complex, and the mechanical considerations related to constructing the device. Exemplary permeable membrane materials include a wide variety of natural and synthetic polymers, such as polydimethylsiloxanes (silicone
rubbers), ethylenevinylacetate copolymer (EVA), polyurethanes, polyurethane- polyether copolymers, polyethylenes, polyamides, polyvinylchlorides (PVC), polypropylenes, polycarbonates, polytetrafluoroethylenes (PTFE), cellulosic materials, e.g., cellulose triacetate and cellulose nitrate/acetate, and hydrogels, e.g., 2- hydroxyethylmethacrylate (HEMA).
Other items may be contained in the device, such as other conventional components of therapeutic products, depending upon the desired device characteristics. For example, the compositions according to this invention may also include one or more
preservatives or bacteriostatic agents, e.g., methyl hydroxybenzoate, propyl hydroxybenzoate, chlorocresol, benzalkonium chlorides, and the like. These pharmaceutical compositions also can contain other active ingredients such as antimicrobial agents, particularly antibiotics, anesthetics, analgesics, and antipruritic agents.
Administration as suppositories
The compositions of the present invention may be formulated for administration as suppositories. A low melting wax, such as a mixture of fatty acid glycerides or cocoa butter is first melted and the active component is dispersed homogeneously, for example, by stirring. The molten homogeneous mixture is then poured into convenient sized molds, allowed to cool, and to solidify.
The active composition may be formulated into a suppository comprising, for example, about 0.5% to about 50% of a composition of the invention, disposed in a polyethylene glycol (PEG) carrier (e.g., PEG 1000 [96%] and PEG 4000 [4%].
The compositions of the present invention may be formulated for vaginal
administration. Pessaries, tampons, creams, gels, pastes, foams or sprays containing in addition to the active ingredient such carriers as are known in the art to be appropriate.
Respiratory tract administration
The compositions of the present invention may be formulated for nasal administration. The solutions or suspensions are applied directly to the nasal cavity by conventional means, for example with a dropper, pipette or spray. The formulations may be provided
in a single or multidose form. In the latter case of a dropper or pipette this may be achieved by the patient administering an appropriate, predetermined volume of the solution or suspension. In the case of a spray this may be achieved for example by means of a metering atomizing spray pump.
The compositions of the present invention may be formulated for aerosol
administration, particularly to the respiratory tract and including intranasal
administration. The composition will generally have a small particle size for example of the order of 5 microns or less. Such a particle size may be obtained by means known in the art, for example by micronization. The active ingredient is provided in a pressurized pack with a suitable propellant such as a chlorofluorocarbon (CFC) for example dichlorodifluoromethane, trichlorofluoromethane, or dichlorotetrafluoroethane, carbon dioxide or other suitable gas. The aerosol may conveniently also contain a surfactant such as lecithin. The dose of drug may be controlled by a metered valve. Alternatively the active ingredients may be provided in a form of a dry powder, for example a powder mix of the composition in a suitable powder base such as lactose, starch, starch derivatives such as hydroxypropylmethyl cellulose and
polyvinylpyrrolidine (PVP). The powder carrier will form a gel in the nasal cavity. The powder composition may be presented in unit dose form for example in capsules or cartridges of e.g., gelatin or blister packs from which the powder may be administered by means of an inhaler.
When desired, formulations can be prepared with enteric coatings adapted for sustained or controlled release administration of the active ingredient.
The pharmaceutical preparations are preferably in unit dosage forms. In such form, the preparation is subdivided into unit doses containing appropriate quantities of the active component. The unit dosage form can be a packaged preparation, the package containing discrete quantities of preparation, such as packeted tablets, capsules, and powders in vials or ampoules. Also, the unit dosage form can be a capsule, tablet, cachet, or lozenge itself, or it can be the appropriate number of any of these in packaged form.
Pharmaceutically acceptable salts
Pharmaceutically acceptable salts of the instant compounds, where they can be prepared, are also intended to be covered by this invention. These salts will be ones which are acceptable in their application to a pharmaceutical use. By that it is meant that the salt will retain the biological activity of the parent compound and the salt will not have untoward or deleterious effects in its application and use in treating diseases.
Pharmaceutically acceptable salts are prepared in a standard manner. If the parent compound is a base it is treated with an excess of an organic or inorganic acid in a suitable solvent. If the parent compound is an acid, it is treated with an inorganic or organic base in a suitable solvent.
The compounds of the invention may be administered in the form of an alkali metal or earth alkali metal salt thereof, concurrently, simultaneously, or together with a pharmaceutically acceptable carrier or diluent, especially and preferably in the form of a pharmaceutical composition thereof, whether by oral, rectal, or parenteral (including subcutaneous) route, in an effective amount.
Examples of pharmaceutically acceptable acid addition salts for use in the present inventive pharmaceutical composition include those derived from mineral acids, such as hydrochloric, hydrobromic, phosphoric, metaphosphoric, nitric and sulfuric acids, and organic acids, such as tartaric, acetic, citric, malic, lactic, fumaric, benzoic, glycolic, gluconic, succinic, p-toluenesulphonic acids, and arylsulphonic, for example. Administration forms
The pharmaceutical composition may be prepared so it is suitable for one or more particular administration methods. Furthermore, the method of treatment described herein may involve different administration methods. In general any administration method, wherein at least one isolated U20 encoding polynucleotide, transcriptional product and/or polypeptide thereof, functional equivalent thereof, variants or fragments thereof may be administered to an individual in a manner so that active an isolated U20 encoding polynucleotide, transcriptional product and/or polypeptide thereof, functional equivalent thereof, variants or fragments thereof may reach the site of disease may be employed with the present invention.
The main routes of drug delivery, in the treatment method are intravenous, oral, and topical, as will be described below. Other drug-administration methods, such as subcutaneous injection or via inhalation, which are effective to deliver the drug to a target site or to introduce the drug into the bloodstream, are also contemplated.
The mucosal membrane to which the pharmaceutical preparation of the invention is administered may be any mucosal membrane of the mammal to which the biologically active substance is to be given, e.g. in the nose, vagina, eye, mouth, genital tract, lungs, gastrointestinal tract, or rectum, preferably the mucosa of the nose, mouth or vagina.
Compositions of the invention may be administered parenterally, that is by intravenous, intramuscular, subcutaneous intranasal, intrarectal, intravaginal or intraperitoneal administration. The subcutaneous and intramuscular forms of parenteral administration are generally preferred. Appropriate dosage forms for such administration may be prepared by conventional techniques. The compositions may also be administered by inhalation, that is by intranasal and oral inhalation administration. Appropriate dosage forms for such administration, such as an aerosol formulation or a metered dose inhaler, may be prepared by conventional techniques.
The compositions according to the invention may be administered with at least one other compound. The compounds may be administered simultaneously, either as separate formulations or combined in a unit dosage form, or administered sequentially.
Combination treatment
It may be advantageously to add additional therapeutics to the pharmaceutical composition or to administer one or more therapeutics. Thus, in one embodiment the invention relates to a pharmaceutical composition according to the invention, further comprising at least one additional therapeutic agent. In analogy, at least one additional therapeutic agent may be used in the methods for treating TNF receptor related diseases.
The isolated polypeptide of the present invention, the isolated nucleic acid sequence of the present invention, the vector of the present invention, the host cell of the present invention may be used in a pharmaceutical composition or in a method comprising
administering an effective amount to a cell, tissue, organ, animal or patient in need. Such a pharmaceutical composition or method can optionally further comprise coadministration or combination therapy for treating such immune diseases, wherein the administering of said composition further comprises administering, before concurrently, and/or after, at least one selected from at least one TNF antagonist (e.g., but not limited to a TNF antibody or fragment, a soluble TNF receptor or fragment, fusion proteins thereof, or a small molecule TNF antagonist), an antirheumatic (e.g., methotrexate, auranofin, aurothioglucose, azathioprine, etanercept, gold sodium thiomalate, hydroxychloroquine sulfate, leflunomide, sulfasalzine), a muscle relaxant, a narcotic, a non-steroid anti-inflammatory drug (NSAID), an analgesic, an anesthetic, a sedative, a local anethetic, a neuromuscular blocker, an antimicrobial (e.g.,
aminoglycoside, an antifungal, an antiparasitic, an antiviral, a carbapenem,
cephalosporin, a flurorquinolone, a macrolide, a penicillin, a sulfonamide, a
tetracycline, another antimicrobial), an antipsoriatic, a corticosteriod, an anabolic steroid, a diabetes related agent, a mineral, a nutritional, a thyroid agent, a vitamin, a calcium related hormone, an antidiarrheal, an antitussive, an antiemetic, an antiulcer, a laxative, an anticoagulant, an erythropieitin (e.g. epoetin alpha), a filgrastim (e.g. G- CSF, Neupogen), a sargramostim (GM-CSF, Leukine), an immunization, an
immunoglobulin, an immunosuppressive (e.g., basiliximab, cyclosporine, daclizumab), a growth hormone, a hormone replacement drug, an estrogen receptor modulator, a mydriatic, a cycloplegic, an alkylating agent, an antimetabolite, a mitoticinhibitor, a radiopharmaceutical, an antidepressant, antimanic agent, an antipsychotic, an anxiolytic, a hypnotic, a sympathomimetic, a stimulant, donepezil, tacrine, an asthma medication, a beta agonist, an inhaled steroid, a leukotriene inhibitor, a methylxanthine, a cromolyn, an epinephrine or analog, dornase alpha (Pulmozyme), a cytokine or a cytokine antagonist. Suitable dosages are well known in the art. See, e.g., Wells et al., eds., Pharmacotherapy Handbook, 2nd Edition, Appleton and Lange, Stamford, CT (2000); PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe Edition, Tarascon Publishing, Loma Linda, CA (2000).
TNF antagonists suitable for compositions, combination therapy, co-administration, devices and/or methods of the present invention include, but are not limited to, anti- TNF antibodies, antigen-binding fragments thereof, and receptor molecules which bind specifically to TNF; compounds which prevent and/or inhibit TNF synthesis, TNF release or its action on target cells, such as thalidomide, tenidap, phosphodiesterase
inhibitors (e.g, pentoxifylline and rolipram), A2b adenosine receptor agonists and A2b adenosine receptor enhancers; compounds which prevent and/or inhibit TNF receptor signalling, such as mitogen activated protein (MAP) kinase inhibitors; compounds which block and/or inhibit membrane TNF cleavage, such as metalloproteinase inhibitors; compounds which block and/or inhibit TNF activity, such as angiotensin converting enzyme (ACE) inhibitors (e.g., captopril); and compounds which block and/or inhibit TNF production and/or synthesis, such as MAP kinase inhibitors.
In another embodiment, the additional therapeutic agent is selected from the group consisting of anti-TNF-alpha monoclonal antibodies: infliximab (Remicade), adalimumab (Humira), certolizumab pegol (Cimzia), golimumab (Simponi), TNF-alpha receptor fusion protein: etanercept (Enbrel), or TNF-alpha inhibitory molecules:
pentoxifylline or bupropion. Thus in an embodiment the invention relates to a pharmaceutical composition, wherein the additional therapeutic is selected from the group consisting of anti-TNF-alpha monoclonal antibodies: infliximab (Remicade), adalimumab (Humira), certolizumab pegol (Cimzia), golimumab (Simponi), TNF-alpha receptor fusion protein: etanercept (Enbrel), or TNF-alpha inhibitory molecules: pentoxifylline or bupropion.
Thus, one aspect of the invention relates to a kit of parts comprising a therapeutic agent as described above and the isolated polypeptide of the invention, or the isolated polynucleotide of the invention, or the vector of the invention, or the host cell of the invention as a combination for the simultaneous, separate or successive administration in TNFR1 related disease therapy.
Dosing regimes
The dosage requirements of monomeric alpha-lactalbuminin complex, preferably LAC to be administered will vary with the particular drug composition employed, the route of administration and the particular subject being treated. Ideally, a patient to be treated by the present method will receive a pharmaceutically effective amount of the compound in the maximum tolerated dose, generally no higher than that required before drug resistance develops.
For all methods of use disclosed herein for the compounds, the daily oral dosage regimen will preferably be from about 0.01 to about 80 mg/kg of total body weight. The daily parenteral dosage regimen may be about 0.001 to about 80 mg/kg of total body weight. The daily topical dosage regimen will preferably be from 0.1 mg to 150 mg, administered one to four, preferably two or three times daily. The daily inhalation dosage regimen will preferably be from about 0.01 mg/kg to about 1 mg/kg per day. It will also be recognized by one of skill in the art that the optimal quantity and spacing of individual dosages of a compound or a pharmaceutically acceptable salt thereof will be determined by the nature and extent of the condition being treated, the form, route and site of administration, and the particular patient being treated, and that such optimums can be determined by conventional techniques. It will also be appreciated by one of skill in the art that the optimal course of treatment, i.e., the number of doses of a compound or a pharmaceutically acceptable salt thereof given per day for a defined number of days, can be ascertained by those skilled in the art using conventional course of treatment determination tests.
The daily dose of the active compound varies and is dependant on the type of administrative route, but as a general rule it is 1 to 100 mg/dose of active compound at personal administration, and 2 to 200 mg/dose in topical administration. The number of applications per 24 hours depend of the administration route, but may vary, e. g. in the case of a topical application in the no. se from 3 to 8 times per 24 hours, i. e. , depending on the flow of phlegm produced by the body treated in therapeutic use.
The compound according to the present invention is given in an effective amount to an individual in need there of. The amount of compound according to the present invention in one preferred embodiment is in the range of from about 0.01 milligram per kg body weight per dose to about 20 milligram per kg body weight per dose, such as from about 0.02 milligram per kg body weight per dose to about 18 milligram per kg body weight per dose, for example from about 0.04 milligram per kg body weight per dose to about 16 milligram per kg body weight per dose, such as from about 0.06 milligram per kg body weight per dose to about 14 milligram per kg body weight per dose, for example from about 0.08 milligram per kg body weight per dose to about 12 milligram per kg body weight per dose, such as from about 0.1 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 0.2 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for
example from about 0.3 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 0.4 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 0.5 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 0.6 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 0.7 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 0.8 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 0.9 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 1 .0 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 1 .2 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 1 .4 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 1 .6 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 1 .8 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 2.0 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 2.2 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 2.4 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 2.6 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 2.8 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 3.0 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 3.2 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 3.4 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 3.6 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 3.8 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 4.0 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 4.2 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 4.4 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 4.6 milligram per kg body weight per
dose to about 10 milligram per kg body weight per dose, for example from about 4.8 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 5.0 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 5.2 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 5.4 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 5.6 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 5.8 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 6.0 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 6.2 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 6.4 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 6.6 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 6.8 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 7.0 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 7.2 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 7.4 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 7.6 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 7.8 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, for example from about 8.0 milligram per kg body weight per dose to about 10 milligram per kg body weight per dose, such as from about 0.2 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, for example from about 0.3 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, such as from about 0.4 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, for example from about 0.5 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, such as from about 0.6 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, for example from about 0.7 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, such as from about 0.8 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, for example from about 0.9 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, such as
from about 1 .0 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, for example from about 1 .2 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, such as from about 1 .4 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, for example from about 1 .6 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, such as from about 1 .8 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, for example from about 2.0 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, such as from about 2.2 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, for example from about 2.4 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, such as from about 2.6 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, for example from about 2.8 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, such as from about 3.0 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, for example from about 3.2 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, such as from about 3.4 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, for example from about 3.6 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, such as from about 3.8 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, for example from about 4.0 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, such as from about 4.2 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, for example from about 4.4 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, such as from about 4.6 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, for example from about 4.8 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, such as from about 5.0 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, for example from about 5.2 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, such as from about 5.4 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, for example from about 5.6 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, such as from about 5.8 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, for example from about 6.0 milligram per kg body weight per dose to about 8 milligram per kg body weight per dose, such as from about 0.2 milligram per kg
body weight per dose to about 6 milligram per kg body weight per dose, for example from about 0.3 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, such as from about 0.4 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, for example from about 0.5 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, such as from about 0.6 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, for example from about 0.7 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, such as from about 0.8 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, for example from about 0.9 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, such as from about 1 .0 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, for example from about 1 .2 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, such as from about 1 .4 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, for example from about 1 .6 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, such as from about 1 .8 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, for example from about 2.0 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, such as from about 2.2 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, for example from about 2.4 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, such as from about 2.6 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, for example from about 2.8 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, such as from about 3.0 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, for example from about 3.2 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, such as from about 3.4 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, for example from about 3.6 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, such as from about 3.8 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, for example from about 4.0 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, such as from about 4.2 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, for example from about 4.4 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, such as from about 4.6 milligram per kg body weight per dose to
about 6 milligram per kg body weight per dose, for example from about 4.8 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose, such as from about 5.0 milligram per kg body weight per dose to about 6 milligram per kg body weight per dose.
The term "unit dosage form" as used herein refers to physically discrete units suitable as unitary dosages for human and animal subjects, each unit containing a
predetermined quantity of a compound, alone or in combination with other agents, calculated in an amount sufficient to produce the desired effect in association with a pharmaceutically acceptable diluent, carrier, or vehicle. The specifications for the unit dosage forms of the present invention depend on the particular compound or compounds employed and the effect to be achieved, as well as the pharmacodynamics associated with each compound in the host. The dose administered should be an "effective amount" or an amount necessary to achieve an "effective level" in the individual patient.
Since the "effective level" is used as the preferred endpoint for dosing, the actual dose and schedule can vary, depending on individual differences in pharmacokinetics, drug distribution, and metabolism. The "effective level" can be defined, for example, as the blood or tissue level desired in the patient that corresponds to a concentration of one or more compounds according to the invention.
Expression vector
Construction of vectors for recombinant expression of HHV U20 polypeptides for use in the invention may be accomplished using conventional techniques which do not require detailed explanation to one of ordinary skill in the art. For review, however, those of ordinary skill may wish to consult Maniatis et al., in Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, (NY 1982). Expression vectors may be used for generating producer cells for recombinant production of NsG33 polypeptides for medical use, and for generating therapeutic cells secreting NsG33 polypeptides for naked or encapsulated therapy.
Briefly, construction of recombinant expression vectors employs standard ligation techniques. For analysis to confirm correct sequences in vectors constructed, the genes are sequenced using, for example, the method of Messing, et al., (Nucleic Acids
Res., 9: 309-, 1981 ), the method of Maxam, et al., (Methods in Enzymology, 65: 499, 1980), or other suitable methods which will be known to those skilled in the art.
Size separation of cleaved fragments is performed using conventional gel
electrophoresis as described, for example, by Maniatis, et al., (Molecular Cloning, pp. 133-134,1982).
For generation of efficient expression vectors, these should contain regulatory sequences necessary for expression of the encoded gene in the correct reading frame. Expression of a gene is controlled at the transcription, translation or post-translation levels. Transcription initiation is an early and critical event in gene expression. This depends on the promoter and enhancer sequences and is influenced by specific cellular factors that interact with these sequences. The transcriptional unit of many genes consists of the promoter and in some cases enhancer or regulator elements (Banerji et al., Cell 27: 299 (1981 ); Corden et al., Science 209: 1406 (1980); and Breathnach and Chambon, Ann. Rev. Biochem. 50: 349 (1981 )). For retroviruses, control elements involved in the replication of the retroviral genome reside in the long terminal repeat (LTR) (Weiss et al., eds., The molecular biology of tumor viruses: RNA tumor viruses, Cold Spring Harbor Laboratory, (NY 1982)). Moloney murine leukemia virus (MLV) and Rous sarcoma virus (RSV) LTRs contain promoter and enhancer sequences (Jolly et al., Nucleic Acids Res. 1 1 : 1855 (1983); Capecchi et al., In :
Enhancer and eukaryotic gene expression, Gulzman and Shenk, eds., pp. 101 -102, Cold Spring Harbor Laboratories (NY 1991 ). Other potent promoters include those derived from cytomegalovirus (CMV) and other wild-type viral promoters.
It may also be advantageously to have the polynucleotide encoding the U20 protein or active derivative thereof positioned in a vector. Thus, in an aspect the invention relates to a recombinant vector comprising the nucleic acid sequence according to the invention. A vector comprising a polynucleotide as defined elsewhere herein.
By having the polynucleotide positioned in a vector the polynucleotide can easily be stored, amplified, modified and expressed. Therefore, in yet an embodiment the invention relates to a vector according to the invention, wherein the vector is selected from the group consisting of plasmids, cosmids, phages, bacterial artificial chromosomes (BAC), Phagemids and P1 -derived artificial chromosomes. It is of course
to be understood that the vector may comprise one or more regulatory sequences e.g. controlling the expression of the U20 protein or active derivative thereof. The regulatory sequence may either form part of the final protein or peptid or it may not form part of the final protein or peptid. The at least one regulatory sequence may be a promoter operably linked to the polynucleotide. Non-limiting examples of promoters are promoters selected from CMV (cytomegalovirus), SV40 (simian adenovirus), AdMLP, elongation factor-1 alpha, Rous sarcoma virus long terminal repeat promoter, T7 promoter. Host cell
The choice of vector may depend on the type of host cell which can be used to amplify the vector and express the polypeptides of the present invention. For cloning and expression purposes it may be useful to have the vector cloned in a host cell. In this way the vector can be maintained, cloned and purified and thus provide a source for almost unlimited numbers of the vectors. Moreover, stable producer cell lines expressing U20 protein or active derivative thereof according to the invention may also be advantageous. In order to maintain a producer cell selection marker may be incorporated in the expression vector. Thus, in yet an aspect the invention relates to an isolated host cell transformed or transduced with the recombinant vector according to the invention. Different types of host cells may be used. Thus, in another embodiment the invention relates to host cells selected from the group consisting of bacteria, fungi and molds. In a more specific embodiment the bacteria are selected from the group consisting of Bacillus licheniformis, Bacillus subtilis and E.coli and the fungi and molds are selected from the group consisting of Aspergillus niger and Aspergillus oryzae.
The host cell may thus be selected from E.coli, Yeast, Saccharomyces cerevisiae, S. pombe or insect cells. The host cells may in one embodiment be a mammalian cell, such as human, feline, porcine, simian, canine, murine, rat, mouse or rabbit. In one particular embodiment the host cell is mammalian. Where the host cell is mammalian, the host cells may be a human embryonal kidney cells (HEK cells), such as the cell lines deposited at the American Type Culture Collection with the numbers CRL-1573 and CRL-10852, chick embryo fibroblast, hamster ovary cells, baby hamster kidney cells, human cervical carcinoma cells, human melanoma cells, human kidney cells, human umbilical vascular endothelium cells, human brain endothelium cells, human
oral cavity tumor cells, monkey kidney cells, mouse fibroblast, mouse kidney cells, mouse connective tissue cells, mouse oligodendritic cells, mouse macrophage, mouse fibroblast, mouse neuroblastoma cells, mouse pre-B cell, mouse B lymphoma cells, mouse plasmacytoma cells, mouse teratocacinoma cells, rat astrocytoma cells, rat mammary epithelium cells, COS, CHO, BHK, 293, VERO, HeLa, MDCK, WI38, HTC1 16 cell or NIH 3T3 cell.
Kit
The different components of the invention could be comprised in one or more types of kits. Thus, in an aspect the invention relates to s kit comprising a) at least one component selected from
a. the isolated polypeptide of the invention b. the isolated nucleic acid sequence of any of the claims of the invention or
c. the vector of the invention or
d. the host cell of any of the claims of the invention or e. the pharmaceutical composition of the invention or f. the kit of parts of the invention or
g. the kit of the present invention and
b) an instruction manual.
The instruction manual provides instructions to how the components should be used, dosing regimes and diseases for which the kit is applicable.
A kit comprising the U20 protein or an active derivative thereof according to the invention may be used in the clinic when U20 or an active derivative thereof is going to be provided to a patient suffereing from disease which may require treatment with U20 or an active derivative thereof. A kit comprising the pharmaceutical composition may be used for similar purposes.
A kit comprising the polynucleotide, the vector or the host cell may be used for production of the U20 protein or an active derivative thereof. The instruction manual provides the user with instruction on how to use the different components.
Diagnosticum
The polypeptides, methods, compositions and kits of the invention may also be used as a diagnosticum, Thus, in an aspect the invention relates to an isolated U20 protein or an active derivative thereof according to the invention for use as a diagnosticum. Since the polypeptides of the invention interacts with the TNF receptor, for example TNFR1 , the polypeptides, methods, compositions or kit may be used for measuring the amount of receptors expressed in a sample in a similar way as a standard antibody. The expression can be detected using a standard sandwich assay known to the person skilled in the art. If no secondary antibody able to bind to the polypeptides of the present invention (after binding to the receptor) a tag may be positioned, in frame, on the polypeptides, thereby allowing binding and detection of the secondary antibody.
Thus in an embodiment the invention relates to the polypeptides for use in determining the expression level of a TNF receptor in a sample. The amount of receptor may be used for determining the amount of a pharmaceutical composition, or polypeptide of the present invention is needed in a specific treatment of an individual.
The polypeptide may also be used as a diagnosticum to provide precise measurements of TNFa in a sample. Since the polypeptides binds to the TNF-alpha receptor, the polypeptide may be used to prevent the binding of TNFa to its receptor in a cellular assay, such as a biological assay where cells secrete or are induced to secrete TNFa to the supernatant. The use of U20 polypeptide or amino acids may prevent the consumption of TNFa from the sample and thus provide precise measurements of amount of secreted TNFa.
The invention will now be described in further details in the following non-limiting examples.
Examples
Example 1
HHV-6B rescues cells from TNF -induced apoptosis
In order to determine whether or not HHV-6B is able to rescue cells from extrinsic induced apoptosis, we first established criteria for rapid induction of apoptosis through TNF receptorl (TNFR1 ). Treatment of HCT1 16 cells with TNFa and cycloheximide (CHX) and subsequent Western blot (WB) analysis of poly (ADP-ribose) polymerase (PARP) cleavage. Activation of caspase 3 leads to cleavage in PARP within its nuclear
localization domain to yield a lower molecular weight (MW) fragment, which can be identified by WB. These analyses showed that apoptosis can be induced rapidly in HCT1 16 cells by a combined TNFoc/CHX treatment (Figure 1 A). CHX treatment blocks NFKB translocation to the nucleus, thereby stimulating complex II formation, which leads to apoptosis. Complex II is a cytosolic complex, which is assembled 30-60 min after TNFoc stimulation. This complex contains TRADD, TRAF2, RIP1 , FADD and caspase 8.
When the cells were infected with HHV-6B for 48 hours post infection (hpi) prior to TNFoc/CHX treatment, the majority of the cells were rescued from apoptosis (Figure
1 B). CHX treatment blocks phosphorylation of ΙκΒ, which directs the TNFR1 response toward formation of complex II, and thereby directing it against apoptosis. Treatment with Camptothecin, which induces activation of the intrinsic apoptotic pathway, caused apoptosis in both wild type (wt) cells and HHV-6B-infected cells.
When TNFoc binds to TNFR1 it can induce several different downstream pathways, depending on intracellular protein complexes that subsequently are formed. In order to examine where HHV-6B might block the induction, we examined several proteins downstream of TNFR1 . The first pathway we analyzed is mediated through FADD and TRADD, which mediates cleavage of pro-caspase-3 into active caspase-3. Caspase-3 is essential for cleavage of a large number of proteins destined for degradation during apoptosis. We analyzed the cleavage of caspase-3 by WB using an antibody specific for cleaved caspase-3. Cleavage was completely blocked in HHV-6B-infected cells (Figure 2A). The first caspase to be activated in this pathway is caspase-8, which upon activation cleaves pro-caspase-3. HHV-6B infection blocked the cleavage of pro- caspase-8 to active caspase-8 (Figure 2A).
Another TNFR1 -induced pathway leads to IKBOC phosphorylation, through TRAF2, which results in NFKB translocation to the nucleus, and subsequent transcription of either pro-apoptotic or anti-apoptotic genes, depending on cell type and cellular gene expression profile. WB analysis of IKBOC phosphorylation showed that HHV-6B infection could block this pathway (Figure 2A).
These results indicate that the HHV-6B infection induces a block of the TNFR1 pathway at or in close proximity to the receptor. To rule out that the observed effects
were due to down regulation or internalization of TNFR1 , we analyzed HHV-6B- infected HCT1 16 cells for the surface expression of TNFR1 . Flowcytometry confirmed comparable levels of TNFR1 on HHV-6B-infected and uninfected cells (Figure 2B).
Example 2
Early protein U20 is sufficient for TNFR1 block
We wanted to identify HHV-6B proteins essential for the block in TNFR1 signaling. We cloned several early gene products and used these to generate stably expressing cell lines in HCT1 16 cells. These clones were screened for resistance to TNFoc-induced apoptosis (data not shown). One gene, termed U20, possessed the ability to inhibit TNFoc-induced apoptosis when it was stably expressed (Figure 3A). Like HHV-6B- infected cells, the U20-expressing clone (termed U20s) was not resistant to
camptothecin-induced apoptosis (Figure 3B). In order to ensure that the HCT1 16-U20S cells expressed sufficient levels of mRNA, we made cDNA from the clone and amplified this using primers specific for U20 (Primers: 5'-ATGATAACTGTTTTTGTCGC-3' and 5'-TTACAAAGGCAACATTTCTA-3', SEQ ID NO: 12 and 13, respectively).
The amplified product gave rise to an intense band on agarose gel electrophoresis, a band of the expected size for U20 (Figure 3C). To verify the results obtained with the stable clone, we conducted experiments with transiently expressing cells. HCT1 16 cells transfected with U20 expression plasmid showed a reduced resistance to TNFoc-induced apoptosis when analyzed by WB for PARP cleavage (Figure 3D). The lower levels of rescue during transient expression could be explained by a transfection efficiency that only approached 50%. To verify this result, we also analyzed HCT1 16 cells, transiently expressing U20, by flow cytometry for the apoptosis marker 7-aminoactinomycin D (7-AAD). This analysis also indicated a reduced resistance to TNFoc-induced apoptosis in U20-expressing cells (Figure 3E-G).
U20 appears to block TNFoc induced apoptosis, and could potentially be the protein solely responsible for this block during infection with HHV-6B. We wanted to test whether U20 blocked TNFR1 signaling similarly to our observations during infection. We conducted WB analysis on wt HCT1 16 cells and HCT1 16-U20s cells treated with TNFoc/CHX and probed with antibodies against active caspase-3 and -8 and ρ-ΙκΒ. In addition, confocal microscopy was used to visualize active caspase-3. These analyses
gave results similar to those obtained with infected cells with no caspase cleavage and no IKB phosphorylation (Figure 4A & B) .
To rule out that the observations was caused by downregulation of TNFR1 or the signaling adaptors TRADD and TRAF2, we conducted WB against TRADD and TRAF2, and flowcytometry against TNFR1 . U20s cells contained similar levels of TNFR1 , TRADD and TRAF2 when compared with wt cells (Figure 4C & D).
Furthermore, confocal microscopy analysis of TNFR1 showed a similar distribution of TNFR1 in U20 cells and wt cells (data not shown). Example 3
Open reading frame (ORF) U20 analysis
In order to determine whether or not the U20 ORF is transcribed during infection, we conducted PCR using U20 specific primers (Primers: 5'-ATGATAACTGTTTTTGTCGC- 3' and 5 ' -TT AC AA AG G C AAC ATTTCT A-3 ' ) spanning the entire U20 ORF on cDNA generated from HCT1 16 cells infected at various time points (Figure 5A). PCR products from infected cells generated with U20 spanning primers were extracted and sequencing was performed to confirm that the bands corresponded to U20. To get a better view of the expression kinetics during non-productive infection, we conducted real-time PCR on cDNA from HHV-6B-infected HCT1 16 cells using real-time PCR primers for U20 (Primers: 5'-CGAAGCTGAAAATCACGACA-3' and 5'-
CCGCAGACAACTGCTATTGA-3', SEQ ID NO:14 and 15, respectively (Figure 5B)). To verify whether U20 is also transcribed during productive infection, we conducted PCR on cDNA generated from RNA from infected MOLT3 cells (data not shown). The PCR results showed an expression profile similar to a recent report in which U20 was designated an early gene.
These analyses demonstrate that U20 is indeed a gene and it is transcribed during both productive and non-productive infections. Theoretical analyses identified U20 as a membrane protein with a single membrane spanning oc-helix (aa 319 - 343) and an N-terminal signal peptide (aa 1 - 15) for ER sorting to the plasma membrane (Figure 5C). The orientation of U20 in the membrane is predicted to be with the N-terminal end at the outside of the cell and the C-terminal end in the cytoplasm. Several highly probable N-glycosylation sites [corresponding to
amino acid position 58, 78, 107, 133, 145, 154, 161 , 227 in SEQ ID NO: 1 ] on the N- terminal side of the membrane and none on the C-terminal side support this
orientation. Theoretic analysis further predicted an immunoglobulin-like fold spanning aa 182 - 300. Furthermore, analysis indicates a highly organized N-terminal part and a highly disordered C-terminal part. A structural model of U20 from HHV-6B is shown in Figure 10. Taken together, these data strongly support that U20 is expressed on the surface of the cells, and oriented with a large N-terminal extracellular part and a small C-terminal intracellular part.
Example 4
U20 disrupts assembly of the TNFR1 complex
When TNFR1 binds TNFa it rapidly forms a signaling complex, which includes the adaptor protein TRADD. To analyze whether or not the signaling complex is formed, we analyzed the subcellular distribution pattern of TRADD, at different time points post TNFa stimulation of U20s and wt HCT1 16 cells. Wt cells stimulated with TNFa, showed a rapid dislocation of TRADD from the ER throughout the cell in a vesicle-like pattern (Figure 6A). HCT1 16-U20S cells, however, did not show any translocation of TRADD upon TNFa treatment, indicating a failed complex I formation (Figure 6B). Complex I is a complex that is formed immediately following TNFa stimulation and contains TNFR1 with the recruited proteins TRADD, TRAF2 and RIP, but not FADD or caspase 8.
This failure of complex I to assemble could be caused by a lack of TNFa binding to TNFR1 .
If U20 interacts directly with TNFR1 , it may prevent binding of anti-TNFR1 antibodies to TNFR1 . We used an antibody (HTR19) (supplied by Dr. B. W. Deleuran) that only recognizes TNFR1 , if TNFa is not bound to the receptor. As expected, we identified by flowcytometry HTR19 binding to wt HCT1 16 cells in the absence of TNFa, whereas binding was virtually absent in the presence of TNFa (Figure 7A). Importantly, when U20s cells were stained with the HTR19 antibody, no signal was observed with or without TNFa (Figure 7B), despite these cells expressed TNFR1 when stained by antibodies (Santa Cruz Biotechnology, Santa Cruz, USA) that binds independently of TNFa. These observations strongly indicate that U20 directly interacts with the TNFR1 .
Example 5
Sequence homology for U20 from HHV-6B
To identify sequence homologies of U20, an alignment of the nucleotide sequence (Figure 1 1 ) and the amino acid sequence of U20 (Figure 9) were done. At the amino acid level the identity between U20 from HHV-6B and HHV-6A was 88.9% and between HHV-6B and HHV-7 it was 24.1 %. The domain structure between HHV-6B and HHV-6A is conserved throughout the protein (Table 1 and 2).
Example 6
HHV-6 infection blocks TNFR1 signalling; cf. Figure 12.
Early protein U20 is sufficient for blocking TNFR1 signalling; cf. Figure 13 U20 inhibit death receptor signalling; cf. Figure 14.
U20 inhibits TNFalpha internalization; cf. Figure 15.
U20 sensitize cells to Etoposide killing through programmed necrosis; cf. Figure 16.
Sequences
SEQ ID No: 1
Hu ma n Herpesvi rus 6B
polypeptide
434 aa
(>U20-HHV-6B)
MITVFVACLFQCVSSLPAKLYIKTTLAEGIGKLQTVIGIDNDIVFAYERLYGDLTLRNHTAVGE TLFDLAGSLEEGKNSTVDRFLGHVVIREFHRLHAGLQYVSVQNFSVSELVCFVNNNTQLSGSYV FLARNTTYVQIDLFNENRSFVHDLINVSSFLQNRSLHVLSFYARRFCVEDILNFYGKVVFGDSK YRPPQVFSKRDTGLLVCTARRYRPIGTNIQWSLHNQTVSDDHTTDDFIRTEISGQLLYSYERAL SRALSMTHREFSCEITHKLLVTPALLTREDAFSFKGFVNPVKESEDTFPRHNFPAPHRKKFNKL QLLWIFIVIPIAAGCMFLYILTRYIQFFVSGGSSSNPNRVLKRRRGNDEVPMVIMEVEYCNYEA ENHDMELHSVQNVRDDS IAVVCGNNSFDIERQS IKSHESFSNVKLEMLPL
SEQ ID No: 2
Hu ma n Herpesvi rus 6B
DNA
1354 nt
>U20 HHV-6B
ATGATAACTGTTTTTGTCGCATGTTTATTTCAATGCGTTTCTTCGTTGCCGGCTAAACTTTACATCAAGA CTACTCTGGCTGAAGGAATCGGTAAACTTCAGACCGTTATCGGCATTGATAACGATATCGTTTTTGCTT ACGAGAGGCTTTATGGAGATTTGACTTTGCGGAACCATACGGCTGTTGGAGAGACGTTGTTTGACCTC GCGGGAAGCCTGGAAGAAGGAAAAAATTCTACCGTAGACCGT I I I I I AGGACATGTGGTAATACGTG
AATTTCATAGATTGCACGCTGGGCTTCAGTATGTCTCAGTTCAAAATTTTTCTGTAAGCGAATTGGTTT GCTTCGTCAACAATAATACGCAGTTGTCTGGCAGCTACG I I I I I I TAGCGAGAAATACAACCTATGTAC AGATAGATCTGTTTAATGAGAATCGTAGTTTTGTTCACGATCTTATAAATGTGAGCAG I I I I I I ACAAA ATCGTTCTTTGCACGTATTGTCGTTTTATGCAAGACGTTTTTGTGTAGAAGACATCTTAAATTTCTATG GTAAGGTTGTTTTCGGAGACTCTAAATATCGTCCTCCGCAAGTTTTCAGCAAGAGAGATACAGGTTTG CTCGTCTGTACAGCTCGACGATATCGGCCCATTGGAACGAACATTCAATGGAGTCTTCACAACCAAAC AGTTTCAGATGATCATACGACTGACGATTTCATTAGAACCGAAATTTCCGGTCAACTTCTATACTCTTA TGAAAGAGCATTGAG
TAGAGCTTTGAGCATGACACATCGTGAGTTTTCATGTGAAATTACGCATAAATTATTAGTTACCCCAGC TCTCTTAACAAGAGAGGATGCTTTTAGTTTCAAAGGTTTTGTGAATCCCGTGAAGGAATCCGAGGACA CGTTTCCTCGTCATAATTTTCCAGCGCCGCACAGAAAAAAATTTAATAAATTGCAGCTGTTGTGGATTT TCATTGTAATTCCAATTGCTGCTGGGTGTATGTTTCTATATATTTTAACGCGGTATATTCAGTTTTTTGT TTCTGGGGGATCTTCATCGAATCCAAATAGGGTGCTAAAAAGGCGTAGAGGGAATGATGAAGTGCCT ATGGTGATTATGGAAGTAGAATACTGCAACTACGAAGCTGAAAATCACGACATGGAACTTCATAGCGT TCAAAACGTCCGTGATGATTCAATAGCAGTTGTCTGCGGAAATAATTCGTTTGATATAGAACGACAGT CAATAAAATCTCACGAAAGTTTTAGCAATGTCAAGTTAGAAATGTTGCCTTTGTAAAATGTTTCCATGA CTGTGAGGTGATTTGAACAGAGA I I I I I I AAATAAA
SEQ ID No: 3
Hu ma n Herpesvi rus 6A
polypeptide
422 aa
(>U20-HHV-6A)
MITVFVACLFQCVSSLPAKLYIKTTLAERIGKLQTVIGIDNDIVFAYERLYEDLTLLNHTVVGE
TLFDLTGSLEEGKNSTVDRFLGHVVIREFHRLHAGLQYVSLRNFSVSELVCFVNNNTQLSGSYV FLAGNTTYVQIDLFNENRGFVHDLINLSSFLQNRSLHVLSFYARRFCVEDILNFYGKVVFGDSR HRPPQVFSKRDTGLLVCTARRYRPIGTNIQWSIQNQTVSDDHMTDDFIRTEIAGQLLYSYERAL SRALSMTQRNFSCEITHKLLVTPALLTREDAFSFKGFVNPVKQSEDMFPRHNFPAPHRKKFNKL QLLWIFTVIPIAAGCMFVYMLTRYILFFVSGGCSLNPNRVLKRRRRNDEVPMVIMEVEYCNYEA
DDYMELHSVQKVRDNSIAVVCGNNSFDIERQSKISRNF
SEQ ID No: 4
Hu ma n Herpesvi rus 6A
DNA
1349 nt
>U20 HHV-6A
ATGATAACTGTCTTTGTCGCATGCTTATTTCAATGCGTTTCTTCGTTGCCGGCTAAACTTTACATCAAG ACTACTCTGGCTGAAAGAATCGGTAAACTTCAGACCGTTATTGGCATTGATAATGATATAG I I I I I GCT
TACGAGAGGCTCTATGAAGATTTGACATTGCTGAACCATACGGTTGTTGGAGAGACGTTGTTTGACCT TACGGGGAGCTTGGAAGAAGGAAAAAATTCTACCGTAGACCGTTTCTTAGGACATGTGGTGATACGT GAATTTCATAGACTGCACGCTGGGCTTCAGTATGTCTCTCTTCGAAA I I I I I CTGTAAGCGAATTGGTT TGCTTCGTTAACAATAATACGCAGTTGTCTGGCAGCTACG I I I I I I TAGCGGGTAATACAACCTATGTA CAGATAGATCTGTTTAATGAAAATCGTGGTTTTGTTCACGATCTTATAAATTTGAGCAGT I I I I I ACAAA
ATCGGTCTTTGCACGTATTGTCGTTTTATGCAAGACGTTTTTGTGTAGAAGACATCTTAAATTTCTATG GTAAGGTTGTTTTCGGGGACTCTAGACATCGTCCTCCGCAAGTTTTCAGCAAGAGAGATACAGGTTTG CTCGTTTGTACAGCTCGGCGATATCGGCCCATTGGAACGAATATTCAATGGAGTATTCAAAACCAGAC AGTTTCAGATGATCATATGACTGACGATTTCATTAGAACCGAAATTGCCGGTCAACTTCTATACTCTTA TGAAAGAGCATTGAGTAGAGCTTTGAGCATGACACAACGTAA I I I I I CATGTGAAATTACGCATAAATT
ATTAGTTACCCCAGCTCTCTTAACAAGAGAGGATGCTTTTAGTTTTAAAGGTTTTGTGAATCCCGTGAA GCAATCCGAAGACATGTTTCCACGTCATAATTTTCCAGCACCGCACAGAAAAAAATTTAATAAATTGCA GCTGCTGTGGA I I I I I ACTGTGATACCAATTGCTGCTGGGTGTATGTTTGTATATATGTTAACGCGGTA TATTCTG I I I I I I GTTTCTGGGGGATGTTCATTGAATCCAAATAGGGTGCTAAAAAGGCGTAGGAGAA ATGATGAAGTGCCTATGGTGATTATGGAAGTGGAATACTGCAATTACGAAGCTGATGATTACATGGAA CTCCATAGCGTTCAAAAGGTCCGTGACAATTCGATAGCGGTTGTCTGCGGAAATAATTCGTTTGATAT AGAACGACAGTCTAAAATCTCACGAAA I I I I I AGCAATGTGAAGTTAGAAATTTTACCTTTGGAAAATA TTTCCATGACTGTGAGGTTATTTGAATAGAGA I I I I I I I AATAAA SEQ ID No: 5
Hu ma n Herpesvi rus 7
polypeptide
391 aa
(>U20-HHV-7)
MFVKKTCLLVFIVHFTKYVAHKLTMSI INKEHLEKYELMMDNYELAHYKNLSVGNTS ISSFLTD FPTKFNALSYVFSTNKDESTLNS IFSYFVWSHRNRSVHYHHNFNAEFICEIDTVLKSHYLLKVH NDTTLNVNLLDFSSSGFDENMYANVELLLILKFYVQQFCVKQLADFSAHVTFGQDGYRPPVVSI TKRGSNITCSAQLYSPLGLSIQWKLENKLVPDDLTEEYLASDILKGKKFYFFFKS INTESANYQ CVVIHQKCNYSFNVLEFNNSDEYFQNLFHKTYKHGNYFEVFWILLI IPILAFIFS I ITVKKMFF
EIWLHVMAVLNKRKDLDRSDSVKVIMEIDPCDYISVENLCATEKMEI SDSDCLSKDSPDLQIHG TLDFMCH
SEQ ID No: 6
Hu ma n Herpesvi rus 7
DNA
1190 nt
Nucleotide sequence of genes
>U20 HHV-7
ATGTTTGTGAAAAAAACATGTCTTTTAGTTTTCATTGTACATTTTACCAAATATGTGGCACATAAGTTGA CAATGTCAATAATCAATAAAGAACATCTTGAGAAATATGAATTGATGATGGATAACTATGAACTGGCAC ATTATAAGAATCTGTCTGTCGGCAATACTAGTATTTCGTCTTTCCTCACAGATTTTCCAACGAAATTTAA TGCTTTGTCTTACGTCTTTTCTACCAACAAGGACGAATCTACATTAAACAGCATTTTCTCTTATTTTGTC TGGTCACATAGGAATCGCAGTGTTCATTATCACCACAATTTTAACGCTGAATTCATTTGTGAAATTGAC ACCGTTTTGAAAAGTCATTATTTGTTAAAAGTTCATAATGACACGACACTGAATGTCAACCTGTTGGAT TTTTCTAGCAGTGGTTTTGACGAAAACATGTATGCTAATGTTGAATTATTACTTATTCTCAAATTTTATG TGCAGCAGTTTTGTGTTAAACAACTCGCAGATTTTTCTGCACATGTGACCTTCGGTCAAGATGGATATC GGCCACCTGTGGTGTCCATCACAAAACGTGGATCTAACATAACGTGTTCGGCGCAGCTTTATAGTCCA CTGGGATTGAGTATTCAATGGAAGCTAGAAAATAAACTGGTTCCTGATGATTTGACAGAAGAATATTT GGCATCGGACATCTTAAAAGGGAAAAAATTCTAC I I I I I I I I I AAATCTATTAACACGGAGTCCGCTAA TTATCAATG
TGTGGTAATTCATCAAAAATGTAATTACTCTTTTAACGTCCTTGAATTTAATAATTCAGATGAATATTTT CAAAATTTGTTTCACAAAACTTACAAACACGGGAATTACTTTGAAGTTTTTTGGATACTTTTAATCATTC CTATACTTGCCTTTATTTTTTCTATAATCACTGTTAAAAAAATG I I I I I I GAAATTTGGCTGCATGTTATG
GCTGTTCTTAATAAGCGTAAGGACCTTGATAGATCTGATAGTGTTAAAGTAATCATGGAAATTGATCCC
TGTG ATTATATATCTGTGG AAAATTTGTGTG CAACTG AAAAAATG G AAATTTCAG ATTCAG ACTG CCTG
TCTAAGGATTCTCCAGATCTTCAAATTCACGGGACCTTAGATTTCATGTGTCATTAATGAGTTTCAATA
AA
SEQ ID NO 7 :
Human Herpesvirus 6B
mRNA
>U20_HHV-6B
AUGAUAACUGUUUU UGUCGCAUGUUUAUUUCAAUGCGUUUCUUCGUUGCCGGCUAAAC UUUACAUCAAGACUACUCUGGCUGAAGGAAUCGGUAAACUUCAGACCGUUAUCGGCAUU GAUAACGAUAUCGUUUUUGCUUACGAGAGGCUUUAUGGAGAUUUGACUUUGCGGAACC AUACGGCUGUUGGAGAGACGUUGUUUGACCUCGCGGGAAGCCUGGAAGAAGGAAAAAA UUCUACCGUAGACCGUU UUUUAGGACAUGUGGUAAUACGUGAAU UUCAUAGAUUGCACG CUGGGCUUCAGUAUGUCUCAGUUCAAAAUUU UUCUGUAAGCGAAUUGGUUUGCUUCGU CAACAAUAAUACGCAGUUGUCUGGCAGCUACGU UUUUUUAGCGAGAAAUACAACCUAUG UACAGAUAGAUCUGUUUAAUGAGAAUCGUAGUUUUGUUCACGAUCUUAUAAAUGUGAGC AGUUUUU UACAAAAUCGUUCUUUGCACGUAUUGUCGUUUUAUGCAAGACGUUUUUGUG UAGAAGACAUCUUAAAUUUCUAUGGUAAGGUUGUUUUCGGAGACUCUAAAUAUCGUCCU CCGCAAGUUUUCAGCAAGAGAGAUACAGGUUUGCUCGUCUGUACAGCUCGACGAUAUC GGCCCAUUGGAACGAACAUUCAAUGGAGUCUUCACAACCAAACAGUUUCAGAUGAUCAU ACGACUGACGAUUUCAUUAGAACCGAAAUUUCCGGUCAACUUCUAUACUCU UAUGAAAG AGCAUUGAGUAGAGCUU UGAGCAUGACACAUCGUGAGUUUUCAUGUGAAAU UACGCAUA AAU UAU UAGUUACCCCAGCUCUCUUAACAAGAGAGGAUGCUUUUAGUUUCAAAGGUUUU GUGAAUCCCGUGAAGGAAUCCGAGGACACGUUUCCUCGUCAUAAUUUUCCAGCGCCGC ACAGAAAAAAAU UUAAUAAAUUGCAGCUGUUGUGGAUUUUCAUUGUAAUUCCAAUUGCU GCUGGGUGUAUGUUUCUAUAUAUUUUAACGCGGUAUAUUCAGUUUUU UGU UUCUGGGG GAUCUUCAUCGAAUCCAAAUAGGGUGCUAAAAAGGCGUAGAGGGAAUGAUGAAGUGCCU AUGGUGAUUAUGGAAGUAGAAUACUGCAACUACGAAGCUGAAAAUCACGACAUGGAACU UCAUAGCGUUCAAAACGUCCGUGAUGAUUCAAUAGCAGUUGUCUGCGGAAAUAAUUCGU UUGAUAUAGAACGACAGUCAAUAAAAUCUCACGAAAGUU UUAGCAAUGUCAAGUUAGAAA UGUUGCCUUUGUAAAAUGUUUCCAUGACUGUGAGGUGAUUUGAACAGAGAUUUUUUAAA UAAA SEQ ID NO 8:
Human Herpesvirus 6A
mRNA
(>U20_HHV-6A)
AUGAUAACUGUCUU UGUCGCAUGCUUAUUUCAAUGCGUUUCUUCGUUGCCGGCUAAAC UUUACAUCAAGACUACUCUGGCUGAAAGAAUCGGUAAACUUCAGACCGUUAUUGGCAUU GAUAAUGAUAUAGUUUU UGCUUACGAGAGGCUCUAUGAAGAUUUGACAUUGCUGAACCA UACGGUUGUUGGAGAGACGUUGUUUGACCU UACGGGGAGCUUGGAAGAAGGAAAAAAU UCUACCGUAGACCGUUUCUUAGGACAUGUGGUGAUACGUGAAUUUCAUAGACUGCACG CUGGGCUUCAGUAUGUCUCUCUUCGAAAUUUUUCUGUAAGCGAAUUGGU UUGCUUCGU UAACAAUAAUACGCAGUUGUCUGGCAGCUACGU UUUUUUAGCGGGUAAUACAACCUAUG UACAGAUAGAUCUGUUUAAUGAAAAUCGUGGUUUUGUUCACGAUCUUAUAAAUUUGAGC AGUUUUU UACAAAAUCGGUCUUUGCACGUAUUGUCGUUUUAUGCAAGACGU UUUUGUG UAGAAGACAUCUUAAAUUUCUAUGGUAAGGUUGUUUUCGGGGACUCUAGACAUCGUCCU CCGCAAGUUUUCAGCAAGAGAGAUACAGGUUUGCUCGUUUGUACAGCUCGGCGAUAUC GGCCCAUUGGAACGAAUAUUCAAUGGAGUAUUCAAAACCAGACAGUUUCAGAUGAUCAU AUGACUGACGAUUUCAUUAGAACCGAAAUUGCCGGUCAACUUCUAUACUCU UAUGAAAG AGCAUUGAGUAGAGCUU UGAGCAUGACACAACGUAAUUUUUCAUGUGAAAUUACGCAUA AAU UAU UAGUUACCCCAGCUCUCUUAACAAGAGAGGAUGCUUUUAGUUUUAAAGGUUUU GUGAAUCCCGUGAAGCAAUCCGAAGACAUGUUUCCACGUCAUAAUU UUCCAGCACCGCA CAGAAAAAAAUUUAAUAAAUUGCAGCUGCUGUGGAUUUUUACUGUGAUACCAAUUGCUG CUGGGUGUAUGUUUGUAUAUAUGUUAACGCGGUAUAUUCUGUUUUUUGUUUCUGGGGG AUGUUCAUUGAAUCCAAAUAGGGUGCUAAAAAGGCGUAGGAGAAAUGAUGAAGUGCCUA UGGUGAUUAUGGAAGUGGAAUACUGCAAU UACGAAGCUGAUGAUUACAUGGAACUCCAU
AGCGUUCAAAAGGUCCGUGACAAUUCGAUAGCGGUUGUCUGCGGAAAUAAU UCGUUUG
AUAUAGAACGACAGUCUAAAAUCUCACGAAAUUUUUAGCAAUGUGAAGUUAGAAAUUUUA
CCUUUGGAAAAUAUUUCCAUGACUGUGAGGUUAUUUGAAUAGAGAUUUUU UUAAUAAA SEQ ID NO 9:
Human Herpesvirus 7
mRNA
(>U20_HHV-7)
AUGUUUGUGAAAAAAACAUGUCUU UUAGUUUUCAUUGUACAUUUUACCAAAUAUGUGGC ACAUAAGUUGACAAUGUCAAUAAUCAAUAAAGAACAUCUUGAGAAAUAUGAAUUGAUGAU GGAUAACUAUGAACUGGCACAUUAUAAGAAUCUGUCUGUCGGCAAUACUAGUAUUUCGU CUUUCCUCACAGAU UUUCCAACGAAAUUUAAUGCUUUGUCUUACGUCUU UUCUACCAAC AAGGACGAAUCUACAUUAAACAGCAUUUUCUCUUAUUUUGUCUGGUCACAUAGGAAUCG CAGUGUUCAUUAUCACCACAAUU UUAACGCUGAAUUCAUUUGUGAAAU UGACACCGUU U UGAAAAGUCAUUAUUUGUUAAAAGUUCAUAAUGACACGACACUGAAUGUCAACCUGUUG GAUUUUUCUAGCAGUGGUUUUGACGAAAACAUGUAUGCUAAUGUUGAAUUAUUACUUAU UCUCAAAUUUUAUGUGCAGCAGUU UUGUGUUAAACAACUCGCAGAUUUUUCUGCACAUG UGACCUUCGGUCAAGAUGGAUAUCGGCCACCUGUGGUGUCCAUCACAAAACGUGGAUC UAACAUAACGUGUUCGGCGCAGCUUUAUAGUCCACUGGGAUUGAGUAUUCAAUGGAAG CUAGAAAAUAAACUGGUUCCUGAUGAUUUGACAGAAGAAUAUUUGGCAUCGGACAUCUU AAAAGGGAAAAAAUUCUACUUU UUUUUUAAAUCUAUUAACACGGAGUCCGCUAAUUAUCA AUGUGUGGUAAUUCAUCAAAAAUGUAAU UACUCUUUUAACGUCCU UGAAUU UAAUAAUU CAGAUGAAUAUUUUCAAAAU UUGUUUCACAAAACUUACAAACACGGGAAUUACUUUGAAG UUUUUUGGAUACUUUUAAUCAUUCCUAUACUUGCCUUUAUUUUUUCUAUAAUCACUGUU AAAAAAAUGUUUUUUGAAAU UUGGCUGCAUGUUAUGGCUGUUCUUAAUAAGCGUAAGGA CCUUGAUAGAUCUGAUAGUGUUAAAGUAAUCAUGGAAAUUGAUCCCUGUGAUUAUAUAU CUGUGGAAAAUUUGUGUGCAACUGAAAAAAUGGAAAUUUCAGAUUCAGACUGCCUGUCU AAGGAUUCUCCAGAUCUUCAAAU UCACGGGACCUUAGAUUUCAUGUGUCAU UAAUGAGU UUCAAUAAA
SEQ ID NO 10
HHV6B extracellular domain (16-319)
LPAKLYIKTTLAEGIGKLQTVIGIDNDIVFAYERLYGDLTLRNHTAVGETLFDLAGSLEEGKNS TVDRFLGHVVIREFHRLHAGLQYVSVQNFSVSELVCFVNNNTQLSGSYVFLARNTTYVQIDLFN ENRSFVHDLINVSSFLQNRSLHVLSFYARRFCVEDILNFYGKVVFGDSKYRPPQVFSKRDTGLL VCTARRYRPIGT IQWSLHNQTVSDDHTTDDFIRTEI SGQLLYSYERALSRALSMTHREFSCEI THKLLVTPALLTREDAFSFKGFVNPVKESEDTFPRHNFPAPHRKKFNK
SEQ ID NO 1 1
HHV6a extracellular domain
LPAKLYIKTTLAERIGKLQTVIGIDNDIVFAYERLYEDLTLLNHTVVGETLFDLTGSLEEGKNS TVDRFLGHVVIREFHRLHAGLQYVSLRNFSVSELVCFVNNNTQLSGSYVFLAGNTTYVQIDLFN ENRGFVHDLINLSSFLQNRSLHVLSFYARRFCVEDILNFYGKVVFGDSRHRPPQVFSKRDTGLL VCTARRYRPIGT IQWS IQNQTVSDDHMTDDFIRTEIAGQLLYSYERALSRALSMTQRNFSCEI THKLLVTPALLTREDAFSFKGFVNPVKQSEDMFPRHNFPAPHRKKFNK
Claims
1 . An isolated polypeptide for use as a medicament or a diagnosticum said polypeptide comprising
a) amino acids consisting of the sequence of SEQ ID No. 1 , 3 or 5;
b) a biologically active sequence variant of the amino acid sequence,
wherein the variant has at least 70% sequence identity to said SEQ ID NO. 1 , 3 or 5; or
c) a biologically active fragment of at least 50 contiguous amino acids of any of a) through b), wherein said fragment is a fragment of SEQ ID NO 1 , 3 or 5.
2. The isolated polypeptide according to claim 1 , wherein said polypeptide is a variant of SEQ ID NO:1 , 3 or 5 having at least 80% sequence identity, such as preferably at least 81 % sequence identity, more preferably e.g. at least 82% sequence identity, such as more preferably at least 83% sequence identity, e.g. more preferably at least 84% sequence identity, more preferably such as at least 85% sequence identity, more preferably e.g. at least 86% sequence identity, more preferably such as at least 87% sequence identity, more preferably e.g. at least 88% sequence identity, more preferably such as at least 89% sequence identity, more preferably e.g. at least 90% sequence identity, more preferably such as at least 91 % sequence identity, more preferably e.g. at least 92% sequence identity, such as at least 93% sequence identity, more preferably e.g. at least 94% sequence identity, more preferably such as at least 95% sequence identity, more preferably e.g. at least 96% sequence identity, more preferably such as at least 97% sequence identity, more preferably e.g. at least 98% sequence identity, more preferably such as at least 99% sequence identity, more preferably e.g. at least 99.5% sequence identity to said SEQ ID NO. 1 , 3 or 5.
3. The isolated polypeptide according to claim 1 , wherein the polypeptide fragment is a contiguous stretch of amino acids containing less than 99.5%, such as less than 98%, e.g. less than 97%, such as less than 96%, e.g. less than 95%, such as less than 94%, e.g. less than 93%, such as less than 92%, e.g. less than 91 %, such as less than 90%, e.g. less than 88%, such as less than 86%, e.g. less than 84%, e.g. less than 82%, such as less than 80%, e.g. less than 75%, such as less than 70%, e.g. less than 65%, such as less than 60%, e.g. less than 55%, such as less than 50%, e.g. less than 45%, such as less than 40%, e.g. less than 35%, such as less than 30%, e.g. less than 25%, such as less than 20%, such as less than 15%, e.g. less than 10% contiguous amino acids of the amino acid sequence of SEQ ID NO. 1 , 3 or 5.
4. The isolated polypeptide according to any of the preceding claims, wherein any amino acid specified in a chosen sequence is changed to provide a conservative substitution,
5. The isolated polypeptide according to any of the preceeding claims attached to a carrier.
6. The isolated polypeptide according to claim 5, wherein the carrier comprises an avidin moiety, such as streptavidin, which is optionally
biotinylated.
7. The isolated polypeptide according to claim 5 attached, such as covalently bound, to a solid support or a semi-solid support.
8. The isolated polypeptide according to any of the preceding claims operably fused to an affinity tag, such as a His-tag.
9. The isolated polypeptide according to any of the preceding claims, wherein said polypeptide is part of a fusion polypeptide operably fused to an N- terminal flanking sequence.
10. The isolated polypeptide according to any of claims 1 -8, wherein said fusion polypeptide comprises the isolated polypeptide operably fused to an C- terminal flanking sequence.
1 1 . The polypeptide according to any of the preceding claims, wherein one or more amino acid residues are modified, said modification(s) preferably being selected from the group consisting of in vivo or in vitro chemical derivatization, such as acetylation or carboxylation, glycosylation, such as glycosylation resulting from exposing the polypeptide to enzymes which affect glycosylation, for example mammalian glycosylating or deglycosylating enzymes,
phosphorylation, such as modification of amino acid residues which results in phosphorylated amino acid residues, for example phosphotyrosine,
phosphoserine and phosphothreonine.
12. The polypeptide according to any of the preceding claims wherein one or more amino acid residues are modified so as to preferably improve the resistance to proteolytic degradation and stability or to optimize solubility properties or to render the polypeptide more suitable as a therapeutic agent.
13. The polypeptide according to claim 12 comprising amino acid residues other than naturally occurring L-amino acid residues.
14. The polypeptide according to claim 13 comprising D-amino acid residues.
15. The polypeptide according any of the preceding claims comprising non- naturally occurring, synthetic amino acids.
16. The polypeptide according to any of the preceding claims further comprising one or more blocking groups, preferably in the form of chemical substituents suitable to protect and/or stabilize the N- and C-termini of the polypeptide from undesirable degradation.
17. The polypeptide according to claim 16, wherein the one or more blocking groups include protecting groups which do not adversely affect in vivo activities of the polypeptide.
18. The polypeptide according to claim 16, wherein the one or more blocking groups are introduced by alkylation or acylation of the N-terminus.
19. The polypeptide according to claim 16, wherein the one or more blocking groups are selected from N-terminal blocking groups comprising Ci to C5 branched or non-branched alkyl groups and acyl groups, such as formyl and acetyl groups, as well as substituted forms thereof, such as the
acetamidomethyl (Acm) group.
20. The polypeptide according to claim 16, wherein the one or more blocking groups are selected from N-terminal blocking groups comprising desamino analogs of amino acids, which are either coupled to the N-terminus of the peptide or used in place of the N-terminal amino acid residue.
21 . The polypeptide according to claim 16, wherein the one or more blocking groups are selected from C-terminal blocking groups wherein the carboxyl group of the C-terminus is either incorporated or not, such as esters, ketones, and amides, as well as descarboxylated amino acid analogues.
22. The polypeptide according to claim 16, wherein the one or more blocking groups are selected from C-terminal blocking groups comprising ester or ketone-forming alkyi groups, such as lower (Ci to C6) alkyi groups, for example methyl, ethyl and propyl, and amide-forming amino groups, such as primary amines (-NH2), and mono- and di-alkylamino groups, such as methylamino, ethylamino, dimethylamino, diethylamino, methylethylamino, and the like.
23. The polypeptide according to claim 16, wherein free amino group(s) at the N-terminal end and free carboxyl group(s) at the termini can be removed altogether from the polypeptide to yield desamino and descarboxylated forms thereof without significantly affecting the biological activity of the polypeptide.
24. An isolated polynucleotide for use as a medicament comprising a nucleic acid or its complementary sequence, said polynucleotide being selected from the group consisting of:
a) i) a polynucleotide encoding an amino acid sequence consisting of SEQ ID No. 1 , 3 or 5;or
ii) a biologically active sequence variant of the amino acid sequence, wherein the variant has at least 70% sequence identity to said SEQ ID NO. 1 , 3 or 5; and iii) a biologically active fragment of at least 50 contiguous amino acids of any of a) through b), wherein said fragment is a fragment of SEQ ID NO 1 , 3 or 5 or
b) SEQ ID NO.: 2, 4, 6, 7, 8 or 9 or
c) a polynucleotide comprising a nucleic acid having 70% sequence identity to SEQ ID NO.: 2, 4, 6, 7, 8 or 9 or
d) a polynucleotide hybridising to SEQ ID NO.: 2, 4, 6, 7, 8 or 9 and e) a polynucleotide complementary to any of a) to d).
25. The polynucleotide according to claim 24, wherein said polynucleotideis i) a polynucleotide encoding an amino acid sequence consisting of SEQ ID No. 1 , 3 or 5; or
ii) a biologically active sequence variant of the amino acid
sequence, wherein the variant has at least 70% sequence identity to said SEQ ID NO. 1 , 3 or 5 and
iii) iii) a biologically active fragment of at least 50 contiguous amino acids of any of a) through b), wherein said fragment is a fragment of SEQ ID NO 1 , 3 or 5.
26. The polynucleotide according to claim 24, wherein said polynucleotide is SEQ ID NO.: 2, 4, 6, 7, 8 or 9.
27. The polynucleotide according to claim 24, wherein said polynucleotideis a polynucleotide comprising a nucleic acid having 70% sequence identity to SEQ ID NO.: 2, 4, 6, 7, 8 or 9.
28. The polynucleotide according to claim 24, wherein said polynucleotide is capable of hybridising to a polynucleotide having the sequence of SEQ ID NO.: 2, 4, 6, 7, 8 or 9.
29. The polypeptide according to claim 24, wherein said polynucleotide is complementary to
i) a polynucleotide encoding an amino acid sequence consisting of SEQ ID No. 1 , 3 or 5; or a biologically active sequence variant of the amino acid sequence, wherein the variant has at least 70% sequence identity to said SEQ ID NO. 1 , 3 or 5 and
iii) a biologically active fragment of at least 50 contiguous acids of any of a) through b), wherein said fragment is a fragment of SEQ ID NO 1 , 3 or 5.
30. The polynucleotide according to claim 24, wherein said polypeptide is complementary to SEQ ID NO.: 2, 4, 6, 7, 8 or 9.
31 . The polynucleotideaccording to claim 24, wherein said polynucleotideis complementary to a polynucleotide comprising a nucleic acid having 70% sequence identity to SEQ ID NO.: 2, 4, 6, 7, 8 or 9
32. The polynucleotideof any of claims 24 to 31 , wherein the nucleic acid sequence of said polynucleotide differs by a single nucleotide from a nucleic acid sequence selected from the group consisting of SEQ ID NO: 2, 4, 6, 7, 8 and 9.
33. The polynucleotide according to any of claims 24 to 31 , wherein said nucleic acid sequence of said polynucleotidehas at least 80% sequence identity, such as preferably at least 81 % sequence identity, more preferably e.g. at least 82% sequence identity, such as more preferably at least 83% sequence identity, e.g. more preferably at least 84% sequence identity, more preferably such as at least 85% sequence identity, more preferably e.g. at least 86% sequence identity, more preferably such as at least 87% sequence identity, more preferably e.g. at least 88% sequence identity, more preferably such as at least 89% sequence identity, more preferably e.g. at least 90% sequence identity, more preferably such as at least 91 % sequence identity, more preferably e.g. at least 92% sequence identity, such as at least 93% sequence identity, more preferably e.g. at least 94% sequence identity, more preferably such as at least 95% sequence identity, more preferably e.g. at least 96% sequence identity, more preferably such as at least 97% sequence identity, more preferably e.g. at least 98% sequence identity, more preferably such as at least 99% sequence identity, more preferably e.g. at least 99.5% sequence identity to a nucleotide sequence selected from the group consisting of SEQ ID No. 2, 4, 6, 7, 8, and 9;
34. The polynucleotide according to any of claims 24 to 31 , wherein said nucleic acid sequence of the polynucleotide, contains a contiguous nucleic acid sequence fragment less than 99.5%, such as less than 98%, e.g. less than 97%, such as less than 96%, e.g. less than 95%, such as less than 94%, e.g. less than 93%, such as less than 92%, e.g. less than 91 %, such as less than 90%, e.g. less than 88%, such as less than 86%, e.g. less than 84%, e.g. less than 82%, such as less than 80%, e.g. less than 75%, such as less than 70%, e.g. less than 65%, such as less than 60%, e.g. less than 55%, such as less than 50%, e.g. less than 45%, such as less than 40%, e.g. less than 35%, such as less than 30%, e.g. less than 25%, such as less than 20%, such as less than 15%, e.g. less than 10% of nucleotide sequence selected from the group consisting of SEQ ID No. 2, 4, 6, 7, 8, and 9.
35. The polynucleotide according to any of claims 24 to 31 , wherein the nucleic acid sequence of the polynucleotide is capable of hybridizing to the nucleic acid sequence selected from the group consisting of SEQ ID NO: SEQ ID NO: 2, 4, 6, 7, 8 and 9, or a fragment thereof of 10 contiguous nucleic acids, under stringent conditions.
36. The polynucleotide according to any of claims 24 to 35, wherein the nucleic acids of said polynucleotide is DNA, RNA, PNA or LNA.
37. An expression vector comprising the polynucleotide of any of claims 24 to 36 for medical use.
38. The vector according to claim 37, further comprising a promoter operably linked to said polynucleotide.
39. The vector of claim 38, wherein the promoter is selected from CMV, SV40, AdMLP and elongation factor-1 alpha, Rous sarcoma virus long terminal repeat promoter.
40. An isolated host cell transfected or transduced with the vector of any of claims 37 to 39 for medical use.
41 . The host cell of claim 40 being selected from E.coli, Yeast,
Saccharomyces cerevisiae, S. pombe, insect cells.
42. The host cell of claim 40, wherein said host cell is a mammalian cell, such as human, feline, porcine, simian, canine, murine, rat, mouse or rabbit.
The host cell of claim 40, wherein said host cell is a mammalian cell.
44. The host cell of claim 42, wherein said host cell is a human embryonal kidney cells (HEK cells), such as the cell lines deposited at the American Type Culture Collection with the numbers CRL-1573 and CRL-10852, chick embryo fibroblast, hamster ovary cells, baby hamster kidney cells, human cervical carcinoma cells, human melanoma cells, human kidney cells, human umbilical vascular endothelium cells, human brain endothelium cells, human oral cavity tumor cells, monkey kidney cells, mouse fibroblast, mouse kidney cells, mouse connective tissue cells, mouse oligodendritic cells, mouse macrophage, mouse fibroblast, mouse neuroblastoma cells, mouse pre-B cell, mouse B lymphoma cells, mouse plasmacytoma cells, mouse teratocacinoma cells, rat astrocytoma cells, rat mammary epithelium cells, COS, CHO, BHK, 293, VERO, HeLa, MDCK, WI38, HTC1 16 cell or NIH 3T3 cell.
45. A pharmaceutical composition comprising
a) the isolated polypeptide of any of claims 1 to 23 or
b) the isolated polynucleotide of any of the claims 24 to 36 or c) the vector of any of the claims 37 to 39 or
d) the host cell of any of the claims 40 to 44
and a pharmaceutical acceptable carrier and/or diluent.
46. The pharmaceutical composition according to claim 45, wherein said pharmaceutical composition further comprises at least one additional therapeutic agent.
47. The pharmaceutical composition according to claim 46, wherein said additional therapeutic agent is selected from the group consisting of anti-TNF antibodies, antigen-binding fragments thereof, receptor molecules which bind specifically to TNF; compounds which prevent and/or inhibit TNF synthesis, TNF release or its action on target cells, such as thalidomide, tenidap, phosphodiesterase inhibitors (e.g, pentoxifylline and rolipram), A2b adenosine receptor agonists and A2b adenosine receptor enhancers; compounds which prevent and/or inhibit TNF receptor signalling, such as mitogen activated protein (MAP) kinase inhibitors; compounds which block and/or inhibit membrane TNF cleavage, such as metalloproteinase inhibitors; compounds which block and/or inhibit TNF activity, such as angiotensin converting enzyme (ACE) inhibitors (e.g., captopril); and compounds which block and/or inhibit TNF production and/or synthesis, such as MAP kinase inhibitors.
48. The pharmaceutical composition according to claim 46, wherein said additional therapeutic agent is selected from the group consisting of anti-TNF- alpha monoclonal antibodies: infliximab (Remicade), adalimumab (Humira), certolizumab pegol (Cimzia), golimumab (Simponi), TNF-alpha receptor fusion protein: etanercept (Enbrel), or TNF-alpha inhibitory molecules: pentoxifylline or bupropion.
49. A kit of parts comprising a therapeutic agent as defined in claims 47 to 48 and the isolated polypeptide of any of claims 1 to 23, or the isolated
polynucleotide of any of the claims 24 to 36, or the vector of any of the claims 37 to 39, or the host cell of any of the claims 40 to 44 as a combination for the simultaneous, separate or successive administration in TNFR1 related disease therapy.
50. A pharmaceutical composition for treating, ameliorating and/or preventing TNF receptor related diseases comprising
a) the isolated polypeptide of any of claims 1 to 23 or
b) the isolated nucleic acid sequence of any of the claims 24 to 36 or c) the vector of any of the claims 37 to 39 or
d) the host cell of any of the claims 40 to 44
e) the kit of parts of claim 49
51 . A kit comprising
a) at least one component selected from
i) the isolated polypeptide of any of claims 1 to 23 or
ii) the isolated polynucleotide of any of the claims 24 to 36 or
iii) the vector of any of the claims 37 to 39 or
iv) the host cell of any of the claims 40 to 44 or
v) the pharmaceutical composition of any of claims 45 to 48 or
vi) the kit of parts of claim 49, and
b) an instruction manual.
52. Method for treating, ameliorating and/or preventing TNF receptor related diseases comprising administration of
a) the isolated polypeptide of any of claims 1 to 23 or
b) the isolated polynucleotide of any of the claims 24 to 36 or c) the vector of any of the claims 37 to 39 or
d) the host cell of any of the claims 40 to 44 or
e) the pharmaceutical composition of any of claims 45 to 48 or f) the kit of parts of claim 49
g) the kit of claim 51
in a therapeutically effective amount to an individual in need thereof
53. An isolated polypeptide of as defined in any of claims 1 to 23 or an isolated nucleic acid sequence as defined in any of claims 24 to 36 or a vector of any of the claims 37 to 39 or
a host cell of any of the claims 40 to 44 or
a pharmaceutical composition of any of claims 45 to 48 or
a kit of parts of claim 49 or
a kit of claim 51 for the treatment of TNF receptor related diseases
54. Use of
a) the isolated polypeptide of any of claims 1 to 23 or
b) the isolated polynucleotide of any of the claims 24 to 36 or c) the vector of any of the claims 37 to 39 or
d) the host cell of any of the claims 40 to 44 or e) the pharmaceutical composition of any of claims 45 to 48 or f) the kit of parts of claim 49
g) the kit of claim 51
for the manufacture of a medicament for treatment of TNF receptor related diseases.
55. A method for decreasing TNF receptor activity comprising
i) contacting a TNF receptor with
a) the isolated polypeptide of any of claims 1 to 23 or
b) the isolated polynucleotide of any of the claims 24 to 36 or c) the vector of any of the claims 37 to 39 or
d) the host cell of any of the claims 40 to 44 or
e) the pharmaceutical composition of any of claims 45 to 48 or f) the kit of parts of claim 49
g) the kit of claim 51
ii) obtaining a decrease in TNF receptor activity compared to a standard level of TNF receptor activity in an individual.
56. The method according to claim 55, wherein said receptor is the TNFR1 and/or TNFR2.
57. The method according to claim 55, wherein said receptor is TNFR1 .
58. The method according to claim 55, wherein said receptor activity is signal transduction.
59. The method according to claim 55, wherein said receptor activity is induction of apoptosis.
60. The method according to claim 59, wherein said receptor activity is apoptosis induced by TNF-alpha activity.
61 . The method according to any of claims 55 to 60, wherein said decreased TNF receptor activity is at least 10%, preferably such as at least 15%, more preferably such as at least 25%, more preferably such as at least 30%, more preferably such as at least 35%, more preferably such as at least 40%, more preferably such as at least 45%, more preferably such as at least 50%, more preferably such as at least 55%, more preferably such as at least 60%, more preferably such as at least 70%, more preferably such as at least 75%, more preferably such as at least 80%, more preferably such as at least 85%, more preferably such as at least 90%, more preferably such as at least 95%, more preferably such as at least 96%, more preferably such as at least 97%, more preferably such as at least 98%, more preferably such as at least 99%, more preferably such as 100% of a standarc I TNF receptor activity in an individual.
62. The methods according to any of claims 54 to 61 wherein said method is performed in vitro.
63. The methods according to any of claims 54 to 61 , wherein said method is performed in vivo.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DKPA201070044 | 2010-02-08 | ||
| DKPA201070044 | 2010-02-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011095174A1 true WO2011095174A1 (en) | 2011-08-11 |
Family
ID=43838033
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/DK2011/050032 WO2011095174A1 (en) | 2010-02-08 | 2011-02-08 | Human herpes virus 6 and 7 u20 polypeptide and polynucleotides for use as a medicament or diagnosticum |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2011095174A1 (en) |
Citations (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4469863A (en) | 1980-11-12 | 1984-09-04 | Ts O Paul O P | Nonionic nucleic acid alkyl and aryl phosphonates and processes for manufacture and use thereof |
| US4476301A (en) | 1982-04-29 | 1984-10-09 | Centre National De La Recherche Scientifique | Oligonucleotides, a process for preparing the same and their application as mediators of the action of interferon |
| US4588580A (en) | 1984-07-23 | 1986-05-13 | Alza Corporation | Transdermal administration of fentanyl and device therefor |
| US4788062A (en) | 1987-02-26 | 1988-11-29 | Alza Corporation | Transdermal administration of progesterone, estradiol esters, and mixtures thereof |
| US4816258A (en) | 1987-02-26 | 1989-03-28 | Alza Corporation | Transdermal contraceptive formulations |
| US4904475A (en) | 1985-05-03 | 1990-02-27 | Alza Corporation | Transdermal delivery of drugs from an aqueous reservoir |
| US4927408A (en) | 1988-10-03 | 1990-05-22 | Alza Corporation | Electrotransport transdermal system |
| US5023243A (en) | 1981-10-23 | 1991-06-11 | Molecular Biosystems, Inc. | Oligonucleotide therapeutic agent and method of making same |
| US5177196A (en) | 1990-08-16 | 1993-01-05 | Microprobe Corporation | Oligo (α-arabinofuranosyl nucleotides) and α-arabinofuranosyl precursors thereof |
| US5188897A (en) | 1987-10-22 | 1993-02-23 | Temple University Of The Commonwealth System Of Higher Education | Encapsulated 2',5'-phosphorothioate oligoadenylates |
| US5264423A (en) | 1987-03-25 | 1993-11-23 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5276019A (en) | 1987-03-25 | 1994-01-04 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5278302A (en) | 1988-05-26 | 1994-01-11 | University Patents, Inc. | Polynucleotide phosphorodithioates |
| US5321131A (en) | 1990-03-08 | 1994-06-14 | Hybridon, Inc. | Site-specific functionalization of oligodeoxynucleotides for non-radioactive labelling |
| US5399676A (en) | 1989-10-23 | 1995-03-21 | Gilead Sciences | Oligonucleotides with inverted polarity |
| US5405939A (en) | 1987-10-22 | 1995-04-11 | Temple University Of The Commonwealth System Of Higher Education | 2',5'-phosphorothioate oligoadenylates and their covalent conjugates with polylysine |
| US5455233A (en) | 1989-11-30 | 1995-10-03 | University Of North Carolina | Oligoribonucleoside and oligodeoxyribonucleoside boranophosphates |
| US5466677A (en) | 1993-03-06 | 1995-11-14 | Ciba-Geigy Corporation | Dinucleoside phosphinates and their pharmaceutical compositions |
| US5476925A (en) | 1993-02-01 | 1995-12-19 | Northwestern University | Oligodeoxyribonucleotides including 3'-aminonucleoside-phosphoramidate linkages and terminal 3'-amino groups |
| US5519126A (en) | 1988-03-25 | 1996-05-21 | University Of Virginia Alumni Patents Foundation | Oligonucleotide N-alkylphosphoramidates |
| US5550111A (en) | 1984-07-11 | 1996-08-27 | Temple University-Of The Commonwealth System Of Higher Education | Dual action 2',5'-oligoadenylate antiviral derivatives and uses thereof |
| US5571799A (en) | 1991-08-12 | 1996-11-05 | Basco, Ltd. | (2'-5') oligoadenylate analogues useful as inhibitors of host-v5.-graft response |
| US5587361A (en) | 1991-10-15 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides having phosphorothioate linkages of high chiral purity |
| US5625050A (en) | 1994-03-31 | 1997-04-29 | Amgen Inc. | Modified oligonucleotides and intermediates useful in nucleic acid therapeutics |
| US5770722A (en) | 1994-10-24 | 1998-06-23 | Affymetrix, Inc. | Surface-bound, unimolecular, double-stranded DNA |
| US6379666B1 (en) | 1999-02-24 | 2002-04-30 | Edward L. Tobinick | TNF inhibitors for the treatment of neurological, retinal and muscular disorders |
| US6509015B1 (en) | 1996-02-09 | 2003-01-21 | Basf Aktiengesellschaft | Human antibodies that bind human TNFa |
| US6524821B1 (en) * | 1998-07-31 | 2003-02-25 | Centre de Recherche du Centre Hospitalier de l'Université de Montréal | Anti-apoptotic compositions comprising the R1 subunit of herpes simplex virus ribonucleotide reductase or its N-terminal portion; and uses thereof |
| US20050191696A1 (en) * | 1998-05-18 | 2005-09-01 | Apoptosis Technology, Inc. | Compounds, methods of screening, and in vitro and in vivo uses involving anti-apoptotic genes and anti-apoptotic gene products |
| WO2006038027A2 (en) | 2004-10-08 | 2006-04-13 | Domantis Limited | SINGLE DOMAIN ANTIBODIES AGAINST TNFRl AND METHODS OF USE THEREFOR |
| WO2006056779A2 (en) | 2004-11-25 | 2006-06-01 | Ucb Pharma S.A. | ANTI-TNF ALPHA ANTIBODIES WHICH SELECTIVELY INHIBIT TNF ALPHA SIGNALLING THROUGH THE p55R |
| EP1681346A1 (en) * | 2003-08-29 | 2006-07-19 | Soiken Inc. | Recombinant virus vector originating in hhv-6 or hhv-7, method of producing the same, method of transforming host cell using the same, host cell transformed thereby and gene therapy method using the same |
| US20080226677A1 (en) * | 2004-05-06 | 2008-09-18 | Yasuko Mori | Recombinant virus vector for gene transfer into lymphoid cells |
| EP1309691B1 (en) | 2000-08-07 | 2009-10-21 | Centocor Ortho Biotech Inc. | Anti-tnf antibodies, compositions, methods and uses |
-
2011
- 2011-02-08 WO PCT/DK2011/050032 patent/WO2011095174A1/en active Application Filing
Patent Citations (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4469863A (en) | 1980-11-12 | 1984-09-04 | Ts O Paul O P | Nonionic nucleic acid alkyl and aryl phosphonates and processes for manufacture and use thereof |
| US5023243A (en) | 1981-10-23 | 1991-06-11 | Molecular Biosystems, Inc. | Oligonucleotide therapeutic agent and method of making same |
| US4476301A (en) | 1982-04-29 | 1984-10-09 | Centre National De La Recherche Scientifique | Oligonucleotides, a process for preparing the same and their application as mediators of the action of interferon |
| US5550111A (en) | 1984-07-11 | 1996-08-27 | Temple University-Of The Commonwealth System Of Higher Education | Dual action 2',5'-oligoadenylate antiviral derivatives and uses thereof |
| US4588580B1 (en) | 1984-07-23 | 1989-01-03 | ||
| US4588580B2 (en) | 1984-07-23 | 1999-02-16 | Alaz Corp | Transdermal administration of fentanyl and device therefor |
| US4588580A (en) | 1984-07-23 | 1986-05-13 | Alza Corporation | Transdermal administration of fentanyl and device therefor |
| US4904475A (en) | 1985-05-03 | 1990-02-27 | Alza Corporation | Transdermal delivery of drugs from an aqueous reservoir |
| US4816258A (en) | 1987-02-26 | 1989-03-28 | Alza Corporation | Transdermal contraceptive formulations |
| US4788062A (en) | 1987-02-26 | 1988-11-29 | Alza Corporation | Transdermal administration of progesterone, estradiol esters, and mixtures thereof |
| US5286717A (en) | 1987-03-25 | 1994-02-15 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5264423A (en) | 1987-03-25 | 1993-11-23 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5276019A (en) | 1987-03-25 | 1994-01-04 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5188897A (en) | 1987-10-22 | 1993-02-23 | Temple University Of The Commonwealth System Of Higher Education | Encapsulated 2',5'-phosphorothioate oligoadenylates |
| US5405939A (en) | 1987-10-22 | 1995-04-11 | Temple University Of The Commonwealth System Of Higher Education | 2',5'-phosphorothioate oligoadenylates and their covalent conjugates with polylysine |
| US5519126A (en) | 1988-03-25 | 1996-05-21 | University Of Virginia Alumni Patents Foundation | Oligonucleotide N-alkylphosphoramidates |
| US5278302A (en) | 1988-05-26 | 1994-01-11 | University Patents, Inc. | Polynucleotide phosphorodithioates |
| US5453496A (en) | 1988-05-26 | 1995-09-26 | University Patents, Inc. | Polynucleotide phosphorodithioate |
| US4927408A (en) | 1988-10-03 | 1990-05-22 | Alza Corporation | Electrotransport transdermal system |
| US5399676A (en) | 1989-10-23 | 1995-03-21 | Gilead Sciences | Oligonucleotides with inverted polarity |
| US5455233A (en) | 1989-11-30 | 1995-10-03 | University Of North Carolina | Oligoribonucleoside and oligodeoxyribonucleoside boranophosphates |
| US5321131A (en) | 1990-03-08 | 1994-06-14 | Hybridon, Inc. | Site-specific functionalization of oligodeoxynucleotides for non-radioactive labelling |
| US5536821A (en) | 1990-03-08 | 1996-07-16 | Worcester Foundation For Biomedical Research | Aminoalkylphosphorothioamidate oligonucleotide deratives |
| US5541306A (en) | 1990-03-08 | 1996-07-30 | Worcester Foundation For Biomedical Research | Aminoalkylphosphotriester oligonucleotide derivatives |
| US5563253A (en) | 1990-03-08 | 1996-10-08 | Worcester Foundation For Biomedical Research | Linear aminoalkylphosphoramidate oligonucleotide derivatives |
| US5177196A (en) | 1990-08-16 | 1993-01-05 | Microprobe Corporation | Oligo (α-arabinofuranosyl nucleotides) and α-arabinofuranosyl precursors thereof |
| US5571799A (en) | 1991-08-12 | 1996-11-05 | Basco, Ltd. | (2'-5') oligoadenylate analogues useful as inhibitors of host-v5.-graft response |
| US5587361A (en) | 1991-10-15 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides having phosphorothioate linkages of high chiral purity |
| US5476925A (en) | 1993-02-01 | 1995-12-19 | Northwestern University | Oligodeoxyribonucleotides including 3'-aminonucleoside-phosphoramidate linkages and terminal 3'-amino groups |
| US5466677A (en) | 1993-03-06 | 1995-11-14 | Ciba-Geigy Corporation | Dinucleoside phosphinates and their pharmaceutical compositions |
| US5625050A (en) | 1994-03-31 | 1997-04-29 | Amgen Inc. | Modified oligonucleotides and intermediates useful in nucleic acid therapeutics |
| US5770722A (en) | 1994-10-24 | 1998-06-23 | Affymetrix, Inc. | Surface-bound, unimolecular, double-stranded DNA |
| US6509015B1 (en) | 1996-02-09 | 2003-01-21 | Basf Aktiengesellschaft | Human antibodies that bind human TNFa |
| US20050191696A1 (en) * | 1998-05-18 | 2005-09-01 | Apoptosis Technology, Inc. | Compounds, methods of screening, and in vitro and in vivo uses involving anti-apoptotic genes and anti-apoptotic gene products |
| US6524821B1 (en) * | 1998-07-31 | 2003-02-25 | Centre de Recherche du Centre Hospitalier de l'Université de Montréal | Anti-apoptotic compositions comprising the R1 subunit of herpes simplex virus ribonucleotide reductase or its N-terminal portion; and uses thereof |
| US6379666B1 (en) | 1999-02-24 | 2002-04-30 | Edward L. Tobinick | TNF inhibitors for the treatment of neurological, retinal and muscular disorders |
| EP1309691B1 (en) | 2000-08-07 | 2009-10-21 | Centocor Ortho Biotech Inc. | Anti-tnf antibodies, compositions, methods and uses |
| EP1681346A1 (en) * | 2003-08-29 | 2006-07-19 | Soiken Inc. | Recombinant virus vector originating in hhv-6 or hhv-7, method of producing the same, method of transforming host cell using the same, host cell transformed thereby and gene therapy method using the same |
| US20080226677A1 (en) * | 2004-05-06 | 2008-09-18 | Yasuko Mori | Recombinant virus vector for gene transfer into lymphoid cells |
| WO2006038027A2 (en) | 2004-10-08 | 2006-04-13 | Domantis Limited | SINGLE DOMAIN ANTIBODIES AGAINST TNFRl AND METHODS OF USE THEREFOR |
| WO2006056779A2 (en) | 2004-11-25 | 2006-06-01 | Ucb Pharma S.A. | ANTI-TNF ALPHA ANTIBODIES WHICH SELECTIVELY INHIBIT TNF ALPHA SIGNALLING THROUGH THE p55R |
Non-Patent Citations (46)
| Title |
|---|
| "Controlled Drug Delivery: Fundamentals and Applications", 1987, MARCEL DEKKER INC. |
| "Current Protocols in Molecular Biology", 2001, JOHN WILEY AND SONS, INC |
| "Merck Manual", 1972, MERCK & COMPANY |
| "Merck Manual", 1992, MERCK & COMPANY |
| "PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000", 2000, TARASCON PUBLISHING |
| "Pharmacotherapy Handbook", 1998, APPLETON AND LANGE |
| "Pharmacotherapy Handbook", 2000, APPLETON AND LANGE |
| "Remington: The Science and Practice of Pharmacy", 1995, MACK PUBLISHING COMPANY |
| "The molecular biology of tumor viruses: RNA tumor viruses", 1982, COLD SPRING HARBOR LABORATORY |
| "Transdermal Delivery of Drugs", vol. 1-3, 1987, CRC PRESS |
| "Transdermal Drug Delivery: Developmental Issues and Research Initiatives", 1989, MARCEL DEKKER, INC. |
| AUSUBEL ET AL.: "Current Protocols in Molecular Biology", 1999 |
| BANERJI ET AL., CELL, vol. 27, 1981, pages 299 |
| BEAUCAGE; CARRUTHERS, TETRAHEDRON LETT., vol. 22, 1981, pages 1859 - 1862 |
| BREATHNACH; CHAMBON, ANN. REV. BIOCHEM., vol. 50, 1981, pages 349 |
| CAPECCHI ET AL.: "Enhancer and eukaryotic gene expression", 1991, COLD SPRING HARBOR LABORATORIES, pages: 101 - 102 |
| CORDEN ET AL., SCIENCE, vol. 209, 1980, pages 1406 |
| CROFT, NATURE REV IMMUNOL, vol. 9, 2009, pages 271 - 285 |
| DATABASE UniProt [Online] 1 May 2000 (2000-05-01), "SubName: Full=U20;", XP002634295, retrieved from EBI accession no. UNIPROT:Q9QJ46 Database accession no. Q9QJ46 * |
| DATABASE UniProt [Online] 1 November 1996 (1996-11-01), "RecName: Full=Glycoprotein U20; Flags: Precursor;", XP002634296, retrieved from EBI accession no. UNIPROT:Q69555 Database accession no. Q69555 * |
| DATABASE UniProt [Online] 1 November 1996 (1996-11-01), "SubName: Full=U20; SubName: Full=U20 protein;", XP002634297, retrieved from EBI accession no. UNIPROT:Q69502 Database accession no. Q69502 * |
| DOMINGUEZ ET AL., J VIROL, vol. 73, 1999, pages 8040 - 8052 |
| E. CREIGHTON: "Proteins-Structure and Molecular Properties", 1993, W. H. FREEMAN AND COMPANY |
| FORD ET AL., PROTEIN EXPRESSION AND PURIFICATION, vol. 2, 1991, pages 95 |
| GRUSSENMEYER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 82, 1985, pages 7952 |
| GUPTA SUDHIR ET AL: "Differential Effect of Human Herpesvirus 6A on Cell Division and Apoptosis among Naive and Central and Effector Memory CD4(+) and CD8(+) T-Cell Subsets", JOURNAL OF VIROLOGY, vol. 83, no. 11, June 2009 (2009-06-01), pages 5442 - 5450, XP002634293, ISSN: 0022-538X * |
| HOPP ET AL., BIOTECHNOLOGY, vol. 6, 1988, pages 1204 |
| J. BIOL. CHEM., vol. 243, 1969, pages 3552 - 59 |
| JOLLY ET AL., NUCLEIC ACIDS RES., vol. 11, 1983, pages 1855 |
| KARIN; GALLAGHER, IMMUNOL REV, vol. 228, 2009, pages 225 - 240 |
| MANIATIS ET AL., MOLECULAR CLONING, 1982, pages 133 - 134 |
| MANIATIS ET AL.: "Molecular Cloning: A Laboratory Manual", 1982, COLD SPRING HARBOR LABORATORY |
| MATTEUCCI ET AL., J. AM. CHEM. SOC., vol. 103, 1981, pages 3185 |
| MAXAM ET AL., METHODS IN ENZYMOLOGY, vol. 65, 1980, pages 499 |
| MESSING ET AL., NUCLEIC ACIDS RES., vol. 9, 1981, pages 309 |
| NEEDLEMAN; WUNSCH, J. MOL. BIOL., vol. 48, 1970, pages 443 |
| NILSSON ET AL., EMBO J., vol. 4, 1985, pages 1075 |
| NILSSON ET AL., METHODS ENZYMOL., vol. 198, no. 3, 1991 |
| OHYASHIKI J H ET AL: "Transcriptional profiling of human herpesvirus type B (HHV-6B) in an adult T cell leukemia cell line as in vitro model for persistent infection", BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS, ACADEMIC PRESS INC. ORLANDO, FL, US, vol. 329, no. 1, 1 April 2005 (2005-04-01), pages 11 - 17, XP004756990, ISSN: 0006-291X, DOI: DOI:10.1016/J.BBRC.2005.01.090 * |
| PEARSON; LIPMAN, PROC. NATL. ACAD. SCI. (U.S.A.), vol. 85, 1988, pages 2444 |
| RATTAN ET AL.: "Protein Synthesis: Posttranslational Modifications and Aging", ANN. N.Y. ACAD. SCI., vol. 663, 1992, pages 48 - 62, XP009082490, DOI: doi:10.1111/j.1749-6632.1992.tb38648.x |
| SEIFTER ET AL., METH. ENZYMOL., vol. 182, 1990, pages 626 - 646 |
| SMITH; JOHNSON, GENE, vol. 67, pages 31 |
| SMITH; WATERMAN, ADV. APPL. MATH., vol. 2, 1981, pages 482 |
| TAKEMOTO MASAYA ET AL: "Productive human herpesvirus 6 infection causes aberrant accumulation of p53 and prevents apoptosis.", THE JOURNAL OF GENERAL VIROLOGY APR 2004 LNKD- PUBMED:15039530, vol. 85, no. Pt 4, April 2004 (2004-04-01), pages 869 - 879, XP002634294, ISSN: 0022-1317 * |
| WOLD, F.: "Posttranslational Covalent Modification of Proteins", 1983, ACADEMIC PRESS, pages: 1 - 12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2458349T3 (en) | Homologous IL-17 polypeptides and therapeutic uses thereof | |
| US7045133B2 (en) | VEGF-D/VEGF-C/VEGF peptidomimetic inhibitor | |
| Tanaka et al. | RES-701-1, a novel, potent, endothelin type B receptor-selective antagonist of microbial origin. | |
| JP2002502589A (en) | 45 human secreted proteins | |
| EA015860B1 (en) | Methods for treatment of autoimmune diseases using neutrokine-alpha antagonist | |
| JP2001520039A (en) | Human tumor necrosis factor receptor-like proteins, TR11, TR11SV1 and TR11SV2 | |
| JP2002518010A (en) | 94 human secreted proteins | |
| JP2001524814A (en) | 28 human secreted proteins | |
| JP2009131263A (en) | 50 human secreted proteins | |
| JP2002533058A (en) | 97 human secreted proteins | |
| JP2002508167A (en) | 110 human secreted proteins | |
| JP2001521383A (en) | 20 human secretory proteins | |
| US20150299304A1 (en) | Modulation of the VPS10P-Domain Receptors for the Treatment of Cardiovascular Disease | |
| JP2002501738A (en) | 67 human secreted proteins | |
| CN101454341B (en) | Use of multivalent synthetic ligands of surface nucleolin for treating cancer or inflammation | |
| JP2003514541A (en) | 18 human secreted proteins | |
| JP2002532083A (en) | 47 human secreted proteins | |
| JP2002512521A (en) | 32 human secreted proteins | |
| US20180335433A1 (en) | Compositions targeting pkc-theta and uses and methods of treating pkc-theta pathologies, adverse immune responses and diseases | |
| JP2002500035A (en) | 36 human secreted proteins | |
| JP2002506625A (en) | Cytokine receptor common γ chain-like | |
| CA2441327A1 (en) | Human arginine-rich protein-related compositions | |
| JP2003508034A (en) | Cytokine receptor common gamma chain analog | |
| US20110124552A1 (en) | GMCSF and Truncated CCL2 Conjugates and Methods and Uses Thereof | |
| JP2003524366A (en) | 64 human secreted proteins |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11704400 Country of ref document: EP Kind code of ref document: A1 |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11704400 Country of ref document: EP Kind code of ref document: A1 |