WO2013108068A1 - Process for the preparation of 2-pyridinylmethylsulfinyl benzimidazoles, their analogs and optically active enantiomers - Google Patents
Process for the preparation of 2-pyridinylmethylsulfinyl benzimidazoles, their analogs and optically active enantiomers Download PDFInfo
- Publication number
- WO2013108068A1 WO2013108068A1 PCT/IB2012/001561 IB2012001561W WO2013108068A1 WO 2013108068 A1 WO2013108068 A1 WO 2013108068A1 IB 2012001561 W IB2012001561 W IB 2012001561W WO 2013108068 A1 WO2013108068 A1 WO 2013108068A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- process according
- solution
- methyl
- group
- analogs
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention discloses a commercially viable, cost effective and energy efficient process for the preparation of 2-pyridinylmethylsulfinyl benzimidazoles, their analogs and optically active enantiomers or pharmaceutically acceptable salts, hydrates or solvates thereof in high purity via application of reactors such as plug flow reactor, microreactor, microfluidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, fluidized bed reactor or fixed bed reactor.
- reactors such as plug flow reactor, microreactor, microfluidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, fluidized bed reactor or fixed bed reactor.
- Benzimidazole a heterocyclic aromatic organic compound, formed by fusion of benzene and imidazole, occurs naturally as N-ribosyl-dimethylbenzimidazole, which serves as axial ligand for cobalt in Vitamin B-12.
- Benzimidazole derivatives particularly, pyridinylmethylsulfinyl benzimidazole compounds are known to have H7K + -ATPase inhibitory action and therefore have considerable importance in the therapy of diseases associated with an increased secretion of gastric acid or used as an anti-ulcerative agent.
- pyridinylmethylsulfinyl substituted benzimidazole compounds or pharmaceutically acceptable salts thereof are known for example, from EP0005129, EP 174726, EP 166287, EP268956 and EP254588 as given in Formula I below:
- Rabeprazole is another compound of the same class and chemically known by 2-[[[(4-(3-methoxypropoxy)-2-methyl-2-pyridinyl]methyl]sulfinyl-lH- benzimidazole. It was reported in US 5,045,552 and marketed as sodium salt in the United States under the brand name Aciphex for healing of erosive or ulcerative GERD, maintenance of healing of GERD and treatment of symptomatic GERD.
- Pantoprazole is the active ingredient of a pharmaceutical product that is marketed as sodium salt in the United States by Wyeth-pharms Inc and was protected by US 4,758,579 and sold under the brand name Protonix. Pantoprazole is chemically represented (5-(difluoromethoxy)-2-[[(3,4-dimethoxy-2-pyridinyl)methyl]sulfinyl]- lH-benzimidazole. Pantoprazole useful for short-term treatment of erosive esophagitis associated with gastroesophageal reflux disease (GERD), maintenance of healing of erosive esophagitis and pathological hypersecretory conditions including Zollinger-Ellison syndrome.
- GSD gastroesophageal reflux disease
- Lansoprazole another compound represented by 2-[[[3-methyl-4(2,2,2,- triflouroethoxy)-2-pyridiyl]methyl]sulfinyl]-lH-benzimidazole and was reported in US 4,628,098. It is marketed under the brand name Prevacid(R) for short-term treatment of duodenal ulcer, ⁇ . Pylori eradication to prevent recurrence of duodenal ulcer and maintenance of healed duodenal ulcers.
- Tenatoprazole represented by 5-methoxy-2-((4-methoxy-3,5-dimethyl-2- pyridylmethyl) sulphinyl)-lH-imidazo (4,5-b) pyridine and was reported in US 4,808,596 and is marketed under brand name Ulsacare. Tenatoprazole is also a proton pump inhibitor indicated for the treatment of reflux oesophagitis and peptic ulcer.
- Ilaprazole another compound represented by 2-[(4-methoxy-3-methylpyridin-2-yl) methylsulfinyl]-6-pyrrol- l-yl- lH-benzimidazole was protected by US 5,703,097. It is used for the treatment of dyspepsia, peptic ulcer disease (PUD), gastroesophageal reflux disease (GORD/GERD) and duodenal ulcer.
- PID peptic ulcer disease
- GORD/GERD gastroesophageal reflux disease
- the preparation of 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazoles Formula (I) by oxidation of compound of Formula II is generally known and discussed in U.S Pat Nos. 4,255,431, 5,045,552, 4,508,905 and 4,628,098.
- CA 1,263,1 19 describes the use of hydrogen peroxide over a vanadium catalyst (such as vanadium pentoxide, sodium vanadate and vanadium acteylacetonate).
- CA 1, 127,158 similarly describes the use of peracids, peresters, ozone, etc.
- EP 533,264 describes the use of magnesium monoperoxyphthalate as the oxidizing agent.
- W091/18895 describes the use of m-chloroperoxy benzoic acid as the oxidizing agent.
- GB 2,069,492 generally describes this m-chloroperoxybenzoic acid along with other peroxy acids for the oxidation of substituted (phenylthiomethyl)pyridines.
- US 5,374,730 relates to omeprazole and lansoprazole, in particular, two novel synthetic methods for their preparation.
- the sulfinyl compounds are produced from the corresponding acetamide sulfide compound using hydrogen peroxide as oxidizing agent to form amide sulfinyl compound, followed by alkaline hydrolysis to the sulfinyl carboxylate or salt, and decarboxylation.
- US 6,313,303 discloses the process for preparing rabeprazole, lansoprazole and other related compounds by oxidation of thioether precursor compound with N- halosuccinamide, l,3-dihalo-5,5-dimethylhydantoin or dichloroisocyanurate in the presence of a base.
- EP 484,265 discloses a process for the preparation of omeprazole (example 32 and 33) by oxidation of 2-(((3,5-dimethyl-4-methoxy-2- pyridinyl)methyl)sulfinyl)-5-methoxy-lH-benzimidazole in suitable organic solvent with 50% H 2 0 2 in presence of catalyst like (P(W 3 Oio)4.xH 2 0; ammonium molybdate, having the formula (NFU ⁇ MoC ⁇ ; sodium tungstate, having the formula Na 2 W04; phosphomolybdic acid, having the formula ⁇ 3 ( ⁇ ( ⁇ 3 ⁇ 0 )4 ⁇ 2 ⁇ ; and silicotungstic acid, having the formula H4(Si(W 3 Oio)4.xH 0 at lower temperatures.
- catalyst like P(W 3 Oio)4.xH 2 0
- ammonium molybdate having the formula (NFU ⁇ MoC ⁇
- sodium tungstate having the formula
- WO2012/004802 discloses a continuous micromixer based process for the synthesis of sulphoxide compounds with a reaction time of less than or equal to one minute having_ selectivity of > 95% towards the sulphoxide compounds,
- the process has several drawbacks with respect to (i)selection of oxidizing agent (ii) selectivity (iii) use of solvent, (iv) micromixer design etc.
- WO'802 refers that the reaction of imidazo[4,5-b]pyridine compound using oxidizing agent dissolved in solvent to obtain sulphoxide, wherein the commonly used oxidizing agents such as H 2 0 2j sodium hypochlorite gives only of 2 to 3% conversion, which is highly undesirable on commercial scale.
- a plug flow reactor is a tubular reactor is fundamentally a continuous reactor where there are no moving parts other than pumps that deliver the reactants.
- static mixing elements such as glass beads inside the tubular reactor is done that provides ideal conditions of radial mixing and near plug flow necessary to perform reactions.
- the flow of reactants pumped in the reactor is laminar and the properties of the reaction medium i.e. pressure, temperature, reactant and product concentrations are the same throughout the entire cross section of flow.
- Plug flow reactors usually operate in adiabatic and non-isothermal conditions. Consequently, from the standpoint of kinetic parameters of a chemical reaction under isothermal conditions, plug flow reactors are more efficient than stirred tank reactors.
- Microreactors have been defined as "Microsystems fabricated, at least partially, by methods of microtechnology and precision engineering. Fluid channels range from about 1 um (nanoreactors) to about 1 mm (minireactors)." See Microreactors, Ehrfeld, Hessel & Lowe 2000, the entire disclosure of which is incorporated herein by reference.
- microreactors consist of miniaturized channels, often imbedded in a flat surface referred to as the "chip.” These flat surfaces can be glass plates or plates of metals such as stainless steel or Hastelloy. Microreactors have proven to be highly valuable tools in organic chemistry due to their wide flexibility of operating conditions with efficient heat transfer, optimized mixing, and high reaction control.
- the continuous mode of operation in microreactor offers several advantages over a batch process such as simplified operations, reduced reaction time, precise process control, higher reproducibility, and in some cases, even enhanced reaction selectivity.
- the immediate separation of products from the reaction mixture eliminates possible side products that may result from secondary reactions.
- the continuous mode may also offer a means to replace large reactors, which are costly and take up expensive laboratory or chemical plant space.
- microreactors offer their own unique advantages over traditional continuous processing systems including the highly efficient heat transfer and small reaction volumes that allow safer handling of exothermic reactions, reactions involving explosive and toxic materials, increased precision to reaction control and other hazardous reactions that are normally difficult to scale up.
- Microreactors are particularly useful for rapid optimization, screening different reaction conditions, catalysts, ligands, bases, and solvents; mechanistic studies; cost effective industrial scale up; and rapid screening for new pharmaceuticals.
- the microreactors can be operated to produce large amounts of the desired end product by using the concept, wherein multiple microreactors are run continuously and in series and/or in parallel to simulate a large scale flow reactor, improving the productivity of the plant.
- the present invention provides an improved method for the preparation of 2- [(pyridinyl)methyl]sulfinyl-substituted benzimidazoles, their analogs and optically active enantiomers or pharmaceutically acceptable salts, hydrates or solvates thereof using reactors such as plug flow reactor, microreactor, microfiuidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, fluidized bed reactor or fixed bed reactor.
- reactors such as plug flow reactor, microreactor, microfiuidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, fluidized bed reactor or fixed bed reactor.
- the principal object of the present invention is to provide a process for the preparation of 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazoles, their analogs and optically active enantiomers or pharmaceutically acceptable salts, hydrates or solvates thereof using reactors such as plug flow reactor, microreactor, microfluidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, fluidized bed reactor or fixed bed reactorwhich alleviates the drawbacks of prior art process.
- reactors such as plug flow reactor, microreactor, microfluidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, fluidized bed reactor or fixed bed reactorwhich alleviates the drawbacks of prior art process.
- the present invention provides a process for preparation of 2-[(pyridinyI)methyl]suIfinyl-substituted benzimidazole of Formula I, their analogs and optically active enantiomers or pharmaceutically acceptable salts, hydrates or solvates thereof,
- Ri, R 2 , R 3 , 4, R 5 , R 6 , R 7 are the same or different and selected from hydrogen, C 1 -7 alkyl, C 1 .7 alkoxy, optionally substituted by halogen, l -aza-2,4- cyclopentadiene, R 8 is hydrogen or nitrogen protecting group, A is carbon or nitrogen.
- the process employs application of reactors such as plug flow reactor, microreactor, microfluidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, fluidized bed reactor or fixed bed reactor, for the preparation of desired product in high yield and high purity with enhanced in-process control on impurities specifically sulphone impurity with shorter reaction time.
- reactors such as plug flow reactor, microreactor, microfluidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, fluidized bed reactor or fixed bed reactor
- Ri , R 2 , R 3 , R4, R 5 , R 6 , R7 are the same or different and selected from hydrogen, Ci. 7 alkyl, C1-7 alkoxy, optionally substituted by halogen, l -aza-2,4- cyclopentadiene, R 8 is hydrogen or nitrogen protecting group, A is carbon or nitrogen comprising the steps of:
- the preparation of solution A is optionally carried out in solvent depending upon the type of reactor selected for the oxidation reaction e.g. microreactor.
- the solvent used for the preparation of solution A is selected from the group comprising of alcohols such as methanol, ethanol, propanol, butanol, ethylene glycol and the like; aromatic hydrocarbon such as toluene, xylene and the like, ethers such as diisopropyl ether, t-butyl methyl ether, tetrahydrofuran, dioxane, monoglyme, diglyme and the like; esters such as ethyl acetate, methyl acetate and the like, halogenated hydrocarbons such as dichloromethane, chloroform, dichloroethane and the like; ketones such as acetone, methyl ethyl ketone, ethyl isobutyl ketone and the like; nitriles such as aceton
- the preparation of solution A is optionally carried out in presence of a base, wherein the base is selected from organic or inorganic.
- the organic base is selected from amines such as triethylamine, diisopropylamine, diisopropylethylamine, piperidine and the like, alkali metal alkoxide such as sodium methoxide, potassium methoxide and the like.
- the inorganic base is selected from the ammonia, alkali and alkaline earth metal carbonate, bicarbonate, hydroxide, hydride and the like wherein alkali and alkaline earth metal is selected from a group comprising of lithium, sodium, potassium, magnesium, calcium, barium and the like.
- the oxidizing agent in solution B is selected from the group comprising of hydrogen peroxide, alkyl hydro peroxide, aryl alkyl hydro peroxide, alkali or alkaline earth metal, hypohalite, wherein the alkali or alkaline earth metal is selected from a group comprising of sodium, lithium, potassium, magnesium, calcium and the halite is selected from the group comprising of fluorite, chlorite, bromite and the like.
- the solvent is selected form the group comprising of alcohols such as methanol, ethanol, propanol, butanol, ethylene glycol and the like; aromatic hydrocarbons such as toluene, xylene and the like, ethers such as diisopropyl ether, t-butyl methyl ether, tetrahydrofuran, dioxane, monoglyme, diglyme and the like; esters such as ethyl acetate, methyl acetate and the like; halogenated hydrocarbons such as dichloromethane, chloroform, dichloroethane and the like; ketones such as acetone, methyl ethyl ketone, ethyl isobutyl ketone and the like; nitriles such as acetonitrile, propionitrile and the like; aprotic polar solvents such as ⁇ , ⁇ -dimethylformamide, dimethylsulphoxide, hexamethylphospho
- the preparation of solution B is optionally carried out in presence of a base, wherein the base is selected from organic or inorganic.
- the organic base is selected from amines such as triethylamine, diisopropylamine, diisopropylethylamine, piperidine and the like, alkali metal alkoxide such as sodium methoxide, potassium methoxide and the like.
- the inorganic base is selected from the ammonia, alkali or alkaline earth metal carbonate, bicarbonate, hydroxide, hydride and the like wherein alkali and alkaline earth metal is selected from a group comprising of lithium, sodium, potassium, magnesium, calcium, barium and the like.
- the oxidation reaction is carried out in a continuous mode, wherein the reactor carries material as a flowing stream and reactants are continuously fed into the reactor and emerge as continuous stream of product.
- the oxidation reaction isoptionally carried out in the presence of a metal catalyst.
- the metal of the metal catalyst is selected from the group comprising of rhenium, vanadium, molybdenum, tungsten, cerium and yettribium.
- the oxidation reaction is optionally carried out under inert atmosphere or under an inert gas stream.
- the inert gas include nitrogen, helium, neon, argon and the like.
- the preparation of solution A is carried out by mixing the chiral transition metal complex with the compound of Formula II or their analogs, wherein the chiral transition metal complex is prepared from a transition metal compound and a chiral ligand.
- the transition metal is selected from the group comprising titanium, vanadium, molybdenum and tungsten, and the like preferably titanium, vanadium and tungsten compound.
- Preferred transition metal compound is titanium (IV) isopropoxide, titanium (IV) propoxide, titanium (IV) ethoxide, titanium (IV) methoxide, vanadium oxy tripropoxide or vanadium oxytriisopropoxide and the like.
- the chiral ligand used herewith is selected from branched or unbranched alkyl diol or an aromatic diol.
- Preferred chiral diols are esters of tartaric acid especially (+)-diethyl L-tartarate or (-)-diethyl D-tartarate, (+)- dimethyl L-tartarate, (-)-dimethyl D-tartarate and the like.
- the asymmetric oxidation is optionally carried out in presence of a catalyst.
- the preferred catalyst used in the process is water.
- the reaction of resulting solution A and solution B from steps (a) and (b), respectively is carried out in a reactor such as plug flow reactor, microreactor, microfluidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, flmdized bed reactor or fixed bed reactor.
- the reaction is carried out by simultaneous feeding of two streams at a temperature of -20 to 100°C.
- the flow rate of streams of solution A and solution B is controlled based upon the design of the reactor.
- the residence time necessary in the method according to the present invention depends on various parameters, such as, for example, the temperature or reactivity of the starting materials, flow rate etc.
- the term “residence time” refers to the internal volume of the reaction zone within the plug flow reactor occupied by the reactant fluid flowing through the space, at the temperature and pressure being used.
- the residence time may be for example, between about 1.2 minutes and about 10 minutes.
- the plug flow reactor used herein is designed in such a way that there is availability of multiple inlet points for feeding a reactant / reagent, while the reaction is in progress.
- the 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazoles of Formula I, their analogs and optically active enantiomers obtained from step (c) are optionally converted to the desired pharmaceutically acceptable salts, hydrates or solvates thereof by following the processes well known in prior art literature.
- Solution A Dissolved 16.4g of sodium hydroxide in 200ml of water at 25-30°C and added 200ml of acetonitrile and l OOg of 5-difluoromethoxy-2-[(3,4- dimethoxy-2-pyridinyl) methylsulphanyl]-lH-benzimidazole to obtain solution A.
- Solution B Dissolved 6.87g of sodium hydroxide in 62.5ml of water at 25-30°C and added to 307.68g of sodium hypochlorite solution (4%) to obtain solution B.
- the solutions A and B prepared above were cooled to -5-0°C.
- the temperature of microreactor was set to -5-0°C.
- the microreactor channels were fed simultaneously with solution A at flow rate 30ml/minutes and solution B at flow rate 22ml/minutes at -5-0°C.
- the resulting reaction mass was treated with an aqueous solution of sodium thiosulphate (8.0g sodium thiosulphate in 20ml water), then the reaction mass was washed with 100ml dichloromethane.
- reaction mass was adjusted to 8.0-8.5 by addition of acetic acid at 25-30°C.
- the reaction mass was extracted with dichloromethane
- the organic layer was then treated with a solution of 9.8g sodium hydroxide in 10ml water at 25-30°C.
- the organic solvent was distilled out under vacuum at 40-45°C.
- To the resulting reaction mass 300ml of acetonitrile and 5.4ml of water was added at 25-30°C.
- the reaction mass was then stirred at 25-30°C for 30 minutes.
- the reaction mass was maintained at 25-30°C for 8- 10hours.
- the reaction mass was then cooled to at 10- 15°C to obtain the solid which was filtered and dried under vacuum to obtain the title compound.
- Solution A Dissolved 16.4g of sodium hydroxide in 200ml water at 25-30°C and added it to a solution of l OOg of 5-(Difluoromethoxy)-2-[[(3,4- dimethoxy-2-pyridinyl)methyl]thio]- l H-benzimidazole in 200ml of acetonitrile at 25-30°C to obtain solution A.
- solution B Dissolved 7.17g of sodium hydroxide in 65ml of water at 25-30°C and added to 708.8g of sodium hypochlorite solution (4%) to obtain solution B.
- the solution of A and B prepared above were cooled to -5 to 0°C.
- the temperature of plug flow reactor was set to -5 to -10°C.
- the plug flows were fed simultaneously with solution A at flow rate 22ml/minutes and solution B at flow rate 32ml/minutes at -5 to -10°C.
- the resulting reaction mass was treated with an aqueous solution of sodium thiosulphate (8.0g sodium thiosulphate in 20ml water), then the reaction mass was washed with 100ml dichloromethane.
- the pH of reaction mass was adjusted to 8.0-8.5 by addition of acetic acid at 25-30°C.
- reaction mass was extracted with dichloromethane The organic layer was then treated with a solution of 9.0g sodium hydroxide in 9.0ml water at 25-30°C. The organic solvent was distilled out under vacuum at 40-45°C. To the resulting reaction mass 200ml of acetonitrile and 5.0ml of water was added at 25-30°C. The reaction mass was then stirred at 25-30°C for 30 minutes. The reaction mass was maintained at 25-30°C for 8-10hours. The reaction mass was then cooled to at 10-15°C to obtain the solid which was filtered and dried under vacuum to obtain the title compound.
- Stage 1 Preparation of rabeprazole
- Solution A Dissolved 1 1.5g of sodium hydroxide in 200ml of water at 20-30°C and added it to a solution of l OOg of 2-[[[(4-(3-methoxypropoxy)-2- methyl-2-pyridinyl]methyl]sulfanyl-lH-benzimidazole in 500ml of methanol at 25- 30°C to obtain solution A.
- Solution B Dissolved 7.25g of sodium hydroxide in 100ml of water at 25-30°C and added to 287.92g of sodium hypochlorite solution to obtain solution B.
- the solutions A and B prepared above were cooled to -5-0°C.
- the temperature of microreactor was set to -2-3°C.
- the microreactor channels were fed simultaneously with solution A at flow rate 30ml/minutes and solution B at flow rate 12.2ml/minutes at -2-3°C.
- the resulting reaction mass was treated with an aqueous solution of sodium thiosulphate (14.0g sodium thiosulphate in 100ml water) followed by addition of dichloromethane.
- the layers were separated and the pH of aqueous layer was adjusted to 8.8-9.2 by addition of acetic acid at 10- 15°C.
- the reaction mass was extracted with 200ml of dichloromethane The organic layer was then treated with a solution of 1 1.64g sodium hydroxide in 600ml water at 25-30°C and stirred. The aqueous layer was washed with dichloromethane. To the resulting aqueous layer 200ml of acetonitrile was added at 25-30°C. The reaction mass was then cooled to 0-5°C and pH of the reaction mass was adjusted to 8.5-8.7 using 30% aqueous acetic acid solution. The reaction mass was then stirred at 0-5°C for 10-12 hours. The solid obtained was filtered and suck dried.
- the wet material obtained from Stage-1 was charged to a solution of 1 1.01 g sodium hydroxide in 500ml water at 25-30°C. The reaction mass was then stirred for about 1 hour and washed with dichloromethane. The aqueous layer was then spray dried to obtain the title compound.
- Solution A Dissolved 1 1 .5g of sodium hydroxide in 200ml water at 20-30°C and added it to a solution of lOOg of 2[[[(4-(3-methoxypropoxy)-2-methyl- 2-pyridinyl]methyl]sulfanyl-lH-benzimidazole in 500ml of methanol at 25-30°C to obtain solution A.
- solution B Dissolved 7.25g of sodium hydroxide in 100ml of water at 25-30°C and added to 650g of sodium hypochlorite (4%) to obtain solution B.
- the solution of A and B prepared above were cooled to -5 to 0°C.
- the temperature of plug flow reactor was set to -2 to -5°C.
- the plug flows were fed simultaneously with solution A at flow rate 31ml/minutes and solution B at flow rate 30ml/minutes at -2 to -5°C.
- the resulting reaction mass was continuously quenched with an aqueous solution of sodium thiosulphate (8.0g sodium thiosulphate in 20ml water) followed by addition of dichloromethane.
- the temperature of microreactor was set to 25°C.
- the microreactor channels were fed simultaneously with solution A at flow rate 20ml/minutes and solution B at flow rate 9.1 ml/minutes at -25-30°C.
- the resulting reaction mass was cooled to 0-5°C and treated with solution of 40g potassium hydroxide in 100ml methanol.
- the reaction mass was then stirred for 1 hour.
- the reaction mass was filtered and suck dried. Charged 500ml water to the wet material at 25-30°C and the resulting reaction mass was stirred for 15minutes.
- the layers were separated and added 500ml of dichloromethane to the aqueous layer and the pH was adjusted to 7.5-8.0 using 10% aqueous acetic acid.
- Solution A To 60.4g of titanium isopropoxide, added 62.59g of (-) diethyl tartrate at 20-25°C. The solution is then stirred for 15minutes and the temperature of reaction mass is raised to 55-58°C. The reaction mass is again stirred for 30-45minutes. To the resulting mass added lOOg of (5-methoxy-2-[[(4-methoxy-3,5-dimethyl-2- pyridyl)methyl]sulfanyl]-lH-benzimidazole), stirred for 30minutes and added 2.1 g of water to obtain solution A.
- Solution B Took 89g of cumene hydroperoxide (70% solution) as solution B.
- the temperature of circulating water on plug flow reactor was set to 35-38°C.
- the plug flow feeding points were fed simultaneously with solution A at flow rate 15.0 g/minutes at 55-58°C and solution B at flow rate 4.0 ml/minutes at 25-30°C.
- the resulting reaction mass from plug flow reactor was directly taken into a solution of 30.7g triethylamine in 500 ml toluene and 400ml water at 0-5°C.
- the organic layer was separated and washed with water, then with brine solution and cooled to 10- 15°C.
- methanolic potassium hydroxide solution was added in 10-15 minutes at 10-15°C.
- the temperature of the reaction mass was raised to 20-25°C.
- reaction mass was seeded with pure crystals of esomeprazole potassium. The reaction mass was then stirred for 8 hours at 20-25°C. The reaction mass was filtered and suck dried. Charged 500ml water to the wet material at 20- 25°C and the resulting reaction mass was stirred. To the reaction mass added 500ml of water and the pH of reaction mass was adjusted to 7.5-8.0 using 5% aqueous acetic acid. Added 500 ml of dichloromethane to the reaction mass. The layers were separated and washed the organic layer with water. The solvent was distilled out under vacuum. The semi solid mass was taken in methyl ethyl ketone and cooled to 10-15°C.
- solution B Took sodium hypochlorite solution (6.0% w/w) solution B.
- the solution A and B prepared above were cooled to 0 to 5°C.
- the temperature of plug flow reactor was set to 0 to -5°C.
- the plug flows were fed simultaneously with solution A at flow rate 21 ml/minutes and solution B at flow rate 12ml/minutes at -5 to 0°C.
- the resulting reaction mass was treated with an aqueous solution of sodium thiosulphate (20.0g sodium thiosulphate in 50ml water) followed by addition of 400ml of water.
- the pH of reaction mass was adjusted to 8.5 - 9 by addition of acetic acid at 20-30°C.
- the precipitated solid material was filtered, washed with water and dried to obtain title compound.
Abstract
The present invention provides a commercially viable, cost effective and energy efficient process for the preparation of 2-pyridinylmethylsulfinyl benzimidazoles, their analogs and optically active enantiomers or pharmaceutically acceptable salts, hydrates or solvates thereof in high purity via application of reactors such as plug flow reactor, microreactor, microfluidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, fluidized bed reactor or fixed bed reactor.
Description
PROCESS FOR THE PREPARATION OF 2-PYRIDINYLMETHYLSULFINYL BENZIMIDAZOLES, THEIR ANALOGS AND OPTICALLY ACTIVE
ENANTIOMERS FIELD OF THE INVENTION
The present invention discloses a commercially viable, cost effective and energy efficient process for the preparation of 2-pyridinylmethylsulfinyl benzimidazoles, their analogs and optically active enantiomers or pharmaceutically acceptable salts, hydrates or solvates thereof in high purity via application of reactors such as plug flow reactor, microreactor, microfluidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, fluidized bed reactor or fixed bed reactor.
BACKGROUND OF THE INVENTION
Benzimidazole, a heterocyclic aromatic organic compound, formed by fusion of benzene and imidazole, occurs naturally as N-ribosyl-dimethylbenzimidazole, which serves as axial ligand for cobalt in Vitamin B-12.
Benzimidazole derivatives, particularly, pyridinylmethylsulfinyl benzimidazole compounds are known to have H7K+-ATPase inhibitory action and therefore have considerable importance in the therapy of diseases associated with an increased secretion of gastric acid or used as an anti-ulcerative agent. Such pyridinylmethylsulfinyl substituted benzimidazole compounds or pharmaceutically acceptable salts thereof are known for example, from EP0005129, EP 174726, EP 166287, EP268956 and EP254588 as given in Formula I below:

I
Omeprazole: Rj = CH3, R2 = OCH3, R3 = CH3, R4=R6=R7=H, R5=OCH3, R8 = H, A = C
Lansoprazole: Ri = H, R2 = OCH2CF3, R3 = CH3, R4=R5=R6=R7=H, R8 = H, A = C Pantoprazole: Ri = H, R2 = OCH3, R3 = OCH3, R4=R6=R7=H, R5=OCHF2, R8 = H, A = C
Rabeprazole: R, = H, R2 = 0(CH2)3OCH3, R3 = CH3,
 R8 = H, A = C
R8 = H, A = C
Tenatoprazole: R, = CH3, R2 -OCH3, R3 = CH3, R4≠ H, R6=R7=H, R5 = OCH3, R8= H, A = N
Ilaprazole: Ri-H, R2=OCH3, R3-CH3, R4=H, R5= l-aza,2,4-cyclopentadiene R6=R7=H,
R8 = H, A = C
The compounds (5-methoxy-2-[[(4-methoxy-3,5-dimethyl-2- pyridyl)methyl]sulfinyl]-lH-benzimidazole) and its optically active analog S-(5- methoxy-2-[[(4-methoxy-3,5-dimethyl-2-pyridyl)methyl]sulfinyl]-lH-benzimidazole) known by generic names omeprazole and esomprazole, respectively are marketed in United States under the brand name Prilosec and Nexium for treatment of duodenal ulcer, gastric ulcer and GERD; maintenance of healing of errosive esophagitis, and long term treatment of pathological hypersecretory conditions. Omeprazole was disclosed in US 4,738,974 and esomeprazole was disclosed in W094/27988.
Rabeprazole is another compound of the same class and chemically known by 2-[[[(4-(3-methoxypropoxy)-2-methyl-2-pyridinyl]methyl]sulfinyl-lH-
benzimidazole. It was reported in US 5,045,552 and marketed as sodium salt in the United States under the brand name Aciphex for healing of erosive or ulcerative GERD, maintenance of healing of GERD and treatment of symptomatic GERD.
Pantoprazole is the active ingredient of a pharmaceutical product that is marketed as sodium salt in the United States by Wyeth-pharms Inc and was protected by US 4,758,579 and sold under the brand name Protonix. Pantoprazole is chemically represented (5-(difluoromethoxy)-2-[[(3,4-dimethoxy-2-pyridinyl)methyl]sulfinyl]- lH-benzimidazole. Pantoprazole useful for short-term treatment of erosive esophagitis associated with gastroesophageal reflux disease (GERD), maintenance of healing of erosive esophagitis and pathological hypersecretory conditions including Zollinger-Ellison syndrome.
Lansoprazole another compound represented by 2-[[[3-methyl-4(2,2,2,- triflouroethoxy)-2-pyridiyl]methyl]sulfinyl]-lH-benzimidazole and was reported in US 4,628,098. It is marketed under the brand name Prevacid(R) for short-term treatment of duodenal ulcer, Η. Pylori eradication to prevent recurrence of duodenal ulcer and maintenance of healed duodenal ulcers.
Tenatoprazole represented by 5-methoxy-2-((4-methoxy-3,5-dimethyl-2- pyridylmethyl) sulphinyl)-lH-imidazo (4,5-b) pyridine and was reported in US 4,808,596 and is marketed under brand name Ulsacare. Tenatoprazole is also a proton pump inhibitor indicated for the treatment of reflux oesophagitis and peptic ulcer.
Ilaprazole another compound represented by 2-[(4-methoxy-3-methylpyridin-2-yl) methylsulfinyl]-6-pyrrol- l-yl- lH-benzimidazole was protected by US 5,703,097. It is used for the treatment of dyspepsia, peptic ulcer disease (PUD), gastroesophageal reflux disease (GORD/GERD) and duodenal ulcer.
The preparation of 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazoles Formula (I) by oxidation of compound of Formula II is generally known and discussed in U.S Pat Nos. 4,255,431, 5,045,552, 4,508,905 and 4,628,098.

Various methods employing different oxidants to perform this. oxidation are reported in the prior art literature.
For example, CA 1,263,1 19 describes the use of hydrogen peroxide over a vanadium catalyst (such as vanadium pentoxide, sodium vanadate and vanadium acteylacetonate). CA 1, 127,158 similarly describes the use of peracids, peresters, ozone, etc. EP 533,264 describes the use of magnesium monoperoxyphthalate as the oxidizing agent. W091/18895 describes the use of m-chloroperoxy benzoic acid as the oxidizing agent. GB 2,069,492 generally describes this m-chloroperoxybenzoic acid along with other peroxy acids for the oxidation of substituted (phenylthiomethyl)pyridines.
US 5,374,730 relates to omeprazole and lansoprazole, in particular, two novel synthetic methods for their preparation. According to the process, the sulfinyl compounds are produced from the corresponding acetamide sulfide compound using hydrogen peroxide as oxidizing agent to form amide sulfinyl compound, followed by alkaline hydrolysis to the sulfinyl carboxylate or salt, and decarboxylation.
US 6,313,303 discloses the process for preparing rabeprazole, lansoprazole and other related compounds by oxidation of thioether precursor compound with N-
halosuccinamide, l,3-dihalo-5,5-dimethylhydantoin or dichloroisocyanurate in the presence of a base.
Similarly, EP 484,265 discloses a process for the preparation of omeprazole (example 32 and 33) by oxidation of 2-(((3,5-dimethyl-4-methoxy-2- pyridinyl)methyl)sulfinyl)-5-methoxy-lH-benzimidazole in suitable organic solvent with 50% H202 in presence of catalyst like (P(W3Oio)4.xH20; ammonium molybdate, having the formula (NFU^MoC^; sodium tungstate, having the formula Na2W04; phosphomolybdic acid, having the formula Η3(Ρ(Μθ3θι0)4 χΗ2Ο; and silicotungstic acid, having the formula H4(Si(W3Oio)4.xH 0 at lower temperatures. Use of base in the oxidation process is essential. The process is suffering from tedious and costlier work up for the isolation of the product. The use of organic solvent like methylene dichloride and ethyl acetate and then washing with the same solvent at - 15°C is very tedious workup procedure.
WO2012/004802 discloses a continuous micromixer based process for the synthesis of sulphoxide compounds with a reaction time of less than or equal to one minute having_ selectivity of > 95% towards the sulphoxide compounds, However, the process has several drawbacks with respect to (i)selection of oxidizing agent (ii) selectivity (iii) use of solvent, (iv) micromixer design etc. WO'802 refers that the reaction of imidazo[4,5-b]pyridine compound using oxidizing agent dissolved in solvent to obtain sulphoxide, wherein the commonly used oxidizing agents such as H202j sodium hypochlorite gives only of 2 to 3% conversion, which is highly undesirable on commercial scale. The only oxidizing agent giving more than 82% conversion is a costly reagent and its use is discouraged on commercial scale and is uneconomical. Further, though the applicant claims the selectivity of >95%, the actual examples shows the selectivity less than 93%.
However, it has been reported in the literature that the respective sulfone compound of Formula (III) is also formed due to over oxidation of thioether compound of Formula (II), as the reaction is not ceased at the stage of sulfoxide production but further proceeds to a side reaction where a part of the produced sulfoxide is furthermore oxidized to sulfone. When sulfone is formed, there is a problem not only that the yield of the objective sulfoxide is reduced, but also that it is difficult to separate and purify them, since there is a close resemblance in physicochemical property between the two.

III
Thus, there exists a need in the art for the development of an industrially feasible, cost effective, economic and simple process capable of controlling the sulphone impurity along with other impurities, for producing 2-[(pyridinyl)methyl]sulfinyl- substituted benzimidazoles, their analogs, and optically active enantiomers or pharmaceutically acceptable salts, hydrates or solvates thereof with high purity and high yield.
Accordingly, as an alternative to the prior art methods, in the present invention new, improved conditions have been developed and optimized for the synthesis of 2- [(pyridinyl)methyl]sulfinyl-substituted benzimidazoles, their analogs and optically active enantiomers or pharmaceutically acceptable salts, hydrates or solvates thereof by application of reactors such as plug flow reactor, microreactor microfluidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, fluidized bed reactor or fixed bed reactor .
Chemical reactors are vessels, wherein chemical reactions are carried out; their performance determines the reliability and suitability of a process, its environment safety, the consumption of energy and the raw materials required. Of all the known chemical reactors, the continuous flow reactors are well suited for carrying out reactions, wherein high yield and purity is desired and best utilized continuous flow reactors are plug flow reactor, microreactor etc. A plug flow reactor is a tubular reactor is fundamentally a continuous reactor where there are no moving parts other than pumps that deliver the reactants. To achieve efficient mixing of reactants the addition of static mixing elements such as glass beads inside the tubular reactor is done that provides ideal conditions of radial mixing and near plug flow necessary to perform reactions. In a plug flow reactor, the flow of reactants pumped in the reactor is laminar and the properties of the reaction medium i.e. pressure, temperature, reactant and product concentrations are the same throughout the entire cross section of flow. Further, all the elemental volumes of the reaction medium remain in the reactor for the same period of time and the change in concentration, temperature, and pressure with time are identical for each elemental volume. Plug flow reactors usually operate in adiabatic and non-isothermal conditions. Consequently, from the standpoint of kinetic parameters of a chemical reaction under isothermal conditions, plug flow reactors are more efficient than stirred tank reactors. Microreactors have been defined as "Microsystems fabricated, at least partially, by methods of microtechnology and precision engineering. Fluid channels range from about 1 um (nanoreactors) to about 1 mm (minireactors)." See Microreactors, Ehrfeld, Hessel & Lowe 2000, the entire disclosure of which is incorporated herein by reference. Typical microreactors consist of miniaturized channels, often imbedded in a flat surface referred to as the "chip." These flat surfaces can be glass plates or plates of metals such as stainless steel or Hastelloy. Microreactors have proven to be highly valuable tools in organic chemistry due to their wide flexibility of operating conditions with efficient heat transfer, optimized mixing, and high reaction control.
The continuous mode of operation in microreactor offers several advantages over a batch process such as simplified operations, reduced reaction time, precise process control, higher reproducibility, and in some cases, even enhanced reaction selectivity. The immediate separation of products from the reaction mixture eliminates possible side products that may result from secondary reactions. The continuous mode may also offer a means to replace large reactors, which are costly and take up expensive laboratory or chemical plant space. In addition to the above benefits, microreactors offer their own unique advantages over traditional continuous processing systems including the highly efficient heat transfer and small reaction volumes that allow safer handling of exothermic reactions, reactions involving explosive and toxic materials, increased precision to reaction control and other hazardous reactions that are normally difficult to scale up. Microreactors are particularly useful for rapid optimization, screening different reaction conditions, catalysts, ligands, bases, and solvents; mechanistic studies; cost effective industrial scale up; and rapid screening for new pharmaceuticals. Furthermore, the microreactors can be operated to produce large amounts of the desired end product by using the concept, wherein multiple microreactors are run continuously and in series and/or in parallel to simulate a large scale flow reactor, improving the productivity of the plant. The present invention provides an improved method for the preparation of 2- [(pyridinyl)methyl]sulfinyl-substituted benzimidazoles, their analogs and optically active enantiomers or pharmaceutically acceptable salts, hydrates or solvates thereof using reactors such as plug flow reactor, microreactor, microfiuidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, fluidized bed reactor or fixed bed reactor. The process has distinct advantages in regard to cycle time, energy consumption, yield and product purity over traditional methods.
OBJECT AND SUMMARY OF THE INVENTION
The principal object of the present invention is to provide a process for the preparation of 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazoles, their analogs and optically active enantiomers or pharmaceutically acceptable salts, hydrates or solvates thereof using reactors such as plug flow reactor, microreactor, microfluidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, fluidized bed reactor or fixed bed reactorwhich alleviates the drawbacks of prior art process.
It is another object of the present invention to provide a cost effective and industrially feasible process for producing 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazoles, their analogs and optically active enantiomers or pharmaceutically acceptable salts, hydrates or solvates thereof, wherein the process provides high yield and high purity of the desired product by reducing the formation of impurity, preferably sulfone impurity in a consistent and reproducible manner. In accordance with an embodiment, the present invention provides a process for preparation of 2-[(pyridinyI)methyl]suIfinyl-substituted benzimidazole of Formula I, their analogs and optically active enantiomers or pharmaceutically acceptable salts, hydrates or solvates thereof,

wherein Ri, R2, R3, 4, R5, R6, R7 are the same or different and selected from hydrogen, C 1 -7 alkyl, C 1 .7 alkoxy, optionally substituted by halogen, l -aza-2,4-
cyclopentadiene, R8 is hydrogen or nitrogen protecting group, A is carbon or nitrogen. comprising the steps of:
(a) preparing solution A by dissolving 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazoles of Formula II or their analogs, optionally in a solvent;

II wherein Ri, R2, R3, R4, R5, Re, R7, R , A are as defined above;
(b) preparing solution B by dissolving oxidizing agent optionally in a solvent;
(c) reacting solution A and solution B in reactor such as plug flow reactor, microreactor, micro fluidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, fluidized bed reactor or fixed bed reactor, to obtain 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazoles of Formula I, their analogs and optically active enantiomers; and
(d) optionally converting 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazole of Formula I, their analogs and optically active enantiomers to their pharmaceutically acceptable salts, hydrates or solvates.
DESCRIPTION OF THE INVENTION
While this specification concludes with claims particularly pointing out and distinctly claiming that, which is regarded as the invention, it is anticipated that the invention can be more readily understood through reading the following detailed description of the invention and study of the included examples. The present invention provides a commercially viable and economical process for producing 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazoles, their analogs and optically active enantiomers or pharmaceutically acceptable salts, hydrates or solvates thereof. The process employs application of reactors such as plug flow reactor, microreactor, microfluidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, fluidized bed reactor or fixed bed reactor, for the preparation of desired product in high yield and high purity with enhanced in-process control on impurities specifically sulphone impurity with shorter reaction time.
The process according to the present invention for producing 2- [(pyridinyl]methyl]sulfinyl-substituted benzimidazoles of Formula I, their analogs or their optically active enantiomers or pharmaceutically acceptable salts, hydrates or solvates thereof

I wherein Ri , R2, R3, R4, R5, R6, R7 are the same or different and selected from hydrogen, Ci.7 alkyl, C1-7 alkoxy, optionally substituted by halogen, l -aza-2,4-
cyclopentadiene, R8 is hydrogen or nitrogen protecting group, A is carbon or nitrogen comprising the steps of:
(a) preparing solution A by dissolving 2-[(pyridinyl)methyl]suIfinyl-substituted benzimidazoles of Formula II or their analogs optionally in a solvent;

II wherein Ri, R , R3, R4, R5, R6, R7, R8, A are as defined above
(b) preparing solution B by dissolving oxidizing agent optionally in a solvent;
(c) reacting solution A and solution B in reactor such as plug flow reactor, microreactor, microfluidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, fluidized bed reactor or fixed bed reactor, to obtain 2-[(pyridinyl)methyI]sulfinyl-substituted benzimidazoles of Formula I, their analogs and optically active enantiomers; and
(d) optionally converting 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazoles of Formula I, their analogs and optically active enantiomers to their pharmaceutically acceptable salts, hydrates or solvates.
According to the present invention, the preparation of solution A is optionally carried out in solvent depending upon the type of reactor selected for the oxidation reaction
e.g. microreactor. The solvent used for the preparation of solution A is selected from the group comprising of alcohols such as methanol, ethanol, propanol, butanol, ethylene glycol and the like; aromatic hydrocarbon such as toluene, xylene and the like, ethers such as diisopropyl ether, t-butyl methyl ether, tetrahydrofuran, dioxane, monoglyme, diglyme and the like; esters such as ethyl acetate, methyl acetate and the like, halogenated hydrocarbons such as dichloromethane, chloroform, dichloroethane and the like; ketones such as acetone, methyl ethyl ketone, ethyl isobutyl ketone and the like; nitriles such as acetonitrile, propionitrile and the like; aprotic polar solvents such as Ν,Ν-dimethylformamide, dimethylsulphoxide, hexamethylphosphoramide, and the like; water or mixtures thereof.
According to the present invention, the preparation of solution A is optionally carried out in presence of a base, wherein the base is selected from organic or inorganic. The organic base is selected from amines such as triethylamine, diisopropylamine, diisopropylethylamine, piperidine and the like, alkali metal alkoxide such as sodium methoxide, potassium methoxide and the like. The inorganic base is selected from the ammonia, alkali and alkaline earth metal carbonate, bicarbonate, hydroxide, hydride and the like wherein alkali and alkaline earth metal is selected from a group comprising of lithium, sodium, potassium, magnesium, calcium, barium and the like.
According to the present invention, the oxidizing agent in solution B is selected from the group comprising of hydrogen peroxide, alkyl hydro peroxide, aryl alkyl hydro peroxide, alkali or alkaline earth metal, hypohalite, wherein the alkali or alkaline earth metal is selected from a group comprising of sodium, lithium, potassium, magnesium, calcium and the halite is selected from the group comprising of fluorite, chlorite, bromite and the like. The solvent is selected form the group comprising of alcohols such as methanol, ethanol, propanol, butanol, ethylene glycol and the like; aromatic hydrocarbons such as toluene, xylene and the like, ethers such as diisopropyl ether, t-butyl methyl ether, tetrahydrofuran, dioxane, monoglyme,
diglyme and the like; esters such as ethyl acetate, methyl acetate and the like; halogenated hydrocarbons such as dichloromethane, chloroform, dichloroethane and the like; ketones such as acetone, methyl ethyl ketone, ethyl isobutyl ketone and the like; nitriles such as acetonitrile, propionitrile and the like; aprotic polar solvents such as Ν,Ν-dimethylformamide, dimethylsulphoxide, hexamethylphosphoramide, and the like; water or mixtures thereof.
According to the present invention, the preparation of solution B is optionally carried out in presence of a base, wherein the base is selected from organic or inorganic. The organic base is selected from amines such as triethylamine, diisopropylamine, diisopropylethylamine, piperidine and the like, alkali metal alkoxide such as sodium methoxide, potassium methoxide and the like. The inorganic base is selected from the ammonia, alkali or alkaline earth metal carbonate, bicarbonate, hydroxide, hydride and the like wherein alkali and alkaline earth metal is selected from a group comprising of lithium, sodium, potassium, magnesium, calcium, barium and the like. According to the present invention, the oxidation reaction is carried out in a continuous mode, wherein the reactor carries material as a flowing stream and reactants are continuously fed into the reactor and emerge as continuous stream of product.
According to the present invention, the oxidation reaction isoptionally carried out in the presence of a metal catalyst. Preferably, the metal of the metal catalyst is selected from the group comprising of rhenium, vanadium, molybdenum, tungsten, cerium and yettribium.
According to the present invention, the oxidation reaction is optionally carried out under inert atmosphere or under an inert gas stream. Examples of the inert gas include nitrogen, helium, neon, argon and the like.
According to the present invention, for asymmetric oxidation of compound of Formula II or their analogs, the preparation of solution A is carried out by mixing the chiral transition metal complex with the compound of Formula II or their analogs, wherein the chiral transition metal complex is prepared from a transition metal compound and a chiral ligand. The transition metal is selected from the group comprising titanium, vanadium, molybdenum and tungsten, and the like preferably titanium, vanadium and tungsten compound. Preferred transition metal compound is titanium (IV) isopropoxide, titanium (IV) propoxide, titanium (IV) ethoxide, titanium (IV) methoxide, vanadium oxy tripropoxide or vanadium oxytriisopropoxide and the like. The chiral ligand used herewith is selected from branched or unbranched alkyl diol or an aromatic diol. Preferred chiral diols are esters of tartaric acid especially (+)-diethyl L-tartarate or (-)-diethyl D-tartarate, (+)- dimethyl L-tartarate, (-)-dimethyl D-tartarate and the like.
The asymmetric oxidation is optionally carried out in presence of a catalyst. The preferred catalyst used in the process is water.
According to the present invention, the reaction of resulting solution A and solution B from steps (a) and (b), respectively, is carried out in a reactor such as plug flow reactor, microreactor, microfluidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, flmdized bed reactor or fixed bed reactor. The reaction is carried out by simultaneous feeding of two streams at a temperature of -20 to 100°C. The flow rate of streams of solution A and solution B is controlled based upon the design of the reactor.
The residence time necessary in the method according to the present invention, depends on various parameters, such as, for example, the temperature or reactivity of the starting materials, flow rate etc. The term "residence time" refers to the internal volume of the reaction zone within the plug flow reactor occupied by the reactant fluid flowing through the space, at the temperature and pressure being used. The
residence time may be for example, between about 1.2 minutes and about 10 minutes.
According to the present invention, the plug flow reactor used herein is designed in such a way that there is availability of multiple inlet points for feeding a reactant / reagent, while the reaction is in progress.
After the reaction is completed, suitable separation/solvent extraction/isolation, filtration and/or purification steps are conducted to isolate the desired sulphoxide compound for example, the resulting 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazoles are subjected to washing, charcoal treatment, filtration steps, etc. The final product is optionally purified for example, by a suitable recrystallization procedure.
According to the present invention, the 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazoles of Formula I, their analogs and optically active enantiomers obtained from step (c) are optionally converted to the desired pharmaceutically acceptable salts, hydrates or solvates thereof by following the processes well known in prior art literature.
The major advantages realized in the present invention as compared to prior art batch processes are high yield, high purity, consistency, absence or least formation of sulphone and/or unknown impurity. These distinctively identified advantages of the reactions in reactor such as plug flow reactor, microreactor, microfluidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, fluidized bed reactor or fixed bed reactorresults from minimized residency time and continuous flow nature of the reaction, which thereby reduces the contact time between desired product and the oxidizing agent.
The present invention is further described in greater detail as illustrated in the non- limiting examples. It should be understood that variation and modification of the process are possible within the ambit of the invention broadly disclosed herein.
EXAMPLES Example 1
Preparation of Sodium 5-difluoromethoxy-2-f(3,4-dimethoxy-2- pyridinvDmethylsulphinvH-IH-benzimidazole sesquihydrate (Pantoprazole sodium sesquihydrate) in microreactor
Preparation of Solution A: Dissolved 16.4g of sodium hydroxide in 200ml of water at 25-30°C and added 200ml of acetonitrile and l OOg of 5-difluoromethoxy-2-[(3,4- dimethoxy-2-pyridinyl) methylsulphanyl]-lH-benzimidazole to obtain solution A.
Preparation of Solution B: Dissolved 6.87g of sodium hydroxide in 62.5ml of water at 25-30°C and added to 307.68g of sodium hypochlorite solution (4%) to obtain solution B. The solutions A and B prepared above were cooled to -5-0°C. The temperature of microreactor was set to -5-0°C. The microreactor channels were fed simultaneously with solution A at flow rate 30ml/minutes and solution B at flow rate 22ml/minutes at -5-0°C. After the completion of reaction, the resulting reaction mass was treated with an aqueous solution of sodium thiosulphate (8.0g sodium thiosulphate in 20ml water), then the reaction mass was washed with 100ml dichloromethane. The pH of reaction mass was adjusted to 8.0-8.5 by addition of acetic acid at 25-30°C. The reaction mass was extracted with dichloromethane The organic layer was then treated with a solution of 9.8g sodium hydroxide in 10ml water at 25-30°C. The organic solvent was distilled out under vacuum at 40-45°C. To the resulting reaction mass 300ml of acetonitrile and 5.4ml of water was added at 25-30°C. The reaction
mass was then stirred at 25-30°C for 30 minutes. The reaction mass was maintained at 25-30°C for 8- 10hours. The reaction mass was then cooled to at 10- 15°C to obtain the solid which was filtered and dried under vacuum to obtain the title compound.
HPLC purity: 99.81 %
Sulphone impurity: 0.02%
Example 2
Preparation of Sodium 5-(difluoromethoxy)-2-fK3, 4-dimethoxy-2-pyridinyI) methyll sulfinyll-lH-benzimidazol-l-ide sesquihvdrate (Pantoprazole sodium sesquihvdrate) in plug flow reactor
Preparation of Solution A: Dissolved 16.4g of sodium hydroxide in 200ml water at 25-30°C and added it to a solution of l OOg of 5-(Difluoromethoxy)-2-[[(3,4- dimethoxy-2-pyridinyl)methyl]thio]- l H-benzimidazole in 200ml of acetonitrile at 25-30°C to obtain solution A.
Preparation of solution B: Dissolved 7.17g of sodium hydroxide in 65ml of water at 25-30°C and added to 708.8g of sodium hypochlorite solution (4%) to obtain solution B.
The solution of A and B prepared above were cooled to -5 to 0°C. The temperature of plug flow reactor was set to -5 to -10°C. The plug flows were fed simultaneously with solution A at flow rate 22ml/minutes and solution B at flow rate 32ml/minutes at -5 to -10°C. After the completion of reaction, the resulting reaction mass was treated with an aqueous solution of sodium thiosulphate (8.0g sodium thiosulphate in 20ml water), then the reaction mass was washed with 100ml dichloromethane. The pH of reaction mass was adjusted to 8.0-8.5 by addition of acetic acid at 25-30°C. The reaction mass was extracted with dichloromethane The organic layer was then
treated with a solution of 9.0g sodium hydroxide in 9.0ml water at 25-30°C. The organic solvent was distilled out under vacuum at 40-45°C. To the resulting reaction mass 200ml of acetonitrile and 5.0ml of water was added at 25-30°C. The reaction mass was then stirred at 25-30°C for 30 minutes. The reaction mass was maintained at 25-30°C for 8-10hours. The reaction mass was then cooled to at 10-15°C to obtain the solid which was filtered and dried under vacuum to obtain the title compound.
HPLC purity: 99.9%
Sulphone impurity: 0.03% Example 3
Preparation of 2-tf[(4-(3-methoxypropoxy)-2-methyl-2- pyridinvHinethyllsulfinyl-lH-benzimidazole sodium (Rabeprazole sodium) in microreactor
Stage 1 : Preparation of rabeprazole Preparation of Solution A: Dissolved 1 1.5g of sodium hydroxide in 200ml of water at 20-30°C and added it to a solution of l OOg of 2-[[[(4-(3-methoxypropoxy)-2- methyl-2-pyridinyl]methyl]sulfanyl-lH-benzimidazole in 500ml of methanol at 25- 30°C to obtain solution A.
Preparation of Solution B: Dissolved 7.25g of sodium hydroxide in 100ml of water at 25-30°C and added to 287.92g of sodium hypochlorite solution to obtain solution B.
The solutions A and B prepared above were cooled to -5-0°C. The temperature of microreactor was set to -2-3°C. The microreactor channels were fed simultaneously with solution A at flow rate 30ml/minutes and solution B at flow rate 12.2ml/minutes at -2-3°C. After the completion of reaction, the resulting reaction mass was treated
with an aqueous solution of sodium thiosulphate (14.0g sodium thiosulphate in 100ml water) followed by addition of dichloromethane. The layers were separated and the pH of aqueous layer was adjusted to 8.8-9.2 by addition of acetic acid at 10- 15°C. The reaction mass was extracted with 200ml of dichloromethane The organic layer was then treated with a solution of 1 1.64g sodium hydroxide in 600ml water at 25-30°C and stirred. The aqueous layer was washed with dichloromethane. To the resulting aqueous layer 200ml of acetonitrile was added at 25-30°C. The reaction mass was then cooled to 0-5°C and pH of the reaction mass was adjusted to 8.5-8.7 using 30% aqueous acetic acid solution. The reaction mass was then stirred at 0-5°C for 10-12 hours. The solid obtained was filtered and suck dried.
HPLC purity: 99.8%
Sulphone impurity: 0.02%
Stage 2: Preparation of Rabeprazole sodium
The wet material obtained from Stage-1 was charged to a solution of 1 1.01 g sodium hydroxide in 500ml water at 25-30°C. The reaction mass was then stirred for about 1 hour and washed with dichloromethane. The aqueous layer was then spray dried to obtain the title compound.
Example 4
Preparation of* 2- f [ f ( 4-(3-methoxypropoxy)-2-methyl-2- pyridinyllmethyllsuirinyl-IH-benzimidazole sodium (Rabeprazole sodium) in plug flow reactor
Stage 1 : Preparation of Rabeprazole
Preparation of Solution A: Dissolved 1 1 .5g of sodium hydroxide in 200ml water at 20-30°C and added it to a solution of lOOg of 2[[[(4-(3-methoxypropoxy)-2-methyl-
2-pyridinyl]methyl]sulfanyl-lH-benzimidazole in 500ml of methanol at 25-30°C to obtain solution A.
Preparation of solution B: Dissolved 7.25g of sodium hydroxide in 100ml of water at 25-30°C and added to 650g of sodium hypochlorite (4%) to obtain solution B. The solution of A and B prepared above were cooled to -5 to 0°C. The temperature of plug flow reactor was set to -2 to -5°C. The plug flows were fed simultaneously with solution A at flow rate 31ml/minutes and solution B at flow rate 30ml/minutes at -2 to -5°C. After the completion of reaction, the resulting reaction mass was continuously quenched with an aqueous solution of sodium thiosulphate (8.0g sodium thiosulphate in 20ml water) followed by addition of dichloromethane. The layers were separated and the pH of aqueous layer was adjusted to 8.8 - 9.2 by addition of acetic acid at 10-15°C. The reaction mass was extracted with 200ml of dichloromethane The organic layer was treated with a solution of 1 1.64g sodium hydroxide in 600ml water at 25-30°C and stirred. The aqueous layer was washed with dichloromethane. To the resulting aqueous layer 200ml of acetonitrile was added at 25-30°C.The reaction mass was then cooled to 0-5°Cand pH of the reaction mass was adjusted to 8.5-8.7 using 30%aqueousacetic acid solution. The reaction mass was then stirred at 0-5°C for 10-12 hours. The solid obtained was filtered and suck dried. Stage 2: Preparation of Rabeprazole sodium
The wet material obtained from stage- 1 was charged to a solution of 11.01 g sodium hydroxide in 500ml water at 25-30°C. The reaction mass was then stirred for about 1 hour and washed with dichloromethane. The aqueous layer was then spray dried to obtain the title compound. HPLC purity: 99.9%
Sulphone impurity: 0.02%
Example 5
Preparation of S-(5-methoxy-2-fK4-methoxy-3,5-dimethyl-2- pyridyDmethynsulfinvH-lH-benzimidazole) (Esomeprazole potassium) in microreactor
Preparation of Solution A : A solution of l OOg of (5-methoxy-2-[[(4-methoxy-3,5- dimethyl-2-pyridyl)methyl]sulfanyl]-lH-benzimidazole) in 400ml toluene was heated to 50-55°C under stirring. To the resulting solution, added 25.9g titanium isopropoxide and 37.6g (-)diethyl tartarate. The reaction mass was then stirred for 15minutes at 50-55°C and added 0.82g of water, thereafter reaction mass was again stirred for 1 hour. The reaction mass was then cooled to 25-30°C and added 1 1.8g of diisopropyl ethyl amine to obtain solution A.
Preparation of Solution B: Dissolved 59.8g of cumene hydroperoxide in 200ml toluene to obtain solution B.
The temperature of microreactor was set to 25°C. The microreactor channels were fed simultaneously with solution A at flow rate 20ml/minutes and solution B at flow rate 9.1 ml/minutes at -25-30°C. After the completion of reaction, the resulting reaction mass was cooled to 0-5°C and treated with solution of 40g potassium hydroxide in 100ml methanol. The reaction mass was then stirred for 1 hour. The reaction mass was filtered and suck dried. Charged 500ml water to the wet material at 25-30°C and the resulting reaction mass was stirred for 15minutes. The layers were separated and added 500ml of dichloromethane to the aqueous layer and the pH was adjusted to 7.5-8.0 using 10% aqueous acetic acid. The layers were separated and washed the organic layer with water and brine solution. The solvent was distilled out under vacuum at 35-40°C. The semi solid mass was taken in methyl ethyl ketone
and cooled to 0-5°C. Added a solution of 20g potassium hydroxide in 100ml methanol to the reaction mass, and raised the temperature to 25-30°C. The reaction mass was then stirred for l hour, filtered the solid and dried under vacuum at 35-40°C to obtain the title compound. HPLC purity: NLT 99%
Sulphone impurity: 0.17%
Example 6
Preparation of S-(5-methoxy-2-[f(4-methoxy-3,5-dimethyl-2- pyridvOmethyllsulfinyll-LH-benzimidazole) (Esomeprazole potassium) in plug flow reactor
Solution A: To 60.4g of titanium isopropoxide, added 62.59g of (-) diethyl tartrate at 20-25°C. The solution is then stirred for 15minutes and the temperature of reaction mass is raised to 55-58°C. The reaction mass is again stirred for 30-45minutes. To the resulting mass added lOOg of (5-methoxy-2-[[(4-methoxy-3,5-dimethyl-2- pyridyl)methyl]sulfanyl]-lH-benzimidazole), stirred for 30minutes and added 2.1 g of water to obtain solution A.
Solution B: Took 89g of cumene hydroperoxide (70% solution) as solution B.
The temperature of circulating water on plug flow reactor was set to 35-38°C. The plug flow feeding points were fed simultaneously with solution A at flow rate 15.0 g/minutes at 55-58°C and solution B at flow rate 4.0 ml/minutes at 25-30°C. The resulting reaction mass from plug flow reactor was directly taken into a solution of 30.7g triethylamine in 500 ml toluene and 400ml water at 0-5°C. The organic layer was separated and washed with water, then with brine solution and cooled to 10- 15°C. To the resulting organic layer, methanolic potassium hydroxide solution was
added in 10-15 minutes at 10-15°C. The temperature of the reaction mass was raised to 20-25°C. The reaction mass was seeded with pure crystals of esomeprazole potassium. The reaction mass was then stirred for 8 hours at 20-25°C. The reaction mass was filtered and suck dried. Charged 500ml water to the wet material at 20- 25°C and the resulting reaction mass was stirred. To the reaction mass added 500ml of water and the pH of reaction mass was adjusted to 7.5-8.0 using 5% aqueous acetic acid. Added 500 ml of dichloromethane to the reaction mass. The layers were separated and washed the organic layer with water. The solvent was distilled out under vacuum. The semi solid mass was taken in methyl ethyl ketone and cooled to 10-15°C. Added a solution of 30g potassium hydroxide in 100ml methanol to the reaction mass, and raised the temperature to 20-25°C. The reaction mass was then stirred for 2-3hour, filtered the solid and dried under vacuum at 45-50°C to obtain the title compound.
HPLC purity: NLT 99%
Sulphone impurity: 0.12%
Example 7
Preparation of 2-(((3-methyl-4-(2,2,2-trifluoroethoxy)pyridin-2- yl)methyl)sulfinyl)-lH-benzo[dlimidazole (Lansoprazole) in plug flow reactor Preparation of Solution A: Dissolved 17.0g of sodium hydroxide in 70ml water at 25-30°C and added it to a solution of lOOg of 2-(((3 -methyl -4-(2,2,2- trifluoroethoxy)pyridin-2-yl) methyl)
sulfanyl)-lH-benzo[d]imidazole in mixture of solvent 400ml of acetonitrile and 300ml of methanol at 25-30°C to obtain solution A.
Preparation of solution B: Took sodium hypochlorite solution (6.0% w/w) solution B.
The solution A and B prepared above were cooled to 0 to 5°C. The temperature of plug flow reactor was set to 0 to -5°C. The plug flows were fed simultaneously with solution A at flow rate 21 ml/minutes and solution B at flow rate 12ml/minutes at -5 to 0°C. After the completion of reaction, the resulting reaction mass was treated with an aqueous solution of sodium thiosulphate (20.0g sodium thiosulphate in 50ml water) followed by addition of 400ml of water. The pH of reaction mass was adjusted to 8.5 - 9 by addition of acetic acid at 20-30°C. The precipitated solid material was filtered, washed with water and dried to obtain title compound.
HPLC purity: 99.6%
Sulphone impurity: 0.01 %
Claims
We Claim:
1. A process for producing 2-[(pyridinyl]methyl]sulfinyl-substituted benzimidazoles of Formula I, their analogs or their optically active enantiomers or pharmaceutically acceptable salts, hydrates or solvates thereof
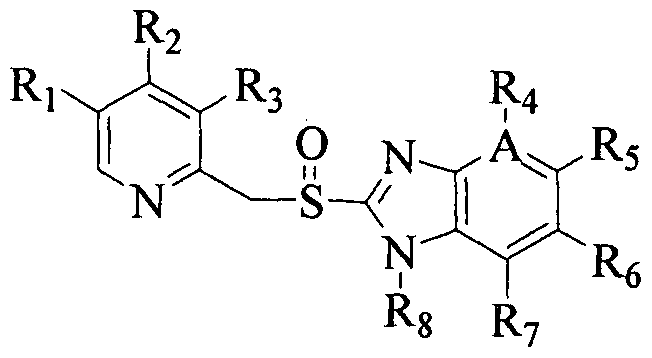
I wherein Ri , R2, R3, R4, R5, R6, R7 are the same or different and selected from hydrogen, C 1.7 alkyl, C 1-7 alkoxy, optionally substituted by halogen, l -aza-2,4- cyclopentadiene, R8 is hydrogen or nitrogen protecting group, A is carbon or nitrogen comprising the steps of:
(a) preparing solution A by dissolving 2-[(pyridinyl)methyl]sulfinyl- substituted benzimidazoles of Formula II or their analogs, optionally in a solvent;
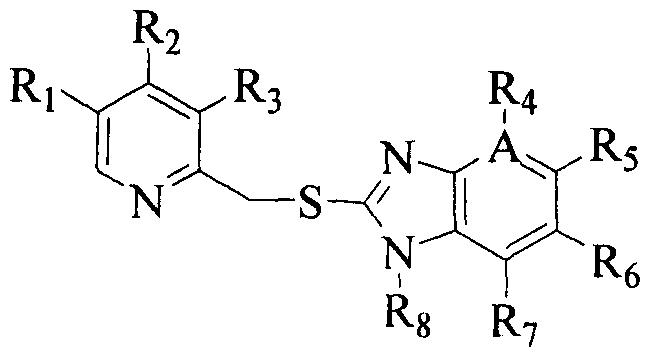
II wherein Ri , R2, R3, R4, R5, i, R?, Rs, A are as defined above
(b) preparing solution B by dissolving oxidizing agent optionally in a solvent;
(c) reacting solution A and solution B in reactor selected from plug flow reactor, microreactor, microfluidic flow reactor, tubular flow reactor, coil-type flow reactor, laminar flow reactor, packed bed reactor, fluidized bed reactor and fixed bed reactor, to obtain 2-[(pyridinyl)methyl]sulfinyl- substituted benzimidazoles of Formula I, their analogs and optically active enantiomers; and
(d) optionally converting 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazoles of Formula I, their analogs and optically active enantiomers to their pharmaceutically acceptable salts, hydrates and solvates.
A process for producing 2-[(pyridinyl]methyl]sulfmyl-substituted benzimidazoles of Formula I, their analogs or their optically active enantiomers or pharmaceutically acceptable salts, hydrates and solvates thereof

I wherein Ri, R , R3, R4, R5, R6, R7 are the same or different and selected from hydrogen, Ci-7 alkyl, Ci-7 alkoxy, optionally substituted by halogen, l -aza-2,4- cyclopentadiene, R8 is hydrogen or nitrogen protecting group, A is carbon or nitrogen
comprising the steps of:
(a) preparing solution A by dissolving 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazoles of Formula II or their analogs optionally in a solvent;

II wherein Ri , R2, R3, R4, R5, R6, R7, R8, A are as defined above,
(b) preparing solution B by dissolving oxidizing agent optionally in a solvent;
(c) reacting solution A and solution B in a plug flow reactor, to obtain 2- [(pyridinyl)methyl]sulfinyl-substituted benzimidazoles of Formula I, their analogs and optically active enantiomers; and
(d) optionally converting 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazoles of Formula I, their analogs and optically active enantiomers to their pharmaceutically acceptable salts, hydrates and solvates.
3. A process for producing 2-[(pyridinyl]methyl]sulfinyl-substituted benzimidazoles, their analogs or their optically active enantiomers of Formula I or pharmaceutically acceptable salts, hydrates or solvates thereof

(a) preparing solution A by dissolving 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazoies of Formula II or their analogs in a solvent;

II wherein Ri, R2, R3, R4, R5, R6, R7, R8, A are as defined above,
(b) preparing solution B by dissolving oxidizing agent optionally in a solvent;
(c) reacting solution A and solution B in a microreactor to obtain 2- [(pyridinyl)methyl]sulfmyl-substituted benzimidazoies of Formula I, their analogs and optically active enantiomers; and
(d) optionally converting 2-[(pyridinyl)methyl]sulfinyl-substituted benzimidazoies of Formula I, their analogs and optically active enantiomers to their pharmaceutically acceptable salts, hydrates and solvates.
4. The process according to claims 1 , 2 or 3, wherein the reaction of solution A with solution B is carried out in a continuous mode.
5. The process according to claims 1, 2 or 3, wherein the solvent used in steps 1(a), 2(a) or 3(a) for the preparation of solution A is selected form the group comprising of alcohols, aromatic hydrocarbon, ethers, esters, halogenated hydrocarbons, ketones, nitriles, aprotic polar solvents, water and mixtures thereof.
6. The process according to claim 5, wherein the solvent used is selected from the group comprising of methanol, ethanol, propanol, butanol, ethylene glycol, toluene, xylene, diisopropyl ether, t-butyl methyl ether, tetrahydrofuran, dioxane, monoglyme, diglyme, ethyl acetate, methyl acetate, dichloromethane, chloroform, dichloroethane, acetone, methyl ethyl ketone, ethyl isobutyl ketone, acetonitrile, propionitrile, N,N-dimethylformamide, dimethylsulphoxide and hexamethylphosphoramide.
7. The process according to claims 1, 2 or 3, wherein the preparation of solution A in steps 1(a), 2(a) or 3(a) is optionally carried out in the presence of a base.
8. The process according to claim 7, wherein the base is selected from organic and inorganic.
9. The process according to claim 8, wherein the organic base is selected from the group comprising of amines and alkali metal alkoxides.
10. The process according to claim 9, wherein the amine is selected from the group comprising of triethylamine, diisopropylamine, diisopropylethylamine and piperidine.
1 1. The process according to claim 9, wherein the alkali metal alkoxide is selected from sodium methoxide and potassium methoxide.
12. The process according to claim 8, wherein the inorganic base is selected from the group comprising of ammonia, alkali and alkaline earth metal carbonate, bicarbonate, hydroxide and hydride.
13. The process according to claim 12, wherein the alkali and alkaline earth metal is selected from the group comprising of lithium, sodium, potassium, magnesium, calcium and barium.
14. The process according to claim 1 , 2 or 3, wherein the oxidizing agent in solution B is selected from the group comprising of hydrogen peroxide, alkyl hydro peroxide, aryl alkyl hydro peroxide, alkali and alkaline earth metal hypohalite.
15. The process according to claim 14, wherein the alkali and alkaline earth metal is selected from the group comprising of sodium, lithium, potassium, magnesium and calcium.
16. The process according to claim 14, wherein the halite is selected from the group comprising of fluorite, chlorite and bromite.
17. The process according to claim 1, 2 or 3, wherein the solvent used for the preparation of solution B in steps 1(b), 2(b) or 3 (b) is selected from the group comprising of alcohols, aromatic hydrocarbon, ethers, esters, halogenated hydrocarbons, ketones, nitriles, aprotic polar solvents, water and mixtures thereof.
18. The process according to claim 17, wherein the solvent used is selected from the group comprising of methanol, ethanol, propanol, butanol, ethylene glycol, toluene, xylene, diisopropyl ether, t-butyl methyl ether, tetrahydrofuran, dioxane, monoglyme, diglyme, ethyl acetate, methyl acetate, dichloromethane, chloroform, dichloroethane, acetone, methyl ethyl ketone,
ethyl isobutyl ketone, acetonitrile, propionitrile, N,N-dimethylformamide, dimethylsulphoxide and hexamethylphosphoramide.
19. The process according to claims 1 , 2 or 3, wherein the preparation of solution B in steps 1(b), 2(b) or 3(b) is optionally carried out in the presence of a base.
20. The process according to claim 19, wherein the base is selected from organic and inorganic.
21. The process according to claim 19, wherein the organic base is selected from the group comprising of amines and alkali metal alkoxide.
22. The process according to claim 21 , wherein the amine is selected from the group comprising of triethylamine, diisopropylamine, diisopropylethylamine and piperidine.
23. The process according to claim 21 , wherein the alkali metal alkoxide is selected from the group comprising of sodium methoxide and potassium methoxide.
24. The process according to claim 20, wherein the inorganic base is selected from the group comprising of ammonia, alkali and alkaline earth metal carbonate, bicarbonate, hydroxide and hydride.
25. The process according to claim 24, wherein the alkali and alkaline earth metal is selected from the group comprising of lithium, sodium, potassium, magnesium, calcium and barium.
26. The process according to claims 1 , 2 or 3, wherein the reaction in steps 1 (c), 2(c) or 3(c) is optionally carried out in the presence of a metal catalyst.
27. The process according to claim 26, wherein the metal of the metal catalyst is selected from the group comprising of rhenium, vanadium, molybdenum, tungsten, cerium and yettribium.
28. The process according to claims 1 , 2 or 3, wherein the oxidation of compound of Formula II to compound of Formula I is asymmetric.
29. The process according to claim 28, wherein the preparation of solution A in steps 1(a), 2(a) or 3(a) is carried out by mixing the chiral transition metal complex with the compound of Formula II, optionally in solvent.
30. The process according to claim 29, wherein the chiral transition metal complex is prepared from a transition metal compound and a chiral ligand.
31. The process according to claim 30, wherein the transition metal compound is selected from the group comprising of titanium (IV) isopropoxide, titanium (IV) propoxide, titanium (IV) ethoxide, titanium (IV) methoxide, vanadium oxy tripropoxide and vanadium oxytriisopropoxide.
32. The process according to claim 30, wherein the chiral ligand is selected from the group comprising (+)-diethyI L-tartarate, (-)-diethyl D-tartarate, (+)- dimethyl L-tartarate and (-)-dimethyl D-tartarate.
33. The process according to claim 28, wherein the asymmetric oxidation is carried out in presence of a catalyst.
34. The process according to claim 33, wherein the catalyst used is water.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201280067674.2A CN104203938A (en) | 2012-01-21 | 2012-08-14 | Process for the preparation of 2-pyridinylmethylsulfinyl benzimidazoles, their analogs and optically active enantiomers |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN186DE2012 | 2012-01-21 | ||
| IN186/DEL/2012 | 2012-01-21 | ||
| IN1334/DEL/2012 | 2012-05-01 | ||
| IN1334DE2012 | 2012-05-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013108068A1 true WO2013108068A1 (en) | 2013-07-25 |
Family
ID=47018258
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2012/001561 WO2013108068A1 (en) | 2012-01-21 | 2012-08-14 | Process for the preparation of 2-pyridinylmethylsulfinyl benzimidazoles, their analogs and optically active enantiomers |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN104203938A (en) |
| WO (1) | WO2013108068A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20190055481A (en) * | 2017-11-15 | 2019-05-23 | 주식회사 다산제약 | Process for preparing high purity ilaprazole crystalline form B |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106554348A (en) * | 2016-11-04 | 2017-04-05 | 扬子江药业集团有限公司 | A kind of method for preparing Levpantoprazole Sodium |
| CN113801096B (en) * | 2020-06-12 | 2023-03-24 | 杭州中美华东制药有限公司 | Preparation method of dexlansoprazole |
Citations (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0005129A1 (en) | 1978-04-14 | 1979-10-31 | Aktiebolaget Hässle | Substituted pyridylsulfinylbenzimidazoles having gastric acid secretion properties, pharmaceutical preparations containing same, and intermediates for their preparation |
| GB2069492A (en) | 1980-02-20 | 1981-08-26 | Wyeth John & Brother Ltd | Novel sulphur compounds |
| EP0166287A1 (en) | 1984-06-16 | 1986-01-02 | Byk Gulden Lomberg Chemische Fabrik GmbH | Dialkoxyridines, process for their preparation, their application and medicaments containing them |
| EP0174726A1 (en) | 1984-08-16 | 1986-03-19 | Takeda Chemical Industries, Ltd. | Pyridine derivatives and their production |
| EP0254588A1 (en) | 1986-07-25 | 1988-01-27 | Tokyo Tanabe Company Limited | Imidazo[4,5-b] pyridine compounds, process for preparing same and pharmaceutical compositions containing same |
| US4738974A (en) | 1983-03-04 | 1988-04-19 | Aktiebolaget Hassle | Base addition salts of omeprazole |
| EP0268956A2 (en) | 1986-11-13 | 1988-06-01 | Eisai Co., Ltd. | Pyridine derivatives, pharmaceutical compositions comprising the same, the use of the same for the manufacture of medicaments having therapeutic or preventative value, and a process for preparing the same |
| CA1263119A (en) | 1987-08-04 | 1989-11-21 | Masayasu Kato | Production of 2-(2- pyridylmethylsulfinyl)benzimidazole compounds |
| WO1991018895A1 (en) | 1990-06-07 | 1991-12-12 | Aktiebolaget Astra | Improved method for synthesis |
| EP0484265A1 (en) | 1990-10-31 | 1992-05-06 | Centro Genesis Para La Investigacion, S.L. | A process for the preparation of omeprazol |
| EP0533264A1 (en) | 1991-09-20 | 1993-03-24 | Merck & Co. Inc. | Process for the preparation of anti-ulcer agents |
| WO1994027988A1 (en) | 1993-05-28 | 1994-12-08 | Astra Aktiebolag | Optically pure salts of pyridinylmethyl sulfinyl-ih-benzimidazole compounds |
| US5374730A (en) | 1993-11-04 | 1994-12-20 | Torcan Chemical Ltd. | Preparation of omeprazole and lansoprazole |
| US5703097A (en) | 1994-02-28 | 1997-12-30 | Il-Yang Pharm. Co., Ltd. | 5-pyrrolyl-2-pyridylmethylsulfinyl benzimidazole derivatives |
| US6313303B1 (en) | 1997-07-11 | 2001-11-06 | Eisai Co., Ltd. | Process for the preparation of pyridine derivatives |
| US20060089376A1 (en) * | 2004-10-27 | 2006-04-27 | Joshi Ramesh A | Tenatoprazole salts and process of preparation thereof |
| WO2007068925A1 (en) * | 2005-12-17 | 2007-06-21 | Pliva Hrvatska D.O.O. | An improved process for preparing of, impurities free, substituted 2-benzimidazolesulfoxide compound |
| WO2008018091A1 (en) * | 2006-08-08 | 2008-02-14 | Jubilant Organosys Limited | Process for producing sulphoxide compounds |
| WO2009066321A2 (en) * | 2007-10-03 | 2009-05-28 | Ipca Laboratories Limited | Process for optically active sulfoxide compounds |
| WO2010049746A1 (en) * | 2008-10-27 | 2010-05-06 | Dynamit Nobel Gmbh Explosivstoff-Und Systemtechnic | Process for the synthesis of fipronil |
| WO2012004802A1 (en) | 2009-07-07 | 2012-01-12 | Council Of Scientific & Industrial Research | Continuous flow process for the preparation of sulphoxide compounds |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102241670B (en) * | 2011-04-28 | 2013-05-29 | 苏州特瑞药业有限公司 | Preparation method of high-purity chiral sulphoxide compound |
-
2012
- 2012-08-14 CN CN201280067674.2A patent/CN104203938A/en active Pending
- 2012-08-14 WO PCT/IB2012/001561 patent/WO2013108068A1/en unknown
Patent Citations (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4255431A (en) | 1978-04-14 | 1981-03-10 | Aktiebolaget Hassle | Gastric acid secretion inhibiting substituted 2-(2-benzimidazolyl)-pyridines, pharmaceutical preparations containing same, and method for inhibiting gastric acid secretion |
| CA1127158A (en) | 1978-04-14 | 1982-07-06 | Ulf K. Junggren | 2-pyridylmethylsulfinyl-benzimidazole compounds |
| US4508905A (en) | 1978-04-14 | 1985-04-02 | Aktiebolaget Hassle | Substituted 2-(-benzimidazolyl)pyridines |
| EP0005129A1 (en) | 1978-04-14 | 1979-10-31 | Aktiebolaget Hässle | Substituted pyridylsulfinylbenzimidazoles having gastric acid secretion properties, pharmaceutical preparations containing same, and intermediates for their preparation |
| GB2069492A (en) | 1980-02-20 | 1981-08-26 | Wyeth John & Brother Ltd | Novel sulphur compounds |
| US4738974A (en) | 1983-03-04 | 1988-04-19 | Aktiebolaget Hassle | Base addition salts of omeprazole |
| EP0166287A1 (en) | 1984-06-16 | 1986-01-02 | Byk Gulden Lomberg Chemische Fabrik GmbH | Dialkoxyridines, process for their preparation, their application and medicaments containing them |
| US4758579A (en) | 1984-06-16 | 1988-07-19 | Byk Gulden Lomberg Chemische Fabrik Gmbh | Fluoroalkoxy substituted benzimidazoles useful as gastric acid secretion inhibitors |
| EP0174726A1 (en) | 1984-08-16 | 1986-03-19 | Takeda Chemical Industries, Ltd. | Pyridine derivatives and their production |
| US4628098A (en) | 1984-08-16 | 1986-12-09 | Takeda Chemical Industries, Ltd. | 2-[2-pyridylmethylthio-(sulfinyl)]benzimidazoles |
| US4808596A (en) | 1986-07-25 | 1989-02-28 | Tokyo Tanabe Company, Ltd. | Imidazo[4,5-b]pyridine compounds and pharmaceutical compositions containing same |
| EP0254588A1 (en) | 1986-07-25 | 1988-01-27 | Tokyo Tanabe Company Limited | Imidazo[4,5-b] pyridine compounds, process for preparing same and pharmaceutical compositions containing same |
| US5045552A (en) | 1986-11-13 | 1991-09-03 | Eisai Co., Ltd. | Pyridine derivatives having anti-ulcerative activity |
| EP0268956A2 (en) | 1986-11-13 | 1988-06-01 | Eisai Co., Ltd. | Pyridine derivatives, pharmaceutical compositions comprising the same, the use of the same for the manufacture of medicaments having therapeutic or preventative value, and a process for preparing the same |
| CA1263119A (en) | 1987-08-04 | 1989-11-21 | Masayasu Kato | Production of 2-(2- pyridylmethylsulfinyl)benzimidazole compounds |
| WO1991018895A1 (en) | 1990-06-07 | 1991-12-12 | Aktiebolaget Astra | Improved method for synthesis |
| EP0484265A1 (en) | 1990-10-31 | 1992-05-06 | Centro Genesis Para La Investigacion, S.L. | A process for the preparation of omeprazol |
| EP0533264A1 (en) | 1991-09-20 | 1993-03-24 | Merck & Co. Inc. | Process for the preparation of anti-ulcer agents |
| WO1994027988A1 (en) | 1993-05-28 | 1994-12-08 | Astra Aktiebolag | Optically pure salts of pyridinylmethyl sulfinyl-ih-benzimidazole compounds |
| US5374730A (en) | 1993-11-04 | 1994-12-20 | Torcan Chemical Ltd. | Preparation of omeprazole and lansoprazole |
| US5703097A (en) | 1994-02-28 | 1997-12-30 | Il-Yang Pharm. Co., Ltd. | 5-pyrrolyl-2-pyridylmethylsulfinyl benzimidazole derivatives |
| US6313303B1 (en) | 1997-07-11 | 2001-11-06 | Eisai Co., Ltd. | Process for the preparation of pyridine derivatives |
| US20060089376A1 (en) * | 2004-10-27 | 2006-04-27 | Joshi Ramesh A | Tenatoprazole salts and process of preparation thereof |
| WO2007068925A1 (en) * | 2005-12-17 | 2007-06-21 | Pliva Hrvatska D.O.O. | An improved process for preparing of, impurities free, substituted 2-benzimidazolesulfoxide compound |
| WO2008018091A1 (en) * | 2006-08-08 | 2008-02-14 | Jubilant Organosys Limited | Process for producing sulphoxide compounds |
| WO2009066321A2 (en) * | 2007-10-03 | 2009-05-28 | Ipca Laboratories Limited | Process for optically active sulfoxide compounds |
| WO2010049746A1 (en) * | 2008-10-27 | 2010-05-06 | Dynamit Nobel Gmbh Explosivstoff-Und Systemtechnic | Process for the synthesis of fipronil |
| WO2012004802A1 (en) | 2009-07-07 | 2012-01-12 | Council Of Scientific & Industrial Research | Continuous flow process for the preparation of sulphoxide compounds |
Non-Patent Citations (3)
| Title |
|---|
| EHRFELD; HESSEL; LOWE, MICROREACTORS, 2000 |
| KLAUS JÄHNISCH ET AL: "Chemistry in Microstructured Reactors", ANGEWANDTE CHEMIE INTERNATIONAL EDITION, vol. 43, no. 4, 16 January 2004 (2004-01-16), pages 406 - 446, XP055032001, ISSN: 1433-7851, DOI: 10.1002/anie.200300577 * |
| TAKUYA NOGUCHI ET AL: "Highly selective 30% hydrogen peroxideoxidation of sulfides to sulfoxides using micromixing", CHEMICAL COMMUNICATIONS - CHEMCOM; [6015D], ROYAL SOCIETY OF CHEMISTRY, GB, 1 January 2008 (2008-01-01), pages 3040 - 3042, XP002602693, ISSN: 1359-7345, [retrieved on 20080422], DOI: 10.1039/B802502A * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20190055481A (en) * | 2017-11-15 | 2019-05-23 | 주식회사 다산제약 | Process for preparing high purity ilaprazole crystalline form B |
| KR102027388B1 (en) * | 2017-11-15 | 2019-10-01 | 주식회사 다산제약 | Process for preparing high purity ilaprazole crystalline form B |
Also Published As
| Publication number | Publication date |
|---|---|
| CN104203938A (en) | 2014-12-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0997461B1 (en) | Processes for the preparation of pyridine derivatives | |
| US20080091024A1 (en) | Processes for the production of substituted 2-(2-pyridylmethyl) sulfinyl-1H-benzimidazoles | |
| KR101432866B1 (en) | A process of sulfoxidation of biologically active compounds | |
| EP2030973A1 (en) | Process for preparing 2-sulfinyl-1H-benzimidazoles | |
| KR101421862B1 (en) | Nitrogen-containing heterocyclic compound and method for producing same | |
| KR20050104349A (en) | Pharmaceutical process and compounds prepared thereby | |
| WO2013108068A1 (en) | Process for the preparation of 2-pyridinylmethylsulfinyl benzimidazoles, their analogs and optically active enantiomers | |
| JP2019194219A (en) | Manufacturing method of optically active proton pump inhibitory compound | |
| AU3410699A (en) | Chemical process for the production of sulphinyl derivatives by oxidation of the corresponding co-derivatives with perborates | |
| US20100210848A1 (en) | Process for optically active sulfoxide compounds | |
| EP1921075B1 (en) | A process for the preparation of pyridine-methylsulfinyl compounds | |
| WO2010134099A1 (en) | One pot process for preparing omeprazole and related compounds | |
| JP2008150361A6 (en) | Method for preparing pyridine-methylsulfinyl compound | |
| US20090005570A1 (en) | Method for preparing 2- (2-pyridinylmethylsulfinyl) benzimidazoles | |
| WO2009066309A2 (en) | Process for preparation of omeprazole | |
| US20040138466A1 (en) | Processes for the production of substituted 2-(2-pyridylmethyl) sulfinyl-1H-benzimidazoles | |
| US20070225500A1 (en) | Process for the Preparation of Pyridin-2-Ylmethylsulphinyl-1H-Benzimidazol Compounds | |
| WO2007129328A2 (en) | Process for preparing 2-[pyridinyl]sulfinyl-substituted benzimidazoles | |
| AU2002240242A1 (en) | Processes for the production of substituted 2-(2-pyridylmethyl) sulfinyl-1H-benzimidazoles | |
| JP2000016992A (en) | Production of pyridine derivative | |
| JPH1171371A (en) | Production of pyridine derivative | |
| KR20070031945A (en) | Process for the preparation of pyridin-2-ylmethylsulphinyl-1h-benzimidazol compounds | |
| KR20090041023A (en) | Process for preparing 2-(2-pyridylmethyl)sulfinyl-1h-benzimidazole | |
| MXPA00008482A (en) | Chemical process for the production of sulphinyl derivatives by oxidation of the corresponding co-derivatives with perborates | |
| KR20070105018A (en) | Method of Preparing Rabeprazole and Its Intermediate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12772401 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |